 Lucas Binnenkade†
Lucas Binnenkade† Maximilian Kreienbaum
Maximilian Kreienbaum Kai M. Thormann
Kai M. Thormann- Institute for Microbiology and Molecular Biology, Justus Liebig University Giessen, Giessen, Germany
Bacterial extracellular nucleases have multiple functions in processes as diverse as nutrient acquisition, natural transformation, biofilm formation, or defense against neutrophil extracellular traps (NETs). Here we explored the properties of ExeM in Shewanella oneidensis MR-1, an extracellular nuclease, which is widely conserved among species of Shewanella, Vibrio, Aeromonas, and others. In S. oneidensis, ExeM is crucial for normal biofilm formation. In vitro activity measurements on heterologously produced ExeM revealed that this enzyme is a sugar-unspecific endonuclease, which requires Ca2+ and Mg2+/Mn2+ as co-factors for full activity. ExeM was almost exclusively localized to the cytoplasmic membrane fraction, even when a putative C-terminal membrane anchor was deleted. In contrast, ExeM was not detected in medium supernatants. Based on the results we hypothesize that ExeM predominantly interacts with DNA in close proximity to the cell, e.g., to promote biofilm formation and defense against NETs, or to control uptake of DNA.
Introduction
Extracellular DNA ubiquitously occurs in terrestrial, marine and fresh-water habitats as the product of passive or active cell lysis or active DNA transport (Vlassov et al., 2007; Ibánez de Aldecoa et al., 2017). The concentration varies substantially between various environments and was demonstrated to reach up to 20 mg g-1 in activated sludge and 0.31 g of total DNA/m2 in the top cm of deep-sea sediments. More than 90% out of these 0.31 g/m2 are thought to be extracellular DNA (Palmgren and Nielsen, 1996; Niemeyer and Gessler, 2002; Dell’Anno and Danovaro, 2005). Thus, extracellular DNA affects bacteria in many different settings and environments. Bacteria were demonstrated to exploit DNA as a source of carbon, nitrogen, and phosphorus. In deep-sea ecosystem functioning extracellular DNA was even found to play a key role in the marine phosphorus cycles (Dell’Anno and Danovaro, 2005; Pinchuk et al., 2008; Mulcahy et al., 2010; Seper et al., 2011; Ibánez de Aldecoa et al., 2017). Extracellular DNA represents an important source for an intra- and interspecies exchange and spread of genetic information by horizontal gene transfer (Molin and Tolker-Nielsen, 2003). In addition, DNA has long been recognized as a highly common and, many times, functionally and structurally important component of the extracellular matrix of bacterial biofilms (reviewed in Flemming and Wingender, 2010; Dragos and Kovacs, 2017).
Given the abundance and various important functions of extracellular DNA, bacteria require appropriate means to degrade and modulate this molecule accordingly. To this end, many bacteria produce extracellular nucleases, which may either be anchored to the cell envelope or completely released into the extracellular space. During biofilm formation, the activity of such nucleases was demonstrated to balance the extent of biofilm formation by promoting biofilm dispersal in various species such as Staphylococcus, Neisseria, Haemophilus, Vibrio, Shewanella, Ralstonia, and others (Mann et al., 2009; Gödeke et al., 2011a; Kiedrowski et al., 2011; Seper et al., 2011; Steichen et al., 2011; Beenken et al., 2012; Kiedrowski et al., 2014; Minh Tran et al., 2016). Extracellular nucleolytic activity prevents DNA to accumulate to levels toxic to the cell, an effect that is due to the cation-chelating properties of DNA (Mulcahy et al., 2008; Heun et al., 2012). In addition, extracellular nucleases are crucial for utilizing DNA as a nutrient (Pinchuk et al., 2008; Mulcahy et al., 2010; Gödeke et al., 2011a; Heun et al., 2012). Moreover, they affect transformation activity and horizontal gene transfer by either degrading extracellular DNA from the environment to prevent entry into the cell (Focareta and Manning, 1987; Wu et al., 2001; Blokesch and Schoolnik, 2008) or as component of the natural DNA-uptake machinery (Berge et al., 2002).
In many pathogenic bacteria, another important function of extracellular nucleases is to promote the degradation of and escape from neutrophil extracellular traps (NETs). Such NETs consist of a DNA-matrix as a scaffold for an arsenal of antimicrobial proteins to degrade potential virulence factors and to immobilize and kill invading bacteria (Brinkmann et al., 2004; Halverson et al., 2015). A role of extracellular nucleases in evading NETs has been shown for a wide range of bacterial pathogens including Vibrio cholerae, Streptococcus sp., Staphylococcus aureus, Prevotella intermedia, and Mycoplasma pneumoniae (Beiter et al., 2006; Buchanan et al., 2006; Berends et al., 2010; Seper et al., 2013; Doke et al., 2017; Liu et al., 2017). Recent work has shown a similar role of extracellular nucleolytic activity for the plant pathogen Ralstonia solanacearum to degrade DNA deposited into the soil by root border cells to trap pathogens (Hawes et al., 2011; Tran et al., 2016).
Taken together, extracellular nucleases play a central role for the modification and/or degradation of eDNA in microbial biofilms and microbial communities and exhibit diverse functions with medical relevance, such as natural transformation, degradation of DNA in NETs, and induction of biofilm dispersal for biofilm control. However, the underlying molecular and regulatory mechanisms are still poorly understood and remain to be elucidated in more detail. In this study we further characterized the extracellular nuclease ExeM from Shewanella oneidensis MR-1.
Generally, bacteria of the genus Shewanella are facultatively anaerobic gammaproteobacteria, which are characterized by their ability to use an enormous arsenal of external electron acceptors for respiration, a high tolerance for different sodium chloride concentrations and the capability to grow in a wide range of temperatures (reviewed in Hau and Gralnick, 2007; Fredrickson et al., 2008). Hence, Shewanella sp. have been isolated from a wide range of habitats from fresh and salt water sediments to rotten fish, and some Shewanella species have been identified as commensal human pathogens (Janda and Abbott, 2014). S. oneidensis has emerged as a model species for Shewanella physiology and biofilm formation. Previous work provided evidence that extracellular DNA is an important structural component of S. oneidensis biofilms and is produced by prophage-induced lysis in particular during cell-surface attachment due to iron-induced intracellular stress (Gödeke et al., 2011b, 2012; Binnenkade et al., 2014). In addition, this species is capable of using DNA as sole source of carbon, nitrogen, and phosphorus (Pinchuk et al., 2008). S. oneidensis MR-1 produces three extracellular nucleases, EndA, ExeS, and ExeM (Gödeke et al., 2011a; Heun et al., 2012). While EndA primarily degrades DNA in culture supernatants and is mainly required to use DNA as source of phosphorus, ExeS, and ExeM contribute only little to the nucleolytic activity in the supernatant. In contrast, ExeM, which was proposed to be associated with the cell envelope, strongly affects biofilm formation and cellular detachment dynamics (Gödeke et al., 2011a; Heun et al., 2012). The putative ortholog of ExeM in V. cholerae, Xds, was, similarly, shown to be involved in normal biofilm formation, nutrient acquisition and escape from NETs (Seper et al., 2011, 2013), strongly indicating an important role of ExeM/Xds for various cellular processes. However, rather little is known about the activity and localization of these nucleases. Here we performed a deeper characterization of ExeM to further elucidate its potential functions in DNA degradation.
Materials and Methods
Strains and Growth Conditions
Supplementary Table 2 shows all bacterial strains used in this study. Routinely, S. oneidensis and E. coli were cultivated in LB medium at 30 and 37°C, respectively. Agar was added to a final concentration of 1.5% (w/v) for solidification purposes. If needed, 2,6-diaminopimelic acid (300 μM), ampicillin (100 μg ml-1) and/or kanamycin (50 μg ml-1) were added. For biofilm assays of S. oneidensis MR-1, cells were grown in LM medium containing 0.5 M lactate (Paulick et al., 2009).
Strain Constructions
Standard protocols (Sambrook et al., 1989) were applied for general DNA manipulations using kits for preparation and purification of DNA (VWR International GmbH, Darmstadt, Germany) and enzymes (Fermentas, Sankt Leon-Rot, Germany) accordingly. Plasmids (Supplementary Table 3) were introduced into S. oneidensis MR-1 by conjugation from E. coli WM3064. For markerless in-frame deletions sequential homologous recombination was applied using suicide vector pNPTS-138-R6K as previously described (Lassak et al., 2010). To construct vectors, either Gibson assembly (Gibson et al., 2009) or standard restriction/ligation methods were used. The oligonucleotides used for the required PCRs are shown in Supplementary Table 4.
Cultivation and Quantification of Biofilms
Static biofilm assays were performed essentially as previously described (Thormann et al., 2004). Briefly, 170 μl LM medium per well on a 96 well plate (Sarstedt, Nümbrecht) was inoculated with 5 μl of a S. oneidensis overnight culture (8 wells per strain) and incubated at 30°C for 24–48 h. Optical density at 600 nm was determined before adding 10 μl 0.5% crystal violet to each well. After incubation for 10 min at RT the whole supernatant was discarded and 200 μl were added to wash the cells. Again, the supernatant was discarded and finally 200 μl 96% EtOH (w/v) were added and incubated for 5 min at RT before determining the absorption at 580 nm. To obtain relative biofilm formation the degree of surface attachment was normalized to that of the wild-type. At least three independent experiments were carried out.
Enrichment of ExeM
For construction of over expression plasmids, vector pMal-P2X (NEB) was altered by exchanging the Factor Xa protease cleavage site for a TEV protease cleavage site, resulting in plasmid pMal-P2-TEV. Then, plasmids encoding ExeM, ExeM-ΔLTD (ExeM lacking the N-terminal LTD domain; Figure 1), ExeM-ΔYhcR (ExeM lacking the YhcR nuclease domain; Figure 1) and ExeM-ΔEEP (ExeM lacking the C-terminal EEP nuclease domain; Figure 1) were constructed by amplifying the appropriate gene regions without both the N-terminal signal sequence and the C-terminal hydrophobic regions and cloning it into pMal-P2-TEV. This resulted in in-frame fusions to malE, encoding the maltose binding protein (MBP) and targeting of the fusion protein to the periplasm. E. coli BL 21 Star (DE3) was transformed with the resulting vectors. 400 ml SOB medium containing 0.2% (w/v) glucose were inoculated with an overnight culture in LB (containing 50 μg ml-1 ampicillin) of the resulting strains. Cells were grown at 37°C to an OD600 of 0.5, rapidly cooled down on ice and IPTG was added to a final concentration of 0.3 mM. After incubating the cultures for 4 h at 25°C, the cells were harvested by centrifugation, frozen in liquid nitrogen and subsequently stored at -20°C. Cell pellets were lysed by resuspending them in 30 ml ice-cold 1× PBS buffer containing 0.5 mM AEBSF-hydrochloride (Carl Roth GmbH + Co. KG, Germany) and subsequently passing them through a cooled “French Press” three times. Centrifugation at 8,000 g for 10 min at 4°C and subsequent ultracentrifugation at 30,000 g at 4°C were used to remove unbroken cells and insoluble cell debris, respectively. At 4°C, the solution was added to a column containing 5 ml amylose resin and eluted in 1.5 ml fractions by adding column buffer (20 mM Tris-HCl, 200 mM NaCl, 10 mM maltose). The elution fractions were then analyzed via SDS-PAGE and pooled if containing high levels of protein. Five hundred microliter of the sample were further purified by size-exclusion chromatography (Superdex 200 HR 10/30, GE healthcare) using Buffer A (20 mM Tris-HCL, 200 mM NaCl, pH 7) for isocratic elution. Protein concentration was measured with a spectrophotometer (NANODROP 1000, Thermo Fisher Scientific). After another analysis by SDS-PAGE, 30% (v/v) glycerol was added to the samples, which were then frozen in liquid nitrogen and stored at -20°C.

FIGURE 1. Domain architecture of ExeM. Schematic illustration of conserved domains identified by sequence comparison (BLASTP). Cysteine residues that are potentially involved in disulfide bond formation are indicated on the upper side. Putative metal binding sites (MB) and putative phosphate binding sites (PB) are indicated on the lower site. Deletions of the domains were designed in a way that the exact amino acids of the respective domain were deleted, except deletion of L-TM, where amino acids 833–869 were deleted.
In vivo and in vitro DNA Degradation Assays
Nuclease Activity in Culture Supernatants
Qualitative nuclease assays in medium supernatants were conducted essentially as previously described (Gödeke et al., 2011a). Appropriate cells from an overnight LB preculture were incubated in fresh medium at an OD600 of 0.05 and were grown to an OD600 of 1.5. 230 μl aliquots of cell-free filter-sterilized supernatant were mixed with an appropriate nucleic acid sample at a final concentration of 5 μg ml-1. The samples were incubated at 30°C, aliquots were removed at regular intervals and the integrity of the DNA was analyzed by agarose gel electrophoresis. The assay was repeated in at least two independent experiments.
Nuclease Activity of Washed Cells
For a comparison of nuclease activity in culture supernatants and washed cells, cultures were grown to an OD600 of 1.5. In order to inhibit further protein synthesis, chloramphenicol was added to a final concentration of 30 μg ml-1 and cultures were incubated for 20 min at 30°C. To determine the nuclease activity of washed cells, 230 μl aliquots were washed three times in LB medium containing chloramphenicol (30 μg ml-1). For a comparable determination of nuclease activity in the respective culture supernatants, 230 μl aliquots of the same culture were used without washing in LB. Subsequently, cell suspensions were mixed with a 20 μl nucleic acid sample (833 bp PCR product) at a final concentration of 5 μg ml-1. The samples were incubated at 30°C, and 20 μl aliquots of each supernatant were removed, centrifuged, and analyzed by agarose gel electrophoresis at regular intervals.
DNA-Agar Assays
Appropriate S. oneidensis strains were inoculated from overnight cultures to an OD600 of 0.05 and incubated for 3 h while shaking. Cell suspensions were diluted 1:10 and 8 μl of the dilution were spotted carefully on DNA-agar plates (Beckton Dickinson GmbH, Heidelberg, Germany). After letting the spots dry, plates were incubated at RT for at least 48 h. 1N HCl was added carefully to plates to precipitate DNA. After incubating for 5 min plates were scanned using an Epson Perfection V700 Photo Scanner (Epson, Japan). Three independent assays were conducted.
GelRed Assays
Determination of the nuclease activity of purified ExeM, MBP-ExeM and its truncated versions was essentially performed as previously described (Heun et al., 2012). Briefly, in a 96-well plate 8 μg of enriched protein (corresponding to ∼25 pmol MBP-ExeM), 250 ng purified pBluescript II KS(+) DNA and 67 μl of 3× GelRed nucleic acid stain (Biotrend, Germany) were added to 10 mM Tris-HCl (pH 7.6) with 12.5 mM Mg2+ and Ca2+ in a final volume of 200 μl. Decrease of fluorescence of the GelRed stain were followed and recorded using a Tecan Infinite M200 microplate reader (Tecan, Switzerland). Most characterizations were carried out using amylose-enriched MBP-ExeM, which allowed using higher amounts of protein even though the amount of intact protein was lower (<50%). At least three independent assays were conducted.
Membrane Separations
Inner and outer membrane fractions were separated and purified according to a sarkosyl-based protocol presented by Brown et al. (2010). S. oneidensis MR-1 was cultured overnight in 10 ml LB medium and reinoculated in 250 ml LB medium at an OD600 of 0.05. Strains with plasmid pBBMT-kan-exeM or derivatives (for the overproduction of ExeM or truncated ExeM variants) were cultured in the presence of 50 μg ml-1 kanamycin and induced with 0.2% (w/v) L-arabinose at an OD600 of 0.6. At an OD600 of 2, the cells were harvested by centrifugation at 10,000 g for 10 min. An 80 ml fraction of the culture was kept for isolation of the periplasmic fraction (see below). Unless stated otherwise, all centrifugations were performed at 4°C. The supernatant (SN) was ultracentrifuged twice at 35,000 g for 1 h and stored at -20°C for further analyzes. The cell pellet was suspended in 30 ml ice-cold 20 mM sodium phosphate buffer (pH 7.5) and passed one time through a prechilled “French press.” The clear lysate was centrifuged at 8,000 g for 10 min to remove unbroken cells, and an aliquot of the supernatant was stored as “whole cell lysate sample.” Ten milliliters of the remaining supernatant were ultracentrifuged at 45,000 g for 1 h. The supernatant was removed and centrifuged again to remove residual membrane fractions and insoluble protein and stored at -20°C as soluble fraction. The tube containing the whole membrane fraction was inverted to drain, and a sample was frozen in liquid nitrogen and stored at -20°C for further analyzes. The remaining whole membrane fraction was suspended in 0.5% sarkosysl (20 mM sodium phosphate) by frequent “pipetting” and orbital shaking at 220 rpm for 30 min at room temperature. The crude membrane suspension was ultracentrifuged at 45,000 rpm for 1 h to sediment the outer membrane. The supernatant containing the inner membrane was removed and the outer membrane sample was washed in ice-cold sodium phosphate buffer, spun down again by ultracentrifugation at 45,000 rpm for 1 h, suspended in 500 μl sodium phosphate, frozen in liquid nitrogen, and stored at -20°C. The supernatant containing the inner membrane was washed and concentrated to 500 μl using Vivaspin®6 centrifugation filter tubes (Sartorius Stedim Biotech GmbH, Germany) with a cutoff of 5 kDa. The inner membrane sample was frozen in liquid nitrogen and stored at -20°C for further analyzes.
The periplasmic protein fraction was isolated by osmotic shock according to Ross and coworkers (Ross et al., 2007). Eighty milliliters of the initial culture were centrifuged at 8,000 g for 10 min and suspended in 10 ml of 50 mM Tris-HCl, pH 8.0, 250 mM sucrose. The suspension was incubated for 5 min at room temperature and centrifuged at 8,000 g for 15 min. The pellet was suspended in ice-cold 5 mM MgSO4 and kept on ice with occasional inversion. The soluble periplasmic fraction was obtained from the supernatant after centrifugation at 8,000 g for 15 min. The periplasmic fraction was concentrated to a final volume of 500 μl using Vivaspin®6 centrifugation filter tubes (Sartorius Stedim Biotech GmbH, Germany) with a 5 kDa cutoff.
Immunoblotting and Antibody Enrichment
Production of ExeM was determined using immunoblot analysis. Samples were taken from exponentially growing cultures and adjusted to an OD600 of 10. Immunoblot detection was essentially carried out as described earlier (Bubendorfer et al., 2012). Samples were separated by electrophoresis using 11% SDS-Polyacrylamide gels (SDS-PAGE) and subsequently transferred to a PVDF membrane by electroblotting. Detection of signals was carried out using the CDP-Star chemiluminescence substrate (Roche, Germany) and imaged using the FUSION-SL chemiluminescence imager (Peqlab, Erlangen, Germany).
The antibody against ExeM was ordered from Eurogentech (Germany) and further purified as follows. For purification of 2 ml of high-titer serum, 1 mg of ExeM was run on a gel and subsequently blotted to a PVDF membrane. The band of interest was cut and washed with acidic glycine buffer (100 mM Glycine, pH 2.5) for 5 min and then again washed twice with TBS [500 mM NaCl, 20 mM Tris, pH 7.4, 0.05% (v/v) Tween-20] for 2 min and blocked by soaking it with TBS-B (3% of fraction V bovine serum albumin in TBS buffer) for 1 h at RT while slightly shaking. The Membrane was washed twice with TBS and 2 ml of serum in 8 ml of TBS were added and incubated overnight at 4°C. Again, the membrane was washed twice with TBS and twice with PBS (0.135 NaCl, 3.5 mM KCl, 8 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) each for 5 min. For elution 1 ml of acidic glycine buffer was added and after 10 min incubation at RT transferred to a new tube containing 1 M Tris, pH 8.0, bringing the final pH to 7.0. Elution was repeated and both fractions were pooled. Finally, antibodies were stored at 4°C with 5 mM sodium azide and 1 mg ml-1 of bovine serum albumin for stabilization. For detection, the antibodies were used in a 1:5,000 dilution. As a second antibody anti-rabbit coupled to goat AP was used in a 1:20,000 dilution. The antibody reliably detected purified MBP-ExeM and ExeM expressed in S. oneidensis MR-1 with only little unspecific binding to proteins in PAGE-separated crude extract (Supplementary Figure 1).
Phylogenetic Analysis of ExeM-Like Nucleases
BLAST analysis was used to identify protein sequences showing high sequence similarities to ExeM (SO_1066) among the gammaproteobacteria. The sequences were aligned by ClustalW2 and subjected to phylogenetic analysis by PhylM using the LG substitution model and an aLRT-SH-like fast likelihood-based method (Larkin et al., 2007; Guindon et al., 2010). Phylogenetic trees were constructed by iTOL (Letunic and Bork, 2007). Branch lengths were disregarded for this analysis.
Results
ExeM Domain Structure
ExeM is a protein of 871 aa with a predicted molecular mass of 93.7 kDa. Protein domain analysis (Figure 1) by InterPro (Finn et al., 2017) suggest the presence of an N-terminal signal sequence (aa 1–24) followed by a lamin tail domain (LTD; aa 28–128) and a stretch (aa 28–128) resembling an oligonucleotide-binding structural motif similar to that of Bacillus subtilis YcgR, an endonuclease cleaving both RNA and DNA (Oussenko et al., 2004). A large domain belonging to the exonuclease-endonuclease-phosphatase (EEP) domain superfamily is predicted to account for almost the entire C-terminal half of the protein (aa 464–830). This EEP domain contains two residues (E521, D822) predicted to be involved in metal co-factor binding and three residues (H642, N699, H823), which are potentially involved in phosphate binding. The EEP domain is followed downstream by a prolin-rich stretch of 13 residues (aa 849–865; PAPVVPPKPQPTP) and a hydrophobic domain (aa 849–865; GGALGYLGLALLSLLGL) followed by four arginine residues (RRRR) at the very C-terminus of ExeM. This arrangement suggests that, while the major part of ExeM is excreted into the periplasm, the protein may remain anchored to the cytoplasmic membrane by a putative C-terminal transmembrane anchor the orientation of which is coordinated by the positively charged cytoplasmic four-arginine stretch (von Heijne, 1989, 1992; van de Vossenberg et al., 1998). Thus, release from the membrane may require proteolytic cleavage within the region linking the C-terminal membrane anchor and the EEP domain. Accordingly, the so-called “Gly-Gly-Cterm motif” consisting of a GG motif followed by a transmembrane helix and a cluster of basic residues has been predicted to constitute a processing signal for intramembrane serine proteases, referred to as rhombosortases. Accordingly, a putative rhombosortase (SO_2504) was also identified in S. oneidensis MR-1 (Haft and Varghese, 2011).
Potential orthologs to SoExeM with a highly similar domain structure were identified in other Shewanella and Vibrio species (such as VcXds), and also in species of Aeromonas, Pseudoalteromonas and, most distantly related, Pseudomonas (Supplementary Figure 2), which also lacks the putative C-terminal membrane anchor and rhombosortase processing site.
In Vitro and in vivo Activity of ExeM
To investigate the nucleolytic activity of ExeM, the role of the different domains and to identify potential co-factors, the protein was heterologously overproduced in E. coli. ExeM is predicted to be exported from the cytoplasm, and several bacterial nucleases have been shown to only fold into their active form after export into the periplasm to inhibit premature activity and to prevent loss of chromosome integrity. This can be conferred by disulfide bridge formation between cysteine residues (Wu et al., 2001; Li et al., 2003; Heun et al., 2012). Accordingly, four cysteine residues are present in ExeM (C187, C272, C650, C670; Figure 1) which may be involved in proper folding. Therefore, for heterologous production ExeM lacking its native signaling sequence as well as the C-terminal transmembrane region was targeted to the periplasm by an N-terminal fusion to maltose-binding protein (MBP) which also drastically increased yield and stability (Supplementary Figure 3). However, purified MBP-ExeM was still prone to rapid decay and aggregation which was further increased upon MBP-tag removal by TEV cleavage. Purified ExeM directly isolated from a gel subsequent to PAGE separation was used for antibody production, and for further in vitro assays MBP-ExeM was employed. In in vitro DNA-degradation assays ExeM readily degraded DNA and RNA as well as linear and plasmid DNA (Figure 2A) and therefore appears to act as a sugar-unspecific endonuclease. The presence of Ca2+ was crucial for enzyme activity and full activity additionally required Mg2+ or Mn2+ (Figure 2B). Other divalent cations (Zn2+, Ni2+, and Cu2+) did not affect nuclease function (data not shown). To determine the optimal concentration for these potential co-factors, a range of different concentrations was applied (Figure 2C). The results indicated that DNA degradation occurred most rapidly at concentration of 12–25 mM for Ca2+, 6–25 mM for Mg2+, and 0.1–6 mM for Mn2+. Highest activity of ExeM was found at an equimolar concentration of ∼12.5 mM for Ca2+ and Mg2+ (Figure 2D). Defining one unit of enzyme activity as the amount of MBP-ExeM required to completely degrade 1 μg of pBluescript vector within 10 min at 30°C in reaction buffer supplemented with 12.5 mM Mg2+ and 12.5 mM Ca2+, the specific activity of purified MBP-ExeM was ∼3 U/mg.
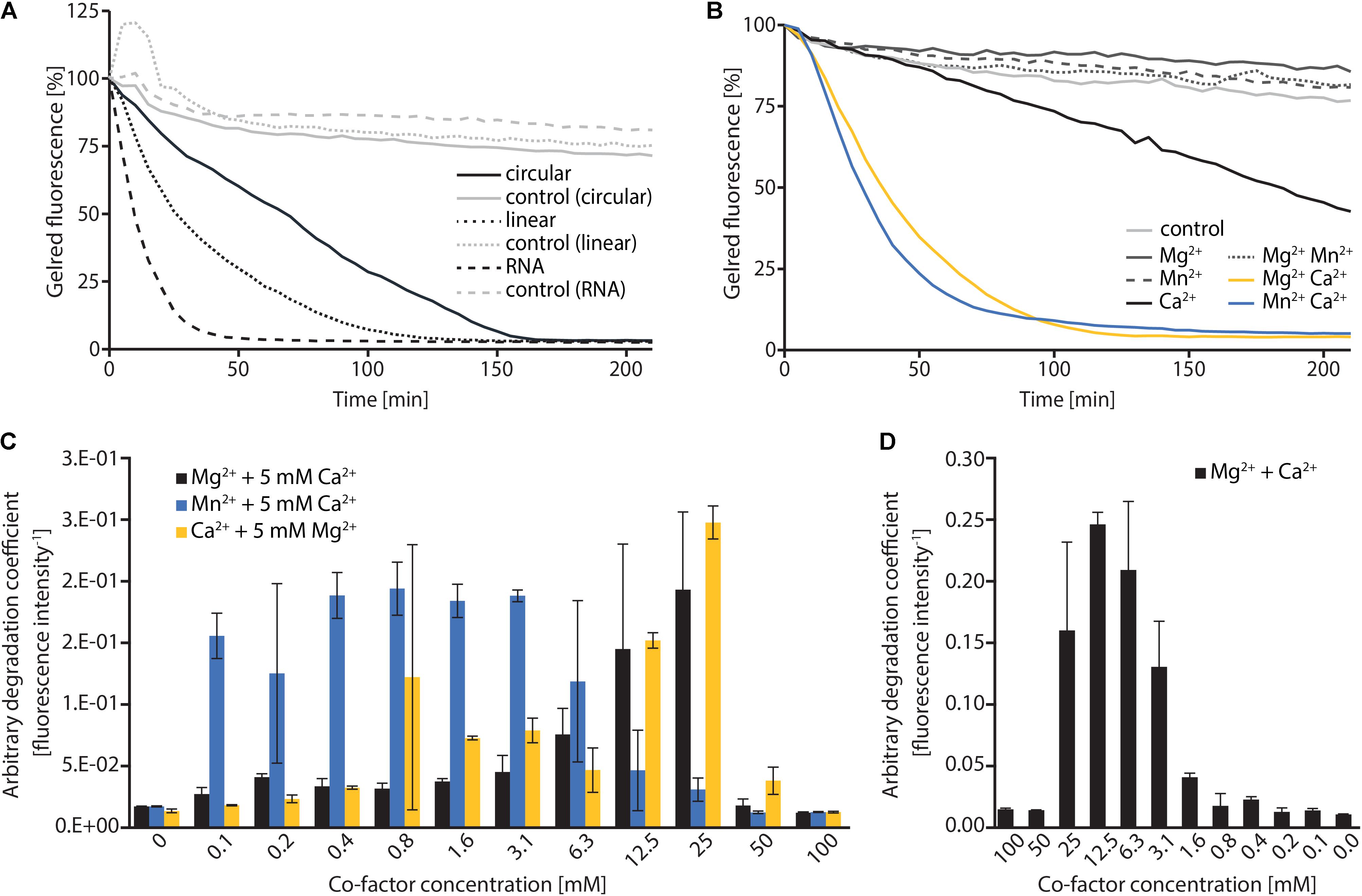
FIGURE 2. In vitro characterization of MBP-ExeM nucleolytic activity. (A) Degradation of pBluescript plasmid DNA (circular and linear; 250 ng) and RNA (1.8 μg) by MBP-ExeM (8 μg) as indicated by a loss in fluorescence of nucleic acid stain GelRedTM. The buffer contained 5 mM Mg2+ and Ca2+ to support ExeM’s nucleolytic activity. Control samples contained no MBP-ExeM. The assay was performed in triplicates in two independent experiments. The curves are based on the mean values of one representative experiment. (B) Degradation of pBluescript plasmid DNA (250 ng) by MBP-ExeM (8 μg; ∼25 pmol MBP-ExeM) as indicated by a loss in fluorescence of nucleic acid stain GelRedTM. The buffer contained 5 mM Mg2+, Mn2+, or Ca2+ (or combinations) to support ExeM’s nucleolytic actvity. The control samples contained no additional metal ions. The assay was performed in triplicates in two independent experiments. The curves are based on the mean values of one representative experiment. (C) Comparison of pBluescript plasmid DNA (250 ng) degradation by MBP-ExeM (8 μg protein; ∼25 pmol MBP-ExeM) in the presence of a range of concentrations of one cofactor, while the indicated second cofactor was kept at 5 mM. (D) Comparison of pBluescript plasmid DNA (250 ng) degradation by MBP-ExeM (8 μg) in the presence of Mg2+ and Ca2+ at different equimolar concentrations. The arbitrary degradation coefficient in (C,D) represents the reciprocal mean value of the fluorescence intensity (in % of the initial value) of nucleic acid stain GelRedTM after 150 min (time point at which the fluorescence of at least one sample approached 0). Error bars represent standard deviations of two independent experiments performed each at least in triplicates.
To further determine the role of the putative ExeM domains we heterologously overproduced versions in which one of the major predicted domains was deleted (MBP-ExeMΔLTD; MBP-ExeMΔYcgR; MBP-ExeMΔEEP) and applied these for in vitro-testing (Figure 3A). Loss of the YcgR-like and the EEP domains resulted in a complete inactivity of the nuclease. The ExeM version truncated by the N-terminal LTD domain showed slow but significant DNA degradation in the fluorescence-based assay, and a visible DNA fragment was absent after agarose separation after incubation with ExeMΔLTD (Figure 3B).
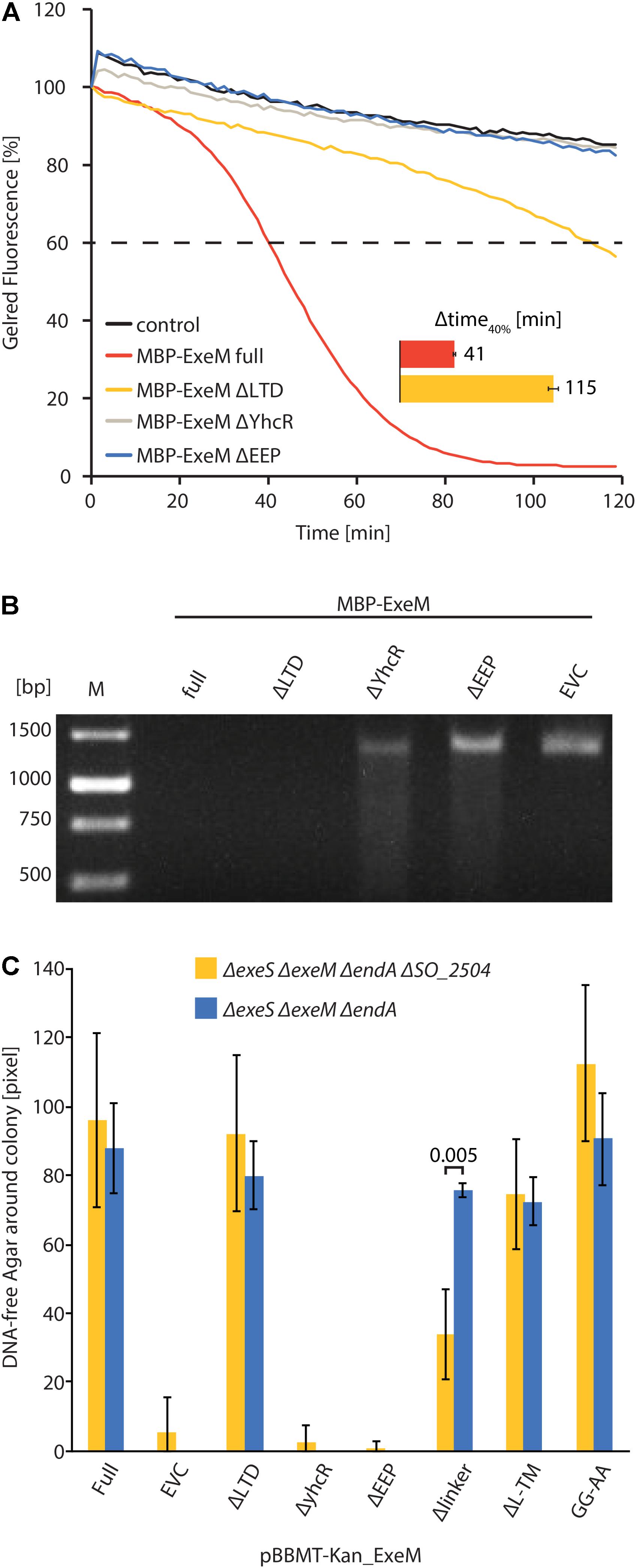
FIGURE 3. Influence of domain deletions of ExeM activity. (A) Degradation of 250 ng circular pBluescript plasmid DNA by different truncated versions of MBP-ExeM as indicated by a loss in fluorescence of nucleic acid stain GelRedTM. The buffer contained 5 mM Mg2+ and Ca2+ to support ExeM’s nucleolytic activity. Control samples contained no MBP-ExeM. The assay was performed in triplicates in three independent experiments. The curves are based on the mean values of one representative experiment. The inset (Δtime40%) indicates the time it took for 40% of the initial fluorescence to dissipate. (B) Degradation of a ∼1,400 bp PCR fragment by different truncated versions of MBP-ExeM over 1 h on a 1% agarose gel. M indicates GeneRulerTM 1 kb DNA Ladder. (C) Degradation of high molecular weight DNA in agar plates displayed as DNA free radius around colonies overexpressing different ExeM versions in pixels. The indicated values are the mean of three independent experiments. Error bars represent the standard deviation. Only for one mutant version of ExeM (Δlinker) a significant difference was seen in the absence of the putative rhombosortase (SO_2504), as indicated by the p-value. The significance threshold was adjusted from p < 0.05 to p < 0.00625 using Bonferroni correction. p-Values are given in Supplementary Table 1.
To test the in vivo activity of ExeM and corresponding mutant versions, the appropriate S. oneidensis strains were incubated on DNA agar and the extracellular nucleolytic activity was determined as a measure of clearance of turbidity around the colonies due to DNA degradation (Figure 3C). Comparison of the clearance area revealed that loss of ExeM resulted in a significant decrease in extracellular degradation, and, accordingly, approximately wild-type levels of extracellular DNA degradation were observed when ExeM was the only remaining nuclease. To determine the activity of the mutated versions of the protein, exeM and the corresponding variants (exeMΔLTD; exeMΔYcgR; exeMΔEEP) were expressed from a plasmid under control of an inducible promoter. To avoid interference with the activity of the native extracellular nucleases, EndA, ExeM, ExeS, the expression was carried out in a strain in which all three nucleases were deleted (ΔendA ΔexeM ΔexeS). The strain expressing wild-type exeM displayed the expected zone of DNA clearance while mutants lacking the ycgR-like and the EEP domains were inactive. In contrast, expression of exeMΔLTD resulted in a clearing zone of almost the size of wild-type exeM. Thus, the YcgR-like and the EEP domain are crucial for ExeM function and/or stability while the LTD domain is dispensible for extracellular DNA degradation.
Previous studies indicated that ExeM is implicated in S. oneidensis MR-1 biofilm formation and benefits biofilm formation under static conditions (Gödeke et al., 2011a; Heun et al., 2012). We therefore determined the effect of externally added ExeM on biofilm formation (Figure 4). We found that, when purified MBP-ExeM was added to attaching cells, biofilm formation was significantly diminished. In contrast, 24 h-old biofilms were no more affected as no significant release of biomass could be detected upon addition of ExeM (Figure 4). This may suggest that ExeM may negatively affect biofilm formation when expressed in incorrect amounts or already in attaching cells.
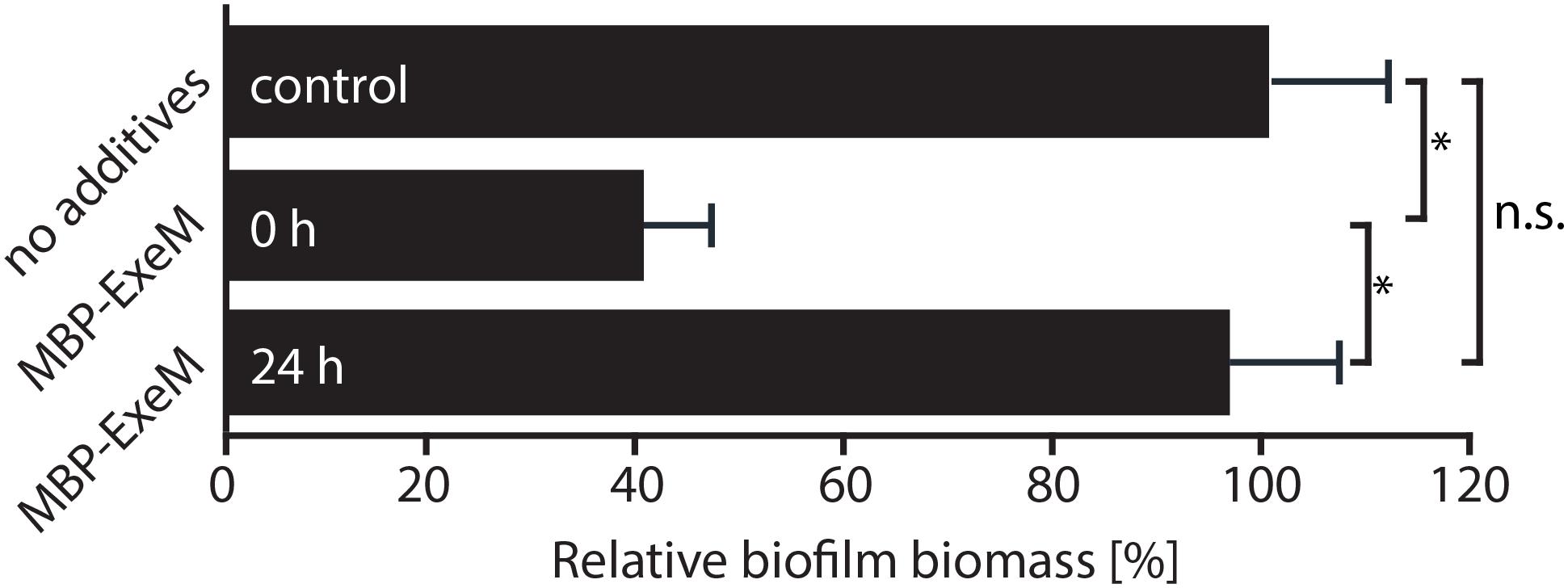
FIGURE 4. Effect of exogenously added MBP-ExeM on biofilm formation. Biofilm formation (static conditions) of the Shewanella oneidensis MR-1 wild-type after addition of MBP-ExeM (0.025 U) compared to an untreated control culture. MBP-ExeM was added prior to inoculation (0 h) or after 24 h of incubation (24 h; and incubated for further 2 h after addition). The values are means of three replicates. Error bars represent standard deviations. Asterisks indicate a significant difference between the two samples as calculated by standard t-tests after Bonferroni correction (p < 0.017). p-values are 1.5 × 10-12 (comparing control and 0 h), 0.43 (control and 24 h), and 2.5 × 10-09 (24 and 0 h), respectively.
Transport, Localization, and Processing of ExeM
The domain structure of ExeM strongly indicates that the protein is transported across the cytoplasmic membrane but may remain tethered by the C-terminal transmembrane domain and requires proteolytic cleavage, e.g., by a rhombosortase, to be released. Earlier proteome studies have identified ExeM within the cell envelope, however, it is unclear whether the nuclease is associated within the inner or outer membrane (Tang et al., 2007; Brown et al., 2010). Earlier studies suggested weak activity of ExeM in the medium supernatant (Gödeke et al., 2011a). Therefore, ExeM transport across the cell envelope and the nuclease’s final destination remained controversial.
To gain a better understanding of the ExeM localization and transport, three mutants of ExeM were produced: ExeMΔlinker, lacking the region between the EEP and the putative C-terminal transmembrane domain, ExeMΔlinker-TM, lacking both C-terminal linker and transmembrane domain, and ExeMGG-AA, in which the putative rhombosortase processing site (GG) was substituted (for an overview see Figure 1). The corresponding genes exeM and the various mutant versions were expressed at low levels from a plasmid under control of an arabinose-inducible promoter as the native production levels turned out to be too low to be reliably detected by the polyclonal antibody raised against purified ExeM. To prevent possible functional interference with the natively expressed extracellular nucleases in S. oneidensis MR-1, the mutant strain S. oneidensis MR-1 ΔendA ΔexeS ΔexeM was used for production. The strains were grown in planktonic culture to exponential growth phase, and the whole cell extract, the outer and inner membrane fractions of the cells were prepared. In addition, the periplasmic and supernatant fractions were harvested and concentrated. Subsequently, the presence of ExeM and the corresponding mutant variations were determined by immunoblot analysis of the different fractions (Figure 5).
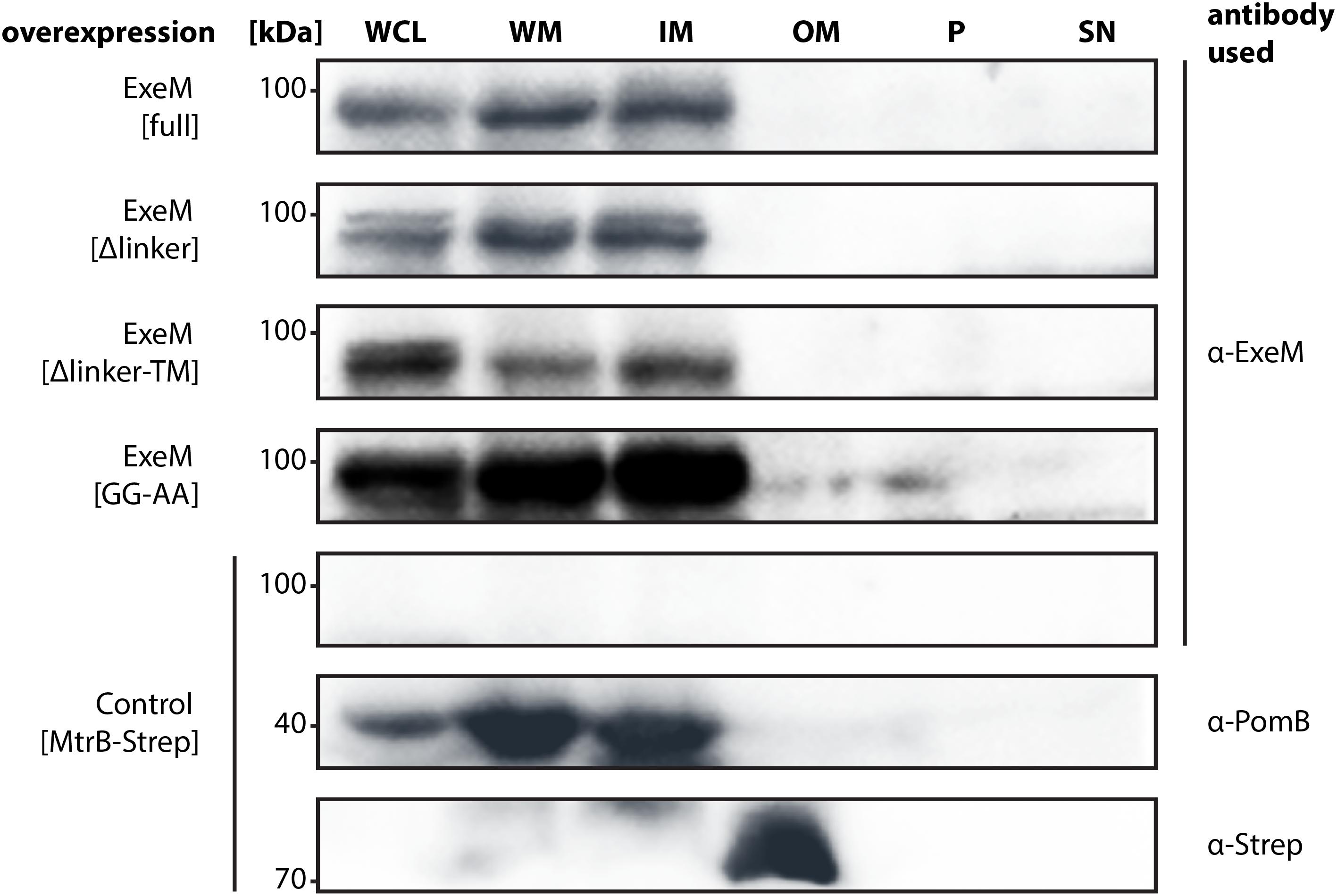
FIGURE 5. Localization of full-length ExeM and truncated ExeM constructs in various cellular fractions of S. oneidensis MR-1. Immunodetection of ExeM (α-ExeM antibody), PomB (α-PomB antibody), and MtrB-Strep (α-Strep antibody) in the whole cell lysate (WCL), the whole membrane fraction (WM), the inner membrane fraction (IM), the outer membrane fraction (OM), the periplasmic fraction (P), and the cell-free culture supernatant (SN). Full-length ExeM and truncated variants were overproduced in the ΔendA ΔexeS ΔexeM nuclease mutant. The ΔendA ΔexeS ΔexeM nuclease mutant overproducing MtrB-Strep was utilized as control for outer-membrane location and PomB as a control for inner-membrane location.
ExeM was almost exclusively detected in the fractions containing the inner membrane, strongly indicating that the nuclease is at least transiently associated with the inner membrane. Notably, this was also observed for some amount of ExeM lacking the C-terminal transmembrane region, which potentially represented ExeM in the process of being transported or associated with the cytoplasmic membrane also in the absence of the C-terminal domain. Substitution of the putative rhombosortase processing site (ExeMGG-AA) resulted in an increase of ExeM abundance in the inner membrane, suggesting that cleavage from the inner membrane may occur. In addition, ExeM variants lacking the linker region occurred as double bands, also indicating processing. However, no ExeM in significant amounts could be detected in the outer membrane fraction but also not within the periplasmic fraction or the supernatant. As earlier experiments have indicated potential ExeM activity in cell free abstracts, these experiments were repeated under conditions of phosphate starvation, which have been shown to increase exeM expression (Gödeke et al., 2011a). Also the analysis of lyophilized supernatant of these cultures did not identify any amounts of ExeM (data not shown). The results suggest that under planktonic conditions at least the major part of ExeM remains associated with the inner membrane after export and may be rather unstable upon release.
To further determine whether rhombosortase-mediated processing of ExeM is required for activity under conditions of surface growth, we measured the zone of clearance of ExeM producing strains on DNA-agar plates (Figure 3C). To this end, ExeM was produced in a strain lacking the three extracellular nucleases (ΔendA ΔexeS ΔexeM) and also in a strain additionally lacking the putative rhombosortase SO_2504 (ΔendA ΔexeS ΔexeM ΔSO_2504). The results clearly demonstrated that, generally, processing by SO_2504 is not required for the extracellular nucleolytic activity of ExeM as the zone of clearance was only little affected in the absence of the putative rhombosortase, and also the GG to AA substitution of the predicted cleaving site motif had little effect. In addition, deletion of the C-terminal transmembrane anchor decreased the extracellular ExeM degradation activity by some 20%, indicating that release of ExeM from the inner membrane does not increase its activity. Thus, processing by a rhombosortase or other proteases is not necessarily required for ExeM function under these conditions. However, in a strain producing an ExeM variant lacking the linker region displayed a pronounced decrease in extracellular nucleolytic activity, suggesting that this linker region affects activity or stability of ExeM.
Discussion
In previous studies, we assigned the primary function of ExeM in S. oneidensis MR-1 to biofilm formation (Gödeke et al., 2011a; Heun et al., 2012). However, molecular features that determine functional specificity in S. oneidensis MR-1 biofilm formation are yet to be elucidated. In particular, some characteristics inherent by ExeM that are required for modulation, processing, and degradation of biofilm-specific eDNA as well as ExeM activation and transport remain elusive. The results obtained in this study represent a first basis for elucidating ExeM’s structure–function relationships and its role in biofilm formation of S. oneidensis MR-1. As the nuclease ExeM is conserved in Shewanellaceae, Vibrionaceae, Aeromonadaceae, and Pseudoalteromonadacae, we expect our findings to, similarly, extent to other bacterial species.
Our in vitro degradation assays with purified MBP-ExeM fusion protein strongly indicated that ExeM is a sugar-unspecific endonuclease, as linear and circular DNA as well as RNA substrates were readily degraded. Notably, previous studies on Xds, the nuclease ortholog to ExeM in V. cholerae, implicated that Xds functions as an exonuclease (Seper et al., 2011). The Xds data were generated using culture supernatants containing Xds instead of enriched protein in in vitro assays, which may explain these functional differences. Protein homology analyzes suggested that ExeM belongs to the diverse superfamiliy of two-metal-ion-dependent nucleases which also include DNaseI (Yang, 2011), and putative metal-binding sites were identified within the EEP domain by in silico analysis (Figure 1). Accordingly, we found that ExeM strictly requires Ca2+ as co-factor, and full activity occurred in the presence of Mg2+ but also Mn2+⋅ Ca2+ and Mg2+ are highly common co-factors for nucleases (Yang, 2011), and also Mn2+ has already been identified previously as functional co-factors for nucleases such as for B. subtilis YcgR as well as for another S. oneidensis extracellular endonuclease, EndA (Oussenko et al., 2004; Heun et al., 2012). Shewanella sp. are well-adapted to redox-stratified environments and are capable of releasing soluble Mn2+ ions from highly abundant, but rather insoluble manganese minerals by respiratory electron transfer (Hau and Gralnick, 2007). Thus, increased local Mn2+ concentrations can occur in appropriate environments and were found to occur in Lake Oneida sediments from which S. oneidensis MR-1 was originally isolated (Myers and Nealson, 1988). However, whether this finding holds any physiological relevance with respect to an environmental adaption of Shewanella sp. or rather is an in vitro effect due to similar properties of Mg2+ and Mn2+ cations is so far unclear.
All three major predicted putative domains (LTD, YcgR-like, EEP) are required for full ExeM activity, however, a truncated version lacking the N-terminal LTD domain still displayed residual activity. LTDs are found in eukaryotic nuclear lamins and several uncharacterized proteins from phylogenetically diverse bacteria and some archaea. In bacteria these domains mainly occur with membrane-associated hydrolases of the metallo-β-lactamase, synaptojanin, calcineurin-like phosphoesterase superfamilies, or in secreted or periplasmic proteins associated with oligosaccharide-binding domains or as multiple tandem repeats in a single protein (Mans et al., 2004; Dittmer and Misteli, 2011). The role of LTD domains has not yet been elucidated, but according to the general predicted function of the prokaryotic proteins harboring LTD domains, a potential role in directing proteins to the membrane or membrane-associated structures was suggested (Mans et al., 2004). Our results are not inconsistent with such a role of the LTD domain in ExeM. However, as ExeM-LTD in vitro function in membrane-free environments is decreased, the LTD domain may also be directly involved in the nucleolytic activity. In contrast to the LTD domain, the presence of the YcgR-like region is crucial for ExeM function as the deletion results in an almost complete loss of nucleolytic activity. The YcgR-like region comprises two putative OB-fold nucleic acid–binding regions and may therefore be involved in substrate interaction (Theobald et al., 2003; Oussenko et al., 2004).
Two previous proteomic studies on S. oneidensis MR-1 have identified ExeM to be associated with the cell envelope. However, it remained unclear whether the nuclease was localized to the cytoplasmic (Brown et al., 2010) or the outer membrane (Tang et al., 2007). In our study, ExeM almost exclusively occurred in the cytoplasmic membrane fraction, independently of production levels. As in vivo assays using DNA plates showed that active ExeM was produced upon ectopic exeM expression, we concluded that this localization pattern was unlikely to be exclusively due to potential effects of the overproduction of a secreted protein and also corresponded to the presence of the putative C-terminal membrane anchor. This and the presence of a putative rhombosortase processing site suggested that ExeM may be released from the membrane upon appropriate proteolytic processing. However, we found that the absence of the rhombosortase or substitutions in the predicted cleavage site did not have a major effect on membrane association or ExeM-mediated DNA-degradation on DNA-agar plates. An exception to this occurred in rhombosortase-free mutants expressing an ExeM variant solely lacking the linker region connecting the protein with the predicted C-terminal membrane anchor. In this case, the extracellular nucleolytic activity was diminished to about 40% of wild-type levels. This may be explained by the finding, that in S. oneidensis MR-1, the rhombosortase is predicted to process a number of extracellular proteases which, in the absence of cleavage, may remain and accumulate in the cytoplasmic membrane (Haft and Varghese, 2011). This may affect the stability of linker-lacking ExeM if the protein remains close to the cytoplasmic membrane.
In addition, association of ExeM with the inner membrane still occurred in the absence of the predicted membrane anchor, and, furthermore, although earlier studies suggested that some ExeM-mediated activity may occur in culture supernatants (Gödeke et al., 2011a), this finding could not be verified in the absence of the other two S. oneidensis MR-1 extracellular nucleases, ExeS and, in particular, EndA (Heun et al., 2012) (this study). Accordingly, we did not detect ExeM in highly concentrated medium supernatants even under phosphate-limiting conditions or when overproduced. Similar results were reported from the proteome analysis of S. oneidensis MR-1 culture supernatants grown under phosphate-limiting conditions in a chemostat (Pinchuk et al., 2008). It may therefore be hypothesized that ExeM remains associated with the cytoplasmic membrane and only small amounts of this nuclease are released, which may also be mediated by cell lysis rather than active transport. It may also be speculated that DNA is transported into the periplasm to be degraded by ExeM, as potential orthologs to DNA import systems, e.g., to that of V. cholerae (Matthey and Blokesch, 2016) can readily be identified also in Shewanella sp. Thus, ExeM may have an additional role in restricting the entry of extracellular DNA into the cells.
Like for many other bacterial species, DNA is a major structural compound of S. oneidensis MR-1 biofilms and assemblages (Pinchuk et al., 2008; Gödeke et al., 2011b; Binnenkade et al., 2014). Under hydrodynamic conditions, loss of ExeM results in the formation of biofilms with tightly packed cells, which contain elevated amounts of extracellular DNA and in which biofilm dispersal is highly reduced (Gödeke et al., 2011a; Heun et al., 2012). In contrast, the presence of ExeM is required for normal biofilm formation of S. oneidensis under static conditions, and in its absence, biofilm formation is significantly decreased (Gödeke et al., 2011a). This is not intuitive, as an accumulation of extracellular DNA would rather be expected to lead to more robust surface–attached communities also under these conditions. In this study we found that externally added purified MBP-ExeM negatively affected biofilm formation but was unable to disperse already formed biofilms. We therefore hypothesize that correct timing and amount of ExeM production is crucial for function in biofilms. Since ExeM was predominantly present in the cell envelope and could not be detected in supernatants, we further hypothesize that ExeM may act on extracellular DNA which is in close proximity to or directly interacting with the cell envelope. ExeM may therefore act at the individual cell level to promote or fine–tune proper cell–cell interactions or detachment. It can also not be excluded that ExeM is involved in supporting biofilms cells with phosphate, although in S. oneidensis this role appears to be rather fulfilled by the nuclease EndA (Heun et al., 2012). Clarification of the role ExeM plays in S. oneidensis MR-1 biofilm formation therefore requires further studies. In addition, the ExeM ortholog of V. cholerae, Xds, was demonstrated to be involved in degrading the structurally crucial DNA-scaffold of eukaryotic NETs, thus lowering the susceptibility for NET-mediated extracellular killing (Seper et al., 2013). If such a role of ExeM also applies to Shewanella sp., in particular S. algae and S. putrefaciens which have been identified as commensal pathogens (Janda and Abbott, 2014), remains to be shown.
Author Contributions
LB, MK, and KT conceived experiments, discussed results, and wrote the manuscript. LB and MK conducted experiments.
Funding
The work was supported by grant TH831/3-2 to KT by the Deutsche Forschungsgemeinschaft DFG.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Parts of this work first appeared in LB’s thesis, which can be accessed online (Binnenkade, 2015). We are grateful to Ulrike Ruppert for excellent technical support and to Tim Rick for assisting with the protein purification. We thank Johannes Gescher for kindly providing pBAD-MtrB-Strep.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01761/full#supplementary-material
References
Beenken, K. E., Spencer, H., Griffin, L. M., and Smeltzer, M. S. (2012). Impact of extracellular nuclease production on the biofilm phenotype of Staphylococcus aureus under in vitro and in vivo conditions. Infect. Immun. 80, 1634–1638. doi: 10.1128/IAI.06134-11
Beiter, K., Wartha, F., Albiger, B., Normark, S., Zychlinsky, A., and Henriques-Normark, B. (2006). An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 16, 401–407. doi: 10.1016/j.cub.2006.01.056
Berends, E. T., Horswill, A. R., Haste, N. M., Monestier, M., Nizet, V., and von Kockritz-Blickwede, M. (2010). Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2, 576–586. doi: 10.1159/000319909
Berge, M., Moscoso, M., Prudhomme, M., Martin, B., and Claverys, J. P. (2002). Uptake of transforming DNA in Gram-positive bacteria: a view from Streptococcus pneumoniae. Mol. Microbiol. 45, 411–421. doi: 10.1046/j.1365-2958.2002.03013.x
Binnenkade, L. (2015). Molecular Control of Extracellular DNA Release and Degradation in Shewanella oneidensis MR-1 Biofilms: The Role of Phages and Nucleases. Ph.D. thesis, Phillips Universtät Marburg, Marburg.
Binnenkade, L., Teichmann, L., and Thormann, L. M. (2014). Iron triggers lambdaSo prophage induction and release of extracellular DNA in Shewanella oneidensis MR-1 biofilms. Appl. Environ. Microbiol. 80, 5304–5316. doi: 10.1128/AEM.01480-14
Blokesch, M., and Schoolnik, G. K. (2008). The extracellular nuclease Dns and its role in natural transformation of Vibrio cholerae. J. Bacteriol. 190, 7232–7240. doi: 10.1128/JB.00959-08
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. doi: 10.1126/science.1092385
Brown, R. N., Romine, M. F., Schepmoes, A. A., Smith, R., and Dand Lipton, M. S. (2010). Mapping the subcellular proteome of Shewanella oneidensis MR-1 using sarkosyl-based fractionation and LC-MS/MS protein identification. J. Proteome Res. 9, 4454–4463. doi: 10.1021/pr100215h
Bubendorfer, S., Held, S., Windel, N., Paulick, A., Klingl, A., and Thormann, K. M. (2012). Specificity of motor components in the dual flagellar system of Shewanella putrefaciens CN-32. Mol. Microbiol. 83, 335–350. doi: 10.1111/j.1365-2958.2011.07934.x
Buchanan, J. T., Simpson, A. J., Aziz, R. K., Liu, G. Y., Kristian, S. A., Kotb, M., et al. (2006). DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr. Biol. 16, 396–400. doi: 10.1016/j.cub.2005.12.039
Dell’Anno, A., and Danovaro, R. (2005). Extracellular DNA plays a key role in deep-sea ecosystem functioning. Science 309:2179. doi: 10.1126/science.1117475
Dittmer, T. A., and Misteli, T. (2011). The lamin protein family. Genome Biol. 12:222. doi: 10.1186/gb-2011-12-5-222
Doke, M., Fukamachi, H., Morisaki, H., Arimoto, T., Kataoka, H., and Kuwata, H. (2017). Nucleases from Prevotella intermedia can degrade neutrophil extracellular traps. Mol. Oral Microbiol. 32, 288–300. doi: 10.1111/omi.12171
Dragos, A., and Kovacs, A. T. (2017). The peculiar functions of the bacterial extracellular matrix. Trends Microbiol. 25, 257–266. doi: 10.1016/j.tim.2016.12.010
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199. doi: 10.1093/nar/gkw1107
Flemming, H. C., and Wingender, J. (2010). The biofilm matrix. Nat. Rev. Microbiol. 8, 623–633. doi: 10.1038/nrmicro2415
Focareta, T., and Manning, P. A. (1987). Extracellular proteins of Vibrio cholerae: molecular cloning, nucleotide sequence and characterization of the deoxyribonuclease (DNase) together with its periplasmic localization in Escherichia coli K-12. Gene 53, 31–40. doi: 10.1016/0378-1119(87)90090-4
Fredrickson, J. K., Romine, M. F., Beliaev, A. S., Auchtung, J. M., Driscoll, M. E., Gardner, T. S., et al. (2008). Towards environmental systems biology of Shewanella. Nat. Rev. Microbiol. 6, 592–603. doi: 10.1038/nrmicro1947
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A. III, and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. doi: 10.1038/nmeth.1318
Gödeke, J., Binnenkade, L., and Thormann, K. M. (2012). Transcriptome analysis of early surface-associated growth of Shewanella oneidensis MR-1. PLoS One 7:e42160. doi: 10.1371/journal.pone.0042160
Gödeke, J., Heun, M., Bubendorfer, S., Paul, K., and Thormann, K. M. (2011a). Roles of two Shewanella oneidensis MR-1 extracellular endonucleases. Appl. Environ. Microbiol. 77, 5342–5351. doi: 10.1128/AEM.00643-11
Gödeke, J., Paul, K., Lassak, J., and Thormann, K. M. (2011b). Phage-induced lysis enhances biofilm formation in Shewanella oneidensis MR-1. ISME J. 5, 613–626. doi: 10.1038/ismej.2010.153
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Haft, D., and Varghese, H. N. (2011). GlyGly-CTERM and rhombosortase: a C-terminal protein processing signal in a many-to-one pairing with a rhomboid family intramembrane serine protease. PLoS One 6:e28886. doi: 10.1371/journal.pone.0028886
Halverson, T. W., Wilton, M., Poon, K. K., Petri, B., and Lewenza, S. (2015). DNA is an antimicrobial component of neutrophil extracellular traps. PLoS Pathog. 11:e1004593. doi: 10.1371/journal.ppat.1004593
Hau, H., and Gralnick, J. A. (2007). Ecology and biotechnology of the genus Shewanella. Annu. Rev. Microbiol. 61, 237–258. doi: 10.1146/annurev.micro.61.080706.093257
Hawes, M. C., Curlango-Rivera, G., Wen, F., White, G. J., Vanetten, H. D., and Xiongm, Z. (2011). Extracellular DNA: the tip of root defenses? Plant Sci. 180, 741–745. doi: 10.1016/j.plantsci.2011.02.007
Heun, M., Binnenkade, L., Kreienbaum, M., and Thormann, K. M. (2012). Functional specificity of extracellular nucleases of Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 78, 4400–4411. doi: 10.1128/AEM.07895-11
Ibánez de Aldecoa, A. L., Zafra, O., and González-Pastor, J. E. (2017). Mechanisms and regulation of extracellular DNA release and its biological roles in microbial communities. Front. Microbiol. 8:1390. doi: 10.3389/fmicb.2017.01390
Janda, J., and Abbott, S. L. (2014). The genus Shewanella: from the briny depths below to human pathogen. Crit. Rev. Microbiol. 40, 293–312. doi: 10.3109/1040841X.2012.726209
Kiedrowski, M. R., Crosby, H. A., Hernandez, F. J., Malone, C. L., McNamara, J. O. II, and Horswill, A. R. (2014). Staphylococcus aureus Nuc2 is a functional, surface-attached extracellular nuclease. PLoS One 9:e95574. doi: 10.1371/journal.pone.0095574
Kiedrowski, M. R., Kavanaugh, J. S., Malone, C. L., Mootz, J. M., Voyich, J. M., Smeltzer, M. S., et al. (2011). Nuclease modulates biofilm formation in community-associated methicillin-resistant Staphylococcus aureus. PLoS One 6:e26714. doi: 10.1371/journal.pone.0026714
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lassak, J., Henche, A. L., Binnenkade, L., and Thormann, K. M. (2010). ArcS, the cognate sensor kinase in an atypical Arc system of Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 76, 3263–3274. doi: 10.1128/AEM.00512-10
Letunic, I., and Bork, P. (2007). Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. doi: 10.1093/bioinformatics/btl529
Li, C. L., Hor, L. I., Chang, Z. F., Tsai, L. C., Yang, W. Z., and Yuan, H. S. (2003). DNA binding and cleavage by the periplasmic nuclease Vvn: a novel structure with a known active site. EMBO J. 22, 4014–4025. doi: 10.1093/emboj/cdg377
Liu, J., Sun, L., Liu, W., Guo, L., Liu, Z., Wei, X., et al. (2017). A nuclease from Streptococcus mutans facilitates biofilm dispersal and escape from killing by neutrophil extracellular traps. Front. Cell. Infect. Microbiol. 7:97. doi: 10.3389/fcimb.2017.00097
Mann, E. E., Rice, K. C., Boles, B. R., Endres, J. L., Ranjit, D., Chandramohan, L., et al. (2009). Modulation of eDNA release and degradation affects Staphylococcus aureus biofilm maturation. PLoS One 4:e5822. doi: 10.1371/journal.pone.0005822
Mans, B. J., Anantharaman, V., Aravind, L., and Koonin, E. V. (2004). Comparative genomics, evolution and origins of the nuclear envelope and nuclear pore complex. Cell Cycle 3, 1612–1637. doi: 10.4161/cc.3.12.1316
Matthey, N., and Blokesch, M. (2016). The DNA-uptake process of naturally competent Vibrio cholerae. Trends Microbiol. 24, 98–110. doi: 10.1016/j.tim.2015.10.008
Minh Tran, T., MacIntyre, A., Khokhani, D., Hawes, M., and Allen, C. (2016). Extracellular DNases of Ralstonia solanacearum modulate biofilms and facilitate bacterial wilt virulence. Environ. Microbiol. 18, 4103–4117. doi: 10.1111/1462-2920.13446
Molin, S., and Tolker-Nielsen, T. (2003). Gene transfer occurs with enhanced efficiency in biofilms and induces enhanced stabilisation of the biofilm structure. Curr. Opin. Biotechnol. 14, 255–261. doi: 10.1016/S0958-1669(03)00036-3
Mulcahy, H., Charron-Mazenod, L., and Lewenza, S. (2008). Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 4:e1000213. doi: 10.1371/journal.ppat.1000213
Mulcahy, H., Charron-Mazenod, L., and Lewenza, S. (2010). Pseudomonas aeruginosa produces an extracellular deoxyribonuclease that is required for utilization of DNA as a nutrient source. Environ. Microbiol. 12, 1621–1629. doi: 10.1111/j.1462-2920.2010.02208.x
Myers, C. R., and Nealson, K. H. (1988). Bacterial manganese reduction and growth with manganese oxide as the sole electron acceptor. Science 240, 1319–1321. doi: 10.1126/science.240.4857.1319
Niemeyer, J., and Gessler, F. (2002). Determination of free DNA in soils. J. Plant Nutr. Soil Sci. 165, 121–124. doi: 10.1002/1522-2624(200204)165:2<121::AID-JPLN1111121>3.0.CO;2-X
Oussenko, I. A., Sanchez, R., and Bechhofer, D. H. (2004). Bacillus subtilis YhcR, a high-molecular-weight, nonspecific endonuclease with a unique domain structure. J. Bacteriol. 186, 5376–5383. doi: 10.1128/JB.186.16.5376-5383.2004
Palmgren, R., and Nielsen, P. H. (1996). Accumulation of DNA in the exopolymeric matrix of activated sludge and bacterial cultures. Water Sci. Technol. 34, 233–240. doi: 10.2166/wst.1996.0555
Paulick, A., Koerdt, A., Lassak, J., Huntley, S., Wilms, I., Narberhaus, F., et al. (2009). Two different stator systems drive a single polar flagellum in Shewanella oneidensis MR-1. Mol. Microbiol. 71, 836–850. doi: 10.1111/j.1365-2958.2008.06570.x
Pinchuk, G. E., Ammons, C., Culley, D. E., Li, S. M., McLean, J. S., Romine, M. F., et al. (2008). Utilization of DNA as a sole source of phosphorus, carbon, and energy by Shewanella spp.: ecological and physiological implications for dissimilatory metal reduction. Appl. Environ. Microbiol. 74, 1198–1208. doi: 10.1128/AEM.02026-07
Ross, D. E., Ruebush, S. S., Brantley, S. L., Hartshorne, R. S., Clarke, T. A., Richardson, D. J., et al. (2007). Characterization of protein-protein interactions involved in iron reduction by Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 73, 5797–5808. doi: 10.1128/AEM.00146-07
Sambrook, K., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Press.
Seper, A., Fengler, V. H., Roier, S., Wolinski, H., Kohlwein, S. D., Bishop, A. L., et al. (2011). Extracellular nucleases and extracellular DNA play important roles in Vibrio cholerae biofilm formation. Mol. Microbiol. 82, 1015–1037. doi: 10.1111/j.1365-2958.2011.07867.x
Seper, A., Hosseinzadeh, A., Gorkiewicz, G., Lichtenegger, S., Roier, S., Leitner, D. R., et al. (2013). Vibrio cholerae evades neutrophil extracellular traps by the activity of two extracellular nucleases. PLoS Pathog. 9:e1003614. doi: 10.1371/journal.ppat.1003614
Steichen, C. T., Cho, C., Shao, J. Q., and Apicella, M. A. (2011). The Neisseria gonorrhoeae biofilm matrix contains DNA and an endogenous nuclease controls its incorporation. Infect. Immun. 79, 1504–1511. doi: 10.1128/IAI.01162-10
Tang, X., Yi, W., Munske, G. R., Adhikari, D. P., Zakharova, N. L., and Bruce, J. E. (2007). Profiling the membrane proteome of Shewanella oneidensis MR-1 with new affinity labeling probes. J. Proteome Res. 6, 724–734. doi: 10.1021/pr060480e
Theobald, D. L., Mitton-Fry, R. M., and Wuttke, D. S. (2003). Nucleic acid recognition by OB-fold proteins. Annu. Rev. Biophys. Biomol. Struct. 32, 115–133. doi: 10.1146/annurev.biophys.32.110601.142506
Thormann, K. M., Saville, R. M., Shukla, S., Pelletier, D. A., and Spormann, A. M. (2004). Initial phases of biofilm formation in Shewanella oneidensis MR-1. J. Bacteriol. 186, 8096–8104. doi: 10.1128/JB.186.23.8096-8104.2004
Tran, T. M., MacIntyre, A., Hawesm, M., and Allen, C. (2016). Escaping underground nets: extracellular DNases degrade plant extracellular traps and contribute to virulence of the plant pathogenic bacterium Ralstonia solanacearum. PLoS Pathog. 12:e1005686. doi: 10.1371/journal.ppat.1005686
van de Vossenberg, J. L., Albers, S. V., van der Does, C., Driessen, A. J., and van Klompenburg, W. (1998). The positive inside rule is not determined by the polarity of the delta psi (transmembrane electrical potential). Mol. Microbiol. 29, 1125–1127. doi: 10.1046/j.1365-2958.1998.01001.x
Vlassov, V. V., Laktionov, P. P., and Rykova, E. Y. (2007). Extracellular nucleic acids. Bioessays 29, 654–667. doi: 10.1002/bies.20604
von Heijne, G. (1989). Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature 341, 456–458. doi: 10.1038/341456a0
von Heijne, G. (1992). Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J. Mol. Biol. 225, 487–494. doi: 10.1016/0022-2836(92)90934-C
Wu, S. I., Lo, S. K., Shao, C. P., Tsai, H. W., and Hor, L. I. (2001). Cloning and characterization of a periplasmic nuclease of Vibrio vulnificus and its role in preventing uptake of foreign DNA. Appl. Environ. Microbiol. 67, 82–88. doi: 10.1128/AEM.67.1.82-88.2001
Keywords: DNA, biofilm, membrane, endonuclease, Shewanella
Citation: Binnenkade L, Kreienbaum M and Thormann KM (2018) Characterization of ExeM, an Extracellular Nuclease of Shewanella oneidensis MR-1. Front. Microbiol. 9:1761. doi: 10.3389/fmicb.2018.01761
Received: 28 March 2018; Accepted: 13 July 2018;
Published: 03 August 2018.
Edited by:
Satoshi Tsuneda, Waseda University, JapanReviewed by:
Yosuke Tashiro, Shizuoka University, JapanJohannes Gescher, Karlsruher Institut für Technologie (KIT), Germany
Nick Stephen Jakubovics, Newcastle University, United Kingdom
Copyright © 2018 Binnenkade, Kreienbaum and Thormann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai M. Thormann, a2FpLnRob3JtYW5uQG1pa3JvLmJpby51bmktZ2llc3Nlbi5kZQ==
†These authors have contributed equally to this work.