 Yupeng Wang
Yupeng Wang Ke Jiang2†
Ke Jiang2† Quan Zhang
Quan Zhang Songshu Meng
Songshu Meng Chan Ding
Chan Ding- 1Department of Dermatology of First Affiliated Hospital, Dalian Medical University, Dalian, China
- 2Cancer Center, Institute of Cancer Stem Cell, Dalian Medical University, Dalian, China
- 3College of Veterinary Medicine, Yangzhou University, Yangzhou, China
- 4Department of Avian Infectious Diseases, Shanghai Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Shanghai, China
Autophagy is a homoeostatic process by which cytoplasmic material is targeted for degradation by the cell. Viruses have learned to manipulate the autophagic pathway to ensure their own replication and survival. Although much progress has been achieved in dissecting the interplay between viruses and cellular autophagic machinery, it is not well understood how the cellular autophagic pathway is utilized by viruses and manipulated to their own advantage. In this review, we briefly introduce autophagy, viral xenophagy and the interaction among autophagy, virus and immune response, then focus on the interplay between NS-RNA viruses and autophagy during virus infection. We have selected some exemplary NS-RNA viruses and will describe how these NS-RNA viruses regulate autophagy and the role of autophagy in NS-RNA viral replication and in immune responses to virus infection. We also review recent advances in understanding how NS-RNA viral proteins perturb autophagy and how autophagy-related proteins contribute to NS-RNA virus replication, pathogenesis and antiviral immunity.
Introduction to Autophagy
Macroautophagy (hereafter called autophagy) is an evolutionarily conserved process that includes the immersion and transport of cytosolic contents to the lysosome for degradation (Mizushima et al., 2002). According to the molecular explanations of autophagy and pertinent processes proposed recently by Galluzzi et al. (2017), there are two primary attributes that distinguish genuine, functional autophagic reactions, regardless of kind: (i) they include cytoplasmic material; and (ii) they peak with (and rigidly depend on) lysosomal degradation. The execution of autophagy involves more than 30 essential autophagy-related (Atg) genes (Mizushima and Klionsky, 2007). These Atg gene products participate in several distinct stages of autophagosome biogenesis: initiation with Atg1 (in yeast) or its equivalent Ulk1 (in mammals) complex, vesicle nucleation with the Atg6 (Beclin-1 in mammals)-class III phosphatidylinositol 3-kinase (PI3K) (hVps34) complex, vesicle elongation with the microtubule-associated protein light chain 3 (LC3) lipidation and vesicle fusion with SNX18 complex (Yin et al., 2016; Anding and Baehrecke, 2017; Davis et al., 2017; Kimmelman and White, 2017). Given that autophagy is a dynamic process, the rate at which lysosomes degrade autophagy substrates is a good indicator of such a global efficiency in autophagic responses, which is commonly known as “autophagic flux” (Loos et al., 2014). Galluzzi et al. (2017) suggested that autophagic flux refers to the rate at which the molecular machinery for autophagy identifies, segregates, and disposes of its substrates (through lysosomal degradation). Bafilomycin A1 (BafA1) is widely used to assess autophagy flux for its ability to prevent the fusion of autophagosomes and lysosomes. Although autophagy has robust cytoprotective functions in the majority of pathophysiological and experimental settings (Menzies et al., 2015), in some cases, excessive or uncontrolled levels of autophagy can trigger autophagy-dependent cell death, termed “autophagic cell death” (Liu and Levine, 2015).
Originally autophagy was identified as a response to starvation, and it was thought of as a non-selective digestion process; However, it is now evident that autophagy specifically degrade aggregated proteins and damaged cellular organelles via autophagy receptors that link cargo to growing autophagosomal membranes (Stolz et al., 2014). An autophagy receptor is defined by its ability to bridge cargo and autophagosomal membrane, leading to the engulfment of cargo by the autophagic membrane (Deng et al., 2017). The selective autophagy is generally mediated by autophagy receptors such as p62 /SQSTM1 for degradation of ubiquitylated protein aggregates (Pankiv et al., 2007; Ichimura et al., 2008), FAM134B for endoplasmic reticulum (ER) turnover (Khaminets et al., 2015; Chiramel et al., 2016), optineurin (OPTN) in xenophagy (Wild et al., 2011), NBR1 (neighbor of BRCA1) acting as an aggrephagy receptor (Kirkin et al., 2009). These autophagy receptors interacts with ATG8/LC3/GABARAP via the presence of a LC3 Interaction Region (LIR), also known as LC3 interaction motif (LIM) or Atg8 interaction motif (AIM), thus determining cargo recognition in selective autophagy (Birgisdottir et al., 2013; Wild et al., 2014). The autophagic destruction of invading pathogens, a process called xenophagy (Levine, 2005), involves autophagy receptors such as p62 and NDP52. Specifically, viral xenophagy (virophagy), as defined recently by Galluzzi et al. (2017) is anautophagic response targeting fully formed cytoplasmic virions or components. Mounting studies have demonstrated that viral infections may have complex interconnections with the autophagic process (Jackson, 2015; Lennemann and Coyne, 2015). Recent studies reported the three following main outcomes of these interactions: (i) autophagic machinery is utilized as a scaffold to promote virus replication; (ii) viruses disrupt or inhibit autophagic machinery to avoid restriction of their replication; and (iii) autophagy limit virus replication. Interestingly, replication of certain virus seems not to be affected either positively or negatively by autophagy, as observed in Drosophila C virus and human rhinovirus (Cherry et al., 2006; Brabec-Zaruba et al., 2007). Additionally, it is possible that certain virus may have different interactions with autophagy, dependent on the cell type. HIV exploits the autophagic machinery during early stages in replication, but in macrophages the viral nef protein blocks the conversion of autophagosomes to autolysosomes, thereby preventing the loss of virus to be proteolyticaly degraded (Kyei et al., 2009). Consequently, the modulation of autophagy by different viruses, virus strains or serotypes, may cause different effects on their host cells, thereby contributing to specific viral pathogenesis.
Modulation of Autophagy by NS-RNA Viruses
Negative-strand (NS)-RNA viruses encompass some of the most significant human and animal pathogens extant, such as Ebola virus (EBOV), influenza virus (IAV), Nipah virus, Newcastle disease virus (NDV) and rabies virus (RABV). Similar to other viral pathogens, NS-RNA viruses overcome the host cell’s defense to ensure their own survival and propagation. Not surprisingly, autophagy plays a critical role in NS-RNA virus replication and/or infection. By dissecting the known interactions between NS-RNA viruses and the cellular autophagy machinery (Table 1), several distinct features are displayed as below:
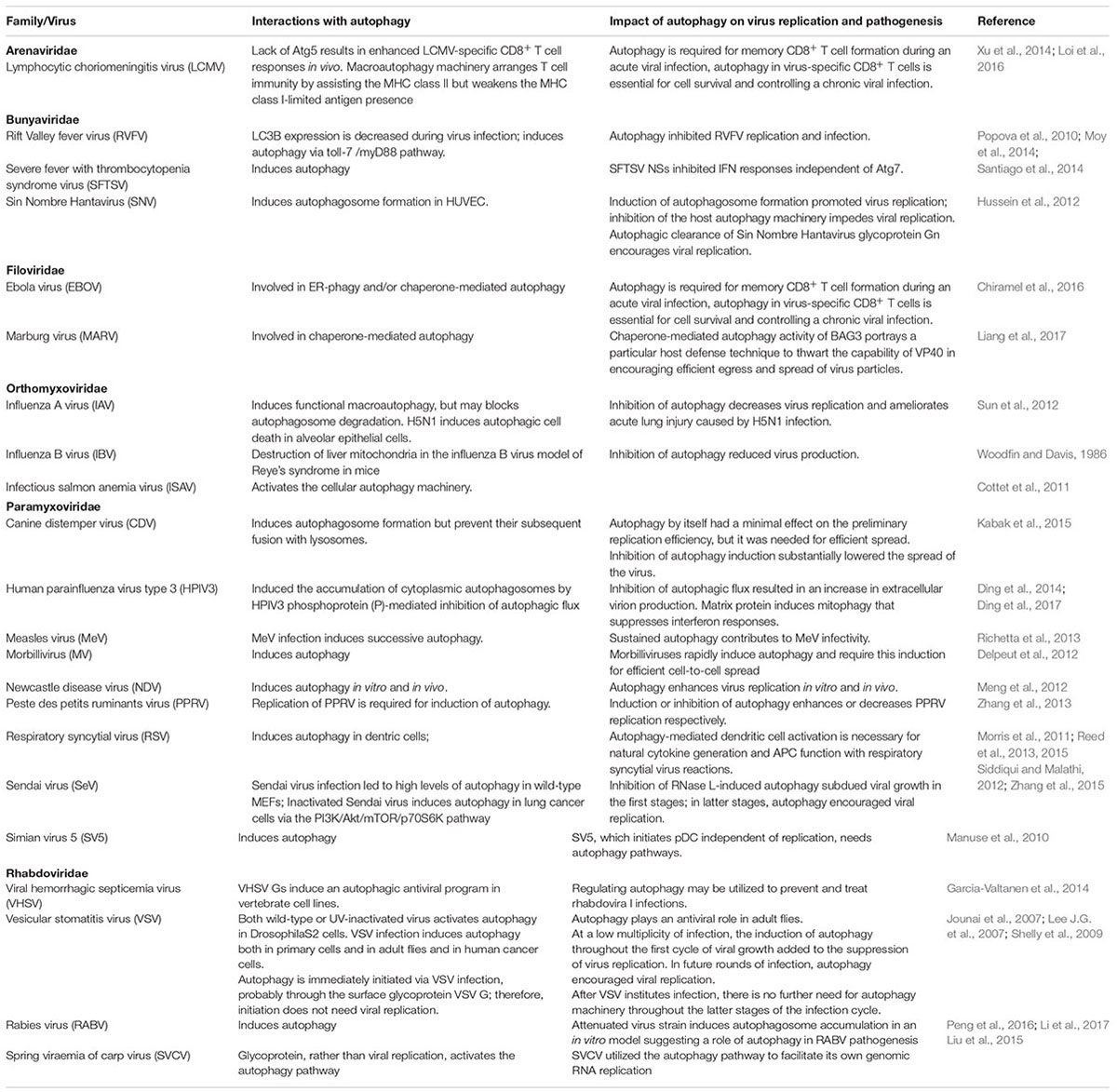
TABLE 1. Summary of known interactions between NS-RNA viruses and autophagy.
(i) Some NS-RNA viruses induce autophagy in the absence of viral replication. It is reported that Vesicular stomatitis virus (VSV), either wild-type or UV-inactivated, activated autophagy in Drosophila S2 cells (Wakana et al., 2013). Further study revealed that such activation of autophagy is most likely via the surface glycoprotein VSV G, and thus not requiring viral replication. In addition, inactivated Sendai virus (HVJ-E) induced autophagy in lung cancer cells via the PI3K/Akt/mTOR/p70S6K signaling pathway, and inducing autophagy enhanced HVJ-E-induced apoptosis (Zhang et al., 2015).
(ii) The autophagic induction pattern may differ between attenuated and virulent NS-RNA virus strains. In the case of measles virus (MeV), attenuated MeV strain induced a first transient wave of autophagy immediately upon infection via a CD46-Cyt-1/GOPC pathway (Joubert et al., 2009; Richetta et al., 2013). Interestingly, a second wave of autophagy was initiated after viral replication by the expression of the non-structural MeV protein C and was sustained overtime within infected cells (Richetta et al., 2013). Importantly, the sustained autophagy played a role in viral infectivity (Richetta et al., 2013). Mechanically, MeV protein C induced the next wave of autophagy throughout the interaction with immunity-associated GTPase family M (IRGM), a mediator of autophagy (Gregoire et al., 2011). Therefore, the reduced MeV strain prompts two waves of autophagy throughout infection in the specific molecular pathways. Of note, the harmful MeV strain was not able to prompt the initial CD46-dependent autophagic wave, but induced and exploited the later autophagic wave to replicate (Richetta et al., 2013). In addition, Xia et al. (2014) reported that MeV strain Edm utilized mitophagy to encourage viral replication by alleviating antiviral natural immune reactions. In the case of another NS-RNA virus, RABV, part of the family Rhabdoviridae, lessened SRV9 prompted more autophagosomes to gather rather than pathogenic CVS-11 in an in vitro model, suggesting a role of autophagy in RABV pathogenesis.
Notably, while the majority of the NS-RNA viruses induce autophagy for their own benefit during virus infection, some of them block autophagy especially at two checkpoints of the process, i.e., early during autophagosome formation and at the stage of autophagosome fusion with late endosomes or lysosomes. One example is the segmented RNA virus IAV which inhibits autophagosome maturation, leading to increased apoptotic cell death of infected cells (Gannage et al., 2009). The inhibition of autophagy flux by IAV is mediated by its matrix protein 2 (M2), which interacts with LC3 via its LIR motif. This interaction leads to the redistribution of LC3-coupled membranes to the cell membrane, which is esscential for IAV budding and transmission (Beale et al., 2014). Interestingly, canine distemper virus (CDV) and human parainfluenza virus type 3 (HPIV3) also block autophagosome fusion with lysosomes (Ding et al., 2014), which may promote different aspects of the viral replication cycle. Further studies revealed that HPIV3 blocks the SNAP29-STX17-mediated autophagosome-lysosome fusion via the HPIV3-P protein, which leads to improved viral release (Ding et al., 2014; Faure, 2014). However, the detailed mechanism needs to be further investigated.
Given that NS-RNA viruses target autophagy during infection, the underlying mechanism(s) by which NS-RNA viruses perturb autophagy remains elusive. A large body of evidence indicates that viral proteins play a critical role in NS-RNA virus-induced autophagy. For paramyxoviruses, viral glycoprotein-mediated membrane fusion triggers autophagy (Delpeut et al., 2012). In addition, in the case of Spring viraemia of carp virus (SVCV), part of the family Rhabdoviridae, SVCV glycoprotein, instead of viral replication, stimulates the autophagy pathway (Liu et al., 2015). Of importance, SVCV used the autophagy pathway to ease its own genomic RNA replication (Liu et al., 2015). A number of viral proteins encoded by separate NS-RNA viruses interact with cellular autophagy-related proteins, thereby contributing to the autophagy induction during virus infection. This will be discussed in detail in the following section.
Role of Autophagy in NS-RNA Viral Replication
Given the known interplay between NS-RNA viruses and autophagy, how does autophagy impact on NS-RNA virus infection? Also, how do NS-RNA viruses benefit from autophagy? Accumulating evidence indicates that for most NS-RNA viruses, autophagy functions as a proviral mechanism in infected cells (Figure 1 the pro-viral and anti-viral functions of autophagy during negative-stranded RNA viral infection), although this conclusion is largely based on in vitro investigations. Therefore in vivo studies on the role of autophagy in NS-RNA virus infection and pathogenesis, especially how autophagy regulates innate and adaptive immune responses to these pathogens, are of great interest and importance, particularly for those most significant human and animal pathogens, such as IAV and NDV. In the following sections, we discuss recent progresses in understanding the role of autophagy in NS-RNA viral replication cycle, ranging from virus entry up to egress.
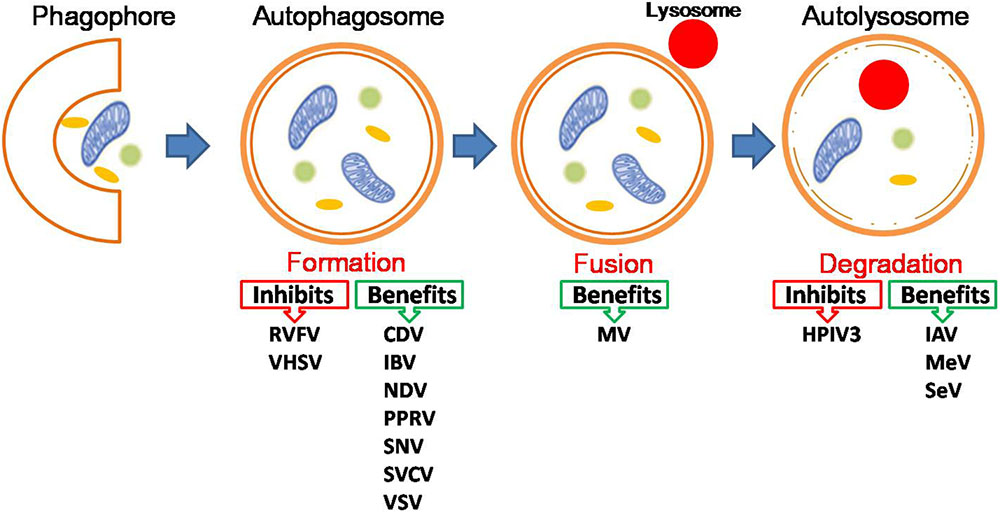
FIGURE 1. The pro-viral and anti-viral functions of autophagy during negative-stranded RNA viral infection. For detailed information, please see the main text. Abbreviations for viruses as below: Canine distemper virus (CDV), Human parainfluenza virus type 3 (HPIV3), Influenza A virus (IAV), Influenza B virus (IBV), Lymphocytic choriomeningitis virus (LCMV), Marburg virus (MARV), Measles virus (MeV), Morbillivirus (MV), Newcastle disease virus (NDV), Peste des petits ruminants virus (PPRV), Rift Valley fever virus (RVFV), Sendai virus (SeV), Sin Nombre Hantavirus (SNV), Spring viraemia of carp virus (SVCV), Vesicular stomatitis virus (VSV), Viral hemorrhagic septicemia virus (VHSV).
IAV a member of the Orthomyxoviridae, causes severe morbidity and mortality in animals and humans. The complex interplay between IAV and host autophagy machinery has been extensively reviewed (Rossman and Lamb, 2009; Dumit and Dengjel, 2012; Yeganeh et al., 2013; Munz, 2014; Zhang et al., 2014; Tripathi et al., 2015). Generally, it is well recognized that IAV infection triggers autophagosome formation, but inhibits the fusion of autophagosomes with lysosomes. IAV proteins M2, hemagglutinin (HA) and non-structural protein 1 (NS1) are involved in initiating the formation of autophagosomes in infected cells (Gannage et al., 2009; Zhirnov and Klenk, 2013). A number of studies indicate that IAV strains such as H1N1, H3N2, H9N2 infection induce autophagy in HEK293 cells (Ren et al., 2015), murine macrophages (Law et al., 2010), monkey and canine kidney cells (Khare et al., 2013), human alveolar epithelial cells (Zhirnov and Klenk, 2013) and mouse dendritic cells (DCs) (Zang et al., 2016), which is reportedly involved in viral replication (Zhou et al., 2009; Lupfer et al., 2013). However, it should be noted that the role of autophagy in IAV replication is still debated in some settings. Law et al. (2014) reported that autophagy was not involved in H9N2/G1 virus replication in primary human blood macrophages, although autophagic responses played a role in IAV-induced CXCL10 and interferon-α expression in human macrophages. Nevertheless, the critical role of autophagy in IAV replication has been documented by a few in vivo studies (Gannage et al., 2009; Hahn et al., 2014). Using mouse models with recombination at the Atg5 locus in the distal respiratory epithelium (Hahn et al., 2014), Hahn et al. (2014) found that a 50% decrease in autophagy in the bronchoalveolar epithelium significantly attenuated influenza A/H3N2 viral replication, leading to improved lung structure and function and reduced morbidity and mortality following infection indicating that the reserve autophagic capacity in alveolar epithelia provides a replicative niche for IAV. However, these studies are not consistent with an early observation by Gannage’ et al. (2009) which showed that IAV replication progresses normally even in an autophagy-deficient cell line. Interestingly, another work in the mouse embryonic fibroblast (MEF) cell line has shown that IAV does not induce autophagy unless apoptosis is first inhibited (McLean et al., 2009), possibly explaining why Gannage’ et al. (2009) did not see any effect on viral replication when autophagy is inhibited in the MEF cell line. Supporting the research by Gannage’ et al. (2009) recently Sun et al. (2012) reported that the autophagy inhibition did not have a notable effect on the viral replication of IAV H5N1 in vitro or in vivo. Thus, the ultimate effect of autophagy on viral replication may be clarified only when examined in the context of the host immune system during an in vivo infection.
Interestingly, while it is generally believed that autophagic membranes might be utilized to provide the necessary means for viral envelope acquisition and/or to export virus particles from infected cells, it seems not in the case of IAV. Although IAV infection results in the accumulation of autophagic LC3-positive membranes (Gannage et al., 2009; Beale et al., 2014; Ren et al., 2015), which is delivered to the cell surface for IAV budding, LC3 is not incorporated into the budding virus particles.
Looking from the cellular autophagy side, it has been demonstrated that Atg genes have been implicated in viral entry, viral release, and cell death during IAV infection (Sun et al., 2012; Beale et al., 2014; Pirooz S. et al., 2014). Antonucci et al. (2015) reported that IAV protein synthesis was markedly reduced in mice pancreatic epithelial cells lacking the essential Atg7. Another study using Atg7-/- MEFs by Liu et al. (2016) also found that autophagy deficiency significantly reduced the levels of IAV proteins, mRNA and genomic RNAs (vRNAs) without affecting viral entry. These studies together with data presented by Gannage et al. (2009) indicated that autophagy is utilized by IAV for accumulation of viral elements during IAV replication without affecting progeny virus production. It seems that autophagy is used by IAV to avoid the host’s defenses. Thus, the ultimate effect of autophagy on viral replication may be clarified only when examined in the context of the host immune system during an in vivo infection.
NDV a member of the Paramyxoviridae, is an important avian pathogen. Our study first reported that NDV FMW strain (NDV/FMW) induced autophagy in cell culture model, which plays a role in NDV replication (Meng et al., 2012). Subsequently we observed the conversion of LC3-I to LC3-II in heart, liver, spleen, lung, and kidney of NDV-infected chickens, confirmed the induction of autophagy in vivo (Sun et al., 2014). Of importance, modulation of the induction of autophagy with wortmannin, chloroquine, or starvation impacts NDV generation and pathogenesis in lung and intestine tissues in chickens (Sun et al., 2014), indicating that autophagy assists the replication of NDV in chicken cells and tissues and affects NDV pathogenesis (Sun et al., 2014). These studies are additionally supported by a current study that revealed that autophagosome formation was a requirement for NDV replication in chick embryo fibroblasts (Kang et al., 2017). In addition, recent work from our laboratory has further demonstrated that NDV nucleocapsid protein (NP) or phosphoprotein (P) was enough to prompt autophagy in vitro, involving activation of the ER stress-related unfolded protein response (UPR) pathways (Cheng et al., 2016). It should be noted that although the above studies indicate that NDV-induced autophagy benefits NDV replication, the detailed mechanism remains elusive.
Given that some NDV strains such as FMW, which is used in our in vitro study, are onlytic, the role of autophagy in oncolytic NDV replication is investigated by several studies. We found that NDV/FMW triggered autophagy in paclitaxel-resistant A549 lung cancer cells via dampening the class I PI3K/Akt/mTOR/p70S6K pathway whereas it attenuated the autophagic process in cisplatin-resistant A549 cells through the activation of the negative regulatory pathway (Jiang et al., 2014). Surprisingly, autophagy modulation does not increase virus progeny in these drug resistant cells, which is in consistent to the observation from NDV-infected parent cells (Jiang et al., 2014). In addition, Wei group found that NDV La Sota strain induced autophagy and preserved autophagic flux in non-small cell lung cancer cells and mitophagy promoted NDV replication by blocking intrinsic apoptosis (Meng et al., 2014). Interestingly, our recent investigation showed that NDV/FMW promotes autophagy flux in lung cancer cell spheroids (Hu et al., 2015). The difference in the autophagy pattern induced by oncolytic NDV may be ascribed to the different virulence between these NDV strains.
VSV a member of the Rhabdoviridae, is a prototypic non-segmented negative-strand RNA virus. In the case of VSV, the role of autophagy in virus replication seems to be controversial. An early study by Jounai et al. (2007) showed that either Atg5- or Atg7-deficient MEFs, in which Atg5-Atg12 conjugation is impaired, were resistant to VSV replication, suggesting that autophagy favors VSV replication. Further analysis indicated that the Atg5-Atg12 conjugate immediately interacts with the retinoic acid-inducible gene I (RIG-I) and IFN-β promoter stimulator 1 (IPS-1); this molecular connection causes the inhibition of type I IFN production and allows VSV replication inside the infected cells. On the contrary, Shelly et al. (2009) observed that VSV infection prompts autophagy in the primary cells and in adult flies, and suppression of autophagy causes greater viral replication and pathogenesis in cells and animals, supporting a an essential anti-viral role of autophagy in Drosophila against VSV (Shelly et al., 2009). The underlying mechanism was investigated in a subsequent study by the same group, who reported that Toll-7 interacted with VSV at the plasma membrane and induced antiviral autophagy in a drosophila model (Nakamoto et al., 2012). Interestingly, early to the investigation by Shelly et al. (2009) and Lee H.K. et al. (2007) observed that pharmacological inhibitors of autophagy, 3-methyladenine (3-MA) and Wortmannin inhibited VSV recognition in plasmacytoid dendritic cells (pDCs) while they specifically inhibited autophagosome formation without affecting viral entry and infection (Lee H.K. et al., 2007). Considering these studies are performed in different animal models, they seemingly indicate that the role of autophagy in VSV replication may be spices dependent. Similar to NDV, VSV is known as an oncolytic virus. The role of autophagy in VSV infection and oncolysis was investigated by several labs. Schache et al. (2009) reported that oncolytic VSV induced autophagy in a variety of cancer cells, and autophagy seems not play a role in VSV virotherapy while the role of autophagy in virus replication has not been investigated in these settings. However, a recent study by Shulak et al. (2014) showed that either 3-MA treatment or genetic ablation of the autophagic marker Atg5 decreased VSV replication and oncolysis in human prostate cancer PC3 cell, consisting with the study by Jounai et al. (2007) in mouse model. Taking together, the precise mechanism underlying the conflicting role of autophagy in VSV infection remains to be clarified, although VSV G protein-mediated autophagy during virus infection has been investigated in a number of studies, which we will discuss below.
While autophagy is shown to promote virus replication for most NS-RNA viruses, for some NS-RNA viruses like CDVand MeV, autophagy may just favor virus spread rather than replication (Richetta et al., 2013; Kabak et al., 2015). CDV and MeV prompt autophagy at the initial infection stages. For attenuated/vaccinal MeV but not virulent MeV, the early wave of autophagy induction upon virus infection may contribute to virus entry (Richetta et al., 2013). However, the late wave of autophagy induced by the virulent but not the non-replicative UV-treated MeV plays a critical role in virus replication (Richetta et al., 2013; Guirimand et al., 2015). Moreover, a very late autophagy induction by the cell–cell fusion also contributes to MeV replication (Herschke et al., 2007). Interestingly, although peste des petits ruminants virus (PPRV), along with CDV and MeV, is classified in the genus Morbillivirus within the family Paramyxoviridae, PPRV uses the autophagy machinery to ease its replication in host cells (Zhang et al., 2013), indicating that the role of autophagy in virus replication and/or spread is virus dependent.
Notably, other patterns of selective autophagy such as ER-phagy also play roles in NS-RNA viral replication. FAM134B is the selective autophagy receptor for endoplasmic reticulum turnover (Khaminets et al., 2015; Mochida et al., 2015). Using FAM134B-/- MEFs, Chiramel et al. (2016) recently demonstrated that ER-phagy limits EBOV replication in mouse cells. Similarly, SQSTM1/p62-mediated mitophagy enhances MeV replication by mitigating DDX58/RIG-I-like receptor signaling (Xia et al., 2014). Altogether, these studies highlights the role of selective autophay in NS-RNA viral replication.
The role of autophagy in NS-RNA viral replication is generally mediated by autophagy-related proteins. Accumulating evidence reveals that autophagy-related proteins are targeted by NS-RNA viruses during infection and importantly these proteins play a role in NS-RNA virus infection and pathogenesis. Using yeast two-hybrid and bioinformatic analysis, Gregoire et al. (2012) determined the molecular interactions between 44 autophagy-associated proteins and 83 viral proteins belonging to five different RNA virus families including two families of NS-RNA viruses, Paramyxoviridae and Orthomyxoviridae (Gregoire et al., 2012). This interactome revealed that IRGM is the most targeted autophagy-associated protein (Gregoire et al., 2012). Downregulation of IRGM expression prevented autophagy induction by MeV, and impaired viral particle production. Interestingly, the expression of IRGM-interacting MeV-C protein was sufficient to induce autophagy through an IRGM dependent pathway, which could contribute to the facilitation of the syncytia formation. Together, the study by Gregoire et al. (2012) demonstrated that IRGM is a common target of RNA viruses that subvert the autophagy network. In addition, the human inhibitory complement receptor CD46, a type I glycoprotein expressed by all nucleated human cells, has been reported to be a direct inducer of autophagy and binds multiple pathogens, including MeV (Persson et al., 2010). It should be pointed out that the effect of certain autophagy-related proteins on NS-RNA viruses may be independent of autophagy. For instance, UV-radiation resistance-associated gene (UVRAG), an autophagic tumor suppressor, is required for the entry of the prototypic negative-strand RNA virus, including IAV and VSV, by a mechanism independent of IFN and autophagy (Pirooz S.D. et al., 2014).
In addition, autophagy-related proteins may directly interact with NS-RNA viral proteins to affect virus spread and/or replication. Among them, Beclin-1 is often targeted to induce autophagy by viral proteins such as IAV M2 protein. BCL2 associated athanogene 3 (BAG3), a regulator chaperone-mediated autophagy, sequestered EBOV and Marburg (MARV) viruses VP40 away from the site of budding at the plasma membrane, thereby counteracting the function of VP40 in promoting efficient egress and spread of virus particles (Liang et al., 2017). In addition, diverse autophagy receptors contribute to NS-RNA virus replication and/or pathogenesis. Chiramel et al. (2016) found that FAM134B, the selective autophagy receptor for endoplasmic reticulum turnover, inhibits replication of Ebola virus strains in MEFs via targeting the production of viral proteins GP and VP40 and nucleocaspid lattices. Interestingly, in the case of MeV, the autophagy receptors NDP52 and T6BP, but not OPTN, impacted MeV replication, although independently, and possibly through physical interaction with MeV proteins including MeV-N, MeV-C or MeV-V (Petkova et al., 2017).
NS-RNA Virus Proteins Target Autophagy Machinery
Growing evidence indicates that both viral proteins and host cellular autophagy-related proteins play critical roles in the interplay between NS-RNA virus and autophagy, thereby contributing to virus replication and/or pathogenesis. In one hand, viral proteins such as IAV proteins including M2 have been shown to interact with autophagy-related proteins to affect the life of virus in infected cells. IAV proteins M2, HA, and NS1 take part in starting the formation of autophagosomes in infected cells (Gannage et al., 2009; Zhirnov and Klenk, 2013). Using yeast two-hybrid technique Gregoire et al. (2011) examined the interactions between 9 IAV proteins and 44 human autophagy-associated proteins and shown the associations of NP protein with Atg4C, BNIP3, and GOPC proteins, NS1with Atg5 and GOPC, NS2 with Atg5, Atg9A, IRGM and UVRAG, PB1-F2 with Atg5 and IRGM, PB2 with SQSTM1, and M2 with Beclin-1. However, given these interactions among IAV proteins and autophagy-related proteins, besides M2, whether other IAV proteins regulate autophagy during IAV infection is largely unknown. Recent study by Zhirnov and Klenk (2013) showed that NS1 expressed alone was unable to upregulate autophagy, whereas HA and M2 were. However, NS1 stimulated autophagy indirectly by up-regulating the synthesis of HA and M2, thus NS1 and HA along with M2 are involved in stimulation of autophagy in infected cells (Zhirnov and Klenk, 2013). Nevertheless, the precise roles of these IAV proteins in the interaction between IAV and autophagy need to be further confirmed in in vivo studies. Below we discussed how three important viral proteins from IAV, MeV and VSV play critical roles in virus replication and/or pathogenesis via the interaction with the cellular autophagic machinery.
Matrix 2 M2 functions as a proton-selective ion channel and regulates IAV assembly and budding. Gannage’ et al. (2009) first reported that the ability of IAV to evade autophagy depends on the M2 ion-channel protein, expression of M2 was enough to reproduce the phenotype of autophagosomes gathering and obstructing autophagosome maturation. The functional result of obstructing autophagosome development by M2 is a larger susceptibility of IAV-infected cells to apoptosis (Gannage et al., 2009). Further analysis demonstrated that the first 60 amino acids of M2 enable associated with Beclin-1 and were sufficient for inhibition of autophagy (Gannage et al., 2009). Interestingly, the role of proton channel activity of M2 in IAV-perturbed autophagy is debated, as Gannage’ et al. (2009) reported that proton channel activity of M2 was not involved in IAV-induced autophagy arrest whereas Ren recently showed that M2 proton channel activity was involved in blocking the fusion of autophagosomes with lysosomes (Gannage et al., 2009; Ren et al., 2015). In addition to the above-mentioned mechanism, another possible molecular mechanism underlying how M2 regulates host cellular autophagy machinery was revealed by Beale et al. (2014). They found that the cytoplasmic tail of IAV M2 interacts directly with the autophagy protein LC3 and promotes LC3 relocalization to the plasma membrane (Beale et al., 2014). Importantly, mutations in M2 that abolish LC3 binding reduced filamentous virion budding and stability in vitro (Beale et al., 2014), although how this process enhances virion stability remains to be explored. Therefore, IAV infection may subvert autophagy to boost transmission to new cells and/or hosts by increasing virion stability.
The well-studied interaction between M2 and LC3 highlights the importance of the role of LIR motif in viral proteins in virus-perturbed autophhagy. In deed, a very recent study revealed that a large number of potential LIR sequences contained within the viral proteins from over 16000 viral sequences and 2500 viral species (Jacomin et al., 2017). However, whether these LIRs are bona fide and functional sequences that are important for the viral life cycle remains to be investigated although some of them have been identified as functional player in viral life.
VSV surface glycoprotein G VSV-G is involved in receptor recognition at the host cell surface and triggers membrane fusion after endocytosis of the viron. Shelly et al. found that UV-inactivated VSV or VSV-G virus-like particles induced autophagy in DrosophilaS2 cells, indicating that VSV-G alone is adequate to induce autophagy without any additional virus components or viral genome replication (Shelly et al., 2009). But how VSV-G is recognized? The authors suggest that either a Drosophila TLR or a viral receptor may be the trigger (Shelly et al., 2009). This was confirmed in a subsequent study which indicated that VSV was recognized by Toll-7, which interacted with VSV virions at the plasma membrane, and this recognition was required for the induction of antiviral autophagy, suggesting that VSV-G may serve as the pathogen-associated molecular pattern (PAMP) to be recognized by Drosophila TLR (Shelly et al., 2009). However, it cannot rule out that VSV-G or VSV may interact with other cell surface receptors in mammalian cells to induce autophagy.
C protein MeV protein C is a non-structural protein not present within the virion and suppresses the host innate immune response by interfering with IFN signaling pathways. Throughout MeV infection, the MeV-C protein induced autophagy in infected cells via IRGM-dependent pathway, which could add to easing of syncytia formation (Gregoire et al., 2011). Importantly, MeV-C expression is a necessary requirement for the effective prompting of another MeV-induced autophagy wave (Richetta et al., 2013). Although MeV was lacking C protein expression, which does not prompt autophagy in infected mononucleated cells, autophagy was still noted in syncytia, suggesting that the expression of MeV-C protein is not critical to prompt autophagy syncytia. Thus, other proteins of MeV may be involved in this process. Indeed, it has recently been suggested that MeV could prompt autophagy via a fusogenic-dependent mechanism that necessitates the coexpression of MeV-F and MeV-H proteins (Delpeut et al., 2012).
Together, although a few viral proteins encoded by NS-RNA viruses have been revealed their roles in regulating autophagy in vitro, further investigation is needed to determine whether these proteins modulate autophagy in vivo infection models in the context of their complete genomes, and importantly, to what extent, viral protein-modulated autophagy contributes to virus replication and/or pathogenesis.
Role of Autophagy in Immune Reponses to NS-RNA Virus Infection and in Pathogenesis
Given that selective autophagy target intracellular pathogens for destruction, it is now regarded as a critical aspect of the innate immune response. In fact, both innate as well as adaptive immune responses to virus infections are modulated by autophagy (Paludan et al., 2005; Schmid and Munz, 2007; English et al., 2009; Kuballa et al., 2012; Richetta and Faure, 2013; Paul and Munz, 2016). Below by dissecting the role of autophagy in IAV pathogenesis, we describe the role of autophagy in immune responses to IAV.
The role of autophagy in IAV pathogenesis has been examined in vivo. Study by Sun et al. (2012) demonstrated that autophagic cell death was responsible for the acute lung injury and the high mortality rate (60%) induced by IAV H5N1 in a mouse model. Importantly, H5N1, but not seasonal H1N1, induced autophagic cell death in alveolar epithelial cells (Sun et al., 2012), suggesting that the interplay between IAV and host autophagy machinery may be virus strain and virus virulence- dependent, since the seasonal H1N1 is a low pathogenic strain. A recent in vivo study by Lu et al. (2016) demonstrated that mice deficient of an Atg gene Epg5 exhibited elevated baseline innate immune cellular and cytokine-based lung inflammation and were resistant to lethal IAV infection. Similar results were observed in myeloid cells deletion of autophagy genes including Atg14, FIP200, Atg5, and Atg7 (Lu et al., 2016). These studies suggest that autophagy can mediate the anti-IAV immunity, thereby contributing to IAV-induced pathogenesis. The underlying mechanism is investigated by several studies. Early studies found that autophagy could ease the effective antigen cross-priming of IAV-specific CD8+ T cells (Uhl et al., 2009), and participated in the generation of CXCL10 and IFN-α via macrophages upon H1N1 virus infection (Law et al., 2010). The latest study found that mice lacking Atg5 or Atg7 have defective effector responses to IAV infection (Puleston et al., 2014). Recently Schlie et al. (2015) showed that in reaction to influenza infection, Atg5-/- CD8+ T cells had a lowered ability to achieve the peak effector reaction and they were not able to sustain cell viability throughout the effector phase, indicating that effector CD8+ T cells need autophagy to subdue apoptosis and retain survival in reaction to a viral infection (Schlie et al., 2015).
In addition to macrophages and T cells, other immune cells such as DCs also contribute to the effect of autophagy in antiviral immunity upon IAV infection. Chen et al. (2014) recently found that mice with B cell-specific removal of Atg7 (B/Atg7-/- mice) demonstrated typical primary antibody reactions following immunization against influenza, but they did not produce protective secondary antibody reactions when challenged with IAV, causing high viral loads, broad lung damage, and raised the fatality rate (Chen et al., 2014), indicating that autophagy has a pivotal part in retaining the memory B cells that protect against influenza virus infection. In addition, Zang recently reported that H1N1 virus infection-induced autophagy in DCs has an intensive part in DC-regulated immune reactions (Zang et al., 2016). They found that autophagosomes delivered H1N1 viruses into lysosomes to initiate the TLR signaling pathway and that autophagic deficiency damaged the antigen-presenting capability of DCs and their ability to prompt Th cell differentiation and inhibit the MHC-I cross-presentation of DCs upon H1N1 infection (Zang et al., 2016). In addition, autophagy-deficient BMDCs were weakened in their capability to prompt H1N1-specific natural and adoptive immune reactions in vivo (Zang et al., 2016).
The induction of the cytokine storm has been believed to be a primary cause of death in IAV H1N1-infected patients (Law et al., 2010). Of note, autophagy has been revealed to be critical for the generation of cytokines from innate immune cells. Using H5N1 pseudotyped viral particles (H5N1pps), Pan H.Y. et al., 2014 showed that obstructing autophagy with 3-MA (an autophagy inhibitor) or siRNA knockdown of autophagy-related genes (Beclin1 and Atg5) greatly lowered H5N1pps-induced proinflammatory cytokines and chemokines, including IL-1β, TNF-α, IL-6, CCL2, and CCL5, both in vitro and in vivo (Pan H. et al., 2014), suggesting the crucial part of autophagy in H5N1pps-triggered inflammatory responses. In line with the above observations, Lu et al. (2016) reported that Epg5 deficiency in mice caused an increase in IL-1β and IL-13 protein in lung macrophages, conferring influenza resistance. Interestingly, IAV could be sensed by NOD2, a member of the NLR family. The NOD2-RIPK2 axis could phosphorylates ULK1, leading to enhanced mitophagy which prevents excessive inflammasome activation (Lupfer et al., 2013). Therefore, these studies suggest that autophagy regulates inflammasome activity, thereby providing new possible targets for immunotherapy to combat IAV infection.
In addition to playing a role in IAV immunity, autophagy-related proteins, such as Atg5 and Atg7, also play a role in the generation of both innate and adaptive immune responses to other NS-RNA viruses (Subauste, 2009). Lee H.K. et al. (2007) demonstrated that in pDCs, Atg5 was required for the transport of VSV and Sendai virus (SV) genetic material starting at the cytoplasm and went into the endosomal compartment in which the ssRNA sensor TLR7 remains (Lee H.K. et al., 2007). This resulted in the production of IFN-αinnate response to the viruses (Lee H.K. et al., 2007), indicating that the generation of type I interferons via pDCs is an important part of the natural immunity to the virus that can be moderated by the autophagy protein Atg5. However, it was documented that autophagy proteins, including Atg5, can negatively moderate type I interferon generation, thereby promoting viral replication (Jounai et al., 2007). Atg5-deficient MEFs were resistant to VSV replication because of raised Type I IFN generation in reaction to VSV genetic material (Jounai et al., 2007). Mechanically, after infecting with VSV, the Atg5-Atg12 conjugate hinders type I interferon generation by attaching toRIG-I and IPS-1 via the caspase recruitment domains (Jounai et al., 2007). Subsequently another study indicated that Atg5 deletion in MEFs increased Type I IFN production, thus conferring resistance to VSV infection (Tal et al., 2009). As for Atg7, Chen et al. (2014) recently showed that when challenging with IAV in mice with B cell-specific deletion of Atg7 (B/Atg7-/- mice) with IAV, these mice failed to generate protective secondary antibody responses although normal primary antibody responses after immunization against IAV was induced, suggesting that autophagy is necessary for the survival of virus-specific memory B cells in mice and the maintenance of protective antibody responses required to combat IAV infection (Chen et al., 2014). Therefore, the role of autophagy-related proteins in the antiviral immunity needs to be further investigated, since increasing evidence indicates that autophagy proteins regulate cellular responses other than autophagy.
Of note, autophagy-related proteins also target the innate immunity components, such as RIG-1, to add to the replication of particular NS-RNA viruses (Lei et al., 2012; Rey-Jurado et al., 2015). Jounai et al. (2007) reported that the overexpression of a mutant of RIG-1 prompted the initiation of NF-κB and IFN-β, which was subdued via the Atg5-Atg12 complex. Interestingly, this suppression appeared to be reliant on the infection with VSV and was documented to prompt a conformational alteration in RIG-1 (Jounai et al., 2007), suggesting that Atgs activated by VSV could modulate RIG-1.
Given the role of autophagy-related proteins in antiviral adaptive immunity during NS-RNA virus infection, autophagy could be exploited by NS-RNA viruses to evade host immunity (Rey-Jurado et al., 2015). Using a Beclin-1+/- mouse model, Reed et al. (2013) revealed that the depletion of autophagy function resulted in lowered MHC-II expression following RSV infection and the inability to generate IFN-γ and IL-17, and as a result, hindering DC maturation and the start of an efficient antiviral adaptive immune response against this virus. In addition, the expression of granzyme B in CD8+ T cells has been demonstrated to be downregulated in the Beclin-1+/- mice (Reed et al., 2013).
Concluding Remarks
The increasing evidence on the role of autophagy in NS-RNA virus replication and pathogenesis suggests that modulation of autophagy may represent a novel therapeutic strategy against virus infection, such as IAV. However, several important issues remained to be explored:
(i) Most of the data regarding the interplay between NS-RNA viruses and host autophagy machinery are obtained in cell cultural model; the question remained whether such interactions are present or relevant during infections in vivo. (ii) How different NS-RNA viruses perturb autophagy to augment replication and pathogenesis? Reversely, how autophagy regulates the various aspects of NS-RNA virus replication and propagation? Obviously, our current understanding of the role of the autophagy machinery in the propagation and control of virus infections, the ability of viruses to co-opt the cellular autophagic pathway to establish virulence in vivo is in its infancy. For example, for NS-RNA viruses, the role of autophagy in the assembly of viral components and budding of viral particles remains largely unknown. (iii) It is not known whether the in vitro or animal trials of autophagy modulators can be translated into useful therapeutics against viral infections in humans. Particularly, such approaches should not only enhance augment direct antiviral activity against NS-RNA virus infection, but could also augment natural acquired immune responses and vaccination strategies. Taking these points into consideration, targeting autophagy may lead to the development of a new class of specific antiviral therapies for the treatment of NS-RNA viruses-related diseases.
Author Contributions
SM designed the study. YW, KJ, and SM wrote the manuscript and reviewed the manuscript. KJ and QZ edited the manuscript. CD participated in developing the hypothesis. All authors listed approved the version to be published and have made a substantial and intellectual contribution to the work.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81372471 to SM, 81502674 to KJ, and 31530074 to CD). National Key Research and Development Program (Grant No. 2017YFD050160504 to QZ), Young and Middle-aged Academic Leaders Plan of Yangzhou University to QZ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to all authors whose primary work could not be cited owing to space constraints or whose work was inadvertently overlooked.
References
Anding, A. L., and Baehrecke, E. H. (2017). Cleaning house: selective autophagy of organelles. Dev. Cell 41, 10–22. doi: 10.1016/j.devcel.2017.02.016
Antonucci, L., Fagman, J. B., Kim, J. Y., Todoric, J., Gukovsky, I., Mackey, M., et al. (2015). Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc. Natl. Acad. Sci. U.S.A. 112, E6166–E6174. doi: 10.1073/pnas.1519384112
Beale, R., Wise, H., Stuart, A., Ravenhill, B. J., Digard, P., and Randow, F. (2014). A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 15, 239–247. doi: 10.1016/j.chom.2014.01.006
Birgisdottir, A. B., Lamark, T., and Johansen, T. (2013). The LIR motif - crucial for selective autophagy. J. Cell Sci. 126(Pt 15), 3237–3247. doi: 10.1242/jcs.126128
Brabec-Zaruba, M., Berka, U., Blaas, D., and Fuchs, R. (2007). Induction of autophagy does not affect human rhinovirus type 2 production. J. Virol. 81, 10815–10817. doi: 10.1128/JVI.00143-07
Chen, M., Hong, M. J., Sun, H., Wang, L., Shi, X., Gilbert, B. E., et al. (2014). Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat. Med. 20, 503–510. doi: 10.1038/nm.3521
Cheng, J. H., Sun, Y. J., Zhang, F. Q., Zhang, X. R., Qiu, X. S., Yu, L. P., et al. (2016). Newcastle disease virus NP and P proteins induce autophagy via the endoplasmic reticulum stress-related unfolded protein response. Sci. Rep. 6:24721. doi: 10.1038/srep24721
Cherry, S., Kunte, A., Wang, H., Coyne, C., Rawson, R. B., and Perrimon, N. (2006). COPI activity coupled with fatty acid biosynthesis is required for viral replication. PLOS Pathog. 2:e102. doi: 10.1371/journal.ppat.0020102
Chiramel, A. I., Dougherty, J. D., Nair, V., Robertson, S. J., and Best, S. M. (2016). FAM134B, the selective autophagy receptor for endoplasmic reticulum turnover, inhibits replication of Ebola virus strains makona and mayinga. J. Infect. Dis. 214(Suppl. 3), S319–S325. doi: 10.1093/infdis/jiw270
Cottet, L., Rivas-Aravena, A., Cortez-San Martin, M., Sandino, A. M., and Spencer, E. (2011). Infectious salmon anemia virus–genetics and pathogenesis. Virus Res. 155, 10–19. doi: 10.1016/j.virusres.2010.10.021
Davis, S., Wang, J., and Ferro-Novick, S. (2017). Crosstalk between the secretory and autophagy pathways regulates autophagosome formation. Dev. Cell 41, 23–32. doi: 10.1016/j.devcel.2017.03.015
Delpeut, S., Rudd, P. A., Labonte, P., and von Messling, V. (2012). Membrane fusion-mediated autophagy induction enhances morbillivirus cell-to-cell spread. J. Virol. 86, 8527–8535. doi: 10.1128/JVI.00807-12
Deng, X., Pan, X., Cheng, C., Liu, B., Zhang, H., Zhang, Y., et al. (2017). Regulation of SREBP-2 intracellular trafficking improves impaired autophagic flux and alleviates endoplasmic reticulum stress in NAFLD. Biochim. Biophys. Acta 1862, 337–350. doi: 10.1016/j.bbalip.2016.12.007
Ding, B., Zhang, G., Yang, X., Zhang, S., Chen, L., Yan, Q., et al. (2014). Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 15, 564–577. doi: 10.1016/j.chom.2014.04.004
Ding, B., Zhang, L., Li, Z., Zhong, Y., Tang, Q., Qin, Y., et al. (2017). The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe 21, 538.e4–547.e4. doi: 10.1016/j.chom.2017.03.004
Dumit, V. I., and Dengjel, J. (2012). Autophagosomal protein dynamics and influenza virus infection. Front. Immunol. 3:43. doi: 10.3389/fimmu.2012.00043
English, L., Chemali, M., Duron, J., Rondeau, C., Laplante, A., Gingras, D., et al. (2009). Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 10, 480–487. doi: 10.1038/ni.1720
Faure, M. (2014). The p value of HPIV3-mediated autophagy inhibition. Cell Host Microbe 15, 519–521. doi: 10.1016/j.chom.2014.04.014
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo-San Pedro, J. M., Cecconi, F., et al. (2017). Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. doi: 10.15252/embj.201796697
Gannage, M., Dormann, D., Albrecht, R., Dengjel, J., Torossi, T., Ramer, P. C., et al. (2009). Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380. doi: 10.1016/j.chom.2009.09.005
Garcia-Valtanen, P., Ortega-Villaizan Mdel, M., Martinez-Lopez, A., Medina-Gali, R., Perez, L., Mackenzie, S., et al. (2014). Autophagy-inducing peptides from mammalian VSV and fish VHSV rhabdoviral G glycoproteins (G) as models for the development of new therapeutic molecules. Autophagy 10, 1666–1680. doi: 10.4161/auto.29557
Gregoire, I. P., Rabourdin-Combe, C., and Faure, M. (2012). Autophagy and RNA virus interactomes reveal IRGM as a common target. Autophagy 8, 1136–1137. doi: 10.4161/auto.20339
Gregoire, I. P., Richetta, C., Meyniel-Schicklin, L., Borel, S., Pradezynski, F., Diaz, O., et al. (2011). IRGM is a common target of RNA viruses that subvert the autophagy network. PLOS Pathog. 7:e1002422. doi: 10.1371/journal.ppat.1002422
Guirimand, T., Delmotte, S., and Navratil, V. (2015). VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 43, D583–D587. doi: 10.1093/nar/gku1121
Hahn, D. R., Na, C. L., and Weaver, T. E. (2014). Reserve autophagic capacity in alveolar epithelia provides a replicative niche for influenza A virus. Am. J. Respir. Cell Mol. Biol. 51, 400–412. doi: 10.1165/rcmb.2013-0437OC
Herschke, F., Plumet, S., Duhen, T., Azocar, O., Druelle, J., Laine, D., et al. (2007). Cell-cell fusion induced by measles virus amplifies the type I interferon response. J. Virol. 81, 12859–12871. doi: 10.1128/JVI.00078-07
Hu, L., Sun, S., Wang, T., Li, Y., Jiang, K., Lin, G., et al. (2015). Oncolytic newcastle disease virus triggers cell death of lung cancer spheroids and is enhanced by pharmacological inhibition of autophagy. Am. J. Cancer Res. 5, 3612–3623.
Hussein, I. T., Cheng, E., Ganaie, S. S., Werle, M. J., Sheema, S., Haque, A., et al. (2012). Autophagic clearance of Sin Nombre hantavirus glycoprotein Gn promotes virus replication in cells. J. Virol. 86, 7520–7529. doi: 10.1128/JVI.07204-11
Ichimura, Y., Kominami, E., Tanaka, K., and Komatsu, M. (2008). Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 4, 1063–1066.
Jackson, W. T. (2015). Viruses and the autophagy pathway. Virology 479–480, 450–456. doi: 10.1016/j.virol.2015.03.042
Jacomin, A. C., Samavedam, S., Charles, H., and Nezis, I. P. (2017). iLIR@viral: a web resource for LIR motif-containing proteins in viruses. Autophagy 13, 1782–1789. doi: 10.1080/15548627.2017.1356978
Jiang, K., Li, Y., Zhu, Q., Xu, J., Wang, Y., Deng, W., et al. (2014). Pharmacological modulation of autophagy enhances Newcastle disease virus-mediated oncolysis in drug-resistant lung cancer cells. BMC Cancer 14:551. doi: 10.1186/1471-2407-14-551
Joubert, P. E., Meiffren, G., Gregoire, I. P., Pontini, G., Richetta, C., Flacher, M., et al. (2009). Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 6, 354–366. doi: 10.1016/j.chom.2009.09.006
Jounai, N., Takeshita, F., Kobiyama, K., Sawano, A., Miyawaki, A., Xin, K. Q., et al. (2007). The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. U.S.A. 104, 14050–14055. doi: 10.1073/pnas.0704014104
Kabak, Y. B., Sozmen, M., Yarim, M., Guvenc, T., Karayigit, M. O., and Gulbahar, M. Y. (2015). Immunohistochemical detection of autophagy-related microtubule-associated protein 1 light chain 3 (LC3) in the cerebellums of dogs naturally infected with canine distemper virus. Biotech. Histochem. 90, 601–607. doi: 10.3109/10520295.2015.1064999
Kang, Y., Yuan, R., Xiang, B., Zhao, X., Gao, P., Dai, X., et al. (2017). Newcastle disease virus-induced autophagy mediates antiapoptotic signaling responses in vitro and in vivo. Oncotarget 8, 73981–73993. doi: 10.18632/oncotarget.18169
Khaminets, A., Heinrich, T., Mari, M., Grumati, P., Huebner, A. K., Akutsu, M., et al. (2015). Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358. doi: 10.1038/nature14498
Khare, D., Godbole, N. M., Pawar, S. D., Mohan, V., Pandey, G., Gupta, S., et al. (2013). Calcitriol [1, 25[OH]2 D3] pre- and post-treatment suppresses inflammatory response to influenza A (H1N1) infection in human lung A549 epithelial cells. Eur. J. Nutr. 52, 1405–1415. doi: 10.1007/s00394-012-0449-7
Kimmelman, A. C., and White, E. (2017). Autophagy and tumor metabolism. Cell Metab. 25, 1037–1043. doi: 10.1016/j.cmet.2017.04.004
Kirkin, V., Lamark, T., Johansen, T., and Dikic, I. (2009). NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 5, 732–733.
Kuballa, P., Nolte, W. M., Castoreno, A. B., and Xavier, R. J. (2012). Autophagy and the immune system. Annu. Rev. Immunol. 30, 611–646. doi: 10.1146/annurev-immunol-020711-074948
Kyei, G. B., Dinkins, C., Davis, A. S., Roberts, E., Singh, S. B., Dong, C., et al. (2009). Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 186, 255–268. doi: 10.1083/jcb.200903070
Law, A. H., Lee, D. C., Leon, T. Y., and Lau, A. S. (2014). Role for autophagy in cellular response to influenza virus infection. Hong Kong Med. J. 20(Suppl. 6), 20–24.
Law, A. H., Lee, D. C., Yuen, K. Y., Peiris, M., and Lau, A. S. (2010). Cellular response to influenza virus infection: a potential role for autophagy in CXCL10 and interferon-alpha induction. Cell. Mol. Immunol. 7, 263–270. doi: 10.1038/cmi.2010.25
Lee, H. K., Lund, J. M., Ramanathan, B., Mizushima, N., and Iwasaki, A. (2007). Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401. doi: 10.1126/science.1136880
Lee, J. G., Yoo, I. D., and Kim, W. G. (2007). Differential antiviral activity of benzastatin C and its dechlorinated derivative from Streptomyces nitrosporeus. Biol. Pharm. Bull. 30, 795–797.
Lei, Y., Wen, H., Yu, Y., Taxman, D. J., Zhang, L., Widman, D. G., et al. (2012). The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 36, 933–946. doi: 10.1016/j.immuni.2012.03.025
Lennemann, N. J., and Coyne, C. B. (2015). Catch me if you can: the link between autophagy and viruses. PLOS Pathog. 11:e1004685. doi: 10.1371/journal.ppat.1004685
Levine, B. (2005). Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120, 159–162. doi: 10.1016/j.cell.2005.01.005
Li, L., Jin, H., Wang, H., Cao, Z., Feng, N., Wang, J., et al. (2017). Autophagy is highly targeted among host comparative proteomes during infection with different virulent RABV strains. Oncotarget 8, 21336–21350. doi: 10.18632/oncotarget.15184
Liang, J., Sagum, C. A., Bedford, M. T., Sidhu, S. S., Sudol, M., Han, Z., et al. (2017). Chaperone-mediated autophagy protein BAG3 negatively regulates Ebola and Marburg VP40-mediated egress. PLOS Pathog. 13:e1006132. doi: 10.1371/journal.ppat.1006132
Liu, G., Zhong, M., Guo, C., Komatsu, M., Xu, J., Wang, Y., et al. (2016). Autophagy is involved in regulating influenza A virus RNA and protein synthesis associated with both modulation of Hsp90 induction and mTOR/p70S6K signaling pathway. Int. J. Biochem. Cell Biol. 72, 100–108. doi: 10.1016/j.biocel.2016.01.012
Liu, L., Zhu, B., Wu, S., Lin, L., Liu, G., Zhou, Y., et al. (2015). Spring viraemia of carp virus induces autophagy for necessary viral replication. Cell. Microbiol. 17, 595–605. doi: 10.1111/cmi.12387
Liu, Y., and Levine, B. (2015). Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 22, 367–376. doi: 10.1038/cdd.2014.143
Loi, M., Muller, A., Steinbach, K., Niven, J., Barreira da Silva, R., Paul, P., et al. (2016). Macroautophagy proteins control MHC class I levels on dendritic cells and shape anti-viral CD8+ T cell responses. Cell Rep. 15, 1076–1087. doi: 10.1016/j.celrep.2016.04.002
Loos, B., du Toit, A., and Hofmeyr, J. H. (2014). Defining and measuring autophagosome flux-concept and reality. Autophagy 10, 2087–2096. doi: 10.4161/15548627.2014.973338
Lu, Q., Yokoyama, C. C., Williams, J. W., Baldridge, M. T., Jin, X., DesRochers, B., et al. (2016). Homeostatic control of innate lung inflammation by Vici Syndrome Gene Epg5 and additional autophagy genes promotes influenza pathogenesis. Cell Host Microbe 19, 102–113. doi: 10.1016/j.chom.2015.12.011
Lupfer, C., Thomas, P. G., Anand, P. K., Vogel, P., Milasta, S., Martinez, J., et al. (2013). Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 14, 480–488. doi: 10.1038/ni.2563
Manuse, M. J., Briggs, C. M., and Parks, G. D. (2010). Replication-independent activation of human plasmacytoid dendritic cells by the paramyxovirus SV5 requires TLR7 and autophagy pathways. Virology 405, 383–389. doi: 10.1016/j.virol.2010.06.023
McLean, J. E., Datan, E., Matassov, D., and Zakeri, Z. F. (2009). Lack of Bax prevents influenza A virus-induced apoptosis and causes diminished viral replication. J. Virol. 83, 8233–8246. doi: 10.1128/JVI.02672-08
Meng, C., Zhou, Z., Jiang, K., Yu, S., Jia, L., Wu, Y., et al. (2012). Newcastle disease virus triggers autophagy in U251 glioma cells to enhance virus replication. Arch. Virol. 157, 1011–1018. doi: 10.1007/s00705-012-1270-6
Meng, G., Xia, M., Wang, D., Chen, A., Wang, Y., Wang, H., et al. (2014). Mitophagy promotes replication of oncolytic Newcastle disease virus by blocking intrinsic apoptosis in lung cancer cells. Oncotarget 5, 6365–6374. doi: 10.18632/oncotarget.2219
Menzies, F. M., Garcia-Arencibia, M., Imarisio, S., O’Sullivan, N. C., Ricketts, T., Kent, B. A., et al. (2015). Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. 22, 433–444. doi: 10.1038/cdd.2014.151
Mizushima, N., and Klionsky, D. J. (2007). Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27, 19–40. doi: 10.1146/annurev.nutr.27.061406.093749
Mizushima, N., Ohsumi, Y., and Yoshimori, T. (2002). Autophagosome formation in mammalian cells. Cell Struct. Funct. 27, 421–429.
Mochida, K., Oikawa, Y., Kimura, Y., Kirisako, H., Hirano, H., Ohsumi, Y., et al. (2015). Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 522, 359–362. doi: 10.1038/nature14506
Morris, S., Swanson, M. S., Lieberman, A., Reed, M., Yue, Z., Lindell, D. M., et al. (2011). Autophagy-mediated dendritic cell activation is essential for innate cytokine production and APC function with respiratory syncytial virus responses. J. Immunol. 187, 3953–3961. doi: 10.4049/jimmunol.1100524
Moy, R. H., Gold, B., Molleston, J. M., Schad, V., Yanger, K., Salzano, M. V., et al. (2014). Antiviral autophagy restrictsRift Valley fever virus infection and is conserved from flies to mammals. Immunity 40, 51–65. doi: 10.1016/j.immuni.2013.10.020
Munz, C. (2014). Influenza A virus lures autophagic protein LC3 to budding sites. Cell Host Microbe 15, 130–131. doi: 10.1016/j.chom.2014.01.014
Nakamoto, M., Moy, R. H., Xu, J., Bambina, S., Yasunaga, A., Shelly, S. S., et al. (2012). Virus recognition by Toll-7 activates antiviral autophagy in Drosophila. Immunity 36, 658–667. doi: 10.1016/j.immuni.2012.03.003
Paludan, C., Schmid, D., Landthaler, M., Vockerodt, M., Kube, D., Tuschl, T., et al. (2005). Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307, 593–596. doi: 10.1126/science.1104904
Pan, H., Zhang, Y., Luo, Z., Li, P., Liu, L., Wang, C., et al. (2014). Autophagy mediates avian influenza H5N1 pseudotyped particle-induced lung inflammation through NF-kappaB and p38 MAPK signaling pathways. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L183–L195. doi: 10.1152/ajplung.00147.2013
Pan, H. Y., Sun, H. M., Xue, L. J., Pan, M., Wang, Y. P., Kido, H., et al. (2014). Ectopic trypsin in the myocardium promotes dilated cardiomyopathy after influenza A virus infection. Am. J. Physiol. Heart Circ. Physiol. 307, H922–H932. doi: 10.1152/ajpheart.00076.2014
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., et al. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145. doi: 10.1074/jbc.M702824200
Paul, P., and Munz, C. (2016). Autophagy and mammalian viruses: roles in immune response, viral replication, and beyond. Adv. Virus Res. 95, 149–195. doi: 10.1016/bs.aivir.2016.02.002
Peng, J., Zhu, S., Hu, L., Ye, P., Wang, Y., Tian, Q., et al. (2016). Wild-type rabies virus induces autophagy in human and mouse neuroblastoma cell lines. Autophagy 12, 1704–1720. doi: 10.1080/15548627.2016.1196315
Persson, B. D., Schmitz, N. B., Santiago, C., Zocher, G., Larvie, M., Scheu, U., et al. (2010). Structure of the extracellular portion of CD46 provides insights into its interactions with complement proteins and pathogens. PLOS Pathog. 6:e1001122. doi: 10.1371/journal.ppat.1001122
Petkova, D. S., Verlhac, P., Rozieres, A., Baguet, J., Claviere, M., Kretz-Remy, C., et al. (2017). Distinct contributions of autophagy receptors in measles virus replication. Viruses 9:E123. doi: 10.3390/v9050123
Pirooz, S., He, S., O’Connell, D., Khalilzadeh, P., Yang, Y., and Liang, C. (2014). Viruses customize autophagy protein for efficient viral entry. Autophagy 10, 1355–1356. doi: 10.4161/auto.29075
Pirooz, S. D., He, S., Zhang, T., Zhang, X., Zhao, Z., Oh, S., et al. (2014). UVRAG is required for virus entry through combinatorial interaction with the class C-Vps complex and SNAREs. Proc. Natl. Acad. Sci. U.S.A. 111, 2716–2721. doi: 10.1073/pnas.1320629111
Popova, T. G., Turell, M. J., Espina, V., Kehn-Hall, K., Kidd, J., Narayanan, A., et al. (2010). Reverse-phase phosphoproteome analysis of signaling pathways induced by Rift valley fever virus in human small airway epithelial cells. PLOS ONE 5:e13805. doi: 10.1371/journal.pone.0013805
Puleston, D. J., Zhang, H., Powell, T. J., Lipina, E., Sims, S., Panse, I., et al. (2014). Autophagy is a critical regulator of memory CD8+ T cell formation. eLife 3:e03706. doi: 10.7554/eLife.03706
Reed, M., Morris, S. H., Jang, S., Mukherjee, S., Yue, Z., and Lukacs, N. W. (2013). Autophagy-inducing protein beclin-1 in dendritic cells regulates CD4 T cell responses and disease severity during respiratory syncytial virus infection. J. Immunol. 191, 2526–2537. doi: 10.4049/jimmunol.1300477
Reed, M., Morris, S. H., Owczarczyk, A. B., and Lukacs, N. W. (2015). Deficiency of autophagy protein Map1-LC3b mediates IL-17-dependent lung pathology during respiratory viral infection via ER stress-associated IL-1. Mucosal Immunol. 8, 1118–1130. doi: 10.1038/mi.2015.3
Ren, Y., Li, C., Feng, L., Pan, W., Li, L., Wang, Q., et al. (2015). Proton channel activity of influenza A virus matrix protein 2 contributes to autophagy arrest. J. Virol. 90, 591–598. doi: 10.1128/JVI.00576-15
Rey-Jurado, E., Riedel, C. A., Gonzalez, P. A., Bueno, S. M., and Kalergis, A. M. (2015). Contribution of autophagy to antiviral immunity. FEBS Lett. 589, 3461–3470. doi: 10.1016/j.febslet.2015.07.047
Richetta, C., and Faure, M. (2013). Autophagy in antiviral innate immunity. Cell. Microbiol. 15, 368–376. doi: 10.1111/cmi.12043
Richetta, C., Gregoire, I. P., Verlhac, P., Azocar, O., Baguet, J., Flacher, M., et al. (2013). Sustained autophagy contributes to measles virus infectivity. PLOS Pathog. 9:e1003599. doi: 10.1371/journal.ppat.1003599
Rossman, J. S., and Lamb, R. A. (2009). Autophagy, apoptosis, and the influenza virus M2 protein. Cell Host Microbe 6, 299–300. doi: 10.1016/j.chom.2009.09.009
Santiago, F. W., Covaleda, L. M., Sanchez-Aparicio, M. T., Silvas, J. A., Diaz-Vizarreta, A. C., Patel, J. R., et al. (2014). Hijacking of RIG-I signaling proteins into virus-induced cytoplasmic structures correlates with the inhibition of type I interferon responses. J. Virol. 88, 4572–4585. doi: 10.1128/JVI.03021-13
Schache, P., Gurlevik, E., Struver, N., Woller, N., Malek, N., Zender, L., et al. (2009). VSV virotherapy improves chemotherapy by triggering apoptosis due to proteasomal degradation of Mcl-1. Gene Ther. 16, 849–861. doi: 10.1038/gt.2009.39
Schlie, K., Westerback, A., DeVorkin, L., Hughson, L. R., Brandon, J. M., MacPherson, S., et al. (2015). Survival of effector CD8+ T cells during influenza infection is dependent on autophagy. J. Immunol. 194, 4277–4286. doi: 10.4049/jimmunol.1402571
Schmid, D., and Munz, C. (2007). Innate and adaptive immunity through autophagy. Immunity 27, 11–21. doi: 10.1016/j.immuni.2007.07.004
Shelly, S., Lukinova, N., Bambina, S., Berman, A., and Cherry, S. (2009). Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 30, 588–598. doi: 10.1016/j.immuni.2009.02.009
Shulak, L., Beljanski, V., Chiang, C., Dutta, S. M., Van Grevenynghe, J., Belgnaoui, S. M., et al. (2014). Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-kappaB-dependent autophagy. J. Virol. 88, 2927–2940. doi: 10.1128/JVI.03406-13
Siddiqui, M. A., and Malathi, K. (2012). RNase L induces autophagy via c-Jun N-terminal kinase and double-stranded RNA-dependent protein kinase signaling pathways. J. Biol. Chem. 287, 43651–43664. doi: 10.1074/jbc.M112.399964
Stolz, A., Ernst, A., and Dikic, I. (2014). Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 16, 495–501. doi: 10.1038/ncb2979
Subauste, C. S. (2009). Autophagy as an antimicrobial strategy. Expert Rev. Anti Infect. Ther. 7, 743–752. doi: 10.1586/eri.09.41
Sun, Y., Li, C., Shu, Y., Ju, X., Zou, Z., Wang, H., et al. (2012). Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Sci. Signal. 5:ra16. doi: 10.1126/scisignal.2001931
Sun, Y., Yu, S., Ding, N., Meng, C., Meng, S., Zhang, S., et al. (2014). Autophagy benefits the replication of Newcastle disease virus in chicken cells and tissues. J. Virol. 88, 525–537. doi: 10.1128/JVI.01849-13
Tal, M. C., Sasai, M., Lee, H. K., Yordy, B., Shadel, G. S., and Iwasaki, A. (2009). Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 2770–2775. doi: 10.1073/pnas.0807694106
Tripathi, S., White, M. R., and Hartshorn, K. L. (2015). The amazing innate immune response to influenza A virus infection. Innate Immun. 21, 73–98. doi: 10.1177/1753425913508992
Uhl, M., Kepp, O., Jusforgues-Saklani, H., Vicencio, J. M., Kroemer, G., and Albert, M. L. (2009). Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ. 16, 991–1005. doi: 10.1038/cdd.2009.8
Wakana, Y., Villeneuve, J., van Galen, J., Cruz-Garcia, D., Tagaya, M., and Malhotra, V. (2013). Kinesin-5/Eg5 is important for transport of CARTS from the trans-Golgi network to the cell surface. J. Cell Biol. 202, 241–250. doi: 10.1083/jcb.201303163
Wild, P., Farhan, H., McEwan, D. G., Wagner, S., Rogov, V. V., Brady, N. R., et al. (2011). Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233. doi: 10.1126/science.1205405
Wild, P., McEwan, D. G., and Dikic, I. (2014). The LC3 interactome at a glance. J. Cell Sci. 127(Pt 1), 3–9. doi: 10.1242/jcs.140426
Woodfin, B. M., and Davis, L. E. (1986). Liver autophagy in the influenza B virus model of Reye’s syndrome in mice. J. Cell. Biochem. 31, 271–275. doi: 10.1002/jcb.240310404
Xia, M., Gonzalez, P., Li, C., Meng, G., Jiang, A., Wang, H., et al. (2014). Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 88, 5152–5164. doi: 10.1128/JVI.03851-13
Xu, X., Araki, K., Li, S., Han, J. H., Ye, L., Tan, W. G., et al. (2014). Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat. Immunol. 15, 1152–1161. doi: 10.1038/ni.3025
Yeganeh, B., Rezaei Moghadam, A., Tran, A. T., Rahim, M. N., Ande, S. R., Hashemi, M., et al. (2013). Asthma and influenza virus infection:focusing on cell death and stress pathways in influenza virus replication. Iran. J. Allergy Asthma Immunol. 12, 1–17.
Yin, Z., Pascual, C., and Klionsky, D. J. (2016). Autophagy: machinery and regulation. Microb. Cell 3, 588–596. doi: 10.15698/mic2016.12.546
Zang, F., Chen, Y., Lin, Z., Cai, Z., Yu, L., Xu, F., et al. (2016). Autophagy is involved in regulating the immune response of dendritic cells to influenza A (H1N1) pdm09 infection. Immunology 148, 56–69. doi: 10.1111/imm.12587
Zhang, Q., Zhu, H., Xu, X., Li, L., Tan, H., and Cai, X. (2015). Inactivated Sendai virus induces apoptosis and autophagy via the PI3K/Akt/mTOR/p70Sr6K pathway in human non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 465, 64–70. doi: 10.1016/j.bbrc.2015.07.130
Zhang, R., Chi, X., Wang, S., Qi, B., Yu, X., and Chen, J. L. (2014). The regulation of autophagy by influenza A virus. Biomed Res. Int. 2014:498083. doi: 10.1155/2014/498083
Zhang, Y., Wu, S., Lv, J., Feng, C., Deng, J., Wang, C., et al. (2013). Peste des petits ruminants virus exploits cellular autophagy machinery for replication. Virology 437, 28–38. doi: 10.1016/j.virol.2012.12.011
Zhirnov, O. P., and Klenk, H. D. (2013). Influenza A virus proteins NS1 and hemagglutinin along with M2 are involved in stimulation of autophagy in infected cells. J. Virol. 87, 13107–13114. doi: 10.1128/JVI.02148-13
Keywords: negative-strand RNA virus, autophagy, virus replication, immune response, selective autophagy
Citation: Wang Y, Jiang K, Zhang Q, Meng S and Ding C (2018) Autophagy in Negative-Strand RNA Virus Infection. Front. Microbiol. 9:206. doi: 10.3389/fmicb.2018.00206
Received: 05 December 2017; Accepted: 30 January 2018;
Published: 13 February 2018.
Edited by:
Hideki Ebihara, Mayo Clinic, United StatesReviewed by:
Abhilash Inasu Chiramel, National Institute of Allergy and Infectious Diseases (NIH), United StatesAsuka Nanbo, Hokkaido University, Japan
Copyright © 2018 Wang, Jiang, Zhang, Meng and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chan Ding, c2hvdmVsZGVlbkBzaHZyaS5hYy5jbg== Songshu Meng, c3NtZW5nQGRtdS5lZHUuY24= Quan Zhang, enF1YW5AeXp1LmVkdS5jbg==
†These authors have contributed equally to this work.