 Agnieszka Włodarczyk1
Agnieszka Włodarczyk1 Maciej Lirski2
Maciej Lirski2 Anna Fogtman2Marta Koblowska2,3Grzegorz Bidziński4
Anna Fogtman2Marta Koblowska2,3Grzegorz Bidziński4 Renata Matlakowska1*
Renata Matlakowska1*- 1Laboratory of Environmental Pollution Analysis, Faculty of Biology, University of Warsaw, Warsaw, Poland
- 2Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland
- 3Laboratory of Systems Biology, Faculty of Biology, University of Warsaw, Warsaw, Poland
- 4KGHM Polska Miedź S.A., Lubin Mine Division, Lubin, Poland
Black shales are one of the largest reservoirs of fossil organic carbon and inorganic reduced sulfur on Earth. It is assumed that microorganisms play an important role in the transformations of these sedimentary rocks and contribute to the return of organic carbon and inorganic sulfur to the global geochemical cycles. An outcrop of deep subterrestrial ~256-million-year-old Kupferschiefer black shale was studied to define the metabolic processes of the deep biosphere important in transformations of organic carbon and inorganic reduced sulfur compounds. This outcrop was created during mining activity 12 years ago and since then it has been exposed to the activity of oxygen and microorganisms. The microbial processes were described based on metagenome and metaproteome studies as well as on the geochemistry of the rock. The microorganisms inhabiting the subterrestrial black shale were dominated by bacterial genera such as Pseudomonas, Limnobacter, Yonghaparkia, Thiobacillus, Bradyrhizobium, and Sulfuricaulis. This study on black shale was the first to detect archaea and fungi, represented by Nitrososphaera and Aspergillus genera, respectively. The enzymatic oxidation of fossil aliphatic and aromatic hydrocarbons was mediated mostly by chemoorganotrophic bacteria, but also by archaea and fungi. The dissimilative enzymatic oxidation of primary reduced sulfur compounds was performed by chemolithotrophic bacteria. The geochemical consequences of microbial activity were the oxidation and dehydrogenation of kerogen, as well as oxidation of sulfide minerals.
Introduction
Sedimentary rocks are one of the largest reservoirs of fossil organic carbon and inorganic reduced sulfur compounds on Earth. The total content of Corganic and Ssulfidic in these rocks is estimated at ~ 1.5 × 1022 g and ~ 39.5 × 1020 g, respectively (Berner, 1989; Rickard, 2012). The richest reservoir of Corganic and Ssulfidic are shales, the most abundant ancient sedimentary rocks on Earth, in which they may constitute up to 20 and 0.4 wt%, respectively (Tourtelot, 1979; Rickard, 2012). The oxidation of these sedimentary rocks is of great significance, because it contributes to the return of organic carbon and inorganic sulfur trapped in the form of refractory fossil organic matter and insoluble sulfides to the global biogeochemical cycles on the Earth's near surface and subsurface.
Microbial processes of sulfide mineral dissolution based on chemolithotrophic oxidation have been known since the 1950s (Bryner et al., 1954), and the heterotrophic processes of assimilation of the fossil organic carbon trapped in black shales were discovered at the beginning of the Twenty first century (Petsch et al., 2001). These processes have always been studied and described independently; however, in the case of sedimentary rocks such as kerogen- and sulfide mineral-bearing black shales, these processes can coexist. There is still very little knowledge about such simultaneous processes and many questions remain unanswered. The taxonomy, physiology and metabolism of microbial communities inhabiting black shales are some of the most important and as yet understudied areas of research. The biochemical pathways of microbial processes cause oxidation, and the way these processes affect the geochemistry of oxidized rocks and waters are largely unknown. In addition, the diversity of sedimentary rocks, not only in terms of their origin and age, location, geochemical composition but also their physicochemical properties, justifies further research toward a better understanding of these unique geomicrobial processes.
In our field studies, we examined the lithobiontic microbial community (LMC) inhabiting the subterrestrial Kupferschiefer black shale at a depth of 777 meters and its role in the oxidative weathering of fossil hydrocarbons and sulfide minerals. Kupferschiefer is a neutral Lopingian sedimentary rock of marine origin, containing large amounts of organic carbon (up to 16 wt%) dominated by hydrocarbons (kerogen type II), and also a high content of sulfur (~2.9 wt%), dominated by reduced sulfur compounds [bornite (Cu5FeS4), chalcocite (Cu2S), digenite (Cu9S5), chalcopyrite (CuFeS2), pyrite (FeS2), sphalerite (ZnS), galena (PbS), covellite (CuS), and minerals belonging to the tennantite-tetrahedrite series; (Cu, Fe)12As4S13 – (Cu, Fe)12Sb4S13] (Sawłowicz, 1989; Speczik, 1994; Oszczepalski, 1999).
In the current studies, the profile of the black shale which had been exposed 12 years ago during mining activity in the area of Lubin copper mine was analyzed (Figure 1). In a subterrestrial mine the processes of the aerobic oxidation of fossil hydrocarbons and sulfide minerals occurring in the Kupferschiefer black shale may be intensive and may be proceed with the participation of either abiotic factors (oxygen, water) or microorganisms inhabiting the black shale. In previous laboratory studies, we described the long-term oxidation of Kupferschiefer kerogen by an indigenous bacterial consortium (Stasiuk et al., 2017). We reported that the bacterial oxidation of fossil hydrocarbons may be one of the processes responsible for kerogen weathering. However, the research so far had been limited to only a bacterial metaproteome analysis and identification of oxidized organic compounds. Metagenomic tools had not been utilized, and the participation of archaea and fungi in these processes had also not been recognized. Similarly, our knowledge about the influence of microorganisms on sulfide minerals deposited in Kupferschiefer black shale is even more limited. The dissolution of these minerals has been observed in laboratory experiments, although this was caused by chemical compounds—intermediates and products of fossil organic matter degradation by heterotrophic bacteria (Włodarczyk et al., 2016). So far, neither bacterial nor archaeal nor fungal enzymatic processes of oxidation of inorganic sulfur compounds have been studied.

Figure 1. Outcrop of subterrestrial black shale - macroscopic (A) and scanning electron microscope (B,C) images.
The aim of our study was to define the microbial metabolic processes of fossil hydrocarbons and sulfide mineral oxidation at the molecular level of resolution. The taxonomic diversity of LMC inhabiting the black shale, including bacteria, archaea and fungi, was identified. Next, protein-encoding genes (PEGs) and enzymes responsible for the oxidative metabolism of hydrocarbons and sulfide minerals were indicated in the metagenome and metaproteome of LMC, respectively. The microorganisms and the metabolic processes crucial in the oxidation of these two components of black shale were indicated. Finally, the geochemical consequences of microbial activity were described.
Materials and Methods
Site and Samples Description
Three samples of black shale were collected from a 12-year-old outcrop at a depth of 777 m in Lubin copper mine (Figure 1). The mine is located in Lubinsko-Głogowski Copper District (51.434015, 16.156069) (SW Poland). The samples were collected aseptically from outcrops (to a depth of 10 cm) and kept at −80 and −4°C until processing in the lab. The unweathered black shale originating from a freshly exposed outcrop (2 days) in Lubin mine has previously been described (Stasiuk et al., 2017).
Microscopic Observations
The samples were fixed in paraformaldehyde vapor, gold coated and observed under a scanning electron microscope (Leo 1430VP, LEO Electron Microscopy Inc., USA).
DNA Isolation
DNA was isolated from 100 g of sample according to a modified procedure of Zhou et al. (1996). The samples were resuspended in 100 ml of DNA extraction buffer (0.1 M Na2EDTA, 0.1 M Tris-HCl, 0.1 M Na2HPO4, 1.5 M NaCl, 1% CTAB (hexa-decylmethylammonium bromide), pH 8.0) containing proteinase K and lysozyme (750 μl; 10 mg/ml) and then were incubated overnight at 37°C with horizontal shaking. Following the addition of 20% SDS, each sample was incubated for 4 h at 65°C and centrifuged (6,000 × g, 10 min). Harvested supernatant was mixed with an equal volume of chloroform/isoamyl alcohol mixture (24:1; v/v) and centrifuged (6,000 × g, 10 min). The aqueous phase was collected and precipitated overnight with 0.6 volume of isopropanol at room temperature. Next, the sample was centrifuged (16,000 × g, 20 min, 4°C), and the pellet was washed with cold 70% ethanol and allowed to dry. Isolated DNA was resuspended in 50 μl of sterile deionized water and stored at −80°C.
DNA Sequencing and Analysis
Isolated DNA was used to prepare barcoded library with Ion Xpress™ Plus Fragment Library Kit (Thermo Fisher) according to manufacturer's manual. Library was subjected to clonal amplification on Ion One Touch 2 system (Thermo Fisher) using Ion PI™ Template OT2 200 Kit v2 (Thermo Fisher) and sequenced on Ion Proton sequencer using Ion PI™ Sequencing 200 Kit v2 (Thermo Fisher) according to manufacturer's manual. Reads were demultiplexed with Torrent Suite software. Fastq file was quality trimmed and filtered for low complexity reads, reads shorter than 34 nucleotides and duplicate reads using PRINSEQ (Schmieder and Edwards, 2011). Filtered reads were aligned to National Center for Biotechnology Information Non-Redundant (NCBI nr) protein database, using DIAMOND-0.8.18 Basic Local Alignment Search Tool X (BLASTX) Alignment Mode (Buchfink et al., 2015) with “more sensitive” option and VTML200 scoring matrix and following parameters: λ = 0.318, K = 0.13, gap open penalty = 15 and gap extend penalty = 1.5. Statistical parameters for VTML200 scoring matrix were computed using fastx36 script from FASTA-36.3.8d suite (Pearson 1988-2014). Resulting alignment file was “meganized” using script supplied with MEGAN 6.6.1 (Huson et al., 2007). Taxonomical analyses were performed in MEGAN using Lowest Common Ancestor (LCA) algorithm on alignments from DIAMOND. To minimize false positive hits a cutoff of minimum bitscore 60 and at least 207 reads to report a taxon was selected. Cutoff parameters were defined by thorough analysis of rarefaction curves generated with different filtering parameters. Other LCA parameters were set to default values. Krona charts were generated with data from tab-separated “read name—taxon id” file exported from MEGAN, using ktImportTaxonomy function from KronaTools (Ondov et al., 2011) with default settings. Coverage of selected proteins was calculated using custom pipeline consisting of bash and python scripts, implementing GNU Parallel (Tange, 2011) and ETE3 (Huerta-Cepas et al., 2016). Briefly, for each read, hits with minimum bitscore 60 were used, and the best hits were selected based on other BLAST parameters. Coverage was calculated for unique best scoring hits. Next, reads having multiple best scoring hits were divided and distributed among references present in unique hits based on weights calculated from unique read coverage of those references. Non unique reads (URs), which had no hits among references present in unique hits, were divided based on coverage of species they derived from. Non URs aligning to species not present in unique hits were dropped. Summed coverage for selected proteins was calculated based on protein descriptions from NCBI nr database. Results were manually supervised and corrected until no erroneous assignments were found.
Isolation of Proteins
Proteins were isolated from 100 g of sample according to the modified procedure of Ram et al. (2005). Each sample was resuspended in 120 ml of 20 mM Tris-HCl, pH 8, shaken for 3 min, and sonicated on ice−10 × 1 min, with 1-min pauses (Sonics Vibracell; LABOPLUS, ModelCV18head). One hundred milliliters of 0.4 M Na2CO3 (pH 11) were added to the suspension of the lysed cells and the sample was centrifuged to remove the unlysed cells and cell membrane fragments (6,000 × g, 20 min, 4°C) and filtered (filter with 0.22 μm pore size). Proteins were precipitated from the solution with trichloroacetic acid (TCA) [1:10(v/v)]. The precipitation was performed overnight at 4°C, and then the sample was centrifuged (20,000 × g, 10 min, 4°C). The aqueous phase was discarded and the protein pellet was resuspended in 0.5 ml of methanol precooled to 4°C, and then centrifuged (20,000 × g, 10 min, 4°C). The resulting precipitate was dried. The proteins were stored at −80°C. The analysis was performed in triplicate.
Identification of Proteins
The identification of proteins was performed by LC-MS-MS/MS (liquid chromatography coupled to tandem mass spectrometry) using a Nano-Acquity (Waters) LC system and Orbitrap Velos mass spectrometer (Thermo Electron Corp., San Jose, CA). Prior to the analysis, proteins were subjected to a standard “in-solution digestion” procedure during which proteins were reduced with 50 mM Tris(2-carboxyethyl)phosphine (for 60 min at 60°C), alkylated with 200 mM S-methyl methanethiosulfonate (45 min at room temperature) and digested overnight with trypsin (Sequencing Grade Modified Trypsin; Promega V5111). The peptide mixture was applied to an RP-18 precolumn (nanoACQUITY Symmetry® C18; Waters 186003514) using water containing 0.1% trifluoroacetic acid as the mobile phase and then transferred to a nano-HPLC RP-18 column (nanoACQUITY BEH C18; Waters 186003545) using an acetonitrile (ACN) gradient (5–35% ACN in 180 min) in the presence of 0.05% formic acid with a flow rate of 250 μl/min. The column outlet was directly coupled to the ion source of the spectrometer working in the regime of data dependent MS to MS/MS switch. A blank run ensuring a lack of cross-contamination from previous samples preceded each analysis. Acquired raw data were processed by Mascot Distiller followed by Mascot Search (Matrix Science, London, UK, on-site license) against the NCBI nr database. Peptides with a Mascot Score exceeding a threshold value corresponding to < 5% expectation value, calculated by Mascot procedure, were considered to have been positively identified.
Pyrolytic Analysis
The pyrolytic analysis of each sample was performed using the Rock-Eval technique. This involved thermal decomposition of rock samples (100 mg) in two cycles, pyrolytic and oxidizing, respectively (Lafargue et al., 1998). In the pyrolysis step, the sample was heated to 300°C in a nitrogen atmosphere to release the S1 fraction (mg HC/g rock) made of volatile compounds. This stage was followed by heating the sample to 650°C to release the S2 (mg HC/g rock), S3CO2 (mg CO2/g rock), and S3CO (mg CO/g rock) fractions. S1 and S2 were measured with a flame ionization detector, while S3 with infrared spectroscopy. The S2 fraction represented the products of kerogen cracking and the S3 fraction derived from oxygen-containing moieties (e.g., aldehydes and alcohols). In the second cycle, the sample was heated to 850°C in an oxygen atmosphere to release carbon monoxide and carbon dioxide from the residual and unproductive organic matter and mineral matter. The obtained results were converted into pyrolyzed organic carbon content (PC), residual organic carbon content (RC), and total organic carbon content (TOC). Other important parameters provided by the method are the hydrogen index (HI) and the oxygen index (OI), which allow the characterization of the type of organic matter and petroleum potential of the rock. The analysis was performed in triplicate.
Extraction of Organic Compounds
The sample was dried at 60°C and powdered. Organic compounds were extracted from 20 g of sample using a mixture of dichloromethane/methanol (9:1; v/v) for 24 h with a Soxhlet extraction apparatus. The solvent was evaporated with an N2 stream and then the sample was derivatized with 0.5 ml of BSTFA:TMCS (N,O-bis(Trimethylsilyl)trifluoroacetamide: Trimethylchlorosilane), 99:1 (Supelco, USA), for 30 min at 70°C. A blank sample was prepared according to the same procedure. All analyses were performed in triplicate.
Analysis of Extractable Organic Compounds
The separation of organic compounds was performed using an Agilent 7890A Series Gas Chromatograph (GC) interfaced to an Agilent 5973c Network Mass Selective Detector and an Agilent 7683 Series Injector (Agilent Technologies, USA). A 5 μl sample was introduced (split by 0.3% SD) to an HP-5MS column (30 m × 0.25 mm I.D., 0.25 μm film thickness, Agilent Technologies, USA) using He as the carrier gas at 1 ml/min. The ion source was maintained at 250°C; the GC oven was programmed with a temperature gradient starting at 100°C (for 3 min) and this was gradually increased to 300°C (for 5 min) at 8°C/min. A mass spectrometry (MS) analysis was performed in electron-impact mode at an ionizing potential of 70 eV. Mass spectra were recorded from m/z 40 to 800 (0–30 min).
Selection, Identification and Classification of Organic Compounds
The identification of organic compounds was performed with an Agilent Technologies Enhanced ChemStation (G1701EA ver. E.02.00.493) and The Wiley Registry of Mass Spectral Data (version 3.2, Copyright 1988–2000 by Palisade Corporation, 8th Edition with Structures, Copyright 2000 by John Wiley and Sons, Inc.) using a 3% cut-off threshold. The selected peaks representing organic compounds whose mass spectra indicated compliance with reference mass spectra equal to or higher than 80% were identified using the mass spectra library. The rest of the organic compounds representing lower compliance (50–79%) were assigned to the major classes of organic compounds based on the presence of characteristic and dominating fragmentation ions (aromatic hydrocarbons–m/z 65, 77, 78, 79; aliphatic hydrocarbons–m/z 43, 57, 71, 85, 99; alcohols–m/z 45, 59, 73, 87; aldehydes–m/z 44, 58, 72; carboxylic acids–m/z 43, 45, 57, 59, 60, 71, 73, 85, 87) (Silverstein et al., 2014).
Sulfur Speciation Analysis
The total sulfur concentration was determined by infrared absorption spectroscopy using the Leco method (Leco Application Notes No ASTM E1915). The mean detection limit for this method is 0.02 wt%.
Sulfur speciation analysis was conducted using the methods described by Shimp et al. (1977) and Tuttle et al. (1986). Sulfates were extracted from the shale with hydrochloric acid (6 M) and precipitated from the obtained solution by barium chloride (10 wt%). The barium sulfate precipitate was filtered, calcined (925°C) and weighed and the content of sulfate sulfur was calculated from its mass. Pyrite included in the shale (after extraction of non-pyrite iron) was extracted with nitric acid (18 wt%). Iron was oxidized and its amount was determined using inductively coupled plasma-atomic emission spectrometry. The content of pyritic sulfur was calculated from the amount of oxidized iron(III). Sulfides were separated in an acid medium and absorbed to a solution of zinc acetate (1.0 wt%). Their amount was determined using the colorimetric method based on the reaction of N,N-dimethyl-p-phenylenediamine dihydrochloride in the presence of ferric chloride(III), which results in the formation of methylene blue. The organic sulfur content was calculated by subtracting the sulfate, pyrite and sulfite sulfur content from the total sulfur content. All analyses were performed in triplicate.
Results and Discussion
Taxonomic Diversity of LMC
The taxonomic composition of LMC inhabiting the studied black shale based on the Ion TorrentTM next-generation sequencing of the metagenome is presented in Figure 2, and in Supplementary Presentation 1. The rarefaction curve tended to approach the saturation plateau (Figure S1). LMC consisted of 23 bacterial phyla accounting for 78% of the total reads, as well as two phyla of archaea (0.4%), and two phyla of fungi (0.1%). Moreover, 0.02% of total reads belonged to viruses and the rest (21%) were unclassified (Figure 2A). Two phyla of bacteria dominated in LMC: Proteobacteria (55%) mostly represented by γ-Proteobacteria class (29%), and Actinobacteria (11%) (Figures 2B,C). Nitrospirae constituted 2% of the total reads, the next two bacterial phyla (Bacteroidetes and Chloroflexi) accounted for 1%, while the remaining 18 phyla represented < 1%. Pseudomonadaceae (20%) was the most abundant family and it was dominated by the Pseudomonas stutzeri group (14%) (Figure 2D). Six other bacterial families represented from 1.9 to 8% of the total reads: Microbacteriaceae (8%), Burkholderiaceae (7%), Acidiferrobacteraceae (4%), Bradyrhizobiaceae (3%), Hydrogenophilaceae (3%), and Nitrospiraceae (1.9%). Besides the genus Pseudomonas, six other genera and one species were dominant: Limnobacter (7%), Yonghaparkia (4%), Thiobacillus (3%), Sulfuricaulis limicola (3%), Bradyrhizobium (3%), Nitrospira (1.8%), and Microbacterium (1%) (Figure 2D). Thaumarchaeota (Nitrososphaeria) and Euryarchaeota (Methanomicrobia, Halobacteria, and Thermoplasmata) were the identified archaeal phyla, accounting for 0.2 and 0.1% of the total reads, respectively (Figures 2B,C). Two fungal phyla, Ascomycota (Eurotiomycetes, Sordariomycetes, and Dothideomycetes) and Basidiomycota (Agaricomycetes), constituting 0.09 and 0.02%, respectively, were also detected (Figures 2B,C).
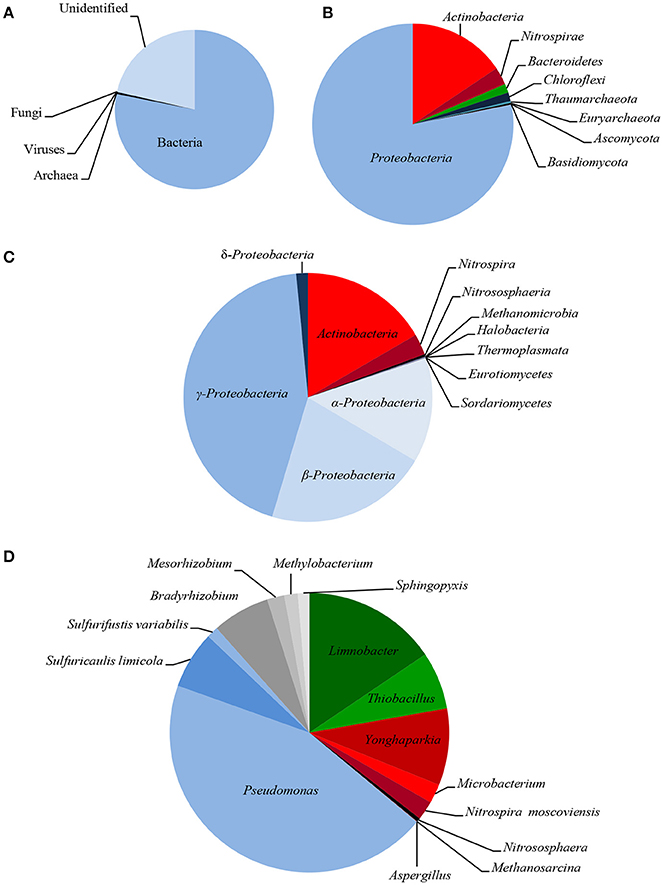
Figure 2. Taxonomic diversity of LMC: microbial superkingdoms (Bacteria, Archaea and Viruses) and kingdom (Fungi) (A); phyla (B); classes (C) and genera/species (D) (bacterial phyla/classes ≥ 1%, genera/species ≥ 0.5%; archaeal and fungal taxons ≥ 0.01%). The detailed taxonomic diversity of LMC is presented in Supplementary Presentation 1.
So far, little is known about the microorganisms inhabiting black shales. The most commonly known cultivable bacteria isolated from terrestrial and subterrestrial black shales have been reported previously. Petsch et al. (2001, 2005) showed that microorganisms inhabiting Late Devonian New Albany Shale (USA) are represented by Pseudomonas spp., Acinetobacter spp., Dechloromonas spp., Clostridium spp., and Comamonas spp. Berlendis et al. (2014) detected Ralstonia picketii in Lower Autunian Autun black shale (France). Only heterotrophic bacteria such as Pseudomonas spp., Bacillus spp., Microbacterium spp. and Acinetobacter spp. as well as Rhodotorulla mucilaginosa yeast have previously been isolated from subterrestrial Kupferschiefer black shale (Matlakowska and Skłodowska, 2009; Rajpert et al., 2013). Only the studies by Li et al. (2014) into the bacterial biodiversity of the terrestrial exposed profile of the Lower Cambrian black shale from Chengkou County (China) have been based on an analysis of the sequence of the amplified 16S rRNA gene. They revealed that the shale was inhabited by bacteria belonging to 33 different groups and here Proteobacteria, Actinobacteria, and Firmicutes were dominant. The presented research is, therefore, the first report on metagenome-based taxonomic bacterial diversity in black shale and the first insight into the diversity of archaea and fungi inhabiting black shale.
General Characteristics of Metagenome and Metaproteome
To identify the metabolic processes of LMC as well as indicate the microorganisms crucial for the oxidation of fossil hydrocarbons and sulfide minerals, an analysis of its metagenome and metaproteome was conducted.
In the metagenome of LCM, 446,915 bacterial, 3,529 archaeal and 1,415 fungal PEGs were identified. Interestingly, among the identified PEGs, 112412 (25%), 1245 (35%), and 866 (61%) bacterial, archeal and fungal proteins, respectively, had no meaningful annotation (described as either hypothetical or unknown protein, or protein of unknown function).
The metaproteome of LMC comprised 788 bacterial, 694 archaeal, and 655 fungal proteins. Among these, 376 bacterial, 503 archaeal, and 455 fungal proteins were identified as high-confidence proteins matching two or more peptides or possessing one characteristic peptide (occurring only in this specific protein). About 60% of bacterial and archaeal high-confidence proteins, as well as 28% of fungal high-confidence proteins, constituted housekeeping proteins. The examined LMC was also enriched in novel proteins of unknown function (hypothetical proteins) representing nearly 70% of fungal and 30–25% of bacterial and archaeal high-confidence proteins.
Oxidative Metabolism of Fossil Hydrocarbons
In total, 5,748 bacterial, 47 archaeal and 8 fungal PEGs identified in the metagenome of LMC were potentially involved in the oxidative metabolism of aromatic and aliphatic hydrocarbons, as well as the metabolism of alcohols, aldehydes and ketones (Figure 3). A detailed summary of hydrocarbon metabolism-related PEGs is presented in Supplementary Data Sheet 1 and Tables S1–S3. Among these, bacterial mono- and dioxygenases (2373 URs), aldehyde dehydrogenases (1106 URs), and alcohol dehydrogenases (912 URs) were dominant (Figure 3A). Bacterial laccase encoding genes (51 URs) were also identified. In the archaeal PEGs the genes encoding alcohol dehydrogenases prevailed (31 URs) (Figure 3B), and in the fungal PEGs the genes encoding alcohol dehydrogenases (3 URs) and cytochrome P450 (3 URs) were predominant (Figure 3C).
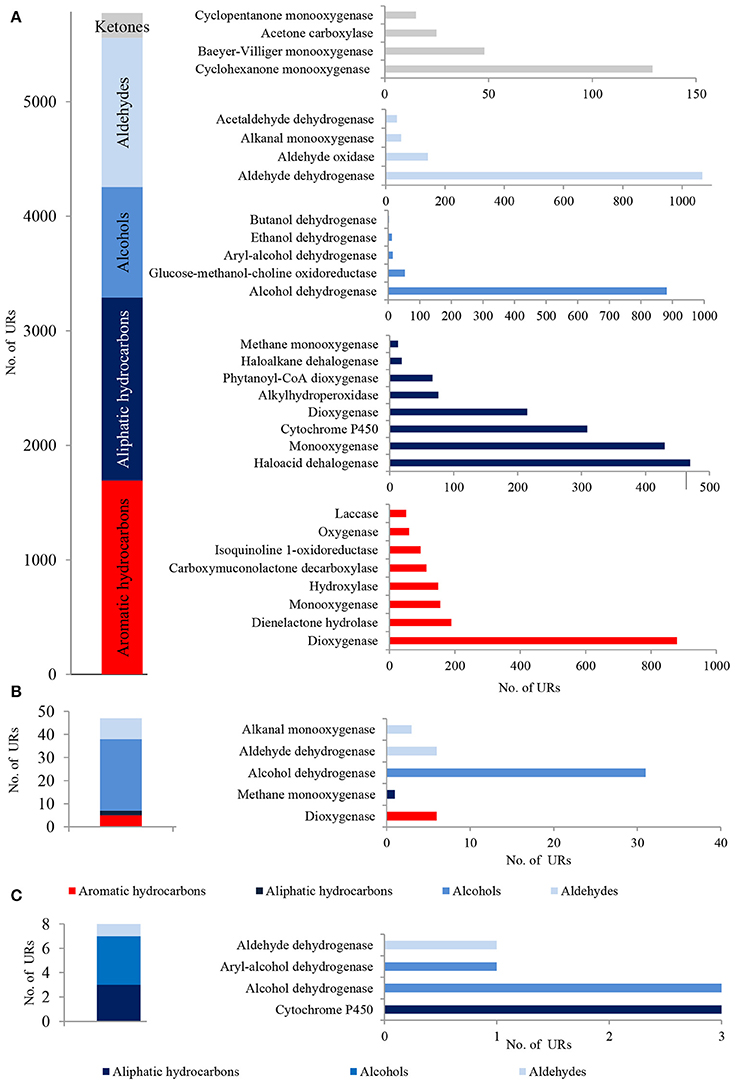
Figure 3. The unique bacterial (A), archaeal (B) and fungal (C) PEGs detected in LMC involved in the oxidative metabolism of hydrocarbons, alcohols, aldehydes and ketones. The detailed list of the PEGs is presented in Tables S1–S3 and in Supplementary Data Sheet 1.
A considerable number of identified PEGs potentially involved in the metabolism of hydrocarbons were derived from Pseudomonas spp. (451 URs) and Bradyrhizobium spp. (447 URs), as well as Sphingopyxis spp. (193 URs), Limnobacter spp. (191 URs), and Microbacterium spp. (133 URs) (Figure 4A; Tables S1–S3). Bradyrhizobium PEGs could play a crucial role in the oxidation of aliphatic (179 URs) and aromatic hydrocarbons (136 URs) present in black shales. Among the Pseudomonas, PEGs were also involved in the oxidative metabolism of aromatic (107 URs) and aliphatic (66 URs) hydrocarbons, as well as alcohols (147 URs) and aldehydes (129 URs) (Figures 4A,B). PEGs of Bradyrhizobium were derived from various strains, whereas most Pseudomonas PEGs originated from bacteria belonging to the P. stutzeri group (Figures 4C,D). Archaeal and fungal PEGs potentially engaged in hydrocarbon metabolism were mainly derived from Nitrososphaera spp. (38 URs) and Aspergillus sp. (4 URs), respectively (Tables S2 and S3). The examined LMC is the first described subsurface community comprised of Limnobacter spp. potentially involved in hydrocarbon metabolism (Figure 4A). The identified Limnobacter genes encoded cytochrome P450, phenol 2-monooxygenases, monooxygenase flavin-binding family proteins, 4-hydroxyacetophenone monooxygenases, quercetin 2,3-dioxygenases, catechol 2,3-dioxygenases, 4-carboxymuconolactone decarboxylases and haloacid dehalogenases (Supplementary Data Sheet 1). Phenol-degrading Limnobacter strains possessing catechol 2,3-dioxygenases had previously been isolated from Baltic Sea surface water (Vedler et al., 2013). Another bacteria potentially participating in hydrocarbon metabolism was Yonghaparkia spp. (Figure 4A). Bacteria belonging to this genus had previously been isolated from microbial communities detected in a gold mine (Linglong, China) (Li et al., 2016) and a uranium mine (Athabasca Basin, Canada) (Bondici et al., 2013), as well as from the black shale mentioned earlier (Chengkou County, China) (Li et al., 2014). Moreover, surprisingly numerous hydrocarbon metabolism-related PEGs, including dioxygenases, laccases, dienelactone hydrolases and haloacid dehalogenases, originated from two chemolithotrophic bacteria, S. limicola and Sulfurifustis variabilis (Figure 4A; Supplementary Data Sheet 1), isolated for the first time by Kojima et al. (2015, 2016).
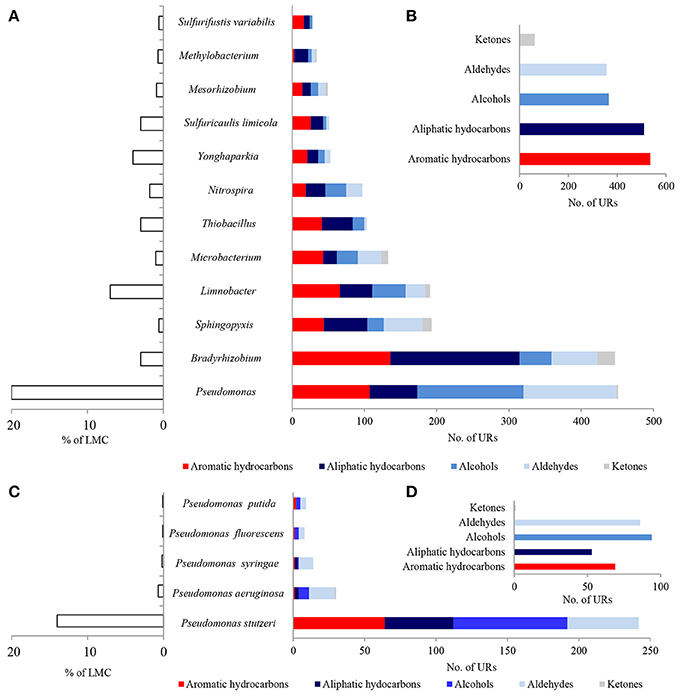
Figure 4. Taxonomic origin (A,C) and number (B,D) of bacterial PEGs detected in the dominating genera and species of bacteria of LMC and in bacteria belonging to genus Pseudomonas related to the oxidative metabolism of hydrocarbons, alcohols, aldehydes and ketones. The detailed list of PEGs is presented in Tables S1–S3 and Supplementary Data Sheet 1; the detailed taxonomic diversity of bacteria is presented in Supplementary Presentation 1.
The metaproteome of LMC comprised 21 bacterial, 13 archaeal and 15 fungal enzymes related to hydrocarbon metabolism (Figure 5; Tables S4–S6). They were dominated by bacterial enzymes participating in the aerobic metabolism of alcohols and aldehydes, including seven alcohol dehydrogenases and six aldehyde dehydrogenases in particular. Four detected dioxygenases were only archaeal enzymes, whereas nine cytochromes P450 were identified primarily among fungal enzymes.
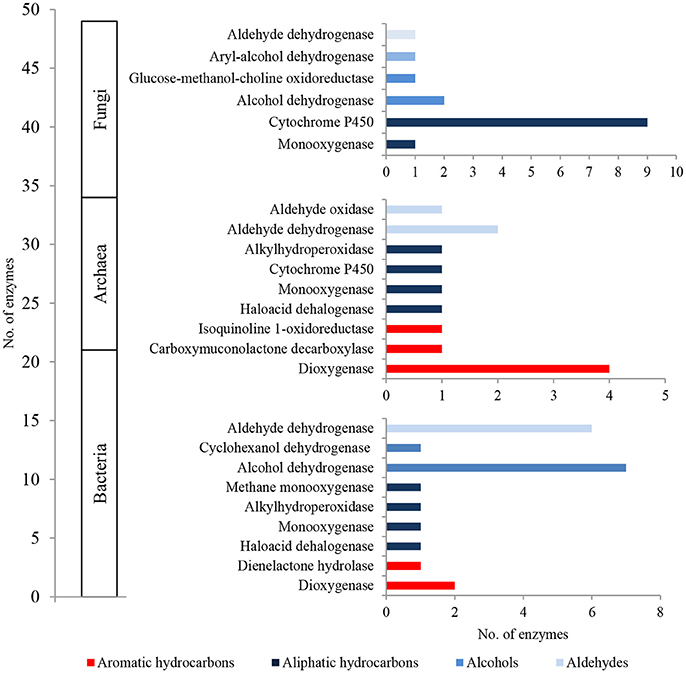
Figure 5. The unique bacterial, archaeal and fungal enzymes detected in the metaproteome of LMC involved in the oxidative metabolism of hydrocarbons, alcohols and aldehydes. The detailed list of the detected enzymes is presented in Tables S4–S6.
Furthermore, we also detected a number of bacterial, archaeal and fungal PEGs, as well as enzymes involved in the utilization of carboxylic acids (e.g., enzymes of the β-oxidation process, such as acyl-CoA dehydrogenase, 3-hydroxyacyl-CoA dehydrogenase and long-chain fatty acid - CoA ligase) and esters (e.g., sulfatase, carboxyesterase, and lipase) (unpresented results).
The presence of the above PEGs, as well as enzymes, demonstrates the potential ability of LMC toward the oxidative metabolism of aromatic and aliphatic hydrocarbons. Most of the identified PEGs and enzymes belong to aliphatic hydrocarbon metabolism pathways which consist of terminal or subterminal oxidation of aliphatic hydrocarbons to carboxylic acids (fatty acids) utilized in the β-oxidation process (Ji et al., 2013).
Investigations into fossil organic matter transformation occurring in black shales are limited to the studies by Petsch et al. (2001, 2005), Seifert et al. (2011), and Berlendis et al. (2014). The first of these proved that microorganisms inhabiting terrestrial Late Devonian New Albany Shale (USA) assimilate ancient organic carbon derived from organic matter in the shale. Berlendis et al. (2014) detected nag-like genes of R. picketii inhabiting terrestrial Lower Autunian Autun black shale (France) which could encode for dioxygenase genes. A research study conducted by Seifert et al. (2011) unequivocally confirmed the ability of indigenous bacteria to mobilize carbon from a Lower Silurian Variscan graptolite shale formation (Germany) and incorporate it into fatty acid metabolism. However, the molecular factors responsible for these processes were not indicated, nor was the influence of these processes on properties of kerogen.
Oxidative Metabolism of Inorganic Sulfur Compounds
796 bacterial PEGs related to the oxidative metabolism of reduced sulfur compounds were detected in the metagenome of the studied LMC (Figure 6). Archaeal and fungal PEGs related to this process were not detected. A detailed summary of PEGs involved in this process is presented in Supplementary Data Sheet 1 and Table S1.
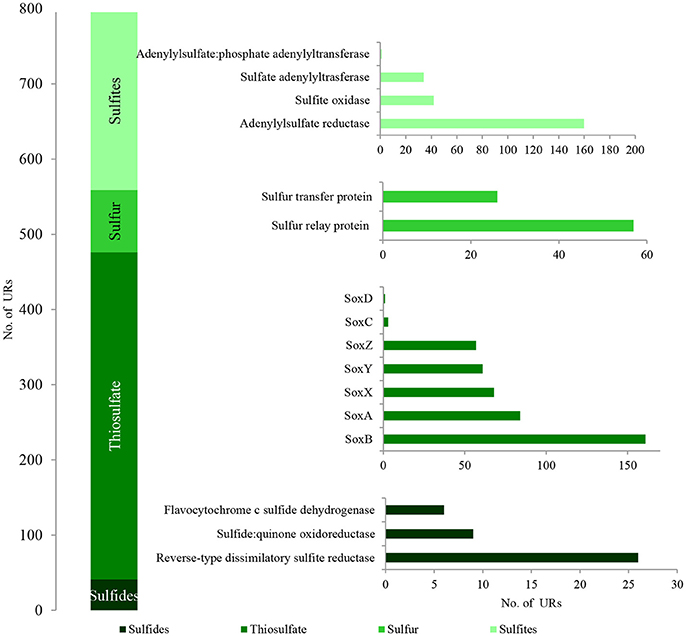
Figure 6. The unique bacterial PEGs detected in LMC involved in the oxidative metabolism of sulfide minerals. The detailed list of PEGs is presented in Table S1 and Supplementary Data Sheet 1.
Among the identified PEGs, the most important were those potentially involved in the dissimilative oxidation of sulfides (Ghosh and Dam, 2009). Sulfide:quinone oxidoreductase (9 URs) involved in sulfide oxidation to sulfur was detected in T. denitrificans, Bradyrhizobium spp., and Nitrospira spp. (Figures 7A,B; Table S1). In addition, flavocytochrome c sulfide dehydrogenase (6 URs) involved in sulfide oxidation was also identified in T. denitrificans (Figures 7C,D). This enzyme catalyzes the conversion of sulfides to elemental sulfur or polysulfide and it is assumed to be the main sulfide oxidizing system in Thiobacillus genus. Furthermore, reverse-type dissimilatory siroheme sulfite reductase (RDsr) (26 URs) participating in the reverse system of dissimilatory sulfite reduction was identified in T. thioparus (Figures 7C,D).
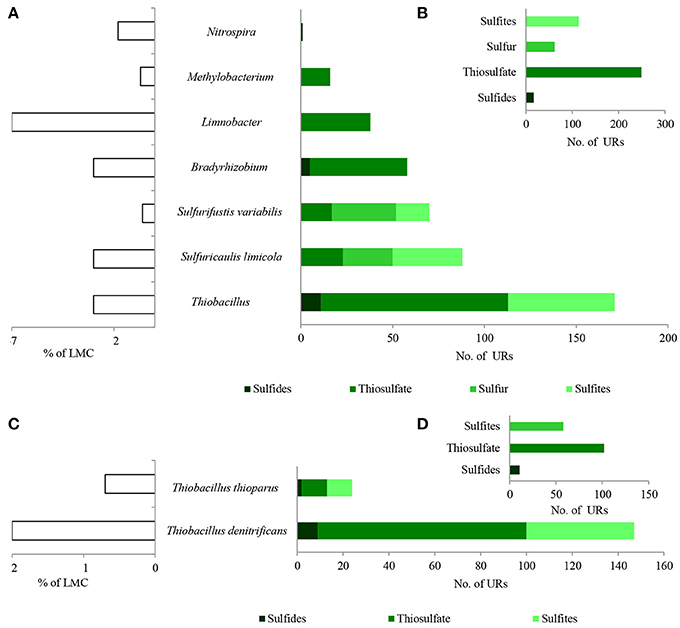
Figure 7. Taxonomic origin (A,C) and number (B,D) of bacterial PEGs detected in the dominating genera and species of bacteria of LMC and two species of Thiobacillus related to the oxidative metabolism of sulfides, thiosulfate, sulfur and sulfites. The detailed list of PEGs is presented in Table S1 and Supplementary Data Sheet 1; the detailed taxonomic diversity of bacteria is presented Supplementary Presentation 1. URs, unique reads.
Direct oxidation of thiosulfate to sulfate catalyzed by the thiosulfate-oxidizing multi-enzyme system, also known as the Sox enzyme system (Friedrich et al., 2001), was also detected in LMC (435 URs) (Figure 6). PEGs for all components of the Sox system (soxX, soxA, soxB, soxC, soxD, soxY, and soxZ) were present in the metagenome of LMC. These were detected mainly in bacteria belonging to genus Thiobacillus (102 URs), especially T. denitrificans (91 URs) as well as Bradyrhizobium (53 URs), Limnobacter (38 URs), S. limicola (23 URs), S. variabilis (17 URs), and Methylobacterium (16 URs) (Figure 7). To conclude, in the studied LMC, thiosulfate oxidation in both known pathways (to sulfates or to sulfates and sulfur) is potentially possible (Wodara et al., 1997). Beside thiosulfate, this molecular mechanism has been proved to oxidize other reduced sulfur species such as sulfide, elemental sulfur, sulfite and tetrathionate (Wodara et al., 1997).
The sulfite oxidation pathway mediated by adenylylsulfate reductase (160 URs), sulfate adenylyltransferase (34 URs) and adenylylsulfate:phosphate adenylyltransferase (1 UR) were detected in S. limicola, T. denitrificans, S. variabilis, and T. thioparus (Kappler and Dahl, 2001) (Figure 7A). Furthermore, a universal system of direct sulfite oxidation by sulfite oxidase (Kappler and Dahl, 2001) (42 URs) was identified in S. limicola, S. variabilis and Rhodoplanes sp. (Table S1).
Finally, PEGs potentially participating in the oxidation of sulfur - sulfur relay proteins (57 URs) and sulfur transfer proteins (26 URs) were detected in S. limicola and S. variabilis (Figure 7A).
In summary, a considerable number of genes potentially involved in inorganic sulfur metabolism originated from Thiobacillus chemolithotrophic neutrophilic bacteria (171 URs), mostly T. denitrificans (147 URs) (Figures 7C,D) and were potentially involved in sulfide, thiosulfate and sulfite oxidation. PEGs related to the oxidation of these three reduced sulfur compounds also originated from two chemolithotrophic bacteria: S. limicola (88 URs), and S. variabilis (70 URs) (Figure 7A). Sulfur compound oxidation PEGs were also identified in Bradyrhizobium spp. (58 URs), Limnobacter spp. (38 URs) and Methylobacterium spp. (16 URs) (Figure 7A), although they were mostly involved in thiosulfate oxidation. Some of these bacteria are known to be capable of chemolithoheterotrophic growth, oxidizing sulfur compounds in the presence of an organic carbon source (Spring et al., 2001; Lu et al., 2011).
In the metaproteome of the tested LMC, only two bacterial enzymes (cytochrome SoxA and DsrH protein) related to the oxidative metabolism of inorganic sulfur compounds were identified (Table S4). Archaeal and fungal enzymes related to this process were not detected.
Taken together, the presence of the described PEGs shows the potential role of LMC in the dissimilative oxidation of reduced sulfur compounds which are used as energy sources and electron donors. Several reduced sulfur compound-oxidizing pathways were detected (Figure 6). The first potential pathway is the most widespread system based on the oxidation of sulfides, sulfur and thiosulfate via sulfites to sulfates, while the second one is direct oxidation of thiosulfate to sulfates (Ghosh and Dam, 2009). Two potential pathways of sulfite oxidation were also detected—direct oxidation to sulfates and indirect oxidation with adenosine phosphosulfate as an intermediate (Kappler and Dahl, 2001). The very small number of enzymes associated with sulfur oxidation is surprising; however, this can be explained firstly by the fact that the content of key sulfur-oxidizing bacteria (Thiobacillus spp., S. limicola, S. variabilis) in LMC is small (about 7%) in comparison to main heterotrophic bacteria participating in hydrocarbon metabolism (Pseudomonas spp., Bradyrhizobium spp., Limnobacter spp., Microbacterium spp., Sphingopyxis spp.) (about 30%). Also the number of detected PEGs potentially involved in sulfur-compound oxidation is about seven times lower (796) than those involved in hydrocarbon metabolism (5,828). Secondly, it is supposed that the lack of expression of these enzymes may be related to the lack of reduced sulfur compounds in the studied weathered black shale, which is described in the next subsection of results.
Geochemistry of the Studied Black Shale
In order to confirm the potential metabolic activity of LMC, we examined the geochemical properties of the 12-year-old weathered black shale and we compared these to the properties of the unweathered shale studied earlier (Stasiuk et al., 2017). Our research included the comparison of: (i) the properties of kerogen and organic carbon content, (ii) the qualitative and semi-quantitative comparative composition of extractable organic matter, and (iii) the sulfur content and its speciation. The potential metabolic activity of LMC described above seems to correlate with geochemical properties of the studied black shale.
The pyrolytic analysis of black shale by the Rock-Eval method showed an unusually high ratio of oxygen to hydrogen in this rock compared with that of the unweathered black shale (Figure 8; Table S7) (Stasiuk et al., 2017), which may confirm the strong oxidation of kerogen. At the same time, we observed a considerably reduced (about 86%) content of Corganic dominated by residual carbon (91%) (Figure 9A). Similarly, a reduced content of free hydrocarbons (about 96%) and hydrocarbon potential (about 91%) confirmed the dehydrogenation of kerogen. All these parameters suggest the change of its type from oil-prone type II to a non-productive, residual and hydrogen-free kerogen IV, which is usually regarded as a product of strong chemical and/or biological degradation of kerogen type III or II.
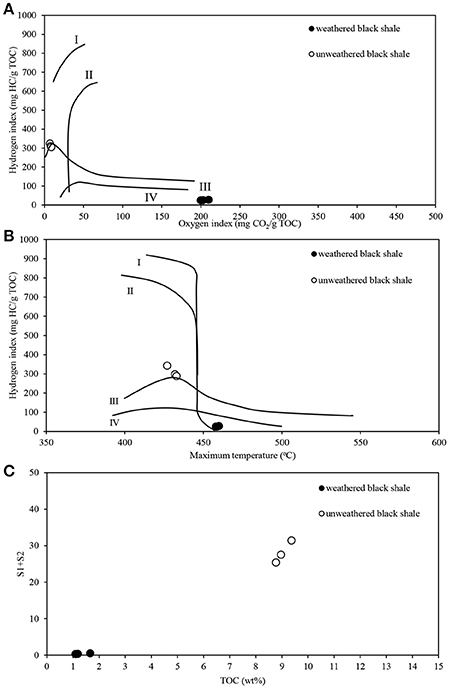
Figure 8. Pyrolytic characteristics of the weathered and unweathered black shale kerogen, obtained during Rock-Eval analysis. (A) Modified Van Krevelen diagram with the correlation of hydrogen (HI) and oxygen (OI) indexes; (B) HI and Tmax crossed-diagram; (C) Correlation of TOC and hydrocarbon potential (S1+S2). The detailed data of Rock-Eval analysis are presented in Table S7. The unweathered black shale is described in detail in Stasiuk et al. (2017).
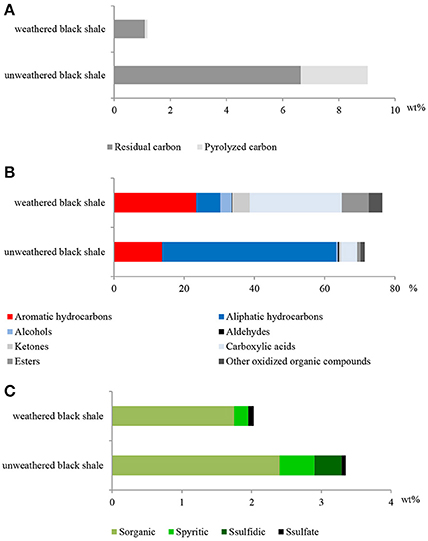
Figure 9. The content of organic carbon represented by pyrolized and residual carbon (A); selected classes of extractable organic compounds (B); content of sulfur and its speciation (C) in the weathered and unweathered black shales. The unweathered black shale is described in detail in Stasiuk et al. (2017).
Also, the composition of extractable organic matter in the studied black shale was unusual and the amount of this mobile matter was also higher compared with that of the unweathered black shale (Figure 9B; Table S7). The content of aromatic hydrocarbons and derivatives increased and at the same moment the content of aliphatic hydrocarbons decreased. These changes could be related to kerogen weathering and partial biodegradation of mainly aliphatic hydrocarbons. About 45% of all extractable organic compounds identified in this rock were oxygenated alcohols, aldehydes, ketones, esters, and above all carboxylic acids (Figure 9, Figure S2, Tables S7, S8). In the unweathered black shale, the content of oxidized organic compounds was estimated at 8–10% (Stasiuk et al., 2017). Numerous oxygenated organic compounds could have been formed with the participation of LMC. The identified carboxylic acids have been created as a result of the oxidative metabolism of fossil hydrocarbons present in black shales. This process can be proved by the detected intermediate products of hydrocarbon degradation, such as alcohols, ketones, aldehydes and esters (Tables S7, S8) (Leahy and Colwell, 1990; Rojo, 2009) as well as by the described PEGs and enzymes (Figures 3, 5). This result is consistent with the results of our previous long-term (365 days) laboratory studies conducted with a consortium of cultivable heterotrophic bacteria isolated from the Kupferschiefer black shale composed of Pseudomonas spp., Microbacterium spp., Bacillus sp., and Acinetobacter sp. In that studies the direct role of bacteria in the oxidation of fossil organic matter and changes of the kerogen type and hydrocarbon potential as well changes in the content and composition of extractable organic matter was showed (Stasiuk et al., 2017). These changes were not observed in the sterile control, where the black shale was exposed to the oxygen and water.
The potential oxidative metabolism of sulfidic minerals by LMC suggested by metagenomic studies (Figures 6, 7) is correlated with the lack of sulfidic sulfur and also loss of pyritic sulfur (0.2 wt%) in the studied black shales, which are normally abundant in the Kupferschiefer (0.39 and 0.50 wt%, respectively) (Figure 9C; Table S7) (Matlakowska et al., 2012). In the tested weathered black shale, no common ore sulfide minerals were detected either using X-ray diffraction or reflected-light microscopy (unpresented results). However, the content of sulfates in the weathered black shale was also very low. This could be related to: (i) the mobilization of sulfates by mine waters, (ii) the dissimilative reduction of sulfates to H2S by sulfate reducing bacteria present in LMC (Supplementary Presentation 1), or (iii) the assimilative reduction of sulfates by microorganisms. The first two processes are the most probable, because in the studied black shale we observed a significant decrease (about 39%) in the total sulfur content and simultaneous loss of organic sulfur (about 27%) compared to the unweathered black shale (Figure 9C).
Conclusions
Two catabolic microbial processes performed by two trophic groups of microorganisms were described in the deep subterrestrial black shale. One is the enzymatic dissimilative oxidation of fossil hydrocarbons mediated mostly by chemoorganotrophs. The other is the dissimilative enzymatic oxidation of primary reduced sulfur compounds conducted by chemolithotrophs. Twelve genera of bacteria dominate in LMC and play a crucial role in these processes (Figures 4, 7). The described microbial processes potentially play a role in the weathering of kerogen and sulfide minerals and cause organic carbon and inorganic sulfur mobilization from black shales.
Data Deposition
The results of metagenomic DNA sequencing are deposited at: http://www.ebi.ac.uk/ena/data/view/PRJEB21522.
Author Contributions
RM: concept of research and funding; GB, AW, and RM: Sampling; AW: DNA isolation protein isolation, and organic compound extraction; MK and AF: DNA sequencing; ML: Bioinformatics analyses; RM and AW: Manuscript preparation.
Funding
This work was supported with a research grant from the National Science Centre (Poland) (2012/07/B/NZ8/01904).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge Dr. Wojciech Kaczmarek (KGHM Polska Miedź S.A.) for organizing an expedition to the mine.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00972/full#supplementary-material
References
Berlendis, S., Beyssac, O., Derenne, S., Benzerara, K., Anquetil, C., Guillaumet, M., et al. (2014). Comparative mineralogy, organic geochemistry and microbial diversity of the Autun black shale and Graissessac coal (France). Int. J. Coal Geol. 132, 147–157. doi: 10.1016/j.coal.2014.07.005
Berner, R. A. (1989). Biogeochemical cycles of carbon and sulfur and their effect on atmospheric oxygen over Phanerozoic time. Global Planet. Change 48, 97–122. doi: 10.1016/0921-8181(89)90018-0
Bondici, V. F., Lawrence, J. R., Khan, N. H., Hill, J. E., Yergeau, E., Wolfaardt, G. M., et al. (2013). Microbial communities in low permeability, high pH uranium mine tailings: characterization and potential effects. J. Appl. Microbiol. 114, 1671–1686. doi: 10.1111/jam.12180
Bryner, L. C., Beck, J. V., Davis, B. B., and Wilson, D. G. (1954). Microorganisms in leaching sulfide minerals. Ind. Eng. Chem. 46, 2587–2592. doi: 10.1021/ie50540a051
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Friedrich, C. G., Rother, D., Bardischewsky, F., Quentmeier, A., and Fischer, J. (2001). Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl. Environ. Microbiol. 67, 2873–2882. doi: 10.1128/AEM.67.7.2873-2882.2001
Ghosh, W., and Dam, B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 33, 999–1043. doi: 10.1111/j.1574-6976.2009.00187.x
Huerta-Cepas, J., Serra, F., and Bork, P. (2016). ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 33, 1635–1638. doi: 10.1093/molbev/msw046
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Ji, Y., Mao, G., Wang, Y., and Bartlam, M. (2013). Structural insights into diversity and n-alkane biodegradation mechanisms of alkane hydroxylases. Front. Microbiol. 4:58. doi: 10.3389/fmicb.2013.00058
Kappler, U., and Dahl, C. (2001). Enzymology and molecular biology of prokaryotic sulfite oxidation. FEMS Microbiol. Lett. 203, 1–9. doi: 10.1111/j.1574-6968.2001.tb10813.x
Kojima, H., Shinohara, A., and Fukui, M. (2015). Sulfurifustis variabilis gen. nov., sp. nov., a sulfur oxidizer isolated from a lake, and proposal of Acidiferrobacteraceae fam. nov. and Acidiferrobacterales ord. nov. Int. J. Syst. Evol. Microbiol. 65, 3709–3713. doi: 10.1099/ijsem.0.000479
Kojima, H., Watanabe, T., and Fukui, M. (2016). Sulfuricaulis limicola gen. nov., sp. nov., a sulfur oxidizer isolated from a lake. Int. J. Syst. Evol. Microbiol. 66, 266–270. doi: 10.1099/ijsem.0.000709
Lafargue, E., Marquis, F., and Pillot, D. (1998). Rock-Eval 6 applications in hydrocarbon exploration, production, and soil contamination studies. Oil Gas Sci. Technol. 53, 421–437. doi: 10.2516/ogst:1998036
Leahy, J. G. (1990). Microbial degradation of hydrocarbons in the environment. Microbial Rev. 54, 305–315.
Li, J., Sun, W., Wang, S., Sun, Z., Lin, S., and Peng, X. (2014). Bacteria diversity, distribution and insight into their role in S and Fe biogeochemical cycling during black shale weathering. Environ. Microbiol. 16, 3533–3547. doi: 10.1111/1462-2920.12536
Li, M., Tianc, H., Wang, L., and Duan, J. (2016). Bacterial diversity in Linglong gold mine, China. Geomicrobiol. J. 34, 267–273. doi: 10.1080/01490451.2016.1186763
Lu, H., Sato, Y., Fujimura, R., Nishizawa, T., Kamijo, T., and Ohta, H. (2011). Limnobacter litoralis sp. nov., a thiosulfate-oxidizing, heterotrophic bacterium isolated from a volcanic deposit, and emended description of the genus Limnobacter. Int. J. Syst. Evol. Microbiol. 61, 404–407. doi: 10.1099/ijs.0.020206-0
Matlakowska, R., and Skłodowska, A. (2009). The culturable bacteria isolated from organic-rich black shale potentially useful in biometallurgical procedures. J. Appl. Microbiol. 107, 858–866. doi: 10.1111/j.1365-2672.2009.04261.x
Matlakowska, R., Skłodowska, A., and Nejbert, K. (2012). Bioweathering of Kupferschiefer black shale (Fore-Sudetic Monocline, SW Poland) by indigenous bacteria: implication for dissolution and precipitation of minerals in deep underground mine. FEMS Microbiol. Ecol. 81, 99–110. doi: 10.1111/j.1574-6941.2012.01326.x
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385
Oszczepalski, S. (1999). Origin of the Kupferschiefer polymetallic mineralization in Poland. Miner. Deposita 34, 599–613. doi: 10.1007/s001260050222
Petsch, S. T., Edwards, K. J., and Eglinton, T. I. (2005). Microbial transformations of organic matter in black shales and implications for global biogeochemical cycles. Palaeogeogr. Palaeoclimatol. Palaeoecol. 219, 157–170. doi: 10.1016/j.palaeo.2004.10.019
Petsch, S. T., Eglinton, T. I., and Edwards, K. J. (2001). 14C-dead living biomass: evidence for microbial assimilation of ancient organic carbon during shale weathering. Science 292, 1127–1131. doi: 10.1126/science.1058332
Rajpert, L., Skłodowska, A., and Matlakowska, R. (2013). Biotransformation of copper from Kupferschiefer black shale (Fore-Sudetic Monocline, Poland) by yeast Rhodotorula mucilaginosa LM9. Chemosphere 91, 1257–1265. doi: 10.1016/j.chemosphere.2013.02.022
Ram, R. J., VerBerkmoes, N. C., Thelen, M. P., Tyson, G. W., Baker, B. J., Blake, R. C., et al. (2005). Community proteomics of a natural microbial biofilm. Science 308, 1915–1920. doi: 10.1126/science.1109070
Rojo, F. (2009). Degradation of alkanes by bacteria. Environ. Microbiol. 11, 2477–2490. doi: 10.1111/j.1462-2920.2009.01948.x
Sawłowicz, Z. (1989). Organic matter in the Zechstein Kupferschiefer from the Fore-Sudetic Monocline. I. Bitumen. Mineral. Polon. 20, 69–86.
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Seifert, A. G., Trumbore, S., Xu, X., Zhang, D., Kothe, E., and Gleixner, G. (2011). Variable effects of labile carbon on the carbon use of different microbial groups in black slate degradation. Geochim. Cosmochim. Acta 75, 2557–2570. doi: 10.1016/j.gca.2011.02.037
Shimp, N. F., Kuhn, J. K., and Helfinstine, R. J. (1977). Determination of forms of sulfur in coal. Energy Sour. 3, 93–109. doi: 10.1080/00908317708945972
Silverstein, R. M., Webster, F. X., Kiemle, D. J., and Bryce, D. L. (2014). Spectrometric Identification of Organic Compounds. Hoboken, NJ: John Wiley and Sons.
Speczik, S. (1994). Kupferschiefer mineralization in the light of organic geochemistry and coal petrology studies. Geol. Q. 38, 639–650.
Spring, S. P., Kämpfer, P., and Schleifer, K. H. (2001). Limnobacter thiooxidans gen. nov., sp. nov., a novel thiosulfate-oxidizing bacterium isolated from freshwater lake sediment. Int. J. Syst. Evol. Microbiol. 51, 1463–1470. doi: 10.1099/00207713-51-4-1463
Stasiuk, R., Włodarczyk, A., Karcz, P., Janas, M., Skłodowska, A., and Matlakowska, R. (2017). Bacterial weathering of fossil organic matter and organic carbon mobilization from subterrestrial Kupferschiefer black shale - long-term laboratory studies. Environ. Microbiol. Rep. 9, 459–466. doi: 10.1111/1758-2229.12559
Tange, O. (2011). GNU parallel: the command-line power tool. USENIX Magaz. 36, 42–47. doi: 10.5281/zenodo.16303
Tourtelot, H. A. (1979). Black shale deposition and diagenesis. Clays Clay Miner. 27, 313–321. doi: 10.1346/CCMN.1979.0270501
Tuttle, M. L., Goldhaber, M. B., and Williamson, D. L. (1986). An analytical scheme for determining forms of sulfur in oil shales and associated rocks. Talanta 33, 953–961. doi: 10.1016/0039-9140(86)80234-X
Vedler, E., Heinaru, E., Jutkina, J., Viggor, S., Koressaar, T., Remm, M., et al. (2013). Limnobacter spp. as newly detected phenol-degraders among Baltic Sea surface water bacteria characterised by comparative analysis of catabolic genes. Syst. Appl. Microbiol.36, 525–532. doi: 10.1016/j.syapm.2013.07.004
Włodarczyk, A., Szymanska, A., Skłodowska, A., and Matlakowska, R. (2016). Determination of factors responsible for the bioweathering of copper minerals from organic-rich copper-bearing Kupferschiefer black shale. Chemosphere 148, 416–425. doi: 10.1016/j.chemosphere.2016.01.062
Wodara, C., Bardischewsky, F., and Friedrich, C. G. (1997). Cloning and characterization of sulfite dehydrogenase, two c-type cytochromes, and a flavoprotein of Paracoccus denitrificans GB17: essential role of sulfite dehydrogenase in lithotrophic sulfur oxidation. J. Bacteriol. 179, 5014–5023. doi: 10.1128/jb.179.16.5014-5023.1997
Keywords: black shale, kerogen, fossil hydrocarbons, sulfide minerals, metagenome, metaproteome
Citation: Włodarczyk A, Lirski M, Fogtman A, Koblowska M, Bidziński G and Matlakowska R (2018) The Oxidative Metabolism of Fossil Hydrocarbons and Sulfide Minerals by the Lithobiontic Microbial Community Inhabiting Deep Subterrestrial Kupferschiefer Black Shale. Front. Microbiol. 9:972. doi: 10.3389/fmicb.2018.00972
Received: 13 February 2018; Accepted: 25 April 2018;
Published: 15 May 2018.
Edited by:
Magdalena R. Osburn, Northwestern University, United StatesReviewed by:
Lily Melinda Momper, Massachusetts Institute of Technology, United StatesMorgan Volker Evans, The Ohio State University, United States
Copyright © 2018 Włodarczyk, Lirski, Fogtman, Koblowska, Bidziński and Matlakowska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renata Matlakowska, cm1hdGxha293c2thQGJpb2wudXcuZWR1LnBs