 Alba Pérez-Cataluña
Alba Pérez-Cataluña Luis Collado
Luis Collado Oscar Salgado
Oscar Salgado Violeta Lefiñanco
Violeta Lefiñanco María J. Figueras
María J. Figueras- 1Unit of Microbiology, Department of Basic Health Sciences, Faculty of Medicine and Health Sciences, IISPV, University Rovira i Virgili, Reus, Spain
- 2Faculty of Sciences, Institute of Biochemistry and Microbiology, Universidad Austral de Chile, Valdivia, Chile
- 3Laboratory of Microbial Ecology of Extreme Systems, Department of Molecular Genetics and Microbiology, Pontificia Universidad Católica de Chile, Santiago, Chile
The species Arcobacter cryaerophilus is found in many food products of animal origin and is the dominating species in wastewater. In addition, it is associated with cases of farm animal and human infectious diseases,. The species embraces two subgroups i.e., 1A (LMG 24291T = LMG 9904T) and 1B (LMG 10829) that can be differentiated by their 16S rRNA-RFLP pattern. However, some authors, on the basis of the shared intermediate levels of DNA-DNA hybridization, have suggested abandoning the subgroup classification. This contradiction indicates that the taxonomy of this species is not yet resolved. The objective of the present study was to perform a taxonomic evaluation of the diversity of A. cryaerophilus. Genomic information was used along with a Multilocus Phylogenetic Analysis (MLPA) and phenotypic characterization on a group of 52 temporally and geographically dispersed strains, coming from different types of samples and hosts from nine countries. The MLPA analysis showed that those strains formed four clusters (I–IV). Values of Average Nucleotide Identity (ANI) and in silico DNA-DNA Hybridization (isDDH) obtained between 13 genomes representing strains of the four clusters were below the proposed cut-offs of 96 and 70%, respectively, confirming that each of the clusters represented a different genomic species. However, none of the evaluated phenotypic tests enabled their unequivocal differentiation into species. Therefore, the genomic delimited clusters should be considered genomovars of the species A. cryaerophilus. These genomovars could have different clinical importance, since only the cluster I included strains isolated from human specimens. The discovery of at least one stable distinctive phenotypic character would be needed to define each cluster or genomovar as a different species. Until then, we propose naming them “A. cryaerophilus gv. pseudocryaerophilus” (Cluster I = LMG 10229T), “A. cryaerophilus gv. crypticus” (Cluster II = LMG 9065T), “A. cryaerophilus gv. cryaerophilus” (Cluster III = LMG 24291T) and “A. cryaerophilus gv. occultus” (Cluster IV = LMG 29976T).
Introduction
The genus Arcobacter, within the family Campylobacteraceae, was proposed by Vandamme et al. (1991) to reclassify two species that were, at that time, assigned to the genus Campylobacter i.e., Campylobacter nitrofigilis (Arcobacter nitrofigilis, that was selected as the representative or the type species of the genus) and Campylobacter cryaerophila (now Arcobacter cryaerophilus). The phenotypic characteristics that differentiate Campylobacter and Arcobacter are the ability of the latter to grow in aerobic conditions and at lower temperatures (Vandamme et al., 1991; Collado and Figueras, 2011).
Using more than 4000 genomes, Waite et al. (2017) recently analyzed the 16S and 23S rRNA genes and 120 protein sequences and as a result they moved the Epsilonproteobacteria to the phylum level with the name Epsilonbacteraeota. In addition, they created a new family Arcobacteraceae that includes only the genus Arcobacter. Currently, the genus Arcobacter includes 27 species (Park et al., 2016; Whiteduck-Léveillée et al., 2016; Diéguez et al., 2017; Figueras et al., 2017; Tanaka et al., 2017; Pérez-Cataluña et al., 2018), four of which have been linked with human disease: Arcobacter butzleri, A. cryaerophilus, A. thereius, and A. skirrowii (Collado and Figueras, 2011; Figueras et al., 2014; Ferreira et al., 2015). The species A. cryaerophilus has been found in many food products of animal origin (like poultry, pork, lamb, and seafood and in dairy food processing facilities (Collado et al., 2008; Collado and Figueras, 2011).
On the basis of the different Restriction Fragment Length Polymorphism (RFLP) of the 16S and 23S rRNA genes, Kiehlbauch et al. (1991) and Vandamme et al. (1992) divided the species A. cryaerophilus into two subgroups, subgroup 1 or 1A and subgroup 2 or 1B (from here on we will call them subgroups 1A and 1B), represented by strains LMG 24291T (= LMG 9904T) and LMG 10829, respectively. Additionally, it was demonstrated that the two subgroups showed different whole-cell protein and fatty acid contents (Vandamme et al., 1992) and clustered apart by their Amplified Fragment Length Polymorphism (AFLP) patterns (On et al., 2003). A 16S rDNA-RFLP identification method established the separation of the subgroups on the basis of their restriction patterns (Figueras et al., 2008). Despite strains belonging to both subgroups having been found at the same time in animal and human clinical samples and in food products, 1B is generally much more frequently found than 1A (Collado and Figueras, 2011 and references therein). In 2010, Debruyne et al. (2010) reassessed the taxonomy of these two subgroups of A. cryaerophilus using 59 strains isolated mainly from aborted animals (74% of the strains) and human faces (19%). The clustering of the strains obtained by AFLP and by the phylogenetic analysis of the cpn60 gene, together with the shared intermediate levels of DNA-DNA hybridization observed between the strains lead the authors to conclude that despite A. cryaerophilus having a complex taxonomy, the subgroup nomenclature should be abandoned (Debruyne et al., 2010). Furthermore, it was considered that the type strain (LMG 24291T = LMG 9904T) of A. cryaerophilus was not representative of the species because it corresponded with the less abundant 1A subgroup. They therefore proposed that it should be changed for the strain LMG 10829, representative of subgroup 1B (Debruyne et al., 2010). However, a recent metagenomic analysis of Arcobacter populations recovered from sewage samples of the wastewater treatment plant in the city of Reus (Spain) and from various cities of the United States gave evidence that both A. cryaerophilus subgroups (1A and 1B) were dominating in this environment (Fisher et al., 2014). In addition, a different prevalence of the two A. cryaerophilus subgroups was found depending on the wastewater temperature, 1B dominating in wastewater samples with temperatures above 20°C. Fisher et al. (2014) concluded that this finding is relevant because understanding the ecological factors that affect the fate of Arcobacter spp. in wastewater may help to better understand the risks associated with these emerging pathogens. The latter study showed that both subgroups of A. cryaerophilus were abundant and represented two different ecotypes. Therefore, based on those findings, a new polyphasic re-evaluation of the taxonomic diversity of this species is required. The aim of the present study was to investigate the taxonomy of A. cryaerophilus, evaluating strains from 9 different countries recovered from wastewater, different types of shellfish, human faces and various types of animal samples (feces, various viscera from fetuses, uterus, and milk). To our knowledge, this is the most diverse collection of strains of this species studied so far. The polyphasic study involved a phylogenetic analysis of the sequences of the 16S and 23S rRNA genes and of several housekeeping genes, an analysis of 13 genomes (7 of which were obtained in this study) from a representative strains and a phenotypic characterization.
Materials and Methods
Strains Used in This Study
The study included a total of 52 strains that were widely distributed, both geographically and by the type of sample from which they were isolated that, included different host species (humans, pigs, cow, deer, clams, etc.) and environments (water, milk, reclaimed water etc.) as show in Table 1. Six strains possessed their genomes available at the GenBank database, 36 were field isolates from different sources and countries collected over a broad time frame (1985–2013) and 10 strains were from the BCCM/LMG Bacteria Culture Collection (Table 1). Among the latter was the type strain of A. cryaerophilus LMG 24291T that corresponds to subgroup 1A and the reference strain LMG 10829 of the subgroup 1B (Table 1). The 46 strains were re-evaluated or ascribed to subgroups 1A or 1B using the 16S rDNA-RFLP method described by Figueras et al. (2008, 2012). The method consists of the digestion of an amplified fragment (1026 bp) of the 16S rRNA gene with the enzyme MseI, which produces a pattern with different band sizes for subgroup 1A (395, 216, 143, 138 bp) and for subgroup 1B (365, 216, 143, and 138 bp). The RFLP patterns of the six genomes from the GenBank database (genomes L397 to L401 and L406) were obtained by an in silico simulation of the enzymatic digestion using GeneQuest software (DNASTAR, USA). When a different pattern from that expected for A. cryaerophilus was obtained, it was compared with those patterns described for the type strains of all the Arcobacter species by Figueras et al. (2008, 2012). In addition the identity of the strains were confirmed by sequencing the rpoB gene using primers and conditions described in other studies (Collado et al., 2009; Levican et al., 2015).
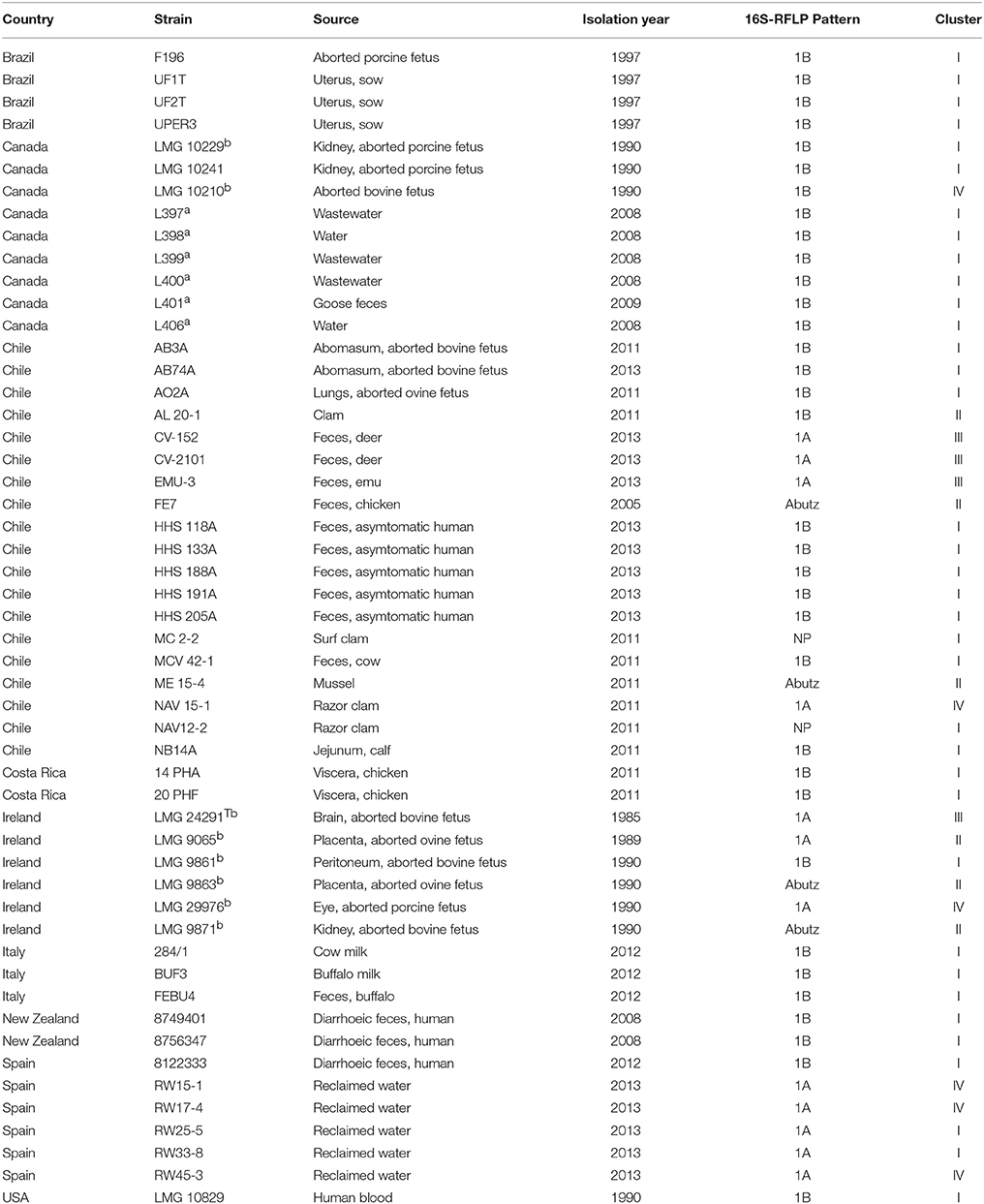
Table 1. Strains used (n = 52) in this study included field isolates, the type and reference collection strains of the species A. cryaerophilus and genomes from the NCBI databasea and 7 obtained in this studyb (accession numbers in Table 2).
Phylogenetic Analysis
A Multilocus Phylogenetic Analysis (MLPA) was carried out by amplifying and sequencing 4 housekeeping genes (gyrB, rpoB, atpA, and cpn60) following protocols described by Levican Asenjo (2013). In addition, these genes and the 16S and 23S rRNA genes were extracted from the 7 obtained genomes and from the 6 downloaded from the GenBank database. Accession number or locus tag of each gene and strain are show in Supplementary Table S1. Genes were aligned (Supplementary Figure S4) using CLUSTALW (Larkin et al., 2007) implemented in MEGA 6 software (Tamura et al., 2013). The same software was used for the phylogenetic analysis using Neighbor-Joining (NJ) algorithm (Kimura, 1980; Saitou and Nei, 1987) and the bootstrap support for individual nodes was calculated with 1,000 replicates.
Whole Genome Sequencing and Analysis
The genome sequence of the type strain of A. cryaerophilus (LMG 24291T) and of six additional strains (LMG 10229T, LMG 9861, LMG 9065T, LMG 9871, LMG 29976T, and LMG 10210) representative of the different MLPA clusters were obtained in the present study using Illumina MiSeq platform (San Diego, CA, USA). The genomic DNA was extracted from pure cultures using the Easy-DNA™ gDNA Purification kit (Invitrogen, Madrid, Spain). Genomic libraries were prepared with the Nextera® XT DNA Sample Preparation Kit (Illumina) following manufacturer's instructions. Genome assembly was carried out with the SPAdes 3.9 (Nurk et al., 2013) and the CGE assemblers (Larsen et al., 2012) and the best results were selected for further analysis. Assembled genomes were annotated using Prokka v1.11 software (Seemann, 2014). Additionally, the protein-encoding sequences (CDS) were annotated using the Rapid Annotation Subsystem Technology (RAST) (Aziz et al., 2008) and the PATRIC server v3.5.2. (Wattam et al., 2017). The general characteristics derived from the NCBI Prokaryotic Genome Automatic Annotation Pipeline (PGAAP) and described for the 13 genomes (6 from the GenBank database and 7 from this study) were: genome size (Mb), number of contigs, N50 (bp), G+C content (%) and the number of predicted CDS. Furthermore, the genomes were compared by the Average Nucleotide Identity (ANI) and the in silico DNA-DNA hybridization (isDDH) indices using OrthoANI (Lee et al., 2015) and Genome-to-Genome Distance Calculator software (Meier-Kolthoff et al., 2013), respectively.
Additionally, a phylogenetic analysis of the 13 genomes (LMG 24291T, LMG 10229T, LMG 9861, L397-L401, L406, LMG 9065T, LMG 9871, LMG 29976T, and LMG 10210) was carried out using the Maximum Likelihood estimation using RAxML (Stamatakis, 2014) with the pipeline implemented in the PATRIC server (Wattam et al., 2017). The genome of A. trophiarum LMG 25534Twas used as outgroup. As a first step, the phylogeny was constructed using a set of homologous proteins identified with BLASTp (Boratyn et al., 2013) and clustered with the Markov Cluster Algorithm (MCL) (Dongen, 2000). The second step was an alignment of the protein set using MUSCLE (Edgar, 2004) and the Hidden Markov Models (HMM) were constructed with HMMER tools (Eddy, 1998).
Virulence and Antibiotic Resistance Genes
Virulence genes were searched by BLASTn analysis with default parameters using the Virulence Factors of Pathogenic Bacteria Database (VFDB) (Chen et al., 2005), Victors Database (University of Michigan, USA) and PATRIC_VF (Wattam et al., 2017). Antibiotic resistance genes were searched using the Antibiotic Resistance Database (ARDB) (Liu and Pop, 2009) and the Comprehensive Antibiotic Resistance Database (CARD) (Jia et al., 2017). The five mentioned databases are included at the Specialty Genes tool available at the PATRIC server (Wattam et al., 2017). Furthermore, the Antibiotic Resistance Gene-Annotation database (ARG-ANNOT) (Gupta et al., 2014) was also used to search antibiotic resistance genes by BLASTp analysis using default parameters and the database ARG-ANNOT AA V3 (March 2017). Virulence and resistance mechanisms were also searched for with RAST (Aziz et al., 2008) and PATRIC servers (Wattam et al., 2017). Additionally, genes related with the virulence of Arcobacter (Collado and Figueras, 2011; Douidah et al., 2012; Levican et al., 2013a) were searched for with BLASTn using sequences obtained from GenBank and from the annotated Arcobacter genomes of A. butzleri RM4018, A. nitrofigilis DSM 7299 and Arcobacter sp. L. The genes studied were cadF and cj1349, which encode two fibronectin binding proteins; ciaB encodes the invasion protein CiaB, mviN gene related to peptidoglycan synthesis; pldA gene encodes a phospholipase; tlyA gene codifies for a hemolysine; hecB related to hemolysis activation; hecA gene that encodes an adhesion protein and finally the gene irgA that codifies an iron-regulated outer membrane protein (Collado and Figueras, 2011; Douidah et al., 2012; Levican et al., 2013a). The accession number or locus tag of those genes are show in Supplementary Table S2. A phylogenetic analysis was conducted using the three virulence genes (cj1349, mviN, and pldA) present in all the studied genomes to evaluate their genetic relatedness and evolution.
Comparison of the Genome Derived Metabolic and Phenotypic Information
The genomes of the seven representative strains from each cluster (LMG 10229T, LMG 9861, LMG 9065T, LMG 9871, LMG 24291T, LMG 29976T, and LMG 10210) were compared using the Functional Comparison Tool implemented in the Seed Viewer (Overbeek et al., 2014). This software uses the protein sequences of each compared genome annotated with RAST (Aziz et al., 2008) and reconstructs the metabolic pathways. On the other hand, the phenotypic traits derived from each genome were obtained with Traitar software (Weimann et al., 2016) using the protein annotations obtained with Prokka v1.2 (Seemann, 2014). This software infers phenotypic traits using data from the Global Infectious Disease and Epidemiology Online Network (GIDEON) and from the Bergey's Systematic Bacteriology (Goodfellow et al., 2012). The software works with a total of 67 traits that embrace different microbiological or biochemical characteristics involved in enzyme activity, growth, oxygen requirements, morphology, and hydrogen sulfide production (Weimann et al., 2016).
Phenotypic Characterization
Phenotypic characterization of the 46 strains included 9 tests recommended in the guidelines for defining new species of the family Campylobacteraceae (Ursing et al., 1994; On et al., 2017) and 7 additional tests used in the description of other Arcobacter spp. (Donachie et al., 2005; Houf et al., 2005). Most of these tests were chosen using as a criterion the biochemical tests that gave variable results for both A. cryaerophilus subgroups in the previous study by On (1996), in which a total of 67 phenotypic tests were analyzed from 9 and 10 strains of subgroups 1A and 1B, respectively. Growth conditions on blood agar were tested (BD Difco, NJ, USA) at 37° and 42°C at three different atmospheres: aerobic, microaerobic, and anaerobic conditions. The biochemical properties were tested at 30°C in aerobic conditions for the 46 strains using positive and negative controls in parallel for each specific test. To evaluate inter laboratory reproducibility, the strains LMG 9065, LMG 9861, LMG 9871, LMG 10229 and LMG 24291Twere tested in parallel in two different laboratories in different countries (Chile and Spain).
Results and Discussion
Molecular Identification and Phylogeny
Table 1 shows that 46 of the 52 strains gave RFLP patterns defined by Figueras et al. (2008) for A. cryaerophilus and 4 showed the one for A. butzleri (FE7, ME15-4, LMG 9863, and LMG 9871). However, strains NAV12-2 and MC2-2 produced a new RFLP pattern different to the described ones (Figueras et al., 2008, 2012). From the 46 strains that gave the pattern of A. cryaerophilus, 34 gave the pattern of the subgroup 1B (including the in silico simulated patterns obtained from the 16S rRNA genes of the 6 GenBank genomes L397- L401 and L406) and 12 the one of the subgroup 1A. This demonstrated once more that subgroup 1B is more abundant than 1A, in agreement with results of previous studies (Debruyne et al., 2010; Collado and Figueras, 2011; Fisher et al., 2014). As Figueras et al. (2012) explained when describing the 16S rDNA-RFLP identification method, different RFLP patterns from those expected for the Arcobacter spp. can obtained for new species or might be due to the existence of a mutation on the targeted site of the endonucleases in a known species. The former occurred for instance in A. mytili (Collado et al., 2009) and A. molluscorum (Figueras et al., 2011a) among other species (Figueras et al., 2011b; Levican et al., 2012, 2013b, 2015). Mutations at the binding site of the endonuclease MseI were described in the strains LMG 9863 and LMG 9871 (used in this study, Table 1), but in this case instead of resulting in a new pattern they were responsible for generating the pattern for A. butzleri instead of A. cryaerophilus (Figueras et al., 2012).
The MLPA with the concatenated sequences (2,408 bp) of the four housekeeping genes (gyrB, rpoB, atpA, and cpn60) of the 52 strains showed that they grouped into four main clusters (Figure 1). Cluster I had 36 strains, most of them (88.8%) from the subgroup 1B, and included the reference strain for the 1B subgroup LMG 10829. The other four strain of this cluster presented the pattern of subgroup 1A (n = 2) and a different pattern to those described (n = 2). Cluster II (n = 6) corresponded to the four strains that showed a 16S rDNA-RFLP pattern similar to the one described for A. butzleri (Figueras et al., 2008) and two other strains with the patterns for the subgroups 1A and 1B. Cluster III, included the type strain of A. cryaerophilus LMG 24291T and three field isolates from Chilean animals all belonging to the subgroup 1A, and Cluster IV comprised six strains, mostly from subgroup 1A (n = 5). Interestingly, strains recovered from human specimens belonged exclusively to Cluster I, suggesting potential host specificity because strains associated with farm animal abortions were present in the four clusters (Figure 1).

Figure 1. Neighbor joining tree based on the concatenated sequences of gyrB, rpoB, atpA, and hsp60 (2,408 bp) genes showing the distribution of the 52 strains in four clusters. Bootstrap values (>50%) based on 1,000 replications are shown at the nodes of the tree. Bar indicates 1 substitutions per 100 bp. Isolation source: •, Human; ◦, Animal; ■, Shellfish; ▴, Water.
A representative type strain was selected from each cluster (I–IV) for further analysis and for constructing a 16S rRNA gene phylogenetic tree (Figure 2). The tree showed that the four strains formed separated branches, strains LMG 24291T and LMG 29976T being the nearest ones. The percentage of similarity of the 16S rRNA gene between the type strains ranged from 99.5% between strains LMG 10229T (Cluster I) and LMG 9065T (Cluster II) to 99.9% between the original type strain of A. cryaerophilus LMG 24291T (Cluster III) and the representative strain of Cluster IV (LMG 29976T). These results agree with what occurs between other species of Arcobacter, such as A. ellisii and A. cloacae (Figueras et al., 2011b; Levican et al., 2013b), where the 16S rRNA gene does not have enough resolution to differentiate the species. The phylogeny of the 23S rRNA gene (Supplementary Figure S1) and the one carried out with the concatenated sequences of the two rRNA genes (Supplementary Figure S2) presented the same topology shown with the 16S rRNA gene (Figure 2) and confirmed that the strains of Cluster III are more closely related to Cluster IV than to the other clusters.
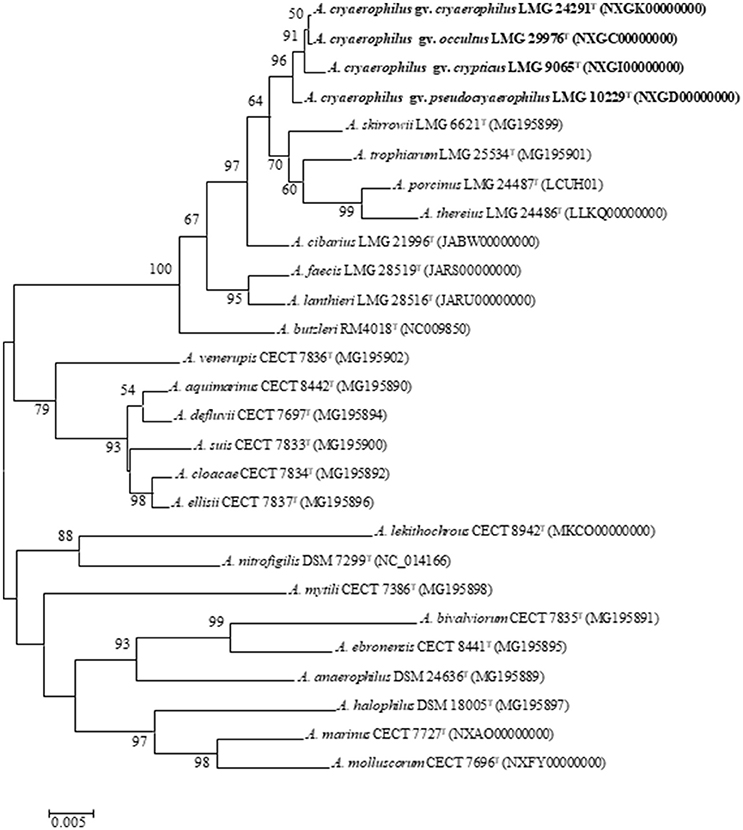
Figure 2. Neighbor joining tree based on 16S rRNA (1,496 bp) sequences showing the phylogenetic position of the representative strains of the four clusters of A. cryaerophilus within the genus Arcobacter. Bootstrap values (>50%) based on 1,000 replications are shown at the nodes of the tree. Bar, 5 substitutions per 1,000 bp.
Genome Analysis
The characteristics of the 13 compared genomes (8 representatives of Cluster I, two of clusters II and IV and one of Cluster III) are shown in Table 2. The quality of the genome sequences was in general in agreement with the minimal standards established for the use of genome data for taxonomical purposes, that embraces characteristics of the sequencing and assembly of the genomes like the depth of coverage, the value of N50 and the number of contigs (Chun et al., 2018). The exceptions were the genome sequence data of strains LMG 10229T, LMG 9871, and LMG 9861 that presented a depth of coverage lower than 50X proposed in the standards (Table 2). Globally, the genomic characteristics of the 13 compared genomes shown in Table 2 were very similar, with sizes that did not differ in more than 0.29 Mb, with a %mol G+C content ranging between 27.0 and 30.0% and with a number of coding sequences or CDS of around 2000 (Table 2). The G+C values were in agreement with those (24.6–31%) described in the recent emended description of the genus Arcobacter (Sasi Jyothsna et al., 2013). Table 3 shows the results from the calculated overall genome related taxonomical indices i.e., ANI and isDDH. For species delineation the generally accepted ANI and isDDH boundary values are 95–96 and 70%, respectively (Goris et al., 2007; Richter and Rossello-Mora, 2009; Meier-Kolthoff et al., 2013; Chun et al., 2018). However, for the genus Arcobacter, ANI values above 96% were the ones that better correlated with isDDH results above 70% in previous studies (Figueras et al., 2017; Pérez-Cataluña et al., 2018) in agreement with what happens in other genera (Beaz-Hidalgo et al., 2015; Figueras et al., 2017; Liu et al., 2017). The ANI values of the representative strains from each of the four different clusters were below the 96% cut-off indicating that the compared genomes belonged to different species, while the intra-cluster ANI values ranged from 96.6 to 98.6%. The isDDH results of <70% found between strains of the four clusters confirmed as the ANI results did that each cluster represented an independent species. The core genome phylogenetic tree inferred from 893 protein sequences of the 13 genomes obtained with PATRIC showed that the genomes also grouped into four different well-supported clusters with bootstraps of 100% (Figure 3). Interestingly, clusters IV and I formed a separate branch from clusters II and III. This indicates that the proteins of the genomes of clusters I and IV are more similar than the nucleotide sequences, and this was also in agreement with the higher values observed with ANI and isDDH for these two clusters.
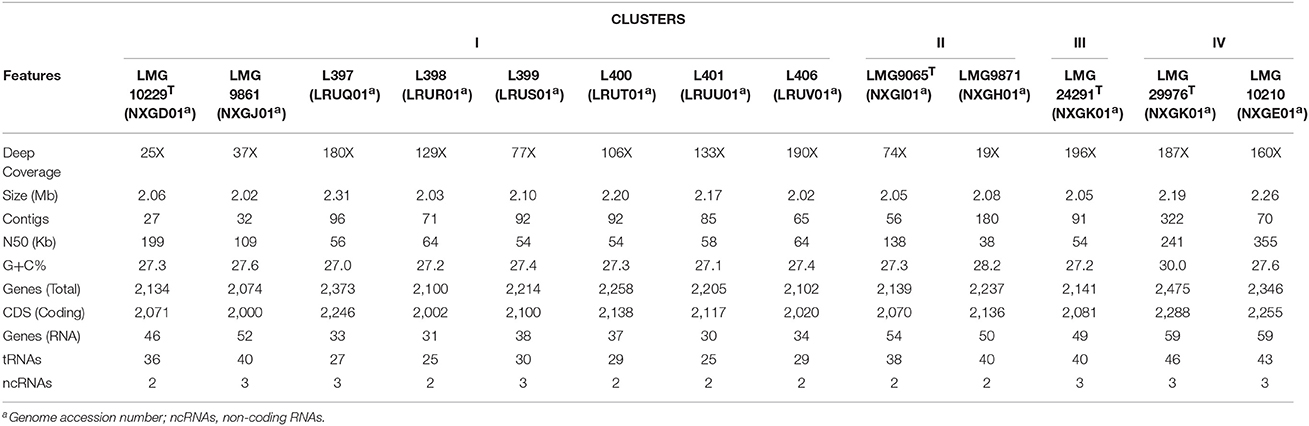
Table 2. Characteristics of the 13 genomes from representative strains from each of the clusters.
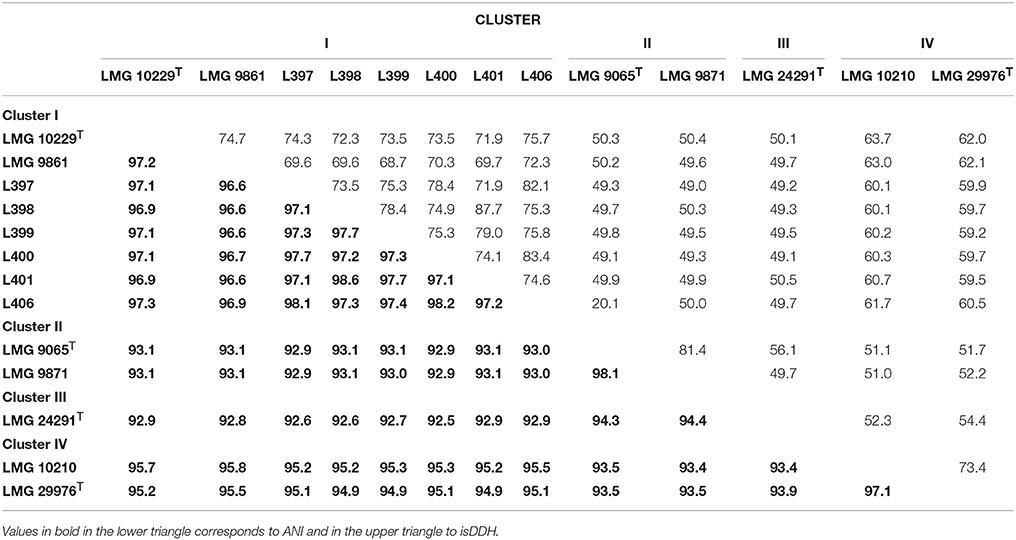
Table 3. Results of Average Nucleotide Identity (ANI) and in silico DNA-DNA hibridization (isDDH) between representative genomes of the four clusters.
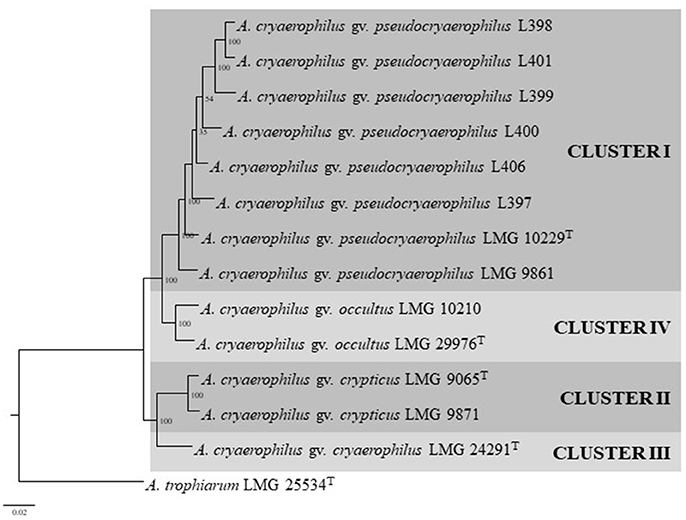
Figure 3. Maximum likelihood tree based on the concatenated sequences of 893 core protein sequences showing the distribution of the 13 genomes used in the same four clusters shown in Figure 1. Boostrap values based on 1,000 replications are shown at the nodes of the tree. Bar indicates 2 substitution per 100 aa.
Virulence and Antibiotic Resistance Genes
Of the different methods and databases used for recognizing virulence factors (Victors, VFDB and PATRIC_VF) none of them were useful for recognizing virulence genes. There were only a few exceptions. The phospholipase C identified with the databases PATRIC_VF and Victors in the genome of the strain LMG 29976T (Cluster IV). The enzyme UDP-N-acetylglucosamine 4-6 dehidratase involved in flagelline glycosylation and identified with the VFDB database in the genomes L397 and L399 (Cluster I). Finally, the Pspa protein (EC 2.3.1.41), essential for gluconeogenesis, identified using PATRIC_VF in the genome LMG 9871 of Cluster II (Table 4). However the BLASTn carried out for the detection of virulence genes showed the presence of different genes related with adhesion (cj1349), invasion (ciaB and mviN) and phospholipase activity (pldA) (Table 4). None of the genomes showed the cadF, hecA and hecB genes that encode a fibronectin binding protein, an adhesion protein and a factor for hemolysis activation, respectively. These results agree with those obtained for the genome of A. thereius LMG 24486T (Rovetto et al., 2017). The irgA and tlyA genes that encode an iron-regulated outer membrane protein and a hemolysine, respectively, were the only ones found i.e. the gene irgA in the genomes L398 and L401 (Cluster I); the gene tlyA in LMG 24291T (Cluster III) and LMG 9065T (Cluster II). The phylogenetic analysis of the concatenated sequences of the four virulence genes present in all the genomes (cj1349, mviN, pldA and ciaB) formed the same four clusters (Supplementary Figure S3). However, the distribution of the clusters was similar to the one obtained with the core genome tree (Figure 3), where clusters I and IV formed a separated branch from clusters II and III.

Table 4. Antibiotic resistant genes and virulence factors.
Regarding the presence of antibiotic resistant mechanisms, all the genomes showed the cmeABC multidrug efflux pump, the MacAB-TolC system for macrolide resistance, the oxqB gene related with quinolone resistance and genes related with the resistance to acriflavine. Resistance to colistin by the genes mcr-1 and mcr-2 were present in 85% of the genomes. The genome L406 was the only one that possessed a β-lactamase gene of class D. Resistance to β-lactamic compounds have been reported in other studies (Atabay and Aydin, 2001; Fera et al., 2003) and the same β-lactamase gene is present in the genome of A. butleri RM4018. However, this gene is absent in the genome of A. thereius LMG 24486T (Rovetto et al., 2017). The genome LMG 29976 was the only one that presented genes for the resistance to streptomycin/spectomycin. The susceptibility of A. cryaerophilus to streptomycin has been previously demonstrated (Kabeya et al., 2004; Rahimi, 2014). However, this is the first report that show the presence of resistance genes to this antimicrobial compound. Mutations on the 23S rRNA (Ren et al., 2011) and the gyrA gene (Carattoli et al., 2002) for erythromycin and quinolone resistance were not detected, despite gyrA mutations have been found in some quinolone-resistant A. cryaerophilus strains (Abdelbaqi et al., 2007; Van den Abeele et al., 2016).
Functional and Phenotypic Inference
Several subsystems where found to be characteristic of each Cluster on the basis of the functional-based comparison between the representative genomes (Figure 4). Cluster I genomes (LMG 10229T and LMG 9861) carry specifically multi-subunit cation antiporters [Na(+) H(+) cation antiporter ABCDEFG] whose function includes sodium tolerance and pH homeostasis in an alkaline environment (Ito et al., 2017). Cluster II genomes (LMG 9065T and LMG 9871) were the only ones that did not show the chromate transport protein ChrA, which confers resistance to chromate compounds present in the other studied genomes. Cluster III (LMG 24291T) was the only one that presented transposable elements as the putative transposase TniA and the Nucleotide Triphosphate binding protein TniB. Finally, the enzyme Adenosine deaminase (EC 3.5.4.4) involved in purine metabolism was only detected in Cluster IV genomes (LMG 29976T and LMG 10210).

Figure 4. Function based comparison between the representative genomes of each cluster using RAST annotation results. Black squares represent de presence and white squares the absence of each subsystem/protein. 1-3 Polar lipids genes: 1. Phosphatidilglycerol phosphatase A (pspA); 2. phosphatidate cytidylyltransferase (cdsA); 3. phosphatidyl serine decarboxylase (psd). Genes pspA and cdsA are involved in the synthesis of phosphatidilglycerol (PG) and psd gene in the synthesis of phosphatidylethanolamine (PE).
From the 67 phenotypic inferred traits analyzed with Traitar 11(16.4%) were found in all the analyzed genomes while 12 were only found in some of them (Figure 5). The genomes of Cluster I (LMG 10229T and LMG 9861) were predicted to produce hydrogen sulfide while those of Cluster IV (LMG 29976T and LMG 10210) showed acetate utilization and bile susceptibility. However, none of these characteristics have been observed when they have been tested in the laboratory on those strains. This might be due to the inability to reproduce the necessary conditions in the laboratory for the expression of these features. None of the other nine traits recognized in some genomes enabled us to differentiate between the IV Clusters.
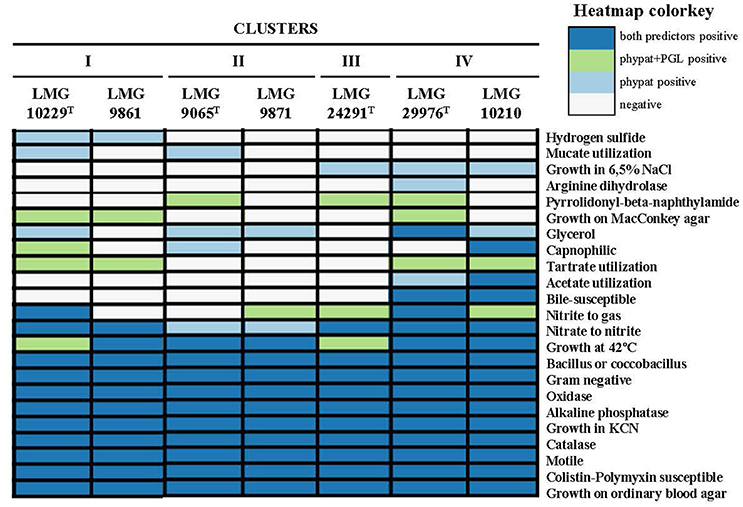
Figure 5. Phenotypic inference using Traitar software for the representative genomes of each cluster. The software uses two prediction models, the phypat model (predicts the presence/absence of proteins found in the phenotype of 234 bacterial species) and a combination of phypat+PGL models (uses the information of phypat combined with the information of the acquisition or loss of protein families and phenotypes through the evolution), to determine the phenotypic characteristics.
Phenotypical Characterization
Table 5 shows the phenotypical results obtained from the strains of each of the four clusters. In agreement with what was found in previous studies where phenotypic test did not differentiate between subgroups 1A and 1B (Neill et al., 1985; Vandamme et al., 1992; On, 1996), none of the performed phenotypic tests enabled to clearly distinguish strains from each of the four phylogenetic clusters. Most of the tests gave variable results except for Cluster IV. However, this might be due to the small number of strains (n = 2) analyzed in this group. Considering these results, each of the three genetically recognized new species (clusters I, II, and IV) should be considered a different genomovar (gv.) of the species A. cryaerophilus. A genomovar is a well-delimited group of strains that correspond to a new species by genomic information but that cannot be phenotypically differentiated (Ursing et al., 1995). Cluster III represents the original species A. cryaerophilus because it embraces the type strain of the species. The value of the phenotypic characterization has already been questioned considering the lack of reproducibility of results between laboratories and some authors have suggested it is now time to base the description of new taxa on the genome sequence analysis (Moore et al., 2010; Sutcliffe, 2015). According to Sutcliffe (2015), phenotypic characterization is harder to evaluate nowadays than the genotype. Considering that genomic characterization is objective and reproducible, we agree with Sutcliffe (2015) that we should be able to define species on the basis of genetic characters like the ones evaluated in this study. This will favor the faster discover of the large number of taxa waiting to be described (Sutcliffe, 2015). However, this will require a modification of the Bacteriological Code, which we hope will happen in the near future.
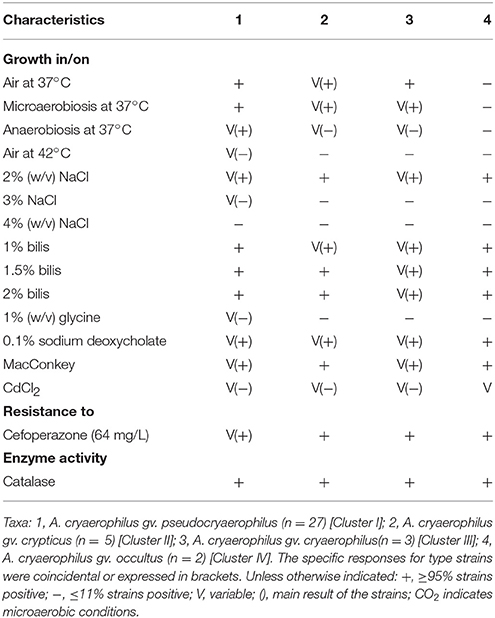
Table 5. Phenotypic characteristics of the four clusters.
Conclusion
The phylogenetic and genomic analysis showed that the strains of the species A. cryaerophilus represent four separated species. In addition, phenotypical and functional traits were in evidence for the genomes selected as representative of each cluster. Despite all the results, phenotypic characterization carried out at the laboratory showed a high inter- and intra-cluster variability that did not allow us to determine specific phenotypic characteristics or therefore to define the three uncovered clusters as three new species. Following current bacterial taxonomic rules, we will not be able to define these species until we find phenotypical characteristics that allow us to discriminate the three new species from each other and from the species A. cryaerophilus. Therefore, we describe them as four genomovars with the names “A. cryaerophilus gv. pseudocryaerophilus” (pseu.do.cry.a.e.ro'phi.lus. Gr. adj. pseudês false, N.L. masc. adj. cryaerophilus specific epithet of an Arcobacter species; N.L. masc. adj. pseudocryaerophilus false cryaerophilus; Cluster I = LMG 10229T), “A. cryaerophilus gv. crypticus” (cryp'ti.cus. L. masc. adj. Crypticus hidden; Cluster II = LMG 9065T), A. cryaerophilus gv. cryaerophilus (Cluster III = LMG 24291T) and “A. cryaerophilus gv. occultus” (oc.cul′tus. L. adj. occultus occulted, hidden; Cluster IV = LMG 29976T). Unfortunately, the phenotype derived from the genome could not be reproduced in the laboratory, either. This might be due to the inability to mimic in vitro the conditions for the expression of these pathways or characteristics. The phenotypic characterization limits a proper description and it might be considered an important shortcoming in the genomic era in which all the molecular and genomic data leave no doubts about the existence of four different species among the investigated A. cryaerophilus strains.
Authors Contributions
LC and MF: designed the work; LC and OS: carried out the phylogenetic analysis; VL and AP-C: carried out the phenotypic characterization of the strains; AP-C: carried out the genome sequencing and analysis; LC, MF, and AP-C: wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Maria Laura Arias (University of Costa Rica), Dr. Mary Nulsen (Massey University), Dr. Andrea Serraino (University of Bologna) and Dr. Sergio Oliveira (ULBRA University of Brazil) for kindly providing Arcobacter strains. We thank Prof. Aharon Oren from the Hebrew University of Jerusalem for supervising and correcting the species name etymology. This work was supported in part by the project DID-UACh S-2013-06 from the Universidad Austral de Chile and by the projects JPIW2013-69 095-C03-03 of MINECO (Spain) and AQUAVALENS of the Seventh Framework Program (FP7/2007-2013) grant agreement 311846 from the European Union. AP-C thanks Institut d'Investigació Sanitária Pere Virgili (IISPV) for her PhD fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00805/full#supplementary-material
Abbreviations
LMG, Laboratorium voor Microbiologie, Universiteit Gent, Belgium Culture Collection; MLPA, Multilocus Phylogenetic Analysis; ANI, Average Nucleotide Identity; isDDH, in silico DNA-DNA hybridization.
References
Abdelbaqi, K., Ménard, A., Prouzet-Mauleon, V., Bringaud, F., Lehours, P., and Mégraud, F. (2007). Nucleotide sequence of the gyrA gene of Arcobacter species and characterization of human ciprofloxacin-resistant clinical isolates. FEMS Immunol. Med. Microbiol. 49, 337–345. doi: 10.1111/j.1574-695X.2006.00208.x
Atabay, H. I., and Aydin, F. (2001). Susceptibility of Arcobacter butzleri isolates to 23 antimicrobial agents. Lett. Appl. Microbiol. 33, 430–433. doi: 10.1046/j.1472-765X.2001.01025.x
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Beaz-Hidalgo, R., Hossain, M. J., Liles, M. R., and Figueras, M.-J. (2015). Strategies to avoid wrongly labelled genomes using as example the detected wrong taxonomic affiliation for Aeromonas genomes in the GenBank database. PLoS ONE 10:e0115813. doi: 10.1371/journal.pone.0115813
Boratyn, G. M., Camacho, C., Cooper, P. S., Coulouris, G., Fong, A., Ma, N., et al. (2013). BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 41, W29–W33. doi: 10.1093/nar/gkt282
Carattoli, A., Dionisi, A., and Luzzi, I. (2002). Use of a LightCycler gyrA mutation assay for identification of ciprofloxacin-resistant Campylobacter coli. FEMS Microbiol. Lett. 214, 87–93. doi: 10.1128/JCM.39.4.1443-1448.2001
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Collado, L., Cleenwerck, I., Van Trappen, S., De Vos, P., and Figueras, M. J. (2009). Arcobacter mytili sp. nov., an indoxyl acetate-hydrolysis-negative bacterium isolated from mussels. Int. J. Syst. Evol. Microbiol. 59, 1391–1396. doi: 10.1099/ijs.0.003749-0
Collado, L., and Figueras, M. J. (2011). Taxonomy, epidemiology, and clinical relevance of the genus Arcobacter. Clin. Microbiol. Rev. 24, 174–192. doi: 10.1128/CMR.00034-10
Collado, L., Inza, I., Guarro, J., and Figueras, M. J. (2008). Presence of Arcobacter spp. in environmental waters correlates with high levels of fecal pollution. Environ. Microbiol. 10, 1635–1640. doi: 10.1111/j.1462-2920.2007.01555.x
Debruyne, L., Houf, K., Douidah, L., De Smet, S., and Vandamme, P. (2010). Reassessment of the taxonomy of Arcobacter cryaerophilus. Syst. Appl. Microbiol. 33, 7–14. doi: 10.1016/j.syapm.2009.10.001
Diéguez, A. L., Balboa, S., Magnesen, T., and Romalde, J. L. (2017). Arcobacter lekithochrous sp. nov., isolated from a molluscan hatchery. Int. J. Syst. Evol. Microbiol. 67, 1327–1332. doi: 10.1099/ijsem.0.001809
Donachie, S. P., Bowman, J. P., On, S. L. W., and Alam, M. (2005). Arcobacter halophilus sp. nov., the first obligate halophile in the genus Arcobacter. Int. J. Syst. Evol. Microbiol. 55, 1271–1277. doi: 10.1099/ijs.0.63581-0
Dongen, S. M. van. (2000). Graph Clustering by Flow Simulation. PhD thesis, University of Utrech, Utrech.
Douidah, L., De Zutter, L., Baré, J., De Vos, P., Vandamme, P., Vandenberg, O., et al. (2012). Occurrence of putative virulence genes in Arcobacter species isolated from humans and animals. J. Clin. Microbiol. 50, 735–741. doi: 10.1128/JCM.05872-11
Eddy, S. R. (1998). Profile hidden Markov models. Bioinformatics 14, 755–763. doi: 10.1093/bioinformatics/14.9.755
Edgar, R. C. (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113
Fera, M., Carbone, M., Maugeri, T. L., Giannone, M., Gugliandolo, C., La Camera, E., et al. (2003). In vitro susceptibility of Arcobacter butzleri and Arcobacter cryaerophilus to different antimicrobial agents. Int. J. Antimicrob. Agents 21, 488–491. doi: 10.1016/S0924-8579(03)00004-9
Ferreira, S., Queiroz, J. A., Oleastro, M., and Domingues, F. C. (2015). Insights in the pathogenesis and resistance of Arcobacter : a review. Crit. Rev. Microbiol. 42, 364–383. doi: 10.3109/1040841X.2014.954523
Figueras, M. J., Collado, L., and Guarro, J. (2008). A new 16S rDNA-RFLP method for the discrimination of the accepted species of Arcobacter. Diagn. Microbiol. Infect. Dis. 62, 11–15. doi: 10.1016/j.diagmicrobio.2007.09.019
Figueras, M. J., Collado, L., Levican, A., Perez, J., Solsona, M. J., and Yustes, C. (2011a). Arcobacter molluscorum sp. nov., a new species isolated from shellfish. Syst. Appl. Microbiol. 34, 105–109. doi: 10.1016/j.syapm.2010.10.001
Figueras, M. J., Levican, A., and Collado, L. (2012). Updated 16S rRNA-RFLP method for the identification of all currently characterised Arcobacter spp. BMC Microbiol. 12:292. doi: 10.1186/1471-2180-12-292
Figueras, M. J., Levican, A., Collado, L., Inza, M. I., and Yustes, C. (2011b). Arcobacter ellisii sp. nov., isolated from mussels. Syst. Appl. Microbiol. 34, 414–418. doi: 10.1016/j.syapm.2011.04.004
Figueras, M. J., Levican, A., Pujol, I., Ballester, F., Rabada Quilez, M. J., and Gomez-Bertomeu, F. (2014). A severe case of persistent diarrhoea associated with Arcobacter cryaerophilus but attributed to Campylobacter sp. and a review of the clinical incidence of Arcobacter spp. New microbes new Infect. 2, 31–37. doi: 10.1002/2052-2975.35
Figueras, M. J., Pérez-Cataluña, A., Salas-Massó, N., Levican, A., and Collado, L. (2017). “Arcobacter porcinus” sp. nov., a novel Arcobacter species uncovered by Arcobacter thereius. New Microbes New Infect. 15, 104–106. doi: 10.1016/j.nmni.2016.11.014
Fisher, J. C., Levican, A., Figueras, M. J., and McLellan, S. L. (2014). Population dynamics and ecology of Arcobacter in sewage. Front. Microbiol. 5:525. doi: 10.3389/fmicb.2014.00525
Goodfellow, M., Kämpfer, P., Busse, H. J., Trujillo, M. E., Suzuki, K. I., Ludwig, W., et al. (2012). Bergey's Manual of Systematic Bacteriology. New York, NY: Springer.
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., et al. (2014). ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 58, 212–220. doi: 10.1128/AAC.01310-13
Houf, K., On, S. L. W., Coenye, T., Mast, J., Van Hoof, J., and Vandamme, P. (2005). Arcobacter cibarius sp. nov., isolated from broiler carcasses. Int. J. Syst. Evol. Microbiol. 55, 713–717. doi: 10.1099/ijs.0.63103-0
Ito, M., Morino, M., and Krulwich, T. A. (2017). Mrp antiporters have important roles in diverse bacteria and archaea. Front. Microbiol. 8:2325. doi: 10.3389/FMICB.2017.02325
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Kabeya, H., Maruyama, S., Morita, Y., Ohsuga, T., Ozawa, S., Kobayashi, Y., et al. (2004). Prevalence of Arcobacter species in retail meats and antimicrobial susceptibility of the isolates in Japan. Int. J. Food Microbiol. 90, 303–308. doi: 10.1016/S0168-1605(03)00322-2
Kiehlbauch, J. A., Plikaytis, B. D., Swaminathan, B., Cameron, D. N., and Wachsmuth, I. K. (1991). Restriction fragment length polymorphisms in the ribosomal genes for species identification and subtyping of aerotolerant Campylobacter species. J. Clin. Microbiol. 29, 1670–1676.
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50, 1355–1361. doi: 10.1128/JCM.06094-11
Lee, I., Kim, Y. O., Park, S.-C., and Chun, J. (2015). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Levican, A., Alkeskas, A., Günter, C., Forsythe, S. J., and Figueras, M. J. (2013a). Adherence to and invasion of human intestinal cells by Arcobacter species and their virulence genotypes. Appl. Environ. Microbiol. 79, 4951–4957. doi: 10.1128/AEM.01073-13
Levican, A., Collado, L., Aguilar, C., Yustes, C., Diéguez, A. L., Romalde, J. L., et al. (2012). Arcobacter bivalviorum sp. nov. and Arcobacter venerupis sp. nov., new species isolated from shellfish. Syst. Appl. Microbiol. 35, 133–138. doi: 10.1016/j.syapm.2012.01.002
Levican, A., Collado, L., and Figueras, M. J. (2013b). Arcobacter cloacae sp. nov. and Arcobacter suis sp. nov., two new species isolated from food and sewage. Syst. Appl. Microbiol. 36, 22–27. doi: 10.1016/j.syapm.2012.11.003
Levican, A., Rubio-Arcos, S., Martinez-Murcia, A., Collado, L., and Figueras, M. J. (2015). Arcobacter ebronensis sp. nov. and Arcobacter aquimarinus sp. nov., two new species isolated from marine environment. Syst. Appl. Microbiol. 38, 30–35. doi: 10.1016/j.syapm.2014.10.011
Levican Asenjo, A. (2013). Sanitary Importance of Arcobacter. PhD thesis, Universitat Rovira I Virgili, Reus.
Liu, B., and Pop, M. (2009). ARDB–Antibiotic Resistance Genes Database. Nucleic Acids Res. 37, D443–D447. doi: 10.1093/nar/gkn656
Liu, Y., Lai, Q., Du, J., and Shao, Z. (2017). Genetic diversity and population structure of the Bacillus cereus group bacteria from diverse marine environments. Sci. Rep. 7:689. doi: 10.1038/s41598-017-00817-1
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Moore, E. R. B., Mihaylova, S. A., Vandamme, P., Krichevsky, M. I., and Dijkshoorn, L. (2010). Microbial systematics and taxonomy: relevance for a microbial commons. Res. Microbiol. 161, 430–438. doi: 10.1016/j.resmic.2010.05.007
Neill, S. D., Campbell, J. N., O'Brien, J. J., Weatherup, S. T. C., and Ellis, W. A. (1985). Taxonomic position of Campylobacter cryaerophila sp. nov. Int. J. Syst. Bacteriol. 35, 342–356. doi: 10.1099/00207713-35-3-342
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 20, 714–737. doi: 10.1089/cmb.2013.0084
On, S. L. (1996). Identification methods for campylobacters, helicobacters, and related organisms. Clin. Microbiol. Rev. 9, 405–422.
On, S. L. W., Harrington, C. S., and Atabay, H. I. (2003). Differentiation of Arcobacter species by numerical analysis of AFLP profiles and description of a novel Arcobacter from pig abortions and turkey faeces. J. Appl. Microbiol. 95, 1096–1105. doi: 10.1046/j.1365-2672.2003.02100.x
On, S. L. W., Miller, W. G., Houf, K., Fox, J. G., and Vandamme, P. (2017). Minimal standards for describing new species belonging to the families Campylobacteraceae and Helicobacteraceae: campylobacter, Arcobacter, Helicobacter and Wolinella spp. Int. J. Syst. Evol. Microbiol. 67, 5296–5311. doi: 10.1099/ijsem.0.002255
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, 206–214. doi: 10.1093/nar/gkt1226
Park, S., Jung, Y. T., Kim, S., and Yoon, J. H. (2016). Arcobacter acticola sp. nov isolated from seawater on the East Sea in South Korea. J. Microbiol. 54, 655–659. doi: 10.1007/s12275-016-6268-4
Pérez-Cataluña, A., Salas-Massó, N., and Figueras, M. J. (2018). Arcobacter canalis sp. nov., isolated from a water canal contaminated with urban sewage. Int. J. Syst. Evol. Microbiol. 68, 1258–1264 doi: 10.1099/ijsem.0.002662
Rahimi, E. (2014). Prevalence and antimicrobial resistance of Arcobacter species isolated from poultry meat in Iran. Br. Poult. Sci. 55, 174–180. doi: 10.1080/00071668.2013.878783
Ren, G. W. N., Wang, Y., Shen, Z., Chen, X., Shen, J., and Wu, C. (2011). Rapid detection of point mutations in domain V of the 23S rRNA gene in erythromycin-resistant Campylobacter isolates by pyrosequencing. Foodborne Pathog. Dis. 8, 375–379. doi: 10.1089/fpd.2010.0676
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rovetto, F., Carlier, A., Van den Abeele, A.-M., Illeghems, K., Van Nieuwerburgh, F., Cocolin, L., et al. (2017). Characterization of the emerging zoonotic pathogen Arcobacter thereius by whole genome sequencing and comparative genomics. PLoS ONE 12:0180493. doi: 10.1371/journal.pone.0180493
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Sasi Jyothsna, T. S., Rahul, K., Ramaprasad, E. V. V., Sasikala, C., and Ramana, C. V. (2013). Arcobacter anaerophilus sp. nov., isolated from an estuarine sediment and emended description of the genus Arcobacter. Int. J. Syst. Evol. Microbiol. 63, 4619–4625. doi: 10.1099/ijs.0.054155-0
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sutcliffe, I. C. (2015). Challenging the anthropocentric emphasis on phenotypic testing in prokaryotic species descriptions: rip it up and start again. Front. Genet. 6:218. doi: 10.3389/fgene.2015.00218
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tanaka, R., Cleenwerck, I., Mizutani, Y., Iehata, S., Bossier, P., and Vandamme, P. (2017). Arcobacter haliotis sp. nov., isolated from abalone species Haliotis gigantea. Int. J. Syst. Evol. Microbiol. 67, 3050–3056. doi: 10.1099/ijsem.0.002080
Ursing, J. B., Lior, H., and Owen, R. J. (1994). Proposal of minimal standards for describing new species of the family Campylobacteraceae. Int. J. Syst. Bacteriol. 44, 842–845. doi: 10.1099/00207713-44-4-842
Ursing, J. B., Rosselló-Mora, R. A., García-Valdes, E., and Lalucat, J. (1995). Taxonomic note: a pragmatic approach to the nomenclature of phenotypically similar genomic groups. Int. J. Syst. Bacteriol. 45:604. doi: 10.1099/00207713-45-3-604
Vandamme, P., Falsen, E., Rossau, R., Hoste, B., Segers, P., Tytgat, R., et al. (1991). Revision of Campylobacter, Helicobacter, and Wolinella taxonomy: emendation of generic descriptions and proposal of Arcobacter gen. nov. Int. J. Syst. Bacteriol. 41, 88–103. doi: 10.1099/00207713-41-1-88
Vandamme, P., Vancanneyt, M., Pot, B., Mels, L., Hoste, B., Dewettinck, D., et al. (1992). Polyphasic taxonomic study of the emended genus Arcobacter with Arcobacter butzleri comb. nov. and Arcobacter skirrowii sp. nov. an aerotolerant bacterium isolated from veterinary specimens. Int. J. Syst. Bacteriol. 42, 344–356. doi: 10.1099/00207713-42-3-344
Van den Abeele, A. M., Vogelaers, D., Vanlaere, E., and Houf, K. (2016). Antimicrobial susceptibility testing of Arcobacter butzleri and Arcobacter cryaerophilus strains isolated from Belgian patients. J. Antimicrob. Chemother. 71, 1241–1244. doi: 10.1093/jac/dkv483
Waite, D. W., Vanwonterghem, I., Rinke, C., Parks, D. H., Zhang, Y., Takai, K., et al. (2017). Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Front. Microbiol. 8:682. doi: 10.3389/fmicb.2017.00682
Wattam, A. R., Davis, J. J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., et al. (2017). Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45, D535–D542. doi: 10.1093/nar/gkw1017
Weimann, A., Mooren, K., Frank, J., Pope, P. B., Bremges, A., and McHardy, A. C. (2016). From genomes to phenotypes: traitar, the microbial trait analyzer. mSystems 1, e00101–e00116. doi: 10.1128/mSystems.00101-16
Keywords: Arcobacter cryaerophilus, isDDH, ANI, MLPA, genomovar
Citation: Pérez-Cataluña A, Collado L, Salgado O, Lefiñanco V and Figueras MJ (2018) A Polyphasic and Taxogenomic Evaluation Uncovers Arcobacter cryaerophilus as a Species Complex That Embraces Four Genomovars. Front. Microbiol. 9:805. doi: 10.3389/fmicb.2018.00805
Received: 28 January 2018; Accepted: 10 April 2018;
Published: 27 April 2018.
Edited by:
Antonio Ventosa, Universidad de Sevilla, SpainReviewed by:
Jason Sahl, Northern Arizona University, United StatesDavid John Studholme, University of Exeter, United Kingdom
Copyright © 2018 Pérez-Cataluña, Collado, Salgado, Lefiñanco and Figueras. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis Collado, bHVpc2NvbGxhZG9AdWFjaC5jbA==
María J. Figueras, bWFyaWFqb3NlLmZpZ3VlcmFzQHVydi5jYXQ=