 Rachel Mackelprang1†
Rachel Mackelprang1† Alyssa M. Grube2†
Alyssa M. Grube2† Regina Lamendella2
Regina Lamendella2 Ederson da C. Jesus3,4
Ederson da C. Jesus3,4 Alex Copeland5
Alex Copeland5 Chao Liang4,6Randall D. Jackson4,7
Chao Liang4,6Randall D. Jackson4,7 Charles W. Rice8Stefanie Kapucija1Bayan Parsa1
Charles W. Rice8Stefanie Kapucija1Bayan Parsa1 Susannah G. Tringe5*
Susannah G. Tringe5* James M. Tiedje3,4*
James M. Tiedje3,4* Janet K. Jansson9*
Janet K. Jansson9*- 1Department of Biology, California State University, Northridge, Northridge, CA, United States
- 2Department of Biology, Juniata College, Huntingdon, PA, United States
- 3Center for Microbial Ecology, Michigan State University, East Lansing, MI, United States
- 4Great Lakes Bioenergy Research Center, U.S. Department of Energy, University of Wisconsin–Madison, Madison, WI, United States
- 5U.S. Department of Energy Joint Genome Institute, Walnut Creek, CA, United States
- 6Institute of Applied Ecology, Chinese Academy of Sciences, Shenyang, China
- 7Department of Agronomy, University of Wisconsin–Madison, Madison, WI, United States
- 8Department of Agronomy, Kansas State University, Manhattan, KS, United States
- 9Earth and Biological Sciences Directorate, Pacific Northwest National Laboratory, Richland, WA, United States
The North American prairie covered about 3.6 million-km2 of the continent prior to European contact. Only 1–2% of the original prairie remains, but the soils that developed under these prairies are some of the most productive and fertile in the world, containing over 35% of the soil carbon in the continental United States. Cultivation may alter microbial diversity and composition, influencing the metabolism of carbon, nitrogen, and other elements. Here, we explored the structure and functional potential of the soil microbiome in paired cultivated-corn (at the time of sampling) and never-cultivated native prairie soils across a three-states transect (Wisconsin, Iowa, and Kansas) using metagenomic and 16S rRNA gene sequencing and lipid analysis. At the Wisconsin site, we also sampled adjacent restored prairie and switchgrass plots. We found that agricultural practices drove differences in community composition and diversity across the transect. Microbial biomass in prairie samples was twice that of cultivated soils, but alpha diversity was higher with cultivation. Metagenome analyses revealed denitrification and starch degradation genes were abundant across all soils, as were core genes involved in response to osmotic stress, resource transport, and environmental sensing. Together, these data indicate that cultivation shifted the microbiome in consistent ways across different regions of the prairie, but also suggest that many functions are resilient to changes caused by land management practices – perhaps reflecting adaptations to conditions common to tallgrass prairie soils in the region (e.g., soil type, parent material, development under grasses, temperature and rainfall patterns, and annual freeze-thaw cycles). These findings are important for understanding the long-term consequences of land management practices to prairie soil microbial communities and their genetic potential to carry out key functions.
Introduction
The original North American prairie was a 3.6 million-km2 expanse of fertile soil (Mollisols). This region is highly productive agriculturally and the majority of the original prairie has been cultivated (Samson and Knopf, 1994). Besides replacing a species-rich plant community with monoculture, land management induces changes in soil physicochemical characteristics. Of particular importance to global biogeochemical cycles is the impact of human activities on nitrogen and carbon storage. In 2016, agriculture was the source of 8.6% of total greenhouse gas emissions in the United States (USEPA, 2018). Thirty one to 39% of the total soil organic carbon (SOC) stocks of the conterminous United States are stored in prairie soils (Guo et al., 2006). Tillage, fertilization, intensive cropping, and erosion causes SOC losses of 20–60% (Stauffer et al., 1940; Mann, 1986; Brye et al., 2001, 2002; Kucharik et al., 2001; Guo and Gifford, 2002; Sanford G.R. et al., 2012; Sanderman et al., 2017). Fertilizer application and other agricultural management practices induce N2O production, making croplands responsible for 76.7% of United States N2O emissions into the atmosphere (USEPA, 2018).
Microbial communities drive carbon and nitrogen cycles in soils. Thus, understanding how agricultural practices impact microbial communities is critical for predicting future greenhouse gas emissions. However, our understanding of microbial diversity within prairie ecosystems and how prairie soil microbiomes contribute to cycling of carbon, nitrogen, and other nutrients is still developing. Disturbances related to agriculture, such as tilling, fertilization, irrigation, and burning change soil properties and alter microbial community structure and functional capacity (Vitousek et al., 1997; Bending et al., 2004; Mao et al., 2011; Orr et al., 2011; Habig and Swanepoel, 2015; Jesus et al., 2015; Oates et al., 2016; Zhang et al., 2017). Fertilization adds mineral N to soils, which is processed by microbial communities through nitrification and denitrification pathways, producing N2O (Firestone and Davidson, 1989). N amendment shifts microbial community composition and functional capacity through changes to taxonomic richness (Coolon et al., 2013), activity (Marx et al., 2001; Ramirez et al., 2012), biomass (Ramirez et al., 2012), increases in active copiotrophic taxa (Fierer et al., 2012), and community composition (Ramirez et al., 2012; Leff et al., 2015). Tillage changes soil physicochemical properties (Phillips et al., 1980; Ismail et al., 1994) and concomitantly alters microbial community structure. It increases the abundance of aerobes, facultative anaerobes, and denitrifiers in near-surface soils (Doran, 1980) and causes changes in biomass (Guo et al., 2006), diversity, and activity (Habig and Swanepoel, 2015; Mbuthia et al., 2015; Nivelle et al., 2016). Monoculture cropping, some pesticide applications, and organic management practices also alter soil microbial community structure and diversity (Bending et al., 2000, 2004; Figuerola et al., 2014; Duncan et al., 2016; Liang et al., 2016; Pose-Juan et al., 2017; Zhang et al., 2017; Zhao et al., 2018).
Broad-scale comparisons between geographic locations, crop types, native prairie, and restored prairie ecosystems capture differences in community structure and function driven by the aggregate effects of cropping. Soil depth, crop systems (crop species, monoculture, and annual versus perennial), and soil variables all contribute to community assemblages and biomass (Acosta-Martinez et al., 2008; Habig and Swanepoel, 2015; Liang et al., 2016; Oates et al., 2016; Zhang et al., 2017). Community composition and function in grassland soils have also been found to vary across the world, likely due to differences in soil pH, climate, and plant communities (Fierer et al., 2013; Leff et al., 2015). Jesus et al. (2015) found that soil type and geographic distance drove community structure in recently established plots but that plant species became a dominant driver over long-term cultivation.
In the interest of restoring native habitat, mitigating biodiversity loss, preserving soil integrity, and investigating sustainable agriculture, grassland restoration is becoming more common in prairie ecosystems (Jangid et al., 2009; Barber et al., 2017). The magnitude and timing of the restoration of microbial community structure remains unclear. Some studies suggest that community response is rapid, occurring in less than a decade after restoration (Herzberger et al., 2014; Duncan et al., 2016; Barber et al., 2017). Other studies suggest that microbial community restoration is a long-term process occurring on the order of decades (Jangid et al., 2009).
Our understanding of microbial diversity within prairie ecosystems and how prairie soil microbial communities contribute to cycling of nutrients is still developing. Here we aimed to gain a better understanding of the effects of land management, specifically long-term cultivation, on soil microbial communities and their potential to carry out key soil processes in the region of the United States that had previously been dominated by prairie. We performed 16S rRNA gene sequencing, lipid analysis, and deep shotgun metagenomic sequencing in cultivated and native prairie soils across a three state transect. We asked how geographic location and cultivation practices influenced microbial community composition and about the capacity of soil microbes to cycle carbon, nitrogen, and other nutrients. An important part of our design was to compare long-term (>50 years) cultivated and never-cultivated sites that were otherwise matched (paired) with respect to soil and landform characteristics. Results from this study serve as a baseline for understanding the impacts of land management on soil communities, and consequently facilitate functional predictions of the impacts of cultivation on carbon and nutrient cycling processes.
Materials and Methods
Sampling Sites
Three native tallgrass prairie sites representative of the U.S. Midwest prairie ecosystem were studied: Manhattan, Kansas (KS); Morris Prairie, Iowa (IA); and Goose Pond Prairie, Wisconsin (WI). These sites constitute a southwest to northeast transect across what was originally tallgrass prairie, but is now mostly converted to highly productive annual crop agriculture. At each location, a nearby long-term agricultural site was selected that matched the never-tilled (remnant) prairie site in soil type, texture, slope, aspect, and drainage. A switchgrass and restored prairie plot adjacent to the corn plot were also included at the Wisconsin site. In the case of non-switchgrass cultivated sites, samples were taken when sites were planted to corn (a complete description of the sites and their history is in Supplementary Methods). All sites were sampled in 2009 during active plant growth and optimum soil moisture conditions: Wisconsin-June 24, Iowa-June 26, and Kansas-August 7. Seven samples were taken at each of the sites with a 1-cm diameter soil corer to a depth of 12 cm. A reference sample (defined as 0 m) and six additional cores were sampled in two directions from the reference (90 degree angle) at 1 cm, 1 m, and 10 m (Supplementary Figure S1). The corn sites were sampled between the rows. At this plant stage few roots were sampled. The litter layer was removed and the soil core extruded into a plastic bag. The eighth sample was a larger volume (500 g) sample taken adjacent to the reference core (the apex) designed for soil chemical and physical analyses. All samples were immediately placed on ice, stored locally under refrigeration, and shipped overnight on blue ice and kept cold until DNA was extracted or frozen until the soil chemistry analyzed. A subsample of approximately 3.2 or 6.4 g (if sufficient DNA was not obtained from the 3.2 g sample) from the reference core (0 m) was used for metagenomic sequencing. Subsamples from all eight cores were used for 16S rRNA gene sequencing and for lipid analysis.
Soil Characterization
All soil chemical and physical attributes were analyzed at the Michigan State University Soil and Plant Nutrient Laboratory except for the boron, sulfur, and aluminum analyses, which were done by A&L Great Lakes Laboratories using the Mehlich 3 method. The chemical analyses were those validated for reflecting bioavailable elements in soils of the North Central region of the United States (46) plus the chemical specific methods of Bradstreet (1965), Huffman and Barbarick (2008), and the United States Environmental Protection Agency (USEPA, 1993). Physical (texture) analysis was by the hydrometer method of Bouyoucos (1951) (see Supplementary Table S1).
DNA Extraction and 16S rRNA Gene Sequencing
DNA was extracted from 250 mg soil portions using the PowerSoil® DNA isolation kit (Mo Bio Laboratories, Carlsbad, CA, United States) according to the manufacturer’s protocol. Multiple extractions were performed for each homogenized soil sample to obtain approximately 10 μg/sample. The V6–V8 region of the small subunit (SSU) rRNA gene was amplified using the primer pair 926f/1392r as described in Kunin et al. (2010). The reverse primer included a 5-bp barcode for multiplexing of samples during sequencing. Sequencing of PCR amplicons was performed at the Joint Genome Institute (JGI) using Roche 454 GS FLX Titanium technology following manufacturer’s instructions with the exception that the final dilution was 1e-8 (Allgaier et al., 2010). Of the 64 total samples, one of the Kansas native prairie samples did not sequence properly, yielding 63 samples for bioinformatics and statistical analysis.
Lipid Analysis
Each core sample was homogenized and a 6-g portion was frozen at -20°C prior to lipid extraction. Membrane lipids were extracted from 3-g lyophilized and milled material in a two-phase aqueous-organic extraction (Bligh and Dyer, 1959). FAME analysis was conducted as described by Microbial ID (Kunitsky et al., 2006). Lipid methyl esters were determined using a Hewlett-Packard 6890 Gas Chromatograph configured and maintained for lipid analysis according to the recommendations of MIDI (Kunitsky et al., 2006). Gas chromatogram parameters were specified and peaks were identified by the MIDI EUYKARY method (MIDI, Newark, DE, United States). Fatty acid concentration was quantified by comparisons of peak areas of the samples compared to two internal standards, 9:0 (nonanoic methyl ester) and 19:0 (nonadecanoic methyl ester) (Sigma, St. Louis, MO, United States), of known concentration. In all subsequent analyses, we excluded fatty acids that were at an average abundance of <0.5 mol% or present in <3 samples.
Total abundance of lipids was used as an index of total microbial biomass. The abundance of indicator lipids for Gram-negative and Gram-positive bacteria, Actinobacteria, and saprophytic and arbuscular mycorrhizal fungi were further analyzed to indicate community response to treatment variables (Vestal and White, 1989; Balser and Firestone, 2005). Lipid data (mol%) were arcsine-transformed for normality and multivariate principal component analysis was carried out using JMP software, version 5.0.
Bioinformatic and Statistical Analyses of 16S rRNA Gene Sequences
16S rRNA gene sequencing resulted in 646,884 16S rRNA gene reads, which were processed in QIIME 1.8.0 (Caporaso et al., 2010). Sequences were denoised, quality filtered, and chimera checked. Clustering was done at a 97% similarity threshold. Operational taxonomic units (OTUs) were assigned to sequences based on 97% identity using the open-reference USEARCH algorithm (Edgar, 2010). Finally, clusters were assigned OTU identifiers, resulting in 8,291 OTUs and a median of 9,776 sequences per sample. The recommended sampling depth of 3,266 sequences/sample for downstream analysis eliminated only one sample (Wisconsin restored prairie, 1 m south from core). OTUs were assigned taxonomy against the May 2013 release of GreenGenes (Lawrence Berkeley National Laboratory, Berkeley, CA, United States) using the RDP classifier method in QIIME.
Alpha Diversity
Alpha and beta diversity metrics were calculated in QIIME. Multiple rarefactions were performed on the OTU table with a minimum and maximum number of sequences of 200 and 3,200, respectively, and a step-size of 500 and 100 iterations, producing 600 rarified tables. Alpha diversity was calculated on the rarified tables using the phylogenetic whole tree method. Rarefaction plots were generated from collated alpha diversity files and each metadata category was plotted. Finally, a student’s t-test was used as implemented in the QIIME script compare_alpha_diversity.py to perform pairwise comparisons between the alpha diversity values of samples of a given metadata category.
To calculate alpha diversity by state when considering only corn and native prairie samples, an OTU table was filtered to exclude the restored prairie and switchgrass samples. Multiple rarefactions were performed on the resulting OTU table with a minimum and maximum number of sequences of 500 and 4,500, respectively, and a step-size of 500 and 50 iterations, producing 400 rarified tables. Alpha diversity was subsequently calculated on the rarified tables using the phylogenetic whole tree method as described above. A student’s t-test was used as described above to calculate pairwise comparisons of alpha diversity by state.
Beta Diversity
Beta diversity was estimated by calculating unweighted UniFrac distances and visualized using principal coordinate analysis. The PERMANOVA test embedded within the QIIME software suite was used to determine the degree to which categorical metadata parameters explained patterns in the UniFrac distance matrix (permutations = 999).
Because our data are not normally distributed, we used the non-parametric Kruskal–Wallis and Mann–Whitney tests implemented in QIIME on the single rarified table to determine which taxa were significantly different in abundance than expected if the OTUs were randomly distributed in the samples. The Mann–Whitney test was used to compare which families differentiate corn and native prairie samples. Taxa with Bonferroni-corrected P-value less than or equal to 0.05 were chosen for visualization. The Kruskal–Wallis test was used to determine which families were significantly different among all four management practices (corn, switchgrass, native prairie, and restored prairie). Similarly, taxa with Bonferroni-corrected P-values less than or equal to 0.05 were selected for visualization.
Correlation of Phylogenetic Profiles and Chemical Metadata
Spearman rank coefficients comparing the relationships between taxa and soil chemical metadata were calculated in R statistical software [R version 3.0.2, Comprehensive R Archive Network (CRAN)]. Briefly, a matrix containing chemical metadata was merged with a matrix containing relative taxonomic abundance, such that the Spearman rank coefficient was calculated between sample-matched chemical metadata and OTU abundance data summarized at the order level. To investigate relationships between bacterial taxa and lipid profiles and to compare the two methods, Spearman rank coefficients were similarly calculated between sample-matched lipid profiles and the relative abundance of OTUs summarized at the order level. Resulting correlation matrices were filtered such that only columns and rows containing at least one correlation ≥ an absolute value of 0.60 were retained. Heatmaps were produced in R statistical software using heatmap.2 (R version 3.0.2, CRAN).
Metagenomic Sequencing, Assembly, and Annotation
We performed shotgun metagenome sequencing using the reference core (0 m) DNA from the native prairie and continuously cultivated sites in each state. Libraries with ∼270 bp inserts were generated with the Illumina TruSeq protocol. Sequencing for each sample was conducted over a period of more than a year and spanned several platform improvements. As a result, for each sample sequence data was a mixture of Illumina GA2 (2 × 76 bp), GAIIx (2 × 100 bp, 2 × 114 bp, and 2 × 150 bp), or HiSeq 2000 (2 × 100 bp). Sequence data were deposited into the NCBI Short Read Archive (Supplementary Table S2).
Due to the large number of reads in these datasets, a Convey (Richardson, TX, United States) HC-1 hybrid core computer was used for preprocessing and roadmap construction. Graph phases of assembly used the Convey implementation (cnygc version 2.0.3208) of Velvet and were run either on the HC-1, a Sun Fire X4600 M2 with 1TB of RAM, a Dell R910 with 1TB of RAM or an IBM 3850 with 1 TB of RAM. Read pre-processing included trimming reads of Illumina quality ‘B’ using the cnygc–trimB operation. Velvet version 1.2.03 was used for contig construction (Zerbino and Birney, 2008). Details for each assembly are included in the Supplementary Methods. Sequencing and assembly statistics are summarized in Supplementary Table S3. Assembled contigs were submitted to Integrated Microbial Genomes (IMG) metagenome annotation pipeline for gene calling and annotation (Markowitz et al., 2014; Huntemann et al., 2015). Predicted protein sequences from IMG were compared to the FOAM database (Prestat et al., 2014) using hmmsearch at default settings (Eddy, 2011). For every sample, the relative abundance of each gene was calculated by dividing the number of hits to that gene by the total number of hits to the FOAM database.
Core Functional Analysis
We defined core genes as those having similar abundances across communities (Shade and Handelsman, 2012). Rare gene families (those observed less than 100 times across all samples) were removed from analysis. Rank abundance curves were generated for each sample and the variance in rank abundance across samples was calculated for each gene. Genes varying the least in rank abundance were considered to represent core genes. To identify functional categories enriched in core genes, we counted the number of core genes (defined as the top 10% of genes varying the least in rank abundance) in each category of the second functional sublevel of the FOAM hierarchy. Permutation tests were performed by randomly assigning different outcome variances to each gene from the observed set of variances 10,000 times to obtain 95% confidence intervals for the number of core genes expected in each category. P-values were corrected for multiple testing using the false discovery rate (Benjamini and Hochberg, 1995).
Phylogenetic and Taxonomic Analysis of Nitrous Oxide Reductase (nosZ)
Phylogenetic diversity of denitrifiers was investigated using the 795 nosZ sequences identified in contigs by IMG/M. To perform multiple sequence alignments, we built a custom HMM profile using nosZ amino acid sequences downloaded from the functional gene pipeline & repository (FunGene) database (Fish et al., 2013) that exceeded 1100 amino acids in length and had a minimum score of 630. The HMM profile was built from the FunGene seed alignment using hmmbuild from the HMMER3 package at default settings (Eddy, 2011). NosZ sequences from our dataset were aligned using hmmalign at default settings. Confidence scores were assigned to each alignment position using Zorro (Wu and Scott, 2012). Residues scoring less than 0.01 were removed from the alignment for tree building purposes. Because genes predicted from metagenome assemblies are often only partial sequences, many do not overlap. Therefore, we selected only sequences greater than 20 amino acids in length that overlapped the region with the highest alignment certainty as determined by Zorro scores. NosZ sequences curated and classified by Sanford R.A. et al. (2012) were added to the alignment using the muscle profile alignment algorithm (Edgar, 2010). A phylogenetic tree was inferred using FastTree (Price et al., 2009) at default settings. The tree was rooted using Haloarcula marismortui and Halorubrum lacusprofundi (Sanford R.A. et al., 2012). We assigned taxonomy to individual nosZ sequences by performing a BLASTP (Altschul et al., 1990) search against the National Center for Biotechnology Information non-redundant (NCBI-NR) database using an E-value cutoff of 1 × 10-5. The resulting file was imported into MEGAN, which performed taxonomic classification (Huson et al., 2016).
Carbohydrate Active Enzymes
Glycoside hydrolase (GH) genes were identified in the raw metagenomic sequence data by comparison to the Carbohydrate Active Enzyme (CAZy) database (Lombard et al., 2014) using the UBLAST algorithm within the USEARCH program1 with an acceleration value 0.2 and an E-value cutoff of 1 × 10-5. GH family assignments were made based on the top hit. We verified the assignments by comparing putative GH sequences to the NCBI-NR database using UBLAST with the same parameters described above.
Results
16S rRNA Gene Sequencing and Diversity Analysis
We sampled native tallgrass prairie (NP) sites from three states (Kansas, Iowa, and Wisconsin) representative of the Midwestern United States (U.S.) prairie ecosystem. At each location, a nearby site was selected that experienced long-term cultivation and was planted to corn (CC) when sampled. At the Wisconsin site, adjacent switchgrass monoculture and restored prairie plots were available and were also sampled. Sequencing of the 16S rRNA gene yielded 645,542 high quality sequences and identified 8291 OTUs.
Computation of alpha diversity metrics revealed significant differences in richness and phylogenetic diversity between cultivated and native prairie sites (Supplementary Figure S2 and Supplementary Table S4). Alpha diversity was significantly higher overall in cultivated soils compared to native prairie soils (P = 0.006) and in switchgrass compared to native prairie soils (P < 0.04), although within-state alpha diversity metrics were not significantly different between management practices (e.g., Iowa CC versus Iowa NP). Alpha diversity was not different between the 250 mg sample taken from the soil core (10 g) and the large scale (500 g) samples, indicating ability to resolve complexity was already saturated with small sample sizes. Alpha diversity differed significantly among states, with Kansas having the highest alpha diversity and Iowa the lowest. All state pairwise comparisons were significant (P < 0.05).
Beta diversity analysis showed clustering of soil samples by management practice across the data set (Figure 1A) and within states (Figures 1B–D). Bacterial communities from cultivated soils clustered together but were separate from native prairie communities (Figure 1A). When comparing samples by state, native prairie, and corn samples formed discrete clusters (Figures 1B–D). At the Wisconsin site, switchgrass, and restored prairie locations were also sampled and the microbial communities in all of the plots that had been cultivated exhibited considerable overlap (Figures 1A,D). State, site, and land management were each found to be significant factors that explained UniFrac distances (P < 0.05). Site was the strongest factor in determining community differences (Pseudo-F 3.814, P = 0.001), closely followed by management practice (Pseudo-F 3.811, P = 0.001). Distance within sites (0, 1 cm, 1 m, and 10 m) was not significant.
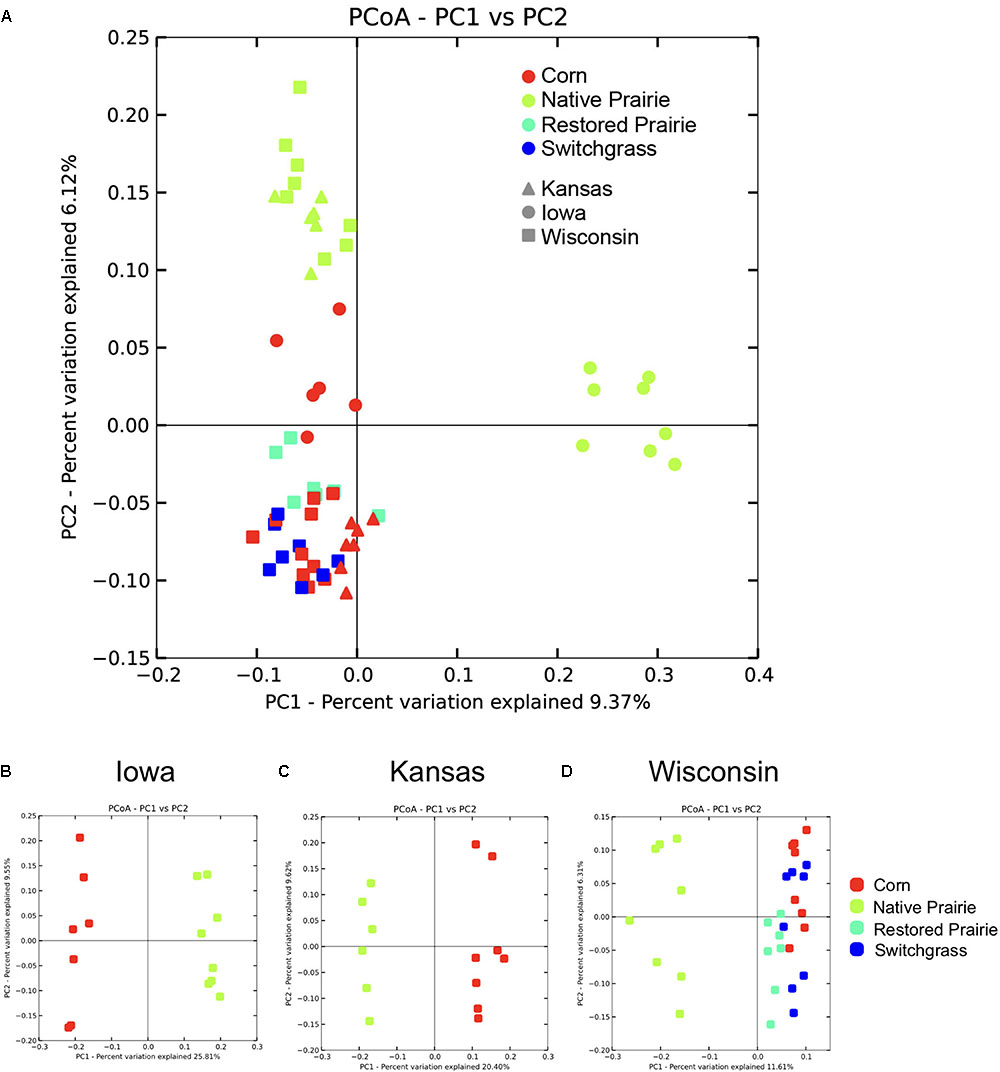
FIGURE 1. Visualization of beta-diversity reveals clustering by management type. Unweighted Unifrac distances were plotted using Principal Coordinate Analysis (PCoA) in QIIME. Each point represents a discrete sample. (A) PCoA plots of all sites and samples. (B–D) PCoA plots by state.
Potential Bioindicators of Land-Management Practice and Soil Chemistry
Twenty-six microbial families differed significantly in abundance between corn and native prairie samples (P < 0.05, Figure 2 and Supplementary Figure S3). Taxa that were more abundant in the cultivated corn soils included the families Nitrosomonadaceae, Nitrospiraceae, three unknown families of class Gemmatimonadetes, and two unknown families of class Anaerolineae. Only seven families were significantly more abundant in native prairie samples, including Rhizobiaceae, Phyllobacteriaceae, Bradyrhizobiaceae, Mycobacteriaceae, and Ktedonobacteraceae. Many of these families have members that carry out key nitrogen cycle processes. The shift from nitrogen fixers in prairie soils to those capable of nitrification in cultivated soils is presumably a response to nitrogen fertilizer application.
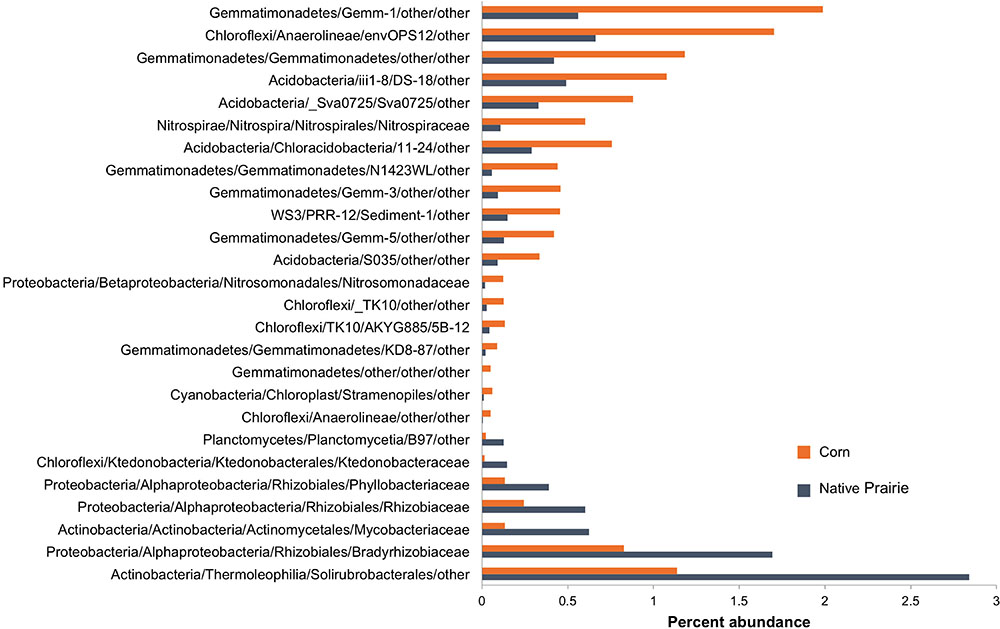
FIGURE 2. Abundance of bacterial families that differentiate corn and native prairie samples. Key OTUs at the family level that are significantly different between corn and native prairie samples, regardless of state (Kansas, Wisconsin, and Iowa). The non-parametric Mann–Whitney t-test (number of permutations = 999) was used to compare relative abundance of families in corn and native prairie samples from a single-rarified OTU table at 3,266 sequences/sample. Families with Bonferroni corrected P-values <0.05 were chosen for visualization.
Spearman correlations between bacterial orders and soil chemical metadata revealed significant trends, specifically in relation to nitrogen (Supplementary Figure S4). Rhizobiales were strongly correlated to ammonium (NH4+) (ρ = 0.61, P < 0.001), while the putative orders Gemm.5, Gemmatimonadetes N1423WL, and Acidobacteria Sva0725 were negatively correlated to ammonium (ρ = -0.67, -0.61, and -0.61, respectively, P < 0.001).
Lipid Profiles
Lipid analysis indicated higher microbial biomass in the native prairie soils compared to the cultivated corn soils, mainly due to lower fungal abundances associated with cultivated corn soils (Figure 3 and Supplementary Tables S5, S6). Arbuscular mycorrhizal fungi, saprotrophic fungi, protozoa, and Actinobacteria-associated lipids were also higher in abundance in native prairie soils than in cultivated corn soils. Both Gram-negative and Gram-positive associated lipids were more abundant in native prairie soils, although the Gram-negative to Gram-positive ratios were not significantly different (Supplementary Figure S5). Notably, 37 of the 50 measured lipids were significantly more abundant in native prairie soils, while only four lipids were slightly, but not significantly, greater in the cultivated corn: 16:1 Cis Alcohol w7, 16:1 ISO G, 16:1 w7c, 18:0 2OH (Supplementary Table S6).
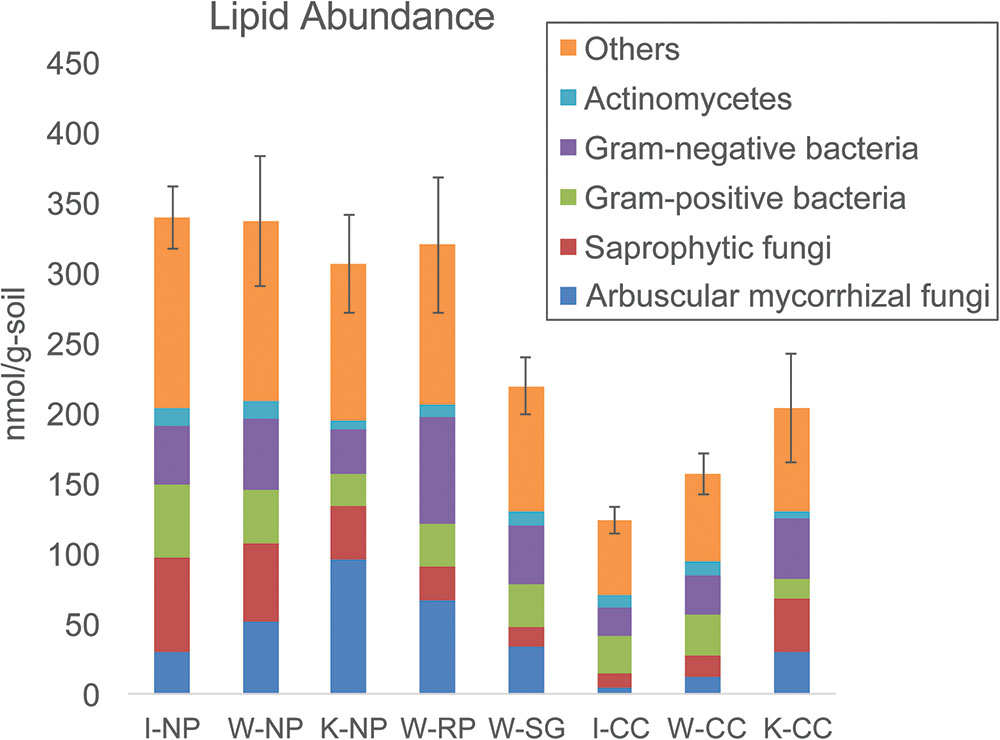
FIGURE 3. Total lipid abundance (microbial biomass nmol/g) and distribution patterns (%) of the main microbial groups. “Others” indicates microbial groups where taxonomic origin cannot be determined. Samples are (from left to right): Wisconsin native prairie, Kansas native prairie, Wisconsin restored prairie, Wisconsin switchgrass, Iowa cultivated corn, Wisconsin cultivated corn, and Kansas cultivated corn. Bar graphs show total lipid abundance and the relative abundance of each group. Error bars are the standard error of the total lipid abundance.
There was a significantly higher ratio of fungal to bacterial lipids in Kansas native prairie compared to the other sampling locations (Supplementary Figure S5). The ratios of lipids corresponding to arbuscular mycorrhizal fungi compared to saprotrophic fungi were consistently lower in cultivated corn, and significantly higher in Kansas native prairie, Wisconsin switchgrass, and Wisconsin restored prairie (Supplementary Figure S5). This suggests that the grasses in these specific fields may be more effectively colonized with symbiotic fungi.
Spearman correlations of lipid profiles to relative abundances of bacterial orders revealed significant relationships between particular taxa and lipids (Supplementary Figure S6). Rhizobiales, Ktedonobacterales, and Planctomycetia order B97 were positively correlated with most measured lipids. Notably, families belonging to these bacterial orders were differentially abundant in corn and native prairie soils, with increased abundances in native prairie soils (Figure 2). Conversely, orders belonging to the Gemmatimonadetes class, Nitrosomonadales, and Anaerolineae order envOPS12 were negatively correlated with most measured lipids (Supplementary Figure S6). Likewise, families of these orders are differentially abundant between corn and native prairie soils, with elevated abundance in the former (Figure 2). Together, these trends are congruent with the general observation of higher microbial biomass in native prairie samples compared to cultivated samples.
Core Functional Gene Analysis
We performed metagenome sequencing on reference cores from the native prairie and cultivated corn soils in each state. Metagenome sequencing resulted in 1.3 terabases (Tb) of sequence data from the six samples (ranging from 159 to 327 Gb per sample). De novo assembly was performed on each of the sequence datasets, yielding 50.8 million contigs >200 bp in length totaling 16.8 Gb of assembled data (Supplementary Table S3).
To explore the functional gene repertoire in our samples, we refrained from between site comparisons because of lack of within site replication. Instead, we identified core functions shared across all samples (Supplementary Table S7). We defined core genes as those varying least in abundance across all sites. These genes may form a backbone supporting ecosystem processes critical in both native and cultivated soil communities. Conceptually, this is similar to taxonomically based core microbiome analyses discussed in Shade and Handelsman (2012). Among the top core genes, we found genes involved in transport, cell regulation and signaling, and nitrogen metabolism. One of the top core genes was adenylate cyclase, which is an essential part of microbial cyclic AMP (cAMP) signaling. Genes related to cAMP signaling have been observed at high frequency in other soil metagenome surveys (Delmont et al., 2012). Two serine/threonine protein kinases, which are widely distributed across bacterial and archaeal phyla and play an important roles in physiology, regulation of cells division and translation, and environmental sensing (Pereira et al., 2011; Shi et al., 2014), were also among the top core genes. Nitrite reductase (nirK), a nitrogen regulatory protein C (ntrC) family gene, and two NitT/Tau family ABC transport genes were among the core genes related to the nitrogen cycle.
To formally identify functional groups enriched in core genes, we combined genes into FOAM ontological groups and found that at functional level 1 (the most general functional level), the prokaryotic type ABC transporters (P = 0.013) and regulation of response to osmotic stress (P = 0.047) categories had more core genes than expected under the null hypothesis that core genes are randomly distributed across FOAM functional categories. Within the prokaryotic type ABC transporters category, the genes detected encoded transporters of a broad range of compounds, including sugars, amino acids, peptides, cell wall components, and metals (Supplementary Figure S7). In the “regulation of response to osmotic stress” FOAM category, genes detected included those encoding sensor kinase proteins of the two-component signal transduction system, two of which belonged to the ompR family (KO: K07636; K02484) that allow bacteria to sense changes in osmolarity (Feng et al., 2003).
Nitrogen Metabolism
Because nitrogen fertilization is a major perturbation to cultivated soils and nitrogen is a key driver of soil microbial community composition (Fierer et al., 2012, 2013), we analyzed nitrogen metabolism pathways that were reconstructed from metagenomic sequence data. Forty-nine genes involved in all major components of the nitrogen cycle were detected across the metagenomes (Figure 4). Genes involved in denitrification were more abundant than nitrification genes. Those involved in the conversion of nitrite to nitrogen gas (nirK: 18%, nirS: 3.1%, norC: 6.5%, norB: 2.0%, and nosZ: 3.4%) made up 33% of all detected nitrogen cycle genes. Genes involved in ammonia assimilation accounted for 39% of all the nitrogen cycle genes. Ammonia monooxygenase, a key gene in the nitrification pathway, was only detected at low levels (<0.3% of nitrogen-cycle genes). NifH, the key marker gene for nitrogen fixation, accounted for 1.7% of the nitrogen-cycle genes.
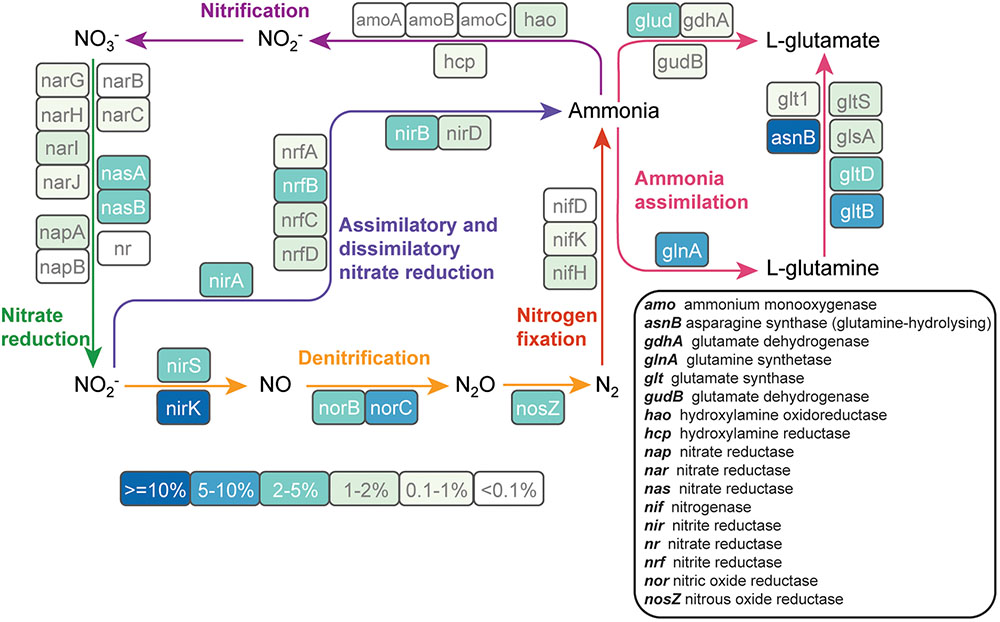
FIGURE 4. The abundance of nitrogen cycle genes in cultivated and never-cultivated tall grass prairie soils in the Midwestern United States. Each gene in the nitrogen cycle is enclosed in a colored box. The color of the box indicates the abundance of each gene relative to all nitrogen cycle genes in the assembled metagenome data. Genes were identified on contigs by comparing predicted protein sequences to the FOAM database. Percentages are averaged across all samples.
Because the denitrification pathway was highly represented in the metagenomes, we focused on the phylogenetic diversity of denitrifiers. Specifically, we focused on nosZ sequences because NosZ is the only enzyme known to catalyze the last step of denitrification: conversion of nitrous oxide (N2O) to nitrogen gas (N2) whereas other steps in the denitrification pathway can be catalyzed by multiple enzymes (Jones et al., 2008). Phylogenetic analysis revealed two distinct clades with strong bootstrap support corresponding to typical (Clade I) and atypical nosZ (Clade II) genes (Figure 5). We were able to assign taxonomy using the lowest common ancestor algorithm (Huson et al., 2016) to 80% of the Clade I sequences at the phylum level, the majority of which were from Proteobacteria. Genes with higher resolution taxonomic assignments were affiliated with Alphaproteobacteria, Betaproteobacteria (primarily Burkholderiales), or Gammaproteobacteria (primarily Pseudomonadales). The 20% of the sequences not assigned to a specific taxonomic group were classified as environmental sequences.
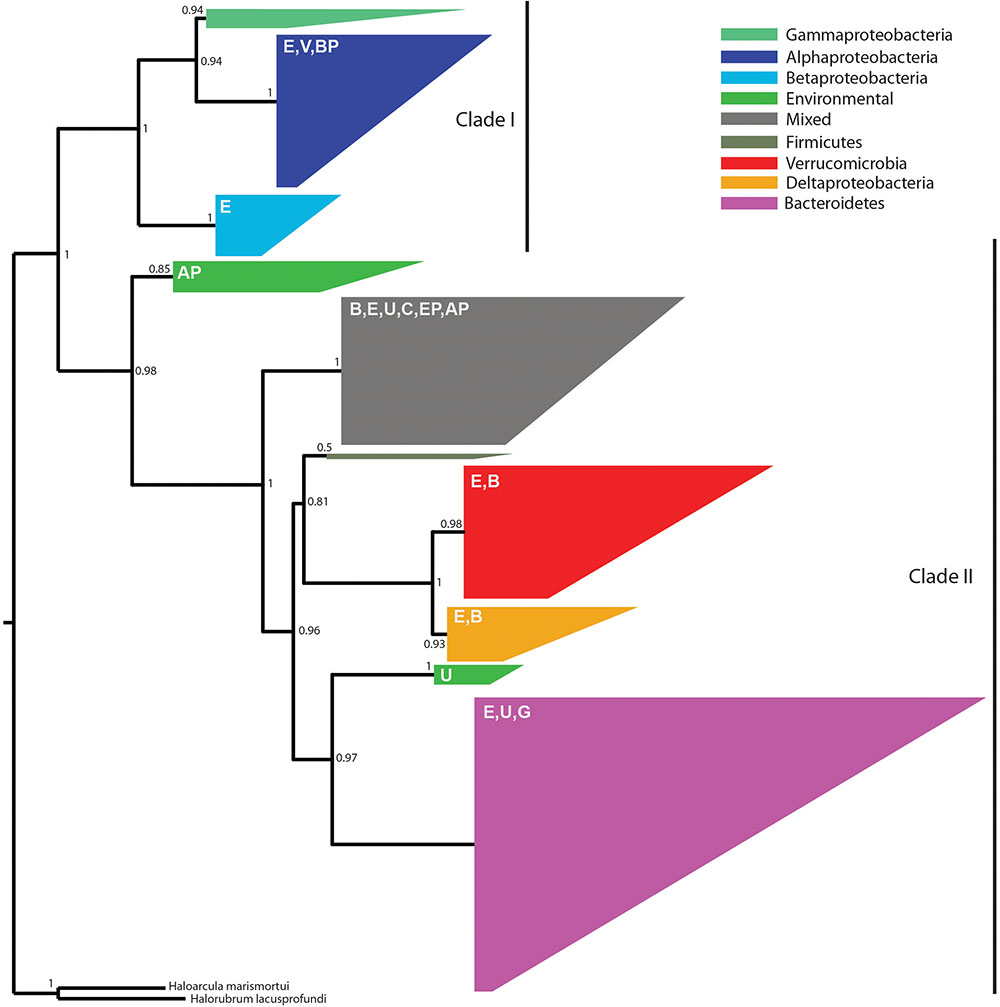
FIGURE 5. An approximate maximum-likelihood phylogenetic tree of 538 nosZ amino acid sequences predicted from metagenome assemblies and 69 sequences previously characterized as typical (Clade I) or atypical (Clade II) (Sanford G.R. et al., 2012). Reliability of each split in the tree was calculated using the Shimodaira–Hasegawa test. The tree was rooted using Haloarcula marismortui and Halorubrum lacusprofundi (Sanford R.A. et al., 2012). Clades are colored according to the majority taxonomic group hosting the majority of sequences. Letters in the upper left of each clade indicate the presence of secondary taxa. E, environmental; V, Verrucomicrobia; BP, Betaproteobacteria; AP, Alphaproteobacteria; B, Bacteroidetes; U, unassigned; C, Chloroflexi; EP, Epsilonproteobacteria; G, Gemmatimonadetes. One group, referred to as mixed in the figure legend, contained no dominant taxonomic group.
Among the two-thirds of nosZ sequences from this study that clustered with Clade II nosZ genes, we were able to classify approximately 75% at the phylum level; the remaining 25% were either environmental or unassigned. We found greater taxonomic diversity among Clade II nosZ genes and they were primarily affiliated with Bacteroidetes (41%), Verrucomicrobia (18%), Deltaproteobacteria (7%), and at lower levels to Chloroflexi, Epsilonproteobacteria, Gemmatimonadetes, Alphaproteobacteria, Betaproteobacteria, and Firmicutes (Figure 5).
Carbohydrate Metabolism
We explored the repertoire of carbohydrate-degrading enzymes in the soil communities by comparing raw reads to GH sequences from the CAZy database (Lombard et al., 2014). The most dominant GH gene families were GH13, representing 36% of all GH sequences, and GH15 at 8% (Supplementary Table S8A). Glucoamylases, which are involved in starch hydrolysis, constituted the bulk of the GH15 enzymes (Cantarel et al., 2009; Marín-Navarro and Polaina, 2011). GH13 also contained a large number of starch-degrading enzymes. Taken together, these data suggest high amylolytic (i.e., starch-degrading) potential in the sampled soil region.
Glycoside hydrolase families targeting plant structural polysaccharides were categorized by function (Pope et al., 2010; Hess et al., 2011) and evaluated separately from other GH families because of their potential to decompose recalcitrant biomass. Endoglucanases (cellulases) were primarily represented by families GH5 and GH9; these two GH families accounted for approximately 9% of all reads within the plant structural polysaccharides category. Other cellulase families were detected at low levels (<1.5%; Supplementary Table S8B). β-glucosidases, primarily GH1, accounted for ∼10% of the plant polysaccharide degrading genes; sequences predicted to be xylanases (GH10 and GH11) made up another 5%. The primary hemicelluloses found in grass cell walls contain L-arabinose side chains (Scheller and Ulvskov, 2010), which may explain the high abundance of α-L-arabinofuranosidases (GH51, GH54, and GH62: 12.9–16.8%) in all of the samples examined.
Discussion
The former tallgrass prairie region of the Midwestern United States is an area of economic and ecological importance for food security, biofuel production, nutrient retention, and is a major terrestrial carbon store, that could be jeopardized with climate change (Boody and DeVore, 2006; Jordan and Warner, 2010; Jokela et al., 2011; Paustian et al., 2016). Predicting the environmental consequences of changes to prairie-derived soils resulting from cultivation practices will likely be improved by understanding the microbial communities involved in carbon and nutrient cycling before and after cultivation.
Here we performed lipid profiling of microbial biomass as an indicator of soil metabolic health and quality (Vestal and White, 1989; Yao et al., 2000). We observed approximately double the microbial (lipid) biomass in native prairie samples compared to their paired cultivated samples (Figure 3 and Supplementary Table S6), suggesting that the prairie is more supportive of microbial biomass likely resulting from higher levels of soil carbon. This observation agrees with the results of Spearman rank correlations of taxa to lipid abundances, in that taxa positively correlated with cultivated corn samples were negatively correlated to total abundance of most measured lipids. Conversely, taxa positively correlated with native prairie samples, such as families of the Rhizobiales order, showed strong positive correlations to most measured lipids (Supplementary Figure S6). The abundance of arbuscular mycorrhizal fungi in native prairie is not surprising as several of the tallgrass prairie grasses are known to be mycorrhizal dependent (Wilson and Hartnett, 1998; Hoeksema et al., 2010; van der Heijden et al., 2015; Koziol and Bever, 2016).
Although the biomass was higher in prairie, alpha diversity was significantly higher in cultivated sites; cultivated corn showed the highest diversity, followed by switchgrass, restored prairie, and native prairie. Evenness did not differ significantly between prairie and cultivated corn. This finding is consistent with the work of Barber et al. (2017) who found that alpha diversity was lowest in native prairie and long-term restoration sites compared to agriculture fields and recently restored sites. Similarly, Acosta-Martinez et al. (2008) found elevated alpha diversity in samples from cultivated cornfields. This increase in alpha diversity, but with a much lower biomass, may be the result of agricultural practices associated with cultivation that provide more microbial niches, such as application of nitrogen-based fertilizer and/or the higher annual fluxes of organic carbon turnover stemming from plant productivity and litter input. Ample provision of otherwise scarce nutrients, such as nitrogen and phosphorus could drive fertilizer-associated increases in diversity (Acosta-Martinez et al., 2008; Mao et al., 2011). Notably, all of the corn plots in our study received a nitrogen-based fertilizer.
Long-term cultivation also resulted in significant changes in bacterial community structure. The soil microbiomes exhibited distinct clustering according to land management practice, with cultivated samples clustering together and separately from the native prairie samples, suggesting evidence of a cultivation-specific microbiome. The native prairie samples not only clustered distinctly from the cultivated samples but also clustered separately by state, suggesting that a combination of local soil history, plant species, and climate influence soil microbial community structures. The similarity of cultivated soil communities–whether from corn, switchgrass (3 years since cultivation), or even restored prairie (10 years since cultivation)–suggests that cultivation in general has a profound influence on the microbial community structure independent of crop species. In a similar vein, Fierer et al. (2012) found that a common practice in cultivation–high levels of N input–changed community structure similarly in both monoculture and grassland sites compared with low and intermediate levels of N addition (Fierer et al., 2012).
The observation that the restored prairie samples have bacterial communities that are intermediate between prairie and cultivated locations (Figure 1D), but more similar to those from the long-term cultivated sites is notable. This finding suggests that the changes associated with agricultural practices endure over long time periods, and that the return of the soil microbiome to the composition found in the native prairie state is a slow process. This is in agreement with previous findings of no difference in bacterial community composition between traditionally managed agricultural fields and a previously cultivated field that was left to recover for 9 years (Buckley and Schmidt, 2003) but in contrast with other data that suggest a faster time-frame for recovery (Herzberger et al., 2014). It is not clear what is driving the differences between these investigations, but it highlights the necessity of further studies to determine of how environment, vegetation, soil physicochemistry, and microbial processes interact in response to land-use changes.
Land-use management not only explained differences in microbial diversity and biomass, but also was correlated with taxonomic changes. In contrast to the native prairie samples, cultivated soil showed significantly higher abundances of the Nitrospiraceae and Nitrosomonadaceae families. The former is involved in ammonia oxidation to nitrite, the rate-limiting step in nitrification (Kowalchuk et al., 2000; Webster et al., 2005), while the latter oxidizes nitrite to nitrate (Lücker et al., 2010). The increased abundance of ammonia oxidizers in the cultivated soils may be a response to the application of ammonia-nitrogen fertilizer for production of corn. Nitrification activity is known to increase with nitrogen fertilizer application (Carey et al., 2016; Ouyang et al., 2016). In native prairie soils, several members of the order Rhizobiales–common rhizosphere-associated microbes–were more abundant than in cultivated soils. In contrast to recent studies (Fierer et al., 2013; Barber et al., 2017), we did not observe significant differences in Verrucomicrobia across sites or treatments.
While 16S rRNA gene sequencing revealed differences in taxa known to perform nitrification (cultivated corn) and nitrogen fixation (native prairie), analysis of the metagenomic data suggests that denitrification was uniformly important in both cultivated and native ecosystems, similar to observations in Nelson et al. (2016). Denitrification returns nitrogen to the atmosphere as inert N2 (complete denitrification) or the potent greenhouse gas N2O (incomplete denitrification). The most abundant nitrogen cycle gene (nirK, which reduces NO2- to NO) encodes one of the first steps in this pathway and was found to be a core functional gene. Clade I and Clade II nosZ genes (encoding the final step in the denitrification pathway), were found in a large number of phyla from both native prairie and cultivated corn samples. Until recently, attenuation of soil N2O emissions was thought to be mediated primarily by members of the Alpha-, Beta-, and Gamma-proteobacteria that are capable of performing all steps in the denitrification pathway (Sanford R.A. et al., 2012). However, bioinformatics analyses have revealed phylogenetically distinct nosZ sequences (Clade II) in a diverse array of organisms lacking other genes in the denitrification pathway (Sanford R.A. et al., 2012). Our observation that approximately 2/3 of prairie soil nosZ genes were atypical corroborates a recent study surveying nosZ in different soil types (Orellana et al., 2014) and suggests a deep reservoir of phylogenetically diverse organisms capable of mitigating N2O emission through N2O reduction to N2.
Soils represent one of the most complex microbial communities on Earth. As such, they present a unique challenge for assembling and analyzing metagenomic data. Prior analysis of the Iowa corn and Iowa prairie metagenomes demonstrated 48 and 31% of contigs (from corn and prairie, respectively) had coverage of less than 10 and that only ∼20% of the sequence data could be assembled (Howe et al., 2014). Full assemblies of soil metagenomes may require many terabases (Gans et al., 2005; Howe et al., 2014; Rodriguez-R et al., 2018). Because soils have substantial spatial heterogeneity, even down to the microstructure scale (Nesme et al., 2016), we designed the study to maximize sequence coverage of a small sample, i.e., to not dilute the community with extraneous DNA from different sites even though they might be local. This design for depth rather than breadth sacrificed the more traditional replicate design for the metagenome samples, limiting our ability to determine how functional genes differ between sites. Our 16S rRNA amplicon data, however, does provide replication for the sites.
Here, we focused on near-surface soils since they are more responsive to land management (Zhang, 2017). However, a significant proportion of biomass resides within deeper soils (Fierer et al., 2003) and deep-soil microbes contribute to long-term carbon sequestration (Rumpel and Kogel-Knabner, 2011). Depth is a major driver of community structure (Pereira et al., 2017; Zhang, 2017). Just as in surface soils, tillage (Sun et al., 2018), and soil physicochemical properties (Zhang et al., 2017) strongly affect microbial communities at depth. To gain a full understanding of how geographic distance, cropping systems, and long-term cultivation influence microbial community structure and function, future studies must consider both horizontal and vertical community distributions.
In summary, we found that cultivation has a significant impact on microbial community biomass, diversity, and composition in soils across the former tallgrass prairie region of the Midwestern United States. However, based on our metagenomic survey, we found that many core functions were conserved, even at small sample scales, across geographic regions and land management practices, suggesting that conditions common to prairie soil, independent of land-use, select for a set of critical features that persist despite perturbation. While DNA sequence information does not reflect current microbial activity since much of it may not be expressed at any given time, it does reflect microbial dynamics over a long time. The paired sites in this case had 50–100 years of cultivation versus none, which resulted in a major microbial biomass change (as documented by lipid data) and major microbial community change (as documented by 16S rRNA data). While the metagenomic portion of this study was designed to a sequence samples deeply because of soil community complexity, further studies are needed to sample more broadly to determine to what extent and which genes are selected under different land management and crop regimes.
Author Contributions
RM, SK, and BP performed the metagenome analyses. AG performed the 16S analyses. RL supervised the bioinformatics. EJ assisted with the sampling and field data. AC performed the sequence data QC and metagenome assemblies. CL performed the lipid analyses and interpretations. RJ and CR the obtained the field samples. ST managed the sequencing and sequence analysis. JT and JJ conceived of study, managed sample preparation and data generation, and coordinated analyses. All authors assisted with writing of the manuscript.
Funding
The work was conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, and was supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 with partial support from the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02-07ER64494). Partial support was also provided under the Laboratory Directed Research and Development Program at PNNL, a multi-program national laboratory operated by Battelle for the U.S. Department of Energy under contract DE-AC05-76RL01830. This research was also supported by a grant to Juniata College from the Howard Hughes Medical Institute (http://www.hmmi.org) through the Precollege and Undergraduate Science Education Program, the National Science Foundation (www.nsf.gov), NSF Award # DBI-1248096, and was informed and disseminated through the Research Coordination Network grant, RCN 1051481.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Phil Hugenholtz, Martin Allgaier, Crystal Wright, Tijana Glavina del Rio, and Krystle Chavarria for assistance in the early phases of the project and Eddy Rubin for supporting the sequencing. We also thank to L. Gary Oates and Harry Read for lipid analyses and Gregg Sanford for field management in Wisconsin.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01775/full#supplementary-material
Footnote
References
Acosta-Martinez, V., Dowd, S., Sun, Y., and Allen, V. (2008). Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol. Biochem. 40, 2762–2770. doi: 10.1016/j.soilbio.2008.07.022
Allgaier, M., Reddy, A., Park, J. I., Ivanova, N., D’haeseleer, P., Lowry, S., et al. (2010). Targeted discovery of glycoside hydrolases from a switchgrass-adapted compost community. PLoS One 5:e8812. doi: 10.1371/journal.pone.0008812
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Balser, T. C., and Firestone, M. K. (2005). Linking microbial community composition and soil processes in a California annual grassland and mixed-conifer forest. Biogeochemistry 73, 395–415. doi: 10.1007/s10533-004-0372-y
Barber, N. A., Chantos-Davidson, K. M., Amel Peralta, R., Sherwood, J. P., and Swingley, W. D. (2017). Soil microbial community composition in tallgrass prairie restorations converge with remnants across a 27-year chronosequence. Environ. Microbiol. 19, 3118–3131. doi: 10.1111/1462-2920.13785
Bending, G. D., Putland, C., and Rayns, F. (2000). Changes in microbial community metabolism and labile organic matter fractions as early indicators of the impact of management on soil biological quality. Biol. Fertil. Soils 31, 78–84. doi: 10.1007/s003740050627
Bending, G. D., Turner, M. K., Rayns, F., Marx, M.-C., and Wood, M. (2004). Microbial and biochemical soil quality indicators and their potential for differentiating areas under contrasting agricultural management regimes. Soil Biol. Biochem. 36, 1785–1792. doi: 10.1016/j.soilbio.2004.04.035
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing on JSTOR. J. R. Stat. Soc. B Stat. Methodol. 57, 289–300. doi: 10.2307/2346101
Bligh, E. G., and Dyer, W. J. (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917. doi: 10.1139/y59-099
Boody, G., and DeVore, B. (2006). Redesigning agriculture. BioScience 56, 839–845. doi: 10.1641/0006-3568(2006)56[839:RA]2.0.CO;2
Bouyoucos, G. J. (1951). A recalibration of the hydrometer method for making mechanical analysis of soils. Agron. J. 43, 434–438. doi: 10.2134/agronj1951.00021962004300090005x
Brye, K. R., Gower, S. T., Norman, J. M., and Bundy, L. G. (2002). Carbon budgets for a prairie and agroecosystems: effects of land use and interannual variability. Ecol. Appl. 12, 962–979. doi: 10.1890/1051-0761(2002)012[0962:CBFAPA]2.0.CO;2
Brye, K. R., Norman, J. M., Bundy, L. G., and Gower, S. T. (2001). Nitrogen and carbon leaching in agroecosystems and their role in denitrification potential. J. Environ. Qual. 30, 58–70. doi: 10.2134/jeq2001.30158x
Buckley, D. H., and Schmidt, T. M. (2003). Diversity and dynamics of microbial communities in soils from agro-ecosystems. Environ. Microbiol. 5, 441–452. doi: 10.1046/j.1462-2920.2003.00404.x
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238. doi: 10.1093/nar/gkn663
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth0510-335
Carey, C. J., Dove, N. C., Beman, J. M., Hart, S. C., and Aronson, E. L. (2016). Meta-analysis reveals ammonia-oxidizing bacteria respond more strongly to nitrogen addition than ammonia-oxidizing archaea. Soil Biol. Biochem. 99, 158–166. doi: 10.1016/j.soilbio.2016.05.014
Coolon, J. D., Jones, K. L., Todd, T. C., Blair, J. M., and Herman, M. A. (2013). Long-term nitrogen amendment alters the diversity and assemblage of soil bacterial communities in tallgrass prairie. PLoS One 8:e67884. doi: 10.1371/journal.pone.0067884
Delmont, T. O., Prestat, E., Keegan, K. P., Faubladier, M., Robe, P., Clark, I. M., et al. (2012). Structure, fluctuation and magnitude of a natural grassland soil metagenome. ISME J. 6, 1677–1687. doi: 10.1038/ismej.2011.197
Doran, J. W. (1980). Soil microbial and biochemical changes associated with reduced tillage. Soil Sci. Soc. Am. J. 4, 765–771. doi: 10.2136/sssaj1980.03615995004400040022x
Duncan, D. S., Jewell, K. A., Suen, G., and Jackson, R. D. (2016). Detection of short-term cropping system-induced changes to soil bacterial communities differs among four molecular characterization methods. Soil Biol. Biochem. 96, 160–168. doi: 10.1016/j.soilbio.2016.02.002
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Feng, X., Oropeza, R., and Walthers, D. (2003). OmpR phosphorylation and its role in signaling and pathogenesis. Science 69, 390–395. doi: 10.3389/fmicb.2013.00331/full
Fierer, N., Ladau, J., Clemente, J. C., Leff, J. W., Owens, S. M., Pollard, K. S., et al. (2013). Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 342, 621–624. doi: 10.1126/science.1243768
Fierer, N., Lauber, C. L., Ramirez, K. S., Zaneveld, J., Bradford, M. A., and Knight, R. (2012). Comparative metagenomic, phylogenetic and physiological analyses of soil microbial communities across nitrogen gradients. ISME J. 6, 1007–1017. doi: 10.1038/ismej.2011.159
Fierer, N., Schimel, J. P., and Holden, P. A. (2003). Variations in microbial community composition through two soil depth profiles. Soil Biol. Biochem. 35, 167–176. doi: 10.1016/S0038-0717(02)00251-1
Figuerola, E. L. M., Guerrero, L. D., Turkowsky, D., Wall, L. G., and Erijman, L. (2014). Crop monoculture rather than agriculture reduces the spatial turnover of soil bacterial communities at a regional scale. Environ. Microbiol. 17, 678–688. doi: 10.1111/1462-2920.12497
Firestone, M. K., and Davidson, E. A. (1989). “Microbial basis of NO and N2O production can consumption in soil,” in Exchange of Trace Gases Between Terrestrial Ecosystems and the Atmosphere, eds M. O. Addreae and D. S. Schimel (Hoboken, NJ: Wiley).
Fish, J. A., Chai, B., Wang, Q., Sun, Y., Brown, C. T., Tiedje, J. M., et al. (2013). FunGene: the functional gene pipeline and repository. Front. Microbiol. 4:291. doi: 10.3389/fmicb.2013.00291
Gans, J., Wolinsky, M., and Dunbar, J. (2005). Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 309, 1387–1390. doi: 10.1126/science.1112665
Guo, L. B., and Gifford, R. M. (2002). Soil carbon stocks and land use change: a meta analysis. Glob. Change Biol. 8, 345–360. doi: 10.1046/j.1354-1013.2002.00486.x
Guo, Y., Amundson, R., Gong, P., and Yu, Q. (2006). Quantity and spatial variability of soil carbon in the conterminous United States. Soil Sci. Soc. Am. J. 70, 590–600. doi: 10.2136/sssaj2005.0162
Habig, J., and Swanepoel, C. (2015). Effects of conservation agriculture and fertilization on soil microbial diversity and activity. Environments 2, 358–384. doi: 10.3390/environments2030358
Herzberger, A. J., Duncan, D. S., and Jackson, R. D. (2014). Bouncing back: plant-associated soil microbes respond rapidly to prairie establishment. PLoS One 9:e115775. doi: 10.1371/journal.pone.0115775
Hess, M., Sczyrba, A., Egan, R., Kim, T.-W., Chokhawala, H., Schroth, G., et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467. doi: 10.1126/science.1200387
Hoeksema, J. D., Chaudhary, V. B., Gehring, C. A., Johnson, N. C., Karst, J., Koide, R. T., et al. (2010). A meta-analysis of context-dependency in plant response to inoculation with mycorrhizal fungi. Ecol. Lett. 13, 394–407. doi: 10.1111/j.1461-0248.2009.01430.x
Howe, A. D., Jansson, J. K., Malfatti, S. A., Tringe, S. G., Tiedje, J. M., and Brown, C. T. (2014). Tackling soil diversity with the assembly of large, complex metagenomes. Proc. Natl. Acad. Sci. U.S.A. 111, 4904–4909. doi: 10.1073/pnas.1402564111
Huffman, S. A., and Barbarick, K. A. (2008). Soil nitrate analysis by cadmium reduction 1. Commun. Soil Sci. Plant Anal. 12, 79–89. doi: 10.1080/00103628109367129
Huntemann, M., Ivanova, N. N., Mavromatis, K., Tripp, H. J., Paez-Espino, D., Palaniappan, K., et al. (2015). The standard operating procedure of the DOE-JGI microbial genome annotation pipeline (MGAP v.4). Stand. Genomic Sci. 10:86. doi: 10.1186/s40793-015-0077-y
Huson, D. H., Beier, S., Flade, I., Gorska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN community edition–interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12:e1004957. doi: 10.1371/journal.pcbi.1004957
Ismail, I., Blevins, R. L., and Frye, W. W. (1994). Long-term no-tillage effects on soil properties and continuous corn yields. Soil Sci. Soc. Am. J. 58, 193–198. doi: 10.2136/sssaj1994.03615995005800010028x
Jangid, K., Williams, M. A., Franzluebbers, A. J., Blair, J. M., Coleman, D. C., and Whitman, W. B. (2009). Development of soil microbial communities during tallgrass prairie restoration. Soil Biol. Biochem. 42, 302–312. doi: 10.1016/j.soilbio.2009.11.008
Jesus, E. D. C., Liang, C., Quensen, J. F., Susilawati, E., Jackson, R. D., Balser, T. C., et al. (2015). Influence of corn, switchgrass, and prairie cropping systems on soil microbial communities in the upper Midwest of the United States. Glob. Change Biol. Bioenergy 8, 481–494. doi: 10.1111/gcbb.12289
Jokela, W., Posner, J., Hedtcke, J., Balser, T., and Read, H. (2011). Midwest cropping system effects on soil properties and on a soil quality index. Agron. J. 103, 1552–1562. doi: 10.2134/agronj2010.0454
Jones, C. M., Stres, B., Rosenquist, M., and Hallin, S. (2008). Phylogenetic analysis of nitrite, nitric oxide, and nitrous oxide respiratory enzymes reveal a complex evolutionary history for denitrification. Mol. Biol. Evol. 25, 1955–1966. doi: 10.1093/molbev/msn146
Jordan, N., and Warner, K. D. (2010). Enhancing the multifunctionality of US agriculture. BioScience 60, 60–66. doi: 10.1525/bio.2010.60.1.10
Kowalchuk, G. A., Stienstra, A. W., Stephen, J. R., and Woldendorp, J. W. (2000). Changes in the community structure of ammonia-oxidizing bacteria during secondary succession of calcareous grasslands. Environ. Microbiol. 2, 99–110. doi: 10.1046/j.1462-2920.2000.00080.x
Koziol, L., and Bever, J. D. (2016). The missing link in grassland restoration: arbuscular mycorrhizal fungi inoculation increases plant diversity and accelerates succession. J. Appl. Ecol. 54, 1301–1309. doi: 10.1111/1365-2664.12843
Kucharik, C. J., Brye, K. R., Norman, J. M., Foley, J. A., Gower, S. T., and Bundy, L. G. (2001). Measurements and modeling of carbon and nitrogen cycling in agroecosystems of southern Wisconsin: potential for SOC sequestration during the next 50 years. Ecosystems 4, 237–258. doi: 10.1007/s10021-001-0007-2
Kunin, V., Engelbrektson, A., Ochman, H., and Hugenholtz, P. (2010). Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12, 118–123. doi: 10.1111/j.1462-2920.2009.02051.x
Kunitsky, C., Osterhout, G., Sasser, M., and Newark, D. E. (2006). “Identification of microorganisms using fatty acid methyl ester (FAME) analysis and the MIDI Sherlock microbial identification system,” in Encyclopedia of Rapid Microbiological Methods, ed. M. J. Miller (Baltimore, MD: PDA), 1–17.
Leff, J. W., Jones, S. E., Prober, S. M., Barberán, A., Borer, E. T., Firn, J. L., et al. (2015). Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc. Natl. Acad. Sci. U.S.A. 112, 10967–10972. doi: 10.1073/pnas.1508382112
Liang, C., Kao-Kniffin, J., Sanford, G. R., Wickings, K., Balser, T. C., and Jackson, R. D. (2016). Microorganisms and their residues under restored perennial grassland communities of varying diversity. Soil Biol. Biochem. 103, 192–200. doi: 10.1016/j.soilbio.2016.08.002
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Lücker, S., Wagner, M., Maixner, F., Pelletier, E., Koch, H., Vacherie, B., et al. (2010). A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc. Natl. Acad. Sci. U.S.A. 107, 13479–13484. doi: 10.1073/pnas.1003860107
Mann, L. K. (1986). Changes in soil carbon storage after cultivation. Soil Sci. 142:279. doi: 10.1097/00010694-198611000-00006
Mao, Y., Yannarell, A. C., and Mackie, R. I. (2011). Changes in N-transforming archaea and bacteria in soil during the establishment of bioenergy crops. PLoS One 6:e24750. doi: 10.1371/journal.pone.0024750
Marín-Navarro, J., and Polaina, J. (2011). Glucoamylases: structural and biotechnological aspects. Appl. Microbiol. Biotechnol. 89, 1267–1273. doi: 10.1007/s00253-010-3034-0
Markowitz, V. M., Chen, I.-M. A., Chu, K., Szeto, E., Palaniappan, K., Pillay, M., et al. (2014). IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 42, D568–D573. doi: 10.1093/nar/gkt919
Marx, M. C., Wood, M., and Jarvis, S. C. (2001). A microplate fluorimetric assay for the study of enzyme diversity in soils. Soil Biol. Biochem. 33, 1633–1640. doi: 10.1016/S0038-0717(01)00079-7
Mbuthia, L. W., Acosta-Martinez, V., DeBruyn, J., Schaeffer, S., Tyler, D., Odoi, E., et al. (2015). Long term tillage, cover crop, and fertilization effects on microbial community structure, activity: implications for soil quality. Soil Biol. Biochem. 89, 24–34. doi: 10.1016/j.soilbio.2015.06.016
Nelson, M. B., Martiny, A. C., and Martiny, J. B. H. (2016). Global biogeography of microbial nitrogen-cycling traits in soil. Proc. Natl. Acad. Sci. U.S.A. 113, 8033–8040. doi: 10.1073/pnas.1601070113
Nesme, J., Achouak, W., Agathos, S. N., Bailey, M., Baldrian, P., Brunel, D., et al. (2016). Back to the future of soil metagenomics. Front. Microbiol. 7:73. doi: 10.3389/fmicb.2016.00073
Nivelle, E., Verzeaux, J., Habbib, H., Kuzyakov, Y., Decocq, G., Roger, D., et al. (2016). Functional response of soil microbial communities to tillage, cover crops, and nitrogen fertilization. Appl. Soil Ecol. 108, 147–155. doi: 10.1016/j.apsoil.2016.08.004
Oates, L. G., Duncan, D. S., Sanford, G. R., Liang, C., and Jackson, R. D. (2016). Bioenergy cropping systems that incorporate native grasses stimulate growth of plant-associated soil microbes in the absence of nitrogen fertilization. Agric. Ecosyst. Environ. 233, 396–403. doi: 10.1016/j.agee.2016.09.008
Orellana, L. H., Rodriguez-R, L. M., Higgins, S., Chee-Sanford, J. C., Sanford, R. A., Ritalahti, K. M., et al. (2014). Detecting nitrous oxide reductase (NosZ) genes in soil metagenomes: method development and implications for the nitrogen cycle. mBio 5:e01193-14. doi: 10.1128/mBio.01193-14
Orr, C. H., James, A., Leifert, C., Cooper, J. M., and Cummings, S. P. (2011). Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Appl. Environ. Microbiol. 77, 911–919. doi: 10.1128/AEM.01250-10
Ouyang, Y., Norton, J. M., Stark, J. M., Reeve, J. R., and Habteselassie, M. Y. (2016). Ammonia-oxidizing bacteria are more responsive than archaea to nitrogen source in an agricultural soil. Soil Biol. Biochem. 96, 4–15. doi: 10.1016/j.soilbio.2016.01.012
Paustian, K., Lehmann, J., Ogle, S., Reay, D., Robertson, G. P., and Smith, P. (2016). Climate-smart soils. Nature 532, 49–57. doi: 10.1038/nature17174
Pereira, A., Andrade, P., Bini, D., Durrer, A., Robin, A., Bouillet, J. P., et al. (2017). Shifts in the bacterial community composition along deep soil profiles in monospecific and mixed stands of Eucalyptus grandis and mixed stands of Eucalyptus grandis and Acacia mangium. PLoS One 12:e0180371. doi: 10.1371/journal.pone.0180371
Pereira, S. F., Goss, L., and Dworkin, J. (2011). Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 75, 192–212. doi: 10.1128/MMBR.00042-10
Phillips, R. E., Thomas, G. W., Blevins, R. L., Frye, W. W., and Phillips, S. H. (1980). No-tillage agriculture. Science 208, 1108–1113. doi: 10.1126/science.208.4448.1108
Pope, P. B., Denman, S. E., Jones, M., Tringe, S. G., Barry, K., Malfatti, S. A., et al. (2010). Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc. Natl. Acad. Sci. U.S.A. 107, 14793–14798. doi: 10.1073/pnas.1005297107
Pose-Juan, E., Igual, J. M., Sanchez-Martin, M. J., and Rodriguez-Cruz, M. S. (2017). Influence of herbicide triasulfuron on soil microbial community in an unamended soil and a soil amended with organic residues. Front. Microbiol. 8:378. doi: 10.3389/fmicb.2017.00378
Prestat, E., David, M. M., Hultman, J., Tas, N., Lamendella, R., Dvornik, J., et al. (2014). FOAM (functional ontology assignments for metagenomes): a Hidden Markov Model (HMM) database with environmental focus. Nucleic Acids Res. 42:e145. doi: 10.1093/nar/gku702
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Ramirez, K. S., Craine, J. M., and Fierer, N. (2012). Consistent effects of nitrogen amendments on soil microbial communities and processes across biomes. Glob. Change Biol. 18, 1918–1927. doi: 10.1111/j.1365-2486.2012.02639.x
Rodriguez-R, L. M., Gunturu, S., Tiedje, J. M., Cole, J. R., and Konstantinidis, K. T. (2018). Nonpareil 3: fast estimation of metagenomic coverage and sequence diversity. mSphere 3:e00039-18. doi: 10.1128/mSystems.00039-18
Rumpel, C., and Kogel-Knabner, I. (2011). Deep soil organic matter–a key but poorly understood component of terrestrial C cycle. Plant Soil 338, 143–159. doi: 10.1007/s11104-010-0391-5
Sanderman, J., Hengl, T., and Fiske, G. J. (2017). Soil carbon debt of 12,000 years of human land use. Proc. Natl. Acad. Sci. U.S.A. 114, 9575–9580. doi: 10.1073/pnas.1706103114
Sanford, G. R., Posner, J. L., Jackson, R. D., Kucharik, C. J., Hedtcke, J. L., and Lin, T.-L. (2012). Soil carbon lost from Mollisols of the North Central U.S.A. with 20 years of agricultural best management practices. Agric. Ecosyst. Environ. 162, 68–76. doi: 10.1016/j.agee.2012.08.011
Sanford, R. A., Wagner, D. D., Wu, Q., Chee-Sanford, J. C., Thomas, S. H., Cruz-García, C., et al. (2012). Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proc. Natl. Acad. Sci. U.S.A. 109, 19709–19714. doi: 10.1073/pnas.1211238109
Scheller, H. V., and Ulvskov, P. (2010). Hemicelluloses. Annu. Rev. Plant Biol. 61, 263–289. doi: 10.1146/annurev-arplant-042809-112315
Shade, A., and Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12. doi: 10.1111/j.1462-2920.2011.02585.x
Shi, L., Pigeonneau, N., Ravikumar, V., Dobrinic, P., Macek, B., Franjevic, D., et al. (2014). Cross-phosphorylation of bacterial serine/threonine and tyrosine protein kinases on key regulatory residues. Front. Microbiol. 5:495. doi: 10.3389/fmicb.2014.00495
Stauffer, R. S., Muckenhirn, R. J., and Odell, R. T. (1940). Organic carbon, pH, and aggregation of the soil of the morrow plats as affected by type of cropping and manurial addition. Agron. J. 32, 819–832. doi: 10.2134/agronj1940.00021962003200110001x
Sun, R., Li, W., Dong, W., Tian, Y., Hu, C., and Liu, B. (2018). Tillage changes vertical distribution of soil bacterial and fungal communities. Front. Microbiol. 9:699. doi: 10.3389/fmicb.2018.0069
USEPA (1993). Methods for the Determination of Inorganic Substances in Environmental Samples. Washington, DC: EPA.
USEPA (2018). Inventory of U.S. Greenhouse Gas Emissions and Sinks: 1990-2016. Available at: https://www.epa.gov/ghgemissions/inventory-us-greenhouse-gas-emissions-and-sinks-1990-2016 [accessed June 15, 2018].
van der Heijden, M. G. A., Martin, F. M., Selosse, M.-A., and Sanders, I. R. (2015). Mycorrhizal ecology and evolution: the past, the present, and the future. New Phytol. 205, 1406–1423. doi: 10.1111/nph.13288
Vestal, J. R., and White, D. C. (1989). Lipid ANALYSIS IN MICROBIAL ECOLOGY. BioScience 39, 535–541. doi: 10.2307/1310976
Vitousek, P. M., Aber, J. D., and Howarth, R. W. (1997). Human alteration of the global nitrogen cycle: sources and consequences. Ecol. Appl. 7, 737–750. doi: 10.1073/pnas.0913658107
Webster, G., Embley, T. M., Freitag, T. E., Smith, Z., and Prosser, J. I. (2005). Links between ammonia oxidizer species composition, functional diversity and nitrification kinetics in grassland soils. Environ. Microbiol. 7, 676–684. doi: 10.1111/j.1462-2920.2005.00740.x
Wilson, G. W., and Hartnett, D. C. (1998). Interspecific variation in plant responses to mycorrhizal colonization in tallgrass prairie. Am. J. Bot. 85, 1732–1738. doi: 10.2307/2446507
Wu, M., and Scott, A. J. (2012). Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28, 1033–1034. doi: 10.1093/bioinformatics/bts079
Yao, H., He, Z., Wilson, M., and Campbell, C. (2000). Microbial biomass and community structure in a sequence of soils with increasing fertility and changing land use. Microb. Ecol. 40, 223–237.
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zhang, B., Penton, C. R., Xue, C., Quensen, J. F., Roley, S. S., Guo, J., et al. (2017). Soil depth and crop determinants of bacterial communities under ten biofuel cropping systems. Soil Biol. Biochem. 112, 140–152. doi: 10.1016/j.soilbio.2017.04.019
Keywords: soil microbiome, land management, metagenomics, native prairie, climate change, carbon cycle, nitrogen cycle
Citation: Mackelprang R, Grube AM, Lamendella R, Jesus EdC, Copeland A, Liang C, Jackson RD, Rice CW, Kapucija S, Parsa B, Tringe SG, Tiedje JM and Jansson JK (2018) Microbial Community Structure and Functional Potential in Cultivated and Native Tallgrass Prairie Soils of the Midwestern United States. Front. Microbiol. 9:1775. doi: 10.3389/fmicb.2018.01775
Received: 30 March 2018; Accepted: 16 July 2018;
Published: 15 August 2018.
Edited by:
Frank Rasche, University of Hohenheim, GermanyReviewed by:
Zhili He, University of Oklahoma, United StatesGwen-Aelle Grelet, Landcare Research, New Zealand
Copyright © 2018 Mackelprang, Grube, Lamendella, Jesus, Copeland, Liang, Jackson, Rice, Kapucija, Parsa, Tringe, Tiedje and Jansson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susannah G. Tringe, c2d0cmluZ2VAbGJsLmdvdg== James M. Tiedje, dGllZGplakBtc3UuZWR1 Janet K. Jansson, amFuZXQuamFuc3NvbkBwbm5sLmdvdg==
†These authors have contributed equally to this work.