 Fan Yang
Fan Yang- State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
Hepatitis B virus infection remains a global healthy issue that needs to be urgently solved. Novel strategies for anti-viral therapy are based on exploring the effective diagnostic markers and therapeutic targets of diseases caused by hepatitis B virus (HBV) infection. It is well-established that not only viral proteins themselves but also key factors from the host control the biological processes associated with HBV, including replication, transcription, packaging, and secretion. Protein post-translational modifications (PTMs), such as phosphorylation, acetylation, methylation, and ubiquitination, have been shown to control protein activity, regulate protein stability, promote protein interactions and alter protein subcellular localization, leading to the modulation of crucial signaling pathways and affected cellular processes. This review focuses on the functions and effects of diverse PTMs in regulating important processes in the HBV life cycle. The potential roles of PTMs in the pathogenesis of HBV-associated liver diseases are also discussed.
Introduction
Despite the available vaccine against hepatitis B virus (HBV) developed in 1981, there are still 364 million people worldwide chronically infected with this hepatotropic virus (Polaris Observatory Collaborators, 2018). The pathogenesis of various severe liver diseases, such as hepatocellular carcinoma, fibrosis, cirrhosis, and acute liver failure, are associated with persistent HBV infection (Lavanchy, 2004; Torres and Davila, 2012; Wu et al., 2018). High morbidity and mortality are attributed to chronic HBV, although the numbers have been decreasing over the past years (Global Global Burden of Disease Study 2013 Collaborators, 2015). Interferons (IFN) and nucleos(t)ide analogs (NAs) are currently the main therapeutic regimens, and the inability to eliminate the virus is attributed not only to drug resistance and side effects but also to the difficulty in eliminating covalently closed circular DNA (cccDNA) and the viral genome integrated in chromosomes (Torres and Davila, 2012). Thus, HBV infection remains a critical global healthy problem that requires the development of novel strategies for anti-HBV therapy.
Hepatitis B virus is a member of the Hepadnaviridae family and mainly consists of its viral envelope and an icosahedral nucleocapsid core with a partially double strand relaxed circular DNA (rcDNA) (Seeger and Mason, 2000). The rcDNA bound with the viral polymerase is released from the capsid, enters into the nucleoplasm and then converts into a cccDNA during infection (Nassal, 2015). The cccDNA, which is formed by a nucleosome-bound minichromosome, is the template for viral messenger RNA (mRNA) transcript synthesis (Bock et al., 2001). The stable existence of cccDNA in infected hepatocytes is one of the key factors for HBV persistent infection.The template for HBV genome replication is pre-genomic RNA (pgRNA), which is one of the transcripts synthesized from the cccDNA (Beck and Nassal, 2007). The viral minus DNA strand is then synthesized from a pgRNA template and serves as the template for the plus DNA strand in the core particle (Seeger and Mason, 2000). After enveloping, some mature core particles are released and others are cycled back to the nucleus (Levrero et al., 2009; Koumbi and Karayiannis, 2015). Four overlapping open reading frames (ORFs) that are partially encoded by the 3.2-kb HBV genome are ultimately translated into necessary proteins, including the HBV core antigen (HBcAg), HBV surface proteins (HBs), HBV X proteins (HBx) and other polymerases, for virus survival (Seeger and Mason, 2000). The regulation of viral replication, transcription and translation not only relies on various viral elements but also depends on many host factors (Moolla et al., 2002).
Post-translational modifications (PTMs) are critical for regulating signal molecule activity in different pathways, controlling protein stability and promoting the interaction between ligands and receptors in various cellular processes (Deribe et al., 2010). Diverse PTMs are known to play a vital role in regulating viral biological processes according to their functions in viral survival and proliferation (Zhang et al., 2013; Chang et al., 2016). Classical PTMs (such as phosphorylation and glycosylation) and increasingly regarded PTMs (such as acetylation, methylation, ubiquitination, SUMOylation, and NEDDylation) have been proven to influence HBV replication, transcription, and immune evasion via their modified targets including viral essential proteins and host-related components, such as transcription factors (Pollicino et al., 2006; Bai et al., 2015; Selzer et al., 2015; Liu et al., 2017). PTMs also control processes in which HBV is deemed to be the major cause of pathogenesis, such as several liver diseases, including hepatocarcinogenesis (Zhang et al., 2016; Gao et al., 2017). In this review, we summarize the positive and negative regulation effects of PTMs in viral biological processes and their roles in the pathogenesis of HBV-associated liver diseases.
The Role of Phosphorylation in the Regulation of HBV Life Cycle
Phosphorylation is one of the most common PTM types in the viral life cycle, and it is regulated by various kinases and phosphatases. Viral protein phosphorylation and dephosphorylation plays a key role in regulating viral activity. The HBV core protein, which is composed of 183 to 185 amino acid residues, can be divided into an N-terminal domain (NTD) for capsid shell formation, a C-terminal domain (CTD) that is essential for HBV replication, and a linker between the NTD and CTD that is crucial for pgRNA packaging, viral DNA synthesis and virion secretion (Yu and Summers, 1991; Nassal, 1992; Liu et al., 2018). The HBc CTD is highlighted for containing several important serine and threonine phosphorylation sites in viral replication (Melegari et al., 2005; Lewellyn and Loeb, 2011; Jung et al., 2014). Three major serine phosphorylation sites (S155, S162, and S170) have been reported for their roles in producing HBV DNA replicative intermediates and rcDNA intermediates in an HBx-dependent manner by mutational analyses (Melegari et al., 2005). A recent study found additional vital phosphorylated sites (T160, S168, and S176) also contributed to pgRNA encapsidation, minus- and plus-strand DNA synthesis, and rcDNA synthesis (Jung et al., 2014). Despite several host cell kinases such as protein kinase C (PKC) (Kann and Gerlich, 1994; Wittkop et al., 2010), a 46-kDa serine protein kinase (Kau and Ting, 1998), and glyceraldehyde-3-phosphate dehydrogenase protein kinase (GAPD-PK) (Duclos-Vallee et al., 1998) have been discovered to phosphorylate the core protein in vitro in the past, none of these papers mapped phosphorylation to specific sites in HBc. Until Daub et al demonstrated that SR protein-specific kinases 1 and 2 (SRPK1 and SRPK2), which are approximately 95 and 115 kDa, respectively, were identified as the important kinases phosphorylating the core protein in vitro, as those serine phosphorylation sites (S155, S162, and S170) are SPRRR motifs in the CTD (Daub et al., 2002). However, the role of the SRPK-regulated phosphorylation to HBV replication is still confused. The reason is that another group found a new HBV replication pathway mediated by SRPK1 and SRPK2, which is independent of core protein phosphorylation (Zheng et al., 2005). These results hint that SRPKs have other functions which may affect HBV replication. Cyclin-dependent kinase 2 (CDK2), another controversial kinase targeting the core protein, was reported by Ludgate et al. (2012) CDK2 can both indeed phosphorylate the HBc CTD in vitro and in vivo because S/T-P motifs, which are specific sites for proline-directed serine-threonine kinases, are present in HBc. Ironically, the inhibitors of CDK2 had no apparent effect on HBV replication (Ludgate et al., 2012). The complicated relationship between host kinases may be the main reason that the inhibitors become invalid. A current report showed that polo-like-kinase 1 (PLK1), a host kinase involved in cell cycle regulation, can effectively promote HBV replication via directly binding with HBc, leading to phosphorylation of the latter (Diab et al., 2017). The authors also mapped the accurate phosphorylated sites to S168, S176, and S178 in the core protein (Diab et al., 2017). Progressive experiments suggested that viral replication can be effectively blocked through inhibiting PLK1 expression in the liver humanized mouse model (Diab et al., 2017). Moreover, they observed that alanine substitutions at the 3 S/P sites induce the loss of PLK1 phosphorylation. This result points out that CDK2 may play a priming role in HBc phosphorylation which promotes the subsequent phosphorylation by PLK1 (Diab et al., 2017). Although the kinases that phosphorylate the core protein are still undetermined, the combination of CDK2 and PLK1 is currently deemed to be more promising based on the above results.
It is well-known that phosphorylation of the HBV core protein (HBc) is a crucial step during HBV lifecycle (Figure 1). HBc phosphorylation can regulate reverse transcription, pgRNA packaging, DNA synthesis, subcellular localization, and virion secretion (Lan et al., 1999; Gazina et al., 2000; Perlman et al., 2005; Basagoudanavar et al., 2007; Chua et al., 2010; Su et al., 2016). Phosphorylation of the core protein is also essential for capsid maturation and stability (Perlman et al., 2005; Basagoudanavar et al., 2007; Ludgate et al., 2016; Su et al., 2016). Moreover, HBc phosphorylation is implicated into regulating the chaperone activity of CTD (Chu et al., 2014).
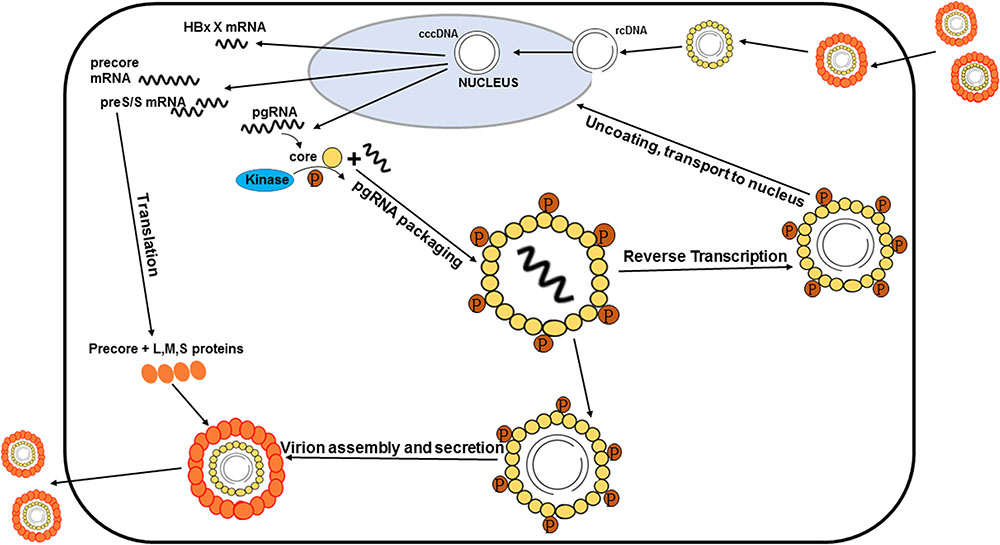
FIGURE 1. Processes of the HBV life cycle regulated by HBc phosphorylation. HBc phosphorylation can regulate reverse transcription, pgRNA packaging, subcellular localization, and virion secretion. Phosphorylation of the core protein is also essential for capsid maturation.
In addition to viral protein phosphorylation, phosphorylation of the host factors can also influence the process of viral replication. For example, Hu et al. (2018) found that the inhibitory effect of SAMHD1 (a restriction factor of HBV) on HBV replication is abrogated when SAMHD1 is phosphorylated by CDK2.
HBc dephosphorylation is thought to be associated with nucleocapsid maturation (Perlman et al., 2005; Basagoudanavar et al., 2007). Perlman et al. (2005) suggested that the nucleocapsid shell is formed by the complete dephosphorylation of the core CTD. Further research by Basagoudanavar et al. (2007) demonstrated that subsequent core protein dephosphorylation at T239, S245, and S259 after minus-strand DNA synthesis can induce nucleocapsid maturation as well as facilitate the synthesis of the plus-strand DNA in duck HBV. A recent study clarified that core protein dephosphorylation appears in pgRNA assembly through a progressive immunoblotting assay (Zhao et al., 2018). A potential reason why core protein allosteric modulators (CpAMs), molecules that bind to the interfaces between the core protein dimer-dimer, can block pgRNA encapsidation is due to interference in core protein dephosphorylation during nucleocapsid assembly (Zhao et al., 2018). Further research demonstrated that core CTD dephosphorylation is not essential for subsequent nucleocapsid and envelope interactions that secrete virions via a mutagenesis assay (Ning et al., 2017). However, evidence that a direct role for triggering core protein dephosphorylation, nucleocapsid maturation and second-strand DNA synthesis is still lacking.
The Role of Glycosylation in the Regulation of HBV Life Cycle
Glycosylation is another classical PTM type in controlling viral biological processes, specifically viral protein stability, virion secretion, and regulating host immune response (Spiro, 2002; Helenius and Aebi, 2004; Vigerust and Shepherd, 2007). Different glycosyltransferases and glycosidases are key factors for regulating protein glycosylation. Previous studies have demonstrated that N-glycosylation of HBV surface proteins, including large, middle, and small proteins, is crucial for virion secretion. In detail, the L and S proteins are necessary for virion formation, and increasing M protein expression levels is beneficial for virion secretion (Garcia T. et al., 2009; Ito et al., 2010). There is a single N-glycosylated site at N146 in the S domain that is essential for virion secretion. Mutations at either N146Q or N146S can block virion secretion, though subviral particle secretion is not affected (Ito et al., 2010). Mutations in N-glycosylated sites contribute to viral immune escape in light of harboring mutations within the HBsAg major hydrophilic region (Lambert and Prange, 2007; Liu et al., 2007; Ito et al., 2010). Progressively, researchers found out that mutations in the S protein provide a higher capacity to regulate virion secretion than those in the L/M protein (Ito et al., 2010; Bi and Tong, 2018). For example, the M133T mutation in the S protein can rescue the inhibition of virion secretion induced by the classic immune escape mutants G145R (El Chaar et al., 2010; Locarnini and Yuen, 2010) and R169H (Ito et al., 2010; Bi and Tong, 2018). To explain this phenomenon, authors found the additional higher molecular weights envelope proteins are associated with the presence of the M113T mutation by Western blot analysis (Ito et al., 2010). They subsequently proved that a new N-linked glycosylation site at N131 (131NST133) is created by the M133T mutation in the S protein. This novel site will produce additional forms of S (gp30), M (gp39), and L (gp45) proteins which are all essential for virion formation and secretion (Ito et al., 2010).
In addition, O-glycosylation of HBV surface proteins has been reported (Tolle et al., 1998; Werr and Prange, 1998; Schmitt et al., 1999; Tai et al., 2002). O-glycosylation of HBV surface proteins varies from different HBV genotypes. Researchers found that O-glycosylation appears in the M protein but not the L protein of HBV genotype D (Tolle et al., 1998; Schmitt et al., 1999). Potential O-glycosylation motifs are also found in HBV genotype B, C, E, and F except genotype A (Tai et al., 2002). Tai et al. (2002) reported that O-glycosylation in the preS2 region can partially cause size heterogeneity of wild type M protein. They suggested that enhanced O-glycosylation may be associated with hypermodification of the internally deleted M protein leading to the immune escaping (Tai et al., 2002).
Glycosylation also affects HBV entry into hepatoma cells. A recent study stated that core-fucosylation plays an important role in HBV infection through HBV-receptor-mediated endocytosis in a pseudovirus HBV model (Takamatsu et al., 2016). From their observations, sodium taurocholate cotransporting polypeptide (NTCP), a classical HBV receptor, is coprecipitated with other proteins and relied on core-fucosylation levels (Takamatsu et al., 2016). In contrast, an opinion from another recent study demonstrated that N-glycosylation is not needed for NTCP to mediate HBV infection in an in vitro experiment (Lee et al., 2018). The authors observed that HepG2 cells are glycosylated at two relative glycosylation sites, N5 and N11, when NTCP is introduced, and mutating either or both sites is unable to block cell surface NTCP availability and its subcellular localization, leading to the inability to inhibit HBV infection (Lee et al., 2018). Thus, further research is essential to explain these controversial results.
The Role of Acetylation in the Regulation of HBV Life Cycle
Acetylation of lysine, which modifies protein primary properties, has a role in various biological processes including viral proliferation and chromatin remodeling (Choudhary et al., 2009; Deribe et al., 2010). It is inversely regulated by acetyltransferases and deacetylases. It has been reported that histone acetylation is implicated in the activation of replication and transcription due to its influences on histones and histone interactions (Strahl and Allis, 2000). The template for the transcription of HBV mRNA, cccDNA, is formed into minichromosomes by histone and non-histone proteins. Researchers found that HBV replication is regulated by the acetylation of cccDNA-bound H3 and H4 histones based on ChIP experiments in 2006 (Pollicino et al., 2006). The application of histone deacetylase inhibitors induces an increase in both HBV replication and cccDNA-bound acetylated H4 (Pollicino et al., 2006). They also revealed that low HBV replication in patients is correlated with the recruitment of p300/CBP and histone deacetylases 1 (HDAC1) to cccDNA (Pollicino et al., 2006). Additional research by another group demonstrated that the processing of relevant enzymes involved in viral minichromosome formation is modulated by HBx (Belloni et al., 2009). The proof that preventing cccDNA deacetylation is regulated by HBx was shown by an evident increase in the histone deacetylases hSirt1 and HDAC1 due to an HBx mutant (Belloni et al., 2009). Previous studies showed that HBx can directly interact with the acetyltransferase p300/CBP complex to promote transcription, causing the activation of the acetylated histone state in HBV minichromosomes (Cougot et al., 2007; Zheng et al., 2009), which indicates that the balance of histone acetylation modifications in HBV is influenced by HBx (Figure 2). A recent study reported that IL6 treatment to NTCP-HepG2 cells causes a decrease in both cccDNA-bound histone acetylation and 3.5-kb pgRNA, leading to replication repression (Palumbo et al., 2015). In detail, the levels of H3 histone-bound cccDNA is slightly increased after IL-6 treatment; however, cccDNA-bound histones are predominantly hypoacetylated (Palumbo et al., 2015). Another new regulator of histone acetylation in HBV replication was discovered by Nishitsuji’s group. They explored how TIP60 (a catalytic subunit in the NuA4 complex localized in the cccDNA chromatin complex) catalyzed the acetylation of histone H4 to recruit Brd4, inducing the inhibition of HBV replication (Nishitsuji et al., 2018).
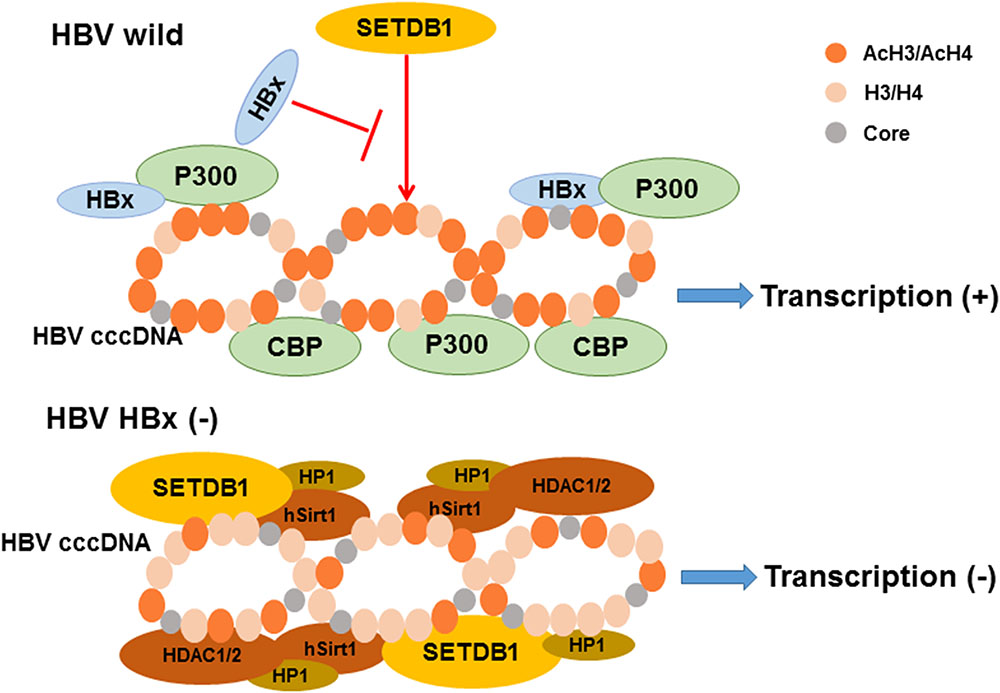
FIGURE 2. The model of relevant chromatin modifying enzymes involved in the regulation of HBV cccDNA transcription influenced by HBx. As indicated, the recruitment of chromatin modifying enzymes onto cccDNA is modulated by HBx expression. The activation of histone acetylation is caused by the interaction of HBx and acetyltransferase p300/CBP complex leading to the cccDNA transcription. In contrast, the recruitment of the histone deacetylases hSirt1 and HDAC1/2 are increased induce silencing of cccDNA transcription in the absence of HBx expression. Besides, hypoacetylation is also accompanied by the recruitment of heterochromatin protein 1 factor (HP1) and SET domain bifurcated 1 (SETDB1). Modified from Riviere et al. (2015).
The Role of Methylation in the Regulation of HBV Life Cycle
Protein methylation, inversely regulated by methyltransferases and demethylases (similar to acetylation), contributes to regulating a series of cellular biological processes, including proliferation, transcription, RNA metabolism, and chromatin remodeling (Nicholson et al., 2009; Deribe et al., 2010). It is well-established that PRMT1, a typical member of the protein arginine methyltransferase (PRMT) family, participates in HBV transcription (Benhenda et al., 2013). Researchers found that PRMT1 can negatively regulate HBV transcription via its methyltransferase activity, which was mainly reflected by histone H4 methylation (Benhenda et al., 2013). However, the inhibitory activity is dependent on the expression of HBx due to the interaction between PRMT1 and HBx (Benhenda et al., 2013). PRMT5, another PRMT, was previously reported in the repression of HIV long terminal repeat transcription through the methylation of SPT5, which is also a crucial HBV transcription regulator (Kwak et al., 2003). A recent study demonstrated that PRMT5 promotes cccDNA-bound H4R3me2s activity to inhibit cccDNA transcription by relying on its methyltransferase activity, while inhibition of core particle DNA production is methyltransferase-independent by preventing interactions with pgRNA (Zhang et al., 2017). Moreover, the interaction between PRMT5 and HBc was also demonstrated by CoIP assay in Huh7 cells (Zhang et al., 2017). Several months later, a group from the Czech Republic showed that PRMT5 not only interacts with HBc, but that PRMT3 also weakly binds with HBc by GST pull-down assay from HEK293T cells (Lubyova et al., 2017). They also found that HBc is methylated on its arginine residues at the C-terminal, with the R150 and R156 methylated sites being detected by mass spectrometry (Lubyova et al., 2017). The potential effect of arginine methylation in HBc is referred to as a regulator for controlling HBc binding to cellular factors, which participate in HBc shuttling between the nucleus and cytoplasm (Lubyova et al., 2017). Some studies have suggested that similar to the influence of histone acetylation, HBV cccDNA transcription is also regulated by histone methylation (Riviere et al., 2015; Ren et al., 2018). Plenty of previous studies have shown that methylation of H3 on lysine 9 (H3K9me) is controlled by histone lysine methyltransferases (HKMT), such as SETDB1 and SETD1A (Wang et al., 2003; Bannister and Kouzarides, 2011; Binda, 2013). A study by a group of French scientists demonstrated that HBV transcriptional silencing is related to H3K9me3 by ChIP-qPCR. The depletion of SETDB1 among the candidate HKMT promotes an obvious decrease in H3K9me3 (Riviere et al., 2015). Moreover, this process is controlled by HBx (Riviere et al., 2015). A similar mechanism was also presented for SETD1A in HBV transcription regulation in a recent paper (Ren et al., 2018).
Despite the number of studies on HBV transcription and replication regulation by methyltransferases, the role of demethylase involvement in the HBV life cycle is still obscure.
The Role of Ubiquitination, Sumoylation and Neddylation in the Regulation of HBV Life Cycle
Proteins are modified by covalent bonding with various short modulated proteins, such as ubiquitin, small ubiquitin-like modifier (SUMO) or other ubiquitin-like proteins, under certain conditions in biological processes, such as transcription, protein secretion, and so on (Hershko and Ciechanover, 1998). Ubiquitination is mainly regulated by E1, E2, and E3 enzymes or negatively controlled by deubiquitination enzymes (DUBs). Focus on ubiquitination involved in the viral life cycle has been increasing. An earlier research reported that γ2-adaptin, a ubiquitin-interacting adaptor, is essential for HBV viral particle release in light of the interaction with the HBV core protein lysine residue 96 in a ubiquitin-dependent manner (Rost et al., 2006), indicating that core protein ubiquitination may be crucial in HBV release. Subsequently, the authors found that Nedd4, an ubiquitin ligating enzyme with C-terminal E3 ubiquitin ligase domains, can partially interact with the core protein due to the PPAY-like motif binding site in the core protein and induce HBV production (Rost et al., 2006). Although two potential ubiquitin acceptor lysine residues (K7 and K96) in the core protein have been discovered, K96, not K7, can serve as the ubiquitin binding site that induces core particle release. The direct evidence that core ubiquitination occurred is still poor and the potential mechanism whereby γ2-adaptin binds Nedd4 to regulate viral particle release has not been fully elucidated (Rost et al., 2006). However, an article suggested that the production of the HBV core particle is not controlled by direct ubiquitin binding to either K7 or K96 (Garcia M.L. et al., 2009). Therefore, no direct evidence of core protein ubiquitination is provided by these studies. Np95/ICBP90-like RING finger protein (NIRF), an E3 ligase, is known as a crucial regulator in cell proliferation (Iwata et al., 2009). A Chinese group discovered that NIRF can directly interact with the HBV core protein in vivo and promote its ubiquitination and degradation through E3 ubiquitin ligase activity (Qian et al., 2012). Additionally, Mature core particle release is also influenced by core protein proteasome degradation regulated by NIRF (Qian et al., 2012).
It is well-established that HBx interacts with damage-specific DNA binding protein 1 (DDB1) to regulate viral replication (Sitterlin et al., 1997, 2000; Bontron et al., 2002). DDB1 is also known as a linker protein for the assembly of Cullin 4-ROC1 RING E3 ubiquitin ligase (CRL4) complexes (Angers et al., 2006; He et al., 2006). In a recent paper, researchers found that the host restriction factor SMC5/6 (a direct ubiquitination substrate of the HBx-CRL4 complex) can inhibit HBV replication. HBx can degrade SMC5/6, leading to enhanced HBV gene expression in a proteasome-dependent manner (Murphy et al., 2016). Therefore, this study provides evidence that HBx may promote HBV replication by degrading host restriction factors through the cullin ubiquitin E3 ligase, which requires interactions between DDB1 and HBx. A contrasting result from a South Korean group was that DDB1 itself can stimulate HBV transcription in an HBx-independent manner (Kim et al., 2016). However, the mechanism by which DDB1 promotes transcription with a cccDNA template has not been elucidated.
SUMOylation is a reversible PTM modulated by small ubiquitin-related modifier (SUMO) proteins, such as SUMO1 and SUMO2/3. SUMOylation is a crucial regulator of protein stability and subcellular localization and interactions (Zhao, 2018). Previous studies have shown that SUMOylation can regulate target protein stability by interfering with ubiquitination (Chen S. et al., 2013; Citro et al., 2013). In a recent study, SUMOylation was reported for its potential influence on HBV ability to evade host immune response (Sengupta et al., 2017). In detail, promyelocytic leukemia nuclear bodies (PML-NB) protein Speckled 110 kDa (Sp110) undergoes a deSUMOylation-driven release from PML-NBs to promote HBV proliferation and can also interact with HBx to maintain HBx stability and modulate several direct target genes of HBx transcription, ultimately inducing persistent viral infection (Sengupta et al., 2017).
NEDDylation is also a reversible PTM that is mainly modulated by the ubiquitin-like molecule neuronal precursor cell-expressed developmentally down regulated protein 8 (NEDD8) via a three-step enzymatic reaction catalyzed by NEDD8-activating enzyme E1 (NAE), NEDD8-conjugating enzyme E2 (UBC12/UBE2M) and NEDD8-E3 ligases (Zhou et al., 2018). Similar to SUMOylation, NEDDylation can also regulate diverse signaling pathways, such as apoptosis, autophagy, and inflammatory responses, by modulating protein stability and interactions (Zhou et al., 2018). A recent study discovered that HBx can be stabilized by the E3 ligase HDM2 in a NEDDylation-dependent manner at Lys91 and Lys95 (Liu et al., 2017). NEDDylated HBx can effectively prevent interactions with the E3 ligase Siah-1, leading to inhibited HBx degradation. Moreover, HBx NEDDylation can promote HBx chromatin localization and regulate the transcription of downstream genes, such as IL-8 and YAP, to promote cell proliferation and hepatocarcinogenesis (Liu et al., 2017).
The Role of PTMs in Hepatocarcinogenesis Due to HBV
It is well-known that major HBV mechanisms related to hepatocarcinogenesis include viral integration and the regulation of trans-activating HBV proteins, such as HBx (Park et al., 2013). HBx can associate with the androgen receptor (AR) signaling pathway, a classical pathway that contributes to HCC, to promote hepatocarcinogenesis (Chiu et al., 2007). Subsequent research has demonstrated that the enhancement of AR transcriptional activity results from AR N-terminal transactivation domain (NTD) phosphorylation by c-Src kinase, which is indirectly activated by HBx (Yang et al., 2009). Moreover, the upstream element cell cycle-related kinase (CCRK) can also control HBx expression through the phosphorylation of glycogen synthase kinase 3β (GSK3β) (Yu et al., 2014).
Regulation by protein phosphorylation, ubiquitination and SUMOylation is also involved in another classical signaling pathway associated with HBx, the NF-κB pathway, in the pathogenesis of HBV-induced HCC. Akt can phosphorylate IKKα at Thr23 and promote its ubiquitination in response to HBx overexpression (Huang et al., 2012). A recent study reported that p65, a crucial NF-κB subunit, can be stabilized through SUMOlyation interactions with SUMO2/3 (Liu J. et al., 2015). In another current study, SUMOylated centrosomal P4.1-associated protein (CPAP) was reported to be implicated in IKK-mediated NF-κB activation, leading to enhanced HBx-induced NF-κB signaling in HBV-related HCC (Yang et al., 2013). HBx also promotes the up regulation of MSL2, which induces APOBEC3B degradation through ubiquitination to maintain HBV cccDNA stability, thus contributing to hepatocarcinogenesis (Gao et al., 2017). Additionally, Li et al. (2018) demonstrated that HBx can induce the phosphorylation of PDK1, in turn to activate the PDK1-WINK1 pathway in HBV-associated HCC tissues. The authors also discussed that activation of the PDK1-WINK1 pathway mediated by HBx may be implicated in the development of HBV-associated HCC (Li et al., 2018).
Although plenty of studies have demonstrated that HBx may indirectly influence protein PTMs leading to hepatocarcinogenesis, HBx can also interfere with protein PTMs through direct interactions. For example, HBx can directly interact with amplified in breast cancer 1 (AIB1), a crucial enhancer required for the activation of specific transcription factors, such as NF-kB and AP-1, preventing interactions between Fbw7α and AIB1 and inhibiting the Fbw7α-mediated ubiquitination and degradation of AIB1, leading to HCC cell invasiveness (Liu et al., 2012). Another example of an HBx direct interaction is that HBx can stabilize the Myc oncoprotein by preventing the interaction between Myc and the Skp/cullin/F-box (SCF) ubiquitin E3 ligase to block Myc oncoprotein ubiquitination and degradation, which ultimately contributes to viral oncogenesis (Lee et al., 2016).
The Role of PTMs in Host Antiviral Resistance Due to HBV
The interferon system is involved in host antiviral responses. Type I interferons (IFNs) exhibit their antiviral functions according to the activation of the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway (Schneider et al., 2014). It is well-established that some host factors, induced after HBV infection, can regulate the activation of STAT1 via diverse PTMs (Christen et al., 2007; Chen J. et al., 2013, Chen K. et al., 2017; Bing et al., 2014). For instance, a previous study discovered that HBV induced the up regulation of serine/threonine phosphatase (PP2A), which can effectively block STAT1 activation due to inhibition of STAT1 arginine methylation by directly interacting with PRMT1 (Christen et al., 2007). Viral proteins also have been testified to inhibit STAT1 activation. A group found that HBV polymerase can repress the activation of STAT1 by blocking the association between STAT1 and protein kinase C-δ (PKC-δ), which can phosphorylate STAT1 Ser727, leading to its activation (Chen J. et al., 2013).
Additionally, modulation of the type I IFN receptor (IFNAR1) promotes its binding with IFN-α to activate the JAK-STAT pathway; this interaction also plays an important role in the interferon-mediated antiviral pathway. Researchers have recently determined that matrix metalloproteinase 9 (MMP-9), which originates from peripheral blood mononuclear cells (PBMCs) and macrophages, can be up regulated by HBV stimulation and interacts with the IFNAR1 extracellular domain to disrupt binding with IFN-α, subsequently facilitating the phosphorylation and ubiquitination of IFNAR1 in a p38 and Ser532 phosphorylation-independent manner (Chen J. et al., 2017).
The HBV-mediated disruption in IFN-β production is also related to PTMs on stimulator of interferon genes (STING), which has been reported to be crucial for the regulation of the immune response to cytosolic DNA (Liu Y. et al., 2015). Previous studies showed that STING ubiquitination is necessary for the induction of IFN-β (Tsuchida et al., 2010; Zhang et al., 2012). The results from Yuan’s group showed that HBV polymerase can physically associate with STING to inhibit STING K63-linked polyubiquitination, while the interactions with the E3 ubiquitin ligases TRIM32, TRIM56, and STING are not influenced (Liu Y. et al., 2015). Additionally, some crucial host factors that are induced by HBV can affect the IFN-β production via different PTMs. NF-κB essential modulator (NEMO) is involved in the process of IFN-β production through phosphorylating IFN regulatory factor 3 (IRF3) in a polyubiquitin-dependent manner (Clark et al., 2013). HBV-induced rubicon can directly bind to NEMO leading to inhibition of NEMO ubiquitination and further influencing the IFN-β production (Wan et al., 2017). Another research reported that parkin, a host factor induced by HBV infection, can modulate the linear ubiquitin chain on the MAVS (a mitochondrial membrane protein) signalosome, which abrogates IFN-β synthesis (Khan et al., 2016).
Conclusion and Perspectives
In the past few decades, a growing number of studies have focused on the diverse functions and complicated mechanisms between PTMs and viral proteins, such as HBc (Table 1); or crucial host factors in controlling viral biological processes. However, the reasons behind specific phenomena and controversial experimental results are still obscure, demonstrating the need for further investigation. Even though phosphorylation, acetylation, methylation, and ubiquitination are increasingly shown to play important roles in HBV replication and transcription, other PTMs including succinylation, hydroxylation, nitration, and carbonylation also potentially participate in this regulation, showing that our understanding of these regulatory processes is still unclear. Aside from the classical function of some PTMs, such as glycosylation, other roles for PTMs in regulating HBV life cycle should be further explored. It has been mentioned that advanced technologies, including new types of mass spectrometry and elaborate analyses that consist of big data analysis, are required to discover novel proteins changes. Additionally, interactomics and fluxomics, new powerful omics studies focused on protein interactions research, should be applied to observe dynamic changes for crucial proteins in the HBV life cycle. In conclusion, further studies need to be performed to address unresolved or controversial problems associated with PTMs regulating mechanisms and potentially unknown PTMs. Identifying new biological functions will not only enrich the theoretical knowledge on PTMs regulating HBV biological processes but also contribute to the identification of effective diagnostic markers and therapeutic targets for diseases caused by HBV infection.
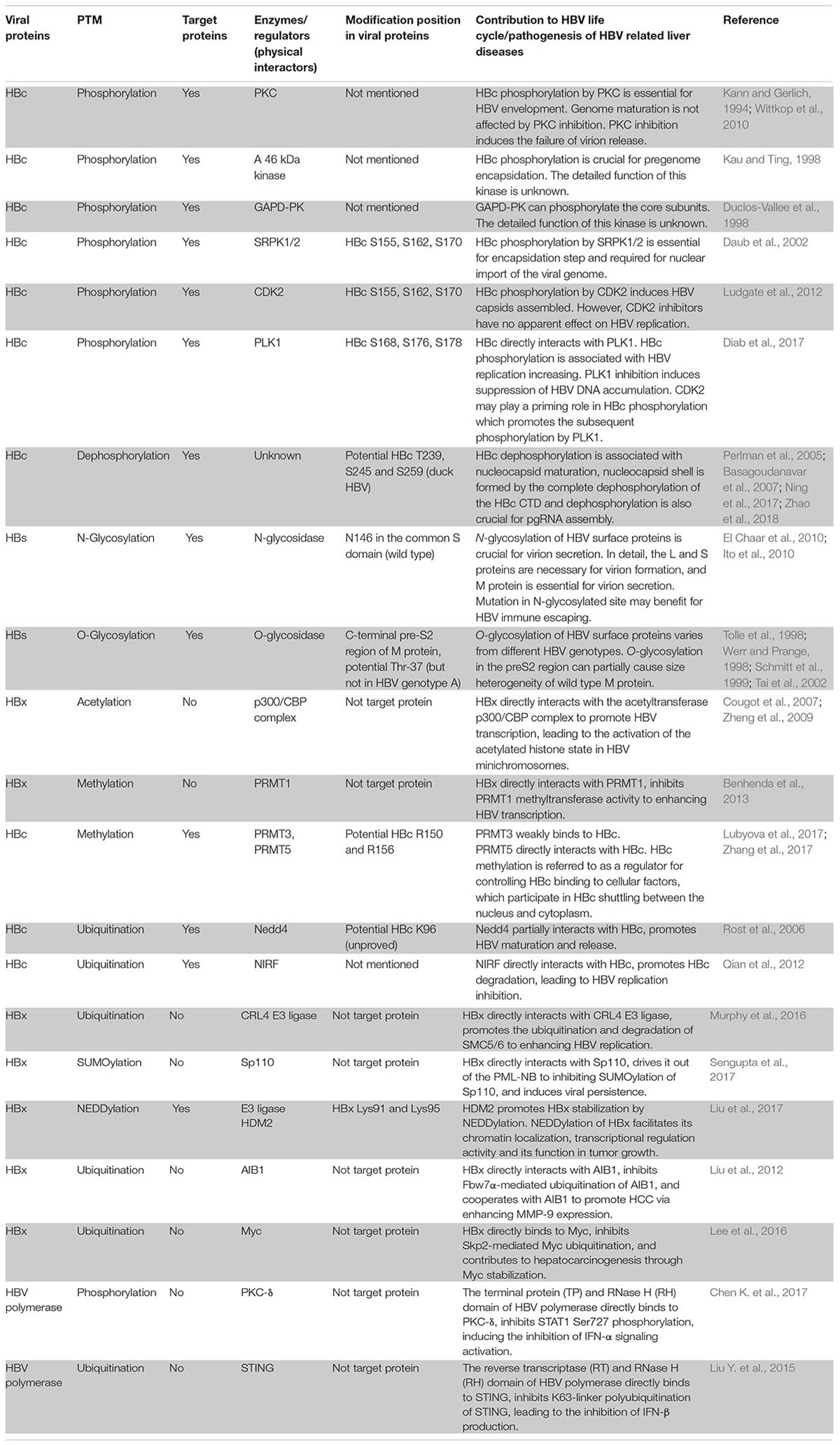
TABLE 1. Viral proteins related post-translational modification (PTM).
Author Contributions
FY wrote the whole manuscript and prepared the table.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81600493).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Angers, S., Li, T., Yi, X., MacCoss, M. J., Moon, R. T., and Zheng, N. (2006). Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593. doi: 10.1038/nature05175
Bai, L., Zhang, W., Tan, L., Yang, H., Ge, M., Zhu, C., et al. (2015). Hepatitis B virus hijacks CTHRC1 to evade host immunity and maintain replication. J. Mol. Cell Biol. 7, 543–556. doi: 10.1093/jmcb/mjv048
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Basagoudanavar, S. H., Perlman, D. H., and Hu, J. (2007). Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J. Virol. 81, 1641–1649. doi: 10.1128/JVI.01671-06
Beck, J., and Nassal, M. (2007). Hepatitis B virus replication. World J. Gastroenterol. 13, 48–64. doi: 10.3748/wjg.v13.i1.48
Belloni, L., Pollicino, T., De Nicola, F., Guerrieri, F., Raffa, G., Fanciulli, M., et al. (2009). Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. U.S.A. 106, 19975–19979. doi: 10.1073/pnas.0908365106
Benhenda, S., Ducroux, A., Riviere, L., Sobhian, B., Ward, M. D., Dion, S., et al. (2013). Methyltransferase PRMT1 is a binding partner of HBx and a negative regulator of hepatitis B virus transcription. J. Virol. 87, 4360–4371. doi: 10.1128/JVI.02574-12
Bi, X., and Tong, S. (2018). Impact of immune escape mutations and N-linked glycosylation on the secretion of hepatitis B virus virions and subviral particles: role of the small envelope protein. Virology 518, 358–368. doi: 10.1016/j.virol.2018.03.011
Binda, O. (2013). On your histone mark, SET, methylate! Epigenetics 8, 457–463. doi: 10.4161/epi.24451
Bing, Y., Zhu, S., Yu, G., Li, T., Liu, W., Li, C., et al. (2014). Glucocorticoid-induced S-adenosylmethionine enhances the interferon signaling pathway by restoring STAT1 protein methylation in hepatitis B virus-infected cells. J. Biol. Chem. 289, 32639–32655. doi: 10.1074/jbc.M114.589689
Bock, C. T., Schwinn, S., Locarnini, S., Fyfe, J., Manns, M. P., Trautwein, C., et al. (2001). Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 307, 183–196. doi: 10.1006/jmbi.2000.4481
Bontron, S., Lin-Marq, N., and Strubin, M. (2002). Hepatitis B virus X protein associated with UV-DDB1 induces cell death in the nucleus and is functionally antagonized by UV-DDB2. J. Biol. Chem. 277, 38847–38854. doi: 10.1074/jbc.M205722200
Chang, P. C., Campbell, M., and Robertson, E. S. (2016). Human oncogenic herpesvirus and post-translational modifications - phosphorylation and SUMOylation. Front. Microbiol. 7:962. doi: 10.3389/fmicb.2016.00962
Chen, J., Wu, M., Zhang, X., Zhang, W., Zhang, Z., Chen, L., et al. (2013). Hepatitis B virus polymerase impairs interferon-alpha-induced STA T activation through inhibition of importin-alpha5 and protein kinase C-delta. Hepatology 57, 470–482. doi: 10.1002/hep.26064
Chen, J., Xu, W., Chen, Y., Xie, X., Zhang, Y., Ma, C., et al. (2017). Matrix metalloproteinase 9 facilitates hepatitis B virus replication through binding with type I interferon (IFN) receptor 1 to repress IFN/JAK/STAT signaling. J. Virol. 91:e01824-16. doi: 10.1128/JVI.01824-16
Chen, K., Liu, J., Liu, S., Xia, M., Zhang, X., Han, D., et al. (2017). Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell 170, 492–506.e14. doi: 10.1016/j.cell.2017.06.042
Chen, S., Yu, X., Lei, Q., Ma, L., and Guo, D. (2013). The sumoylation of zinc-fingers and homeoboxes 1 (ZHX1) by Ubc9 regulates its stability and transcriptional repression activity. J. Cell. Biochem. 114, 2323–2333. doi: 10.1002/jcb.24579
Chiu, C. M., Yeh, S. H., Chen, P. J., Kuo, T. J., Chang, C. J., Chen, P. J., et al. (2007). Hepatitis B virus X protein enhances androgen receptor-responsive gene expression depending on androgen level. Proc. Natl. Acad. Sci. U.S.A. 104, 2571–2578. doi: 10.1073/pnas.0609498104
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. doi: 10.1126/science.1175371
Christen, V., Duong, F., Bernsmeier, C., Sun, D., Nassal, M., and Heim, M. H. (2007). Inhibition of alpha interferon signaling by hepatitis B virus. J. Virol. 81, 159–165. doi: 10.1128/JVI.01292-06
Chu, T. H., Liou, A. T., Su, P. Y., Wu, H. N., and Shih, C. (2014). Nucleic acid chaperone activity associated with the arginine-rich domain of human hepatitis B virus core protein. J. Virol. 88, 2530–2543. doi: 10.1128/JVI.03235-13
Chua, P. K., Tang, F. M., Huang, J. Y., Suen, C. S., and Shih, C. (2010). Testing the balanced electrostatic interaction hypothesis of hepatitis B virus DNA synthesis by using an in vivo charge rebalance approach. J. Virol. 84, 2340–2351. doi: 10.1128/JVI.01666-09
Citro, S., Jaffray, E., Hay, R. T., Seiser, C., and Chiocca, S. (2013). A role for paralog-specific sumoylation in histone deacetylase 1 stability. J. Mol. Cell Biol. 5, 416–427. doi: 10.1093/jmcb/mjt032
Clark, K., Nanda, S., and Cohen, P. (2013). Molecular control of the NEMO family of ubiquitin-binding proteins. Nat. Rev. Mol. Cell Biol. 14, 673–685. doi: 10.1038/nrm3644
Cougot, D., Wu, Y., Cairo, S., Caramel, J., Renard, C. A., Levy, L., et al. (2007). The hepatitis B virus X protein functionally interacts with CREB-binding protein/p300 in the regulation of CREB-mediated transcription. J. Biol. Chem. 282, 4277–4287. doi: 10.1074/jbc.M606774200
Daub, H., Blencke, S., Habenberger, P., Kurtenbach, A., Dennenmoser, J., Wissing, J., et al. (2002). Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J. Virol. 76, 8124–8137. doi: 10.1128/JVI.76.16.8124-8137.2002
Deribe, Y. L., Pawson, T., and Dikic, I. (2010). Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672. doi: 10.1038/nsmb.1842
Diab, A., Foca, A., Fusil, F., Lahlali, T., Jalaguier, P., Amirache, F., et al. (2017). Polo-like-kinase 1 is a proviral host factor for hepatitis B virus replication. Hepatology 66, 1750–1765. doi: 10.1002/hep.29236
Duclos-Vallee, J. C., Capel, F., Mabit, H., and Petit, M. A. (1998). Phosphorylation of the hepatitis B virus core protein by glyceraldehyde-3-phosphate dehydrogenase protein kinase activity. J. Gen. Virol. 79(Pt 7), 1665–1670. doi: 10.1099/0022-1317-79-7-1665
El Chaar, M., Candotti, D., Crowther, R. A., and Allain, J. P. (2010). Impact of hepatitis B virus surface protein mutations on the diagnosis of occult hepatitis B virus infection. Hepatology 52, 1600–1610. doi: 10.1002/hep.23886
Gao, Y., Feng, J., Yang, G., Zhang, S., Liu, Y., Bu, Y., et al. (2017). Hepatitis B virus X protein-elevated MSL2 modulates hepatitis B virus covalently closed circular DNA by inducing degradation of APOBEC3B to enhance hepatocarcinogenesis. Hepatology 66, 1413–1429. doi: 10.1002/hep.29316
Garcia, M. L., Byfield, R., and Robek, M. D. (2009). Hepatitis B virus replication and release are independent of core lysine ubiquitination. J. Virol. 83, 4923–4933. doi: 10.1128/JVI.02644-08
Garcia, T., Li, J., Sureau, C., Ito, K., Qin, Y., Wands, J., et al. (2009). Drastic reduction in the production of subviral particles does not impair hepatitis B virus virion secretion. J. Virol. 83, 11152–11165. doi: 10.1128/JVI.00905-09
Gazina, E. V., Fielding, J. E., Lin, B., and Anderson, D. A. (2000). Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J. Virol. 74, 4721–4728. doi: 10.1128/JVI.74.10.4721-4728.2000
Global Burden of Disease Study 2013 Collaborators (2015). Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386, 743–800.
He, Y. J., McCall, C. M., Hu, J., Zeng, Y., and Xiong, Y. (2006). DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 20, 2949–2954. doi: 10.1101/gad.1483206
Helenius, A., and Aebi, M. (2004). Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi: 10.1146/annurev.biochem.67.1.425
Hu, J., Qiao, M., Chen, Y., Tang, H., Zhang, W., Tang, D., et al. (2018). Cyclin E2-CDK2 mediates SAMHD1 phosphorylation to abrogate its restriction of HBV replication in hepatoma cells. FEBS Lett. 592, 1893–1904. doi: 10.1002/1873-3468.13105
Huang, W. C., Chen, W. S., Chen, Y. J., Wang, L. Y., Hsu, S. C., Chen, C. C., et al. (2012). Hepatitis B virus X protein induces IKKalpha nuclear translocation via Akt-dependent phosphorylation to promote the motility of hepatocarcinoma cells. J. Cell. Physiol. 227, 1446–1454. doi: 10.1002/jcp.22860
Ito, K., Qin, Y., Guarnieri, M., Garcia, T., Kwei, K., Mizokami, M., et al. (2010). Impairment of hepatitis B virus virion secretion by single-amino-acid substitutions in the small envelope protein and rescue by a novel glycosylation site. J. Virol. 84, 12850–12861. doi: 10.1128/JVI.01499-10
Iwata, A., Nagashima, Y., Matsumoto, L., Suzuki, T., Yamanaka, T., Date, H., et al. (2009). Intranuclear degradation of polyglutamine aggregates by the ubiquitin-proteasome system. J. Biol. Chem. 284, 9796–9803. doi: 10.1074/jbc.M809739200
Jung, J., Hwang, S. G., Chwae, Y. J., Park, S., Shin, H. J., and Kim, K. (2014). Phosphoacceptors threonine 162 and serines 170 and 178 within the carboxyl-terminal RRRS/T motif of the hepatitis B virus core protein make multiple contributions to hepatitis B virus replication. J. Virol. 88, 8754–8767. doi: 10.1128/JVI.01343-14
Kann, M., and Gerlich, W. H. (1994). Effect of core protein phosphorylation by protein kinase C on encapsidation of RNA within core particles of hepatitis B virus. J. Virol. 68, 7993–8000.
Kau, J. H., and Ting, L. P. (1998). Phosphorylation of the core protein of hepatitis B virus by a 46-kilodalton serine kinase. J. Virol. 72, 3796–3803.
Khan, M., Syed, G. H., Kim, S. J., and Siddiqui, A. (2016). Hepatitis B virus-induced parkin-dependent recruitment of linear ubiquitin assembly complex (LUBAC) to mitochondria and attenuation of innate immunity. PLoS Pathog. 12:e1005693. doi: 10.1371/journal.ppat.1005693
Kim, W., Lee, S., Son, Y., Ko, C., and Ryu, W. S. (2016). DDB1 stimulates viral transcription of hepatitis B virus via HBx-independent mechanisms. J. Virol. 90, 9644–9653. doi: 10.1128/JVI.00977-16
Koumbi, L., and Karayiannis, P. (2015). The epigenetic control of hepatitis B virus modulates the outcome of infection. Front. Microbiol. 6:1491. doi: 10.3389/fmicb.2015.01491
Kwak, Y. T., Guo, J., Prajapati, S., Park, K. J., Surabhi, R. M., Miller, B., et al. (2003). Methylation of SPT5 regulates its interaction with RNA polymerase II and transcriptional elongation properties. Mol. Cell 11, 1055–1066. doi: 10.1016/S1097-2765(03)00101-1
Lambert, C., and Prange, R. (2007). Posttranslational N-glycosylation of the hepatitis B virus large envelope protein. Virol. J. 4:45. doi: 10.1186/1743-422X-4-45
Lan, Y. T., Li, J., Liao, W., and Ou, J. (1999). Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259, 342–348. doi: 10.1006/viro.1999.9798
Lavanchy, D. (2004). Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 11, 97–107. doi: 10.1046/j.1365-2893.2003.00487.x
Lee, J., Zong, L., Krotow, A., Qin, Y., Jia, L., Zhang, J., et al. (2018). N-linked glycosylation is not essential for sodium taurocholate cotransporting polypeptide to mediate hepatitis b virus infection in vitro. J. Virol. 92:e0732-18. doi: 10.1128/JVI.00732-18
Lee, S., Kim, W., Ko, C., and Ryu, W. S. (2016). Hepatitis B virus X protein enhances Myc stability by inhibiting SCF(Skp2) ubiquitin E3 ligase-mediated Myc ubiquitination and contributes to oncogenesis. Oncogene 35, 1857–1867. doi: 10.1038/onc.2015.251
Levrero, M., Pollicino, T., Petersen, J., Belloni, L., Raimondo, G., and Dandri, M. (2009). Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 51, 581–592. doi: 10.1016/j.jhep.2009.05.022
Lewellyn, E. B., and Loeb, D. D. (2011). The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J. Virol. 85, 1298–1309. doi: 10.1128/JVI.01957-10
Li, C., Lin, C., Cong, X., and Jiang, Y. (2018). PDK1-WNK1 signaling is affected by HBx and involved in the viability and metastasis of hepatic cells. Oncol. Lett. 15, 5940–5946. doi: 10.3892/ol.2018.8001
Liu, J., Sha, M., Wang, Q., Ma, Y., Geng, X., Gao, Y., et al. (2015). Small ubiquitin-related modifier 2/3 interacts with p65 and stabilizes it in the cytoplasm in HBV-associated hepatocellular carcinoma. BMC Cancer 15:675. doi: 10.1186/s12885-015-1665-3
Liu, K., Luckenbaugh, L., Ning, X., Xi, J., and Hu, J. (2018). Multiple roles of core protein linker in hepatitis B virus replication. PLoS Pathog. 14:e1007085. doi: 10.1371/journal.ppat.1007085
Liu, N., Zhang, J., Yang, X., Jiao, T., Zhao, X., Li, W., et al. (2017). HDM2 promotes NEDDylation of hepatitis B virus HBx to enhance its stability and function. J. Virol. 91:e00340-17. doi: 10.1128/JVI.00340-17
Liu, Y., Li, J., Chen, J., Li, Y., Wang, W., Du, X., et al. (2015). Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J. Virol. 89, 2287–2300. doi: 10.1128/JVI.02760-14
Liu, Y., Simsek, E., Norton, P., Sinnathamby, G., Philip, R., Block, T., et al. (2007). The role of the downstream signal sequences in the maturation of the HBV middle surface glycoprotein: development of a novel therapeutic vaccine candidate. Virology 365, 10–19. doi: 10.1016/j.virol.2007.03.042
Liu, Y., Tong, Z., Li, T., Chen, Q., Zhuo, L., Li, W., et al. (2012). Hepatitis B virus X protein stabilizes amplified in breast cancer 1 protein and cooperates with it to promote human hepatocellular carcinoma cell invasiveness. Hepatology 56, 1015–1024. doi: 10.1002/hep.25751
Locarnini, S. A., and Yuen, L. (2010). Molecular genesis of drug-resistant and vaccine-escape HBV mutants. Antivir. Ther. 15(3 Pt B), 451–461. doi: 10.3851/IMP1499
Lubyova, B., Hodek, J., Zabransky, A., Prouzova, H., Hubalek, M., Hirsch, I., et al. (2017). PRMT5: a novel regulator of Hepatitis B virus replication and an arginine methylase of HBV core. PLoS One 12:e0186982. doi: 10.1371/journal.pone.0186982
Ludgate, L., Liu, K., Luckenbaugh, L., Streck, N., Eng, S., Voitenleitner, C., et al. (2016). Cell-free hepatitis B virus capsid assembly dependent on the core protein c-terminal domain and regulated by phosphorylation. J. Virol. 90, 5830–5844. doi: 10.1128/JVI.00394-16
Ludgate, L., Ning, X., Nguyen, D. H., Adams, C., Mentzer, L., and Hu, J. (2012). Cyclin-dependent kinase 2 phosphorylates s/t-p sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J. Virol. 86, 12237–12250. doi: 10.1128/JVI.01218-12
Melegari, M., Wolf, S. K., and Schneider, R. J. (2005). Hepatitis B virus DNA replication is coordinated by core protein serine phosphorylation and HBx expression. J. Virol. 79, 9810–9820. doi: 10.1128/JVI.79.15.9810-9820.2005
Moolla, N., Kew, M., and Arbuthnot, P. (2002). Regulatory elements of hepatitis B virus transcription. J. Viral Hepat. 9, 323–331. doi: 10.1046/j.1365-2893.2002.00381.x
Murphy, C. M., Xu, Y., Li, F., Nio, K., Reszka-Blanco, N., Li, X., et al. (2016). Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. 16, 2846–2854. doi: 10.1016/j.celrep.2016.08.026
Nassal, M. (1992). The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 66, 4107–4116.
Nassal, M. (2015). HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64, 1972–1984. doi: 10.1136/gutjnl-2015-309809
Nicholson, T. B., Chen, T., and Richard, S. (2009). The physiological and pathophysiological role of PRMT1-mediated protein arginine methylation. Pharmacol. Res. 60, 466–474. doi: 10.1016/j.phrs.2009.07.006
Ning, X., Basagoudanavar, S. H., Liu, K., Luckenbaugh, L., Wei, D., Wang, C., et al. (2017). Capsid phosphorylation state and hepadnavirus virion secretion. J. Virol. 91:e00092-17. doi: 10.1128/JVI.00092-17
Nishitsuji, H., Ujino, S., Harada, K., and Shimotohno, K. (2018). TIP60 complex inhibits hepatitis B virus transcription. J. Virol. 92:e01788-17. doi: 10.1128/JVI.01788-17
Palumbo, G. A., Scisciani, C., Pediconi, N., Lupacchini, L., Alfalate, D., Guerrieri, F., et al. (2015). IL6 inhibits HBV transcription by targeting the epigenetic control of the nuclear cccDNA minichromosome. PLoS One 10:e0142599. doi: 10.1371/journal.pone.0142599
Park, E. S., Park, Y. K., Shin, C. Y., Park, S. H., Ahn, S. H., Kim, D. H., et al. (2013). Hepatitis B virus inhibits liver regeneration via epigenetic regulation of urokinase-type plasminogen activator. Hepatology 58, 762–776. doi: 10.1002/hep.26379
Perlman, D. H., Berg, E. A., O’Connor, P. B., Costello, C. E., and Hu, J. (2005). Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc. Natl. Acad. Sci. U.S.A. 102, 9020–9025. doi: 10.1073/pnas.0502138102
Polaris Observatory Collaborators (2018). Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol. Hepatol. 3, 383–403. doi: 10.1016/S2468-1253(18)30056-6
Pollicino, T., Belloni, L., Raffa, G., Pediconi, N., Squadrito, G., Raimondo, G., et al. (2006). Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 130, 823–837. doi: 10.1053/j.gastro.2006.01.001
Qian, G., Jin, F., Chang, L., Yang, Y., Peng, H., and Duan, C. (2012). NIRF, a novel ubiquitin ligase, interacts with hepatitis B virus core protein and promotes its degradation. Biotechnol. Lett. 34, 29–36. doi: 10.1007/s10529-011-0751-0
Ren, J. H., Hu, J. L., Cheng, S. T., Yu, H. B., Wong, V. K. W., Law, B. Y. K., et al. (2018). SIRT3 restricts HBV transcription and replication via epigenetic regulation of cccDNA involving SUV39H1 and SETD1A histone methyltransferases. Hepatology 68, 1260–1276. doi: 10.1002/hep.29912
Riviere, L., Gerossier, L., Ducroux, A., Dion, S., Deng, Q., Michel, M. L., et al. (2015). HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J. Hepatol. 63, 1093–1102. doi: 10.1016/j.jhep.2015.06.023
Rost, M., Mann, S., Lambert, C., Doring, T., Thome, N., and Prange, R. (2006). Gamma-adaptin, a novel ubiquitin-interacting adaptor, and Nedd4 ubiquitin ligase control hepatitis B virus maturation. J. Biol. Chem. 281, 29297–29308. doi: 10.1074/jbc.M603517200
Schmitt, S., Glebe, D., Alving, K., Tolle, T. K., Linder, M., Geyer, H., et al. (1999). Analysis of the pre-S2 N- and O-linked glycans of the M surface protein from human hepatitis B virus. J. Biol. Chem. 274, 11945–11957. doi: 10.1074/jbc.274.17.11945
Schneider, W. M., Chevillotte, M. D., and Rice, C. M. (2014). Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 32, 513–545. doi: 10.1146/annurev-immunol-032713-120231
Seeger, C., and Mason, W. S. (2000). Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64, 51–68. doi: 10.1128/MMBR.64.1.51-68.2000
Selzer, L., Kant, R., Wang, J. C., Bothner, B., and Zlotnick, A. (2015). Hepatitis B virus core protein phosphorylation sites affect capsid stability and transient exposure of the c-terminal domain. J. Biol. Chem. 290, 28584–28593. doi: 10.1074/jbc.M115.678441
Sengupta, I., Das, D., Singh, S. P., Chakravarty, R., and Das, C. (2017). Host transcription factor speckled 110 kDa (Sp110), a nuclear body protein, is hijacked by hepatitis B virus protein X for viral persistence. J. Biol. Chem. 292, 20379–20393. doi: 10.1074/jbc.M117.796839
Sitterlin, D., Bergametti, F., and Transy, C. (2000). UVDDB p127-binding modulates activities and intracellular distribution of hepatitis B virus X protein. Oncogene 19, 4417–4426. doi: 10.1038/sj.onc.1203771
Sitterlin, D., Lee, T. H., Prigent, S., Tiollais, P., Butel, J. S., and Transy, C. (1997). Interaction of the UV-damaged DNA-binding protein with hepatitis B virus X protein is conserved among mammalian hepadnaviruses and restricted to transactivation-proficient X-insertion mutants. J. Virol. 71, 6194–6199.
Spiro, R. G. (2002). Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12, 43R–56R. doi: 10.1093/glycob/12.4.43R
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi: 10.1038/47412
Su, P. Y., Yang, C. J., Chu, T. H., Chang, C. H., Chiang, C., Tang, F. M., et al. (2016). HBV maintains electrostatic homeostasis by modulating negative charges from phosphoserine and encapsidated nucleic acids. Sci. Rep. 6:38959. doi: 10.1038/srep38959
Tai, P. C., Suk, F. M., Gerlich, W. H., Neurath, A. R., and Shih, C. (2002). Hypermodification and immune escape of an internally deleted middle-envelope (M) protein of frequent and predominant hepatitis B virus variants. Virology 292, 44–58. doi: 10.1006/viro.2001.1239
Takamatsu, S., Shimomura, M., Kamada, Y., Maeda, H., Sobajima, T., Hikita, H., et al. (2016). Core-fucosylation plays a pivotal role in hepatitis B pseudo virus infection: a possible implication for HBV glycotherapy. Glycobiology 26, 1180–1189. doi: 10.1093/glycob/cww067
Tolle, T. K., Glebe, D., Linder, M., Linder, D., Schmitt, S., Geyer, R., et al. (1998). Structure and glycosylation patterns of surface proteins from woodchuck hepatitis virus. J. Virol. 72, 9978–9985.
Torres, H. A., and Davila, M. (2012). Reactivation of hepatitis B virus and hepatitis C virus in patients with cancer. Nat. Rev. Clin. Oncol. 9, 156–166. doi: 10.1038/nrclinonc.2012.1
Tsuchida, T., Zou, J., Saitoh, T., Kumar, H., Abe, T., Matsuura, Y., et al. (2010). The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776. doi: 10.1016/j.immuni.2010.10.013
Vigerust, D. J., and Shepherd, V. L. (2007). Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 15, 211–218. doi: 10.1016/j.tim.2007.03.003
Wan, Y., Cao, W., Han, T., Ren, S., Feng, J., Chen, T., et al. (2017). Inducible Rubicon facilitates viral replication by antagonizing interferon production. Cell. Mol. Immunol. 14, 607–620. doi: 10.1038/cmi.2017.1
Wang, H., An, W., Cao, R., Xia, L., Erdjument-Bromage, H., Chatton, B., et al. (2003). mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol. Cell 12, 475–487. doi: 10.1016/j.molcel.2003.08.007
Werr, M., and Prange, R. (1998). Role for calnexin and N-linked glycosylation in the assembly and secretion of hepatitis B virus middle envelope protein particles. J. Virol. 72, 778–782.
Wittkop, L., Schwarz, A., Cassany, A., Grun-Bernhard, S., Delaleau, M., Rabe, B., et al. (2010). Inhibition of protein kinase C phosphorylation of hepatitis B virus capsids inhibits virion formation and causes intracellular capsid accumulation. Cell. Microbiol. 12, 962–975. doi: 10.1111/j.1462-5822.2010.01444.x
Wu, W., Yan, H., Zhao, H., Sun, W., Yang, Q., Sheng, J., et al. (2018). Characteristics of systemic inflammation in hepatitis B-precipitated ACLF: differentiate it from No-ACLF. Liver Int. 38, 248–257. doi: 10.1111/liv.13504
Yang, S. T., Yen, C. J., Lai, C. H., Lin, Y. J., Chang, K. C., Lee, J. C., et al. (2013). SUMOylated CPAP is required for IKK-mediated NF-kappaB activation and enhances HBx-induced NF-kappaB signaling in HCC. J. Hepatol. 58, 1157–1164. doi: 10.1016/j.jhep.2013.01.025
Yang, W. J., Chang, C. J., Yeh, S. H., Lin, W. H., Wang, S. H., Tsai, T. F., et al. (2009). Hepatitis B virus X protein enhances the transcriptional activity of the androgen receptor through c-Src and glycogen synthase kinase-3beta kinase pathways. Hepatology 49, 1515–1524. doi: 10.1002/hep.22833
Yu, M., and Summers, J. (1991). A domain of the hepadnavirus capsid protein is specifically required for DNA maturation and virus assembly. J. Virol. 65, 2511–2517.
Yu, Z., Gao, Y. Q., Feng, H., Lee, Y. Y., Li, M. S., Tian, Y., et al. (2014). Cell cycle-related kinase mediates viral-host signalling to promote hepatitis B virus-associated hepatocarcinogenesis. Gut 63, 1793–1804. doi: 10.1136/gutjnl-2013-305584
Zhang, H., Xing, Z., Mani, S. K., Bancel, B., Durantel, D., Zoulim, F., et al. (2016). RNA helicase DEAD box protein 5 regulates Polycomb repressive complex 2/Hox transcript antisense intergenic RNA function in hepatitis B virus infection and hepatocarcinogenesis. Hepatology 64, 1033–1048. doi: 10.1002/hep.28698
Zhang, J., Hu, M. M., Wang, Y. Y., and Shu, H. B. (2012). TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 287, 28646–28655. doi: 10.1074/jbc.M112.362608
Zhang, W., Chen, J., Wu, M., Zhang, X., Zhang, M., Yue, L., et al. (2017). PRMT5 restricts hepatitis B virus replication through epigenetic repression of covalently closed circular DNA transcription and interference with pregenomic RNA encapsidation. Hepatology 66, 398–415. doi: 10.1002/hep.29133
Zhang, X., Hou, J., and Lu, M. (2013). Regulation of hepatitis B virus replication by epigenetic mechanisms and microRNAs. Front. Genet. 4:202. doi: 10.3389/fgene.2013.00202
Zhao, Q., Hu, Z., Cheng, J., Wu, S., Luo, Y., Chang, J., et al. (2018). Hepatitis B virus core protein dephosphorylation occurs during pregenomic RNA encapsidation. J. Virol. 92:e02139-17. doi: 10.1128/JVI.02139-17
Zhao, X. (2018). SUMO-mediated regulation of nuclear functions and signaling processes. Mol. Cell 71, 409–418. doi: 10.1016/j.molcel.2018.07.027
Zheng, D. L., Zhang, L., Cheng, N., Xu, X., Deng, Q., Teng, X. M., et al. (2009). Epigenetic modification induced by hepatitis B virus X protein via interaction with de novo DNA methyltransferase DNMT3A. J. Hepatol. 50, 377–387. doi: 10.1016/j.jhep.2008.10.019
Zheng, Y., Fu, X. D., and Ou, J. H. (2005). Suppression of hepatitis B virus replication by SRPK1 and SRPK2 via a pathway independent of the phosphorylation of the viral core protein. Virology 342, 150–158. doi: 10.1016/j.virol.2005.07.030
Keywords: HBV, post-translational modifications, cccDNA, replication, transcription, hepatocarcinogenesis
Citation: Yang F (2018) Post-translational Modification Control of HBV Biological Processes. Front. Microbiol. 9:2661. doi: 10.3389/fmicb.2018.02661
Received: 14 August 2018; Accepted: 18 October 2018;
Published: 01 November 2018.
Edited by:
Akio Adachi, Kansai Medical University, JapanReviewed by:
Masaya Sugiyama, National Center for Global Health and Medicine, JapanKohji Moriishi, University of Yamanashi, Japan
Copyright © 2018 Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fan Yang, emp1eWFuZ2ZhbkB6anUuZWR1LmNu