 Marcos H. de Moraes1*
Marcos H. de Moraes1* Emanuel Becerra Soto2†Isai Salas González2,3,5†
Emanuel Becerra Soto2†Isai Salas González2,3,5† Prerak Desai6‡Weiping Chu6
Prerak Desai6‡Weiping Chu6 Steffen Porwollik6Michael McClelland6
Steffen Porwollik6Michael McClelland6 Max Teplitski1
Max Teplitski1- 1Soil and Water Sciences Department, University of Florida, Gainesville, FL, United States
- 2Center for Genomic Sciences, National Autonomous University of Mexico, Cuernavaca, Mexico
- 3Department of Biology, The University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 4Howard Hughes Medical Institute, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 5Curriculum in Bioinformatics and Computational Biology, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 6Department of Microbiology and Molecular Genetics, University of California, Irvine, Irvine, CA, United States
Outbreaks of salmonellosis linked to the consumption of vegetables have been disproportionately associated with strains of serovar Newport. We tested the hypothesis that strains of sv. Newport have evolved unique adaptations to persistence in plants that are not shared by strains of other Salmonella serovars. We used a genome-wide mutant screen to compare growth in tomato fruit of a sv. Newport strain from an outbreak traced to tomatoes, and a sv. Typhimurium strain from animals. Most genes in the sv. Newport strain that were selected during persistence in tomatoes were shared with, and similarly selected in, the sv. Typhimurium strain. Many of their functions are linked to central metabolism, including amino acid biosynthetic pathways, iron acquisition, and maintenance of cell structure. One exception was a greater need for the core genes involved in purine metabolism in sv. Typhimurium than in sv. Newport. We discovered a gene, papA, that was unique to sv. Newport and contributed to the strain’s fitness in tomatoes. The papA gene was present in about 25% of sv. Newport Group III genomes and generally absent from other Salmonella genomes. Homologs of papA were detected in the genomes of Pantoea, Dickeya, and Pectobacterium, members of the Enterobacteriacea family that can colonize both plants and animals.
Introduction
Salmonellosis outbreaks linked to the consumption of fruits (tomatoes, cucumbers, cantaloupes), leafy green vegetables and sprouts became an important public health issue over the last decade, defying the traditional notion that this pathogen is only associated with products of animal origin (Teplitski et al., 2009; Hernandez-Reyes and Schikora, 2013; Jackson et al., 2013; Wiedemann et al., 2014; Bennett et al., 2015). A CDC study identified Salmonella sv. Newport as the predominant serovar involved in outbreaks traced to vegetables. It was responsible for 57% of outbreaks associated with all fresh vegetables, and for 29% of outbreaks associated with vine-stalk vegetables (such as tomatoes and cucumbers). Tomatoes were a major source of salmonellosis outbreaks, and were implicated in up to 90% of outbreaks linked to vine-stalk vegetables, with sv. Newport responsible for 32% of them (Jackson et al., 2013). An analysis of outbreak occurrence from 1990 to 2010 identified 15 outbreaks that were associated with fresh tomatoes, and sv. Newport was responsible for six of them (Bennett et al., 2015). The association of salmonellosis outbreaks with sv. Newport and fresh vegetables reported in independent geographical locations suggests that this serovar may have a close relationship with plants and/or has evolved to persist in the vegetable production environment. At least three hypotheses can explain this phenomenon.
One hypothesis for the overrepresentation of sv. Newport in produce-associated outbreaks is that it evolved functions that make it more fit in plants. This hypothesis is supported by the evidence that sv. Newport outcompetes other serovars during plant colonization. When tested for proliferation in tomatoes, Salmonella sv. Newport reached higher cell numbers in green and pink tomatoes than sv. Typhimurium, Braenderup, and Montevideo (Marvasi et al., 2013b). Cells of serovar Newport were also recovered at higher rates from tomato rhizosphere than those of the serovars Saintpaul, Typhimurium, and Montevideo (Zheng et al., 2013). At least in part, the ability of some strains of sv. Newport to be more competitive within tomatoes could be due to spontaneous non-rdar mutations, which increased Salmonella fitness inside tomatoes (Zaragoza et al., 2012).
Secondly, strains of serovar Newport may be more fit in plants than those of other serovars because they are better able to adapt their physiology to enter and proliferate in tomato tissues (Teplitski and de Moraes, 2018). Studies with regulatory mutants and mutants in metabolic pathways suggest that during tomato colonization, Salmonella utilizes carbohydrates and inorganic sources of nitrogen and then uses the acquired nutrients to synthesize amino acids, LPS, and capsule. For example, genes involved in amino acid biosynthesis and iron acquisition were important for Salmonella growth in tomato pericarps. Inside tomato fruits, at least 51 Salmonella genes were differently regulated, including fadH, involved in fatty acid degradation, and cysB, the regulator for cysteine biosynthesis and acquisition. Changes in surface structures are also part of the Salmonella strategy for proliferation within plant tissues. In unripe tomatoes, the yihT gene, involved in the synthesis of O-antigen, is required for successful colonization of pericarps (Noel et al., 2010; Marvasi et al., 2013a; Nugent et al., 2015; de Moraes et al., 2017). These observations suggest that the environment in plant hosts is nutritionally unbalanced, and Salmonella has to employ and coordinate a diverse set of functions to thrive and establish itself within a niche already occupied by the native microbiota. In many respects, these observations are in line with the requirements of other phytobacteria during their interactions with plants (Lindow and Leveau, 2002; Lindow and Brandl, 2003). However, an earlier analysis of the totality of functions used by S. Typhimurium to colonize tomatoes revealed that, despite some similarities with phytobacteria in its plant colonization strategies, it relies on a distinct set of functions to establish itself within tomatoes (de Moraes et al., 2017).
Lastly, the advantage of sv. Newport over other serovars in colonization of plants could be enhanced by the presence of intrinsic colonization factors in this Salmonella clade. Studies of Salmonella virulence in animals show how the serovars’ genetic diversity can affect interactions with the hosts. For example, distribution of virulence plasmids and pathogenicity islands is serovar-specific, and the presence of these genetic determinants appears to impact host range (Barrow et al., 1987; Barrow and Lovell, 1988; Libby et al., 1997; Desai et al., 2013). Is it possible that sv. Newport has functions, not commonly present in other serovars of Salmonella, that allow it to colonize plants more efficiently?
In this study, we applied comparative genomics combined with transposon insertion screening analysis to identify unique features that distinguish sv. Newport from sv. Typhimurium in the Salmonella-tomato interaction model.
Materials and Methods
Bacterial Strains and Culture Conditions
Bacterial strains and plasmids used in this study are listed in Table 1. Salmonella enterica and Escherichia coli strains were propagated in LB (Luria Bertani) broth (Fisher Scientific) at 37 or 30°C, as specified in the text. When necessary, bacteria were plated onto LB agar (Fisher Scientific) or XLD (Oxoid) agar plates. As appropriate, growth media were supplemented with 100 μg/ml ampicillin, 60 μg/ml kanamycin or 20 μg/ml chloramphenicol. Construction of mutants was done using λ Red mutagenesis (Datsenko and Wanner, 2000). Typically, entire ORFs (from the start to the stop codon) were excised and replaced with a frt-kan-frt or a frt-cat-frt cassette, and the kanamycin (or chloramphenicol) resistance gene was flipped out as in Datsenko and Wanner (2000).
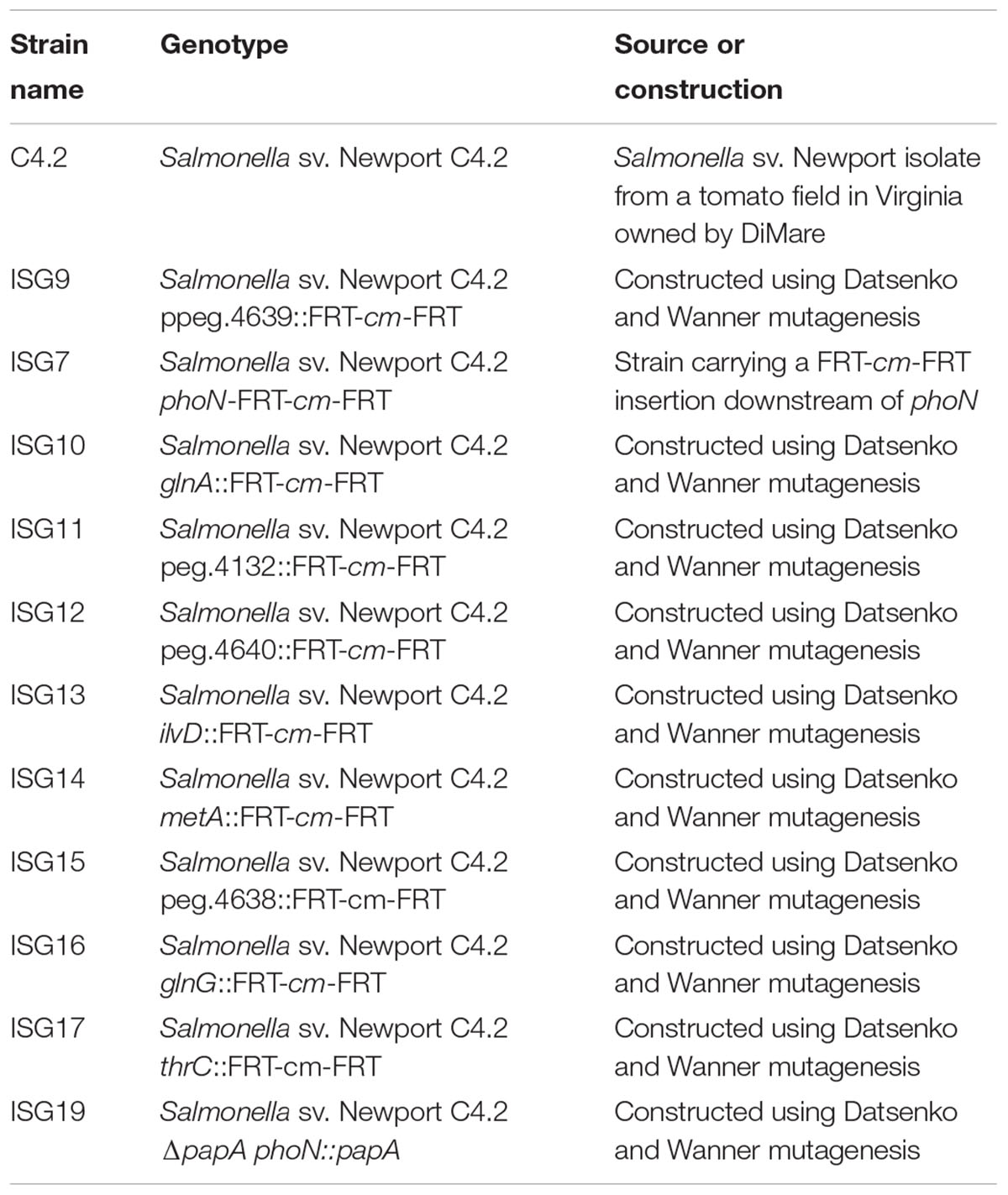
TABLE 1. Strains used in this study.
Transposon Insertion Library Screening in Tomato Pericarps
Tomato inoculation with a S. enterica Newport C4.2 transposon library was performed as previously done with S. enterica Typhimurium ATCC14028 (de Moraes et al., 2017). Tomatoes (cultivar Campari) were obtained from a local grocery store, where they are sold on the vine in a clam shell plastic container. The S. enterica sv. Newport transposon library was grown overnight in LB with the addition of kanamycin at 37°C, and the resulting cultures were resuspended in PBS. Approximately 108 CFU/ml in 3 μL of PBS were inoculated into three shallow wounds in tomato pericarps, and six fruits were used. Tomato fruits were incubated at 22°C and a relative humidity of ∼60% for 7 days. Under these conditions, no gross changes in the appearance of the fruit were observed, although ripening clearly progressed. Salmonella was recovered by collecting 1 g samples of tomato tissue around ∼1 cm of the inoculation site; samples from the same fruit were combined and homogenized in a stomacher (Sevard). Salmonella cells were recovered by centrifugation and were then resuspended and cultured in 50 ml of LB broth for 6 h at 37°C and 250 rpm.
Transposon Insertion Library Construction and Analysis
The transposon insertion library construction and analysis in sv. Newport were performed as described before (de Moraes et al., 2017). Briefly, a library of S. enterica serovar Newport C4.2 Tn5 insertion mutants was constructed with a mini-Tn5 derivative into which we inserted an N18 random barcode using PCR. This derivative was integrated into the genome using the EZ-Tn5 < T7/KAN-2 > promoter insertion kit (Epicentre Biotechnologies, Madison, WI, United States). The transposome complex was dialyzed against water before electroporation into fresh electrocompetent cells of S. enterica sv. Newport C4.2. Transformed cells were recovered on LB agar with kanamycin after overnight growth at 37°C.
Mapping of barcoded transposons to specific locations in the genome was performed as described before (de Moraes et al., 2017). Briefly, genomic DNA from the library was extracted using the GenElute bacterial genomic DNA kit (Sigma-Aldrich). DNA was fragmented by sonication and ligated to Illumina primers. This product was used to amplify the regions, including the N18 barcode and the genomic DNA adjacent to the transposon insertion, using a stepwise nested PCR (de Moraes et al., 2017). Resulting amplicons were purified with the QIAquick PCR Product Purification kit (Qiagen), and 150-base reads were obtained at both ends. The resulting reads were mapped against the de novo assembled sv. Newport C4.2 genome (see below) using Bowtie2. The N18 barcode tag for each mapped read was identified using custom Perl scripts. The same reads, trimmed to remove Tn5 sequences, were also employed to assemble the sv. Newport C4.2 genome, which was then annotated using the RAST package1.
For experiments in tomatoes, transposons were quantified as before (de Moraes et al., 2017). In brief, bacteria were recovered from tomatoes and grown in LB+60 μg/ml kanamycin. Bacteria were pelleted, lysed and subjected to PCR using primers directly flanking the N18 barcode. The frequency of each barcode was enumerated by Illumina sequencing of 20 bases. The aggregated abundances for the input and output libraries were statistically analyzed using edgeR, and the log2-fold changes and FDRs were reported.
Genome Sequence Retrieval, Quality Control and Assembly
Raw reads from Illumina sequencing of S. enterica sv. Newport and sv. Typhimurium strains were recovered from the NCBI Sequence Read Archive (SRA). The SRA identifiers of the strains used are listed in Supplementary Table S1. We opted to assemble genomes de novo to remove biases associated with different assembly methods and to employ the same quality standards. The genomes of the type strains (GCA_000022165.1, GCA_000016045.1, and GCA_000171415.1) were recovered from the NCBI Genome databases. Read quality control and visualization were done using the package Trim Galore (Andrews et al., 2015). Genome assembly was done using SPAdes, using default parameters (Bankevich et al., 2012). Assessment of the genome assembly quality was done with CheckM (Parks et al., 2015). Genomes with more than 1% of contamination and less than 99% of completeness were excluded. Prokka was used to annotate genomes (Seemann, 2014). The resulting.gff files were fed into Roary to build the pan-genome matrix (Page et al., 2015). The analysis of the pan-genome matrix was performed using ad hoc R scripts. We used power-law regression to model the total size of the Salmonella pan-genome. To that end, analysis of random permutations of the addition of new genomes was performed and the number of new genes found per addition was recorded, and used in the regression to estimate the expansion of the pan-genome.
Competition Assays
To confirm the results from the transposon insertion sequencing analysis, the fitness of individual isogenic mutants in relation to the wild-type was estimated. The bacterial population densities of overnight cultures of the wild type strain sv. Newport C4.2 and an isogenic mutant built in this background were set to similar numbers by adjusting their OD600 to the same level. Cells were then spun and resuspended in PBS to the original volume and mixed in a 1:1 ratio, followed by a 10,000-fold dilution in PBS. 3 μl of this mix were inoculated into the tomato pericarp in three separate wounds resulting in ∼103 CFU per tomato fruit. To get an accurate count of the wild type and mutant cells in the inoculum, an aliquot was serially diluted and plated onto XLD agar and incubated overnight at 37°C; fifty randomly picked colonies were then patched onto LB agar with chloramphenicol and the initial mutant:wild-type ratio was determined.
The inoculated tomatoes were incubated at 22°C for 7 days. Wound sites were then sampled using a sterile loop and Salmonella cells were recovered from a streak on XLD plates. Fifty colonies were patched onto LB agar with chloramphenicol to determine the mutant:wild-type ratio in the recovered sample. The competition index (CI) was calculated using the formula (MUTout:WTout)/(MUTin:WTin) (Noel et al., 2010). Statistical significance was determined using ANOVA against the CI of the neutral mutant ISG7. This neutral mutant was constructed with a FRT-cm-FRT insertion downstream of phoN known to not affect Salmonella fitness in tomatoes (Cox et al., 2013). The neutral phenotype of ISG7 was confirmed by competitions against the wild type. The software JMP version 12 was used for all CI analyses.
Growth Curve and rdar Phenotype Characterization
Responses of the strains to oxidative stress were compared by diluting overnight cultures to OD600 = 0.01 in 3 ml of LB with or without 0.5 mM paraquat. The cultures were incubated at 37°C with shaking at 200 rpm. 100 μl aliquots were collected hourly to estimate culture concentrations by serial dilution and plating onto LB agar. All experiments were replicated three times. The rdar phenotype was evaluated by spotting 5 μl of an overnight culture onto salt-less LB with Congo Red as described by Zaragoza et al. (2012).
Results and Discussion
Clade Separation Using Phylogenetic Analysis
In this study, we compared the genes required for growth of an S. enterica sv. Newport strain in tomatoes to previous results (de Moraes et al., 2017) obtained for a Typhimurium strain. We first used comparative genomics analysis to determine whether the strain of sv. Newport recovered from a tomato-linked outbreak and used in this study is an outlier within serovar Newport or whether it is a typical representative. A phylogenetic analysis, constructed with minimum-evolution using SNP distances obtained from 1526 Salmonella genomes (Supplementary Table S1), formed distinct clades for sv. Typhimurium and sv. Newport and displayed a small number of fast evolving genomes with no clear grouping (Figure 1A and Supplementary Figure S1). This result is consistent with previous Salmonella phylogenetic analyses, supporting the notion that sv. Typhimurium strains group as one clade and serovar Newport strains are divided into three different clades (Group I, Group II, Group III) (Sangal et al., 2010).
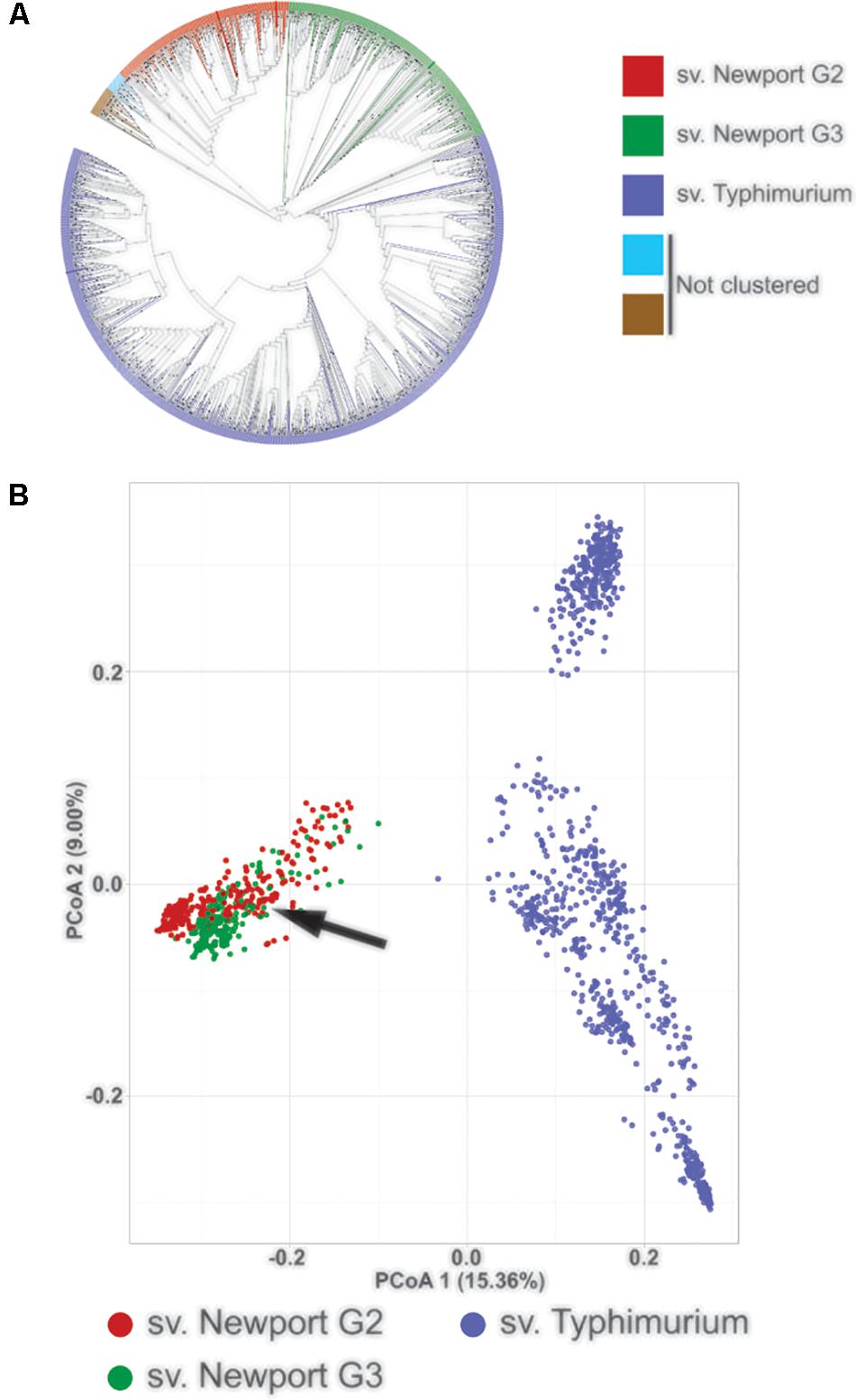
FIGURE 1. Phylogenetic and comparative genomic analysis of the sv. Typhimurium and sv. Newport genomes. (A) Phylogenetic tree for 1,597 genomes of sv Typhimurium and sv Newport isolates, constructed using SNPs and minimal evolution over raw distance. Colors represent the major clades identified and dots represent branches with bootstrap values higher than 0.85. iTOL v4 (Letunic and Bork, 2016) was used to visualize the tree. (B) Principal Coordinate Analysis of the gene presence and absence profile of the same genomes. Salmonella sv. Typhimurium strains are represented in purple, sv. Newport Group II isolates in red and sv. Newport Group III strains in green. The arrow indicates strain C4.2 of sv. Newport, recovered from a tomato-linked outbreak of human salmonellosis and used in this study.
We identified the sv. Newport clades obtained in our analysis by placing the sv. Newport type strains SL257, known to be in Group II, and SL317, known to be in Group III, in the phylogeny tree. The tomato outbreak strain C4.2 used in this study was placed in sv. Newport Group III, consistent with a previous phylogenetic analysis (Cao et al., 2013). Based on its position in the phylogenetic tree, it appears to be a typical representative of the sv. Newport Group III.
Comparison of the Salmonella sv. Typhimurium and sv. Newport Pan-Genomes
The association of multiple strains of sv. Newport with recurrent produce-related outbreaks and the lack of such a strong association for sv. Typhimurium, coupled with the availability of parallel tools for the functional genomics characterization of the interactions of these organisms with diverse hosts, offers an opportunity to address the hypothesis that sv. Newport strains might have additional genes associated with success in tomatoes. Within 1526 Salmonella genomes that passed phylogeny quality controls, gene prediction using Prokka identified 31,675 gene orthologs. These data were used to build a pan-genome matrix (Supplementary Table S1) containing gene orthologs present in each genome. While sv. Typhimurium exhibited a small standard deviation in the number of genes per genome, the sv. Newport strains contained genomes that had up to 2,000 additional genes, primarily due to prophage and plasmids.
Salmonella sv. Typhimurium had an average of 4,628 genes per genome, while sv. Newport Group II had 4,554 genes per genome and sv. Newport Group III had 4,413 genes per genome (Supplementary Figure S1). The presence and absence gene profiles of each group were visualized by Principal Coordinate Analysis (Figure 1B). Principal Coordinate Analysis clustered the sv. Newport Groups II/III and sv. Typhimurium in two distinct groups separated by the first principal coordinate, showing that the difference among serovars corresponded to the presence and absence of genes, not only to serological or SNPs differences. The strain Newport C4.2, used in our model for tomato colonization, was within the cluster of sv. Newport Group III, confirming that its genetic profile is similar to most sv. Newport isolates.
Core and accessory genome analyses revealed similar genome structures for sv. Newport and Typhimurium. To further characterize the differences between Salmonella genomes, we assessed their core and accessory genome content. We used the definition of “core genome” as the genes shared by more than 95% of all the genomes per group, and “accessory genome” as the genes shared by less than 5% of the genomes per group. All the genes between these two classifications are defined as “shell genome.” In sv. Newport Group II and sv. Newport Group III, 61–68% of all identified gene orthologs were part of the accessory genome and 11–12% of all identified gene orthologs were in the core genome (Figure 2A). The relative abundance of genes in the accessory genome in relation to the core genome was similar in the three Salmonella groups tested in this study. Moreover, these data showed that most of the Salmonella genes identified in the groups investigated here were in the accessory genome. The number of genes in the core genome was similar within all groups, ranging from 3,489 to 3,820 genes. All groups shared 3,155 genes (90%) of their core genomes. The sv. Newport groups shared consistently more of their core genomes than they shared with sv. Typhimurium (Figure 2B).
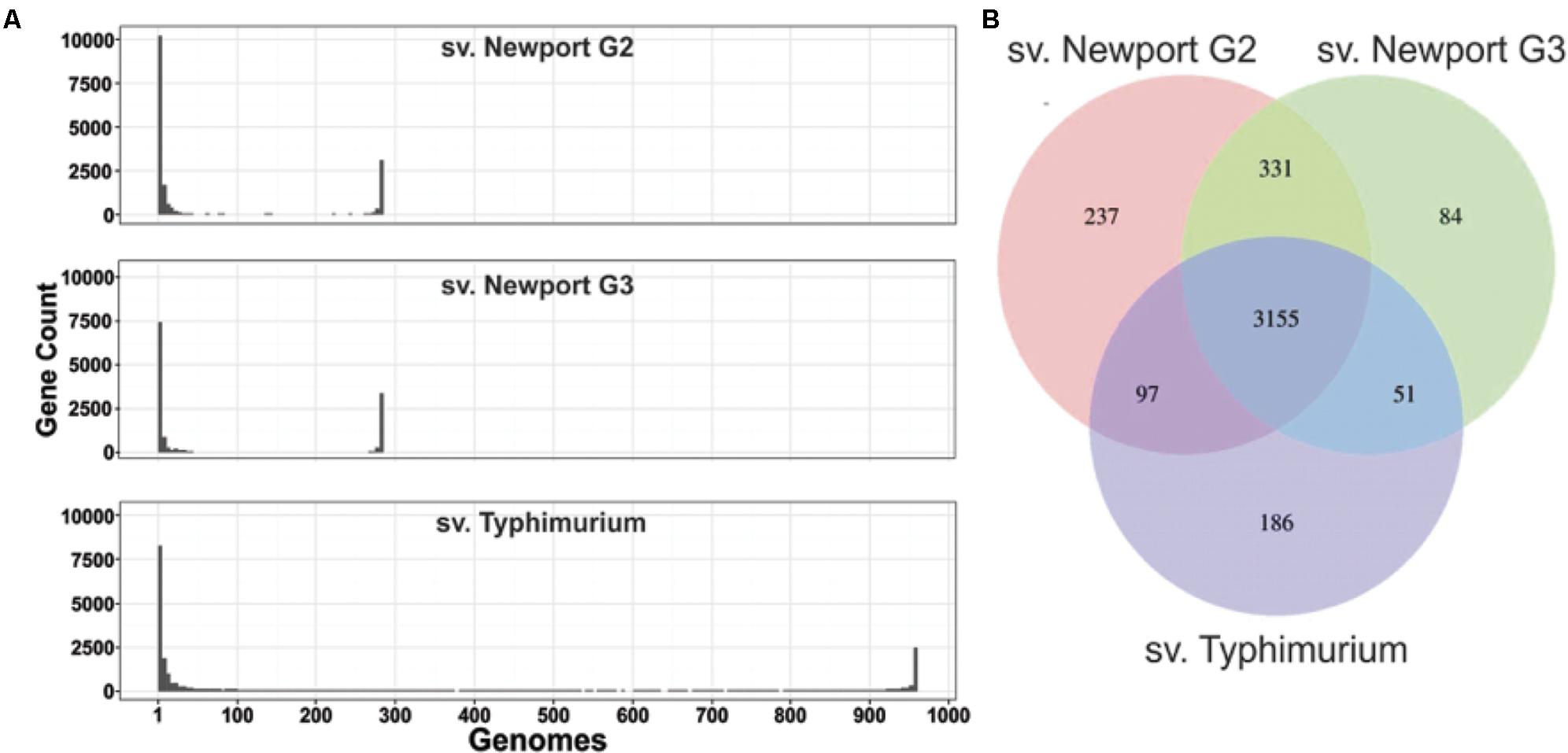
FIGURE 2. Salmonella core, shell and accessory genomes. (A) Number of genes per number of Salmonella sv. Newport Group II, sv. Newport Group III and sv. Typhimurium genomes. Many genes were found in only one or a few genomes (peaks on the left of each plot). The core genes, present in almost all genomes of each group, are represented by the peak on the right of each plot. (B) Venn diagram representing the shared and unique elements of Salmonella core genes (shared by >95% of all members of each group) among the groups studied.
We also investigated whether the serovars’ pan-genomes are open or closed according to the definition of (Medini et al., 2005; Tettelin et al., 2005). Using power-law regression, we found that all pan-genomes of Salmonella serovars analyzed in this study are closed, with an α parameter of 2.92, 2.94, and 2.39 (Supplementary Figure S2) for sv. Newport Group II, sv. Newport Group III and sv. Typhimurium, respectively. All regression curves exhibited similar slopes, although they reached saturation of new genes per genome at different points (around 20 for sv. Newport Group II and Group III, and 10 for sv. Typhimurium, Supplementary Figure S2). The genus Salmonella was previously identified as a closed genome species (Jacobsen et al., 2011).
The characterization of the presence/absence profile of genes in these genomes allowed the identification of 724 group-specific genes in the sv. Newport Group III shell genome and 84 group-specific genes unique to its core genome, compared with sv. Typhimurium. Further analysis was performed to determine if any of these genes were part of the genetic basis of the adaptation of the Group III Newport C4.2 strain to plant colonization.
Transposon Insertion Sequencing Identifies Functions Required for Persistence in Tomatoes
To identify genome regions that encode functions important in colonization of tomato pericarps, we employed a library of transposon insertion mutants in sv. Newport C4.2, using the same screening methods applied before for sv. Typhimurium ATCC 14028 (de Moraes et al., 2017). The reads obtained from the transposon library screening were used to reconstruct the sv. Newport C4.2 genome (NCBI) using the RAST package. The fully annotated genome was deposited (see foot note text 1, username:guest, password:guest), and a comprehensive CSV file with locus tags, transposon insertion counts and orientation, and read counts is presented in Supplementary Table S2.
We mapped 4,811 coding sequences disrupted by the transposon, and mutants in 796 of these had a significant change in their abundance after growth in tomato wounds (FDR < 0.1). 781 of these mutants displayed reduced fitness and 15 had increased fitness (Figure 3). Most of the genes (689) whose disruption led to a reduction of fitness were shared by sv. Newport C4.2 and sv. Typhimurium ATCC 14028.
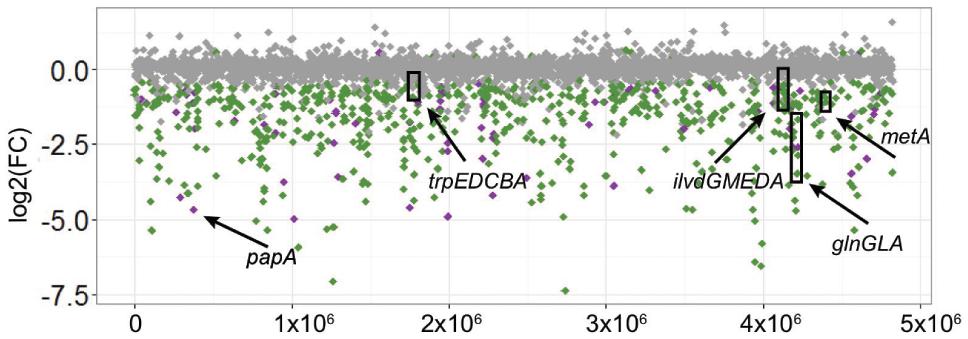
FIGURE 3. Change in abundance of Salmonella C4.2 loci after screening in tomatoes. Relative abundance of transposon insertions in loci after the incubation was compared to the initial inoculum. The position of each locus on the y-axis represents log2(Fold Change) of relative abundance and on the x-axis represents a physical position on the Salmonella chromosome. Loci with a significant change in abundance (FDR < 0.1) are shown in green when shared between S. Newport C4.2 and S. Typhimurium ATCC 14028 and purple when unique to S. Newport C4.2. Loci with an FDR > 0.1 are shown in gray. Arrows point to loci and operons targeted for further experiments.
To understand the cellular functions employed by sv. Newport C4.2 to colonize tomatoes, we identified the metabolic functions involved in this interaction. We mapped the genes identified as required for full fitness in pericarps and retrieved their COGs using the BlastKOALA web interface. Results were plotted against the sv. Newport C4.2 metabolic map. These results were compared with the outcomes of the sv. Typhimurium ATCC 14028 transposon insertion screening in a meta-analysis, with the goal of identifying potential serovar-specific factors. The comparison was done by overlapping the sv. Newport C4.2 and sv. Typhimurium ATCC 14028 metabolic maps. The main functions required for sv. Newport C4.2 colonization of tomato pericarps involved biosynthetic pathways such as amino acid and LPS biosynthesis, fatty acid catabolism and glycolysis. These same pathways were also required by sv. Typhimurium ATCC 14028 (Figure 4). The shared metabolic requirements by sv. Newport C4.2 and sv. Typhimurium ATCC 14028 corroborate the earlier conclusion that Salmonella relies on its robust and diverse metabolism to fully colonize tomatoes (de Moraes et al., 2017).
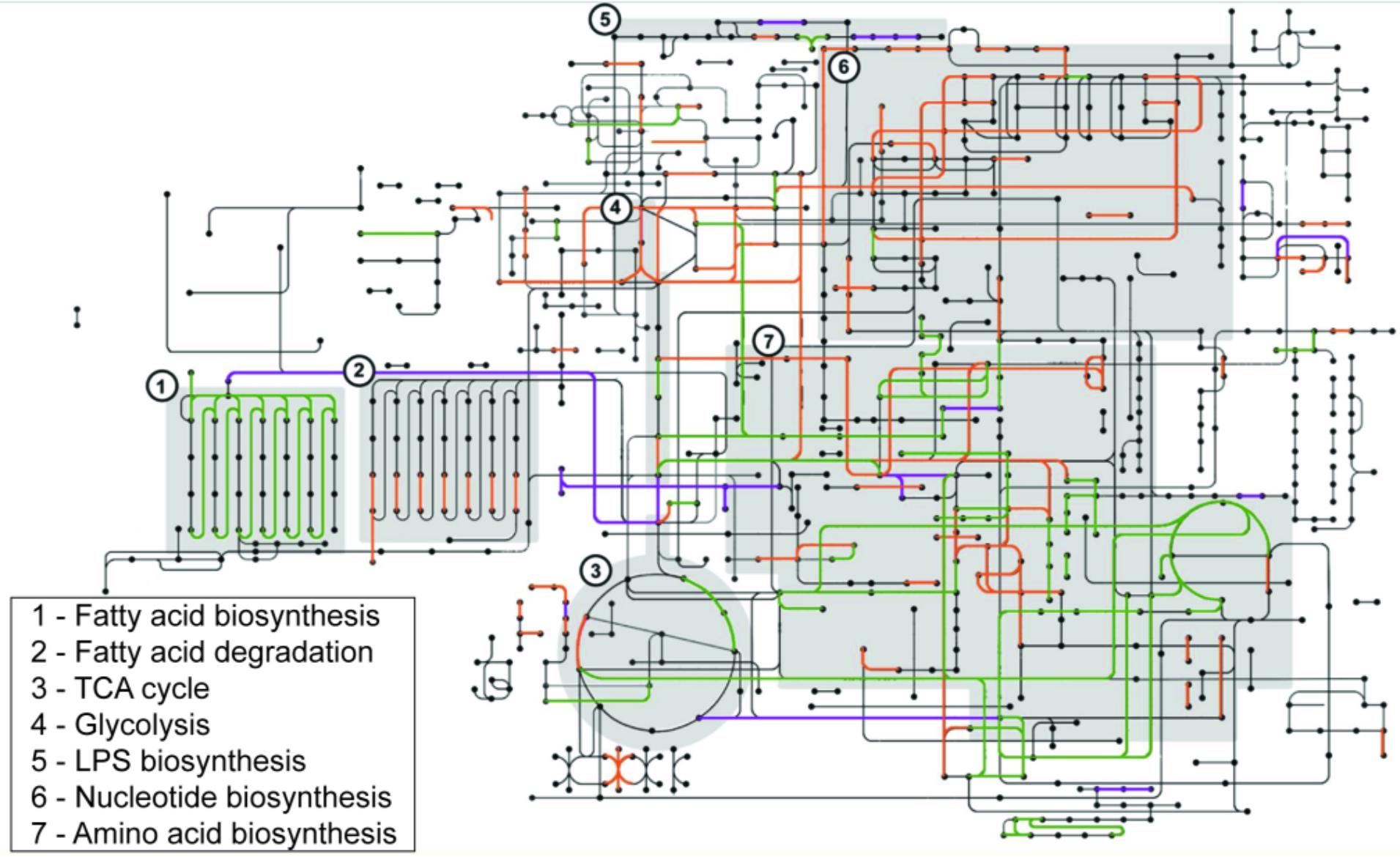
FIGURE 4. Comparison of functions required for colonization of tomatoes between S. Newport C4.2 and S. Typhimurium ATCC 14028. S. Newport C4.2 metabolic pathways involved in tomato colonization are shown in purple, S. Typhimurium ATCC 14028 metabolic pathways involved in tomato colonization are shown in orange, and required pathways shared by both strains are shown in green. Pathways were identified as required for tomato colonization if the corresponding mutants were less represented in the output pool [log2(FC) < -0.5, FDR < 0.1]. Key metabolic steps are shown as shaded boxes. KEGG Orthology terms for the protein sequences corresponding to the underrepresented mutants were retrieved using BlastKOALA and mapped using KEGG Mapper. False color overlays were imposed in Adobe Photoshop 2014.
Biosynthesis of amino acids by Salmonella during its interaction with tomatoes is of special interest for food safety. Different tomato cultivars are known to differ in the amounts of amino acids that accumulate within fruit during ripening (DiLeo et al., 2011; Osorio et al., 2011), and tomato genotypes with different amino acid profiles were shown to support different levels of Salmonella growth (Marvasi et al., 2014). To confirm the results obtained from the transposon insertion screening, we constructed isogenic mutants in the genes associated with amino acid biosynthesis and used these mutants to evaluate their fitness during tomato colonization in competition experiments against their parental strain. The genes selected were thrC, metA, ilvD, trpC, and glnA, involved in the biosynthesis of threonine, methionine, branched amino acids, tryptophan, and glutamine (respectively), and glnG, which codes for the master nitrogen regulator. Competition assays confirmed the results obtained from the transposon library screening. All isogenic mutants tested had a severe defect in fitness as estimated by their competitive indices (CI). The glnA isogenic mutant had the most severe defect in fitness [log2(CI) = -4.4], while the strain lacking the global nitrogen regulator had the smallest reduction of fitness [log2(CI) = -2.4] (Figure 5). The competition assays established that amino acid biosynthesis is a fundamental feature that confers an advantage for Salmonella to colonize tomato pericarps.
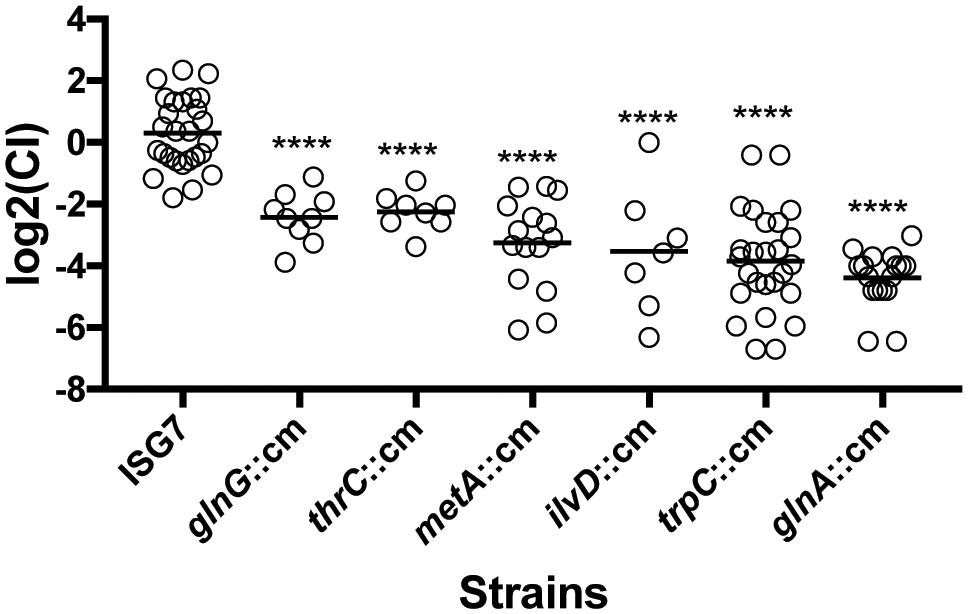
FIGURE 5. Competitive fitness of isogenic mutants involved in amino acid biosynthesis. Approximately 103 CFUs of a mix of the wild type and an isogenic mutant (1:1) were inoculated into each tomato, followed by 7-day incubation at 22°C. The Competitive Index CI was calculated using the formula (MUTout:WTout)/(MUTin:WTin). The replicates were obtained from six tomato fruits with three inoculation sites per fruit. Each dot represents a single replicate and the black bar represents the mean log2(CI) of all replicates. Statistical significance was established by comparing CI’s using ANOVA and Tukey’s post hoc test. Asterisks represent strains that are statistically different from the neutral mutant ISG7 (P-value < 0.05).
The main point of divergence between the sv. Newport C4.2 and sv. Typhimurium ATCC 14028 metabolic requirements for tomato colonization was the biosynthesis of nucleotides (Figure 4). Both purine and pyrimidine synthesis pathways identified as needed for efficient colonization by sv. Typhimurium ATCC 14028 at high titer were not under selection in the sv. Newport C4.2 transposon insertion screening at similar titers, indicating that S. Newport obtains sufficient purines and pyrimidines by scavenging in this environment. Notably, purine and pyrimidine biosynthesis was required by sv. Typhimurium ATCC 14028 even when inoculation was performed with a 10,000-fold lower titer (de Moraes et al., 2017). This result suggests that sv. Newport C4.2 could have a more efficient scavenging system for purines and pyrimidines, which could be advantageous for this strain during plant colonization.
Phenotypic Analysis of the Genes Unique to sv. Newport Reveals Potential Adaptions to Persistence in Plants
The main objective of this work was to explore the hypothesis that the sv. Newport C4.2 isolate from a tomato outbreak has additional genetic features not present in Typhimurium that enable it to colonize plants. Many sv. Newport-specific genes under selection had functions that could not be identified by BlastKOALA or Pfam. A few of those genes had putative metabolic functions. The locus peg.4637-peg.4641 is composed of genes that are absent from other Salmonella genomes and code for predicted proteins with unknown functions and a D-glucuronate permease (peg.4640). Since D-glucuronate is present in tomato fruit, we hypothesized that the reduction of fitness resulting from transposon insertions in this region indicates that Salmonella may be using D-glucuronate permease to scavenge D-glucuronate to proliferate in tomato pericarps. We therefore explored the role of this region during tomato colonization using a competition assay with isogenic mutants for the genes peg.4638, peg.4639, and peg.4640. The competitive indices for these isogenic mutants were not reduced when compared to ISG7, a strain that carries a neutral mutation (Figure 6A). However, this competition assay was performed with an inoculation titer thousands of fold lower than when the phenotype was observed in the transposon screen. It remains possible that this nutrient is used by Salmonella when the bacterial population is high.
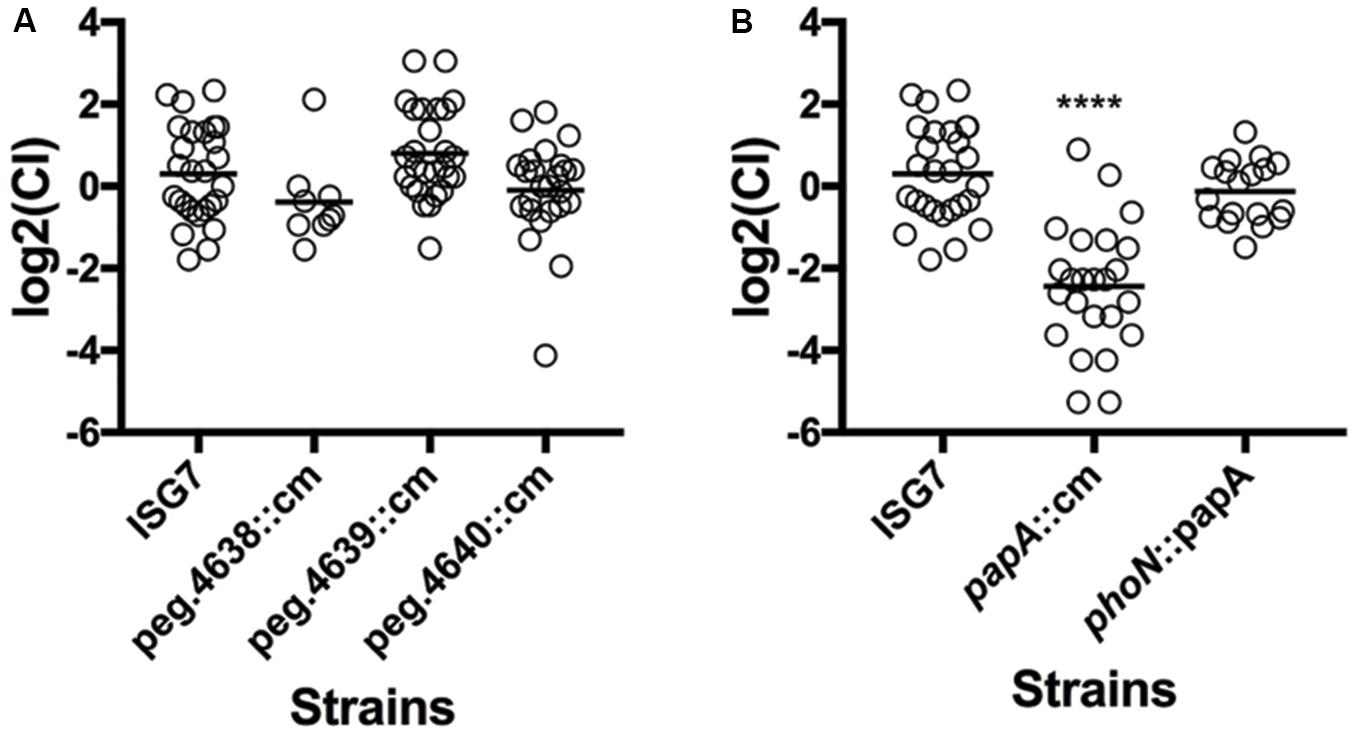
FIGURE 6. Competitive fitness of isogenic mutants of genes unique to S. Newport C4.2. The Competitive Index CI was calculated using the formula (MUTout:WTout)/(MUTin:WTin) and inoculation and statistical analysis was done as described before. (A) CI of isogenic mutants for peg.4638, peg.4639, and peg.4640. (B) CI of isogenic mutants for papA and the complemented isogenic mutant ΔpapA phoN::papA.
Another gene (peg.4132), coding for a small putative protein (44 amino acids), was present in the shell genome of S. enterica sv. Newport C4.2, and a corresponding mutant had a strong reduction of fitness [log2(FC) = -4.66] (Figure 6B). This gene was probably horizontally acquired: it is associated with a region containing mobile elements that includes remnants of phage genes, and it is located near a tRNA gene. The gene has a GC content of 40%, in contrast to the genome average of 52%. We further investigated the role of this gene during tomato colonization using competition assays with an isogenic mutant. The peg4132 gene was required for fitness of the strain S. enterica sv. Newport C4.2 in tomatoes [(log2(CI) = -2.44] (Figure 6). The mutant did not have a growth defect, displayed normal resistance to oxidative stress and was indistinguishable from the wild type when tested for the rdar phenotype (Figure 7). Due to its potential requirement for the colonization of plants, we named peg4132 “papA” (Plant Associated Protein A).
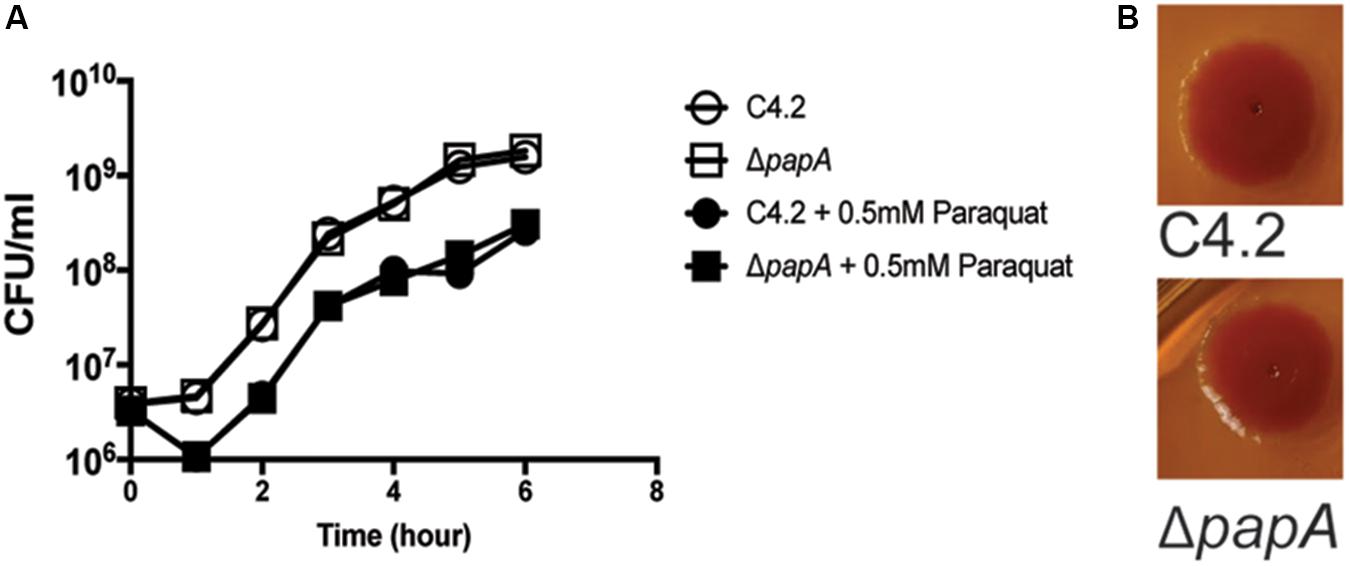
FIGURE 7. Comparison of growth and colony morphology between S. Newport C4.2 and its papA mutant. (A) Growth of strains in LB medium at 250 rpm and 37°C in the presence and absence of 0.5 mM paraquat. Three independent replicates were used per condition. (B) Colony morphology of S. Newport C4.2 and its papA mutant on Congo Red indicator plates. The smooth surface and edges show the absence of the rough and dry phenotype.
To exclude the possibility that the reduction of fitness in this mutant in tomatoes was a result of a polar mutation, papA was cloned with its native promoter region in the vector pKD3 and then reinserted in the phoN locus in the ΔpapA strain, creating a complemented papA strain with a single copy of the gene, stably maintained on the chromosome. The phoN locus was previously shown to be neutral during tomato colonization (Cox et al., 2013). The colonization fitness measured by the competitive index (CI) was not significantly different between the wild type and complemented papA strains (Figure 6B), corroborating that papA is a factor required for full fitness in sv. Newport C4.2 colonization of tomato pericarps.
We analyzed the distribution of papA in the Salmonella genomes, and determined that this gene was exclusive to Newport Group III, where it was found in 71 genomes out of 306, most of them closely associated in one clade. Using BlastP over the NCBI microbial database, we found potential orthologs of papA. Interestingly, many of these orthologs were in other members of the Enterobacteriaceae family that have a lifestyle associated with soil and plants, including Pectobacterium ssp. and Dickeya ssp, suggesting that this gene may be involved in persistence in or on plants.
Author Contributions
MdM, MM, and MT conceived the study and designed the experiments. MdM, EBS, ISG, and PD conducted the computational analysis. MM conceived, designed and oversaw Tn-Seq experiments and the Tn-Seq data analyses. MdM and WC conducted experimental work. MdM, MT, SP, and MM wrote the paper.
Funding
This work was supported by the Center for Produce Safety grant 2016CPS09.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Julia McGuinness and Eric Solomon for assistance with competition experiments (Figures 5, 6).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00877/full#supplementary-material
FIGURE S1 | Number of genes per genome. The total number of genes per genome was plotted for the groups sv. Newport Gil, sv. Newport Group TIT and sv. Typhimurium. The upper whisker extends to the highest value within 1.5 times the interquartile range. The lower whisker extend to the lowest value within 1.5 times the interquartile range. Outliers were defined as values higher than 1.5× interquartile range ± third quartile, or lower than 1.5 ± first quartile, and were plotted as dots. The thick black line within the boxes represents the mean.
FIGURE S2 | Total number of genes and new genes per genome obtained using random permutations. (A,C,E) Total number of genes per genome added. (B,D,F) New genes per genome and α and 𝜃 values from power law regression. (A,B) sv. Newport Group II. (C,D) sv. Newport Group III. (E,F) sv. Typhimurium.
TABLE S1 | Pangenome matrix containing genomes ID, metadata, and presence/absence profile of genes.
TABLE S2 | Genome Annotation of Newport C4.2 plus Tn5 fitness screening data compared to Typhimurium.
Footnotes
References
Andrews, S., Krueger, F., Segonds-Pichon, A., Biggins, L., Virk, B., Dalle-Pezze, P., et al. (2015). Trim Galore! Babraham: Babraham Institute.
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barrow, P. A., and Lovell, M. A. (1988). The association between a large molecular mass plasmid and virulence in a strain of Salmonella-Pullorum. J. Gen. Microbiol. 134, 2307–2316. doi: 10.1099/00221287-134-8-2307
Barrow, P. A., Simpson, J. M., Lovell, M. A., and Binns, M. M. (1987). Contribution of Salmonella Gallinarum large plasmid toward virulence in fowl typhoid. Infect. Immun. 55, 388–392.
Bennett, S. D., Littrell, K. W., Hill, T. A., Mahovic, M., and Behravesh, C. B. (2015). Multistate foodborne disease outbreaks associated with raw tomatoes, United States, 1990-2010: a recurring public health problem. Epidemiol. Infect. 143, 1352–1359. doi: 10.1017/S0950268814002167
Cao, G., Meng, J., Strain, E., Stones, R., Pettengill, J., Zhao, S., et al. (2013). Phylogenetics and differentiation of Salmonella Newport lineages by whole genome sequencing. PLoS One 8:e55687. doi: 10.1371/journal.pone.0055687
Cox, C. E., Mcclelland, M., and Teplitski, M. (2013). Consequences of disrupting Salmonella AI-2 signaling on interactions within soft rots. Phytopathology 103, 352–361. doi: 10.1094/PHYTO-09-12-0237-FI
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
de Moraes, M. H., Desai, P., Porwollik, S., Canals, R., Perez, D. R., Chu, W., et al. (2017). Salmonella persistence in tomatoes requires a distinct set of metabolic functions identified by transposon insertion sequencing. Appl. Environ. Microbiol. 83, e3028-16. doi: 10.1128/AEM.03028-16
Desai, P. T., Porwollik, S., Long, F., Cheng, P., Wollam, A., Bhonagiri-Palsikar, V., et al. (2013). Evolutionary genomics of Salmonella enterica subspecies. mBio 4:e198-13. doi: 10.1128/mBio.00198-13
DiLeo, M. V., Strahan, G. D., Den Bakker, M., and Hoekenga, O. A. (2011). Weighted correlation network analysis (WGCNA) applied to the tomato fruit metabolome. PLoS One 6:e26683. doi: 10.1371/journal.pone.0026683
Hernandez-Reyes, C., and Schikora, A. (2013). Salmonella, a cross-kingdom pathogen infecting humans and plants. FEMS Microbiol. Lett. 343, 1–7. doi: 10.1111/1574-6968.12127
Jackson, B. R., Griffin, P. M., Cole, D., Walsh, K. A., and Chai, S. J. (2013). Outbreak-associated Salmonella enterica serotypes and food commodities, United States, 1998-2008. Emerg. Infect. Dis. 19, 1239–1244. doi: 10.3201/eid1908.121511
Jacobsen, A., Hendriksen, R. S., Aaresturp, F. M., Ussery, D. W., and Friis, C. (2011). The Salmonella enterica pan-genome. Microb. Ecol. 62, 487–504. doi: 10.1007/s00248-011-9880-1
Letunic, I., and Bork, P., (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Libby, S. J., Adams, L. G., Ficht, T. A., Allen, C., Whitford, H. A., Buchmeier, N. A., et al. (1997). The spv genes on the Salmonella Dublin virulence plasmid are required for severe enteritis and systemic infection in the natural host. Infect. Immun. 65, 1786–1792.
Lindow, S. E., and Brandl, M. T. (2003). Microbiology of the phyllosphere. Appl. Environ. Microbiol. 69, 1875–1883. doi: 10.1128/AEM.69.4.1875-1883.2003
Lindow, S. E., and Leveau, J. H. (2002). Phyllosphere microbiology. Curr. Opin. Biotechnol. 13, 238–243. doi: 10.1016/S0958-1669(02)00313-0
Marvasi, M., Cox, C. E., Xu, Y., Noel, J. T., Giovannoni, J. J., and Teplitski, M. (2013a). Differential regulation of Salmonella Typhimurium genes involved in O-antigen capsule production and their role in persistence within tomato fruit. Mol. Plant. Microbe. Interact. 26, 793–800. doi: 10.1094/MPMI-09-12-0208-R
Marvasi, M., Hochmuth, G. J., Giurcanu, M. C., George, A. S., Noel, J. T., Bartz, J., et al. (2013b). Factors that affect proliferation of Salmonella in tomatoes post-harvest: the roles of seasonal effects, irrigation regime, crop and pathogen genotype. PLoS One 8:e80871. doi: 10.1371/journal.pone.0080871
Marvasi, M., Noel, J. T., George, A. S., Farias, M. A., Jenkins, K. T., Hochmuth, G., et al. (2014). Ethylene signalling affects susceptibility of tomatoes to Salmonella. Microb. Biotechnol. 7, 545–555. doi: 10.1111/1751-7915.12130
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Noel, J. T., Arrach, N., Alagely, A., Mcclelland, M., and Teplitski, M. (2010). Specific responses of Salmonella enterica to tomato varieties and fruit ripeness identified by in vivo expression technology. PLoS One 5:e12406. doi: 10.1371/journal.pone.0012406
Nugent, S. L., Meng, F., Martin, G. B., and Altier, C. (2015). Acquisition of iron is required for growth of Salmonella spp. in tomato fruit. Appl. Environ. Microbiol. 81, 3663–3670. doi: 10.1128/AEM.04257-14
Osorio, S., Alba, R., Damasceno, C. M., Lopez-Casado, G., Lohse, M., Zanor, M. I., et al. (2011). Systems biology of tomato fruit development: combined transcript, protein, and metabolite analysis of tomato transcription factor (nor, rin) and ethylene receptor (Nr) mutants reveals novel regulatory interactions. Plant. Physiol. 157, 405–425. doi: 10.1104/pp.111.175463
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Sangal, V., Harbottle, H., Mazzoni, C. J., Helmuth, R., Guerra, B., Didelot, X., et al. (2010). Evolution and population structure of Salmonella enterica serovar Newport. J. Bacteriol. 192, 6465–6476. doi: 10.1128/JB.00969-10
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Teplitski, M., Barak, J. D., and Schneider, K. R. (2009). Human enteric pathogens in produce: un-answered ecological questions with direct implications for food safety. Curr. Opin. Biotechnol. 20, 166–171. doi: 10.1016/j.copbio.2009.03.002
Teplitski, M., and de Moraes, M. (2018). Of mice and men....and plants: comparative genomics of the dual lifestyles of enteric pathogens. Trends Microbiol. doi: 10.1016/j.tim.2018.02.008 [Epub ahead of print].
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Wiedemann, A., Virlogeux-Payant, I., Chausse, A. M., Schikora, A., and Velge, P. (2014). Interactions of Salmonella with animals and plants. Front. Microbiol. 5:791. doi: 10.3389/fmicb.2014.00791
Zaragoza, W. J., Noel, J. T., and Teplitski, M. (2012). Spontaneous non-rdar mutations increase fitness of Salmonella in plants. Environ. Microbiol. Rep. 4, 453–458. doi: 10.1111/j.1758-2229.2012.00364.x
Keywords: tomato, plant-microbe interactions, comparative genomics, pan-genome, vegetable safety
Citation: de Moraes MH, Becerra Soto E, Salas González I, Desai P, Chu W, Porwollik S, McClelland M and Teplitski M (2018) Genome-Wide Comparative Functional Analyses Reveal Adaptations of Salmonella sv. Newport to a Plant Colonization Lifestyle. Front. Microbiol. 9:877. doi: 10.3389/fmicb.2018.00877
Received: 23 December 2017; Accepted: 16 April 2018;
Published: 18 May 2018.
Edited by:
Adam Schikora, Julius Kühn-Institut, GermanyReviewed by:
Young Min Kwon, University of Arkansas, United StatesYufeng Yao, Shanghai Jiao Tong University, China
Copyright © 2018 de Moraes, Becerra Soto, Salas González, Desai, Chu, Porwollik, McClelland and Teplitski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcos H. de Moraes, bWhkbW9yYWVzQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work.
‡Present address: Prerak Desai, Janssen Pharmaceuticals, Inc., Spring House, PA, United States