 Florian Tagini1
Florian Tagini1 Trestan Pillonel1
Trestan Pillonel1 Antony Croxatto1
Antony Croxatto1 Claire Bertelli1
Claire Bertelli1 Angela Koutsokera2Alban Lovis2
Angela Koutsokera2Alban Lovis2 Gilbert Greub1,3*
Gilbert Greub1,3*- 1Institute of Microbiology, Department of Laboratory Medicine, Lausanne University Hospital, Lausanne University, Lausanne, Switzerland
- 2Division of Pulmonology, Department of Medicine, Lausanne University Hospital, Lausanne, Switzerland
- 3Division of Infectious Diseases, Department of Medicine, Lausanne University Hospital, Lausanne, Switzerland
Corynebacterium diphtheriae is the etiological agent of diphtheria, a disease caused by the presence of the diphtheria toxin. However, an increasing number of records report non-toxigenic C. diphtheriae infections. Here, a C. diphtheriae strain was recovered from a patient with a past history of bronchiectasis who developed a severe tracheo-bronchitis with multiple whitish lesions of the distal trachea and the mainstem bronchi. Whole-genome sequencing (WGS), performed in parallel with PCR targeting the toxin gene and the Elek test, provided clinically relevant results in a short turnaround time, showing that the isolate was non-toxigenic. A comparative genomic analysis of the new strain (CHUV2995) with 56 other publicly available genomes of C. diphtheriae revealed that the strains CHUV2995, CCUG 5865 and CMCNS703 share a lower average nucleotide identity (ANI) (95.24 to 95.39%) with the C. diphtheriae NCTC 11397T reference genome than all other C. diphtheriae genomes (>98.15%). Core genome phylogeny confirmed the presence of two monophyletic clades. Based on these findings, we propose here two new C. diphtheriae subspecies to replace the lineage denomination used in previous multilocus sequence typing studies: C. diphtheriae subsp. lausannense subsp. nov. (instead of lineage-2), regrouping strains CHUV2995, CCUG 5865, and CMCNS703, and C. diphtheriae subsp. diphtheriae subsp. nov, regrouping all other C. diphtheriae in the dataset (instead of lineage-1). Interestingly, members of subspecies lausannense displayed a larger genome size than subspecies diphtheriae and were enriched in COG categories related to transport and metabolism of lipids (I) and inorganic ion (P). Conversely, they lacked all genes involved in the synthesis of pili (SpaA-type, SpaD-type and SpaH-type), molybdenum cofactor and of the nitrate reductase. Finally, the CHUV2995 genome is particularly enriched in mobility genes and harbors several prophages. The genome encodes a type II-C CRISPR-Cas locus with 2 spacers that lacks csn2 or cas4, which could hamper the acquisition of new spacers and render strain CHUV2995 more susceptible to bacteriophage infections and gene acquisition through various mechanisms of horizontal gene transfer.
Introduction
Classical diphtheria is due to the production of a toxin during C. diphtheriae infections by strains lysogenized by a bacteriophage (corynephage) holding the toxin gene. Thanks to vaccination programs, the incidence of toxigenic diphtheria has dramatically decreased during the past century in industrialized countries (Kitchin, 2011). However, there has been a recent increase in non-toxigenic C. diphtheriae infections reported with various atypical clinical presentations including pharyngitis, respiratory tract infections, endocarditis, osteomyelitis, septic arthritis or cutaneous infections (Gubler et al., 1998; Romney et al., 2006; Hirata Jr et al., 2008; Edwards et al., 2011; Zasada, 2013; FitzGerald et al., 2015; Kolios et al., 2017; Okamoto et al., 2018). As a potential public health threat, toxigenic C. diphtheriae infections need to be detected. PCRs targeting the toxin encoding gene and the Elek test remain the standard to quickly characterize the toxigenic potential of an isolate (Efstratiou et al., 1994; Public Health England, 2015; De Zoysa et al., 2016). In Switzerland, clustered cases of cutaneous (toxigenic and non-toxigenic) diphtheria were recently reported in the migrant population and whole-genome sequencing was useful to rule out recent direct transmission of a clone (Meinel et al., 2016).
Besides the toxin, other virulence factors such as the three operons encoding for pili (SpaA cluster, SpaD cluster and SpaH cluster) and genes related to iron-uptake may play a role in C. diphtheriae infections (Trost et al., 2012). The regulation of virulence is mainly due to the Diphtheria toxin Repressor (DtxR), which binds the promoter and represses in an iron-dependant manner the transcription of the toxin gene as well as numerous genes involved in iron homeostasis (Schmitt and Holmes, 1991b; Lee et al., 1997; Schmitt et al., 1997). In low-iron conditions, such as in the human host, the repression of the DtxR is released, leading to the transcription of the toxin (Boyd et al., 1990; Schmitt and Holmes, 1991a,b).
C. diphtheriae was historically classified into four biovars—gravis, mitis, intermedius, and belfanti—based on biochemical phenotypic testing (Funke et al., 1997; Goodfellow et al., 2012). However, C. diphtheriae strains within a certain biovar can be genetically more distant than between biovars (Trost et al., 2012; Sangal et al., 2014). Thus, genomics does not support the use of biovars to reliably classify C. diphtheriae isolates (Sangal and Hoskisson, 2016). In addition, there is a lack of correlation between biovar determination and pathogenicity (Bolt et al., 2010). Multilocus sequence typing (MLST), based on the allelic determination of 7 house-keeping genes, has recently been used to separate two distinct lineages, called lineage-1 (comprising most strains) and lineage-2 (regrouping only biovar belfanti strains) (Bolt et al., 2010). A third lineage was described by Farfour et al. but currently only one strain is known to belong to this sequence-type (Farfour et al., 2013).
Confronted with a very particular clinical presentation and bronchoscopy findings in a patient, WGS was applied to a C. diphtheriae isolate, of strain CHUV2995, to exclude the presence of the diphtheria toxin in a clinically relevant turnaround time, in parallel to a specific PCR for the toxin gene and an Elek test. Then, a comparative genomic analysis was performed to investigate the particular genomic features of strain CHUV2995 as well as the presence of virulence factors. The biochemical phenotype was also characterized to better describe this C. diphtheriae strain.
Materials and Methods
Bacterial Strain and Growth Conditions
C. diphtheriae CHUV2995 was isolated from the bronchoalveolar lavage (BAL) of a patient hospitalized in Lausanne University Hospital and subsequently identified using a matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Bruker, US). For Ion Torrent as well as for API Coryne (bioMérieux) identification tests (see below), bacteria were grown on blood agar plates at 37°C in a 5% CO2 humidified atmosphere for 24–48 h. For PacBio sequencing (see below), bacteria were grown in Todd-Hewith Broth (THB) at 37°C in ambient atmosphere for 48–72 h.
DNA Extraction and Sequencing
Genome sequencing was done using two different technologies: PGM Ion Torrent (Life Technologies, Carlsbad, US) technology was used in order to exclude the presence of the toxin in a clinically relevant time frame. The genome was then re-sequenced using a PacBio RSII (Pacific Biosciences, Menlo Park, CA, US) to assemble the numerous repetitive regions of the genome that could not be properly assembled from short read data. Only the bioinformatics analyses performed on the PacBio data are detailed and reported here.
Ion Torrent PGM Sequencing
Genomic DNA extraction and purification were performed using the protocol for Gram-positive bacteria with the Wizard Genomic DNA Purification Kit (Promega, ref. A1120). Libraries were prepared using the Ion Xpress Plus Fragment Library Kit (Life Technologies, ref. 4471269) and the Ion Xpress Barcode Adapters 1–16 Kit (Life Technologies, ref. 4471250). Sequencing of 100 base pairs (bp) paired-reads was done using a PGM Ion Torrent (Life Technologies). Five independent runs were performed in order to achieve a sufficient theoretical coverage for the analysis.
Pacific Biosciences RS II Sequencing (Pacific Biosciences)
Hundred milliliters of culture in the exponential phase (THB medium) were used to obtain enough good quality DNA. Each culture was centrifuged for 2 min at 16,000 g and resuspended in 600 μl of a 4 mg/ml lysozyme solution diluted in EDTA 50 mM. Samples were incubated for 2 h and centrifuged 2 min at 16,000 g. The next purification steps were performed using the Wizard SV Genomic DNA Purification System (Promega, ref. A2361). DNA was finally eluted in 10 mM TRIS pH 8.0. Sequencing was performed on a Pacific Biosciences RS II sequencer using one SMRT cell of chemistry version P6-C4 (Pacific Biosciences, Menlo Park, CA, US). The 131,813 reads obtained presented a mean length of 10,577 bp.
Assembly
De novo assembly of the PacBio sequences of CHUV2995 was carried out using the Hierarchical Genome Assembly Process (HGAP) workflow (PacBio DevNet; Pacific Biosciences, Menlo Park, CA, US), as available in SMRT Analysis v2.3.0. The assembly contained 3 contigs: 1 main circularized contig of 3,088,235 bp, 1 small circularized contig of 22,088 bp and 1 small linear contig of 29,039 bp. Pacbio reads were mapped on the assembly using Burrows-Wheeler Aligner (BWA-SW) v0.7.12 and SAMtools v1.2 (Li et al., 2009; Li and Durbin, 2010). Mapping quality and coverage were assessed using Qualimap v2.2. A coverage drop could be seen in the main contig between positions 2,903,566 and 2,960,750 as well as on the small contigs. Since BWA assigns randomly reads that can equally match different locations in a genome, coverage drop can be seen in wrongly duplicated regions in an assembly. Wrongly duplicated regions were identified using Genome Pair Rapid Dotter (GEPARD) (Krumsiek et al., 2007), and the sequence similarity was further confirmed using Mafft v7.187 (no SNPs could be seen between the duplicated sequences) (Katoh and Standley, 2013). Bases between position 2,931,515 and 2,959,386 of the main contig, as well as the two small contigs, were removed from the assembly. Following that, read mapping showed a uniform coverage across the final 3,060,363 bp chromosome.
Genomes Included in the Analysis
All the C. diphtheriae strains indicated in Table S1, as well as C. ulcerans BR-AD22, were included for the core genome phylogeny and the subsequent comparative genomic analysis. The Average Nucleotide Identity (ANI) was calculated between all pairs of genomes.
Annotation
The CHUV2995 genomic sequence, all the genomes from the University of Basel and the strain TH2031 were annotated using Prokka v1.11 (Seemann, 2014). For all the other genomes, annotation was already provided on RefSeq database (or Genbank if RefSeq annotation was not available). Protein domains were predicted using InterProScan v5.18-57.0 (Jones et al., 2014) and Pfam (Finn et al., 2014). A BLASTP search for every protein sequence was performed against the Clusters of Orthologous Groups database (Galperin et al., 2015), as available on the National Center for Biotechnology Information (NCBI) server (ftp://ftp.ncbi.nih.gov/pub/COG/COG2014/data/prot2003-2014.fa.gz). BLASTP algorithm v2.3.0+ was used with cut-offs of 10−5 for e-value, 20% for amino acid identity and 50% of query coverage (Altschul et al., 1997). In addition, KEGG Orthology (KO) numbers were assigned using GhostKOALA v2.0 (Kanehisa et al., 2016). DOOR 2.0 database was used to look for gene operons (Mao et al., 2014).
Average Nucleotide Identity
The average nucleotide identity (ANI) was calculated using NUCmer v3.1, a tool of the MUMmer software (Kurtz et al., 2004). CHUV2995 strain and NCTC 11397T were both used as a reference for the pairwise calculation. All the genomes of the strains described in Table S1, except the two references (CHUV2995 and NCTC11397T, respectively), were used as input to calculate the ANI.
Prediction and Comparison of Groups of Orthologs/Paralogs
Orthofinder v1.1.4 was used to predict and to cluster orthologs and paralogs into so-called “orthogroups” (Emms and Kelly, 2015). In order to compare the presence/absence of orthogroups in the genomes of all C. diphtheriae and C. ulcerans strains, data was loaded and compared in MySQL 5.7.18 using homemade scripts that could query the tables of orthogroups, InterPro domains, Pfam motifs, COG and KO using MySQL syntax. Classical virulence factors, as previously described in (Allen and Schmitt, 2011; Trost et al., 2012; Sangal et al., 2015) were analyzed (Data Sheet 1).
Core Genome Phylogeny
Core genome alignment was built in two steps: first, amino acid sequences of single-copy orthologous genes of all the C. diphtheriae and C. ulcerans strains were aligned using Mafft v7.187 (Katoh and Standley, 2013) and concatenated. Then, FastTree v2.1.8 (Price et al., 2010) was used to generate the core genome phylogeny (parameters: “–gamma –spr 4 –mlacc 2 –slownni”).
Prophages, Genomic Islands and Crisprs Regions
PHASTER (Arndt et al., 2016) and IslandViewer 4 (Bertelli et al., 2017) were used to predict prophages and genomic islands, respectively. Genomic islands of CHUV2995 were also detected using a homemade script highlighting genomic regions larger than 4,500 bp that did not align with NUCmer in more than 80% of strains. In addition, these genomic regions were merged when less than 2,000 bp apart to tackle the issue of small repeated genomic regions. Finally, CRISPRfinder was used (Grissa et al., 2007) to identify Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR).
Multilocus Sequence Typing
Multilocus sequence typing was performed on all included strains using the mlst software (Seemann T, mlst, Github https://github.com/tseemann/mlst) based on the C. diphtheriae scheme as available from PubMLST (https://pubmlst.org) (Jolley and Maiden, 2010). To evaluate the worldwide distribution of lineage-2 (Bolt et al., 2010), the concatenated sequences of the seven housekeeping genes of each C. diphtheriae ST were retrieved from pubMLST (n = 541, May 2018). All sequences were aligned using mafft v7.187 and a phylogeny reconstructed with FastTree v2.1.8 (with parameters “–nt”) (Price et al., 2010; Katoh and Standley, 2013).
Phenotypic Testing
API Coryne (bioMérieux) identification test was used to test the metabolic features of the strain CHUV2995 according to the manufacturer's instructions.
Results
Clinical Case Report
A young adult patient originally from South-East Asia, in Switzerland for several years, was admitted to the Lausanne University Hospital for persistent painful cervical lymphadenopathies without fever or weight loss. He had a medical history of lymph node and pulmonary tuberculosis complicated by apical bronchiectasis, chronically colonized by Pseudomonas aeruginosa. The patient presented with a 2-week history of asthenia, odynodysphagia, purulent rhinorrhea, dry cough, hemoptoic sputa and reported four episodes of loss of consciousness.
Clinical examination was normal except for a modest pharyngeal hyperemia and a submandibular painful lymphadenopathy (2 × 2 cm). A few other smaller cervical lymphadenopathies were present.
Laboratory analyses revealed a normal C-reactive protein (CRP) (5 mg/l) and normal complete blood count. Renal and hepatic functions were also normal. A chest X-ray was similar to the one performed 1 year before and a thoracic CT-scan confirmed the presence of right upper lobe bronchiectasis without any additional findings. A fine needle biopsy of the submandibular lymphadenopathy showed a non-specific inflammatory process, without granulomas; PCR and cultures for Mycobacterium tuberculosis and other mycobacteria were negative. Serologies for HIV, CMV, EBV, Toxoplasma gondii, Bartonella spp. were also negative. To exclude tuberculosis, a bronchoscopy was performed, that showed normal features up to the proximal trachea (Figure 1A). The carina and both mainstem bronchi had unusual multiple adherent whitish lesions (Figure 1B). The lobar and segmental bronchi exhibited no lesions. Histologic examination of bronchial biopsies revealed chronic inflammation (malpighian hyperkeratotic mucosa). The BAL showed alveolar lymphocytosis (65%). Gram staining of the BAL revealed more than 25 leucocytes per microscopic field and some Gram-positive bacilli. C. diphtheriae (103 CFU/ml) was recovered after culture of the BAL sample and identified by MALDI-TOF (Croxatto et al., 2012) together with, as expected, colonizing P. aeruginosa (104 CFU/ml).
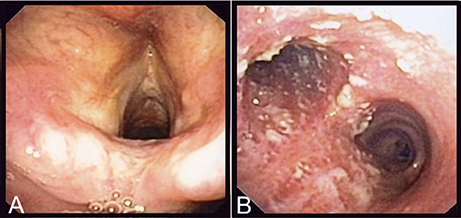
Figure 1. Endoscopic findings of the patient. On (A), are shown the normal epiglottis and larynx of the patient and on (B), the adherent whitish lesions of the distal trachea and both mainstem bronchi.
The C. diphtheriae strain isolated, CHUV2995, was found to be susceptible to all antimicrobial agents tested (penicillin, amoxicillin, clindamycin, levofloxacin, ciprofloxacin, erythromycin and azithromycin). The patient was successfully treated with erythromycin for 14 days. Cardiac investigations did not reveal arrhythmia nor conduction disturbances, precluding the presence of cardiotoxicity. Given the unusual clinical presentation with chronic severe tracheobronchitis, the bacterial genome was sequenced for characterization and exclusion of the presence of the toxin gene for both clinical and epidemiological reasons (the patient came from a center for migrants). The genome analysis, specific PCR and Elek test all confirmed that the C. diphtheriae strain was non-toxigenic.
Here, the acute clinical manifestations of this patient were attributed to C. diphtheriae considering (1) the bronchoscopic findings, (2) the previously documented presence of P. aeruginosa indicating colonization rather than de novo infection, and (3) the clinical improvement after macrolide use (an antibiotic class lacking significant antipseudomonal properties).
Strain CHUV2995 Is Part of a Distinct C. diphtheriae Clade With Particular Genomic Features
To investigate the similarity of C. diphtheriae strain CHUV2995 to other strains available in sequence databases, pairwise ANI calculations were performed with the references CHUV2995 and NCTC 11397T genomes (median alignment coverage 79.89 and 82.44%, respectively). Strain CHUV2995 shared a median ANI of 95.25% (Figure 2) with all strains included in the analysis (Table S1), except CCUG 5865 and CMCNS703 that were more closely related (>99% ANI). By contrast, NCTC 11397T shared more than 98.15% ANI with all C. diphtheriae strains except CHUV2995, CMCNS703, and CCUG 5865. Thus, CHUV2995, CCUG 5865, and CMCNS703 are closely related, and cluster separately from all other C. diphtheriae strains. Despite the relatively large differences in ANI between groups, 16S rRNA gene conservation between all C. diphtheriae strains was above 99% identity, suggesting that the new clade should be classified as part of the C. diphtheriae species (data not shown). Interestingly, CCUG 5865 was recently used by Grosse-Kock et al. (2017) as an outgroup for their core genome phylogeny because it belonged to lineage-2. The multilocus sequence types of CHUV 2995 and CMCNS703 also cluster with lineage-2 (Figure S1 and Data Sheet 3). Given the large genomic distances, we propose to rename lineage-2 as C. diphtheriae subsp. lausannense subsp. nov. (regrouping C. diphtheriae strains CHUV2995, CMCNS703, and CCUG 5865) and lineage-1 by C. diphtheriae subsp. diphtheriae subsp. nov. (regrouping the other genome sequenced C. diphtheriae strains analyzed).
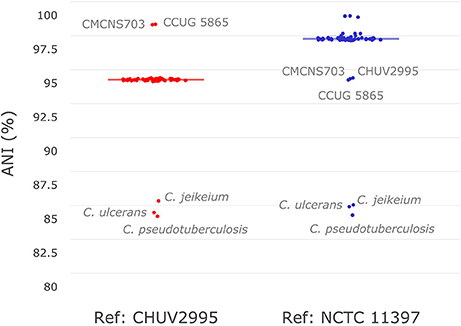
Figure 2. Average nucleotide identity. Pair-wise average nucleotide identities (ANI) with all C. diphtheriae strains as well as other closely related species of the same genus (C. ulcerans, C. pseudotuberculosis, and C. jeikeium) were calculated using C. diphtheriae strain CHUV2995 and C. diphtheriae NCTC 11397T as reference. As shown here in this boxplot (very condensed around the median), all C. diphtheriae strains but CCUG 5865 and CMCNS703 share an ANI between 95 and 96% when compared to strain CHUV2995. Conversely, when using C. diphtheriae strain NCTC 11397T as reference, the ANI shared with all C. diphtheriae is above 98%, except for CHUV2995, CCUG 5865 and CMCNS703. Therefore, two groups of strains were observed, one with CHUV2995, CCUG 5865, and CMCNS703 and the other with the rest of C. diphtheriae strains.
To confirm the monophyly of the new subspecies, the amino acid translation of core genes common to all C. diphtheriae strains as well as C. ulcerans—used as an outgroup—were aligned (total length 323,259 amino acids) and used to reconstruct a maximum-likelihood tree. The phylogenetic tree is consistent with the ANI calculations and shows that strain CHUV2995 forms a monophyletic clade with CCUG 5865 and CMCNS703, whereas members of subspecies diphtheriae form another distinct clade (Figure 3). Interestingly, subspecies lausannense genomes are significantly larger (CHUV2995, 3.06 Mb; CCUG 5865, 2.6 Mb; CMCNS703, 2.73 Mb) than subspecies diphtheriae genomes (p = 0.0040, Wilcoxon rank sum test was used because data distributions were asymetrical in both groups). Two-sample t-test comparing GC content between the two clades was not significant (p = 0.1555). However, the genome of strain CHUV2995 displayed the highest GC content (53.94%), whereas the mean GC content of all other C. diphtheriae strains in the dataset is 53.54 ± 0.16%.

Figure 3. Core genome phylogeny. A maximum likelihood phylogenetic tree was reconstructed based on the concatenated alignment of single-copy orthologous genes belonging to the C. diphtheriae and C. ulcerans core genome. The scale bar represents the number of amino acid substitutions per site alongside the branches. Nodes supports are based on the Shimodaira-Hasegawa (SH) test. Black dots indicate when node values are below 1 (lines are shifted to the right to accommodate the presence of the dots, which should not be considered as phylogenetic distances). CHUV2995 clusters with two closely related isolates, defining the monophyletic subspecies lausannense, whereas the other strains cluster in subspecies diphtheriae. Interestingly, CHUV2995 displays the largest genome of the dataset.
Virulence Factors of Subspecies lausannense
Orthologs of known virulence factors were identified in the genomes of the dataset (Figure 4 and Data Sheet 1). Interestingly, the strains belonging to subspecies lausannense had no orthologs of the pili-associated genes (located on gene operons encoding for SpaA-type, SpaD-type and SpaH-type pili) based on OrthoFinder analysis and additional tBLASTN searches, which is consistent with a previous study (Grosse-Kock et al., 2017). No InterPro domain or Pfam motifs related to these pili-associated genes were found in subspecies lausannense genomes (except for domains that are not pili-specific), thus further supporting the lack of pilus (Data Sheet 1). Similarly, no pili-associated sortases (srtA, srtB, srtC, srtD, and srtE) could be identified in subspecies lausannense (Data Sheet 1). Only class E sortases (such as srtF) which act as housekeeping sortases in C. diphtheriae (Swaminathan et al., 2007; Spirig et al., 2011) were identified in subspecies lausannense. CHUV2995 has a supplementary sortase-family protein (CHUV2995_00246) that shared high amino acid identity (96%) with other pili-associated sortases (class C) but had a small size of 114 amino acids and only one transmembrane domain as compared to a size of ~300 amino acids and two transmembrane domains for classical pili-associated sortases of C. diphtheriae. Unlike pili-associated sortases, this gene was not located in a gene cluster related to pili and its function remains uncertain.
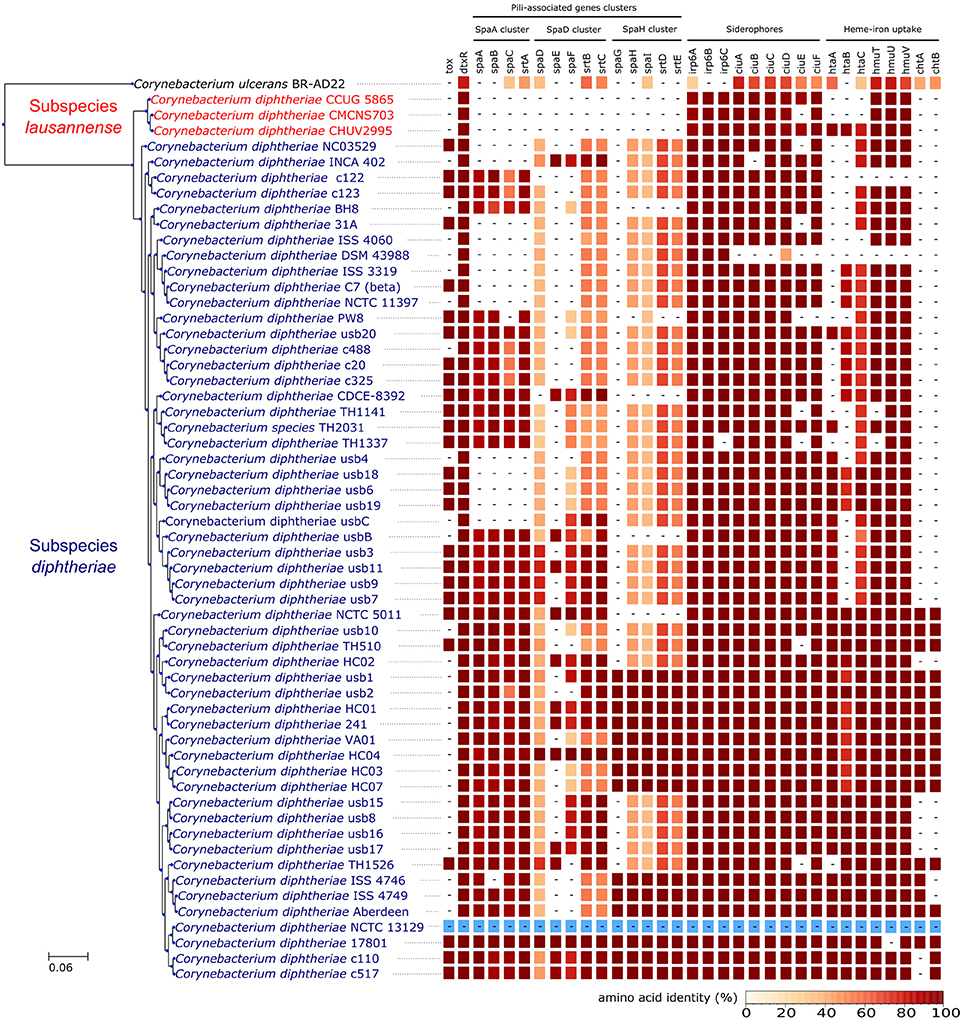
Figure 4. Virulence factors. CHUV2995, CUG 5865, and CMCNS703 (Subspecies lausannense) present no gene encoding for—or associated to—the classical C. diphtheriae pili operons. Interestingly, even C. ulcerans encodes homologs of these genes. CHUV2995 genome encodes more genes related to heme-iron uptake than the other strains of subspecies lausannense. NCTC13129 sequences were used as a reference to compute the amino acid identity (blue).
Regarding iron uptake, the CHUV2995 genome encodes orthologs of htaA, htaB, and htaC (Figure 4 and Data Sheet 1), genes that are involved in heme-associated acquisition of iron (Allen and Schmitt, 2009). These genes shared a high amino acid sequence identity to homologs in strain NCTC 13129, ranging from 80.62% (htaC) to 98.39% (hmuT). Orthologs of those genes were not identified in the two other subspecies lausannense strains.
The three genomes of subspecies lausannense did not harbor the complete sequence of the toxin gene based on orthologs prediction and on the absence of complete Pfam motifs and InterPro domains corresponding to the toxin (Data Sheet 1). However, two overlapping fragments of the R domain of the diphtheria toxin (101 and 38 amino acids, respectively, and overlapping on 34 amino acids), were detected on two small contigs of the strain CMCNS703 genome. Sequence identity with the toxin of NCTC 1329 using BLASTP was 100 and 87.17% for the large and the small fragments, respectively. The presence of a complete but not well-assembled toxin in CMCNS703 genome is unlikely since we did not find another fragment of the toxin in the assembly. Alternatively, remnants of the toxin may be present if the gene is undergoing pseudogenisation and progressive gene loss. Finally, the NCTC 13129 toxin region was compared to the corresponding CHUV2995 region, which lacks the toxin but harbors sequences of another prophage inserted at the same genomic position (Figure S2).
The translated sequence of the dtxR gene was completely identical in the three strains of subspecies lausannense and shared 97.79% amino-acid identity with those of subspecies diphtheriae. It was also identical to that described by Dinu et al. (2014), who reported this allele in both toxigenic and non-toxigenic, toxin gene-bearing isolates, suggesting that this allele could efficiently regulate the toxin gene, if the strain gets lysogenized.
The large genome size could be explained by the large number of genomic islands identified and in particular those formed by prophages in the genome of CHUV2995 (Figure 5). Some of them were conserved in the genomes of the other strains of subspecies lausannense. Several genes encoding for putative siderophores or putative heme-iron uptake systems were detected on the predicted genomic islands. These genes could thus indicate potential pathogenicity islands.
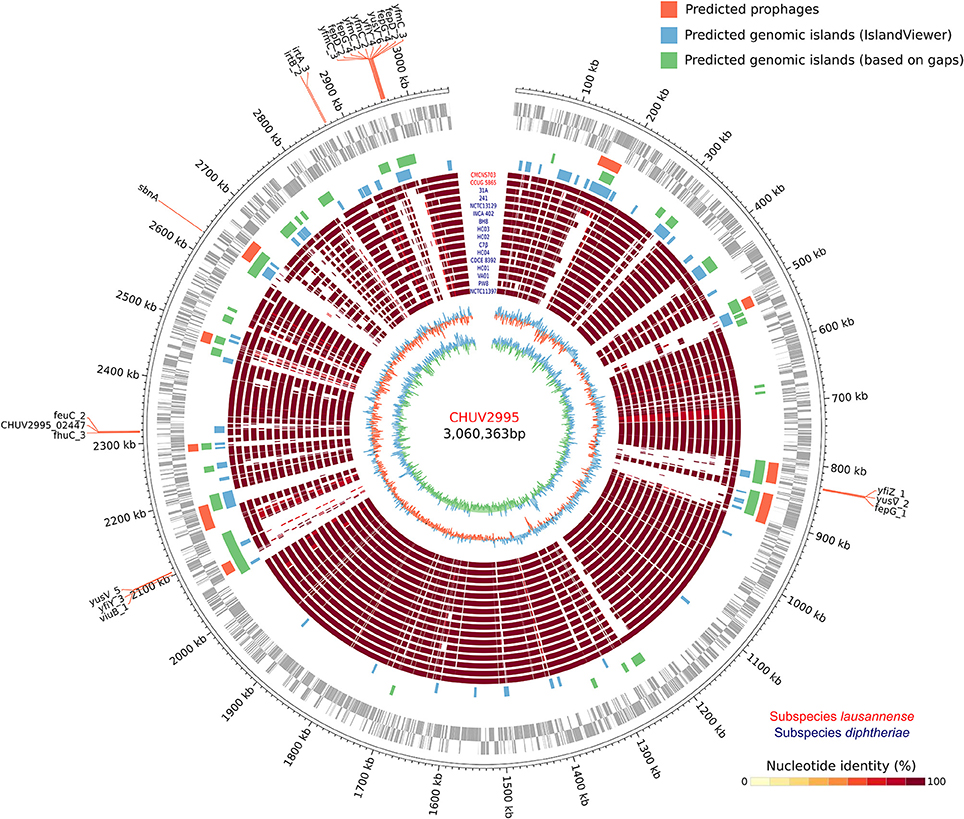
Figure 5. Circular genome representation. Circles from inside to outside represent the GC content, GC skew, 15 complete genome sequences of C. diphtheriae strains as well as the two genome sequences of subspecies lausannense (red), genomic islands (GIs) predicted using IslandViewer 4 (blue), a homemade script (green; regions of ≥4.5 Kb not aligning with NUCmer in more than 80% of the strains), PHASTER (orange; prophages), open reading frames (ORFs) on the lagging strand and on the leading strand (gray) and chromosomal genomic positions. Genes probably involved in the iron uptake (based on automatic annotation) present on the GIs were annotated, thus delineating putative pathogenicity islands.
Differences in Metabolic Capabilities
In order to identify differences in metabolic capabilities between subspecies lausannense and subspecies diphtheriae, we compared KO entries found only in subspecies lausannense or subspecies diphtheriae. Fourteen KO entries were specific to subspecies lausannense (Table 1) and among them, four were related to ABC transporters (3 zinc and 1 cobalt/nickel transport system). The other KO entries were related to amino acid, sugar or fat metabolism. Conversely, 9 KO entries were absent from subspecies lausannense and present in subspecies diphtheriae (Table 2). These KO were related to either nitrate reduction or molybdenum cofactor biosynthesis. Interestingly, all genes annotated with these KO entries were encoded in the same genomic region (Figure 6). More specifically, genes encoding for nitrate reductase subunits alpha (narG), beta (narH), gamma (narI) and delta (narJ) encoded in an operon together with narK (a major facilitator superfamily transporter) and modA-B (a molybdenum ABC transporter) were absent from subspecies lausannense. In addition, the adjacent operons implicated in molybdenum import and cofactor biosynthesis moeBR-moaE(moaB) and mobA-moaC-moeA-moaA were also absent from subspecies lausannense (Figure 6), similarly to previous observations in C. pseudotuberculosis (Viana et al., 2017).
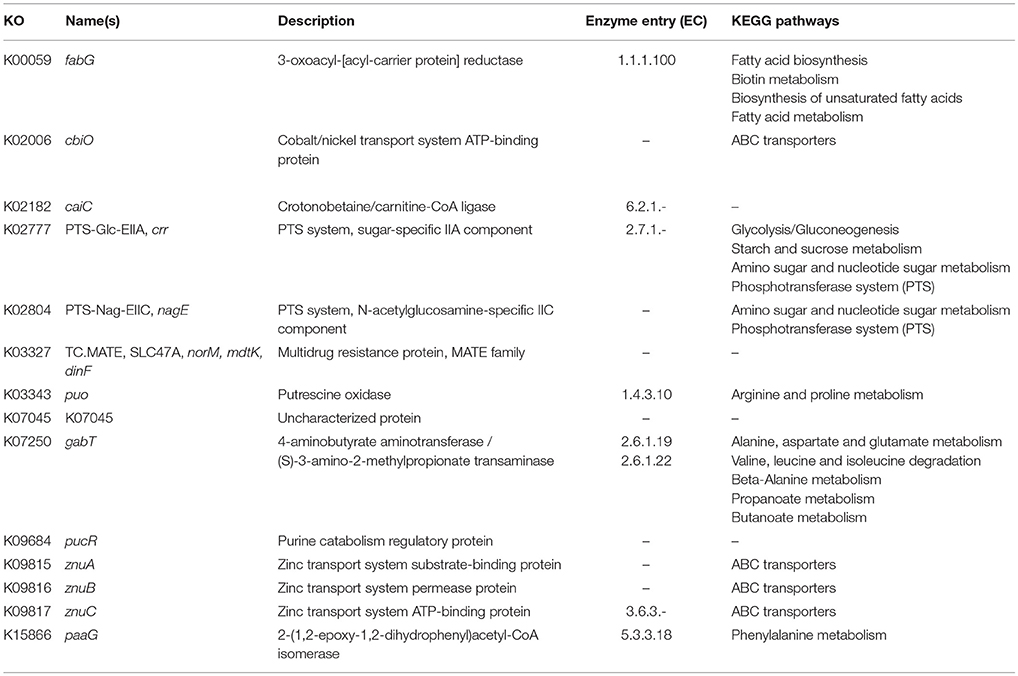
Table 1. KO entries specific to subspecies lausannense and absent from subspecies diphtheriae.
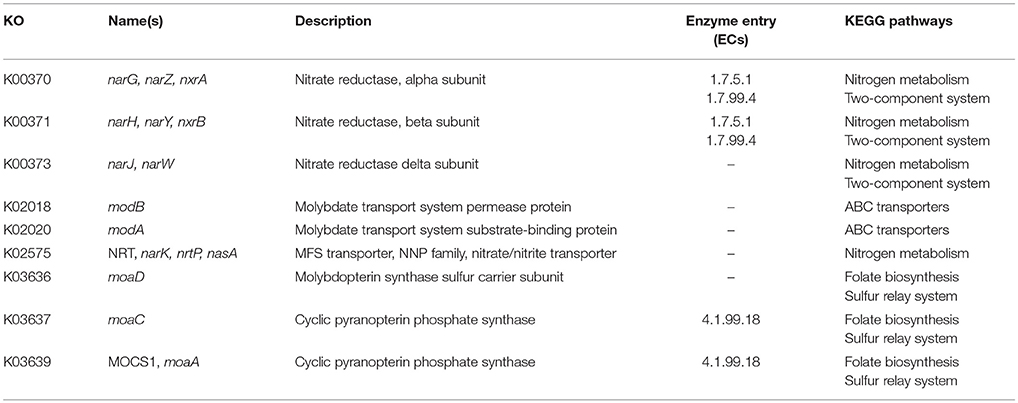
Table 2. KO entries present in subspecies diphtheriae and absent from subspecies lausannense.
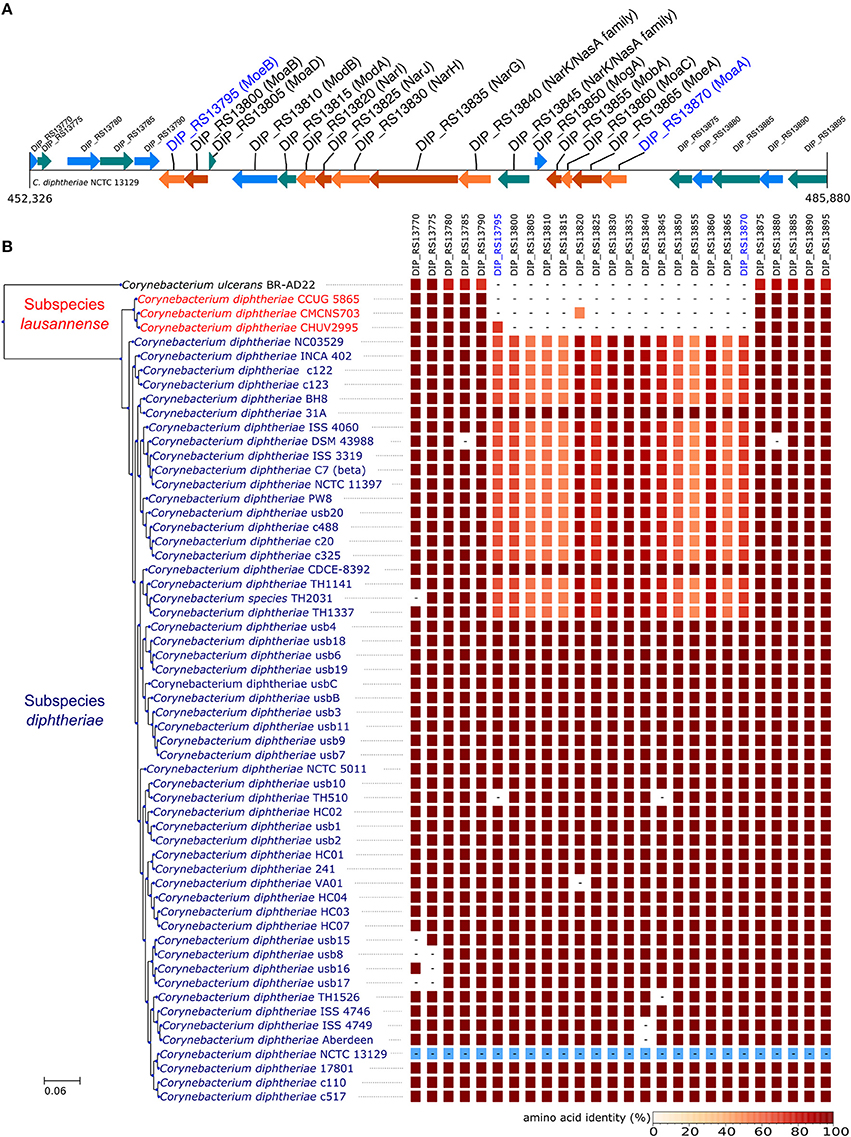
Figure 6. Missing nitrate reductase and molybdenum cofactor biosynthesis genes in subspecies lausannense. (A) The genomic region of the NCTC13129 genome involved in nitrate reduction and molybdenum cofactor biosynthesis is shown. Sequences in brown and orange indicate genes functioning in operon (predicted using DOOR 2.0), reference locus tags are indicated above the coding sequences and the predicted products are written between parenthesis, locus tags in blue indicate the border of the genomic region (corresponding to the locus tags in blue of B); (B) NCTC13129 genomic region (in light blue) was used as a reference to look for orthologs of genes of the genomic region of interest. Each box represents the presence/absence of an ortholog in each genome of the dataset. The color gradient indicates the amino-acid identity as compared to the reference sequence. The putative ortholog of the gene encoding for the nitrate reductase subunit beta in CMCNS703 is either due to a wrong clustering of orthologous proteins or to an assembly artifact (it is located on a small contig). The sequence alignment covers less than 50% of the reference sequence DIP_RS13820 (locus tag of strain NCTC13129) and could be a remnant of the nitrate reductase subunit beta. Similarly, the presence of an ortholog to DIP_RS13795, encoding for MoeB, in CHUV2995 is questionable: amino acid identity was 72.49% but with a coverage of less than 60% of the reference sequence. Therefore, we concluded that the genomic region encoding for the nitrate reducase and the molybdenum cofactor biosynthesis was absent in subspecies lausannense.
CHUV2995 is Enriched in Many COG Categories
The gene content of CHUV2995 was enriched (defined as being above the 90th percentile of the distribution) in many COG categories: E, Amino acid transport and metabolism, G, Carbohydrate transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; J, Translation, ribosomal structure and biogenesis; K, Transcription; L, Replication, recombination and repair, P, Inorganic ion transport and metabolism; R, General function prediction only; V, Defense mechanisms and X, Mobilome: prophages, transposons (Figure 7). Surprisingly, the number of mobility genes (category X) is 8 fold higher than the median of all genomes and far above that of the 15 complete genome sequences included in the dataset. Indeed, repeated elements, such as mobility genes, can be underestimated in draft genomes due to the difficulty in assembling repeats. Categories C and D, related to energy production/conversion and cell cycle/division, respectively, are also slightly enriched in the CHUV2995 genome. Overall, subspecies lausannense is only enriched in categories related to lipid transport and metabolism (I) as well as to inorganic ion transport and metabolism (P) (Figure 7 and Figure S3). No more enrichment in most COG categories genes could be observed when subtracting COGs identified in CHUV2995 genomic islands (Figure S4 and Data Sheet 2), showing the large influence of additional genetic material acquired by CHUV2995 (Figure S5). Only categories G and I (carbohydrate and lipid metabolim) remained slightly enriched.
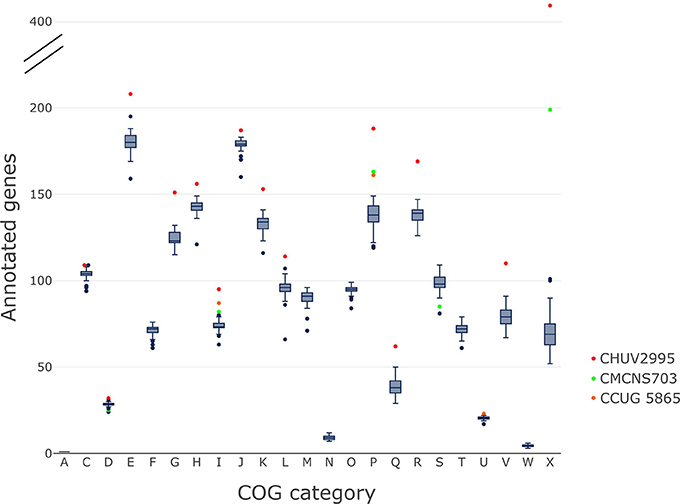
Figure 7. COG categories. The distribution of the number of genes per genome assigned to each COG category is shown on these boxplots.We indicated in red, CHUV2995; in green, CMCNS703 and in orange, CCUG 5865 when they are outliers. CHUV2995 was enriched in many COG categories and overall, subspecies lausannense was enriched in COG categories I and P. COG categories: A, Processing and modification; C, Energy production and conversion; D, Cell cycle control; cell division, chromosome partitioning; E, Amino acid transport and metabolism; F, Nucleotide transport and metabolism; G, Carbohydrate transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; J, Translation, ribosomal structure and biogenesis; K, Transcription; L, Replication, recombination and repair; M, Cell wall/membrane/envelope biogenesis; N, Cell motility; O, Posttranslational modification, protein turnover, chaperones; P, Inorganic ion transport and metabolism; Q, Secondary metabolites biosynthesis, transport and catabolism; R, General function prediction only; S, Function unknown; T, Signal transduction mechanisms; U, Intracellular trafficking, secretion, and vesicular transport; V, Defense mechanisms; W, Extracellular structures; X, Mobilome: prophages, transposons.
Given the enrichment of CHUV2995 in genes related to gene mobility and bacteriophages, it was also interesting to assess the presence of CRISPR-Cas systems in subspecies lausannense genomes, which could be involved in bacterial immunity against bacteriophages. A type II-C CRISPR-Cas system was identified in the CHUV2995 genome and included one single CRISPR region with only two spacers. The region is flanked by genes coding for a hydroxy/phosphor-methyl pyrimidine kinase and a hypothetical protein on one side, and an integrase core domain protein (a mobility gene) on the other side. Strain CCUG 5865 displayed two large CRISPR-Cas systems, one type I-E-a (19 spacers) and one type I-E-b system (also 19 spacers), whereas CMCNS703 only had 1 type I-E-a (3 spacers, and possibly more because the region is located at the border of a contig) (Figure S6).
Phenotypic Testing
The API Corynebacterium revealed a profile compatible with C. diphtheriae biovar mitis or belfanti. However, the alpha-glucosidase test was negative (Table S2), which is rather unusual because it was indicated as positive in 96% of strains of C. diphtheriae biovar mitis and belfanti in the table of identification provided by the manufacturer. Testing more strains belonging to subspecies lausannense would further indicate if the negativity of the alpha-glucosidase activity is systematic. Biovar belfanti strains are usually negative for the nitrate reductase activity despite the presence of the nitrate reductase operon. CHUV2995 was also negative for the nitrate reductase activity (Table S2), which could surprisingly be explained by the lack of the nitrate reductase operon (Figure 6), thus providing a genomic basis for the phenotypic results in this case.
Subspecies lausannense Has a Worldwide Distribution and Is Mostly Isolated From the Respiratory Tract
Out of the 541 ST types available, 78 ST types can be identified as members of lineage-2 (the new subspecies lausannense) (Figure S7 and Data Sheet 3). By looking at the metadata of the 76 corresponding isolates (some ST types did not have corresponding isolates), 69 were recovered in Europe, 2 in Mayotte island, 3 in Algeria, 1 in Russia and 1 in Bangladesh. Interestingly, 27 isolates had a documented isolation site. The two most common isolation sites were upper (n = 10) and lower (n = 7) respiratory tract samples. Skin and wounds (n = 4) were the third most common isolation sites (Data Sheet 3).
Discussion
In this study, we described the isolate CHUV2995 recovered from a patient with tracheobronchitis and propose to classify it in a new subspecies named Corynebacterium diphtheriae subsp. lausannense subsp. nov. For consistency, we propose the name C. diphtheriae subsp. diphtheriae subsp. nov. to regroup the clade containing most of C. diphtheriae isolates. Subspecies diphtheriae and lausannense correspond to lineage-1 and 2, respectively (Bolt et al., 2010; Grosse-Kock et al., 2017). Since the term “lineage” was also used for other ST types that are part of lineage-1 (du Plessis et al., 2017), we think that the use of subspecies could reduce confusion in the literature and help the reader to recognize clades showing these large genomic differences. An ANI cutoff of 95-96% has been proposed to distinguish between two bacterial species (Kim et al., 2014) and ANI values between subspecies lausannense and subspecies diphtheriae fall exactly within this range (Figure 2). However, the 99% nucleotide similarity of 16S rRNA shared between all C. diphtheriae isolates suggests the occurrence of a new subspecies rather than a new species. Indeed, accepted cutoffs for 16S rRNA nucleotide identity between strains of the same species range from 98.2 to 99% (Meier-Kolthoff et al., 2013; Kim et al., 2014). Lastly, core genome phylogeny confirmed the monophyly of the two subspecies.
The two other strains of subspecies lausannense, CCUG 5865 and CMCNS703, were isolated from nasal swabs in the United Kingdom and in India, respectively. Isolates belonging to lineage-2 were previously documented on 4 continents, showing a worldwide distribution of subspecies lausannense (Bolt et al., 2010; Farfour et al., 2013) (Data Sheet 3). Interestingly, several studies reported only lineage-1 (subspecies diphtheriae) isolates, suggesting that lineage-2 is less frequently encountered (Zasada, 2013; du Plessis et al., 2017; Grosse-Kock et al., 2017), as does the observation that ST affiliated to subspecies lausannense represent <15% of known ST.
Although all three subspecies lausannense strains studied encode hmuTUV genes involved the synthesis of the hemin ABC-transporter, CHUV2995 additionally encoded htaA, htaB, and htaC, associated to the binding and uptake of hemin (Allen and Schmitt, 2009). This suggests that CHUV2995 strain exhibits more pathogenic capabilities than the other subspecies lausannense strains.
Interestingly, CHUV2995 (together with the other strains of subspecies lausannense) presented no gene encoding for—or associated with—the classical C. diphtheriae pili operons despite the fact that the bronchoscopy showed adherent whitish lesions suggesting increased adhesive capabilities. However, the patient suffered from bronchiectasis, which is associated with retention of pulmonary secretions and could have promoted the adhesion of a strain with lower adhesive capabilities. Overall, subspecies lausannense might be less virulent than subspecies diphtheriae due to the lack of pili-associated (Broadway et al., 2013) as well as the nitrate reductase encoding genes, which is also known to promote virulence in other bacteria (Vázquez-Torres and Bäumler, 2016).
The genome of CHUV2995 was surprisingly highly enriched in most of the COG categories, suggesting that it has additional metabolic capabilities. All strains of subspecies lausannense had genes with KEGG Orthology functions that were not present in subspecies diphtheriae: namely functions involved in zinc and cobalt/nickel transport system or related to amino acid, sugar or lipid metabolism. In addition, COG category X, related to gene mobility and horizontal gene transfer, was notably enriched in CHUV2995, in line with the elevated number of predicted prophages and genomic islands (Figure 5). Interestingly, CHUV2995 harbored very few CRISPR spacers and encoded a type II-C Cas system lacking cas4 or csn2 (as discussed by Sangal et al., 2013; Jackson et al., 2017; Mir et al., 2018), both involved in the acquisition of new spacers in type II-A and II-B CRISPR-Cas systems. It remains unknown whether spacers may be acquired by II-C CRISPR-Cas systems on their own (Sangal et al., 2013; Jackson et al., 2017). The individual CRISPR-Cas systems identified in genomes of subspecies lausannense were previously described in C. diphtheriae by Sangal et al. (2013), but the concomitance of two type I-E (a and b type) CRISPR-Cas systems, as seen in CCUG 5865, was never reported before (Sangal et al., 2013; Hong et al., 2017).
Interestingly, subspecies lausannense specifically lacks genes encoding the nitrate reductase as well as genes involved in molybdenum cofactor biosynthesis. This genomic region was also found to be present in biovar equi and absent in biovar ovis of C. pseudotuberculosis and explains a positive nitrate reductase test for biovar equi (Viana et al., 2017). There is currently no genomic basis for the biovar classification of C. diphtheriae (Sangal and Hoskisson, 2016). Indeed, some strains, such as INCA 402, are classified as biovar belfanti, which are nitrate reducase negative although they contain the genomic region encoding the nitrate reductase enzymes. This suggests that nitrate reductase activity can depend on a difference at the transcriptional level for subspecies diphtheriae isolates. For subspecies lausannense, the lack of nitrate reductase activity can be explained by the absence of the genomic region involved in the synthesis of the nitrate reductase. Concerning the rest of the phenotypic findings, the API Coryne revealed CHUV2995 to be unusually negative for the alpha-glucosidase activity, which is rare for biovar belfanti (96% positive). However, no genomic explanation could be found since a gene encoding for alpha-glucosidase is present in CHUV2995 genome but may not have been expressed in the culture conditions tested. Practically, our data demonstrate that it is impossible to differentiate between the subspecies based on phenotypic data.
Since most analyses are based on the prediction of groups of orthologous proteins using OrthoFinder, the accuracy of protein clustering might have impacted our ability to identify orthologs in the two clades. Indeed, closely related genes of subspecies lausannense could have been clustered into a single group whereas the gene of interest of the reference strain could have clustered into a different group. This issue was tackled by performing searches for Pfam motifs and InterPro domains as well as BLAST searches of the protein of interest, which did not allow us to identify split groups of orthologs for the proteins of interest.
Our laboratory is not a reference center for C. diphtheriae and PCR for the toxin or Elek test is not routinely implemented in our diagnostic laboratories. Therefore, rapid microbial genomics provided information on the absence of the toxin in a short time frame, impacting on patient care and to prevent unnecessary isolation. It was particularly important to exclude the presence of the toxin gene given the fact that the patient was living in close proximity with many individuals with possible low vaccination rates, which would increase the risk of transmission. Currently, genomics represents an interesting alternative method to answer requests from clinicians, for instance when other simple tests are not available. Using genomics, the global gene content of an isolate—in this case virulence factors—can be quickly assessed, which has also proven useful in selected cases for other bacterial species (Tagini and Greub, 2017). In addition, whole genome sequencing prevents false negative results that can occur due to mutations in PCR target genes (Jaton et al., 2010). Overall, whole-genome sequencing provided interesting insights into C. diphtheriae strain diversity, unraveling large genomic differences between the two subspecies, and enabled us to provide the clinicians with meaningful clinical results in a short turnaround time.
Description of Corynebacterium Diphtheriae Subsp. Lausannense Subsp. Nov.
Corynebacterium diphtheriae subsp. lausannense (lau.san.nen'se. N.L. neut. adj. lausannense, of Lausanne, a city in Switzerland, where the strain was isolated from a patient of the local University Hospital).
This subspecies was previously identified as lineage-2 in multilocus sequence typing (MLST) studies (Bolt et al., 2010; Farfour et al., 2013). Members of subspecies lausannense share an average nucleotide identity ranging from 95.24 to 95.39% with subspecies diphtheriae. Subspecies lausannense regroups only biovar belfanti strains. The type strain is CHUV2995T = CCUG 72509T = DSMZ 107520T, and its complete genome sequence can be found under the bioproject accession number PRJEB24256.
Description of Corynebacterium Diphtheriae Subsp. Diphtheriae Subsp. Nov.
Corynebacterium diphtheriae subsp. diphtheriae (diph.the'ri.ae. Gr. fem. n. diphthera, piece of leather; N.L. fem. n. diphtheria, a disease in which leathery membranes form in the throat; N.L. gen. n. diphtheriae, of diphtheria).
Corynebacterium diphtheriae subsp. diphtheriae corresponds to lineage-1 in multilocus sequence typing studies (Bolt et al., 2010; Farfour et al., 2013) and was historically described as Corynebacterium diphtheriae by Kruse (1886) and Lehmann and Neumann (1896). The type strain is NCTC 11397T = ATCC 27010T = CIP 100721T = DSM 44123T.
Ethics Statement
For single case report, we are exempted from ethical committee approval based on the rules from our local ethical committee.
Author Contributions
FT designed the study, performed the analyses and wrote the manuscript. TP contributed to part of the analysis. AC performed the initial WGS investigation in a short turnaround time, the phenotypic tests and contributed to the redaction of the manuscript. FT, TP, and CB contributed to the interpretation of the results and the redaction of the manuscript. AK and AL followed the patient and contributed to the manuscript. GG contributed to the design of the study, the interpretation of the results and the redaction of the manuscript.
Funding
All the funding came from institutional funds (Institute of Microbiology and Division of Pulmonology).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer VS and handling Editor declared their shared affiliation.
Acknowledgments
We thank Sébastien Aeby, Maria Senra-Ortiz, and France Dusserre for their technical help, Prof. Thierry Calandra and his colleagues of the Division of Infectious Diseases for the clinical management of the patient and finally, Dominik Meinel and Adrian Egli for providing the assemblies of their study (Meinel et al., 2016). We thank Reinhard Zbinden and the technicians of the Institute of Medical Microbiology of the University of Zurich for their expert help and assistance. We thank Androulla Efstratiou and her colleagues of Public Health England for performing the Elek test. The computations were performed at the Vital-IT (http://www.vital-it.ch) Center for high-performance computing of the SIB Swiss Institute of Bioinformatics.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01743/full#supplementary-material
References
Allen, C. E., and Schmitt, M. P. (2009). HtaA is an iron-regulated hemin binding protein involved in the utilization of heme iron in Corynebacterium diphtheriae. J. Bacteriol. 191, 2638–2648. doi: 10.1128/JB.01784-08
Allen, C. E., and Schmitt, M. P. (2011). Novel hemin binding domains in the Corynebacterium diphtheriae HtaA protein interact with hemoglobin and are critical for heme iron utilization by HtaA. J. Bacteriol. 193, 5374–5385. doi: 10.1128/JB.05508-11
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bolt, F., Cassiday, P., Tondella, M. L., DeZoysa, A., Efstratiou, A., Sing, A., et al. (2010). Multilocus sequence typing identifies evidence for recombination and two distinct lineages of Corynebacterium diphtheriae. J. Clin. Microbiol. 48, 4177–4185. doi: 10.1128/JCM.00274-10
Boyd, J., Oza, M. N., and Murphy, J. R. (1990). Molecular cloning and DNA sequence analysis of a diphtheria tox iron-dependent regulatory element (dtxR) from Corynebacterium diphtheriae. Proc. Natl. Acad. Sci. U.S.A. 87, 5968–5972. doi: 10.1073/pnas.87.15.5968
Broadway, M. M., Rogers, E. A., Chang, C., Huang, I.-H., Dwivedi, P., Yildirim, S., et al. (2013). Pilus gene pool variation and the virulence of Corynebacterium diphtheriae clinical isolates during infection of a nematode. J. Bacteriol. 195, 3774–3783. doi: 10.1128/JB.00500-13
Croxatto, A., Prod'hom, G., and Greub, G. (2012). Applications of MALDI-TOF mass spectrometry in clinical diagnostic microbiology. FEMS Microbiol. Rev. 36, 380–407. doi: 10.1111/j.1574-6976.2011.00298.x
De Zoysa, A., Efstratiou, A., Mann, G., Harrison, T. G., and Fry, N. K. (2016). Development, validation and implementation of a quadruplex real-time PCR assay for identification of potentially toxigenic corynebacteria. J. Med. Microbiol. 65, 1521–1527. doi: 10.1099/jmm.0.000382
Dinu, S., Damian, M., Badell, E., Dragomirescu, C. C., and Guiso, N. (2014). New diphtheria toxin repressor types depicted in a Romanian collection of Corynebacterium diphtheriae isolates. J. Basic Microbiol. 54, 1136–1139. doi: 10.1002/jobm.201300686
du Plessis, M., Wolter, N., Allam, M., de Gouveia, L., Moosa, F., Ntshoe, G., et al. (2017). Molecular characterization of Corynebacterium diphtheriae Outbreak Isolates, South Africa, March-June 2015. Emerg. Infect. Dis. 23, 1308–1315. doi: 10.3201/eid2308.162039
Edwards, B., Hunt, A. C., and Hoskisson, P. A. (2011). Recent cases of non-toxigenic Corynebacterium diphtheriae in Scotland: justification for continued surveillance. J. Med. Microbiol. 60, 561–562. doi: 10.1099/jmm.0.025643-0
Efstratiou, A., Maple, C. P. A., and Europe, W. H. O. R. O. (1994). Laboratory Diagnosis of Diphtheria. Manual for the Laboratory Diagnosis of Diphtheria. Available online at: http://www.who.int/iris/handle/10665/108108 (Accessed July 19, 2017).
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157. doi: 10.1186/s13059-015-0721-2
Farfour, E., Badell, E., Dinu, S., Guillot, S., and Guiso, N. (2013). Microbiological changes and diversity in autochthonous non-toxigenic Corynebacterium diphtheriae isolated in France. Clin. Microbiol. Infect. 19, 980–987. doi: 10.1111/1469-0691.12103
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
FitzGerald, R. P., Rosser, A. J., and Perera, D. N. (2015). Non-toxigenic penicillin-resistant cutaneous C. diphtheriae infection: a case report and review of the literature. J. Infect. Public Health 8, 98–100. doi: 10.1016/j.jiph.2014.05.006
Funke, G., von Graevenitz, A., Clarridge, J. E., and Bernard, K. A. (1997). Clinical microbiology of coryneform bacteria. Clin. Microbiol. Rev. 10, 125–159.
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Goodfellow, M., Kämpfer, P., Busse, H.-J., Trujillo, M. E., Suzuki, K., Ludwig, W., et al. (2012). “The actinobacteria, part A,” in Bergey's Manual® of Systematic Bacteriology (New York, NY: Springer), 1034.
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Grosse-Kock, S., Kolodkina, V., Schwalbe, E. C., Blom, J., Burkovski, A., Hoskisson, P. A., et al. (2017). Genomic analysis of endemic clones of toxigenic and non-toxigenic Corynebacterium diphtheriae in Belarus during and after the major epidemic in 1990s. BMC Genomics 18:873. doi: 10.1186/s12864-017-4276-3
Gubler, J., Huber-Schneider, C., Gruner, E., and Altwegg, M. (1998). An outbreak of nontoxigenic Corynebacterium diphtheriae infection: single bacterial clone causing invasive infection among swiss drug users. Clin. Infect. Dis. 27, 1295–1298. doi: 10.1086/514997
Hirata, R. Jr., Pereira, G. A., Filardy, A. A., Gomes, D. L. R., Damasco, P. V., Rosa, A. C. P., et al. (2008). Potential pathogenic role of aggregative-adhering Corynebacterium diphtheriae of different clonal groups in endocarditis. Braz. J. Med. Biol. Res. 41, 986–991. doi: 10.1590/S0100-879X2008001100007
Hong, K.-W., Asmah Hani, A. W., Nurul Aina Murni, C. A., Pusparani, R. R., Chong, C. K., Verasahib, K., et al. (2017). Comparative genomic and phylogenetic analysis of a toxigenic clinical isolate of Corynebacterium diphtheriae strain B-D-16-78 from Malaysia. Infect. Genet. Evol. 54, 263–270. doi: 10.1016/j.meegid.2017.07.015
Jackson, S. A., McKenzie, R. E., Fagerlund, R. D., Kieper, S. N., Fineran, P. C., and Brouns, S. J. J. (2017). CRISPR-Cas: adapting to change. Science 356:eaal5056. doi: 10.1126/science.aal5056
Jaton, K., Ninet, B., Bille, J., and Greub, G. (2010). False-negative PCR result due to gene polymorphism: the example of Neisseria meningitidis. J. Clin. Microbiol. 48, 4590–4591. doi: 10.1128/JCM.01766-10
Jolley, K. A., and Maiden, M. C. J. (2010). BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinforma. Oxf. Engl. 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, M., Oh, H.-S., Park, S.-C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Kitchin, N. R. (2011). Review of diphtheria, tetanus and pertussis vaccines in clinical development. Expert Rev. Vaccines 10, 605–615. doi: 10.1586/erv.11.60
Kolios, A. G. A., Cozzio, A., Zinkernagel, A. S., French, L. E., and Kündig, T. M. (2017). Cutaneous Corynebacterium infection presenting with disseminated skin nodules and ulceration. Case Rep. Dermatol. 9, 8–12. doi: 10.1159/000476054
Krumsiek, J., Arnold, R., and Rattei, T. (2007). Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinforma. Oxf. Engl. 23, 1026–1028. doi: 10.1093/bioinformatics/btm039
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Lee, J. H., Wang, T., Ault, K., Liu, J., Schmitt, M. P., and Holmes, R. K. (1997). Identification and characterization of three new promoter/operators from Corynebacterium diphtheriae that are regulated by the diphtheria toxin repressor (DtxR) and iron. Infect. Immun. 65, 4273.
Lehmann, K., and Neumann, R. (1896). Atlas und Grundriss der Bakteriologie und Lehrbuch der speciellen bakteriologischen Diagnostik. 1st ed. München: J. F. Lehmann.
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Mao, X., Ma, Q., Zhou, C., Chen, X., Zhang, H., Yang, J., et al. (2014). DOOR 2.0: presenting operons and their functions through dynamic and integrated views. Nucleic Acids Res. 42, D654–D659. doi: 10.1093/nar/gkt1048
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Meinel, D. M., Kuehl, R., Zbinden, R., Boskova, V., Garzoni, C., Fadini, D., et al. (2016). Outbreak investigation for toxigenic Corynebacterium diphtheriae wound infections in refugees from Northeast Africa and Syria in Switzerland and Germany by whole genome sequencing. Clin. Microbiol. Infect. 22, 1003.e1–1003.e8. doi: 10.1016/j.cmi.2016.08.010
Mir, A., Edraki, A., Lee, J., and Sontheimer, E. J. (2018). Type II-C CRISPR-Cas9 biology, mechanism, and application. ACS Chem. Biol. 13, 357–365. doi: 10.1021/acschembio.7b00855
Okamoto, K., Hatakeyama, S., Sugita, C., Ogura, K., Ueda, R., Kouda, H., et al. (2018). Nasal diphtheria (chronic carriage) caused by nontoxigenic Corynebacterium diphtheriae. J. Infect. Chemother. doi: 10.1016/j.jiac.2018.01.015. [Epub ahead of print].
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Public Health England, A. (2015). Public Health Control and Management of Diphtheria (in England and Wales) 2015 Guidelines. Available online at: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/416108/Diphtheria_Guidelines_Final.pdf
Romney, M. G., Roscoe, D. L., Bernard, K., Lai, S., Efstratiou, A., and Clarke, A. M. (2006). Emergence of an invasive clone of nontoxigenic Corynebacterium diphtheriae in the urban poor population of Vancouver, Canada. J. Clin. Microbiol. 44, 1625–1629. doi: 10.1128/JCM.44.5.1625-1629.2006
Sangal, V., Blom, J., Sutcliffe, I. C., von Hunolstein, C., Burkovski, A., and Hoskisson, P. A. (2015). Adherence and invasive properties of Corynebacterium diphtheriae strains correlates with the predicted membrane-associated and secreted proteome. BMC Genomics 16:765. doi: 10.1186/s12864-015-1980-8
Sangal, V., Burkovski, A., Hunt, A. C., Edwards, B., Blom, J., and Hoskisson, P. A. (2014). A lack of genetic basis for biovar differentiation in clinically important Corynebacterium diphtheriae from whole genome sequencing. Infect. Genet. Evol. 21, 54–57. doi: 10.1016/j.meegid.2013.10.019
Sangal, V., Fineran, P. C., and Hoskisson, P. A. (2013). Novel configurations of type I and II CRISPR-Cas systems in Corynebacterium diphtheriae. Microbiol. Read. Engl. 159, 2118–2126. doi: 10.1099/mic.0.070235-0
Sangal, V., and Hoskisson, P. A. (2016). Evolution, epidemiology and diversity of Corynebacterium diphtheriae: New perspectives on an old foe. Infect. Genet. Evol. 43, 364–370. doi: 10.1016/j.meegid.2016.06.024
Schmitt, M. P., and Holmes, R. K. (1991a). Characterization of a defective diphtheria toxin repressor (dtxR) allele and analysis of dtxR transcription in wild-type and mutant strains of Corynebacterium diphtheriae. Infect. Immun. 59, 3903–3908.
Schmitt, M. P., and Holmes, R. K. (1991b). Iron-dependent regulation of diphtheria toxin and siderophore expression by the cloned Corynebacterium diphtheriae repressor gene dtxR in C. diphtheriae C7 strains. Infect. Immun. 59, 1899–1904.
Schmitt, M. P., Talley, B. G., and Holmes, R. K. (1997). Characterization of lipoprotein IRP1 from Corynebacterium diphtheriae, which is regulated by the diphtheria toxin repressor (DtxR) and iron. Infect. Immun. 65, 5364.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Spirig, T., Weiner, E. M., and Clubb, R. T. (2011). Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 82, 1044–1059. doi: 10.1111/j.1365-2958.2011.07887.x
Swaminathan, A., Mandlik, A., Swierczynski, A., Gaspar, A., Das, A., and Ton-That, H. (2007). Housekeeping sortase facilitates the cell wall anchoring of pilus polymers in Corynebacterium diphtheriae. Mol. Microbiol. 66, 961–974. doi: 10.1111/j.1365-2958.2007.05968.x
Tagini, F., and Greub, G. (2017). Bacterial genome sequencing in clinical microbiology: a pathogen-oriented review. Eur. J. Clin. Microbiol. Infect. Dis. 36, 2007–2020. doi: 10.1007/s10096-017-3024-6
Trost, E., Blom, J., Soares Sde, C., Huang, I.-H., Al-Dilaimi, A., Schröder, J., et al. (2012). Pangenomic study of Corynebacterium diphtheriae that provides insights into the genomic diversity of pathogenic isolates from cases of classical diphtheria, endocarditis, and pneumonia. J. Bacteriol. 194, 3199–3215. doi: 10.1128/JB.00183-12
Vázquez-Torres, A., and Bäumler, A. J. (2016). Nitrate, nitrite and nitric oxide reductases: from the last universal common ancestor to modern bacterial pathogens. Curr. Opin. Microbiol. 29, 1–8. doi: 10.1016/j.mib.2015.09.002
Viana, M. V. C., Figueiredo, H., Ramos, R., Guimarães, L. C., Pereira, F. L., Dorella, F. A., et al. (2017). Comparative genomic analysis between Corynebacterium pseudotuberculosis strains isolated from buffalo. PLoS ONE 12:e0176347. doi: 10.1371/journal.pone.0176347
Keywords: non-toxigenic diphtheria, comparative genomics, virulence, mobile genetic elements, CRISPR, pili, sortase
Citation: Tagini F, Pillonel T, Croxatto A, Bertelli C, Koutsokera A, Lovis A and Greub G (2018) Distinct Genomic Features Characterize Two Clades of Corynebacterium diphtheriae: Proposal of Corynebacterium diphtheriae Subsp. diphtheriae Subsp. nov. and Corynebacterium diphtheriae Subsp. lausannense Subsp. nov. Front. Microbiol. 9:1743. doi: 10.3389/fmicb.2018.01743
Received: 13 April 2018; Accepted: 12 July 2018;
Published: 17 August 2018.
Edited by:
Iain Sutcliffe, Northumbria University, United KingdomReviewed by:
Michael Peter Schmitt, United States Food and Drug Administration, United StatesVartul Sangal, Northumbria University, United Kingdom
Andreas Burkovski, Friedrich-Alexander-Universität Erlangen-Nürnberg, Germany
Aharon Oren, Hebrew University of Jerusalem, Israel
Copyright © 2018 Tagini, Pillonel, Croxatto, Bertelli, Koutsokera, Lovis and Greub. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gilbert Greub, Z2lsYmVydC5ncmV1YkBjaHV2LmNo