 Laura M. Carroll1
Laura M. Carroll1 Martin Wiedmann1
Martin Wiedmann1 Manjari Mukherjee2
Manjari Mukherjee2 David C. Nicholas3
David C. Nicholas3 Lisa A. Mingle4Nellie B. Dumas4Jocelyn A. Cole4
Lisa A. Mingle4Nellie B. Dumas4Jocelyn A. Cole4 Jasna Kovac2*
Jasna Kovac2*- 1Department of Food Science, Cornell University, Ithaca, NY, United States
- 2Department of Food Science, The Pennsylvania State University, University Park, PA, United States
- 3New York State Department of Health, Corning Tower, Empire State Plaza, Albany, NY, United States
- 4New York State Department of Health, Wadsworth Center, Albany, NY, United States
The Bacillus cereus group comprises multiple species capable of causing emetic or diarrheal foodborne illness. Despite being responsible for tens of thousands of illnesses each year in the U.S. alone, whole-genome sequencing (WGS) is not yet routinely employed to characterize B. cereus group isolates from foodborne outbreaks. Here, we describe the first WGS-based characterization of isolates linked to an outbreak caused by members of the B. cereus group. In conjunction with a 2016 outbreak traced to a supplier of refried beans served by a fast food restaurant chain in upstate New York, a total of 33 B. cereus group isolates were obtained from human cases (n = 7) and food samples (n = 26). Emetic (n = 30) and diarrheal (n = 3) isolates were most closely related to B. paranthracis (group III) and B. cereus sensu stricto (group IV), respectively. WGS indicated that the 30 emetic isolates (24 and 6 from food and humans, respectively) were closely related and formed a well-supported clade distinct from publicly available emetic group III genomes with an identical sequence type (ST 26). The 30 emetic group III isolates from this outbreak differed from each other by a mean of 8.3 to 11.9 core single nucleotide polymorphisms (SNPs), while differing from publicly available emetic group III ST 26 B. cereus group genomes by a mean of 301.7–528.0 core SNPs, depending on the SNP calling methodology used. Using a WST-1 cell proliferation assay, the strains isolated from this outbreak had only mild detrimental effects on HeLa cell metabolic activity compared to reference diarrheal strain B. cereus ATCC 14579. We hypothesize that the outbreak was a single source outbreak caused by emetic group III B. cereus belonging to the B. paranthracis species, although food samples were not tested for presence of the emetic toxin cereulide. In addition to showcasing how WGS can be used to characterize B. cereus group strains linked to a foodborne outbreak, we also discuss potential microbiological and epidemiological challenges presented by B. cereus group outbreaks, and we offer recommendations for analyzing WGS data from the isolates associated with them.
Introduction
The Bacillus cereus (B. cereus) group, also known as B. cereus sensu lato (s.l.) is a complex of closely related species that vary in their ability to cause disease in humans. Foodborne illness caused by members of the group primarily manifests itself in one of two forms: (i) emetic intoxication that is caused by cereulide, a heat-stable toxin produced by B. cereus within a food matrix prior to consumption, or (ii) a diarrheal toxicoinfection, caused by enterotoxins produced by bacteria in the small intestine of the host (Ehling-Schulz et al., 2004; Schoeni and Wong, 2005; Stenfors Arnesen et al., 2008). Here we refer to isolates that carry ces genes encoding the cereulide biosynthetic pathway as emetic isolates, and isolates that lack ces genes but carry either hbl or cytK-2 genes that encode diarrheal enterotoxins as diarrheal isolates. The gene variant cytK-2 was included in this definition, as it was previously found in non-B. cytotoxicus isolates associated with diarrheal illness (Castiaux et al., 2015; Miller et al., 2018). The presence of nhe genes was not included in our present definition of diarrheal isolates, due to the fact that nhe genes are ubiquitously found in the majority of the B. cereus group population (Carroll et al., 2017; Miller et al., 2018), including all isolates in the present study, and their contribution to diarrheal toxicoinfection is not yet fully understood (Doll et al., 2013).
As foodborne pathogens, members of the B. cereus group are estimated to cause 63,400 foodborne disease cases per year in the U.S. (Scallan et al., 2011) and are confirmed or suspected to have been responsible for 235 outbreaks reported in the U.S. between 1998 and 2008 (Bennett et al., 2013). Due in part to its typically self-limiting nature, foodborne illness caused by members of the B. cereus group is under-reported (Granum and Lund, 1997; Stenfors Arnesen et al., 2008), although severe infections resulting in patient death have been reported (Naranjo et al., 2011; Sanaei-Zadeh, 2012; Lotte et al., 2017). Furthermore, B. cereus group isolates that have been linked to human clinical cases of foodborne disease rarely undergo whole-genome sequencing (WGS), as is becoming the norm for other foodborne pathogens (Joensen et al., 2014; Ashton et al., 2015; Moura et al., 2017).
Here, we describe a foodborne outbreak caused by members of the B. cereus group in which WGS was implemented to characterize isolates from human clinical cases and food. To our knowledge, this is the first description of a B. cereus outbreak in which WGS was employed to characterize isolates. By testing various combinations of variant calling methodologies, we showcase how different bioinformatics pipelines can yield vastly different results when pairwise SNP differences are the desired metric for determining whether an isolate is part of an outbreak or not. In addition to discussing the bioinformatic challenges, we examine potential microbiological and epidemiological obstacles that can hinder characterization of B. cereus group isolates from suspected foodborne outbreaks, and we offer recommendations to guide the characterization of future B. cereus group outbreaks using WGS.
Materials and Methods
Collection of Epidemiological Data
Epidemiological investigations were coordinated by the New York State Department of Health (NYSDOH), and the outbreak was reported to the U.S. Centers for Disease Control and Prevention (CDC). Investigation methods included (i) a cohort study, (ii) food preparation review, (iii) an investigation at a factory/production/treatment plant, (iv) food product traceback, and (v) environment/food/water sample testing.
Isolation and Initial Characterization of B. cereus Strains
Stool specimens were plated directly onto mannitol-egg yolk-polymyxin (MYP) agar and incubated aerobically at 37°C for 24 h. Food samples were diluted 1:10 in 1 X PBS, pH 7.4 in a filter bag for homogenizer blenders and homogenized for 2 min. One hundred μl of each homogenized sample were plated onto MYP agar and incubated aerobically at 37°C for 24 h. The MYP agar plates for both the stool specimens and food samples were observed after the 24 h incubation period. Individual B. cereus-like colonies (i.e., pink colored and lecithinase positive) were subcultured on trypticase soy agar (TSA) plates supplemented with 5% sheep blood and incubated aerobically at 37°C for 18–24 h. These isolates were identified as B. cereus using the following conventional microbiological techniques: Gram stain, colony morphology, hemolysis, motility, and spore stain. To test for the presence of parasporal crystals often associated with B. thuringiensis, isolates were cultured for 48 h at 37°C on sporulation agar slants. Smears were prepared, and slides were heat fixed and then stained using malachite green and counter stained with carbol fuchsin (Tallent et al., 2012b). Slides were then observed for the presence or absence of parasporal crystals.
rpoB Allelic Typing
The 33 outbreak isolates were streaked onto brain heart infusion (BHI) agar from their respective cryo stocks stored at −80°C and incubated overnight at 37°C. Single isolated colonies were inoculated in 5 ml BHI broth and incubated overnight at 32°C and used for genomic DNA extraction using Qiagen DNeasy blood and tissue kits (Qiagen). Extracted DNA was used as a template in a PCR reaction using primers targeting a 750 bp sequence of the rpoB gene (RzrpoBF: AARYTIGGMCCTGAAGAAAT and RZrpoBR: TGIARTTRTCATCAACCATGTG) (Ivy et al., 2012). PCR was carried out in 25 μl reactions using GoTaq Green Master Mix (Promega Corporation) under the following thermal cycling conditions: 3 min at 94°C, followed by 40 cycles of 30 s at 94°C, 30 s at 55–45°C (in the first 20 cycles the temperature was reduced for 0.5°C per cycle and then kept at 45°C in the following 20 cycles), followed by 1 min at 72°C, and a final hold at 4°C. The resulting PCR product was used for genotyping and preliminary species identification using the rpoB allele type database available in Food Microbe Tracker (Ivy et al., 2012; Vangay et al., 2013).
Bacterial Growth Conditions and Collection of Bacterial Supernatants
The 33 outbreak isolates, as well as B. cereus s.s. type strain ATCC 14579 and B. cereus emetic reference strain DSM 4312 (Food Microbe Tracker ID FSL M8-0547; Vangay et al., 2013) were streaked onto BHI agar from their respective cryo stocks stored at −80°C. For immunoassays and cytotoxicity assays (see sections Hemolysin BL and Non-hemolytic Enterotoxin Detection and WST-1 Metabolic Activity Assay), cultures grown from single isolated colonies for 18 h at 37°C without shaking were used for inoculation of fresh BHI broth. Fresh cultures were grown to early stationary phase as determined by an OD600 of ~1.5, which equals ~108 CFU/ml. After incubation, growth was quenched by placing cultures on ice. The cultures were then spun down at 16,000 g for 2 min, and the supernatants were collected, aliquoted in duplicate, and stored at −80°C until further use in cytotoxicity assays.
Hemolysin BL and Non-hemolytic Enterotoxin Detection
Diarrheal strains grown as described above were used for qualitative detection of hemolysin BL (Hbl) and non-hemolytic enterotoxins (Nhe) with the Duopath Cereus Enterotoxins immunoassay (Merck). Only select representatives of emetic outbreak strains were tested (i.e., FSL R9-6381, FSL R9-6382, FSL R9-6384, FSL R9-6389, FSL R9-6395, and FSL R9-6399), as they did not carry genes encoding Hbl and were therefore not expected to produce Hbl. Briefly, the temperatures of the cultures and immunoassay kits were adjusted to room temperature. 150 μl of each isolate culture were added to the immunoassay port, following the manufacturer's instructions. The results were read as positive if a red test line was visible after a 30-min incubation at room temperature. Tests were considered valid only when control lines were visible.
WST-1 Metabolic Activity Assay
HeLa cells were seeded in 96-well plates at a seeding density of 8 × 104 cells/cm2 (Fisichella et al., 2009) in Eagle's minimum essential medium (EMEM) supplemented with 10% fetal bovine serum (FBS) and allowed to grow for 18–24 h at 37°C, 5% CO2. After incubation, the medium in each well was replaced with 100 μl of fresh medium containing 5% v/v of bacterial supernatants (prepared as described in section Bacterial Growth Conditions and Collection of Bacterial Supernatants) that were thawed and pre-warmed to 37°C. The combined medium and supernatants were added to the cells using a multichannel pipettor to minimize the variability in the duration of cell exposure to the toxin amongst wells of a 96-well plate. Medium containing 5% BHI was used as a negative control. Medium containing 5% v/v of 1% Triton X-100 dissolved in BHI (final concentration in the test well was 0.05%) was used as a positive control expected to significantly reduce the viability of HeLa cells. After 15 min of intoxication at 37°C, 5% CO2 (Miller et al., 2018), 10 μl of WST-1 dye solution (Roche) was added to each well of the plate, and the plate was incubated for 25 min at 37°C, 5% CO2, resulting in a total of 40 min exposure of cells to the supernatants. After 30 s of orbital shaking at 600 rpm, the absorbances were read by a microplate reader (Thermo Scientific Multiskan GO, Thermo Fisher Scientific) in a precision mode at 450 and 690 nm, the latter being subtracted from the former to account for the background signal (i.e., corrected absorbances) (Fisichella et al., 2009). Each test, including 0.05% Triton X-100, was conducted with six technical replicates and on two different HeLa passages using supernatants from single biological replicates, resulting in a total of 12 technical replicates per isolate. The viability of cells was determined by calculating a ratio of corrected absorbances to that of BHI, converting to percentages, and calculating the mean of the technical replicates for each isolate. The results were compared to the results for cells treated with (i) 0.05% Triton X-100, (ii) B. cereus s.s. type strain ATCC 14579 supernatant (i.e., reference for diarrheal strains), and (iii) B. cereus group strain DSM 4312 supernatant (i.e., reference for emetic strains).
Statistical Analysis of Cytotoxicity Data
A Welch's test and the Games-Howell post-hoc test that are appropriate for analyses of data with non-homogeneous variances were performed using results of all 12 technical replicates of each outbreak-associated isolate, as well as the reference strains and the positive control. For the Games-Howell test, a Bonferroni correction was applied to correct for multiple comparisons. Statistical analyses were carried out in R version 3.4.3 (R Core Team., 2018).
Whole-Genome Sequencing
Genomic DNA was extracted from overnight cultures (~18 h) grown in BHI at 32°C using Qiagen DNeasy blood and tissue kits (Qiagen) or the Omega E.Z.N.A. Bacterial DNA kit (Omega) following the manufacturers' instructions. For the E.Z.N.A. Bacterial DNA kit, the additional steps recommended for difficult-to-lyse bacteria were taken to obtain sufficient DNA yield. Briefly, one ml of an overnight culture was additionally treated with glass beads provided in the E.Z.N.A. kit. DNA was quantified using Qubit 3 and used for Nextera XT library preparation (Illumina). Pooled libraries were sequenced in two Illumina sequencing runs with either 2 × 250 or 2 × 300 bp reads at the Penn State Genomics Core Facility and at the Cornell Animal Health Diagnostic Center.
Initial Data Processing and Genome Assembly
Illumina adapters and low-quality bases were trimmed using Trimmomatic version 0.36 (Bolger et al., 2014) and the default parameters for Nextera paired-end reads, and FastQC version 0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to confirm that read quality was adequate (e.g., no reads flagged as poor quality, no Illumina adapters present). Genomes listed in Supplementary Table S1 were assembled de novo using SPAdes version 3.11.0 (Bankevich et al., 2012), and average per-base coverage was calculated using Samtools version 1.6 (Li et al., 2009) after mapping reads to their respective de novo assemblies using BWA MEM version 0.7.13 (default parameters) (Li and Durbin, 2010).
In silico Typing and Virulence Gene Detection
BTyper version 2.2.0 (Carroll et al., 2017) was used to perform in silico virulence gene detection, multi-locus sequence typing (MLST), panC group assignment (as defined by Guinebretiere et al., 2010), and rpoB allelic typing, as well as to extract the gene sequences for all detected loci. For virulence gene detection, the default settings were used (i.e., 50% amino acid sequence identity, 70% query coverage), as these cut-offs have been shown to correlate with PCR-based detection of virulence genes in B. cereus group isolates (Kovac et al., 2016; Carroll et al., 2017). BMiner version 2.0.2 (Carroll et al., 2017) was used to aggregate the output files from BTyper and create a virulence gene presence/absence matrix.
Construction of k-mer Based Phylogeny Using Outbreak Strains and Genomes of 18 B. cereus Group Species
kSNP version 3.1 (Gardner and Hall, 2013; Gardner et al., 2015) was used to produce a set of core SNPs among the 33 outbreak genomes, plus a type strain or RefSeq reference genome assembly from each of the 18 B. cereus group species listed in Supplementary Table S2 (Stenfors Arnesen et al., 2008; Guinebretière et al., 2013; Jiménez et al., 2013; Miller et al., 2016; Liu et al., 2017), using the optimal k-mer size as determined by Kchooser (k = 21). The resulting core SNPs were used in conjunction with RAxML version 8.2.11 (Stamatakis, 2014) to construct a maximum likelihood (ML) phylogeny using the GTRCAT model with a Lewis ascertainment bias correction (Lewis, 2001) to account for the use of solely variant sites, and 500 bootstrap replicates. The resulting phylogenetic tree was formatted using the phylobase (Hackathon et al., 2017), ggtree (Guangchuang et al., 2017), phytools (Revell, 2012), and ape (Paradis et al., 2004) packages in R version 3.4.3.
Variant Calling and Phylogeny Construction Using Outbreak Isolates
Combinations of five reference-based variant calling pipelines (Table 1) and reference genomes (Table 2), as well as one reference-free SNP calling pipeline (Table 1), were used to separately identify core and total SNPs among (i) all 33 outbreak-related isolates (30 emetic group III isolates and three group IV isolates) and (ii) the subset of 30 emetic group III isolates. For the subset of 30 emetic group III isolates, all reference-based variant calling pipelines described below were additionally run with dustmasked versions of the reference genomes listed in Table 2, in which DustMasker version 1.0.0 (part of BLAST version 2.6.0) (Morgulis et al., 2006) was used to mask low-complexity portions (i.e., intervals with highly biased nucleotide distributions which can bias sequence similarity searches) in each reference genome (Ye et al., 2006).
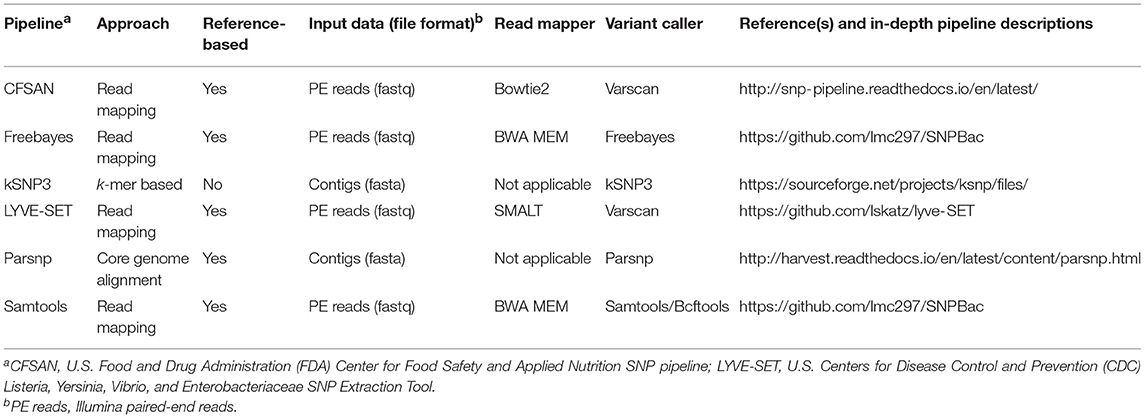
Table 1. Variant calling pipelines tested in this study.
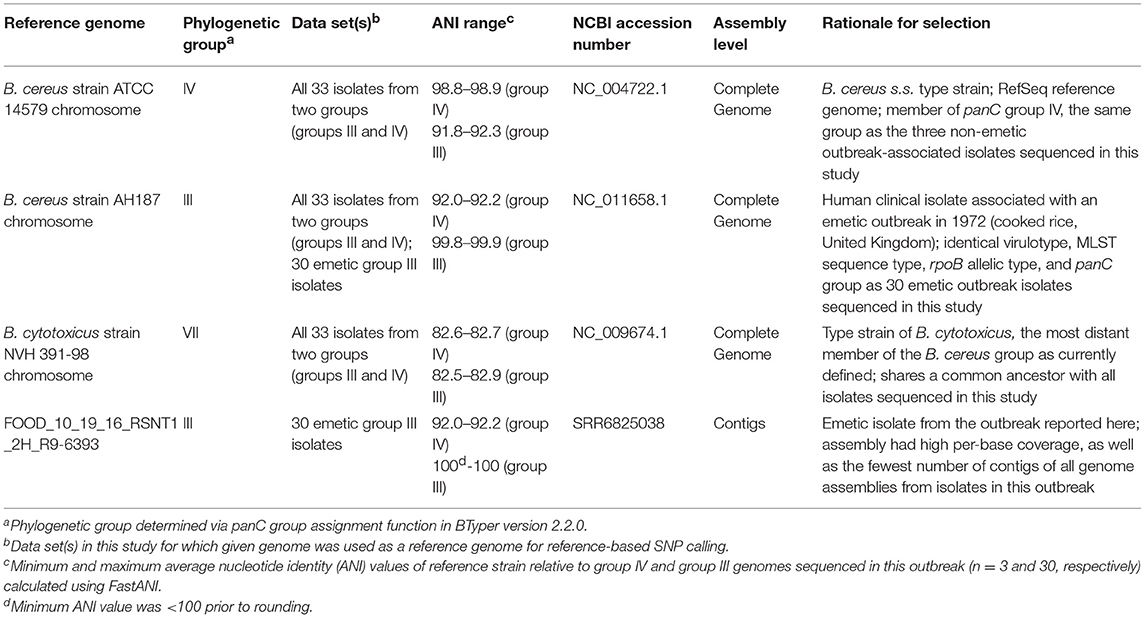
Table 2. Reference genomes used for reference-based variant calling in this study.
For the Samtools and Freebayes pipelines (Table 1), trimmed Illumina paired-end reads from the queried isolates were mapped to the appropriate reference genome using BWA mem version 0.7.13 (Li, 2013) and either Samtools/Bcftools version 1.6 (Li et al., 2009) or Freebayes version 1.1.0 (Garrison and Marth, 2012), respectively, were used to call variants. Vcftools version 0.1.14 (Danecek et al., 2011) was used to remove indels and SNPs with a SNP quality score <20, as well as to construct consensus sequences. For both variant calling pipelines, Gubbins version 2.2.0 (Croucher et al., 2015) was used to remove recombination events from the consensus sequences, and the Neighbor Similarity Score (NSS) (Jakobsen and Easteal, 1996), Maximum Chi-Squared (Smith, 1992), and Pairwise Homoplasy Index (PHI) (Bruen et al., 2006) tests implemented in PhiPack version 1.0 (Bruen et al., 2006) were used to assess whether recombination and homoplasies were present in sequence alignments before and after recombination was removed, using 1,000 permutations each and a window size of 100 (Supplementary Table S3). Both of these pipelines are publicly available and can be reproduced in their entirety (SNPBac version 1.0.0; https://github.com/lmc297/SNPBac).
For the CFSAN (Davis et al., 2015) and LYVE-SET (Katz et al., 2017) pipelines (versions 1.0.1 and 1.1.4 g, respectively; Table 1), trimmed Illumina paired-end reads were used as input, and all default pipeline steps were run as outlined in the manuals. For the Parsnp pipeline (Treangen et al., 2014) (Table 1), assembled genomes of the outbreak isolates were used as input, and Parsnp's implementation of PhiPack (Bruen et al., 2006) was used to filter out recombination events. For kSNP3 (Table 1), assembled genomes of the outbreak isolates were used as input, and Kchooser was used to determine the optimum k-mer size for the full 33-isolate data set and the 30 emetic group III isolate set (k = 21 and 23, respectively).
For all variant calling and filtering pipelines, RAxML version 8.2.10 was used to construct ML phylogenies using the resulting SNPs under the GTRGAMMA model with a Lewis ascertainment bias correction and 1,000 bootstrap replicates. Phylogenetic trees were annotated using FigTree version 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
Variant Calling and Statistical Comparison of Emetic Outbreak Isolates to Publicly Available Genomes
To compare emetic group III isolates from this outbreak to other emetic group III isolates, BTyper version 2.2.1 was used to query all 2,156 B. cereus group genome assemblies available in NCBI's RefSeq database (downloaded March 2018) (Pruitt et al., 2007) and identify all genome assemblies that (i) belonged to group III based on panC sequence, (ii) belonged to ST 26 based on in silico MLST, and (iii) were found to possess the ces operon in its entirety (cesABCD) at the default coverage and identity thresholds. This search produced 25 genome assemblies in addition to the 30 emetic group III genomes sequenced here. Only three of the 25 RefSeq genome assemblies had Sequence Read Archive (SRA) data linked to their BioSample accession numbers, making short read data readily available only for these three isolates. Consequently, only Parsnp version 1.2 and kSNP version 3.1 were used to identify SNPs in all 55 group III emetic genomes (25 from NCBI RefSeq and 30 sequenced here), as these approaches can be used with assembled genomes and do not require short reads as input. For Parsnp, the chromosome of B. cereus AH187 was used as a reference genome. For kSNP3, Kchooser was used to select the optimal k-mer size (k = 21), and the chromosome of B. cereus AH187 was included for k-mer based SNP calling.
RAxML version 8.2.10 was used to construct ML phylogenies using the resulting core SNPs for each of the Parsnp and kSNP3 pipelines under the GTRCAT model with a Lewis ascertainment bias correction and 1,000 bootstrap replicates. Pairwise core SNP differences between all 55 isolates were obtained using the dist.gene function in R's ape package. The permutest and betadisper functions in R's vegan package (Oksanen et al., 2018) were used to conduct an ANOVA-like permutation test to test if publicly available genomes were more variable than isolates from this outbreak based on pairwise core SNP differences and 5 independent trials using 100,000 permutations each. Analysis of similarity (ANOSIM; Clarke, 1993) using the anosim function in the vegan package in R was used to determine if the average of the ranks of within-group distances was greater than or equal to the average of the ranks of between-group distances (Anderson and Walsh, 2013), where groups were defined as (i) the 30 emetic isolates from this outbreak, and (ii) the 25 external emetic ST 26 isolates (downloaded from RefSeq). ANOSIM tests were conducted using pairwise core SNP differences and five independent runs of 10,000 permutations each. For both the ANOVA-like permutation tests and the ANOSIM tests, Bonferroni corrections were used to correct for multiple comparisons at the α = 0.05 level.
Statistical Comparison of Phylogenetic Trees
The Kendall-Colijn (Kendall and Colijn, 2015) test described by Katz et al. (2017) was used to compare the topologies of trees, using the treespace (Jombart et al., 2017), ips (Heibl, 2008), phangorn (Schliep, 2011), docopt (de Jonge, 2016), and stringr (Wickham, 2018) packages in R version 3.4.3. The phylogenies that underwent pairwise testing were constructed using (i) either core or total SNPs identified in 30 emetic group III genomes via all six SNP calling pipelines (Table 1), using either an unmasked or dustmasked closed reference genome (B. cereus AH187; Table 2), and (ii) SNPs identified in 55 emetic ST 26 genomes (25 publicly available genomes and the 30 emetic isolates sequenced here) using the kSNP3 (core and total SNPs) and Parsnp (core SNPs, as Parsnp queries the core genome by definition) pipelines. For all pairwise tree comparisons, a lambda value of 0 (to give weight to tree topology rather than branch lengths; Katz et al., 2017) was used along with 100,000 random trees as a background distribution, and a Bonferroni correction was used to correct for multiple comparisons. Pairs of trees were considered to be more topologically similar than would be expected by chance if a significant P-value (P < 0.05) resulted after correcting for multiple testing (Katz et al., 2017).
Calculation of Average Nucleotide Identity Values
FastANI version 1.0 (Jain et al., 2018) was used to calculate average nucleotide identity (ANI) values between assembled genomes of isolates sequenced in this study and selected reference genomes (Table 2), as well as the genomes of 18 currently published B. cereus group species (Supplementary Table S2).
Availability of Data
Trimmed Illumina reads for all 33 isolates sequenced in this study have been made publicly available (NCBI BioProject Accession PRJNA437714), with NCBI BioSample and SRA accession numbers for all isolates listed in Supplementary Table S1. All figures have been deposited in FigShare (DOI https://doi.org/10.6084/m9.figshare.7001525.v1), and records of all isolates are available in Food Microbe Tracker (Vangay et al., 2013).
Results
Both Emetic and Diarrheal Symptoms Were Reported Among Cases Associated With the B. cereus Foodborne Outbreak
Between September 30th and October 6th, 2016, local health departments in upstate New York's Niagara and Erie counties reported a total of 179 estimated foodborne illness cases among customers of a Mexican fast-food restaurant chain in eight towns/cities. Among these cases, laboratory results were available for ten cases. For seven of these cases, B. cereus group species were isolated from patient stool samples. While no deaths, hospitalizations, or emergency room visits were reported from 169 cases from which information was obtained, 4 resulted in a visit to a health care provider (not including emergency room visits). More than 2/3 of 179 cases were female (69%), and 61% of cases fell within the 20–74 age group. In 156 of 179 total cases (87%), refried beans had been consumed.
Of 169 cases from which information was obtained, 88% reported vomiting, and more than half reported nausea and abdominal cramps (95 and 65%, respectively). However, in addition to vomiting, 38% of cases also reported diarrhea. Additional symptoms reported included (i) weakness (43%), (ii) chills (40%), (iii) dehydration (35%), (iv) headache (28%), (v) myalgia (muscle ache/pain; 16%), (vi) fever (16%), (vii) sweating (16%), and (viii) sore throat (3%). The incubation period observed for all cases ranged from 0.25 to 24 h, with a median of 2 h. The duration of illness ranged from 0.25 to 144 h, with a median estimate of 6 h.
A traceback was conducted, with the source of the outbreak determined to be a processing plant in Pennsylvania. The distributor in Pennsylvania packaged the refried beans specifically for the chain establishment where the outbreak occurred. The establishments where the outbreak occurred received 5 lb trays of pre-cooked, sealed, and frozen refried beans from the production/packaging facility. The refried beans would undergo cooking and a hot hold prior to consumption at the establishments where the outbreak occurred. It was determined that the refried beans were contaminated prior to preparation at the chain establishment.
Stool samples from suspect cases were cultured on MYP agar and B. cereus-like colonies were isolated from seven stool samples. Additionally, B. cereus-like colonies were isolated from nine food samples that were collected from five restaurants. In total, seven isolates from stool samples and 26 isolates from foods were confirmed to belong to the B. cereus group using standard microbiological methods. Isolates that were large Gram-positive rods, beta-hemolytic, and motile were presumptively identified as B. cereus-like. Additionally, spore staining was performed to test for the presence of parasporal crystals associated with B. thuringiensis, for which all isolates were negative. All 33 B. cereus group isolates underwent preliminary molecular characterization by Sanger sequencing of rpoB, which revealed two distinct allelic types belonging to phylogenetic groups III (rpoB allelic type AT 125) and IV (AT 92).
WGS Confirms Presence of Multiple B. cereus Group Species Represented Among Outbreak Strains
rpoB allelic types (ATs) assigned in silico were identical to those obtained using Sanger sequencing for all 33 isolates (Table 3). panC group assignment confirmed the presence of B. cereus s.l. isolates from multiple phylogenetic groups (Table 3), with panC group III (n = 30) and panC group IV (n = 3) represented among the 33 isolates. In silico MLST further resolved the group IV isolates into two sequence types (STs): the two strains isolated from refried beans served at two different restaurants had identical STs, while the single human isolate belonging to group IV had a unique ST (Table 3). All 30 panC group III isolates belonged to ST 26, including the remaining six human clinical isolates (Table 3).
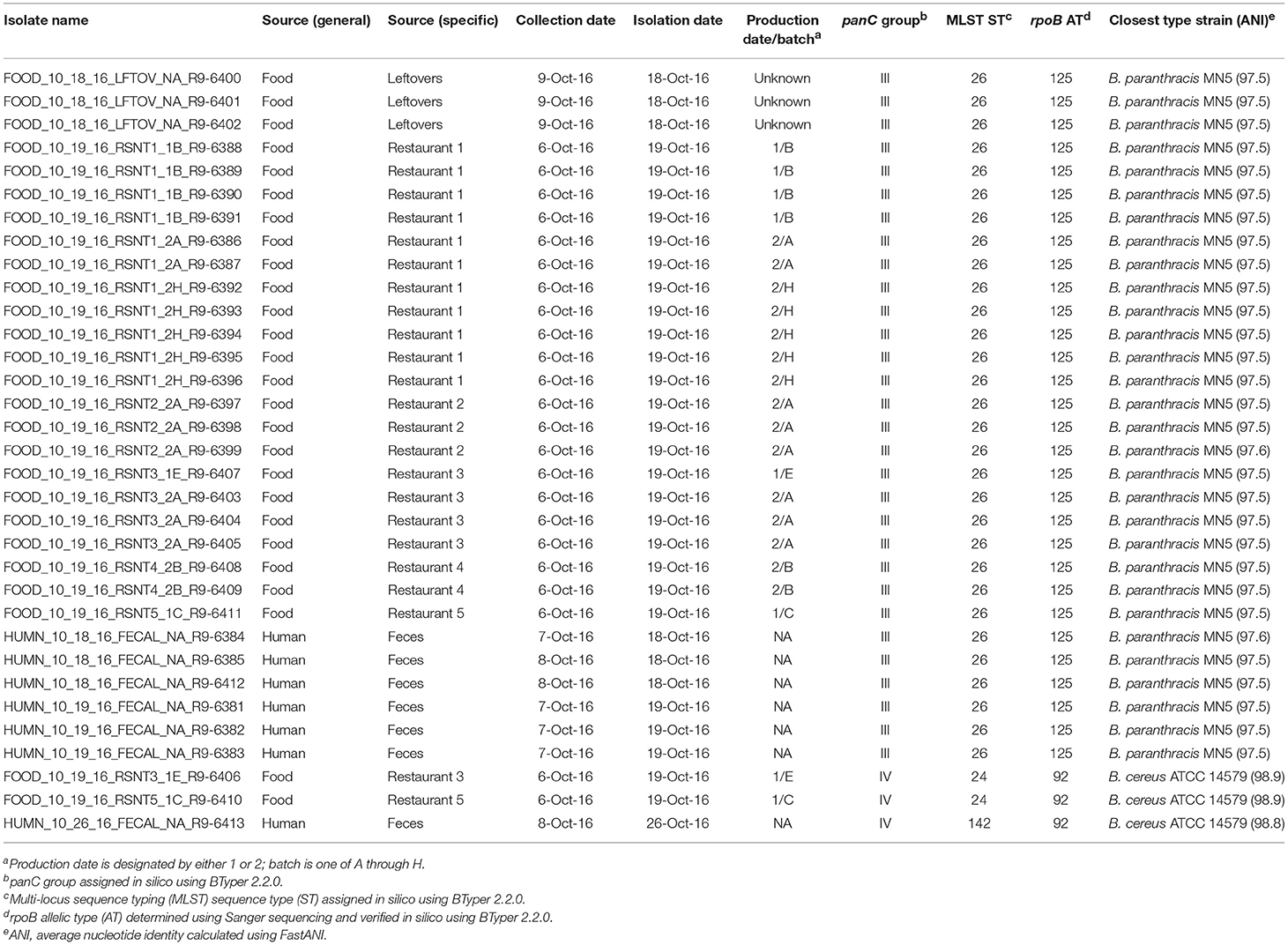
Table 3. List of outbreak isolates and corresponding metadata, single- and multi-locus sequence types, and species.
The presence of isolates from multiple B. cereus s.l. phylogenetic groups, as suggested by the rpoB, panC, and MLST loci among isolates sequenced in conjunction with this outbreak, was confirmed using core SNPs detected in all outbreak isolates, as well as the genomes of 18 currently recognized B. cereus group species (Figure 1). The three isolates assigned to panC group IV using a 7-group scheme (Guinebretière et al., 2008) were most closely related to the B. cereus s.s. type strain (Figure 1). All three group IV B. cereus isolates possessed diarrheal toxin genes hblABCD and cytK-2 at high identity and coverage (Figure 1), which code for enterotoxins hemolysin BL (Hbl) and cytotoxin K variant 2 (CytK-2), respectively. The 30 isolates assigned to panC group III, however, were most closely related to the type strain of B. paranthracis (Figure 1). Unlike the B. paranthracis type strain, all of the group III isolates investigated here were motile and possessed the cesABCD operon (Figure 1), which codes for emetic toxin-producing cereulide synthetase. In the case of isolate HUMN_10_18_16_FECAL_NA_R9-6384, cesD was split onto two contigs.
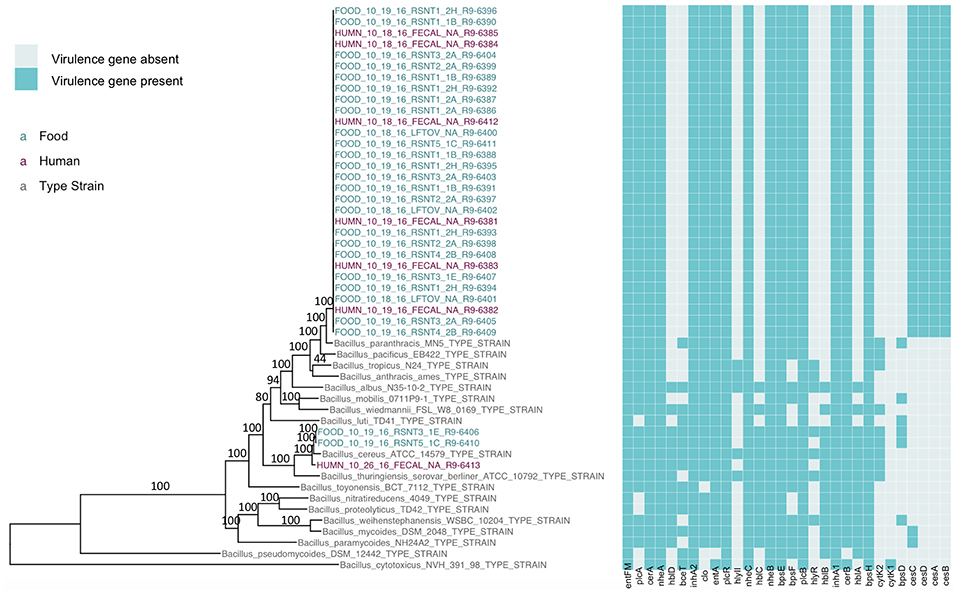
Figure 1. Maximum likelihood phylogeny of core SNPs identified in 33 isolates sequenced in conjunction with a B. cereus outbreak, as well as genomes of the 18 currently recognized B. cereus group species (shown in gray). Core SNPs were identified in all genomes using kSNP3. Heatmap corresponds to presence/absence of B. cereus group virulence genes detected in each sequence using BTyper. Tip labels in maroon and teal correspond to the seven human clinical isolates and 26 isolates from food sequenced in conjunction with this outbreak, respectively. Phylogeny is rooted at the midpoint, and branch labels correspond to bootstrap support percentages out of 500 replicates. Due to the short lengths and low bootstrap support (all values <10) of branches within the outbreak clade, bootstrap support percentages are not shown on branches within the outbreak clade.
Based on average nucleotide identity (ANI) values, the three diarrheal group IV isolates were classified as B. cereus s.s. (ANI > 95; Table 3). The 30 emetic group III isolates from this outbreak, however, most closely resembled the type strain of B. paranthracis (ANI > 95; Table 3), indicating that the emetic group III and diarrheal group IV isolates from this outbreak are different B. cereus group species.
Emetic and Diarrheal B. cereus Isolates Associated With the Foodborne Outbreak do Not Differ in Cytotoxicity
All three diarrheal strains isolated in conjunction with the outbreak (FSL R9-6406, FSL R9-6410, and FSL R9-6413) were found to produce Hbl, as well as non-hemolytic enterotoxin (Nhe). Characterization of six representatives of the emetic isolates tested (i.e., FSL R9-6381, FSL R9-6382, FSL R9-6384, FSL R9-6389, FSL R9-6395, and FSL R9-6399) revealed that they produced Nhe, but not Hbl. The supernatant of diarrheal B. cereus s.s. ATCC 14579 showed a stronger inhibitory effect on the viability of HeLa cells compared to supernatants of the 33 outbreak-associated isolates (Games-Howell P < 0.05; Figure 2). Furthermore, the viability of HeLa cells treated with 0.05% Triton X-100, the positive control, was significantly lower compared to viability of HeLa cells treated with bacterial supernatants (Games-Howell P < 0.05; Figure 2). Among all pairs of emetic isolates, only the viabilities of HeLa cells exposed to the supernatants of isolates FSL R9-6409 and FSL R9-6387 were found to differ (Games-Howell P < 0.05; Figure 2). The differences in HeLa cell viability after treatment with supernatants of these two emetic outbreak-associated strains are likely due to biological variability among replicates, as outbreak-associated emetic isolates were shown to be clonal (Figure 1). Taken together, the emetic group (represented by 30 emetic outbreak-associated isolates) had a mean cell viability of 97.5 ± 5.1%, while the diarrheal group (represented by three diarrheal outbreak-associated isolates) gave a mean cell viability of 101.4 ± 7.9%, as compared to the HeLa cells treated with BHI (i.e., negative control).
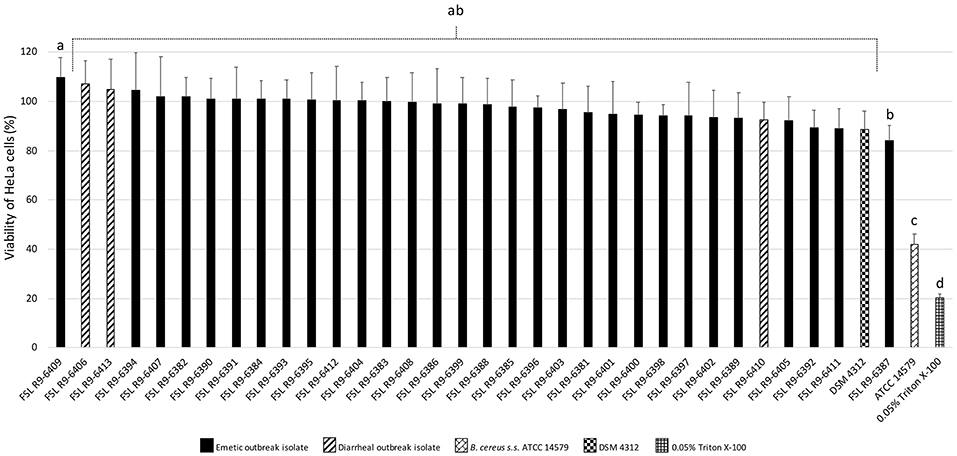
Figure 2. Percentage viability of HeLa cells when treated with supernatants of each isolate as determined by the WST-1 assay. Viability was calculated as ratio of corrected absorbance of solution when HeLa cells were treated with supernatants to the ratio of corrected absorbance of solution when HeLa cells were treated with BHI (i.e., negative control), converted to percentages. The columns represent the mean viabilities, while the error bars represent standard deviations for 12 technical replicates. Any two bars that do not share a common alphabetic character had significantly different percentage viability values (P < 0.05).
Core SNPs Identified Among B. cereus Group Outbreak Isolates From Two Phylogenetic Groups Are Dependent on Variant Calling Pipeline and Reference Genome Selection
To simulate a scenario in which genomes from a B. cereus outbreak spanning multiple phylogenetic groups were analyzed in aggregate, core SNPs were identified in all 33 outbreak isolates from groups III and IV (n = 30 and three isolates, respectively) using (i) combinations of five reference-based variant calling pipelines (Table 1) and three different reference genomes (Table 2) and (ii) a reference-free SNP calling method (Table 1). When genomes from all 33 isolates were analyzed together, the number of core SNPs identified by each pipeline and reference combination varied by up to several orders of magnitude (Figure 3), often with little agreement between pipelines in terms of the core SNPs they reported (Figure 4). Independent of reference genome, the CFSAN pipeline was the most conservative, consistently identifying the fewest number of core SNPs when all 33 isolates were queried in aggregate (50, 27, and 0 core SNPs using reference genomes from groups III, IV, and VII, respectively) (Figure 3). This can be contrasted with the Samtools, Freebayes, and Parsnp pipelines, which produced upwards of 100,000 core SNPs when the selected reference genome was a member of one of the groups being queried in the outbreak isolate set (group III and IV; Figure 3). In cases where a distant genome was used as the reference (group VII B. cytotoxicus type strain chromosome), all reference-based pipelines reported fewer core SNPs than kSNP3's reference-free k-mer based SNP calling approach (Figure 3).
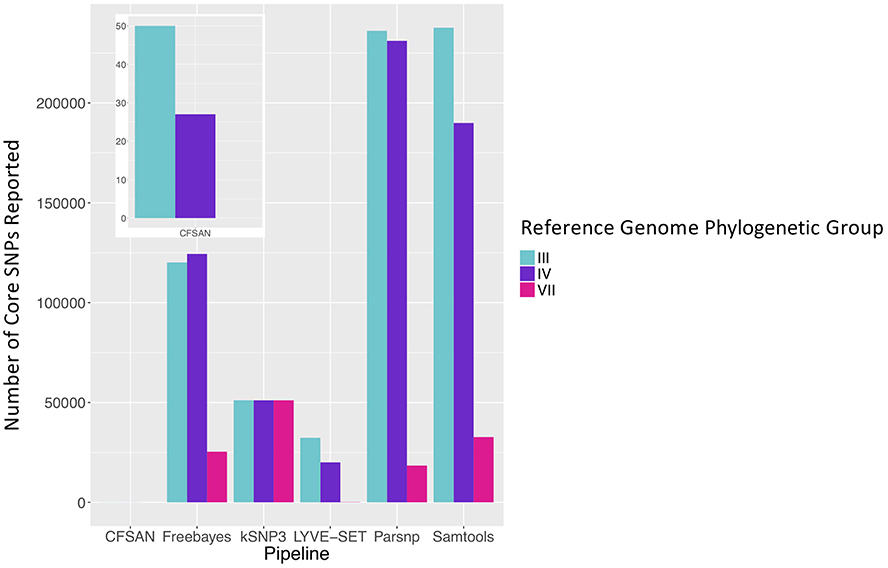
Figure 3. Number of core SNPs identified in 33 B. cereus group isolates from two phylogenetic groups (30 and 3 isolates from groups III and IV, respectively), sequenced in conjunction with a foodborne outbreak. Combinations of five reference-based variant calling pipelines and three reference genomes, as well as one reference-free SNP calling method (kSNP3), were tested.
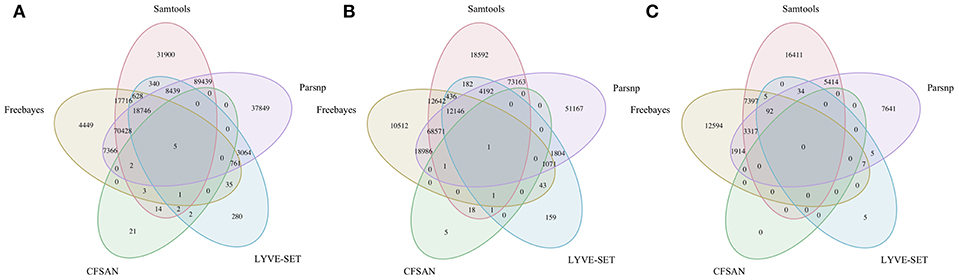
Figure 4. Comparison of core SNP positions reported by five reference-based variant-calling pipelines for 33 B. cereus group strains isolated in association with a foodborne outbreak, with the chromosomes of (A) B. cereus AH187 (group III), (B) B. cereus s.s. ATCC 14579 (group IV), and (C) B. cytotoxicus NVH 391-98 (group VII) used as reference genomes. Ellipses represent each pipeline.
Choice of Variant Calling Pipeline Has Greater Influence on Core SNP Identification Than Choice of Closely Related Closed or Draft Reference Genome for Emetic Group III B. cereus Group Isolates
The 30 emetic group III isolates were queried in the absence of their group IV counterparts using combinations of five reference-based variant calling pipelines (Table 1) and two reference genomes (the closed chromosome of B. cereus AH187, with and without dustmasking, and contigs of one of the isolates identified in this outbreak, with and without dustmasking; Table 2) and one reference-free SNP calling method (Table 1). In this scenario, the choice of variant calling pipeline had a greater effect on the number of core SNPs obtained than the choice of reference genome, as both reference genomes possessed the same virulence gene profile (virulotype), rpoB AT, panC group, MLST sequence type, and were of the same species (B. paranthracis; ANI > 95%) as the 30 emetic isolates (Figure 5A). Congruent with this, the number of pairwise core SNP differences between emetic isolates sequenced in this outbreak varied more with the selection of variant calling pipeline than with reference genome (Figure 6A). When the unmasked closed chromosome of B. cereus AH187 was used as a reference, pairwise core SNP differences among emetic isolates from this outbreak ranged from 0 to 8 (mean of 2.9; CFSAN), 7 to 29 (mean of 16.1; Freebayes), 0 to 8 (mean of 2.8; LYVE-SET), 0 to 64 (mean of 23.6; Parsnp), and 1 to 16 SNPs (mean of 8.2; Samtools) (Figure 5A). Using the reference-free kSNP3 pipeline, this range was 1–46 SNPs (mean of 16.7; Figure 5A). The CFSAN and LYVE-SET pipelines produced nearly identical results in terms of the number and identity of the core SNPs called (23 and 22 SNPs, respectively, 20 of which were detected by both pipelines; Figure 7), as well as the topologies of the phylogenies those SNPs produced: all CFSAN and LYVE-SET phylogenies were more similar to each other than what would be expected by chance (Table 4 and Supplementary Table S4). Additionally, the two methods that relied on assembled genomes rather than short reads for SNP calling (kSNP3 and Parsnp) produced the greatest numbers of core SNPs (Figure 5A).
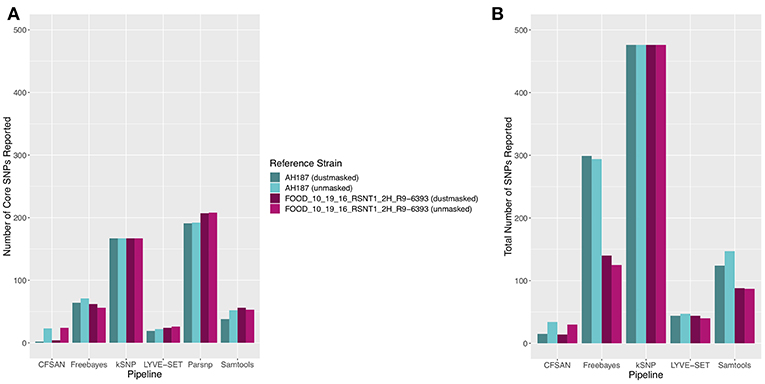
Figure 5. (A) Number of core SNPs and (B) total number of SNPs identified in 30 emetic B. cereus group III strains isolated in association with a foodborne outbreak. Combinations of (A) five and (B) four reference-based variant calling pipelines and two reference genomes (either dustmasked or unmasked) were tested, along with one reference-free SNP calling method (kSNP3). Because the Parsnp pipeline reports core SNPs by definition, it was excluded from Figure 5B (total SNPs). For quantification of the total number of SNPs (Figure 5B), all sites with more than one unique character were counted.
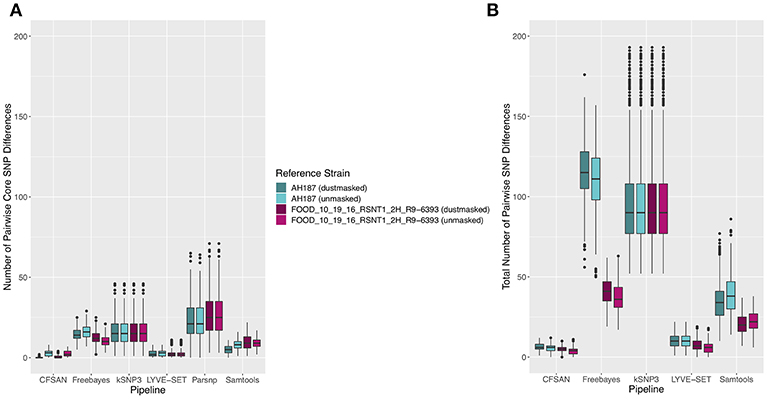
Figure 6. Ranges of pairwise (A) core SNP differences and (B) total SNP differences between 30 emetic group III B. cereus group strains isolated in conjunction with a foodborne outbreak. Combinations of (A) five and (B) four reference-based variant calling pipelines and two reference genomes (either dustmasked or unmasked), as well as one reference-free SNP calling method (kSNP3) were tested. Lower and upper box hinges correspond to the first and third quartiles, respectively. Lower and upper whiskers extend from the hinge to the smallest and largest values no more distant than 1.5 times the interquartile range from the hinge, respectively. Points represent pairwise distances that fall beyond the ends of the whiskers. Because the Parsnp pipeline reports core SNPs by definition, it was excluded from Figure 6B (pairwise differences in total SNPs). For quantification of pairwise differences in the total number of SNPs (Figure 6B), all sites with more than one unique character were included.
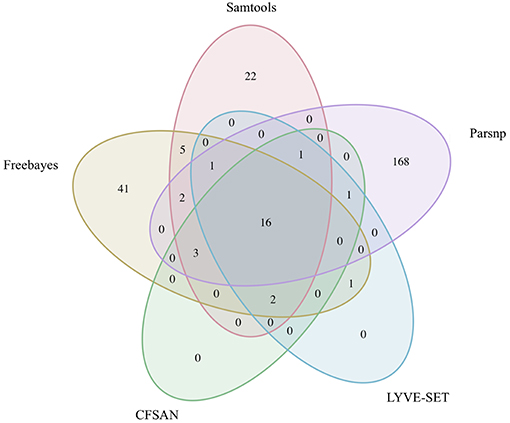
Figure 7. Comparison of core SNP positions reported by five variant-calling pipelines for 30 emetic group III B. cereus group outbreak isolates. Ellipses represent each pipeline, all of which used the chromosome of emetic group III B. cereus AH187 as a reference for variant calling.

Table 4. Maximum likelihood phylogenies of 30 emetic group III outbreak isolates considered to be more topologically similar than would be expected by chance (P < 0.05)a.
Within the emetic group III isolates associated with this outbreak, a total of 32 core SNPs were identified by two or more of the reference-based variant calling pipelines when the unmasked B. cereus AH187 genome was used as a reference, half of which were identified by all five pipelines (Figure 7). Out of these 32 SNPs, 23 were identified in protein coding genes, 14 of which produced non-synonymous amino acid changes (Supplementary Table S5). Genes with non-synonymous changes were involved in molybdopterin biosynthesis (WP_000544623.1), proteolysis (WP_000215096.1 and WP_000857793.1), chitin binding (WP_000795732.1), iron-hydroxamate transport (WP_000728195.1), DNA repair (WP_000947749.1 and WP_000867556.1), DNA replication (WP_000867556.1 and WP_000435993.1), protein transport and insertion into the membrane (WP_000727745.1), and glyoxylase/bleomycin resistance (WP_000800664.1).
In addition to detecting core SNPs in the genomes of the 30 emetic group III isolates, total (core and accessory) SNPs were detected in the 30 emetic group III genomes using combinations of four reference-based variant calling pipelines (Parsnp, which only reports core SNPs, was excluded; Table 1) and two reference genomes (the closed chromosome of B. cereus AH187 and contigs of one of the isolates identified in this outbreak, with and without dustmasking; Table 2) and one reference-free SNP calling method (Table 1). When total SNPs were accounted for, rather than solely core SNPs, all pipeline/reference genome combinations showed increases in the number of SNPs detected and the range of pairwise SNP differences between genomes (Figures 5B, 6B). Whether the addition of accessory SNPs translated into a significant difference in phylogenetic topology, however, depended on the variant calling pipeline used. When the B. cereus AH187 closed chromosome was used as a reference, SNPs detected using the LYVE-SET pipeline produced phylogenies considered to be more topologically similar than would be expected by chance (Kendall-Colijn test P < 0.05), regardless of whether core SNPs or total SNPs were used to construct the phylogeny, and regardless of whether the B. cereus AH187 reference genome was dustmasked or not (Table 4 and Supplementary Table S4). Additionally, all phylogenies produced using the LYVE-SET pipeline and the B. cereus AH187 reference genome (i.e., each combination of core SNPs, total SNPs, dustmasked reference, and unmasked reference) were topologically similar to those produced using the CFSAN pipeline and the unmasked B. cereus AH187 reference genome, regardless of whether all SNPs were included or solely core SNPs (Table 4 and Supplementary Table S4). Other topologically similar phylogeny pairs included phylogenies constructed using (i) core SNPs identified with Freebayes, regardless of whether a dustmasked reference genome was used or not, and (ii) core SNPs identified with Parsnp, regardless of whether a dustmasked reference was used or not (Kendall-Colijn test P < 0.05; Table 4 and Supplementary Table S4).
Phylogenies Constructed Using Core SNPs Identified in 55 Emetic ST 26 B. cereus Genomes by kSNP3 and Parsnp Yield Similar Topologies
To compare the 30 emetic strains from this outbreak to other emetic group III isolates, all emetic group III assembled genomes with ST 26 were downloaded from NCBI. This produced a total of 55 emetic group III isolates with ST 26 (30 isolates from this outbreak and 25 from NCBI RefSeq). Among the 55 emetic ST 26 genomes, Parsnp identified almost twice as many core SNPs as kSNP3 (4,597 and 2,593 core SNPs, respectively). However, the topologies of phylogenies produced using the core SNPs identified by each pipeline were found to be more similar than would be expected by chance (Kendall-Colijn test P < 0.05; Figure 8).

Figure 8. Maximum likelihood phylogenies of 30 emetic group III isolates (ST 26) sequenced in conjunction with a B. cereus outbreak, as well as all other emetic group III ST 26 genomes available in NCBI (n = 25; shown in black). Trees were constructed using core SNPs identified using (A) kSNP3 or (B) Parsnp. Tip labels in maroon and teal correspond to the six human clinical isolates and 24 isolates from food sequenced in conjunction with this outbreak, respectively. Branch labels correspond to bootstrap support percentages out of 1,000 replicates. Due to the short lengths and low bootstrap support of branches within the outbreak clade, bootstrap support percentages are not shown on branches within the outbreak clade.
Based on pairwise core SNP differences, the publicly available genomes showed greater variability than the outbreak isolates described here, regardless of whether kSNP3 or Parsnp was used for variant calling (ANOVA-like permutation test P < 0.05; Supplementary Figure S1). Pairwise core SNP differences of the 30 emetic group III isolates from this outbreak ranged from 0 to 25 SNPs (mean of 8.3) and 0 to 44 SNPs (mean of 11.9) when the kSNP3 and Parsnp pipelines were used, respectively (Supplementary Figure S1). For external ST 26 isolates not associated with this outbreak, pairwise core SNP differences ranged from 0 to 1,474 SNPs (mean of 425.7) and 0 to 3,111 SNPs (mean of 828.3) when kSNP3 and Parsnp were used, respectively (Supplementary Figure S1). Between these two groups (the 30 emetic isolates from this outbreak and the 25 external emetic ST 26 isolates), pairwise core SNP differences ranged from 73 to 1,258 SNPs (mean of 301.7; kSNP3) and 74 to 2,709 SNPs (mean of 528.0; Parsnp) (Supplementary Figure S1). Reflecting this, the average of the ranks of pairwise SNP distances within emetic isolates from this outbreak was less than the average of the ranks of pairwise SNP distances between the emetic isolates from this outbreak and the external ST 26 isolates (ANOSIM P < 0.05). This is likely a result of the differences in variance between the outbreak and external ST 26 isolates, as supported by the results of the ANOVA-like permutation test (Anderson and Walsh, 2013).
Discussion
While B. cereus causes a considerable number of foodborne illness cases annually, outbreaks are rarely investigated with the methodological rigor (e.g., use of WGS) that is increasingly used for surveillance and outbreak investigations targeting other foodborne pathogens. A specific challenge in the U.S. is that, unlike for some other diseases, disease cases caused by B. cereus are typically not reportable, even though foodborne illnesses, regardless of etiology, are reportable in some states, including NY. This, combined with the typically mild course of B. cereus infection, means that human B. cereus isolates are rarely available for WGS. Furthermore, even if clinical B. cereus group isolates are available, WGS may not be used for isolate characterization in cases where infections are mild. Due to the availability of B. cereus isolates for seven human cases, the outbreak reported here presented a unique opportunity to pilot the use of WGS for investigation of B. cereus outbreaks. The data and approaches presented here will not only facilitate future investigation of other B. cereus outbreaks but will also help with application of WGS for investigation of other foodborne disease outbreaks where limited reference WGS data and information on genomic diversity are available.
Addressing the Microbiological and Epidemiological Challenges Associated With Determining the Causative Agent of an Emetic Foodborne Outbreak
The agar MYP used for isolation of strains from food and human clinical samples in the outbreak reported here is one of the two selective differential agars recommended in the FDA BAM protocol for the isolation of B. cereus group strains (Tallent et al., 2012b). The second recommended agar, Bacara, has been shown to be more selective and more effective in suppressing the growth of other Gram-positive microorganisms that may be present in tested samples (e.g., other Bacillus species, Listeria, Staphylococcus) (Tallent et al., 2012a; Kabir et al., 2017). Since Bacara medium has a proprietary formula and cannot be purchased in a dehydrated powder form (Tallent et al., 2012b), it is less likely to be readily available for use in labs that do not routinely test for B. cereus group species. Use of both types of media may increase the success of B. cereus group isolation from food and clinical samples, especially isolation of emetic strains (Ehling-Schulz et al., 2005; Ceuppens et al., 2013). Furthermore, the isolation of B. cereus group strains associated with this outbreak was carried out at 37°C, which is higher than the temperature of 30°C that is recommended by the FDA BAM (Tallent et al., 2012b). Nevertheless, while incubation at this temperature may inhibit the growth of psychrotolerant species of the B. cereus group (e.g., B. weihenstephanensis), it is not expected to interfere with the isolation of B. cereus group strains that are able to grow at human body temperature and cause toxicoinfection. It is also not expected to compromise isolation of emetic isolates with the capacity to cause intoxication, as emetic strains have been previously found primarily in phylogenetic group III, which does not contain psychrotolerant strains (Carroll et al., 2017). Overall, use of both types of isolation media and a moderate incubation temperature of 30°C may minimize the isolation bias.
While the isolation of B. cereus group strains from food and clinical samples is essential for linking them to a potential foodborne outbreak, further information is needed to definitively prove that an outbreak was caused by B. cereus. Emetic disease caused by members of the B. cereus group can be attributed to the production of the highly heat- and pH-resistant toxin cereulide in food prior to ingestion (Ehling-Schulz et al., 2004, 2015; Stenfors Arnesen et al., 2008). Because cereulide is produced within the food matrix itself, prior to consumption, the mere presence of emetic B. cereus group strains in food or human clinical samples cannot definitively prove that an outbreak was caused by a member of the B. cereus group; rather, the presence of cereulide itself is essential for linking food and clinical samples to an outbreak with high confidence (Andersson et al., 2004; Stenfors Arnesen et al., 2008). For this outbreak, the presence of cereulide in food and human clinical samples linked to the outbreak was not assessed, as testing for cereulide is not currently included in the BAM protocol as a routine method for the detection and enumeration of B. cereus in food. Ergo, there is no definitive proof that the outbreak was caused by cereulide-producing emetic group III B. cereus and not a similar foodborne pathogen (e.g., enterotoxins produced by Staphylococcus aureus, which manifest in similar symptoms to those associated with cereulide) (Messelhäusser et al., 2014). However, due to the presence of highly clonal, ces-positive group III ST 26 B. cereus group isolates among food and clinical samples linked to the outbreak, as well as epidemiological data that support this, the emetic strain is the most probable causative agent. While it is not currently included in the BAM protocol for B. cereus isolation (Tallent et al., 2012b), testing for the presence of cereulide in food and clinical samples linked to potential outbreaks caused by emetic B. cereus can aid in providing a definitive link between illness and causative agent.
Considerations for Addressing the Unique Challenges Associated With Characterization of Foodborne Outbreaks Linked to the B. cereus Group Using WGS
In B. cereus outbreaks, interpretation of WGS data can be challenging, especially in cases where strains of multiple closely related species or subtypes appear to be associated with an outbreak. B. cereus outbreaks—particularly emetic outbreaks caused by cereulide-producing B. cereus group isolates—are often associated with improper handling of food (e.g., temperature abuse) (Ehling-Schulz et al., 2004; Stenfors Arnesen et al., 2008). This, and their ubiquitous presence in the environment, make it important to consider the possibility of a multi-strain or multi-species outbreak in addition to a single-source outbreak caused by a single strain. In the outbreak characterized here, B. cereus group strains from two phylogenetic groups, III and IV, were isolated from both human clinical stool samples, as well as refried beans linked to the outbreak. The separation of outbreak-related isolates into three diarrheal group IV isolates (representing two distinct STs) and 30 emetic isolates may be explained by one of the following scenarios: (i) the outbreak was caused by refried beans contaminated with multiple B. cereus group species (isolates from groups III and IV), both of which caused illness in humans, (ii) in addition to housing emetic outbreak strains that belonged to group III, samples of refried beans and patient stool samples harbored group IV B. cereus s.l. isolates that were not part of the outbreak but were incidentally isolated from stool and food samples, or (iii) a subset of patient stool samples and food samples did not harbor B. cereus s.l. group III isolates belonging to the outbreak, but did harbor group IV strains that were isolated and sequenced. In order to determine which of these scenarios explains the presence of multiple B. cereus group species among isolates sequenced in conjunction with a foodborne outbreak, additional epidemiological and microbiological data are needed.
Valuable metrics for inclusion/exclusion of B. cereus group cases in a foodborne outbreak include patient exposure, patient symptoms (e.g., vomiting, diarrhea, onset and duration of illness), levels of B. cereus present in implicated food and patient samples (CFU/g or CFU/ml), cytotoxicity of isolates, and the approach used to select bacterial colonies to undergo WGS (Glasset et al., 2016). However, some of these data may be more valuable than others. In their characterization of 564 B. cereus group strains associated with 140 strong-evidence foodborne outbreaks in France between 2007 and 2014, Glasset et al. (2016) found that patient symptoms could not be associated with the presence of emetic and diarrheal strains. More than half (57%) of the B. cereus outbreaks queried in their study included patients exhibiting both emetic and diarrheal symptoms. Similar results were observed here, as emetic and diarrheal symptoms were reported in 88 and 38% of cases, respectively, with both vomiting and diarrhea reported by multiple patients. All emetic isolates associated with this outbreak carried nhe genes and also produced Nhe enterotoxin, as determined using the immunoassay. While it has been proposed that a combination of emetic and diarrheal symptoms may be due to the fact that emetic group III isolates have been shown to produce diarrheal enterotoxin Nhe at high levels (Glasset et al., 2016), incongruences between isolate virulotype and patient symptoms may still exist. Importantly, this indicates the need for further investigation of factors affecting the expression of B. cereus group virulence genes, as well as their potential synergistic activities (Doll et al., 2013).
Another metric that can be used for determining whether B. cereus group isolates are part of an outbreak or not is the level of B. cereus present in the implicated food. Like patient symptoms, B. cereus counts from implicated foods may aid in an outbreak investigation, but likely cannot definitively prove whether an isolate is part of an outbreak or not. For example, outbreaks caused by implicated foods with B. cereus counts of <103 CFU/g and as low as 400 CFU/g for diarrheal and emetic diseases, respectively, have been described (Glasset et al., 2016), despite levels of at least 105 CFU/g often being detected in implicated foods (Stenfors Arnesen et al., 2008). The levels of B. cereus present in refried beans in the outbreak described here were not determined. However, like patient symptoms, B. cereus count data may be a useful supplemental metric for investigating B. cereus group outbreaks in the future.
In addition to patient symptoms and pathogen load in the food, incubation period can be used to determine whether an isolate is part of an outbreak or not, as it is significantly shorter for emetic strains than diarrheal strains (Ehling-Schulz et al., 2004; Stenfors Arnesen et al., 2008; Glasset et al., 2016). In the outbreak described here, the patient from which a non-emetic group IV B. cereus group strain was isolated reported an incubation time of 1 h, the lowest incubation time of all seven confirmed human clinical cases. However, this is still within the observed range of incubation times for emetic B. cereus disease (0.5–6 h) (Stenfors Arnesen et al., 2008). Although no emetic group III B. cereus s.l. strain was isolated from the clinical sample, it is possible that the patient could have been intoxicated with cereulide produced in the food by the emetic B. cereus strain that caused the outbreak. However, it is also possible that a pathogen which causes similar symptoms to foodborne illness caused by emetic B. cereus was responsible for the patient's illness (e.g., Staphylococcus aureus).
Lastly, cytotoxicity data may also be leveraged to include/exclude outbreak-associated B. cereus group isolates. In the outbreak described here, the patient from which a non-emetic group IV B. cereus group strain was isolated reported vomiting and nausea and no diarrheal symptoms, despite the clinical isolate's possession of multiple diarrheal toxin genes and no emetic toxin genes. This could suggest that the patient was intoxicated with the cereulide, but the isolate itself did not survive the passage through the patient's gastrointestinal tract, or that it survived in a low concentration that resulted in failure of isolation on MYP. It is also possible that our understanding of the specific virulence genes responsible for different B. cereus-associated disease symptoms is still incomplete and that the diarrheal isolate obtained from the clinical sample was in fact responsible for symptoms of vomiting and nausea. To further investigate this, we carried out immunoassay-based detection of Hbl and Nhe enterotoxins, as well as a WST-1 proliferation assay with HeLa cells exposed to bacterial supernatants presumably containing toxins. The results of Hbl and Nhe immunodetection and cytotoxicity revealed that diarrheal isolates only had mild detrimental effects on HeLa cell viability, despite the fact that they produced both hemolysin BL and non-hemolytic enterotoxins. This can be contrasted with the B. cereus s.s. type strain, which substantially reduced the viability of the HeLa cells.
For the outbreak described here, results obtained using a combination of microbiological, epidemiological, and bioinformatic methods indicate that hypothesis (i), in which the diarrheal strains were part of a multi-species outbreak, can likely be excluded. Evidence supporting the conclusion that the human clinical diarrheal isolate was not part of the outbreak described here include: (i) the emetic symptoms reported by the patient were incongruent with the virulotype of the isolate, (ii) the incubation time was typical for intoxication, (iii) the human clinical diarrheal isolate had a different MLST ST compared to all other isolates sequenced in this outbreak, and (iv) the human diarrheal isolate did not exhibit substantial cytotoxicity against HeLa cells (Figure 2). This may be due to the fact that this case was not part of the outbreak and was due to an infection or intoxication caused by another pathogen that leads to disease symptoms similar to B. cereus (e.g., Staphylococcus aureus), or that a group IV B. cereus strain was isolated and sequenced in lieu of the group III emetic outbreak isolate. There is limited evidence as to whether humans can be asymptomatic carriers of group IV B. cereus (Ghosh, 1978; Turnbull and Kramer, 1985), making it likely that isolation and sequencing of a group IV B. cereus strain could be due to the use of MYP agar as the sole selective agar, which has been shown to hinder detection of emetic B. cereus group isolates (Ehling-Schulz et al., 2005; Ceuppens et al., 2013). In future outbreaks, the use of additional selective media (e.g., Bacara agar), enrichment media, and isolation temperatures may aid in isolation of the causative B. cereus group strain.
While we have shown here that WGS data can be a valuable tool for characterizing B. cereus group isolates from a foodborne outbreak, our results also showcase the importance of supplementing WGS data with epidemiological and microbiological metadata to draw meaningful conclusions from B. cereus group genomic data. Furthermore, the availability of WGS and cytotoxicity data from a larger set of B. cereus isolates from symptomatic patients may also provide an opportunity to use comparative genomics approaches to further explore virulence genes that are linked to different disease outcomes in the future.
Recommendations for Analyzing Illumina WGS Data From B. cereus Group Isolates Potentially Linked to a Foodborne Outbreak
WGS is being used increasingly to characterize isolates associated with foodborne disease cases and outbreaks, and rightfully so, as it offers the ability to characterize foodborne pathogens at unprecedented resolution, and it has been able to improve outbreak and cluster detection for numerous foodborne pathogens (Allard et al., 2017; Kovac et al., 2017; Moran-Gilad, 2017; Taboada et al., 2017), including Salmonella enterica (Taylor et al., 2015; Hoffmann et al., 2016; Gymoese et al., 2017), Escherichia coli (Grad et al., 2012; Holmes et al., 2015; Rusconi et al., 2016), and Listeria monocytogenes (Jackson et al., 2016; Kwong et al., 2016; Chen et al., 2017a,b; Moura et al., 2017). However, as demonstrated here and elsewhere, variant calling pipelines and the various mapping/alignment, SNP calling, and SNP filtering practices that they employ (e.g., removal of recombination and clustered SNPs) can influence the identification of SNPs in WGS data and, thus, the topology of a resulting phylogeny (Pightling et al., 2014, 2015; Croucher et al., 2015; Hwang et al., 2015; Katz et al., 2017; Sandmann et al., 2017). This can be particularly problematic for outbreak and cluster detection in bacterial pathogen surveillance: pairwise SNP thresholds are currently widely used to make initial decisions regarding the inclusion or exclusion of isolates in a given outbreak (Taylor et al., 2015; Gymoese et al., 2017; Mair-Jenkins et al., 2017; McCloskey and Poon, 2017; Walker et al., 2018). In such scenarios, just a few SNPs can be the deciding factor in whether a bacterial pathogen is included or excluded as part of an outbreak or cluster (Katz et al., 2017), rendering the choice of variant calling method as non-trivial. Furthermore, choosing an appropriate variant calling pipeline can be particularly challenging for pathogens where there are limited data and expertise with WGS, as is currently the case with B. cereus.
As demonstrated here, the choice of variant calling pipeline can greatly influence the number of core SNPs identified in B. cereus group isolates associated with a foodborne outbreak. In the case of a multi-group outbreak, this effect can be magnified. Naively calling variants in isolates that span multiple B. cereus s.l. phylogenetic groups in aggregate can lead to orders of magnitudes of difference in the number of core SNPs identified by different variant calling pipelines/reference genome combinations. In a multi-group outbreak scenario, it is essential to note that one is effectively dealing with genomic data from multiple species (i.e., ANI < 95), making it impossible to find a reference genome that is closely related to all isolates in a putative outbreak. In the case of some reference-based pipelines that are specifically tailored to identify variants in bacterial isolates from outbreaks (e.g., CFSAN, which is not suited for bacteria differing by more than a few hundred SNPs), calling variants in multiple groups or within a distant reference genome is inappropriate (Davis et al., 2015). Thus, querying outbreak isolates from multiple groups in aggregate using reference-based variant calling methods should be avoided. Furthermore, the results presented here showcase the value of employing single- and/or multi-locus typing approaches prior to variant calling, either via Sanger sequencing or in silico using tools, such as BTyper, as they can aid the design of downstream bioinformatics analyses, including reference genome selection and data partitioning by phylogenetic group.
When the three phylogenetic group IV isolates were excluded from analyses, leaving only the emetic group III isolates, the selection of reference genome caused fewer core SNP discrepancies than choice of variant calling pipeline, provided the reference genome was “similar” to the genomes analyzed. While the selection of a reference genome for reference-based variant calling is not trivial (Pightling et al., 2014; Olson et al., 2015), reference-based variant calling using a closed chromosome (B. cereus AH187) and a draft genome (FOOD_10_19_16_RSNT1_2H_R9-6393) from two isolates that were closely related to, or among the emetic group III isolates sequenced in this outbreak produced nearly identical results in terms of the number and identity of core SNPs detected. Both reference genomes were identical to the emetic group III outbreak isolates sequenced here in terms of panC group, rpoB AT, MLST ST, and virulotype. Additionally, the closed chromosome and draft genome had ANI values of >99.8 and 99.9, respectively, relative to all emetic group III outbreak isolates in this study, which can be considered highly similar. Comparable findings have been observed in analyses of Salmonella enterica serovar Heidelberg WGS data (Usongo et al., 2018), suggesting that either closed genomes or high-quality draft genomes are adequate for reference-based SNP calling, provided both are similar enough to the outbreak strains being queried. While the thresholds at which reference genomes become “similar enough” and of sufficient quality for reference-based SNP calling for outbreak detection warrant further investigation, we have demonstrated here that, for emetic group III ST 26 B. cereus group genomes, the publicly available closed chromosome of B. cereus AH187 can serve as an adequate standard.
With regard to differences in the number of core SNPs identified in the 30 emetic group III isolates using different variant calling pipelines, the pipelines that used assembled genomes as input (kSNP3 and Parsnp) produced higher numbers of core SNPs than their counterparts that relied on short Illumina reads. Additionally, when used to query core SNPs in 55 emetic group III ST 26 B. cereus group genomes, both kSNP3 and Parsnp produced core SNPs that yielded topologically similar phylogenies. kSNP3 employs a reference-free k-mer based SNP calling approach (Gardner and Hall, 2013; Gardner et al., 2015), while Parsnp uses a reference-based core genome alignment approach (Treangen et al., 2014), and both are useful for calling variants in large data sets. These approaches are also valuable when reads are not available for SNP calling (Olson et al., 2015), as demonstrated here by the comparison of outbreak genomes with publicly available genomes: core SNPs obtained using both kSNP3 and Parsnp were able to consistently produce phylogenies in which the 30 emetic isolates from this outbreak formed a well-supported clade among all emetic group III ST 26 B. cereus group genomes. However, kSNP3 has been shown to lack specificity relative to other pipelines (i.e., CFSAN, LYVE-SET) when differentiating outbreak isolates from non-outbreak isolates for L. monocytogenes, E. coli, and S. enterica (Katz et al., 2017). Here, the CFSAN and LYVE-SET pipelines identified similar SNPs that produced highly congruent phylogenies. This is unsurprising, considering that both the CFSAN and LYVE-SET pipelines were designed specifically for identifying SNPs in closely related strains from outbreaks (Katz et al., 2017), and both employ the most stringent filtering criteria of all pipelines tested here.
As WGS Becomes Routinely Integrated Into Food Safety, Clinical, and Epidemiological Realms, It Is Likely That the Number of Illnesses Attributed to B. cereus Will Increase
Here, we offer the first description of a foodborne outbreak caused by B. cereus group species to be characterized using WGS, and we provide a glimpse into the genomic variation one might expect within an emetic group III B. cereus outbreak using several different variant calling pipelines. However, our ability to query emetic group III genomes outside of this outbreak is limited by the lack of publicly available genomic data and metadata from emetic isolates. Of the 2,156 B. cereus group genomes available in NCBI's RefSeq database as of March 2018, only 29 were from group III and possessed the cesABCD operon, 25 of which belonged to MLST ST 26. While not ideal, this is an improvement, as there were only 19 emetic group III genomes available in NCBI's Genbank database in April 2017 (Carroll et al., 2017). As more B. cereus group WGS data—particularly, data from emetic B. cereus group isolates—become publicly available, more outbreaks and clusters are likely to be resolved in tandem, a phenomenon that has been observed for L. monocytogenes (Jackson et al., 2016). Additionally, variant calling and cluster/outbreak detection methods for characterizing B. cereus group isolates from foodborne outbreaks can be further refined and optimized as more WGS, metadata and epidemiological data become available for clinical and non-clinical isolates.
Author Contributions
LC performed computational analyses. MM, LM, ND, and JC performed microbiological experiments. DN provided and interpreted epidemiological data. MW and JK conceived the study. LC, MW, and JK co-wrote the manuscript.
Funding
This material is based on work supported by the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1144153. This work was supported also by the USDA National Institute of Food and Agriculture Hatch Appropriations under Project #PEN04646 and Accession #1015787, and Penn State Huck Institutes of the Life Sciences that supported the whole-genome sequencing through the Penn State Genomics Core Facility.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the Wadsworth Center Tissue Culture & Media Core for providing the media used in this work, and Dr. Joshua Lambert from The Pennsylvania State University for providing tissue culture laboratory facility and advising.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00144/full#supplementary-material
References
Allard, M. W., Bell, R., Ferreira, C. M., Gonzalez-Escalona, N., Hoffmann, M., Muruvanda, T., et al. (2017). Genomics of foodborne pathogens for microbial food safety. Curr. Opin. Biotechnol. 49, 224–229. doi: 10.1016/j.copbio.2017.11.002
Anderson, M. J., and Walsh, D. C. I. (2013). PERMANOVA, ANOSIM, and the mantel test in the face of heterogeneous dispersions: what null hypothesis are you testing? Ecol. Monographs 83, 557–574. doi: 10.1890/12-2010.1
Andersson, M. A., Jääskeläinen, E. L., Shaheen, R., Pirhonen, T., Wijnands, L. M., and Salkinoja-Salonen, M. S. (2004). Sperm bioassay for rapid detection of cereulide-producing Bacillus cereus in food and related environments. Int. J. Food Microbiol. 94, 175–183. doi: 10.1016/j.ijfoodmicro.2004.01.018
Ashton, P., Nair, S., Peters, T., Tewolde, R., Day, M., Doumith, M., et al. (2015). Revolutionising public health reference microbiology using whole genome sequencing: Salmonella as an exemplar. bioRxiv 033225. doi: 10.1101/033225
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bennett, S. D., Walsh, K. A., and Gould, L. H. (2013). Foodborne disease outbreaks caused by Bacillus cereus, Clostridium perfringens, and Staphylococcus aureus–United States, 1998–2008. Clin. Infect. Dis. 57, 425–433. doi: 10.1093/cid/cit244
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bruen, T. C., Philippe, H., and Bryant, D. (2006). A simple and robust statistical test for detecting the presence of recombination. Genetics 172, 2665–2681. doi: 10.1534/genetics.105.048975
Carroll, L. M., Kovac, J., Miller, R. A., and Wiedmann, M. (2017). Rapid, high-throughput identification of anthrax-causing and emetic Bacillus cereus group genome assemblies using BTyper, a computational tool for virulence-based classification of Bacillus cereus group isolates using nucleotide sequencing data. Appl. Environ. Microbiol. 83:e01096-17. doi: 10.1128/AEM.01096-17
Castiaux, V., Liu, X., Delbrassinne, L., and Mahillon, J. (2015). Is cytotoxin K from Bacillus cereus a bona fide enterotoxin? Int. J. Food Microbiol. 211, 79–85. doi: 10.1016/j.ijfoodmicro.2015.06.020
Ceuppens, S., Boon, N., and Uyttendaele, M. (2013). Diversity of Bacillus cereus group strains is reflected in their broad range of pathogenicity and diverse ecological lifestyles. FEMS Microbiol. Ecol. 84, 433–450. doi: 10.1111/1574-6941.12110
Chen, Y., Luo, Y., Curry, P., Timme, R., Melka, D., Doyle, M., et al. (2017a). Assessing the genome level diversity of Listeria monocytogenes from contaminated ice cream and environmental samples linked to a listeriosis outbreak in the United States. PLoS ONE 12:e0171389. doi: 10.1371/journal.pone.0171389
Chen, Y., Luo, Y., Pettengill, J., Timme, R., Melka, D., Doyle, M., et al. (2017b). Singleton sequence type 382, an emerging clonal group of Listeria monocytogenes associated with three multistate outbreaks linked to contaminated stone fruit, caramel apples, and leafy green salad. J. Clin. Microbiol. 55, 931–941. doi: 10.1128/JCM.02140-16
Clarke, K. R. (1993). Non-parametric multivariate analysis of changes in community structure. Aust. J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Davis, S., Pettengill, J. B., Luo, Y., Payne, J., Shpuntoff, A., Rand, H., et al. (2015). CFSAN SNP Pipeline: an automated method for constructing SNP matrices from next-generation sequence data. PeerJ Comput. Sci. 1:e20. doi: 10.7717/peerj-cs.20
de Jonge, E. (2016). Docopt: Command-Line Interface Specification Language. R package version 0.4.5. Available online at: https://CRAN.R-project.org/package=docopt
Doll, V. M., Ehling-Schulz, M., and Vogelmann, R. (2013). Concerted action of sphingomyelinase and non-hemolytic enterotoxin in pathogenic Bacillus cereus. PLoS ONE 8:e61404. doi: 10.1371/journal.pone.0061404
Ehling-Schulz, M., Frenzel, E., and Gohar, M. (2015). Food-bacteria interplay: pathometabolism of emetic Bacillus cereus. Front. Microbiol. 6:704. doi: 10.3389/fmicb.2015.00704
Ehling-Schulz, M., Fricker, M., and Scherer, S. (2004). Bacillus cereus, the causative agent of an emetic type of food-borne illness. Mol. Nutr. Food Res. 48, 479–487. doi: 10.1002/mnfr.200400055
Ehling-Schulz, M., Svensson, B., Guinebretiere, M. H., Lindbäck, T., Andersson, M., Schulz, A., et al. (2005). Emetic toxin formation of Bacillus cereus is restricted to a single evolutionary lineage of closely related strains. Microbiology 151(Pt 1), 183–197. doi: 10.1099/mic.0.27607-0
Fisichella, M., Dabboue, H., Bhattacharyya, S., Saboungi, M. L., Salvetat, J. P., Hevor, T., et al. (2009). Mesoporous silica nanoparticles enhance MTT formazan exocytosis in HeLa cells and astrocytes. Toxicol. In Vitro 23, 697–703. doi: 10.1016/j.tiv.2009.02.007
Gardner, S. N., and Hall, B. G. (2013). When whole-genome alignments just won't work: kSNP v2 software for alignment-free SNP discovery and phylogenetics of hundreds of microbial genomes. PLoS ONE 8:e81760. doi: 10.1371/journal.pone.0081760
Gardner, S. N., Slezak, T., and Hall, B. G. (2015). kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv271
Garrison, E., and Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. arXiv preprint arXiv:1207.3907 [q-bio.GN].
Ghosh, A. C. (1978). Prevalence of Bacillus cereus in the faeces of healthy adults. J. Hyg. 80, 233–236. doi: 10.1017/S0022172400053572
Glasset, B., Herbin, S., Guillier, L., Cadel-Six, S., Vignaud, M. L., Grout, J., et al. (2016). Bacillus cereus-induced food-borne outbreaks in France, 2007 to 2014: epidemiology and genetic characterisation. Euro. Surveill. 21:30413. doi: 10.2807/1560-7917.ES.2016.21.48.30413
Grad, Y. H., Lipsitch, M., Feldgarden, M., Arachchi, H. M., Cerqueira, G. C., Fitzgerald, M., et al. (2012). Genomic epidemiology of the Escherichia coli O104:H4 outbreaks in Europe, 2011. Proc. Natl. Acad. Sci. U.S.A. 109, 3065–3070. doi: 10.1073/pnas.1121491109
Granum, P. E., and Lund, T. (1997). Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Lett. 157, 223–228. doi: 10.1111/j.1574-6968.1997.tb12776.x
Guangchuang, Y., Smith, D. K., Huachen, Z., Yi, G., and Tsan-Yuk, L.T. (2017). ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evolut. 8, 28–36. doi: 10.1111/2041-210X.12628
Guinebretière, M. H., Auger, S., Galleron, N., Contzen, M., De Sarrau, B., De Buyser, M. L., et al. (2013). Bacillus cytotoxicus sp. nov. is a novel thermotolerant species of the Bacillus cereus group occasionally associated with food poisoning. Int. J. Syst. Evol. Microbiol. 63(Pt 1), 31–40. doi: 10.1099/ijs.0.030627-0
Guinebretière, M. H., Thompson, F. L., Sorokin, A., Normand, P., Dawyndt, P., Ehling-Schulz, M., et al. (2008). Ecological diversification in the Bacillus cereus group. Environ. Microbiol. 10, 851–865. doi: 10.1111/j.1462-2920.2007.01495.x
Guinebretiere, M. H., Velge, P., Couvert, O., Carlin, F., Debuyser, M. L., and Nguyen-The, C. (2010). Ability of Bacillus cereus group strains to cause food poisoning varies according to phylogenetic affiliation (groups I to VII) rather than species affiliation. J. Clin. Microbiol. 48, 3388–3391. doi: 10.1128/JCM.00921-10
Gymoese, P., Sørensen, G., Litrup, E., Olsen, J. E., Nielsen, E. M., and Torpdahl, M. (2017). Investigation of outbreaks of Salmonella enterica serovar typhimurium and its monophasic variants using whole-genome sequencing, Denmark. Emerg. Infect. Dis. 23, 1631–1639. doi: 10.3201/eid2310.161248
Hackathon, R., Bolker, B., Butler, M., Cowan, P., de Vienne, D., Eddelbuettel, D., et al. (2017). Phylobase: Base Package for Phylogenetic Structures and Comparative Data. R package version 0.8.4. Available online at: https://CRAN.R-project.org/package=phylobase
Heibl, C. (2008). PHYLOCH: R Language Tree Plotting Tools and Interfaces To Diverse Phylogenetic Software Packages. Avaliable online at: http://www.christophheibl.de/Rpackages.html.
Hoffmann, M., Luo, Y., Monday, S. R., Gonzalez-Escalona, N., Ottesen, A. R., Muruvanda, T., et al. (2016). Tracing origins of the Salmonella bareilly strain causing a food-borne outbreak in the United States. J. Infect. Dis. 213, 502–508. doi: 10.1093/infdis/jiv297
Holmes, A., Allison, L., Ward, M., Dallman, T. J., Clark, R., Fawkes, A., et al. (2015). Utility of whole-genome sequencing of Escherichia coli O157 for outbreak detection and epidemiological surveillance. J. Clin. Microbiol. 53, 3565–3573. doi: 10.1128/JCM.01066-15
Hwang, S., Kim, E., Lee, I., and Marcotte, E. M. (2015). Systematic comparison of variant calling pipelines using gold standard personal exome variants. Sci. Rep. 5:17875. doi: 10.1038/srep17875
Ivy, R. A., Ranieri, M. L., Martin, N. H., den Bakker, H. C., Xavier, B. M., Wiedmann, M., et al. (2012). Identification and characterization of psychrotolerant sporeformers associated with fluid milk production and processing. Appl. Environ. Microbiol. 78, 1853–1864. doi: 10.1128/AEM.06536-11
Jackson, B. R., Tarr, C., Strain, E., Jackson, K. A., Conrad, A., Carleton, H., et al. (2016). Implementation of nationwide real-time whole-genome sequencing to enhance listeriosis outbreak detection and investigation. Clin. Infect. Dis. 63, 380–386. doi: 10.1093/cid/ciw242
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jakobsen, I. B., and Easteal, S. (1996). A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Comput. Appl. Biosci. 12, 291–295. doi: 10.1093/bioinformatics/12.4.291
Jiménez, G., Urdiain, M., Cifuentes, A., López-López, A., Blanch, A. R., Tamames, J., et al. (2013). Description of Bacillus toyonensis sp. nov., a novel species of the Bacillus cereus group, and pairwise genome comparisons of the species of the group by means of ANI calculations. Syst. Appl. Microbiol. 36, 383–391. doi: 10.1016/j.syapm.2013.04.008
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Kaas, R. S., Nielsen, E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. doi: 10.1128/JCM.03617-13
Jombart, T., Kendall, M., Almagro-Garcia, J., and Colijn, C. (2017). Treespace: statistical exploration of landscapes of phylogenetic trees. Mol. Ecol. Resour. 17, 1385–1392. doi: 10.1111/1755-0998.12676
Kabir, M. S., Hsieh, Y. H., Simpson, S., Kerdahi, K., and Sulaiman, I. M. (2017). Evaluation of two standard and two chromogenic selective media for optimal growth and enumeration of isolates of 16 unique Bacillus species. J. Food Prot. 80:952–962. doi: 10.4315/0362-028X.JFP-16-441
Katz, L. S., Griswold, T., Williams-Newkirk, A. J., Wagner, D., Petkau, A., Sieffert, C., et al. (2017). A comparative analysis of the lyve-set phylogenomics pipeline for genomic epidemiology of foodborne pathogens. Front. Microbiol. 8:375. doi: 10.3389/fmicb.2017.00375
Kendall, M., and Colijn, C. (2015). A tree metric using structure and length to capture distinct phylogenetic signals. arXiv:1507.05211v3 [q-bio.PE].
Kovac, J., Bakker, H. D., Carroll, L. M., and Wiedmann, M. (2017). Precision food safety: a systems approach to food safety facilitated by genomics tools. TrAC Trends Analy. Chem. 96(Suppl. C), 52–61. doi: 10.1016/j.trac.2017.06.001
Kovac, J., Miller, R. A., Carroll, L. M., Kent, D. J., Jian, J., Beno, S. M., et al. (2016). Production of hemolysin BL by Bacillus cereus group isolates of dairy origin is associated with whole-genome phylogenetic clade. BMC Genomics 17:581. doi: 10.1186/s12864-016-2883-z
Kwong, J. C., Mercoulia, K., Tomita, T., Easton, M., Li, H. Y., Bulach, D. M., et al. (2016). Prospective whole-genome sequencing enhances national surveillance of Listeria monocytogenes. J. Clin. Microbiol. 54, 333–342. doi: 10.1128/JCM.02344-15
Lewis, P. O. (2001). A likelihood approach to estimating phylogeny from discrete morphological character data. Syst. Biol. 50, 913–925. doi: 10.1080/106351501753462876
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v1 [q-bio.GN].
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liu, Y., Du, J., Lai, Q., Zeng, R., Ye, D., Xu, J., et al. (2017). Proposal of nine novel species of the Bacillus cereus group. Int. J. Syst. Evol. Microbiol. 67, 2499–2508. doi: 10.1099/ijsem.0.001821
Lotte, R., Hérissé, A. L., Berrouane, Y., Lotte, L., Casagrande, F., Landraud, L., et al. (2017). Virulence analysis of bacillus cereus isolated after death of preterm neonates, nice, France, 2013. Emerg. Infect. Dis. 23, 845–848. doi: 10.3201/eid2305.161788
Mair-Jenkins, J., Borges-Stewart, R., Harbour, C., Cox-Rogers, J., Dallman, T., Ashton, P., et al. (2017). Investigation using whole genome sequencing of a prolonged restaurant outbreak of Salmonella Typhimurium linked to the building drainage system, England, February 2015 to March 2016. Euro. Surveill. 22:17-00037. doi: 10.2807/1560-7917.ES.2017.22.49.17-00037
McCloskey, R. M., and Poon, A. F. Y. (2017). A model-based clustering method to detect infectious disease transmission outbreaks from sequence variation. PLoS Comput. Biol. 13:e1005868. doi: 10.1371/journal.pcbi.1005868
Messelhäusser, U., Frenzel, E., Blöchinger, C., Zucker, R., Kämpf, P., and Ehling-Schulz, M. (2014). Emetic Bacillus cereus are more volatile than thought: recent foodborne outbreaks and prevalence studies in Bavaria (2007–2013). Biomed. Res. Int. 2014:465603. doi: 10.1155/2014/465603
Miller, R. A., Beno, S. M., Kent, D. J., Carroll, L. M., Martin, N. H., Boor, K. J., et al. (2016). Bacillus wiedmannii sp. nov., a psychrotolerant and cytotoxic bacillus cereus group species isolated from dairy foods and dairy environments. Int. J. Syst. Evol. Microbiol. 66, 4744–4753. doi: 10.1099/ijsem.0.001421
Miller, R. A., Jian, J., Beno, S. M., Wiedmann, M., and Kovac, J. (2018). Intraclade variability in toxin production and cytotoxicity of bacillus cereus group type strains and dairy-associated isolates. Appl. Environ. Microbiol. 84:e02479–17. doi: 10.1128/AEM.02479-17
Moran-Gilad, J. (2017). Whole genome sequencing (WGS) for food-borne pathogen surveillance and control–taking the pulse. Euro. Surveill. 22:30547. doi: 10.2807/1560-7917.ES.2017.22.23.30547
Morgulis, A., Gertz, E. M., Schäffer, A. A., and Agarwala, R. (2006). A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J. Comput. Biol. 13, 1028–1040. doi: 10.1089/cmb.2006.13.1028
Moura, A., Tourdjman, M., Leclercq, A., Hamelin, E., Laurent, E., Fredriksen, N., et al. (2017). Real-time whole-genome sequencing for surveillance of Listeria monocytogenes, France. Emerg. Infect. Dis. 23, 1462–1470. doi: 10.3201/eid2309.170336
Naranjo, M., Denayer, S., Botteldoorn, N., Delbrassinne, L., Veys, J., Waegenaere, J., et al. (2011). Sudden death of a young adult associated with Bacillus cereus food poisoning. J. Clin. Microbiol. 49, 4379–4381. doi: 10.1128/JCM.05129-11
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2018). Vegan: Community Ecology Package. R package version 2.5-2. Available online at: https://CRAN.R-project.org/package=vegan
Olson, N. D., Lund, S. P., Colman, R. E., Foster, J. T., Sahl, J. W., Schupp, J. M., et al. (2015). Best practices for evaluating single nucleotide variant calling methods for microbial genomics. Front. Genet. 6:235. doi: 10.3389/fgene.2015.00235
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Pightling, A. W., Petronella, N., and Pagotto, F. (2014). Choice of reference sequence and assembler for alignment of Listeria monocytogenes short-read sequence data greatly influences rates of error in SNP analyses. PLoS ONE 9:e104579. doi: 10.1371/journal.pone.0104579
Pightling, A. W., Petronella, N., and Pagotto, F. (2015). Choice of reference-guided sequence assembler and SNP caller for analysis of Listeria monocytogenes short-read sequence data greatly influences rates of error. BMC Res. Notes 8:748. doi: 10.1186/s13104-015-1689-4
Pruitt, K. D., Tatusova, T., and Maglott, D. R. (2007). NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65. doi: 10.1093/nar/gkl,842
R Core Team. (2018). R: A language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: https://www.R-project.org,
Revell, L. J. (2012). phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Rusconi, B., Sanjar, F., Koenig, S. S., Mammel, M. K., Tarr, P. I., and Eppinger, M. (2016). Whole genome sequencing for genomics-guided investigations of Escherichia coli O157:H7 outbreaks. Front. Microbiol. 7:985. doi: 10.3389/fmicb.2016.00985
Sanaei-Zadeh, H. (2012). Can Bacillus cereus food poisoning cause sudden death? J. Clin. Microbiol. 50, 3816–3817. doi: 10.1128/JCM.00059-12
Sandmann, S., de Graaf, A. O., Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Evaluating variant calling tools for non-matched next-generation sequencing data. Sci. Rep. 7:43169. doi: 10.1038/srep43169
Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M. A., Roy, S. L., et al. (2011). Foodborne illness acquired in the United States–major pathogens. Emerg. Infect. Dis. 17, 7–15. doi: 10.3201/eid1701.P11101
Schliep, K. P. (2011). Phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593. doi: 10.1093/bioinformatics/btq706
Schoeni, J. L., and Wong, A. C. (2005). Bacillus cereus food poisoning and its toxins. J. Food Prot. 68, 636–648. doi: 10.4315/0362-028X-68.3.636
Smith, J. M. (1992). Analyzing the mosaic structure of genes. J. Mol. Evol. 34, 126–129. doi: 10.1007/BF00182389
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stenfors Arnesen, L. P., Fagerlund, A., and Granum, P. E. (2008). From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Rev. 32, 579–606. doi: 10.1111/j.1574-6976.2008.00112.x
Taboada, E. N., Graham, M. R., Carriço, J. A., and Van Domselaar, G. (2017). Food safety in the age of next generation sequencing, bioinformatics, and open data access. Front. Microbiol. 8:909. doi: 10.3389/fmicb.2017.00909
Tallent, S. M., Kotewicz, K. M., Strain, E. A., and Bennett, R. W. (2012a). Efficient isolation and identification of Bacillus cereus group. J. AOAC Int. 95, 446–451. doi: 10.5740/jaoacint.11-251
Tallent, S. M., Rhodehamel, E. J., Harmon, S. M., and Bennett, R. W. (2012b). “Chapter 14: Bacillus cereus,” in Bacteriological Analytical Manual (Silver Spring, MD: U.S. Food and Drug Administration). Available online at: https://www.fda.gov/Food/FoodScienceResearch/LaboratoryMethods/ucm070875.htm
Taylor, A. J., Lappi, V., Wolfgang, W. J., Lapierre, P., Palumbo, M. J., Medus, C., et al. (2015). Characterization of foodborne outbreaks of Salmonella enterica serovar enteritidis with whole-genome sequencing single nucleotide polymorphism-based analysis for surveillance and outbreak detection. J. Clin. Microbiol. 53, 3334–3340. doi: 10.1128/JCM.01280-15
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome. Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Turnbull, P. C., and Kramer, J. M. (1985). Intestinal carriage of Bacillus cereus: faecal isolation studies in three population groups. J. Hyg. (Lond). 95, 629–638. doi: 10.1017/S0022172400060733
Usongo, V., Berry, C., Yousfi, K., Doualla-Bell, F., Labbe, G., Johnson, R., et al. (2018). Impact of the choice of reference genome on the ability of the core genome SNV methodology to distinguish strains of Salmonella enterica serovar Heidelberg. PLoS ONE 13:e0192233. doi: 10.1371/journal.pone.0192233
Vangay, P., Fugett, E. B., Sun, Q., and Wiedmann, M. (2013). Food microbe tracker: a web-based tool for storage and comparison of food-associated microbes. J. Food. Prot. 76, 283–294. doi: 10.4315/0362-028X.JFP-12-276
Walker, T. M., Merker, M., Knoblauch, A. M., Helbling, P., Schoch, O. D., van der Werf, M. J., et al. (2018). A cluster of multidrug-resistant Mycobacterium tuberculosis among patients arriving in Europe from the Horn of Africa: a molecular epidemiological study. Lancet Infect. Dis. 18, 431–440. doi: 10.1016/S1473-3099(18)30004-5
Wickham, H. (2018). Stringr: Simple, Consistent Wrappers for Common String Operations. R package version 1.3.1. Available online at: https://CRAN.R-project.org/package=stringr
Keywords: Bacillus cereus, foodborne outbreak, whole-genome sequencing, emetic disease, cytotoxicity, SNP calling, genomic epidemiology
Citation: Carroll LM, Wiedmann M, Mukherjee M, Nicholas DC, Mingle LA, Dumas NB, Cole JA and Kovac J (2019) Characterization of Emetic and Diarrheal Bacillus cereus Strains From a 2016 Foodborne Outbreak Using Whole-Genome Sequencing: Addressing the Microbiological, Epidemiological, and Bioinformatic Challenges. Front. Microbiol. 10:144. doi: 10.3389/fmicb.2019.00144
Received: 24 August 2018; Accepted: 21 January 2019;
Published: 12 February 2019.
Edited by:
Sophia Johler, University of Zurich, SwitzerlandReviewed by:
Adam Lee Bazinet, National Biodefense Analysis and Countermeasures Center (NBACC), United StatesJason Sahl, Northern Arizona University, United States
Ute Messelhäusser, Bayerisches Landesamt für Gesundheit und Lebensmittelsicherheit, Germany
Copyright © 2019 Carroll, Wiedmann, Mukherjee, Nicholas, Mingle, Dumas, Cole and Kovac. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jasna Kovac, anprMzAzQHBzdS5lZHU=