 Rubén Díaz1*
Rubén Díaz1* Gerardo Díaz-Godínez1
Gerardo Díaz-Godínez1 Miguel Angel Anducho-Reyes2
Miguel Angel Anducho-Reyes2 Yuridia Mercado-Flores2
Yuridia Mercado-Flores2 Leonardo David Herrera-Zúñiga3,4*
Leonardo David Herrera-Zúñiga3,4*- 1Laboratory of Biotechnology, Research Center for Biological Sciences, Autonomous University of Tlaxcala, Tlaxcala, Mexico
- 2Agrobiotechnology Laboratory, Polytechnic University of Pachuca, Hidalgo, Mexico
- 3Division of Environmental Engineering Technology of Higher Studies of East Mexico State, Mexico City, Mexico
- 4Area of Biophysical Chemistry, Department of Chemistry, Metropolitan Autonomous University-Iztapalapa, Mexico City, Mexico
Fungal laccase enzymes have a great biotechnological potential for bioremediation processes due to their ability to degrade compounds such as ρ-diphenol, aminophenols, polyphenols, polyamines, and aryldiamines. These enzymes have activity at different pH and temperature values, however, high temperatures can cause partial or total loss of enzymatic activity, so it is appropriate to do research to modify their secondary and/or tertiary structure to make them more resistant to extreme temperature conditions. In silico, a structure of the Lacc 6 enzyme of Pleurotus ostreatus was constructed using a laccase of Trametes versicolor as a template. From this structure, 16 mutants with possible resistance at high temperature due to ionic interactions, salt bridges and disulfide bonds were also obtained in silico. It was determined that 12 mutants called 4-DB, 3-DB, D233C-T310C, F468P, 3-SB, L132T, N79D, N372D, P203C, P203V, T147E, and W85F, presented the lowest thermodynamic energy. Based on the previous criterion and determining the least flexibility in the protein structures, three mutants (4-DB, 3-DB, and P203C) were selected, which may present high stability at high temperatures without affecting their active site. The obtained results allow the understanding of the molecular base that increase the structural stability of the enzyme Lacc 6 of Pleurotus ostreatus, achieving the in silico generation of mutants, which could have activity at high temperatures.
Introduction
One of the greatest qualities studied and demanded by protein engineering in the environmental and biotechnological fields is the search for conformational and structural stability of proteins when subjected to high temperatures, due to the fact that a high percentage of industrial processes are carried out in such conditions.
The native structure of a protein is stabilized by physicochemical interactions (Djikaev and Ruckenstein, 2008; Das et al., 2018); nevertheless, the free energy between the folded and unfolded states has been estimated in an order of 5–20 kcal/mol, which would be equivalent to breaking a small number of such interactions (Venkataramani et al., 2013; Nawaz et al., 2015; Dubey et al., 2017), in this way, structural stability turns out to be a delicate balance of interactions. Proteins can lose their stability and be denatured by various physicochemical treatments such as: heat, pressure, pH, urea or guanidine chloride, among others (Govardhan, 1999). When the bonds that maintain the native structure of a protein are broken, most of the time it loses its functionality due to structural destabilization. One of the processes of protein denaturation most studied is by heat, which occurs in a relatively narrow temperature range. That is why there are multiple proposals to increase their structural resistance at high temperatures (Gao et al., 2016; Kumar et al., 2016; Kean et al., 2017). An example is the hydrophobic packaging where the amino acid residues are oriented toward the protein core, being the main stability factors (Reed et al., 2013; Luna-Martínez et al., 2016); It should be mentioned that studies on directed mutations carried out for the filling of hydrophobic cavities have resulted in an improvement of stability by hydrophobic packaging (Christensen and Kepp, 2013a; Oda and Kinoshita, 2015). Likewise, electrostatic interactions that occur between amino acids of opposite charge, also have great importance to maintain the stability of the conformation of the protein (Sohini and Srikanta, 2009; Christensen and Kepp, 2013a).
In studies on protein flexibility, it has been found that mutations through which conformational degrees of freedom are reduced confer structural resistance to proteins (Zhou et al., 2008; Gribenko et al., 2009). A type of covalent bond that allows the reduction of flexibility is formed by the union of two cysteines, called disulfide bridge and is considered as a primary factor for stability (Radestock and Gohlke, 2011). However, the success in stabilization by disulfide bridges in protein engineering is dependent on the conformational change caused by the themselves folding and the effects of substituting the native residues with cysteines (Pappenberger et al., 1997). Other tactics to reduce flexibility is the substitution of free chain amino acids for prolines and the shortening in the length of loops, which can increase the stability of proteins by compaction. Although today part of the knowledge about flexibility, mobility and protein dynamics are derived from experimental data, it is true that currently there is no experimental technique to monitor conformational changes at the molecular level and atomic resolution as a function of time. For this reason, the molecular, conformational, flexibility and mobility details are often studied by computational simulation techniques providing a possibility to obtain dynamic information on the functionality of proteins (Anbar et al., 2012; Yakimov et al., 2016).
On the other hand, the laccase enzymes (EC.1.10.3.2) that belong to the family of multicopper-oxidases, can oxidize numerous compounds with an oxide-reduction potential that varies between 500 and 800 mV, such as the ρ- diphenol, aminophenols, polyphenols, polyamines and aryl diamines (Xu, 2005). They are N-glycosylated proteins (10–45% of their molecular mass), in general, they are in monomeric form with molecular weight of 20–80 kDa, their pI is between 2.6 and 4.5, being biologically active at pH 2.0–8.5 (Wesenberg et al., 2003; Díaz et al., 2013). These enzymes have been described in bacteria, fungi, plants and insects and more than 100 different proteins have been identified (Shuresh-Kumar et al., 2003). Its physiological function in plants is in the lignification process and in fungi in morphogenesis processes (formation of spores, pigments of fruiting bodies), pathogenesis, virulence and degradation of lignin, but it has also been reported that in some insects act in the formation of cuticle in the process of sclerotization (Kramer et al., 2001) and in bacteria have the function of monomer cross-linking, polymer degradation and the breakdown of aromatic rings (Sharma et al., 2007). The laccases have a great biotechnological potential, for example in the whitening of paper pulp, in the stabilization of wines and beers, in the food industry, in the cosmetic and pharmaceutical industries and in various bioremediation processes including discoloration of textile dyes and degradation of xenobiotic compounds (Mayer and Staples, 2002).
The laccase enzyme Lacc 6 produced by Pleurotus ostreatus has shown a high level of expression under different development conditions and has been partially characterized, determining its molecular weight, pI, optimum pH of activity, Km on different substrates, etc. (Baldrian, 2006; Díaz et al., 2013), because of the above, it was considered to be modified through protein engineering obtaining in silico mutants with greater thermodynamic stability, for its possible application in the degradation of polluting compounds of phenolic origin and recalcitrants that are in extreme temperature conditions.
In this research, a structure of the Lacc 6 enzyme of Pleurotus ostreatus was constructed in silico, using a laccase of Trametes versicolor as a template. From this structure, 16 mutants with possible resistance at high temperature due to ionic interactions, salt bridges and disulfide bonds were also obtained in silico.
Materials and Methods
Modeling of Lacc 6 of Pleurotus ostreatus
The initial alignment was made using ClustalO1 and the secondary structure prediction with Predictprotein program2. The template structure to Lacc 6 model with the highest degree of homology, was search (non-redundant) in BLAST server (Basic Local Alignment Search Tool) through PDB3 (Boratyn et al., 2012). The structural model of the native Lacc 6 laccase from Pleurotus ostreatus was carried out using the Modeller computer package4 (Webb and Sali, 2016).
Preparation of Mutant Proteins of Lacc 6
The thermostable Lacc 6 quimeric protein was determinated by two ways: 1) making an exhaustive literature search to find all laccases with t1/2 over 55°C (Hildén et al., 2009) to structure multiple models of them whith Modweb-ModBase server5 to find the invariant structural motif-interaccions, and 2) finding the most favorable and thermodynamic residue shift in Lacc 6 by PopMusic server6 (Dehouck et al., 2011), each of any shitf were constructed using UCSF CHIMERA7 with SWAPAA algorithm that performs a side chain replacement by rotamers search selecting the lowest score and shocks between the new side chain and chains of its neighborhood (Fersht et al., 1986).
Minimization of Thermodynamic Energy of Lacc 6 Mutants
Once the mutants were built, they were minimized by the steepest descent protocol up to 0.01 kcal/mol, with the NAMD 2.12 package (Scalable Molecular Dynamics) (Phillips et al., 2005), using the CHARMM36 (Chemistry at Harvard Macromolecular Mechanics) force field (Huang et al., 2017), it was performed with explicit solvent at a physiological sodium chloride (NaCl) ion concentration of 0.15 M, the parameters for the copper atoms were taken from Ungar et al. (1997).
Normal Mode Analysis
After the minimization, the structures with the lowest energy, these assumed as the most stable energetically (thermostable mutants) (Burkoff et al., 2012), to later be studied through by the Normal Modes theory with the elNémo server, available at http://www.sciences.univ-nantes.fr/elnemo/ (Suhre and Sanejouand, 2004). The thermostable mutants were always contrasted with Lacc 6 throughout all the work, and all images were constructed using Visual Molecular Dynamics (VMD) Sofware (Humphrey et al., 1996).
Results
The Lacc 6 sequence of Pleurotus ostreatus shown below (UniProtKB: A0A067NQH1_PLEOS), which is constituted by 533 amino acids, where the first 21 residues correspond to the signal peptide.
The secondary structure prediction of Lacc 6 was performed in Predictprotein program8 (Yachdav et al., 2014). The initial alignment was performed using 142 laccase sequences from Trametes using the ClustalO program8 (Sievers et al., 2011) and the best homologous structure obtained with the BLAST server was the laccase of Trametes versicolor 1GYC (Protein Data Bank, PBD), it is worth mentioning that the alignment was consistent with the template structure and the active sites were perfectly aligned between them. Consequently, 1GYC structure was used as a template to make the model of Lacc 6, the homology between its sequence and 1GYC was close to 62% (UniProt Consortium 2012; Coluzza, 2017). The model of Lacc 6 (Figure 1) was built through Modeller computer package with a DOPE score of −1.88. On the other hand, the Lacc 6 model and the prediction of its secondary structure were highly related. The motifs of the Lacc 6 model were characterized by the PDBsum program, by which it was determined that the scaffolding of Lacc 6 consists of: 7 β-sheets, 9 β-hairpin, 8 β-bulge, 28 β-sheets, 10 α-helices (2 of them with helix-helix interactions), 44 β-turns, 8 γ-turns, and 2 disulfide bridges. Once the model was constructed and verified, it was proceeded to be minimized with the purpose of eliminating the steric shocks.
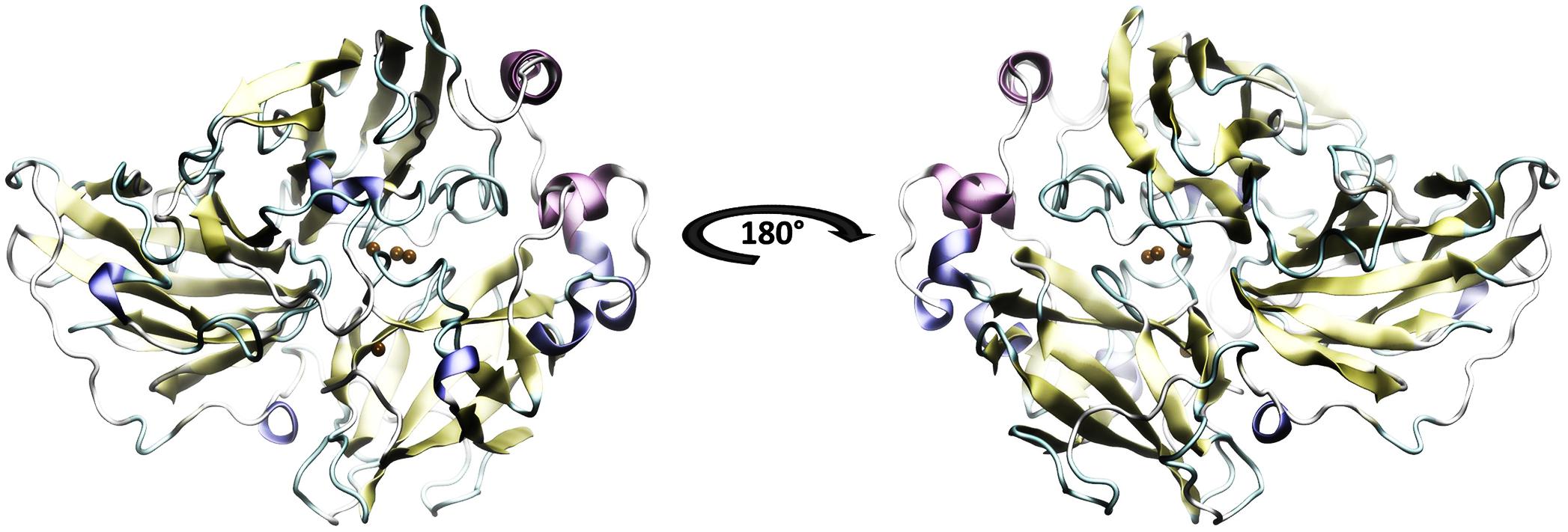
FIGURE 1. Model of the laccase enzyme of Pleurotus ostreatus. In color-coded the secondary structure is presented: yellow, beta sheets; purple and blue, alpha helix; in green and white, loops and non-structured areas. In ocher color the copper atoms belonging to the active site are represented. The 180 degrees rotation allows to appreciate the three structural domains which are defined by Domain A of amino acids 1–131, Domain B of amino acids 132–301 and Domain C the last 194 amino acids.
To generate a thermostable mutant of Lacc 6 according to structural evolution, an exhaustive search of literature on laccases with thermal resistance was carried out (Pezeshgi-Modarres et al., 2016; Coluzza, 2017), of which almost all those found were modeled and reported in Table 1.

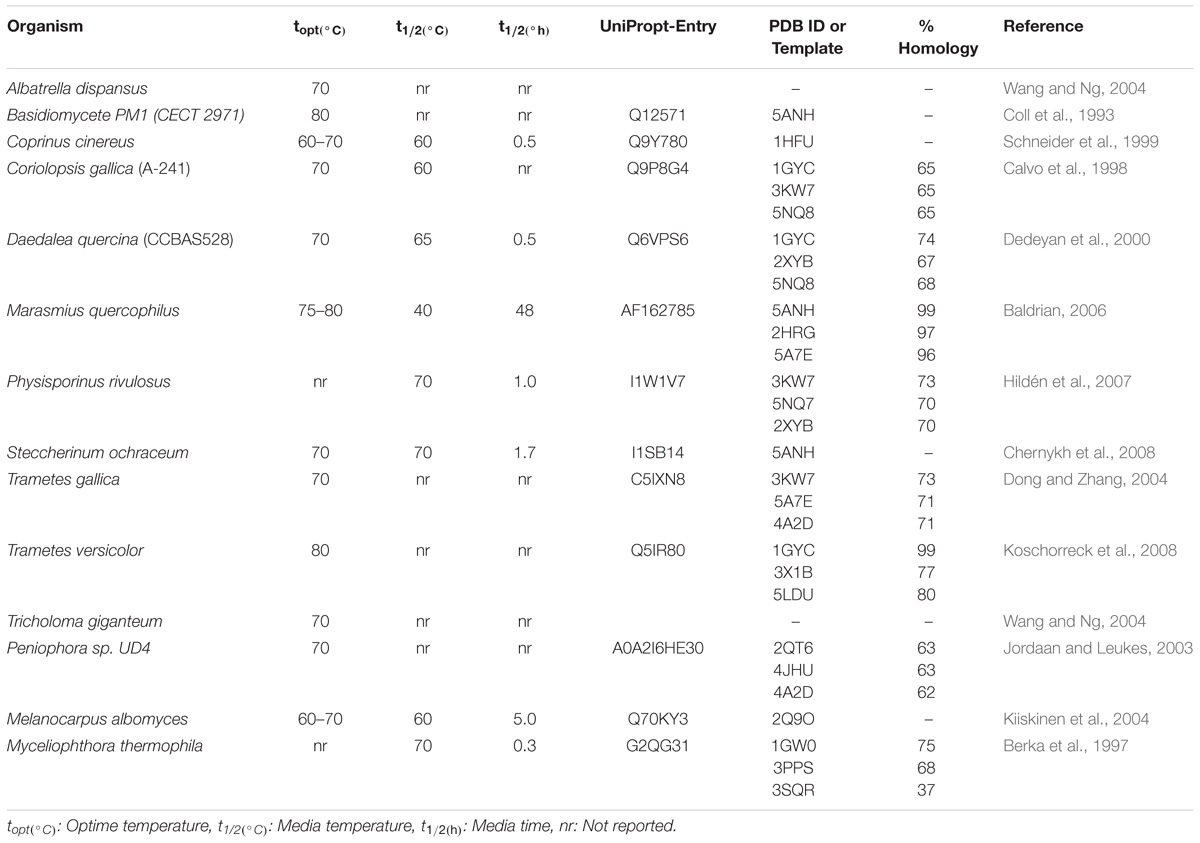
TABLE 1. Catalytic properties at high temperatures of laccase enzymes and accessions data.
Once discovered the thermostable laccases and built their models, this type of proteins was studied with the purpose to find their structural motifs invariants such as salt bridges, ionic interactions and disulfide bonds (Fersht et al., 1986). The structural invariant interactions between Lacc 6 and models or structures of thermostable laccases can be see in Table 2. Location of each mutation was obtained by Lacc 6 model overlap whit any thermostable laccases. Were determined structurally the networks of conserved electrostatic interactions of thermal laccases (Table 2, Columns 2 and 3).
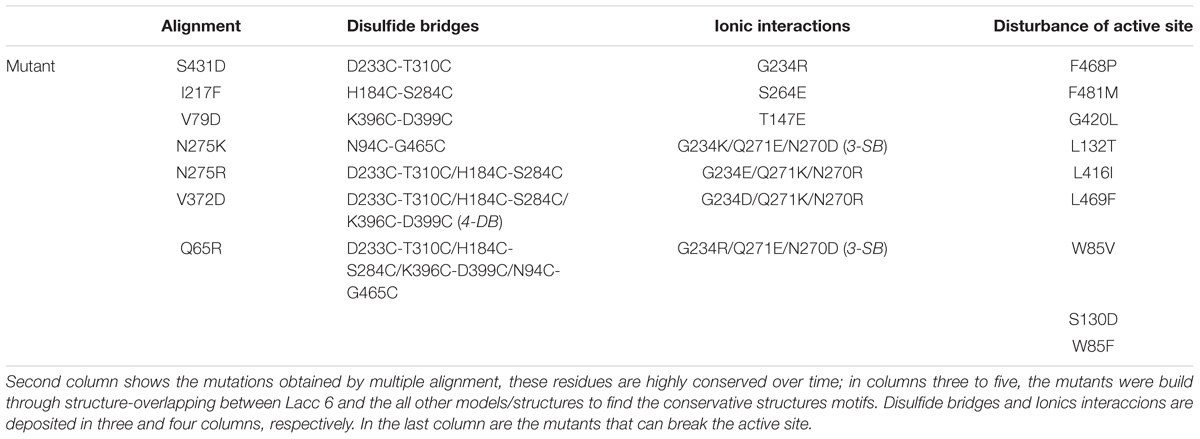
TABLE 2. Mutants with potential to confer thermal stability or disturbance the active site of the enzyme Lacc 6.
By another way, the PoPMuSic hot points (most favorable thermodynamic residue shift) with the ability to confer structural stability to Lacc 6 at high temperatures can be see in Table 3.
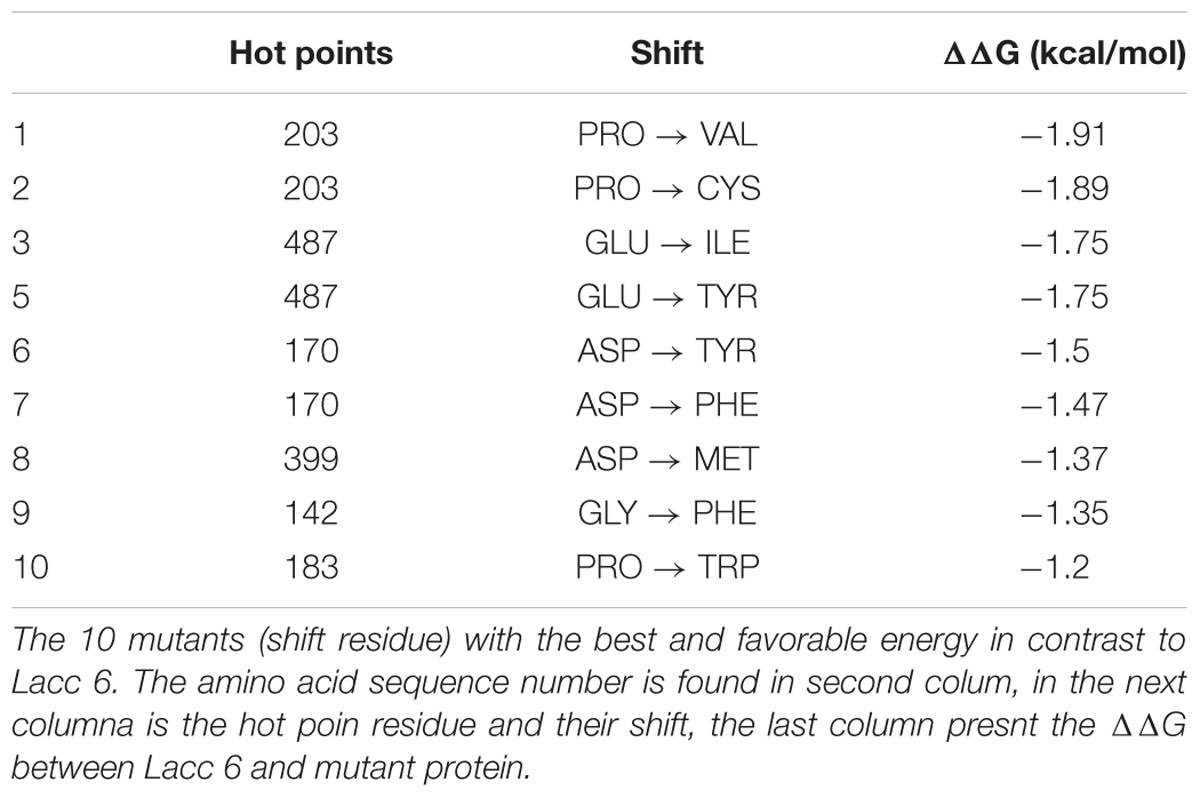
TABLE 3. Main mutations obtained by PoPMuSiC program.
Table 3 contains the shift residue information, the evolutionary invariants with high capacity of confer thermoresistant in Lacc 6 enzyme sequences are reflected on this. The alignment (Figure 2) of the 16 structures, created by WebLogo program10, capture the conservation propensity of each mutate residue set in column 3, with this approach we can see that the best single mutant is P203V showing a ΔΔG = −1.91 kcal/mol below the Lacc 6.
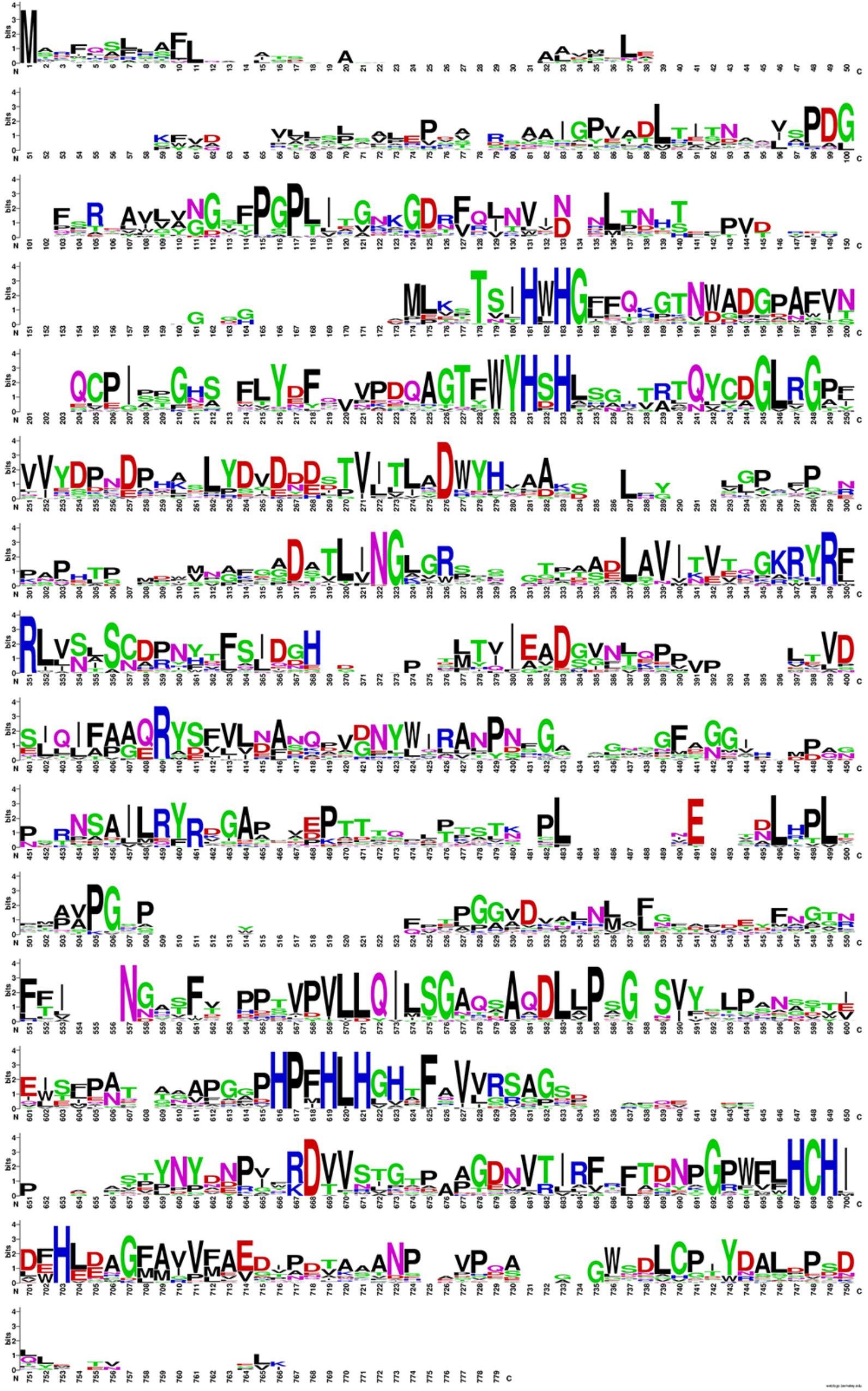
FIGURE 2. Propensity diagram for the alignment of the 16 thermoresistant sequences. In blue color the conservation of Histidines that conform the active site for this class of enzyme is shown, the positions of greater conservation (greater amplitude) generate clues of evolutionary invariants and important for the structural stability of these enzymes.
Based on the Tables 2, 3, a mutant energy minimization was performed in each one, to define the most suitable shift that could confer structural resistance to Lacc 6 (Liwo et al., 2002; Nakagawa and Peyrard, 2006). The mutant energy ponderation on the Table 4 shows that 4-DB, 3-DB, and D233C-T310C (highlighted in gray), are the most favorable energy mutants with the ability to confer structural thermal-resistance. In this singular way, was appreciate that the best designed laccases by the evolution are those with a disulfide bridges networks such as the mutants mentioned above.
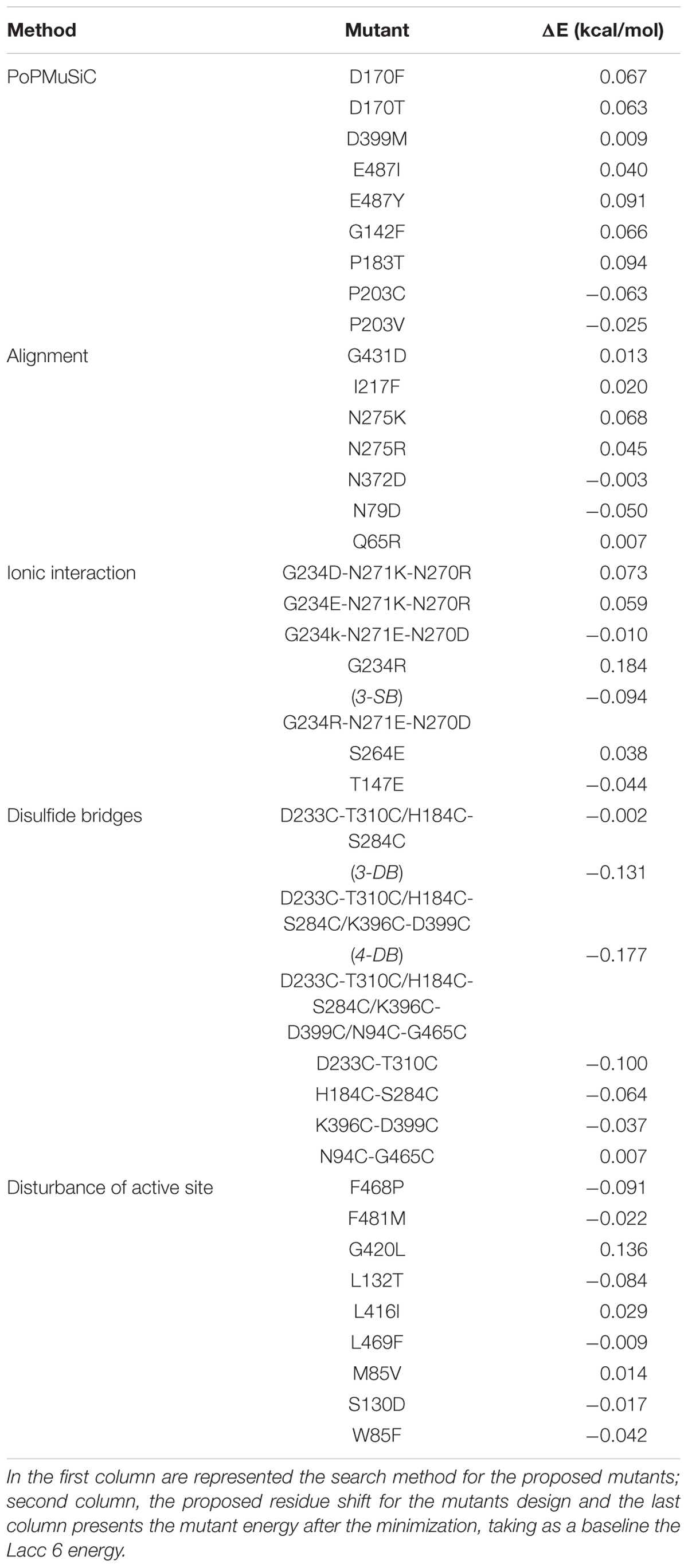
TABLE 4. Energy difference between the native enzyme and their mutants.
Once the energy differences were quantified and analyzed, was continued with a Normal Modes study of the thermostable mutants (4-DB, 3-DB, D233C-T310C, 3-SB, F468P, L132T, and others). The intention of Normal Modes Study is know the collective and correlated movements of the lower energy mutant modes (Tama and Sanejouand, 2001). The modes emitted by the ElNemo program include six trivial zero order frequencies. This study focused on mode 7, corresponds to the protein internal movements identified in direction of maximum movement; 10 snapshots of any structure (referring to the minimized initial structure) were obtained to display and represent the mode dynamics. The difference in the mean squared value of the normalized displacement (R2) of each alpha carbon (Cα) is plotted as a function of the number of residues, where for all residues, significantly mobile regions are seen above and still below the line in the graph of Figure 3.
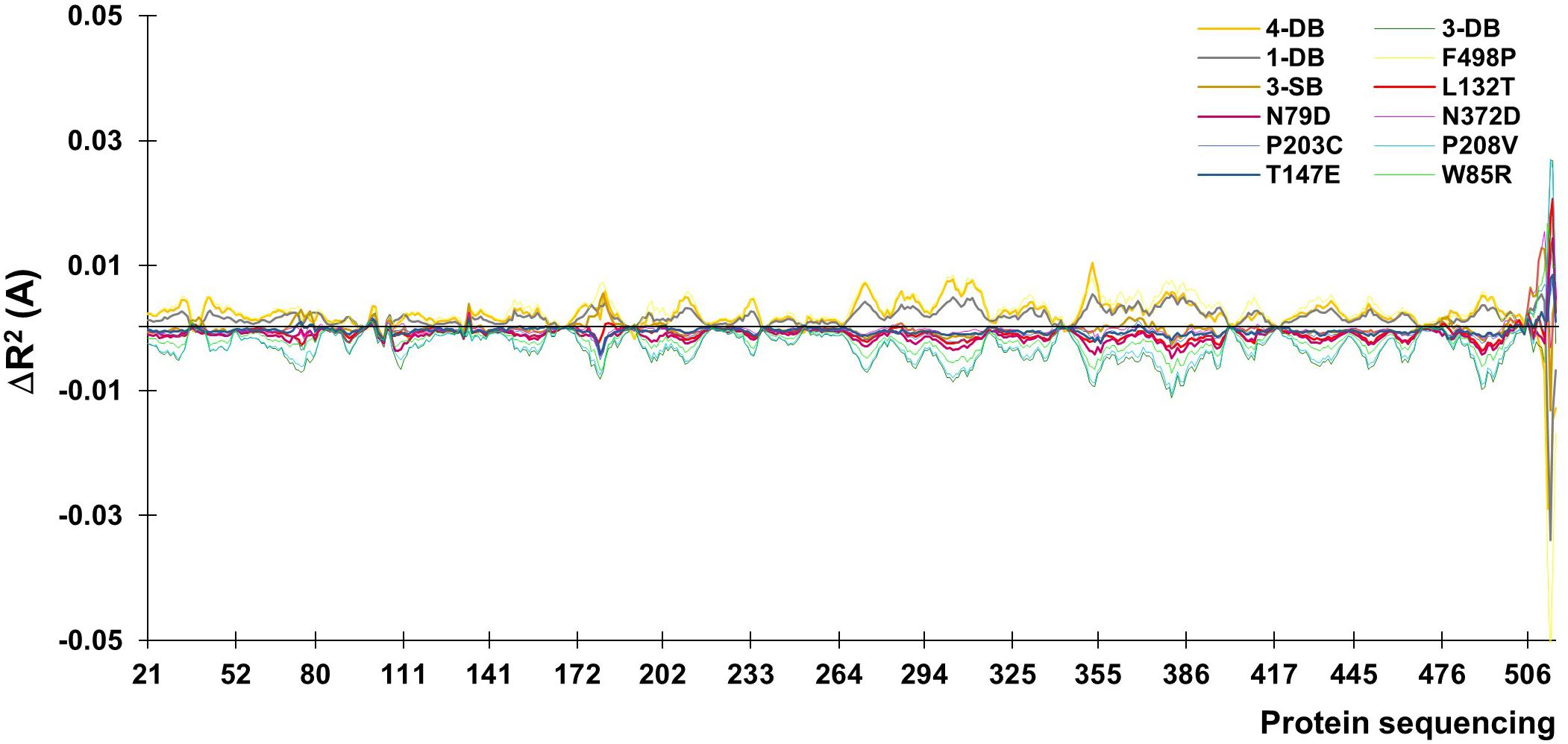
FIGURE 3. Values of mean displacement for the difference between the mutated enzyme and its native counterpart.
The movements of the Cα for normal mode 7 are plotted as a function of the number of residues. The residue with the least displacement appears with a minimum value of R2 below the line at the origin. The higher amplitude values are due to the zones near to the cavity and to the terminal helix, the latter usually being a meaningless section since it is usually an artifact of the software itself, it is usually eliminated before calculations (Altschul et al., 1990).
At a higher energy as shown in Table 4, but still moderate, with a change in the higher R2 for the 4-DB mutant, it exhibits vibrational movements and a flexibility more intense compared to Lacc 6, on the other hand, the 3-DB mutant also shows an even more moderate energy decrease than 4-DB, and also leads to a reduction in the change of R2, becoming a more rigid and less fluctuating structure as shown in Figure 4. The results show that for most of the mutants in which the change in the neighborhoods of the active site was made, it was disturbed as shown in Figure 5, where the L132T mutant is observed. These results indicate that most of the Lacc 6 regions and their mutants behave as a coupled structure that is mutually flexible or rigid, regardless of the energy calculated by the minimization. Inside the protein, displacement is close to 0. At the ends was observed a projection of displacement in the neighborhoods of the mutations; Left upper panel, D233C/T310C mutation; Right upper panel, K396C/D399C mutation and lower right panel, H184C/S284C mutation. On the mode 7, the native protein has greater variation compared to the mutant protein (Figure 6).
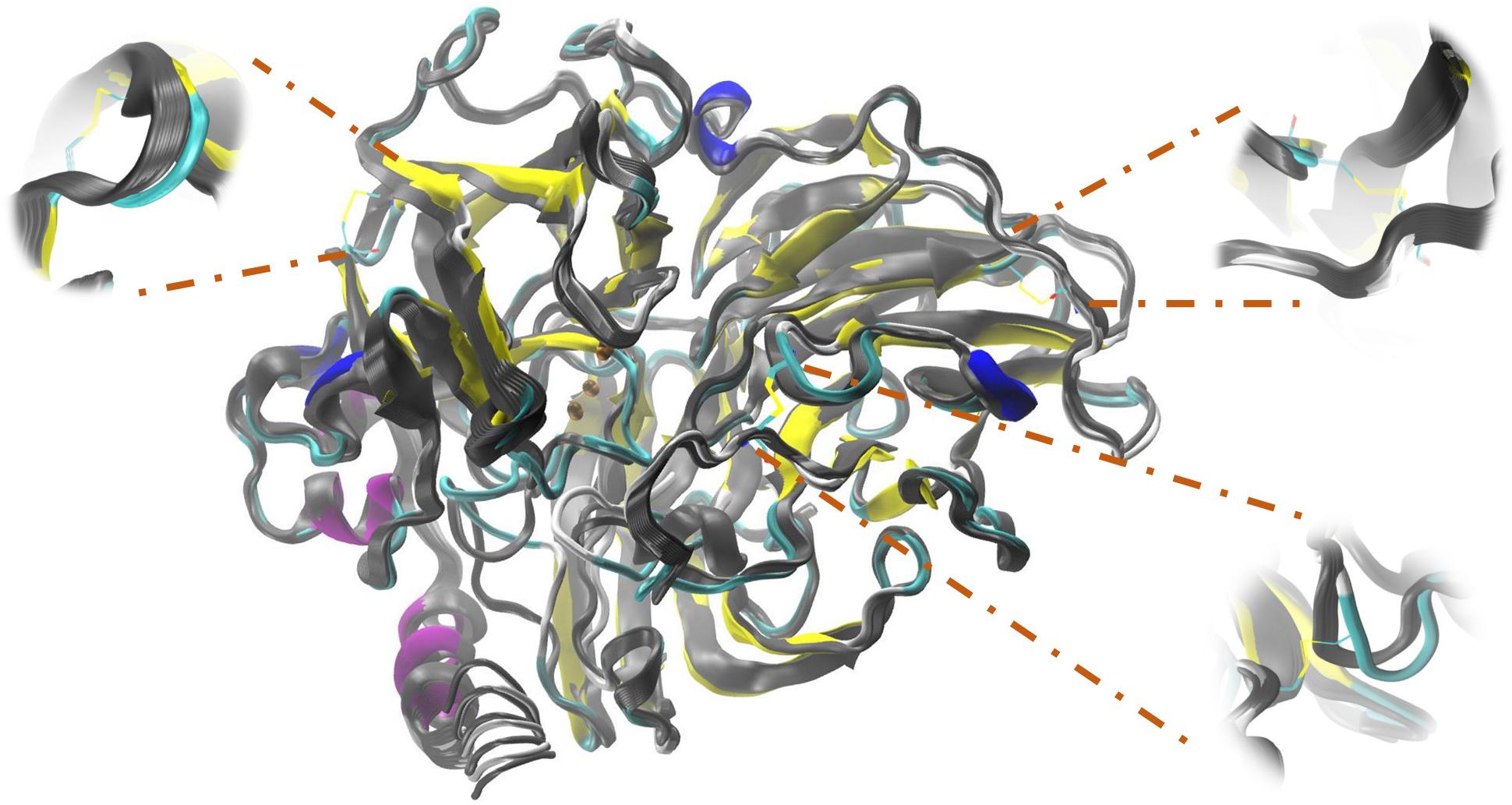
FIGURE 4. Simulation performed in ElNemo of low frequency movements for Lacc 6 and mutant 3-DB. An overlap of the 10 snapshots calculated within mode 7 of the mutated enzyme (in colors according to its secondary structure) on the native enzyme (gray scale), the low/high frequency of the structure indicated by the movement of the Cα which suffer a displacement of between 1.00 and 1.20 Å.
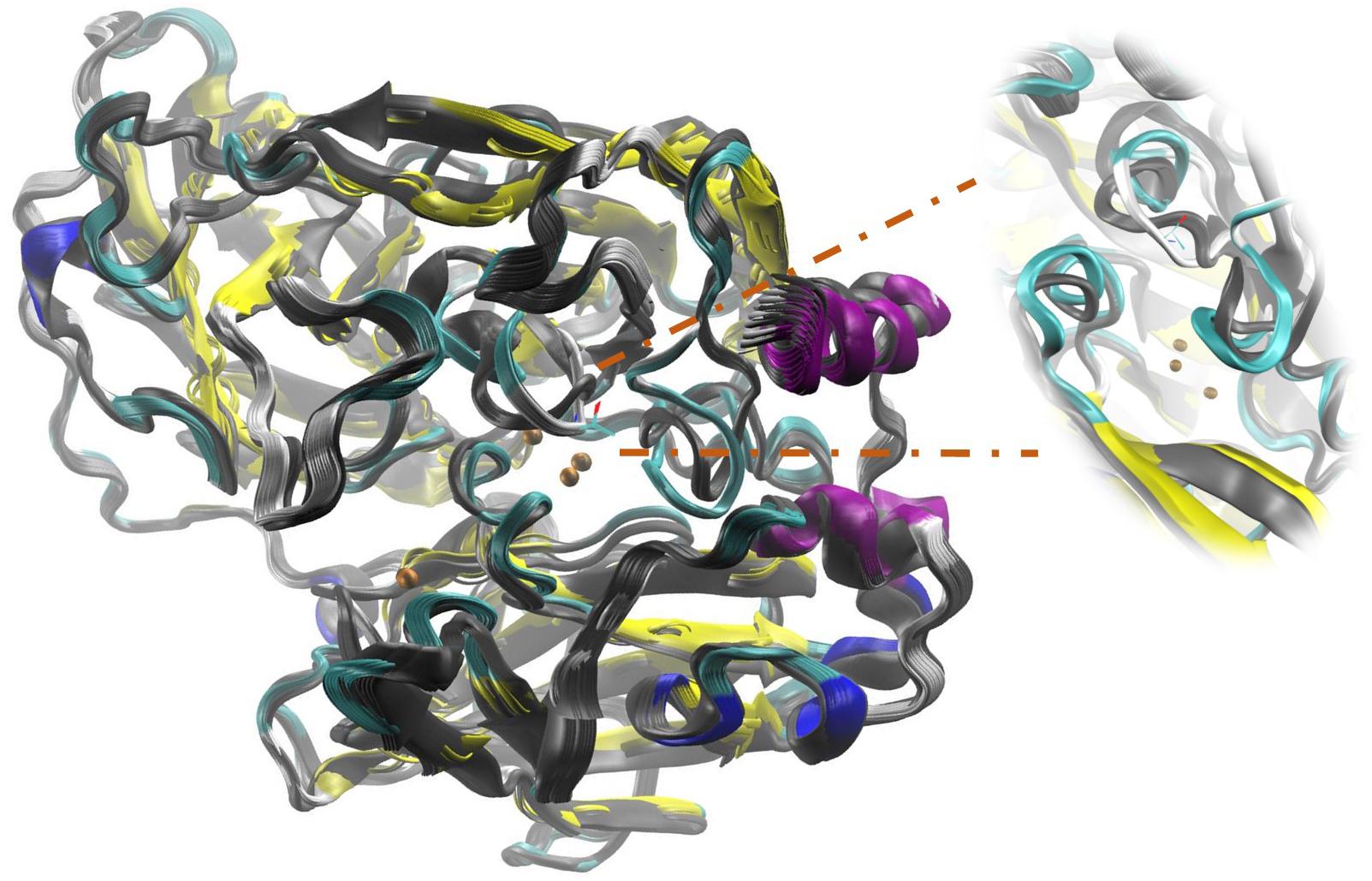
FIGURE 5. Structural overlap illustrating Active Site disturbances due to the mutation performed by replacing side chain Lysine 132 with a Threonine (L132T). The color image corresponds to the mutant enzyme and in grayscale to the native enzyme.
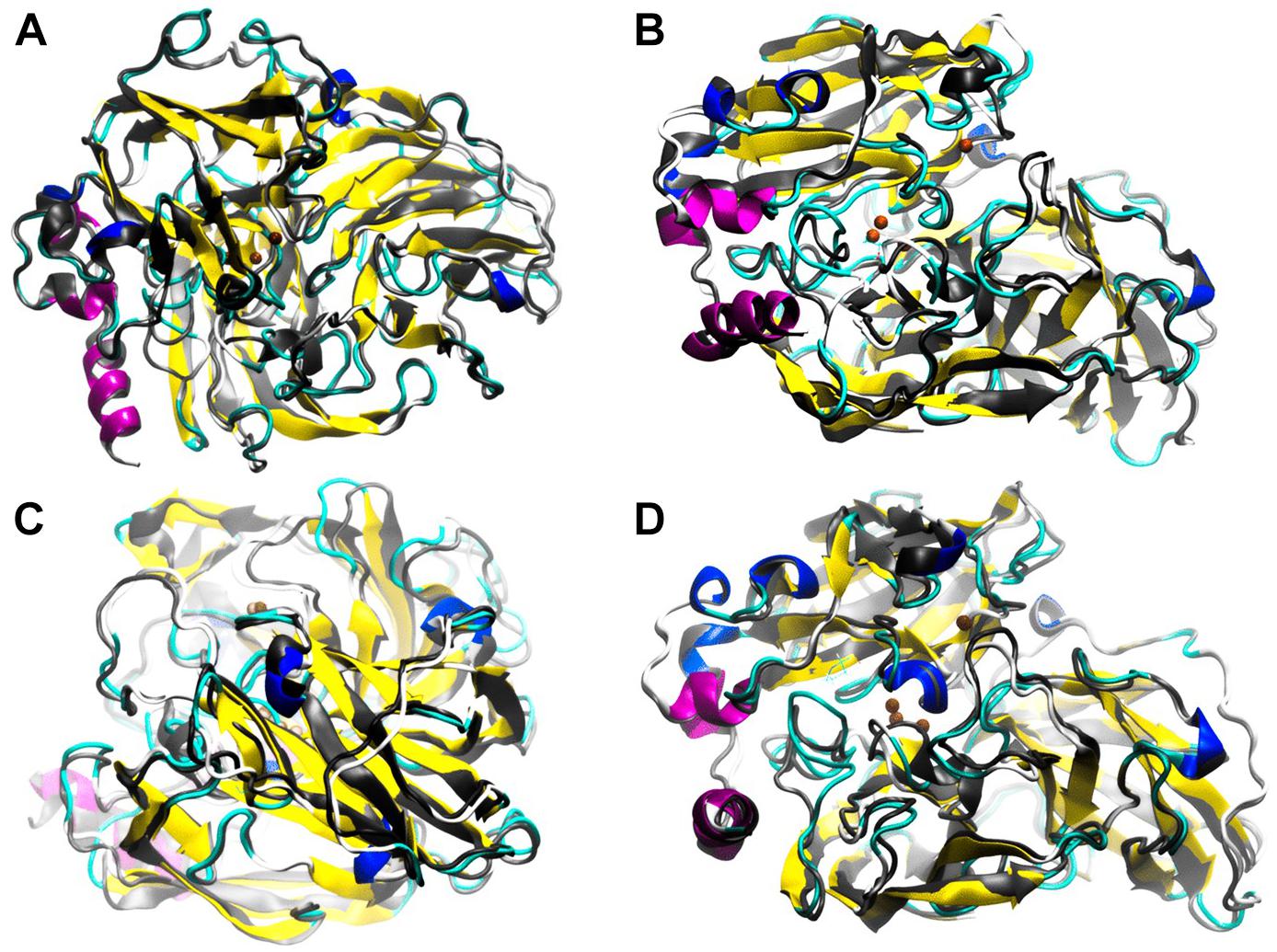
FIGURE 6. Low frequency motions corresponding to mode 7 calculated by ElNemo show a counter-rotation torsion between the domain for the distant copper and the binding domains to trinuclear site. In general, the perturbation to the global structure generated by the mutants: 3-SB (A), L132T (B), P203C (C), and F468P (D) is appreciated. ElNemo, overlapping of low energy motions on the minimized conformation between the native enzyme (grayscale) and the mutant (color coding according to secondary structure). These animations circulate around the 36 conformational snapshots, stored for low-frequency mode 7, centered on the minimized conformation of each type of enzyme. The regions of the most frequent proteins are highlighted given the counter-rotation movements between structural domains.
The three lower frequency movements, 7, 8, and 9, were selected as global representations of the movements (Table 5). Normal mode 7 was selected because values close to zero allow localized movements to be visualized, since normal low-frequency modes are expected to have collective characters, especially those related to the functional conformations of the protein (Tama and Sanejouand, 2001). In addition, as shown in Figure 7, the residues underwent significant displacement, although the structural deviation is small since the differences in RMSD (Root Mean Square Deviation) between the native protein and its mutants are about 1 Å, as can be seen in Table 5. Mode 7 shows a counter-rotation torsion between the structural domains, as can be seen in Figure 6, which would imply inclination and helical flexion, also one can observe that one of the domains rotate around an axis perpendicular to the plane of the membrane, while the others rotate around an inclined axis, this type of movements have not been described for this type of proteins.
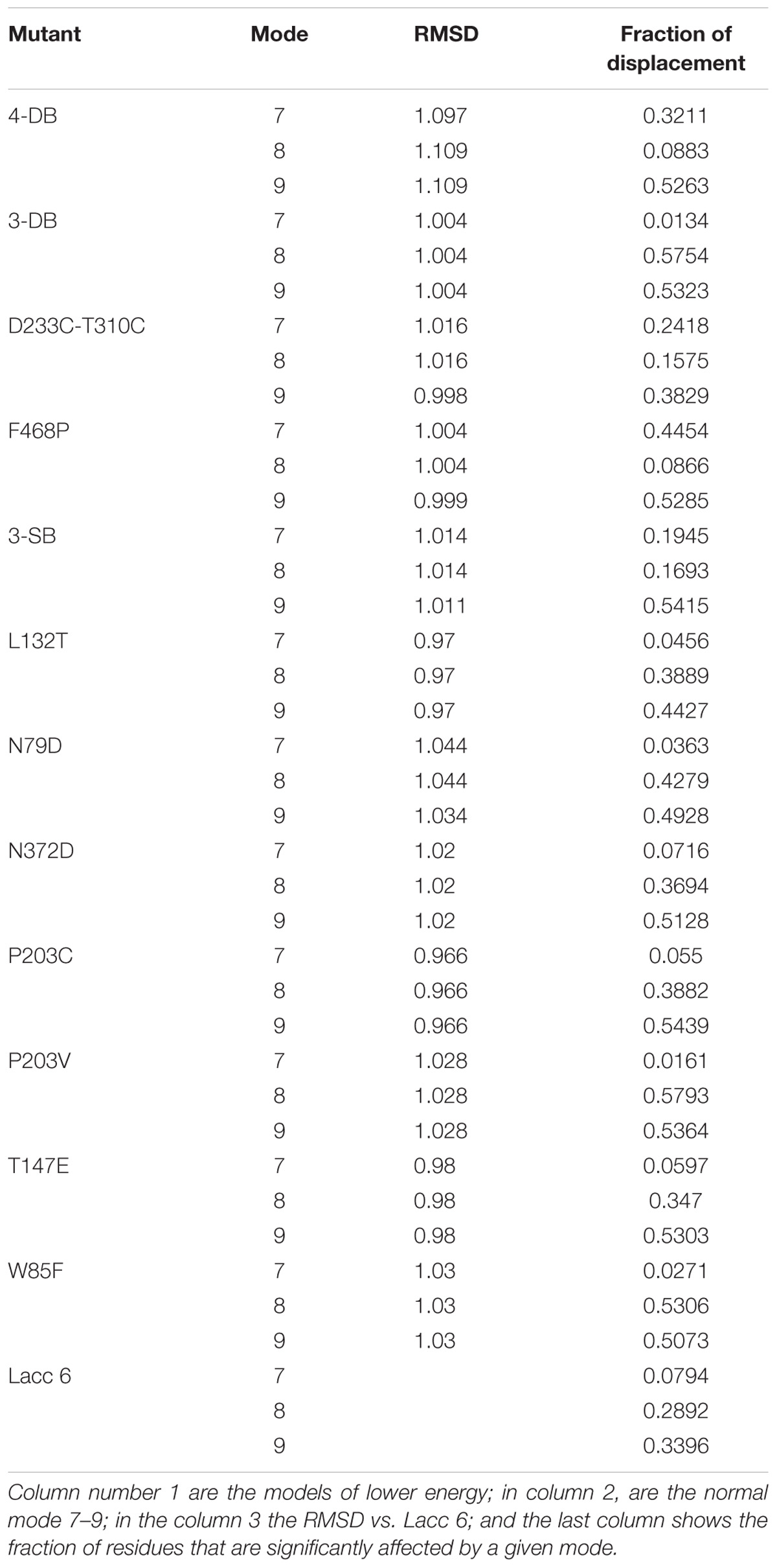
TABLE 5. Root Mead Squarer Deviation (RMSD) values and displacement (fraction) for the mutant enzymes compared to Lacc 6.
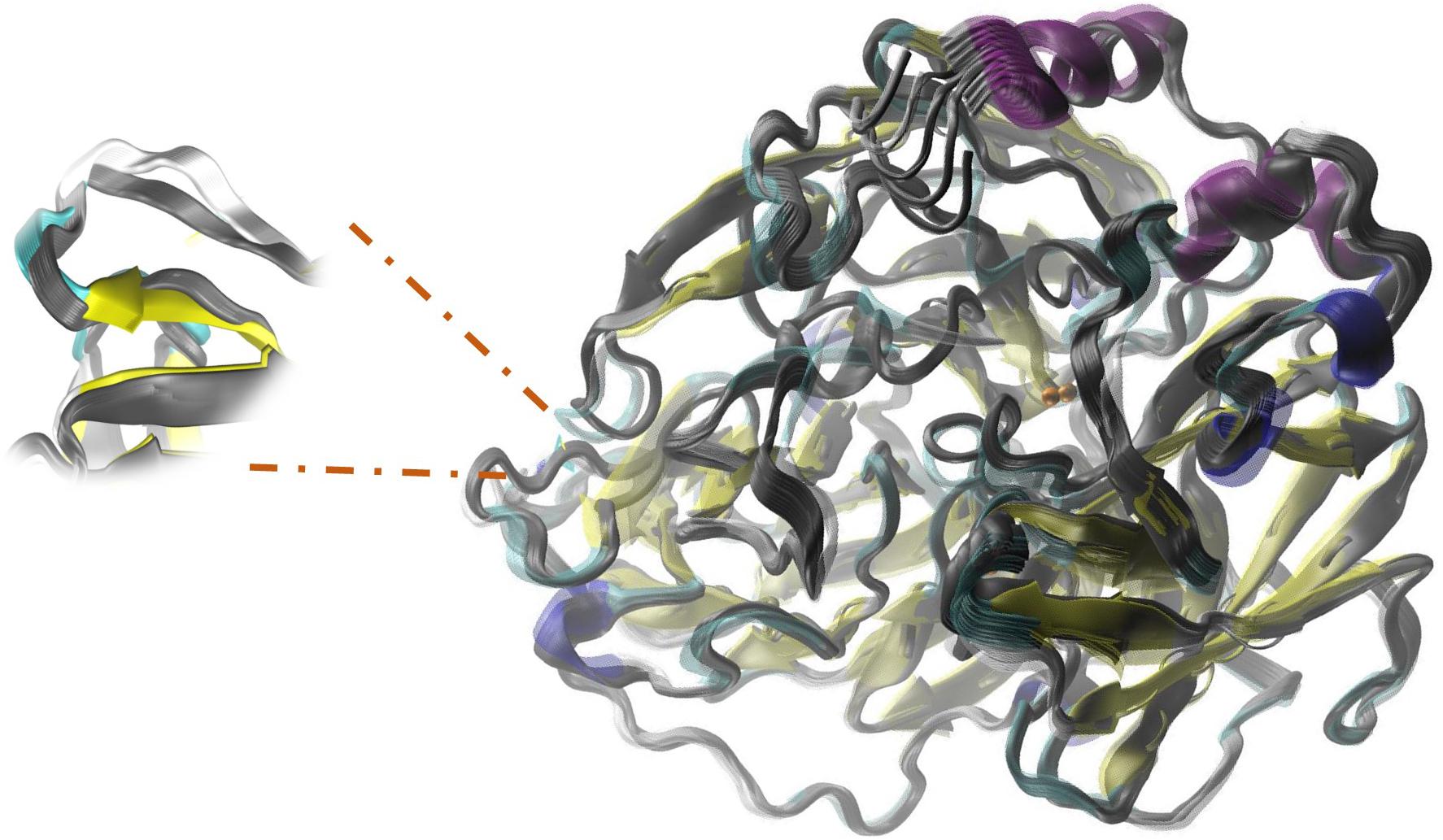
FIGURE 7. Relative movements in ElNemo mode7 between the mutant P203C and the native enzyme. The proteins are represented in the form of slats. The color-coded mutant depending on its secondary structure and native enzyme in grayscale. Also, it is shown an increase in fluctuation of native enzyme and the interconversion of non-random secondary structure for the formation of a beta-sheet on the opposite side to the mutation.
Flexibility and low energy motions were compared between the Lacc 6 and its mutants, ElNemo indicated that the low energy motions between the enzymes for their mutant and native form contain a nucleus in the protein that is immobile except for the L132T and F468P mutants (Figures 6B,D) for which the disturbance of the active site underwent significant alterations, in addition to some mobile external secondary structures.
Figures 4–7 make it possible to appreciate that the more flexible regions act as an interdomain hinge which allows coordinated movements of counter-rotational of each of these. ElNemo mode 7 shows the near domain to remote copper from the trinuclear copper site, it curves and straightens significantly according to the swinging of the contiguous domains, because the copper binding site implies a face of this domain (Figure 8 and Figure 1), these motions could be coupled to binding to the substrate or affect their affinity, especially in mutants designed to disturb the active site.
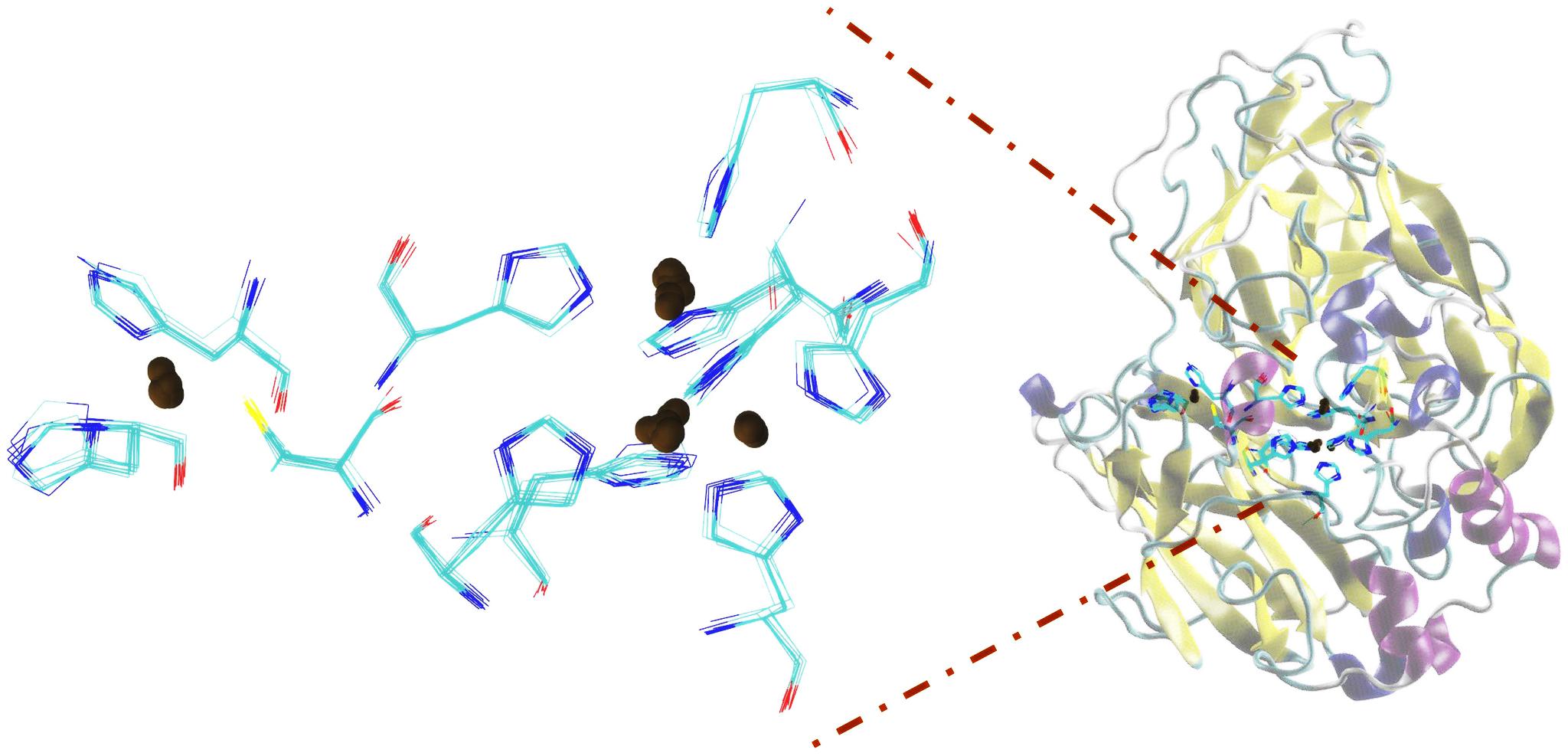
FIGURE 8. Domains close to the copper atom away from the trinuclear site of copper, showing the detail of the splicing of histidines bound to copper atoms.
Discussion
In this study, a bioinformatic analysis of enzyme Lacc 6 of Pleurotus ostreatus and minimization of energy of mutants of this same enzyme, to improve its stability in non-optimal conditions of activity for its possible synthesis and application in the degradation of agroindustrial residues that have been underutilized.
Agroindustrial wastes are a source of environmental pollution, since its composition is rich in lignin, cellulose and hemicellulose that require enzymatic complexes for its decomposition. For example, in 2006, Mexico produced about 76 million tons of agroindustrial waste from 20 types of cultivation and there are no physical, chemical, or enzymatic transformation techniques for their use (Valdez-Vazquez et al., 2010). Laccases have been associated with various biological functions, depending on the organism and its developmental conditions, are secreted into multiple isoforms and its most well-known and studied function is the degradation of lignin (Téllez-Téllez et al., 2008; Díaz et al., 2013).
On the other hand, these types of enzymes have applications of great interest in the industry and within their characteristics must be produced at low cost, be stable to the working conditions in wide ranges of pH and temperature for the treatment of pollutants (Téllez-Téllez et al., 2008; Miele et al., 2010; Díaz et al., 2013). In this study a proposal is made of an improved laccase that shows a minimum activation energy that allows it to be stable and active under adverse reaction conditions.
Several studies have been carried out on the generation of Pleurotus ostreatus laccase enzyme mutants as an alternative to be applied in the biodegradation of xenobiotic and/or recalcitrant compounds as reported by Miele et al. (2010), who randomly performed the generation of 2300 mutants of the POXA1b (Lacc 6) isoenzyme of Pleurotus ostreatus to develop biocatalysts from this enzyme, but no proposal has been made to know how the possible mutations occurred on the active site of this enzyme and to obtain models that would show the effect on the trinuclear copper region by modifying amino acids adjacent to the histidines bound to these metals as is done in this research.
On the other hand, Prins et al. (2015), observed the effect of mutations near the Cu 1 copper site of Streptomyces coelicolor A3 (2), to evaluate their biochemical characteristics against the native enzyme, obtaining three mutations (M298F, V290N, and V290A), the V290N mutant showed approximately double the activity, four to five times greater was the activity of the mutant M298F, however, the thermostability was decreased. In contrast to this study, where the proposed mutations (3-DB, 3-SB, P203C) allowed us to see the effect on the secondary structure of the protein; the changes occurring in the active site region and the energy minimization suggests an increase in the thermostability of the protein and that could tolerate higher to optimal temperatures for prolonged times. Such mutants may be candidates for in vitro expression, as shown in multiple works (Christensen and Kepp, 2013a; Ghosh et al., 2016), in which in silico studies allow the generation and identification of molecular bases in an expeditious manner which are difficult to quantify experimentally, and in vitro studies are feasibly reproduced by in silico studies (Zou et al., 2016), showing that computational tools are a primary aid in the structural study of proteins. Given the results, the production of mutant enzymes is suggested through genetic engineering to be used in bioremediation processes.
In recent years, computational design has been successfully applied for the thermostabilization of enzymes with potential use in biotechnological processes, mimicking the evolution in the laboratory to develop more stable enzyme variants and, more recently, using rational strategies of computer-assisted enzymatic engineering. Festa et al. (2008) performed one of the first notable works in the computational stabilization of a laccase, presented a screening library of 1100 clones of the mutant 1M9B obtained from Pleurotus ostreatus, and showed a single mutation (L112F) giving rise to a more active enzyme but less stable with respect to the native enzyme (POXA1b or Lacc 6), same enzyme used in our research.
On the other hand, Christensen and Kepp (2013a,b), exhaustively studied the isoforms of the laccase of Tramentes versicolor by molecular dynamics and other computational techniques in which they predict and rationalize a laccase to provide stability to mutations of multiple sites in its structure, showing that it is possible to generate stable mutants through computational studies, although to carry it out it turned out to be expensive. Ferrario et al. (2015), tested a thermal treatment with simulations of molecular dynamics on a laccase enzyme, observed that the enzyme conserved its geometry even making conformational changes in the labile parts. In contrast to our research in which through the protocol used to increase the thermal stability of Lacc 6 based on a rational approach by means of multiple methods including phylogenetic analysis, comparison with homologous proteins (particularly thermophilic), optimization of charged interactions (saline bridges and hydrogen bonds), optimization of waste and loops that show unfavorable Ramachandran angles as well as high B factors and computational design based on the structure, suggest a fast and inexpensive computational alternative.
Conclusion
Normal modes indicate that the nucleus of the mutants is found without significant motions, however, the exposed sections and in particular the domain of the distant copper undergo movements counter-rotation and global compression. Likewise, it is predicted that lower energy mutants such as 3-DB, 3-SB, P203C minimize fluctuations by making the skeleton less flexible in the vicinity of them, decreasing the range of motions. In the same way it is found that the mutants L132T and D468P generate a conformational change in the vicinity of the active site. This work predicts the global movements of the enzyme laccase, while providing a new perspective of these enzymes. This allows us to discuss the character of the movement in multiple structures on the same basis and as expected the normal low frequency modes from the modeling of the elastic network provide a good description of the global movements of this enzyme, which allows us to understand the molecular basis of the structural stability of the Lacc 6 enzyme of Pleurotus ostreatus and its mutants. This suggests that it will have greater stability in its structure that will allow it to have oxidase activity at temperatures above the optimum.
Author Contributions
RD performed the experimental design, directed the over-all study, and also drafted the manuscript. YM-F, GD-G and MA-R helped perform the analysis of results and revised the manuscript. LH-Z performed the protein modeling and revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was funded by the Mexican Council of Science and Technology (CONACyT) for supporting this research (National sabbatical stay of Rubén Díaz Godínez) and the Polytechnic University of Pachuca.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^ http://www.clustal.org/omega/
- ^ https://www.predictprotein.org/
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^ https://salilab.org/modeller/
- ^ https://modbase.compbio.ucsf.edu/modweb/
- ^ http://dezyme.com/
- ^ http://www.rbvi.ucsf.edu/chimera
- ^ https://www.predictprotein.org/
- ^ http://www.clustal.org/omega/
- ^ http://weblogo.berkeley.edu/logo.cgi
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anbar, M., Gul, O., Lamed, R., Sezerman, U. O., and Bayer, E. A. (2012). Improved thermostability of Clostridium thermocellum endoglucanase Cel8A by using consensus-guided mutagenesis. Appl. Environ. Microbiol. 78, 3458–3464. doi: 10.1128/aem.07985-11
Baldrian, P. (2006). Fungal laccases-occurrence and properties. FEMS. Microbiol. Rev. 30, 215–242. doi: 10.1111/j.1574-4976.2005.00010.x
Berka, R. M., Schneider, P., Golightly, E. J., Brown, S. H., Madden, M., Brown, K. M., et al. (1997). Characterization of the gene encoding an extracellular laccase of Myceliophthora thermophila and analysis of the recombinant enzyme expressed in Aspergillus oryzae. Appl. Environ. Microbiol. 63, 3151–3157.
Boratyn, G. M., Schäffer, A. A., Agarwala, R., Altschul, S. F., Lipman, D. J., and Madden, T. L. (2012). Domain enhanced lookup time accelerated BLAST. Biol. Direct. 7:12. doi: 10.1186/1745-6150-7-12
Burkoff, N. S., Várnai, C., Wells, S. A., and Wild, L. D. (2012). Exploring the energy landscapes of protein folding simulations with bayesian computation. Biophys. J. 102, 878–886. doi: 10.1016/j.bpj.2011.12.053
Calvo, A. M., Copa-Patiño, J. L., Alonso, O., and Gonzáles, A. E. (1998). Studies of the production and characterization of laccase activity in the basidiomycete Coriolopsis gallica, an efficient decolorizer of alkaline effluents. Arch. Microbiol. 171, 31–36. doi: 10.1007/s002030050674
Chernykh, A., Myasoedova, N., Kolomitseva, M., Ferraroni, M., Briganti, F., Scozzafava, A., et al. (2008). Laccase isoforms with unusual properties from the basidiomycete Steccherinum ochraceum 1833. J. Appl. Microbiol. 105, 2065–2075. doi: 10.1111/j.1365-2672.2008.03924.x
Christensen, N. J., and Kepp, K. P. (2013a). Stability mechanisms of a thermophilic laccase probed by molecular dynamics. PLoS One 8:e61985. doi: 10.1371/journal.pone.0061985
Christensen, N. J., and Kepp, K. P. (2013b). Stability mechanisms of laccase isoforms using a modified FoldX protocol applicable to widely different proteins. J. Chem. Theory Comput. 9, 3210–3223. doi: 10.1021/ct4002152
Coll, P. M., Fernández-Abalos, J. M., Villanueva, J. R., Santamaría, R., and Pérez, P. (1993). Purification and characterization of a phenoloxidase (laccase) from the lignin-degrading basidiomycete PM1 (CECT 2971). Appl. Environ. Microbiol. 59, 2607–2613.
Coluzza, I. (2017). Computational protein design: a review. J. Phys. Condens. Matter. 29:143001. doi: 10.1088/1361-648X/aa5c76
Das, S., Eisen, A., Lin, Y. H., and Chan, H. S. (2018). A lattice model of charge-pattern-dependent polyampholyte phase separation. J. Phys. Chem. B. 122, 5418–5431. doi: 10.1021/acs.jpcb.7b11723
Dedeyan, B., Klonowska, A., Tagger, S., Tron, T., Lacazio, G., Gil, G., et al. (2000). Biochemical and molecular characterization of a laccase from Marasmius quercophilus. Appl. Environ. Microb. 66, 925–929. doi: 10.1128/AEM.66.3.925-929.2000
Dehouck, Y., Kwasigroch, J. M., Gilis, D., and Rooman, M. (2011). PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics 12:151. doi: 10.1186/1471-2105-12-151
Díaz, R., Téllez-Téllez, M., Sánchez, C., Díaz-Godínez, G., and Soriano-Santos, J. (2013). Influence of initial pH of the growing medium on the activity, production and genes expression profiles of laccase of Pleurotus ostreatus in submerged fermentations. Electron. J. Biotechnol. 16. doi: 10.2225/vol16-issue4-fulltext-6
Djikaev, Y. S., and Ruckenstein, E. (2008). Temperature effects on the nucleation mechanism of protein folding and on the barrierless thermal denaturation of a native protein. Phys. Chem. Chem. Phys. 10, 6281–6300. doi: 10.1039/b807399f
Dong, J. L., and Zhang, Y. Z. (2004). Purification and characterization of two laccase isoenzymes from a ligninolytic fungus Trametes gallica. Prep. Biochem. Biotechnol. 34, 179–194. doi: 10.1081/PB-120030876
Dubey, M. K., Zehra, A., Aamir, M., Meena, M., Ahirwal, L., Singh, S., et al. (2017). Improvement strategies, cost effective production, and potential applications of fungal glucose oxidase (GOD): current updates. Front. Microbiol. 8:1032. doi: 10.3389/fmicb.2017.01032
Ferrario, V., Chernykh, A., Fiorindo, F., Kolomytseva, M., Sinigoi, L., Myasoedova, N., et al. (2015). Investigating the role of conformational effects on laccase stability and hyperactivation under stress conditions. Chembiochem. 16, 2365–2372. doi: 10.1002/cbic.201500339
Fersht, A. R., Leatherbarrow, R. J., and Wells, T. N. C. (1986). Quantitative analysis of structure–activity relationships in engineered proteins by linear free-energy relationships. Nature 322, 284–286. doi: 10.1038/322284a0
Festa, G., Autore, F., Fraternali, F., Giardina, P., and Sannia, G. (2008). Development of new laccases by directed evolution: functional and computational analyses. Proteins 72, 25–34. doi: 10.1002/prot.21889
Gao, X., Zeng, J., Yi, H., Zhang, F., Tang, B., and Tang, X. F. (2016). Four inserts within the catalytic domain confer extra stability and activity to hyperthermostable pyrolysin from Pyrococcus furiosus. Appl. Environ. Microbiol. 83:e03228-16. doi: 10.1128/aem.03228-16
Ghosh, M., Sodhi, S. S., Sharma, N., Mongre, R. K., Kim, N., Singh, A. K., et al. (2016). An integrated in silico approach for functional and structural impact of non- synonymous SNPs in the MYH1 gene in Jeju Native Pigs. BMC Genetics 17:35. doi: 10.1186/s12863-016-0341-1
Govardhan, C. (1999). Crosslinking of enzymes for improved stability and performance. Curr. Opin. Biotechnol. 10, 331–335. doi: 10.1016/S0958-1669(99)80060-3
Gribenko, A. V., Patel, M. M., Liu, J., McCallum, S. A., Wang, C., and Makhatadze, G. I. (2009). Rational stabilization of enzymes by computational redesign of surface charge-charge interactions. Proc. Natl. Acad. Sci. U.S.A. 106, 2601–2606. doi: 10.1073/pnas.0808220106
Hildén, K., Hakala, T. K., and Lundell, T. (2009). Thermotolerant and thermostable laccases. Biotechnol. Lett. 31, 1117–1128. doi: 10.1007/s10529-009-9998-0
Hildén, K., Hakala, T. K., Maijala, P., Lundell, T. K., and Hatakka, A. (2007). Novel thermotolerant laccases produced by the white-rot fungus Physisporinus rivulosus. Appl. Microbiol. Biotechnol. 77, 301–309. doi: 10.1007/s00253-007-1155-x
Huang, J., Rauscher, S., Nawrocki, G., Ran, T., Feig, M., de Groot, B. L., et al. (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. doi: 10.1016/j.bpj.2016.11.971
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Jordaan, J., and Leukes, W. D. (2003). Isolation of a thermostable laccase with DMAB and MBTH oxidative coupling activity from a mesophilic white rot fungi. Enzyme. Microb. Technol. 33, 212–219. doi: 10.1016/S0141-0229(03)00116-9
Kean, K. M., Porter, J. J., Mehl, R. A., and Karplus, P. A. (2017). Structural insights into a thermostable variant of human carbonic anhydrase II. Protein Sci. 27, 573–577. doi: 10.1002/pro.3347
Kiiskinen, L. L., Rättö, M., and Kruus, K. (2004). Screening for novel laccase-producing microbes. J. Appl. Microbiol. 97, 640–646. doi: 10.1016/S0141-0229(03)00116-9
Koschorreck, K., Richter, S. M., Swierczek, A., Beifuss, U., Schmid, R. D., and Urlacher, V. B. (2008). Comparative characterization of four laccases from Trametes versicolor concerning phenolic C-C coupling and oxidation of PAHs. Arch. Biochem. Biophys. 474, 213–219. doi: 10.1016/j.abb.2008.03.009
Kramer, K. J., Kanost, M. R., Hopkins, T. L., Jiang, H., Cheng-Zhu, Y., Xu, R., et al. (2001). Oxidative conjugation of chatecols with proteins in insect skeletal system. Tetrahedron 57, 385–392. doi: 10.1016/S0040-4020(00)00949-2
Kumar, S., Prakash, S., Gupta, K., Dongre, A., Balaram, P., and Balaram, H. (2016). Unexpected functional implication of a stable succinimide in the structural stability of Methanocaldococcus jannaschii glutaminase. Nat. Commun. 7:12798. doi: 10.1038/ncomms12798
Liwo, A., Arlukowicz, P., Czaplewski, C., Ołdziej, S., Pillardy, J., and Scheraga, H. A. (2002). A method for optimizing potential-energy functions by a hierarchical design of the potential-energy landscape: application to the UNRES force field. Proc. Natl. Acad. Sci. U.S.A. 99, 1937–1942. doi: 10.1073/pnas.032675399
Luna-Martínez, O. D., Vidal-Limón, A., Villalba-Velázquez, M. I., Sánchez-Alcalá, R., Garduño-Juárez, R., Uversky, V. N., et al. (2016). Simple approach for ranking structure determining residues. PeerJ 4:e2136. doi: 10.7717/peerj.2136
Mayer, A. M., and Staples, R. C. (2002). Laccase: new functions for an old enzyme. Phytochemistry 60, 551–565. doi: 10.1016/S0031-9422(02)00171-1
Miele, A., Giardina, P., Sannia, G., and Faraco, V. (2010). Random mutants of a Pleurotus ostreatus laccase as new biocatalysts for industrial effluents bioremediation. J. Appl. Microbiol. 108, 998–1006. doi: 10.1111/j.1365-2672.2009.04505.x
Nakagawa, N., and Peyrard, M. (2006). The inherent structure landscape of a protein. Proc. Natl. Acad. Sci. U.S.A. 103, 5279–5284. doi: 10.1073/pnas.0600102103
Nawaz, M. A., Rehman, H. U., Bibi, Z., Aman, A., and Ul Qader, S. A. (2015). Continuous degradation of maltose by enzyme entrapment technology using calcium alginate beads as a matrix. Biochem. Biophys. Rep. 4, 250–256. doi: 10.1016/j.bbrep.2015.09.025
Oda, K., and Kinoshita, M. (2015). Physicochemical origin of high correlation between thermal stability of a protein and its packing efficiency: a theoretical study for staphylococcal nuclease mutants. Biophys. Physicobiol. 12, 1–12. doi: 10.2142/biophysico.12.0_1
Pappenberger, G., Schurig, H., and Jaenicke, R. (1997). Disruption of an ionic network leads to accelerated thermal denaturation of D-glyderaldehyde-3-phosphate dehydrogenase from the hyperthermolphilic bacterium Thermotoga maritima. J. Mol. Biol. 274, 676–683. doi: 10.1006/jmbi.1997.1421
Pezeshgi-Modarres, H. P., Mofradab, M. R., and Sanati-Nezhad, A. (2016). Protein thermostability engineering. RSC. Adv. 6, 115252–115270. doi: 10.1039/C6RA16992A
Phillips, J. C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., et al. (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. doi: 10.1002/jcc.20289
Prins, A., Kleinsmidt, L., Khan, N., Kirby, B., Kudanga, T., Vollmer, J., et al. (2015). The effect of mutations near the T1 copper site on the biochemical characteristics of the small laccase from Streptomyces coelicolor A3(2). Enzyme. Microb. Tech. 68, 23–32. doi: 10.1016/j.enzmictec.2014.10.003
Radestock, S., and Gohlke, H. (2011). Protein rigidity and thermophilic adaptation. Proteins 79, 1089–1108. doi: 10.1002/prot.22946
Reed, C. J., Lewis, H., Trejo, E., Winston, V., and Evilia, C. (2013). Protein Adaptations in Archaeal Extremophiles. Archaea 2013:373275. doi: 10.1155/2013/373275
Schneider, P., Caspersen, M. B., Mondorf, K., Halkier, T., Skov, L. K., Østergaard, P. R., et al. (1999). Characterization of a Coprinus cinereus laccase. Enzyme Microb. Technol. 25, 502–508. doi: 10.1016/S0141-0229(99)00085-X
Sharma, P., Goel, R., and Capalash, N. (2007). Bacterial laccase. World J. Microb. Biot. 23, 823–832. doi: 10.1007/s11274-006-9305-3
Shuresh-Kumar, S. V., Prashant, S., Durani, S., and Pramod, W. (2003). Combined sequence and structure analysis of the fungal laccase family. Biotechnol. Bioeng. 83, 386–394. doi: 10.1002/bit.10681
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Sohini, B., and Srikanta, S. (2009). Turning a mesophilic protein into a thermophilic one: a computational approach based on 3D structural features. J. Chem. Inf. Model. 49, 1741–1750. doi: 10.1021/ci900183m
Suhre, K., and Sanejouand, Y. H. (2004). ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic. Acids. Res. 32, W610–W614. doi: 10.1093/nar/gkh368
Tama, F., and Sanejouand, Y. H. (2001). Conformational change of proteins arising from normal mode calculations. Protein Eng. 14, 1–6. doi: 10.1093/protein/14.1.1
Téllez-Téllez, M., Fernández, F. J., Montiel-González, A. M., Sánchez, C., and Díaz-Godínez, G. (2008). Growth and laccase production by Pleurotus ostreatus in submerged and solid-state fermentation. Appl. Microbiol. Biot. 81, 675–679. doi: 10.1007/s00253-008-1628-6
Ungar, L. W., Scherer, N. F., and Voth, G. A. (1997). Classical molecular dynamics simulation of the photoinduced electron transfer dynamics of plastocyanin. Biophys. J. 72, 5–17. doi: 10.1016/S0006-3495(97)78642-9
UniProt Consortium. (2012). Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic. Acids. Res. 40, D71–D75. doi: 10.1093/nar/gkr981
Valdez-Vazquez, I., Acevedo-Benitez, J. A., and Hernandez-Santiago, C. (2010). Distribution and potential of bioenergy resources from agricultural activities in Mexico. Renew. Sust. Energy Rev. 14, 2147–2153. doi: 10.1016/j.rser.2010.03.034
Venkataramani, S., Truntzer, J., and Coleman, D. (2013). Thermal stability of high concentration lysozyme across varying pH: a fourier Transform Infrared study. J. Pharm. Bioallied Sci. 5, 148–153. doi: 10.4103/0975-7406.111821
Wang, H. X., and Ng, T. B. (2004). A novel laccase with fair thermostability from the edible wild mushroom (Albatrella dispansus). Biochem. Biophys. Res. Commun. 319, 381–385. doi: 10.1016/j.bbrc.2004.05.011
Webb, B., and Sali, A. (2016). Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 54:05:Unit–5.6. doi: 10.1002/cpbi.3
Wesenberg, D., Kyriakides, I., and Agathos, S. N. (2003). White-rot fungi and their enzymes for the treatment of industrial dye effluents. Biotechnol. Adv. 22, 161–187. doi: 10.1016/j.biotechadv.2003.08.011
Xu, F. (2005). Applications of oxidoreductases: recent progress. Indust. Biotechnol. 1, 38–50. doi: 10.1089/ind.2005.1.38
Yachdav, G., Kloppmann, E., Kajan, L., Hecht, M., Goldberg, T., Hamp, T., et al. (2014). PredictProtein–an open resource for online prediction of protein structural and functional features. Nucleic. Acids. Res. 42, W337–W343. doi: 10.1093/nar/gku366
Yakimov, A. P., Afanaseva, A. S., Khodorkovskiy, M. A., and Petukhov, M. G. (2016). Design of Stable α-Helical Peptides and Thermostable Proteins in Biotechnology and Biomedicine. Acta Nat. 8, 70–81.
Zhou, X. X., Wang, Y. B., Pan, Y. J., and Li, W. F. (2008). Differences in amino acids composition and coupling patterns between mesophilic and thermophilic proteins. Amino Acids 34, 25–33. doi: 10.1007/s00726-007-0589-x
Keywords: laccase, Pleurotus ostreatus, Lacc 6, mutants, energy minimization
Citation: Díaz R, Díaz-Godínez G, Anducho-Reyes MA, Mercado-Flores Y and Herrera-Zúñiga LD (2018) In silico Design of Laccase Thermostable Mutants From Lacc 6 of Pleurotus Ostreatus. Front. Microbiol. 9:2743. doi: 10.3389/fmicb.2018.02743
Received: 26 July 2018; Accepted: 26 October 2018;
Published: 14 November 2018.
Edited by:
Zofia Piotrowska-Seget, University of Silesia of Katowice, PolandReviewed by:
Mirosława Słaba, University of Lodz, PolandGuiying Li, Guangdong University of Technology, China
Zhen Gao, Nanjing Tech University, China
Copyright © 2018 Díaz, Díaz-Godínez, Anducho-Reyes, Mercado-Flores and Herrera-Zúñiga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rubén Díaz, cmRpYXpnb2RAaG90bWFpbC5jb20= Leonardo David Herrera-Zúñiga, bGRoejA0QGdtYWlsLmNvbQ==