 Byron C. Crump
Byron C. Crump John M. Wojahn
John M. Wojahn Fiona Tomas
Fiona Tomas Ryan S. Mueller
Ryan S. Mueller- 1College of Earth, Ocean, and Atmospheric Sciences, Oregon State University, Corvallis, OR, United States
- 2Department of Fisheries and Wildlife, Oregon State University, Corvallis, OR, United States
- 3Instituto Mediterráneo de Estudios Avanzados (IMEDEA), Universitat de les Illes Balears (UIB) – Consejo Superior de Investigaciones Científicas (CSIC), Esporles, Spain
- 4Department of Microbiology, Oregon State University, Corvallis, OR, United States
Terrestrial plants benefit from many well-understood mutualistic relationships with root- and leaf-associated microbiomes, but relatively little is known about these relationships for seagrass and other aquatic plants. We used 16S rRNA gene amplicon sequencing and metatranscriptomics to assess potential mutualisms between microorganisms and the seagrasses Zostera marina and Zostera japonica collected from mixed beds in Netarts Bay, OR, United States. The phylogenetic composition of leaf-, root-, and water column-associated bacterial communities were strikingly different, but these communities were not significantly different between plant species. Many taxa present on leaves were related to organisms capable of consuming the common plant metabolic waste product methanol, and of producing agarases, which can limit the growth of epiphytic algae. Taxa present on roots were related to organisms capable of oxidizing toxic sulfur compounds and of fixing nitrogen. Metatranscriptomic sequencing identified expression of genes involved in all of these microbial metabolic processes at levels greater than typical water column bacterioplankton, and also identified expression of genes involved in denitrification and in bacterial synthesis of the plant growth hormone indole-3-acetate. These results provide the first evidence using metatranscriptomics that seagrass microbiomes carry out a broad range of functions that may benefit their hosts, and imply that microbe–plant mutualisms support the health and growth of aquatic plants.
Introduction
Bacteria and Archaea associated with plant leaves (phyllosphere microbiome) and roots (rhizosphere microbiome) can have positive impacts on the health of terrestrial plants (Kent and Triplett, 2002; Vorholt, 2012; Turner et al., 2013), but relatively little is known about how microbiomes impact the health of seagrass and other aquatic plants (Turner et al., 2013; Fahimipour et al., 2016). In terrestrial plants, these mutualistic functions include outcompeting pathogenic soil microbes, modulating plant immunity, fixing nitrogen for use by plants (Gruber and Galloway, 2008), and neutralizing harmful products (e.g., methanol and ethanol) exuded from leaves and roots (Abanda-Nkpwatt et al., 2006; Turner et al., 2013).
Terrestrial plant rhizosphere and phyllosphere microbiomes are generally composed of different organisms with different relationships to their host plant. Rhizosphere microbiomes are mainly derived from soil microbiota (Kent and Triplett, 2002) and influenced by chemicals exuded by the plant roots (Haichar et al., 2008). In contrast, terrestrial phyllosphere microbiome source communities are unclear (Vorholt, 2012), but their compositions are strongly influenced by abiotic environmental factors, such as precipitation and light exposure (Turner et al., 2013). Similar contrasts between rhizosphere and phyllosphere microbiomes likely exist for aquatic plants, but relationships with their hosts may involve different processes or use different mechanisms because aquatic plant leaves are often submerged in water, and roots are anchored in water-saturated sediments.
Seagrasses are aquatic flowering plants that form the base of productive coastal ecosystems and provide habitat for many marine organisms such as fish, shellfish, crabs, and algae (Boström et al., 2006). Seagrass beds contribute to important ecosystem services, including nutrient cycling (McGlathery et al., 2007), storm-surge damping (Spalding et al., 2014), water clarification, and by acting as a global carbon sink (Orth et al., 2006). Also, decomposition of eelgrass detritus fuels a variety of food webs, both local and distal to the beds (Hemminga and Duarte, 2000).
Zostera marina (eelgrass) is the most widespread species, present throughout the coasts of the North Atlantic and North Pacific oceans (Green and Short, 2003). In estuaries on the United States Pacific coast, seagrass beds are typically dominated by Z. marina, but, since 1957, an invasive seagrass from Japan, Zostera japonica (Japanese eelgrass), has become established (Baldwin and Lovvorn, 1994). Z. japonica has smaller, thinner leaves and rhizomes than Z. marina (Harrison, 1982). Z. japonica usually colonizes mid-to-low intertidal mudflats and Z. marina is dominant in low to subtidal areas, but both species often co-exist in some locations (Shafer et al., 2014) forming mixed beds that are useful for comparing microbiomes between plants and investigating potential microbiome mutualisms across plant species.
Currently, there are two well-described seagrass–microbe mutualistic relationships. First, sulfur-oxidizing bacteria in seagrass root microbiomes use oxygen provided by the plant roots (Pedersen et al., 2004; Jensen et al., 2007; Hasler-Sheetal and Holmer, 2015) to oxidize sulfide as part of their metabolism (acting alone or in cooperation with root-associated clams; van der Heide et al., 2012). Sulfide is toxic to seagrasses (Goodman et al., 1995; Koch, 2001; Baden et al., 2003) and, thus, the growth of sulfur-oxidizing organism on eelgrass root surfaces benefits seagrass by detoxifying sediments (Jensen et al., 2007; Crump and Koch, 2008; Fahimipour et al., 2016). Second, seagrass microbiomes fix nitrogen, which likely supports plant growth (Capone et al., 1979; Lipschultz et al., 1979; Capone, 1983; Pereg et al., 1994; Lehnen et al., 2016). Several studies have identified diverse nitrogenase genes (Bagwell et al., 2002; Lehnen et al., 2016) and 16S rRNA genes of potential diazotrophs (i.e., nitrogen fixing organisms) associated with eelgrass leaves and roots (Jensen et al., 2007; Crump and Koch, 2008).
There are also several other potential seagrass–microbe symbioses that are relatively well-known in land plants. For instance, land–plant microbiomes degrade a broad range of plant exudates and waste products (Dourado et al., 2013) including methanol (Jourand et al., 2005; Abanda-Nkpwatt et al., 2006) and ethanol (Williams and Yavitt, 2010). Plant microbiomes also produce and degrade many different plant hormones and growth regulators (Glick, 1995; Dodd et al., 2010), including auxins (e.g., indole-3-acetic acid; Patten and Glick, 1996; Omer et al., 2004), cytokinins (e.g., zeatin; Akiyoshi et al., 1984; Ivanova et al., 2000; Mok and Mok, 2001), ethylene (Glick et al., 1998; Meldau et al., 2012), and nitric oxide (Stohr and Ullrich, 2002). Plant microbiomes also control pathogens and other organisms that may cause harm to their host plants (Andrews, 1992; Whipps, 2001).
We investigated potential eelgrass–microbiome mutualisms in two co-occurring species of eelgrass (Z. marina and Z. japonica) using 16S rRNA gene amplicons sequencing and metatranscriptomics. We hypothesized that Z. marina and Z. japonica would share similar leaf and root microbiomes despite morphological differences between the plants. We tested this hypothesis by comparing the phylogenetic composition of leaf and root microbial communities between the two species and with bacterioplankton collected from nearby, but independent, sampling sites. Additionally, we used metatranscriptomic data from these microbiomes to investigate a broad range of potential mutualistic processes between seagrasses and microbes, including sulfur oxidation and nitrogen fixation. Potential mutualisms were hypothesized based on the analysis of relative gene-expression patterns between leaves and roots of the two plant species with metatranscriptomic sequencing, and comparison of these results with typical coastal bacterioplankton metatranscriptomes from a prior study of the nearby Columbia River estuary and river plume.
Materials and Methods
Sample Collection
Samples were collected during low tides on July 28 and September 8, 2014 from two shallow pools (∼10 cm) within tidal mudflats harboring mixed beds of Z. marina and Z. japonica in Netarts Bay, OR, United States (Latitude: 45.394139° and Longitude: -123.939172°). This is a marine-dominated, strongly tidally flushed estuary in which approximately 75% of the water in the bay is replaced with each tidal cycle (Glanzman et al., 1971).
Whole plants were gently removed from sediment with gloved fingers and rinsed with filter-sterilized (Sterivex-GP 0.22 μm, EMD Millipore, Darmstadt, Germany) seawater. Roots and leaves were separated from rhizomes, placed in 50 ml sterile tubes, and rinsed 3 to 5 times with gentle inversion. Samples were fixed in RNAlater, placed on dry ice, and stored at -20°C until extraction. July samples were rinsed with filter-sterilized brackish water collected in a nearby tidal pool at the mouth of a small stream (Latitude: 45.394877° and Longitude: -123.938005°; Figure 1), and September samples were rinsed with filter-sterilized seawater collected from the mouth of the estuary (Latitude: 45.439343° and Longitude: -123.955221°; Figure 1). Microbes captured on the Sterivex filters used to prepare rinse water were preserved with 1 mL of filter-sterilized DNA extraction buffer [DEB; 0.1 M Tris-HCL (pH 8), 0.1 M Na-EDTA (pH 8), 1.5 M NaCl, 5% Cetyltrimethyl ammonium bromide], and stored at -80°C.
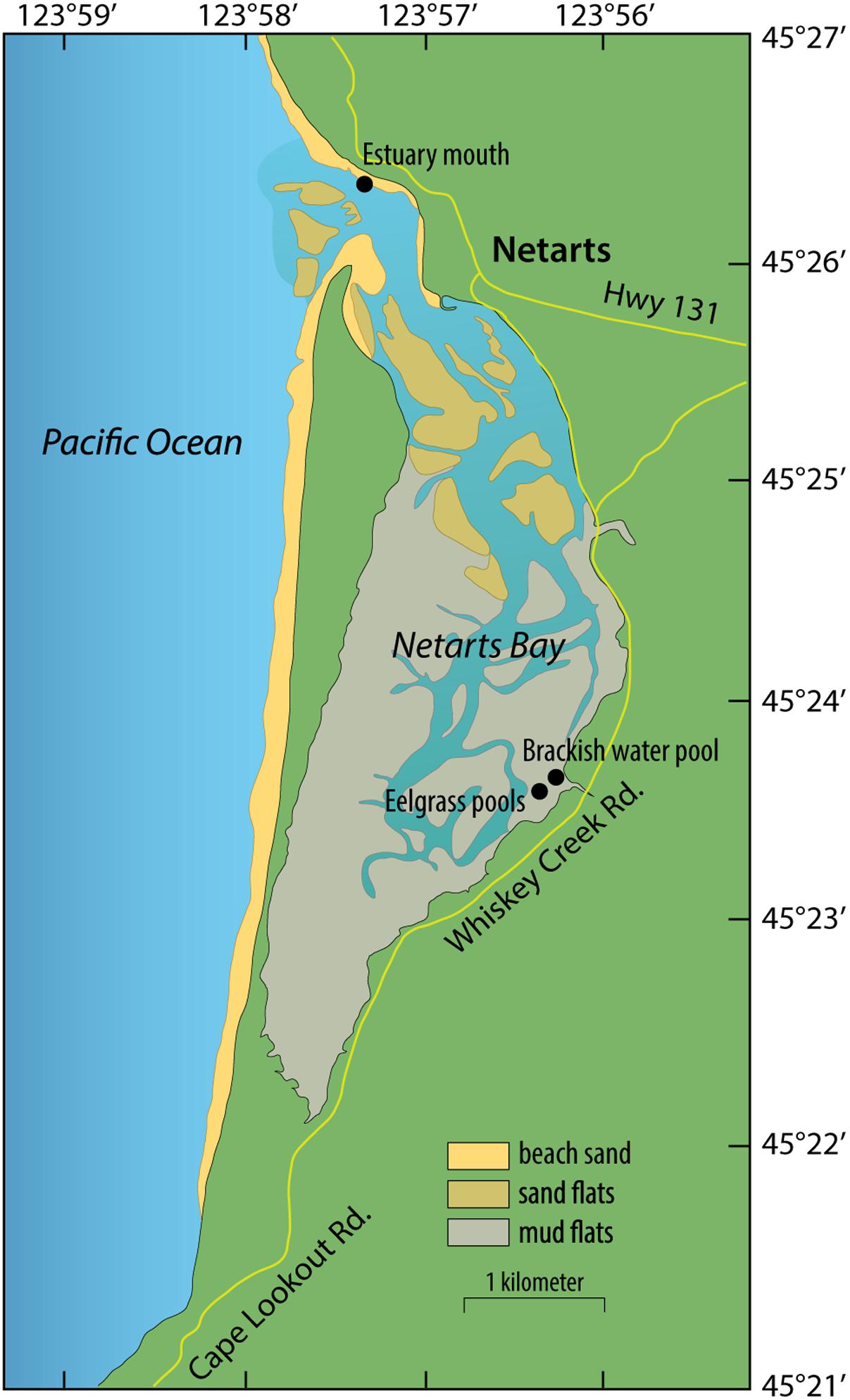
FIGURE 1. Map of sampling sites in Netarts Bay, OR, United States.
Microbial Community Composition
DNA was extracted from leaf and root tissues (triplicate samples of leaf and root tissues from each plant species on each sampling date) and from filtered seawater samples to measure the phylogenetic composition of plant-associated and planktonic bacterial communities. Thawed root and leaf samples were soaked in sterile ultrapure water at 4°C for 20 min to remove RNAlater, and cut into small pieces. DNA was extracted with the PowerBiofilm DNA Isolation Kits (MO BIO Laboratories, Inc.) following manufacturer’s instructions. DNA from water-column microbes collected on Sterivex filters was isolated using a phenol–chloroform extraction method (Crump et al., 2003). Bacterial community composition was determined with PCR amplicon sequencing of 16S rRNA genes following Kozich et al. (2013), with dual-barcoded versions of the PCR primers from Caporaso et al. (2012). The V4 hypervariable region of 16S rRNA genes were PCR amplified with 250 nM primers (final concentration) and HotMasterMix (5 Prime) under the following conditions (94°C for 3 min; 30 cycles of 94°C for 45 s, 50°C for 60 s, 72°C for 90 s; 72°C for 10 min). Three technical PCR replicates were performed for each sample, pooled, and quantified using Picogreen. Amplicons were then pooled at equimolar concentrations, cleaned using a MoBio Ultraclean PCR Clean-Up Kit, quantified using Picogreen, and sequenced at the Oregon State University Center for Genome Research and Biocomputing (CGRB) using the Illumina MiSeq platform and v2 chemistry (2 × 251 base long, paired-end reads).
Amplicon sequences were paired using make.contigs from the ‘mothur’ package (v.1.32.1) (Schloss et al., 2009), converted to QIIME format with split.groups from ‘mothur’ and add_qiime_labels.py from the QIIME software package (Caporaso et al., 2010). Sequences were quality filtered with an expected error rate of 0.5, dereplicated (derep_fulllength), and abundance sorted (sortbysize) using USEARCH (v.7.0.1001_i86linux64) (Edgar, 2013). Singleton sequences were removed and reads were clustered into operational taxonomic units (OTUs) at 97% similarity (cluster_otus). A de novo chimera check is inherent in the cluster_otus algorithm, but a reference-based chimera filtering was also performed (uchime_ref) with the Gold Database1. All reads (including singletons) were subsequently mapped back to representative OTU sequences using UPARSE (usearch_global), and an OTU table listing relative abundances between samples was created. Taxonomy of the representative sequences was assigned in QIIME (assign_taxonomy.py) using the RDP classifier trained to the SILVA database (v.111 database clustered to 97% OTUs). Alpha-diversity was calculated using Catchall within ‘mothur,’ and calculations of similarity matrices, MDS diagrams, ANOSIM, and SIMPER analyses were carried out using PRIMER v6 (PRIMER-E Ltd., Plymouth, United Kingdom).
Metatranscriptomics
Gene-expression patterns of microbial communities were assessed in four tissue samples (Z. marina leaf, Z. japonica leaf, Z. marina root, and Z. japonica root) using metatranscriptomic sequencing of mRNA. These sequences were compared to previously published metatranscriptomic sequences collected from planktonic microbes in the nearby Columbia River estuary and river plume (Fortunato and Crump, 2015). To extract RNA, thawed tissues were removed from RNAlater and cut into small pieces, and cells suspended in RNAlater were captured on Sterivex-GP 0.22-micron filters. The filter material was removed from the Sterivex-GP filter capsule, sliced into pieces, and combined with the tissue fragments. Total RNA was isolated with the MoBio PowerSoil Total RNA Isolation Kit (MO BIO Laboratories, Inc.) following manufacturer’s instructions, DNA was removed using the Ambion TURBO DNA-free kit, and rRNA was depleted with sequential use of two Ribo-Zero Gold kits (Bacterial rRNA, Plant rRNA; Illumina, Inc.) following Benes et al. (2011). Sequencing libraries made from rRNA-depleted samples were constructed with a Wafergen Apollo 324 robot using the PrepX RNA-Seq for Illumina library prep kit. Sequencing was carried out on an Illumina HiSeq 3000 sequencer using paired-end 150 base long reads at CGRB (total 8.4 GB). Metatranscriptome sequence data was deposited with links to BioProject accession number PRJNA419030 in the NCBI BioProject database under accession numbers SRR6310506-SRR63105092.
Metatranscriptomes from the water column of the Columbia River estuary and river plume (Fortunato and Crump, 2015) were downloaded from the European Nucleotide Archive (accessions ERS709858 to ERS709862), quality controlled as above, mapped to the published contigs downloaded from the Integrated Microbial Genomes (IMGs) database (GOLD study ID Gs0084963). These five metatranscriptomes were from 2.0 μm pre-filtered water collected at salinities 0, 5, 15, 25, and 33 PSU in August 2010. RNA was extracted with the RNeasy kit (Qiagen) following Poretsky et al. (2009), rRNA depleted with subtractive hybridization (Stewart et al., 2010), and sequenced as 100 base long paired-end reads on an Illumina HiSeq 1000 system.
Metatranscriptome sequences were co-assembled with Megahit (Li et al., 2015) and CDS sequences were identified and annotated through the Microbial Genome Annotation Pipeline of the IMG online system (Chen et al., 2016). Raw paired-end sequences from each sample were quality controlled (trimfq command in seqtk v.1.0-r72-dirty, default settings) and mapped to CDS sequences with Bowtie2. CDS sequences assigned to eukaryotes and viruses were excluded from analysis. CDS sequences assigned to Cyanobacteria and Fusobacteria included a large proportion of photosynthesis gene transcripts, and so were also excluded because it was unclear whether these were mis-assigned chloroplast transcripts. Abundances of transcripts that mapped to KEGG-annotated CDS sequences were normalized as transcripts per million (TPM) following Wagner (Wagner et al., 2012) to account for variations in sequence length and template length, and were analyzed using tools in MEGAN V.5 (Huson et al., 2011).
DNA sequences from this study are available from NCBI under accession numbers SRP125305 (Amplicon sequences) and SRP125305 (metatranscriptomes). Metatranscriptome assembled contigs and annotations are available from IMG/M ER3 under Taxon ID 3300008055.
Results
Seagrass-associated bacterial community composition was not significantly different between plant species for leaf (ANOSIM, P < 0.199) or root (ANOSIM, P < 0.091) microbiomes (Figure 2A). On the other hand, leaf, root, and water column communities were significantly different from one another (ANOSIM, P < 0.001) (Figure 2A). Leaf microbiomes (Figure 2B) were dominated by Bacteroidetes, Alphaproteobacteria, and Gammaproteobacteria, the last of which included a large proportion of Granulosicoccus representing on average 13% of phyllosphere communities. Root microbiomes were dominated by Bacteroidetes, Deltaproteobacteria, and Gammaproteobacteria, with smaller proportions of Spirochaetes, Firmicutes, and the Epsilonproteobacteria genus Arcobacter. Bacterioplankton communities in water samples were dominated by Bacteroidetes, Alphaproteobacteria, and Gammaproteobacteria, with elevated proportions of Actinobacteria and Betaproteobacteria in brackish water samples from July (Figure 2B).
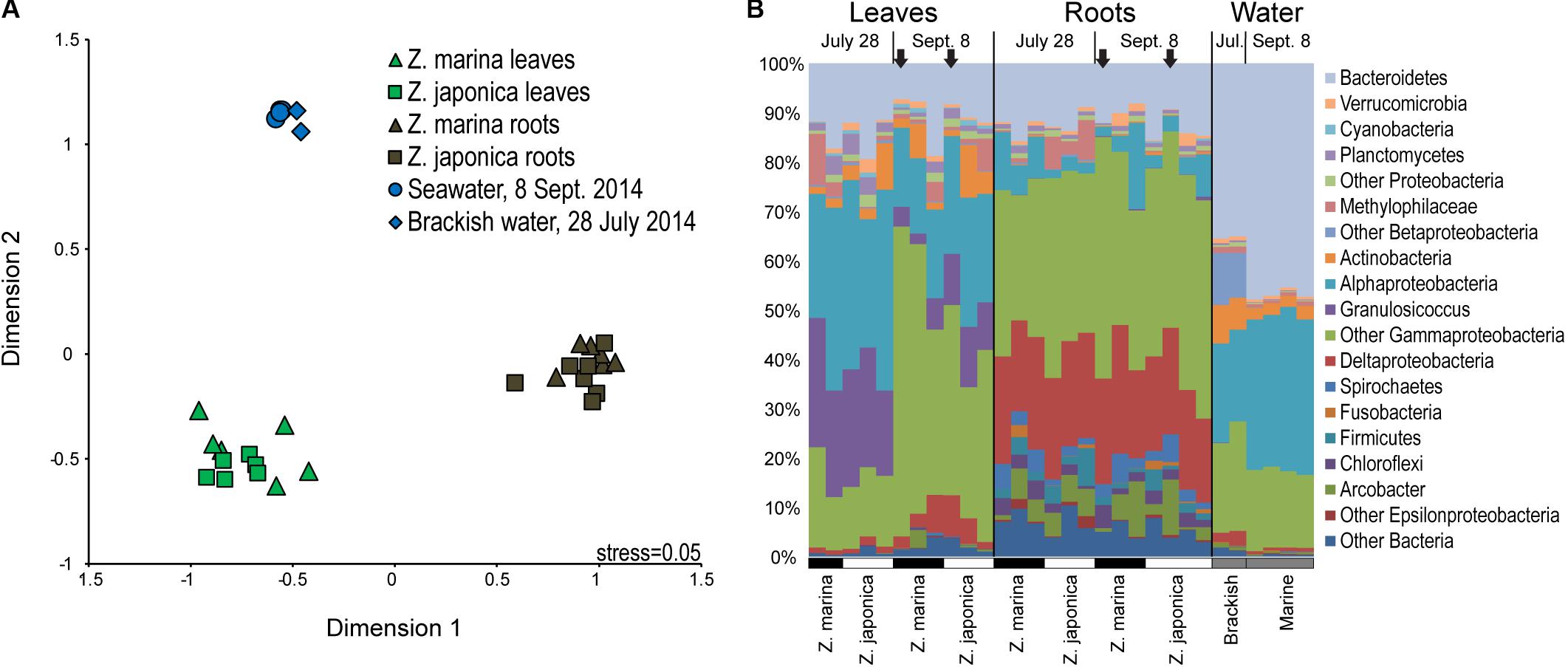
FIGURE 2. Multidimensional scaling diagram (A) showing beta-diversity of plant-associated and water column bacterial communities based on Bray–Curtis similarity calculated from the relative abundances of 16S rRNA gene amplicon sequences grouped into operational taxonomic units (OTUs) based on 97% DNA sequences similarity. (B) Taxonomic diversity of plant-associated and water-column bacterial communities based on 16S rRNA gene amplicon sequences grouped by major taxa with samples labeled by tissue type, date, and plant species. The black arrows indicate samples used for metatranscriptomics.
SIMPER analysis identified indicator taxa differentiating eelgrass leaf communities from root communities (Supplemental Table S1). Leaf indicators were from the Alphaproteobacteria family Rhodobacteraceae, the Betaproteobacteria family Methylophilaceae, the Bacteroidetes genus Polaribacter, and the Gammaproteobacteria genera Granulosicoccus, Simiduia, and Marinomonas. Also, one dominant indicator classified to class Gammaproteobacteria (OTU 69, 3.2% of leaf microbiome), was 93% similar to the methylotrophic organism Methylobacter marinus. Root indictors were classified to the Epsilonproteobacteria genus Arcobacter, the Betaproteobacteria genus Methylotenera, the Bacteroietes family Marinilabiaceae, the Deltaproteobacteria families Desulfobacteraceae and Desulfobulbaceae, and the Gammaproteobacteria genera Sedimenticola, Reinekea, and Vibrio. Root indicators also included three very abundant OTUs (>1% of root microbiome) that were only classified to class Gammaproteobacteria: OTU 6 (11.6% of root microbiome) was 100% similar to Spongiibacter marinus DSM 19753 (GI: 523385909); OTU 30 (3.0% of root microbiome), was 92% similar to the sulfur-oxidizer Thiomicrospira chilensis DSM 12352; and OTU 75 (1.6% of root microbiome) was 98% similar to sulfur oxidizing symbionts from Ridgeia piscesae (GI: 410699265) and Riftia pachyptila (GI: 28913259).
Co-assembly of 5.6 million metatranscriptome sequence reads (Table 1) produced 1.8 million contigs (N50 = 1,058) containing 2.4 million CDS (median length 315 bp). For the metatranscriptome sequences collected from each sample, 13–37% of sequences were assigned to CDS annotated to KEGG functions, of which 15–81% were assigned to Eukaryotes, and 9–80% (0.5–2.1 million reads) were assigned to Bacteria and Archaea other than Cyanobacteria and Fusobacteria (Table 2). Recovery of seagrass microbiome mRNA sequences was similar to other plant microbiome metatranscriptome studies (Cao et al., 2015; Marzano and Domier, 2016) in which a large percentage of mRNA sequences were derived from the host plant.

TABLE 1. Megahit co-assembly statistics for metatranscriptomes sequences.
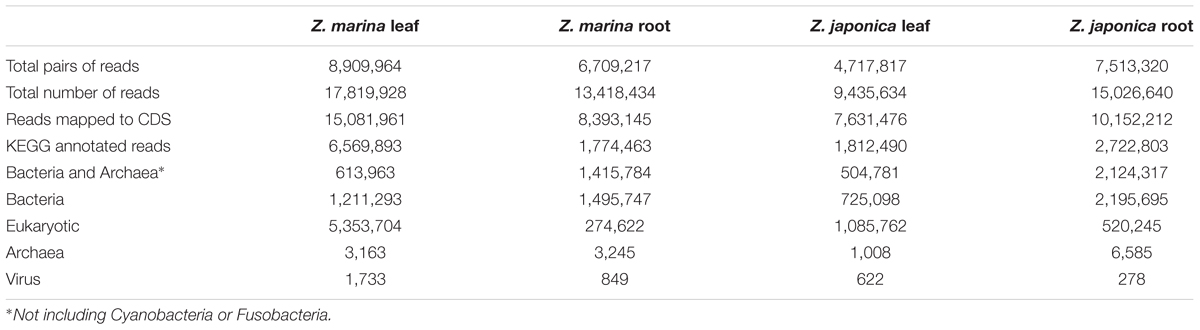
TABLE 2. Metatranscriptome mapping and annotation results from eelgrass microbiomes.
For the metatranscriptome sequences collected from the nearby Columbia River estuary (Fortunato and Crump, 2015), 1–11% mapped to KEGG-annotated CDS, of which 1–7% were assigned to Eukaryotes, and 90–99% (1.1–10.3 million reads) mapped to Bacteria and Archaea other than Cyanobacteria and Fusobacteria (Table 3).
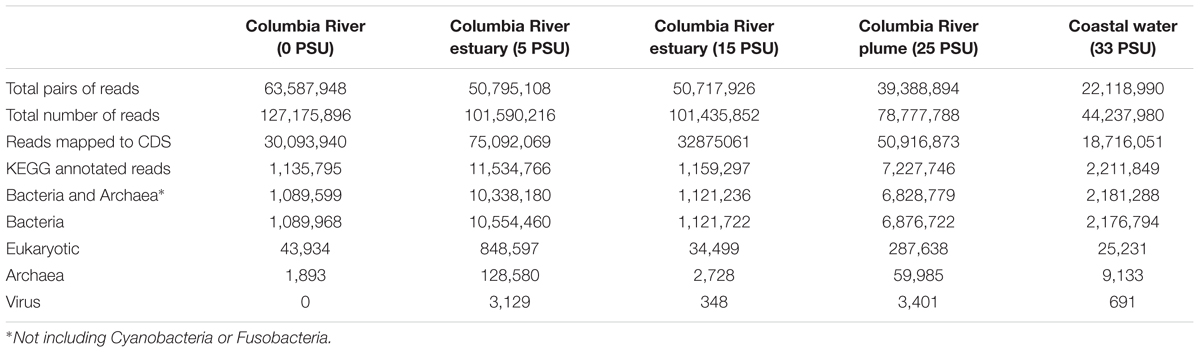
TABLE 3. Metatranscriptome mapping and annotation results from the water column of the Columbia River estuary.
The taxonomic composition of microbial communities based on metratranscriptome sequences was similar to that based on 16S gene amplicon sequences (Figure 3). Transcripts were detected for genes encoding functions potentially involved in plant–microbe mutualisms, including sulfur oxidation, nitrogen fixation, denitrification, methanol and ethanol consumption, beta-agarase production, and plant hormone synthesis (Figure 4). Sulfur oxidation genes on roots and leaves (soxABCXYZ) were expressed by Arcobacter sp., and genes involved in sulfur oxidation and reduction (aprAB, dsrAB) were expressed by both sulfur-oxidizing Gammaproteobacteria and sulfate-reducing Deltaproteobacteria (Figure 5). Nitrogen fixation genes (nifHDK) were primarily expressed by sulfur-oxidizing Gammaproteobacteria, Arcobacter sp., and a known nitrogen-fixing Bacteroidetes. Methanol consumption genes (mtaABC) were expressed primarily by Deltaproteobacteria on roots and by Euryarchaeota on leaves. Denitrification genes were expressed by Gammaproteobacteria, Arcobacter sp., and Thaumarchaeota, and beta-agarase was expressed by Gammaproteobacteria.
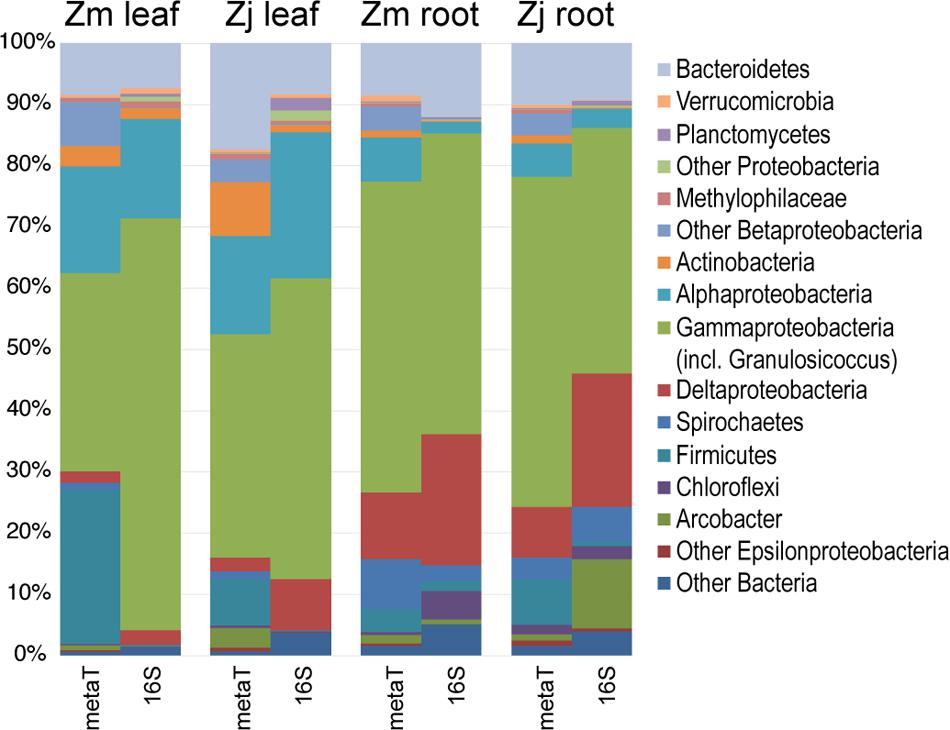
FIGURE 3. Comparison of the taxonomy of KEGG-annotated genes from metatranscriptomes with 16S rRNA gene amplicon sequences. 16S genes assigned to the genus Granulosicoccus were grouped with Gammaproteobacteria. Transcripts and 16S genes assigned to Cyanobacteria and Fusobacteria were omitted. (Zm: Z. marina; Zj: Z. japonica)
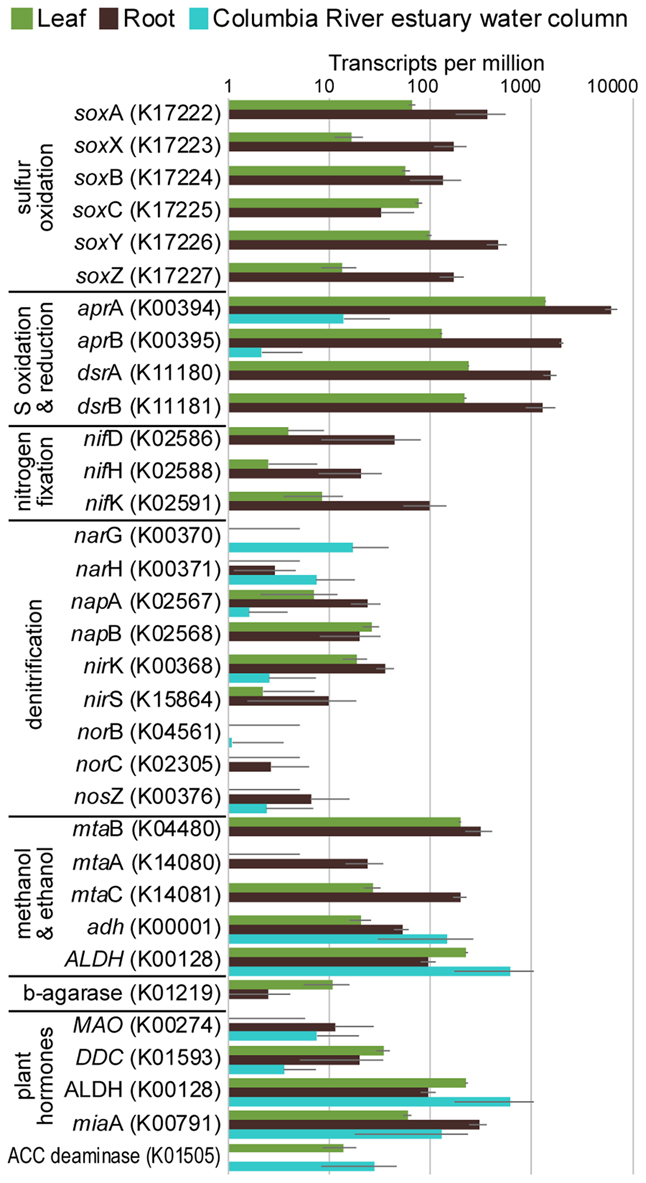
FIGURE 4. Average transcripts per million (TPM) for select genes from metatranscriptomes of eelgrass leaves and roots, and from the water column of the Columbia River estuary. Error bars indicate standard deviation.
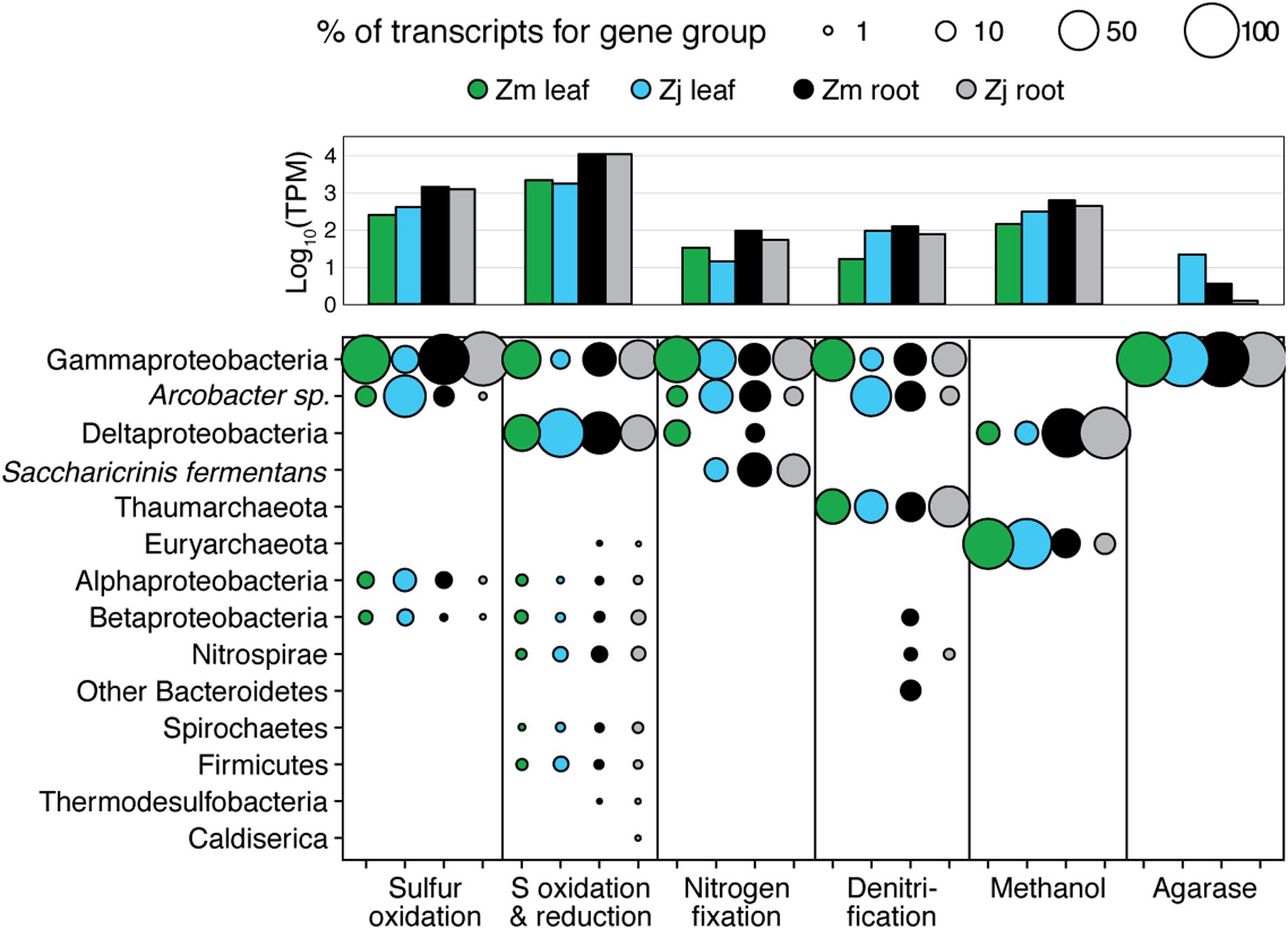
FIGURE 5. Taxonomy of gene transcripts for gene sets from Figure 3. Column graph shows the sums of relative expression (log10 of transcripts per million) of genes in each category for each sample type. Bubble size indicates percent of TPM for each sample within each gene set.
Discussion
The recent expansion of microbiome research demonstrates the close relationships between microbes and their hosts including terrestrial plants (Berendsen et al., 2012), insects (Husnik et al., 2013), and human beings (Huttenhower et al., 2012). Microbiomes of terrestrial plants have been extensively studied (e.g., Kloepper et al., 1980), but microbiomes of aquatic plants have only recently begun to be explored (Uku et al., 2007; Crump and Koch, 2008; Gordon-Bradley et al., 2014; Cucio et al., 2016; Mejia et al., 2016; Ettinger et al., 2017). Several of these studies show that the composition of these microbiomes differs from those in sediment and the water column, but no studies have used metatranscriptomics to describe how these microbiomes interact with their host plants.
In addition to the major physical and chemical controls of seagrass production, such as light and nutrient availability (Touchette and Burkholder, 2000; Ralph et al., 2007), associated microbiomes may also influence plant productivity and health by performing mutualistic functions. Our metatranscriptome results, which captured a snapshot of microbiome gene expression during the day at low tide, suggested that eelgrass microbiomes detoxify compounds that inhibit plant growth (e.g., sulfide, methanol, ethanol), fix nitrogen, and produce agarases that may cause disease and die-off in competitive algal epibionts. These metatranscriptomic results also suggest that eelgrass microbiomes carry out all of the steps involved in denitrification, and in the production of the plant-growth hormone indole-3-acetate (IAA).
The beta-diversity of Z. marina and Z. japonica microbiomes was consistent with several earlier seagrass studies showing many differences between leaf and root communities (Crump and Koch, 2008; Fahimipour et al., 2016; Ettinger et al., 2017), but few discernable differences in the microbiomes of different plant species collected at the same time and location (Cucio et al., 2016). Some earlier studies disagree with this second result and suggest that microbiomes vary with host plant species. However, these studies sampled plants with different structure or physical characteristics (He et al., 2012; Gordon-Bradley et al., 2014), or sampled plants from separate monospecific stands (Uku et al., 2007), or during different times of the year (Crump and Koch, 2008), all of which may influence microbiome composition. Cucio et al. (2016) sampled several co-occurring seagrass species, and found no differences in rhizosphere microbial communities in particles rinsed off roots despite the fact that each species was rooted in different types of sediment within the same habitat. These results suggest that seagrass microbiomes are not co-evolved with their hosts or controlled by sediment type, but instead are controlled by general seagrass metabolic processes (Cucio et al., 2016).
We found large differences between leaf microbiomes and water-column communities, which is inconsistent with a recent global study of eelgrass microbiomes (Fahimipour et al., 2016). In this global study, Z. marina leaf microbiomes were similar to local bacterioplankton, and a source-tracking analysis suggested leaf microbiomes were inoculated from the water column. Water samples for this global study were collected above submerged eelgrass beds, potentially capturing organisms dislodged from leaves. Also, leaf samples were not rinsed with sterilized water, which may have retained water column microbes in leaf samples. In contrast, our sampling approach aimed at limiting cross-contamination of leaf and water communities by washing leaf surfaces with filter-sterilized seawater, and by collecting water samples from nearby, but independent, sites. Differences in these two studies suggests that there is a dynamic exchange between eelgrass leaf microbiomes and the water column when plants are submerged, and that leaf microbiomes may be significant sources of organisms to local bacterioplankton.
The dominant taxa associated with leaves and roots generally coincided with earlier studies of seagrass microbiomes, but with several notable exceptions. For leaf microbiomes, Ettinger et al. (2017) found higher proportions of typically anaerobic Clostridiales in Z. marina leaf microbiomes from Bodega Harbor in California (included in the Firmicutes group in Figure 2), and Mejia et al. (2016) and Weidner et al. (2000) found lower proportions of Bacteroidetes and Gammaproteobacteria associated with the tropical seagrass Halophila stipulacea in the highly saline Gulf of Elat (Aqaba). For root microbiomes, Ettinger et al. (2017) found higher proportions of Epsilonproteobacteria (average ∼18%), but Jensen et al. (2007) found these bacteria in proportions similar to our study (1–12%). Differences among these studies may be driven by differences in regional environmental conditions, but could also be caused by differences in sampling techniques.
Seagrass microbiome studies have used DNA extracted from un-rinsed tissues (Fahimipour et al., 2016; Ettinger et al., 2017), from tissues rinsed with sterile water (this study; Jensen et al., 2007; Crump and Koch, 2008; Jiang et al., 2015), from sediment washed off of tissues (Cucio et al., 2016), and from material scraped or sonicated off the surfaces of un-rinsed and rinsed tissues (Weidner et al., 1996, 2000; Mejia et al., 2016; Bengtsson et al., 2017). These methodological differences limit our ability to compare results among studies, and favor the unified approach of Fahimipour et al. (2016). We extracted DNA from rinsed tissues in order to target the plant microbiomes that were firmly attached to tissues, and we found that the taxonomy and gene expression of these organisms provided information about known and novel interactions between seagrasses and their microbiomes.
Sulfide Detoxification
Sulfide is highly toxic to eelgrass photosynthetic pathways (Goodman et al., 1995), and high sediment sulfide concentrations can lead to a decrease in the ATP available to plant cells (Koch, 2001). Oxidation of toxic sulfide by sulfur-oxidizing bacteria within the plant rhizosphere is a well-known mutualism in seagrasses, which transport oxygen to their roots via aerenchyma to limit tissue anaerobiosis (Hasler-Sheetal and Holmer, 2015), and, in turn, provide oxygen for use as the terminal electron acceptor by sulfur-oxidizing bacteria (van der Heide et al., 2012). We identified several sulfur-oxidizing taxa on roots (Flood et al., 2015; Han and Perner, 2015; Hansen and Perner, 2015), including members of the genera Sedimenticola, Arcobacter, Thiomicrospira, and Sulfuromonas. These taxa averaged 11% of root microbiomes (primarily the root indicator taxa Sedimenticola and Arcobacter) and less than 2% of leaf and water column microbiomes. Similarly, typical sulfate-reducing taxa belonging to the families Desulfobacteraceae, Desulfobulbaceae, Desulfuromonadaceae, and Desulfovibrionaceae accounted for 16% of root microbiomes but less than 2% of leaf and water-column microbiomes, confirming the importance of sulfur cycling in eelgrass rhizosphere (Pedersen et al., 2004; Fahimipour et al., 2016).
Metatranscriptomic analysis identified expression of many genes involved in sulfur oxidation including a sulfur-oxidizing enzyme complex (soxABCXYZ) used by many chemo- and photolithoautotrophic sulfur-oxidizing organisms (Friedrich et al., 2005). In root microbiomes, these genes were almost exclusively assigned to several Gammaproteobacteria genera including Sedimenticola, Thiohalomonas, Dechloromarinus, an endosymbiont of Riftia pachyptila, and to the Epsilonproteobacteria Arcobacter nitrofigilis (Figure 5).
We also identified expression of genes involved in both the oxidation of sulfide and reduction of sulfate, including dissimilatory sulfite reductase genes dsrA and dsrB, and adenylyl-sulfate reductase genes aprA and aprB (Watanabe et al., 2016). These genes were assigned to both sulfur-oxidizing Gammaproteobacteria (e.g., Sedimenticola, Thiohalomonas, Dechloromarinus) and sulfate-reducing Deltaproteobacteria (e.g., Desulfococcus, Desulfospira, Desulfopila, Desulfobacula). Expression of these genes was always much greater for root microbiomes than leaf or water column microbiomes (Figure 4), and accounted for >1% of transcripts from root samples. These results suggest that a large and diverse portion of the eelgrass root microbiome is dedicated to sulfur oxidation, which likely benefits both microbes and plants.
Nitrogen Fixation and Denitrification
Rates of nitrogen fixation by seagrass leaf and root microbiomes were first published in Goering and Parker (1972) and Patriquin and Knowles (1972), and although these rates are sometimes highly variable (Capone et al., 1979; Lehnen et al., 2016), they are thought to account for a significant portion of plant nitrogen demand (Lipschultz et al., 1979; Capone, 1983; Pereg et al., 1994). Nitrogen-fixing bacteria have been identified on seagrass leaves and roots using 16S rRNA gene sequencing (Jensen et al., 2007; Crump and Koch, 2008), and nitrogenase (nifH) gene sequencing (Bagwell et al., 2002; Lehnen et al., 2016). One study sequenced nifH genes in sediments of a tropical seagrass bed of Thalassia testudinum and Syringodium filiforme and identified genes assigned to several classes of Proteobacteria (Bagwell et al., 2002). More recently, nifH sequences from Posidonia oceanica leaves, roots, and rhizomes confirmed that a high diversity of nitrogen-fixing bacteria is associated with plant surfaces, with many of these sequences being assigned to sulfate-reducing Deltaproteobacteria (Lehnen et al., 2016).
Our 16S sequencing identified a high proportion of organisms related to Arcobacter nitrofigilis (1% of leaf and 4% of root), which is a nitrogen-fixing symbiont on the roots of the marsh grass Spartina alterniflora (Pati et al., 2010). Expression of nifD, nifE, and nifH genes was much higher on roots than on leaves or in the water column, likely reflecting the oxygen sensitivity of nitrogenase (Figure 4; Gallon, 1992). Some of these transcripts were assigned to Arcobacter nitrofigilis, but most were assigned to sulfur-oxidizing Gammaproteobacteria and to the nitrogen-fixing Bacteroidetes, Saccharicrinis fermentans (Inoue et al., 2015). In contrast, very few nitrogenase transcripts were assigned to sulfate-reducing Deltaproteobacteria (Figure 5) suggesting that, despite their relative abundance, these organisms are not the main nitrogen fixers in the seagrass microbiome.
Microbially-mediated denitrification is an important ecosystem service of estuarine seagrass beds (Reynolds et al., 2016), which acts to remove excess nitrogen from eutrophic systems, and may influence plant growth through the generation of nitric oxide (NO; Stohr and Ullrich, 2002), an important regulator of development in plants (Domingos et al., 2015). Denitrification rates are variable in seagrass beds (Risgaard-Petersen and Ottosen, 2000; Eyre et al., 2011; Piehler and Smyth, 2011), and often lower than nitrogen fixation rates (Welsh et al., 2000; Russell et al., 2016). We detected higher expression of most denitrification genes in root versus leaf metatranscriptomes (Figure 4) with the exception of the norB gene coding for part of the NO reductase complex, suggesting that NO may be an important end product of denitrification in root microbiomes. Transcripts for each gene mapped to different organisms (e.g., nirK to Thaumarchaeota, nirS to Gammaproteobacteria and Betaproteobacteria, napB to Arcobacteria sp., nosZ to Bacteroidetes; Supplementary Table S2), suggesting that denitrification on plant roots involves the cooperation of several different denitrifying species.
Microbial Consumption of Plant Exudates
Angiosperms produce methanol as a by-product of cell-wall synthesis (Nemecek-Marshall et al., 1995), yet this same product can inhibit germination and retard the growth of angiosperm seedlings (Abanda-Nkpwatt et al., 2006). In strawberry plants, this negative effect is mitigated by methanol-consuming Methylobacterium extorquens (Abanda-Nkpwatt et al., 2006; Kurilenko et al., 2010). Methanol-consuming bacteria are common in marine environments, and include members of the genera Methylophaga and Methylobacter.
The family Methylophilaceae averaged 7% of the leaf microbiome and 4% of the root microbiome in our samples, compared to 1% in water samples, and belonged to four OTUs (Figure 2). However, we detected almost no expression of the methanol dehydrogenase genes (mdh and mxaIF) that are commonly involved in methanol oxidation. Instead, our metatranscriptomic analysis found expression of the genes mtaABC by leaf and root microbiomes (Figure 4). These genes code for a protein complex that irreversibly transforms methanol into methyl-CoM, which is the first step in the disproportionation of methanol to CO2 and methane (Sauer and Thauer, 1999). These genes were expressed mainly by Euryarchaea on leaves, and by Deltaproteobacteria on roots (Figure 5). Expression was also detected for heterodisulfide reductase genes (hdrABC and mvhADG), but expression was very low or absent for other genes involved in methane production including methyl-coenzyme M reductase genes (mcrABCDG) and tetrahydromethanopterin S-methyltransferase genes (mtrABCDEFGH), suggesting that methane is not produced by the eelgrass microbiome.
Terrestrial plants release methanol in large quantities, rivaling the release of other volatile organic compounds (monoterpenes and isoprene), but they release much of this material via stomata to the atmosphere (Nemecek-Marshall et al., 1995). Seagrasses do not have stomata (Kuo and den Hartog, 2007), and likely release plant-produced methanol and other volatile organics via diffusion through root tissue and through their thin leaf cuticle.
Ethanol and acetaldehyde are produced by terrestrial plant roots when soils become anoxic following flooding, causing plants to switch from an aerobic to a fermentative metabolism (Kreuzwieser et al., 1999). Release of these compounds generally follows a diurnal pattern, with no release at night and a burst in the morning when stomata open (Rottenberger et al., 2008). Seagrasses are often rooted in anoxic sediments and use fermentation at night when their photosynthetic oxygen pool is depleted (Touchette and Burkholder, 2000; Pedersen et al., 2004). Under anoxic conditions Z. marina roots can produce ethanol, lactate, and other metabolites, and may release over 95% of the ethanol as exudate (Smith et al., 1988). Ethanol is thought to be toxic to plants only at high concentrations (Tadege et al., 1999), and its oxidation product acetaldehyde is considered highly toxic (Perata and Alpi, 1991). Exacerbating this stress is the fact that these compounds are released into anoxic sediments where they may be used as carbon sources by sulfate-reducing bacteria to generate toxic levels of sulfide (Widdel, 1988).
Many microbes are capable of metabolizing ethanol and acetaldehyde, and we found expression of alcohol dehydrogenase (adh) and aldehyde dehydrogenase (ALDH) genes in our samples (Figure 4). Each of these enzymes can broadly act on a range of alcohols and aldehydes as substrates, indicating that expression here may not be exclusively linked to ethanol and acetaldehyde metabolism. These genes were expressed by a broad range of taxa including Alpha-, Beta-, Gamma- and Delta-proteobacteria, Firmicutes, and Actinobacteria. Low expression of alcohol consumption genes is consistent with low daytime production of ethanol and acetaldehyde during our sampling, and thus, expression of these genes may increase at night when roots undergo anaerobiosis.
Control of Epiphyte Community
As eelgrass leaves age, they accumulate epibiotic algal biofilms that can compete with the eelgrass leaves for light (Wahl, 1989). Epibiotic bacteria have been shown to influence the composition of biofilms on marine macroalgae, and, in doing so, provide protection to their hosts against extensive biofouling (Armstrong et al., 2001; Rao et al., 2005). One study showed that early colonization of P. oceanica seedlings by the bacteria Marinomonas posidonica influenced the composition of the epiphyte community and significantly increased leaf growth (Celdran et al., 2012). Another study showed that Z. marina leaves host high densities of algicidal bacteria (Inaba et al., 2017). One way for bacteria to influence algal growth is through the production of agarases and carrageenases that degrade galactose-based algal polymers and, in the case of agarases, can cause disease and die-off of red seaweed (Schroeder et al., 2003). We found that leaf microbiomes included a high proportion (average 3.3% leaf and 1.8% root) of organisms belonging to the Gammaproteobacteria genus Simiduia, which is associated with agarose hydrolysis (Park et al., 2014; Tawara et al., 2015). We also found expression of beta-agarase genes and several galactosidases were assigned to Gammaproteobacteria in the Z. japonica leaf microbiome (Figure 5). These organisms may be involved in regulating epiphyte communities, and, thus, could have important implications for epiphyte–seagrass competitive interactions.
Plant Hormone Production
Auxins, such as IAA, are key regulators of plant growth and development. A binding site for an auxin response factor was detected in the Z. marina genome (Olsen et al., 2016), suggesting that this class of hormones might be used by seagrasses. However, one study of Posidonia australis found mixed effects of auxins on seedling survival (Glasby et al., 2015), and studies of Halophila decipiens and Cymodocea nodosa found no effect of auxin exposure on growth (Munoz, 1995; Bird et al., 1998).
We detected expression of three genes involved in bacterial conversion of tryptophan to IAA: MAO, which codes for tryptophan dehydrogenase (tryptophan to tryptamine; Shih et al., 1999), DDC, which codes for tryptamine oxidase (tryptamine to indole-3-acetaldehyde; Chassande et al., 1994), and ALDH, which is a family of enzymes that includes IAA dehydrogenases (indole-3-acetaldehyde to IAA; Basse et al., 1996). However, we did not find expression of the KEGG enzyme classified as IAA dehydrogenase (K11817), or of several genes involved in the more common indole-3-acetamide (Amin et al., 2015) and indole-3-pyruvate pathways for IAA production (Spaepen et al., 2007). Expression of a complete tryptamine pathway for IAA production suggests that seagrass microbiomes produce IAA and potentially influence plant growth through regulation of this compound.
Expression of genetic pathways for other plant hormones were limited and incomplete. For example, the cytokinin zeatin (Takei et al., 2004; Dodd et al., 2010) is present in the leaves and roots of the seagrass P. oceanica (L.) Delile, and shows a dynamic distribution in shoot tissues in relation to environmental stress factors (Bruno et al., 2009). Cytokinin is also produced by some bacteria using the ipt gene for cytokinin synthase (Mok and Mok, 2001). However, expression of this gene and all others in the zeatin biosynthesis pathway were not detected except the miaA gene coding for tRNA dimethylallyltransferase.
Another example of a plant signaling molecule is ethylene, which is a growth regulator produced by soil bacteria and plants (Zechmeister-Boltenstern and Smith, 1998; Hayat et al., 2010). Genomic analysis of the seagrasses Z. marina (Olsen et al., 2016) and Z. muelleri (Golicz et al., 2015) showed that genes for ethylene biosynthesis and signaling are missing from these genomes, suggesting that there is no need for microbes to participate in the regulation of ethylene levels. Consistent with this finding, no expression was detected of prokaryotic efe genes (K21815) that act in the synthesis of ethylene (Fukuda et al., 1992; Chen et al., 2010), and expression of ACC deaminase genes (K01505) for regulating plant synthesis of ethylene (Glick et al., 2007) was limited to Z. marina leaf and was lower than in bacterioplankton in the Columbia River estuary. These results suggest that seagrass microbiomes do not produce cytokinins or manipulate ethylene levels in Z. marina or Z. japonica.
Conclusion
The phylogenetic composition of plant-associated bacterial communities was not significantly different between seagrass species for leaf microbiomes or root microbiomes. However, leaf-, root-, and water, column-associated bacterial communities were significantly different from one another. The taxonomy and gene expression of these microbiomes suggest that these communities detoxify sulfide using multiple metabolic pathways (e.g., soxABCXYZ and dsrAB), fix nitrogen, metabolize methanol and ethanol potentially released by eelgrass as waste products, produce agarases that may limit growth of competitive algal epiphytes, and influence plant growth by producing nitric oxide and the hormone IAA.
Author Contributions
BC: project leadership, intellectual contributions, data analysis, and manuscript preparation. JW: laboratory analysis, intellectual contributions, data analysis, and manuscript preparation. FT: intellectual contributions and manuscript preparation. RM: intellectual contributions, data analysis, and manuscript preparation.
Funding
Funding for this undergraduate research project was provided by a DeLoach Work Scholarship to JW from the Oregon State University Honors College, by the OSU Department of Fisheries and Wildlife, and by the Science and Technology Center for Coastal Margin Observation & Prediction (CMOP) supported by the U.S. National Science Foundation Grant No. OCE-0424602.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mark Dasenko and Christopher M. Sullivan at the Oregon State University Center for Genome Research and Biocomputing for assistance with metatranscriptome sample processing and bioinformatics analyses. We also thank scientists at the United States Department of Energy Joint Genome Institute for assistance with metatranscriptome annotations.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00388/full#supplementary-material
Footnotes
References
Abanda-Nkpwatt, D., Musch, M., Tschiersch, J., Boettner, M., and Schwab, W. (2006). Molecular interaction between Methylobacterium extorquens and seedlings: growth promotion, methanol consumption, and localization of the methanol emission site. J. Exp. Bot. 57, 4025–4032. doi: 10.1093/jxb/erl173
Akiyoshi, D. E., Klee, H., Amasino, R. M., Nester, E. W., and Gordon, M. P. (1984). T-DNA of Agrobacterium tumefaciens encodes an enzyme of cytokinin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 81, 5994–5998. doi: 10.1073/pnas.81.19.5994
Amin, S. A., Hmelo, L. R., Van Tol, H. M., Durham, B. P., Carlson, L. T., Heal, K. R., et al. (2015). Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522, 98–101. doi: 10.1038/nature14488
Andrews, J. H. (1992). Biological control in the phyllosphere. Annu. Rev. Phytopathol. 30, 603–635. doi: 10.1146/annurev.py.30.090192.003131
Armstrong, E., Yan, L. M., Boyd, K. G., Wright, P. C., and Burgess, J. G. (2001). The symbiotic role of marine microbes on living surfaces. Hydrobiologia 461, 37–40. doi: 10.1023/a:1012756913566
Baden, S., Gullström, M., Lundén, B., Pihl, L., and Rosenberg, R. (2003). Vanishing seagrass (Zostera marina, L.) in Swedish coastal waters. Ambio 32, 374–377. doi: 10.1579/0044-7447-32.5.374
Bagwell, C. E., La Rocque, J. R., Smith, G. W., Polson, S. W., Friez, M. J., Longshore, J. W., et al. (2002). Molecular diversity of diazotrophs in oligotrophic tropical seagrass bed communities. FEMS Microbiol. Ecol. 39, 113–119. doi: 10.1111/j.1574-6941.2002.tb00912.x
Baldwin, J. R., and Lovvorn, J. R. (1994). Expansion of seagrass habitat by the exotic Zostera japonica, and its use by dabbling ducks and brant in Boundary Bay, British Columbia. Mar. Ecol. Progr. Ser. 103, 119–127. doi: 10.3354/meps103119
Basse, C. W., Lottspeich, F., Steglich, W., and Kahmann, R. (1996). Two potential indole-3-acetaldehyde dehydrogenases in the phytopathogenic fungus Ustilago maydis. Eur. J. Biochem. 242, 648–656. doi: 10.1111/j.1432-1033.1996.0648r.x
Benes, V., Blake, J., and Doyle, K. (2011). Ribo-Zero Gold Kit: improved RNA-seq results after removal of cytoplasmic and mitochondrial ribosomal RNA. Nat. Meth. 8:11. doi: 10.1038/nmeth.f.352
Bengtsson, M. M., Buhler, A., Brauer, A., Dahlke, S., Schubert, H., and Blindow, L. (2017). Eelgrass leaf surface microbiomes are locally variable and highly correlated with epibiotic eukaryotes. Front. Microbiol. 8:11. doi: 10.3389/fmicb.2017.01312
Berendsen, R. L., Pieterse, C. M. J., and Bakker, P. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Bird, K. T., Johnson, J. R., and Jewett-Smith, J. (1998). In vitro culture of the seagrass Halophila decipiens. Aquat. Bot. 60, 377–387. doi: 10.1016/S0304-3770(97)00093-4
Boström, C., Jackson, E. L., and Simenstad, C. A. (2006). Seagrass landscapes and their effects on associated fauna: a review. Estuar. Coast. Shelf Sci. 68, 383–403. doi: 10.1016/j.ecss.2006.01.026
Bruno, A., Petrarulo, M., Salimonti, A., Chiappetta, A., and Bitonti, M. B. (2009). Distinct pattern of zeatin distribution in the shoot apical meristem marks stress conditions in Posidonia oceanica (L.) Delile plants. Plant Biosyst. 143, 25–33. doi: 10.1080/11263500802633220
Cao, H. X., Schmutzer, T., Scholz, U., Pecinka, A., Schubert, I., and Vu, G. T. (2015). Metatranscriptome analysis reveals host-microbiome interactions in traps of carnivorous Genlisea species. Front. Microbiol. 6:526. doi: 10.3389/fmicb.2015.00526
Capone, D. G., Penhale, P. A., Oremland, R. S., and Taylor, B. F. (1979). Relationship between productivity and N2 (C2h2) fixation in a Thalassia testudinum community. Limnol. Oceanogr. 24, 117–125. doi: 10.4319/lo.1979.24.1.0117
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Celdran, D., Espinosa, E., Sanchez-Amat, A., and Marin, A. (2012). Effects of epibiotic bacteria on leaf growth and epiphytes of the seagrass Posidonia oceanica. Mar. Ecol. Progr. Ser. 456, 21–27. doi: 10.3354/meps09672
Chassande, O., Renard, S., Barbry, P., and Lazdunski, M. (1994). Human gene for diamine oxidase, an amiloride binding-protein. Molecular cloning, sequencing, and characterization of the promoter. J. Biol. Chem. 269, 14484–14489.
Chen, I. M. A., Markowitz, V. M., Palaniappan, K., Szeto, E., Chu, K., Huang, J. H., et al. (2016). Supporting community annotation and user collaboration in the integrated microbial genomes (IMG) system. BMC Genomics 17:16. doi: 10.1186/s12864-016-2629-y
Chen, X., Liang, Y., Hua, J., Tao, L., Qin, W. S., and Chen, S. F. (2010). Overexpression of bacterial ethylene-forming enzyme gene in Trichoderma reesei enhanced the production of ethylene. Int. J. Biol. Sci. 6, 96–106. doi: 10.7150/ijbs.6.96
Crump, B. C., Kling, G. W., Bahr, M., and Hobbie, J. E. (2003). Bacterioplankton community shifts in an arctic lake correlate with seasonal changes in organic matter source. Appl. Environ. Microbiol. 69, 2253–2268. doi: 10.1128/AEM.69.4.2253-2268.2003
Crump, B. C., and Koch, E. W. (2008). Attached bacterial populations shared by four species of aquatic angiosperms. Appl. Environ. Microbiol. 74, 5948–5957. doi: 10.1128/aem.00952-08
Cucio, C., Engelen, A. H., Costa, R., and Muyzer, G. (2016). Rhizosphere microbiomes of European seagrasses are selected by the plant, but are not species specific. Front. Microbiol. 7:440. doi: 10.3389/fmicb.2016.00440
Dodd, I. C., Zinovkina, N. Y., Safronova, V. I., and Belimov, A. A. (2010). Rhizobacterial mediation of plant hormone status. Ann. Appl. Biol. 157, 361–379. doi: 10.1111/j.1744-7348.2010.00439.x
Domingos, P., Prado, A. M., Wong, A., Gehring, C., and Feijo, J. A. (2015). Nitric oxide: a multitasked signaling gas in plants. Mol. Plant 8, 506–520. doi: 10.1016/j.molp.2014.12.010
Dourado, M. N., Bogas, A. C., Pomini, A. M., Andreote, F. D., Quecine, M. C., Marsaioli, A. J., et al. (2013). Methylobacterium-plant interaction genes regulated by plant exudate and quorum sensing molecules. Braz. J. Microbiol. 44, 1331–1339. doi: 10.1590/S1517-83822013000400044
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Ettinger, C. L., Voerman, S. E., Lang, J. M., Stachowicz, J. J., and Eisen, J. A. (2017). Microbial communities in sediment from Zostera marina patches, but not the Z. marina leaf or root microbiomes, vary in relation to distance from patch edge. Peerj 5, e3246. doi: 10.7717/peerj.3246
Eyre, B. D., Ferguson, A. J. P., Webb, A., Maher, D., and Oakes, J. M. (2011). Denitrification, N-fixation and nitrogen and phosphorus fluxes in different benthic habitats and their contribution to the nitrogen and phosphorus budgets of a shallow oligotrophic sub-tropical coastal system (southern Moreton Bay, Australia). Biogeochemistry 102, 111–133. doi: 10.1007/s10533-010-9425-6
Fahimipour, A. K., Kardish, M. R., Eisen, J. A., Lang, J. M., Green, J. L., and Stachowicz, J. J. (2016). Global-scale structure of the eelgrass microbiome. Appl. Environ. Microbiol. 83:e03391-16. doi: 10.1101/089797
Flood, B. E., Jones, D. S., and Bailey, J. V. (2015). Sedimenticola thiotaurini sp nov., a sulfur-oxidizing bacterium isolated from salt marsh sediments, and emended descriptions of the genus Sedimenticola and Sedimenticola selenatireducens. Int. J. Syst. Evolut. Microbiol. 65, 2522–2530. doi: 10.1099/ijs.0.000295
Fortunato, C. S., and Crump, B. C. (2015). Microbial gene abundance and expression patterns across a river to ocean salinity gradient. PLoS One 10:e0140578. doi: 10.1371/journal.pone.0140578
Friedrich, C. G., Bardischewsky, F., Rother, D., Quentmeier, A., and Fischer, J. (2005). Prokaryotic sulfur oxidation. Curr. Opin. Microbiol. 8, 253–259. doi: 10.1016/j.mib.2005.04.005
Fukuda, H., Ogawa, T., Ishihara, K., Fujii, T., Nagahama, K., Omata, T., et al. (1992). Molecular cloning in Escherichia coli, expression, and nucleotide sequence of the gene for the ethylene-forming enzyme of Pseudomonas syringae pv. phaseolicola PK2. Biochem. Biophys. Res. Commun. 188, 826–832. doi: 10.1016/0006-291x(92)91131-9
Gallon, J. R. (1992). Reconciling the incompatible: N2 fixation and O2. New Phytol. 122, 571–609. doi: 10.1111/j.1469-8137.1992.tb00087.x
Glanzman, C., Glenne, B., and Burgess, F. (1971). Tidal hydraulics, flushing characteristics and water quality of Netarts Bay. Oreg. State Univ. Eng. Exp. Stn. Bull. 45:33.
Glasby, T. M., Taylor, S. L., and Housefield, G. P. (2015). Factors influencing the growth of seagrass seedlings: a case study of Posidonia australis. Aquat. Bot. 120, 251–259. doi: 10.1016/j.aquabot.2014.09.003
Glick, B. R. (1995). The enhancement of plant-growth by free-living bacteria. Can. J. Microbiol. 41, 109–117. doi: 10.1139/m95-015
Glick, B. R., Penrose, D. M., and Li, J. P. (1998). A model for the lowering of plant ethylene concentrations by plant growth-promoting bacteria. J. Theor. Biol. 190, 63–68. doi: 10.1006/jtbi.1997.0532
Glick, B. R., Todorovic, B., Czarny, J., Cheng, Z. Y., Duan, J., and Mcconkey, B. (2007). Promotion of plant growth by bacterial ACC deaminase. Crit. Rev. Plant Sci. 26, 227–242. doi: 10.1080/07352680701572966
Goering, J. J., and Parker, P. L. (1972). Nitrogen fixation by epiphytes on sea grasses. Limnol. Oceanogr. 17, 320–323. doi: 10.4319/lo.1972.17.2.0320
Golicz, A. A., Schliep, M., Lee, H. T., Larkum, A. W. D., Dolferus, R., Batley, J., et al. (2015). Genome-wide survey of the seagrass Zostera muelleri suggests modification of the ethylene signalling network. J. Exp. Bot. 66, 1489–1498. doi: 10.1093/jxb/eru510
Goodman, J. L., Moore, K. A., and Dennison, W. C. (1995). Photosynthetic responses of eelgrass (Zostera marina L) to light and sediment sulfide in a shallow barrier island lagoon. Aquat. Bot. 50, 37–47. doi: 10.1016/0304-3770(94)00444-q
Gordon-Bradley, N., Lymperopoulou, D. S., and Williams, H. N. (2014). Differences in bacterial community structure on Hydrilla verticillata and Vallisneria americana in a freshwater spring. Microb. Environ. 29, 67–73. doi: 10.1264/jsme2.ME13064
Green, E. P., and Short, F. T. (2003). World Atlas of Seagrasses. Berkeley, CA: University of California Press.
Gruber, N., and Galloway, J. N. (2008). An Earth-system perspective of the global nitrogen cycle. Nature 451, 293–296. doi: 10.1038/nature06592
Haichar, F. E., Marol, C., Berge, O., Rangel-Castro, J. I., Prosser, J. I., Balesdent, J., et al. (2008). Plant host habitat and root exudates shape soil bacterial community structure. ISME J. 2, 1221–1230. doi: 10.1038/ismej.2008.80
Han, Y. C., and Perner, M. (2015). The globally widespread genus Sulfurimonas: versatile energy metabolisms and adaptations to redox clines. Front. Microbiol. 6:989. doi: 10.3389/fmicb.2015.00989
Hansen, M., and Perner, M. (2015). A novel hydrogen oxidizer amidst the sulfur-oxidizing Thiomicrospira lineage. ISME J. 9, 696–707. doi: 10.1038/ismej.2014.173
Harrison, P. G. (1982). Spatial and temporal patterns in abundance of two intertidal seagrasses, Zostera americana den hartog and Zostera marina L. Aquat. Bot. 12, 305–320. doi: 10.1016/0304-3770(82)90024-9
Hasler-Sheetal, H., and Holmer, M. (2015). Sulfide intrusion and detoxification in the seagrass Zostera marina. PLoS One 10:e0129136. doi: 10.1371/journal.pone.0129136
Hayat, R., Ali, S., Amara, U., Khalid, R., and Ahmed, I. (2010). Soil beneficial bacteria and their role in plant growth promotion: a review. Ann. Microbiol. 60, 579–598. doi: 10.1007/s13213-010-0117-1
He, D., Ren, L. J., and Wu, Q. L. (2012). Epiphytic bacterial communities on two common submerged macrophytes in Taihu Lake: diversity and host-specificity. Chin. J. Oceanol. Limnol. 30, 237–247. doi: 10.1007/s00343-012-1084-0
Husnik, F., Nikoh, N., Koga, R., Ross, L., Duncan, R. P., Fujie, M., et al. (2013). Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell 153, 1567–1578. doi: 10.1016/j.cell.2013.05.040
Huson, D. H., Mitra, S., Ruscheweyh, H. J., Weber, N., and Schuster, S. C. (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560. doi: 10.1101/gr.120618.111
Huttenhower, C., Gevers, D., Knight, R., Abubucker, S., Badger, J. H., Chinwalla, A. T., et al. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Inaba, N., Trainer, V. L., Onishi, Y., Ishii, K.-I., Wyllie-Echeverria, S., and Imai, I. (2017). Algicidal and growth-inhibiting bacteria associated with seagrass and macroalgae beds in Puget Sound, WA, USA. Harmful Algae 62, 136–147. doi: 10.1016/j.hal.2016.04.004
Inoue, J., Oshima, K., Suda, W., Sakamoto, M., Iino, T., Noda, S., et al. (2015). Distribution and evolution of nitrogen fixation genes in the phylum bacteroidetes. Microb. Environ. 30, 44–50. doi: 10.1264/jsme2.ME14142
Ivanova, E. G., Doronina, N. V., Shepelyakovskaya, A. O., Laman, A. G., Brovko, F. A., and Trotsenko, Y. A. (2000). Facultative and obligate aerobic methylobacteria synthesize cytokinins. Microbiology 69, 646–651. doi: 10.1023/A:1026693805653
Jensen, S. I., Kuhl, M., and Prieme, A. (2007). Different bacterial communities associated with the roots and bulk sediment of the seagrass Zostera marina. FEMS Microbiol. Ecol. 62, 108–117. doi: 10.1111/j.1574-6941.2007.00373.x
Jiang, Y. F., Ling, J., Dong, J. D., Chen, B., Zhang, Y. Y., Zhang, Y. Z., et al. (2015). Illumina-based analysis the microbial diversity associated with Thalassia hemprichii in Xincun Bay, South China Sea. Ecotoxicology 24, 1548–1556. doi: 10.1007/s10646-015-1511-z
Jourand, P., Renier, A., Rapior, S., De Faria, S. M., Prin, Y., Galiana, A., et al. (2005). Role of methylotrophy during symbiosis between Methylobacterium nodulans and Crotalaria podocarpa. Mol. Plant Microbe Interact. 18, 1061–1068. doi: 10.1094/mpmi-18-1061
Kent, A. D., and Triplett, E. W. (2002). Microbial communities and their interactions in soil and rhizosphere ecosystems. Annu. Rev. Microbiol. 56, 211–236. doi: 10.1146/annurev.micro.56.012302.161120
Kloepper, J. W., Leong, J., Teintze, M., and Schroth, M. N. (1980). Enhanced plant-growth by siderophores produced by plant growth-promoting Rhizobacteria. Nature 286, 885–886. doi: 10.1038/286885a0
Koch, E. W. (2001). Beyond light: physical, geological, and geochemical parameters as possible submersed aquatic vegetation habitat requirements. Estuaries 24, 1–17. doi: 10.2307/1352808
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/aem.01043-13
Kreuzwieser, J., Schnitzler, J. P., and Steinbrecher, R. (1999). Biosynthesis of organic compounds emitted by plants. Plant Biol. 1, 149–159. doi: 10.1111/j.1438-8677.1999.tb00238.x
Kuo, J., and den Hartog, C. (2007). “Seagrass morphology, anatomy, and ultrastructure,” in Seagrasses: Biology, Ecology and Conservation, eds A. W. D. Larkum, R. J. Orth, and C. M. Duarte (Dordrecht: Springer), 51–87.
Kurilenko, V. V., Christen, R., Zhukova, N. V., Kalinovskaya, N. I., Mikhailov, V. V., Crawford, R. J., et al. (2010). Granulosicoccus coccoides sp nov., isolated from leaves of seagrass (Zostera marina). Int. J. Syst. Evolut. Microbiol. 60, 972–976. doi: 10.1099/ijs.0.013516-0
Lehnen, N., Marchant, H. K., Schwedt, A., Milucka, J., Lott, C., Weber, M., et al. (2016). High rates of microbial dinitrogen fixation and sulfate reduction associated with the Mediterranean seagrass Posidonia oceanica. Syst. Appl. Microbiol. 39, 476–483. doi: 10.1016/j.syapm.2016.08.004
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Lipschultz, F., Cunningham, J. J., and Stevenson, J. C. (1979). Nitrogen-fixation associated with 4 species of submerged angiosperms in the central chesapeake bay. Estuar. Coast. Mar. Sci. 9, 813–818.
Marzano, S.-Y. L., and Domier, L. L. (2016). Novel mycoviruses discovered from metatranscriptomics survey of soybean phyllosphere phytobiomes. Virus Res. 213, 332–342. doi: 10.1016/j.virusres.2015.11.002
McGlathery, K. J., Sundback, K., and Anderson, I. C. (2007). Eutrophication in shallow coastal bays and lagoons: the role of plants in the coastal filter. Mar. Ecol. Progr. Ser. 348, 1–18. doi: 10.3354/meps07132
Mejia, A. Y., Rotini, A., Lacasella, F., Bookman, R., Thaller, M. C., Shem-Tov, R., et al. (2016). Assessing the ecological status of seagrasses using morphology, biochemical descriptors and microbial community analyses. A study in Halophila stipulacea (Forsk.) Aschers meadows in the northern Red Sea. Ecol. Indicat. 60, 1150–1163. doi: 10.1016/j.ecolind.2015.09.014
Meldau, D. G., Long, H. H., and Baldwin, I. T. (2012). A native plant growth promoting bacterium, Bacillus sp B55, rescues growth performance of an ethylene-insensitive plant genotype in nature. Front. Plant Sci. 3:112. doi: 10.3389/fpls.2012.00112
Mok, D. W. S., and Mok, M. C. (2001). Cytokinin metabolism and action. Ann. Rev. Plant Physiol. Plant Mol. Biol. 52, 89–118. doi: 10.1146/annurev.arplant.52.1.89
Munoz, J. T. (1995). Effects of some plant growth regulators on the growth of the seagrass Cymodocea nodosa (Ucria) Ascherson. Aquat. Bot. 51, 311–318. doi: 10.1016/0304-3770(95)00481-E
Nemecek-Marshall, M., Macdonald, R. C., Franzen, F. J., Wojciechowski, C. L., and Fall, R. (1995). Methanol emission from leaves - Enzymatic detection of gas-phase methanol and relation of methanol fluxes to stomatal conductance and leaf development. Plant Physiol. 108, 1359–1368. doi: 10.1104/pp.108.4.1359
Olsen, J. L., Rouze, P., Verhelst, B., Lin, Y. C., Bayer, T., Collen, J., et al. (2016). The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530, 331–335. doi: 10.1038/nature16548
Omer, Z. S., Tombolini, R., Broberg, A., and Gerhardson, B. (2004). Indole-3-acetic acid production by pink-pigmented facultative methylotrophic bacteria. Plant Growth Regulat. 43, 93–96. doi: 10.1023/B:GROW.0000038360.09079.ad
Orth, R. J., Carruthers, T. J. B., Dennison, W. C., Duarte, C. M., Fourqurean, J. W., Heck, K. L., et al. (2006). A global crisis for seagrass ecosystems. Bioscience 56, 987–996.
Park, S., Kim, S. I., Jung, Y. T., and Yoon, J. H. (2014). Simiduia curdlanivorans sp nov., a curdlan-degrading bacterium isolated from the junction between the ocean and a freshwater spring, and emended description of the genus Simiduia. Int. J. Syst. Evolut. Microbiol. 64, 3695–3700. doi: 10.1099/ijs.0.065334-0
Pati, A., Gronow, S., Lapidus, A., Copeland, A., Del Rio, T. G., Nolan, M., et al. (2010). Complete genome sequence of Arcobacter nitrofigilis type strain (CIT). Stand. Genom. Sci. 2, 300–308. doi: 10.4056/sigs.912121
Patriquin, D., and Knowles, R. (1972). Nitrogen fixation in rhizosphere of marine angiosperms. Mar. Biol. 16, 49–58. doi: 10.1007/bf00347847
Patten, C. L., and Glick, B. R. (1996). Bacterial biosynthesis on indole-3-acetic acid. Can. J. Microbiol. 42, 207–220. doi: 10.1139/m96-032
Pedersen, O., Binzer, T., and Borum, J. (2004). Sulphide intrusion in eelgrass (Zostera marina L.). Plant Cell Environ. 27, 595–602. doi: 10.1111/j.1365-3040.2004.01173.x
Perata, P., and Alpi, A. (1991). Ethanol-induced injuries to carrot cells: the role of acetaldehyde. Plant Physiol. 95, 748–752. doi: 10.1104/pp.95.3.748
Pereg, L. L., Lipkin, Y., and Sar, N. (1994). Different niches of the Halophila-Stipulacea Seagrass bed harbor distinct populations of nitrogen-fixing bacteria. Mar. Biol. 119, 327–333. doi: 10.1007/BF00347529
Piehler, M. F., and Smyth, A. R. (2011). Habitat-specific distinctions in estuarine denitrification affect both ecosystem function and services. Ecosphere 2, 1–17. doi: 10.1890/es10-00082.1
Poretsky, R., Gifford, S., Rinta-Kanto, J., Vila-Costa, M., and Moran, M. (2009). Analyzing gene expression from marine microbial communities using environmental transcriptomics. J. Vis. Exp. 24:e1086. doi: 10.3791/1086
Ralph, P. J., Durako, M. J., Enríquez, S., Collier, C. J., and Doblin, M. A. (2007). Impact of light limitation on seagrasses. J. Exp. Mar. Bio. Ecol. 350, 176–193. doi: 10.1016/j.jembe.2007.06.017
Rao, D., Webb, J. S., and Kjelleberg, S. (2005). Competitive interactions in mixed-species biofilms containing the marine bacterium Pseudoalteromonas tunicata. Appl. Environ. Microbiol. 71, 1729–1736. doi: 10.1128/aem.71.4.1729-1736.2005
Reynolds, L. K., Waycott, M., Mcglathery, K. J., and Orth, R. J. (2016). Ecosystem services returned through seagrass restoration. Restor. Ecol. 24, 583–588. doi: 10.1111/rec.12360
Risgaard-Petersen, N., and Ottosen, L. D. M. (2000). Nitrogen cycling in two temperate Zostera marina beds: seasonal variation. Mar. Ecol. Progr. Ser. 198, 93–107. doi: 10.3354/meps198093
Rottenberger, S., Kleiss, B., Kuhn, U., Wolf, A., Piedade, M. T. F., Junk, W., et al. (2008). The effect of flooding on the exchange of the volatile C-2-compounds ethanol, acetaldehyde and acetic acid between leaves of Amazonian floodplain tree species and the atmosphere. Biogeosciences 5, 1085–1100. doi: 10.5194/bg-5-1085-2008
Russell, D. G., Warry, F. Y., and Cook, P. L. M. (2016). The balance between nitrogen fixation and denitrification on vegetated and non-vegetated intertidal sediments. Limnol. Oceanogr. 61, 2058–2075. doi: 10.1002/lno.10353
Sauer, K., and Thauer, R. K. (1999). Methanol: coenzyme M methyltransferase from Methanosarcina barkeri - substitution of the corrinoid harbouring subunit MtaC by free cob(I)alamin. Eur. J. Biochem. 261, 674–681. doi: 10.1046/j.1432-1327.1999.00355.x
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-09
Schroeder, D. C., Jaffer, M. A., and Coyne, V. E. (2003). Investigation of the role of a β(1–4) agarase produced by Pseudoalteromonas gracilis B9 in eliciting disease symptoms in the red alga Gracilaria gracilis. Microbiology 149, 2919–2929. doi: 10.1099/mic.0.26513-0
Shafer, D. J., Kaldy, J. E., and Gaeckle, J. L. (2014). Science and management of the introduced seagrass Zostera japonica in North America. Environ. Manag. 53, 147–162. doi: 10.1007/s00267-013-0172-z
Shih, J. C., Chen, K., and Ridd, M. J. (1999). Monoamine oxidase: from genes to behavior. Annu. Rev. Neurosci. 22, 197–217. doi: 10.1146/annurev.neuro.22.1.197
Smith, R. D., Pregnall, A. M., and Alberte, R. S. (1988). Effects of anaerobiosis on root metabolism of Zostera marina (eelgrass): implications for survival in reducing sediments. Mar. Biol. 98, 131–141. doi: 10.1007/bf00392668
Spaepen, S., Vanderleyden, J., and Remans, R. (2007). Indole-3-acetic acid in microbial and microorganism-plant signaling. FEMS Microbiol. Rev. 31, 425–448. doi: 10.1111/j.1574-6976.2007.00072.x
Spalding, M. D., Mcivor, A. L., Beck, M. W., Koch, E. W., Moller, I., Reed, D. J., et al. (2014). Coastal ecosystems: a critical element of risk reduction. Conserv. Lett. 7, 293–301. doi: 10.1111/conl.12074
Stewart, F. J., Ottesen, E. A., and Delong, E. F. (2010). Development and quantitative analyses of a universal rRNA-subtraction protocol for microbial metatranscriptomics. ISME J. 4, 896–907. doi: 10.1038/ismej.2010.18
Stohr, C., and Ullrich, W. R. (2002). Generation and possible roles of NO in plant roots and their apoplastic space. J. Exp. Bot. 53, 2293–2303. doi: 10.1093/jxb/erf110
Tadege, M., Dupuis, I., and Kuhlemeier, C. (1999). Ethanolic fermentation: new functions for an old pathway. Trends Plant Sci. 4, 320–325. doi: 10.1016/s1360-1385(99)01450-8
Takei, K., Ueda, N., Aoki, K., Kuromori, T., Hirayama, T., Shinozaki, K., et al. (2004). AtIPT3 is a key determinant of nitrate-dependent cytokinin biosynthesis in Arabidopsis. Plant Cell Physiol. 45, 1053–1062. doi: 10.1093/pcp/pch119
Tawara, M., Sakatoku, A., Tiodjio, R. E., Tanaka, D., and Nakamura, S. (2015). Cloning and characterization of a novel agarase from a newly isolated Bacterium Simiduia sp strain TM-2 able to degrade various seaweeds. Appl. Biochem. Biotechnol. 177, 610–623. doi: 10.1007/s12010-015-1765-1
Touchette, B. W., and Burkholder, J. M. (2000). Overview of the physiological ecology of carbon metabolism in seagrasses. J. Exp. Mar. Biol. Ecol. 250, 169–205. doi: 10.1016/s0022-0981(00)00196-9
Turner, T. R., James, E. K., and Poole, P. S. (2013). The plant microbiome. Genome Biol. 14:209. doi: 10.1186/gb-2013-14-6-209
Uku, J., Bjork, M., Bergman, B., and Diez, B. (2007). Characterization and comparison of prokaryotic epiphytes associated with three east African seagrasses. J. Phycol. 43, 768–779.
van der Heide, T., Govers, L. L., De Fouw, J., Olff, H., Van Der Geest, M., Van Katwijk, M. M., et al. (2012). A three-stage symbiosis forms the foundation of seagrass ecosystems. Science 336, 1432–1434. doi: 10.1126/science.1219973
Vorholt, J. A. (2012). Microbial life in the phyllosphere. Nat. Rev. Microbiol. 10, 828–840. doi: 10.1038/nrmicro2910
Wagner, G. P., Kin, K., and Lynch, V. J. (2012). Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 131, 281–285. doi: 10.1007/s12064-012-0162-3
Wahl, M. (1989). Marine epibiosis. 1. Fouling and antifouling: some basic aspects. Mar. Ecol. Progr. Ser. 58, 175–189. doi: 10.3354/meps058175
Watanabe, T., Kojima, H., and Fukui, M. (2016). Identity of major sulfur-cycle prokaryotes in freshwater lake ecosystems revealed by a comprehensive phylogenetic study of the dissimilatory adenylylsulfate reductase. Sci. Rep. 6:36262. doi: 10.1038/srep36262
Weidner, S., Arnold, W., and Puhler, A. (1996). Diversity of uncultured microorganisms associated with the seagrass Halophila stipulacea estimated by restriction fragment length polymorphism analysis of PCR-amplified 16S rRNA genes. Appl. Environ. Microbiol. 62, 766–771.
Weidner, S., Arnold, W., Stackebrandt, E., and Puhler, A. (2000). Phylogenetic analysis of bacterial communities associated with leaves of the seagrass Halophila stipulacea by a culture-independent small-subunit rRNA gene approach. Microb. Ecol. 39, 22–31.
Welsh, D. T., Bartoli, M., Nizzoli, D., Castaldelli, G., Riou, S. A., and Viaroli, P. (2000). Denitrification, nitrogen fixation, community primary productivity and inorganic-N and oxygen fluxes in an intertidal Zostera noltii meadow. Mar. Ecol. Progr. Ser. 208, 65–77. doi: 10.3354/meps208065
Whipps, J. M. (2001). Microbial interactions and biocontrol in the rhizosphere. J. Exp. Bot. 52, 487–511. doi: 10.1093/jxb/52.suppl_1.487
Widdel, F. (1988). “Microbiology and ecology of sulfate- and sulfur-reducing bacteria,” in Biology of Anaerobic Microorganisms, ed. A. J. B. Zehnder (New York, NY: John Wiley), 469–585.
Williams, C. J., and Yavitt, J. B. (2010). Temperate Wetland Methanogenesis: the Importance of vegetation type and root ethanol production. Soil Sci. Soc. Am. J. 74, 317–325. doi: 10.2136/sssaj2008.0395
Keywords: symbiosis, estuary, marine, microbiology, DNA, eelgrass, diazotroph, PCR
Citation: Crump BC, Wojahn JM, Tomas F and Mueller RS (2018) Metatranscriptomics and Amplicon Sequencing Reveal Mutualisms in Seagrass Microbiomes. Front. Microbiol. 9:388. doi: 10.3389/fmicb.2018.00388
Received: 21 November 2017; Accepted: 20 February 2018;
Published: 15 March 2018.
Edited by:
Pilar Martínez-Hidalgo, Universidad de Salamanca, SpainReviewed by:
Luisa I. Falcon, Universidad Nacional Autónoma de México, MexicoMathieu Pernice, University of Technology Sydney, Australia
Susana Enríquez, Universidad Nacional Autónoma de México, Mexico
Copyright © 2018 Crump, Wojahn, Tomas and Mueller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Byron C. Crump, YmNydW1wQGNlb2FzLm9yZWdvbnN0YXRlLmVkdQ==