 Julien Briffotaux
Julien Briffotaux Shengyuan Liu
Shengyuan Liu Brigitte Gicquel1,2,3
Brigitte Gicquel1,2,3- 1Department of Tuberculosis Control and Prevention, Shenzhen Nanshan Center for Chronic Disease Control, Shenzhen, China
- 2Emerging Bacterial Pathogens Unit, CAS Key Laboratory of Molecular Virology and Immunology, Institut Pasteur of Shanghai, Chinese Academy of Sciences, Shanghai, China
- 3Mycobacterial Genetics Unit, Institut Pasteur, Paris, France
Antibiotics can stimulate or depress gene expression in bacteria. The analysis of transcriptional responses of Mycobacterium to antimycobacterial compounds has improved our understanding of the mode of action of various drug classes and the efficacy and effect of such compounds on the global metabolism of Mycobacterium. This approach can provide new insights for known antibiotics, for example those currently used for tuberculosis treatment, as well as help to identify the mode of action and predict the targets of new compounds identified by whole-cell screening assays. In addition, changes in gene expression profiles after antimycobacterial treatment can provide information about the adaptive ability of bacteria to escape the effects of antibiotics and allow monitoring of the physiology of the bacteria during treatment. Genome-wide expression profiling also makes it possible to pinpoint genes differentially expressed between drug sensitive Mycobacterium and multidrug-resistant clinical isolates. Finally, genes involved in adaptive responses and drug tolerance could become new targets for improving the efficacy of existing antibiotics.
Introduction
Tuberculosis (TB) is an infectious disease that typically attacks the lungs, it is caused by the bacteria Mycobacterium tuberculosis. This infection has possibly been present in humans since the Neolithic period (Comas et al., 2013). Today, TB is a re-emerging disease and a major public health problem in both developing and developed countries. According to the WHO, 10.4 million people fell ill with TB and 1.4 million died from the disease in 2016 (WHO, 2017). This pathogen can invade the pulmonary alveoli, where it is engulfed by macrophages. The success of this pathogen is due to its ability to survive inside macrophages, evade host defense mechanisms, and manipulate phagosome maturation (Ehrt and Schnappinger, 2009).
Public hygiene and the discovery of streptomycin and para-aminosalicylic acid (PAS) in the 1940s advanced the treatment of TB. In order to improve the efficiency of the treatment, other molecules have been used in combination, such as isoniazid from 1952, ethambutol from the early 1960s, and rifampin from the 1970s (Murray et al., 2015). Current TB treatment requires patients to take a combination of four drugs, called first-line TB drugs: isoniazid (INH), rifampicin (RIF), ethambutol (EMB), and pyrazinamide (PZA), for 2 months (initial phase) and two of them (INH and RIF) for an additional 4 months (continuation phase). Although very efficient, such treatment is long and very demanding. In many settings, the absence of a sufficient healthcare infrastructure and shortage of antibiotics can promote the appearance of resistance. In recent decades, multidrug-resistant TB (MDR-TB), extremely drug-resistant TB (XDR-TB) strains have emerged (Almeida Da Silva and Palomino, 2011; Udwadia et al., 2012). Treating and curing drug-resistant TB is complicated and more than 12 anti-TB drugs can be used for second-line regimens. These drugs are used in varying combinations depending on the circumstances; they include the aminoglycosides (streptomycin, kanamycin, amikacin, and paromomycin), the fluoroquinolones (moxifloxacin, ciprofloxacin, ofloxacin, and levofloxacin), the rifamycins (rifampin, rifabutin, and rifapentine), the beta-lactams (imipenem, tebipenem, and amoxicillin-clavulanic acid), oxazolidinone (linezolid), riminophenazine (clofazimine), ethionamide, PAS, bedaquiline, and SQ109. The targets and drug resistance mechanisms of these compounds have been recently reviewed (Islam et al., 2017), as well as the state of current drug development for the treatment of TB (Hoagland et al., 2016; Tandon and Nath, 2017).
The emergence of resistant strains of M. tuberculosis highlights the need to discover and develop new antimycobacterial compounds and approaches. It is equally important to understand the mode of action of these molecules, their effect on the cell, and the mechanisms by which bacteria can develop resistance.
The study of the biology of M. tuberculosis has been facilitated during the last 20 years by the availability of genome sequencing and genetic tools that allow the deciphering of major, specific metabolic pathways. For example, genetic and biochemical approaches resulted in the discovery of genes carrying mutations that confer isoniazid, ethambutol, ethionamide, and pyrazinamide resistance (Palomino and Martin, 2014). Microbial whole-genome sequencing allows the rapid detection of antibiotic susceptibility and resistance by the identification of resistance mutations (Takiff and Feo, 2015). However, this approach provides no information about the physiological state of the Mycobacterium or antibiotic tolerance due to changes in the transcriptional profile. In addition to the acquired resistance caused by target mutations, several distinctive mechanisms of antimycobacterial resistance have been described (Nasiri et al., 2017): the prevention of access to the target due to impermeability of the mycobacterial cell wall, transport of antimycobacterial compounds out of the cell by efflux pumps, modification of antibiotics by mycobacterial enzymes, and the modulation of gene expression, all leading to antibiotic tolerance.
Antibiotics can affect bacteria at many levels in addition to their direct effects on the target. These include effects on their morphology, metabolism, gene expression, stress response, and mutation rate (Nonejuie et al., 2013; Mitosch and Bollenbach, 2014; Tsai et al., 2015). Moreover, Mycobacterium can tolerate antibiotics due to their ability to reduce their intracellular accumulation by increasing active efflux of these molecules (Poole, 2007; Balganesh et al., 2012).
New knowledge concerning metabolic changes and adaptive responses of Mycobacterium after antibiotic exposure would help us to better understand both the mechanism of action of the antibiotics and the mechanisms of antibiotic resistance. Understanding how antimycobacterial compounds kill bacteria and the cellular response of the bacteria to such compounds is crucial to improving the efficacy and reducing the cytotoxicity of these drugs.
Altering transcription and adjusting physiology are amongst the main mechanisms in the initiation of adaptive processes in a cell (Cases et al., 2003; Perez and Groisman, 2009; Brooks et al., 2011). In Mycobacterium, the transcriptional response to a perturbation, such as antibiotic exposure, nutrient starvation (Betts et al., 2002), or limited oxygen (Bacon et al., 2004; Voskuil et al., 2004), can be assessed by transcriptomic techniques, such as microarrays and RNA sequencing (RNA-seq). DNA microarrays (DNA biochip), invented in the 1990s, are a collection of nucleotide probes or complementary DNA (cDNA) fixed on the surface that can hybridize to RNA, making it possible to analyze gene expression levels. However, the more recent development of RNA-seq, a method in which RNA is purified and cDNAs directly sequenced, enables rapid analysis, offering more flexibility (Mutz et al., 2013). Real-time quantitative reverse transcriptase-PCR (RT-qRT-PCR) is used to confirm changes in gene expression for both microarrays and RNA-seq.
This review provides an analysis of published transcriptional-profiling data of Mycobacterium exposed to various antimycobacterial compounds (Table 1). Overall, theses microarrays or RNA-seq analyses can be used in various ways, depending on the question asked. It can be used to investigate changes in the gene-expression profile of bacteria following antibiotic exposure compared to that of untreated cells (Figure 1), the gene-expression profile of mutants relative to that of wild type cells treated with an antibiotic, or transcriptional profiles of clinical strains, especially MDR strains. Genome-wide expression profiles facilitate the characterization of both the mechanisms of action and the mechanisms of resistance to antimicrobial agents.
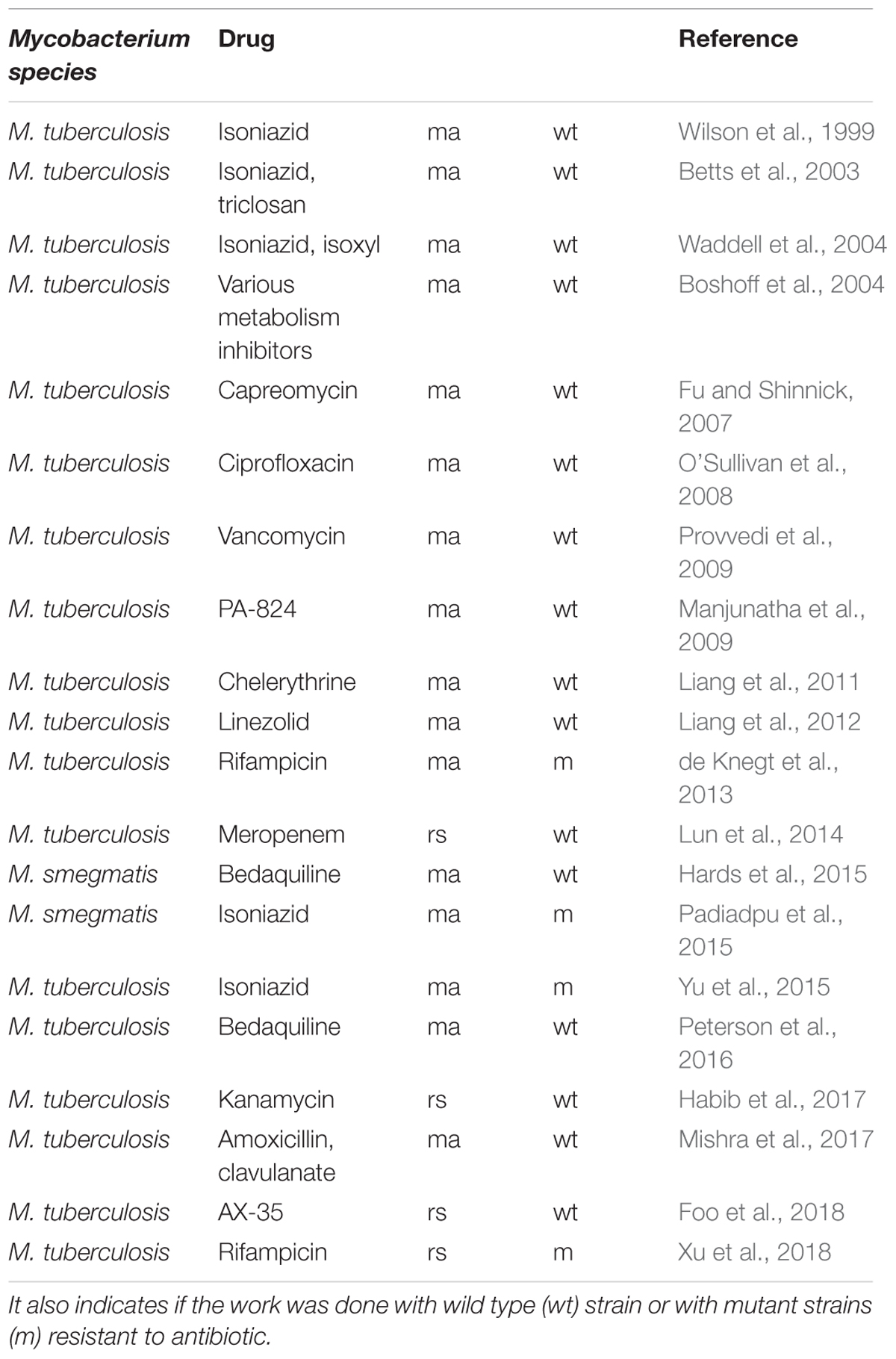
Table 1. Chronology of publications cited in this review on transcriptomic profiling by microarray (ma) or RNA-seq (rs) after anti-bacterial compound treatment.
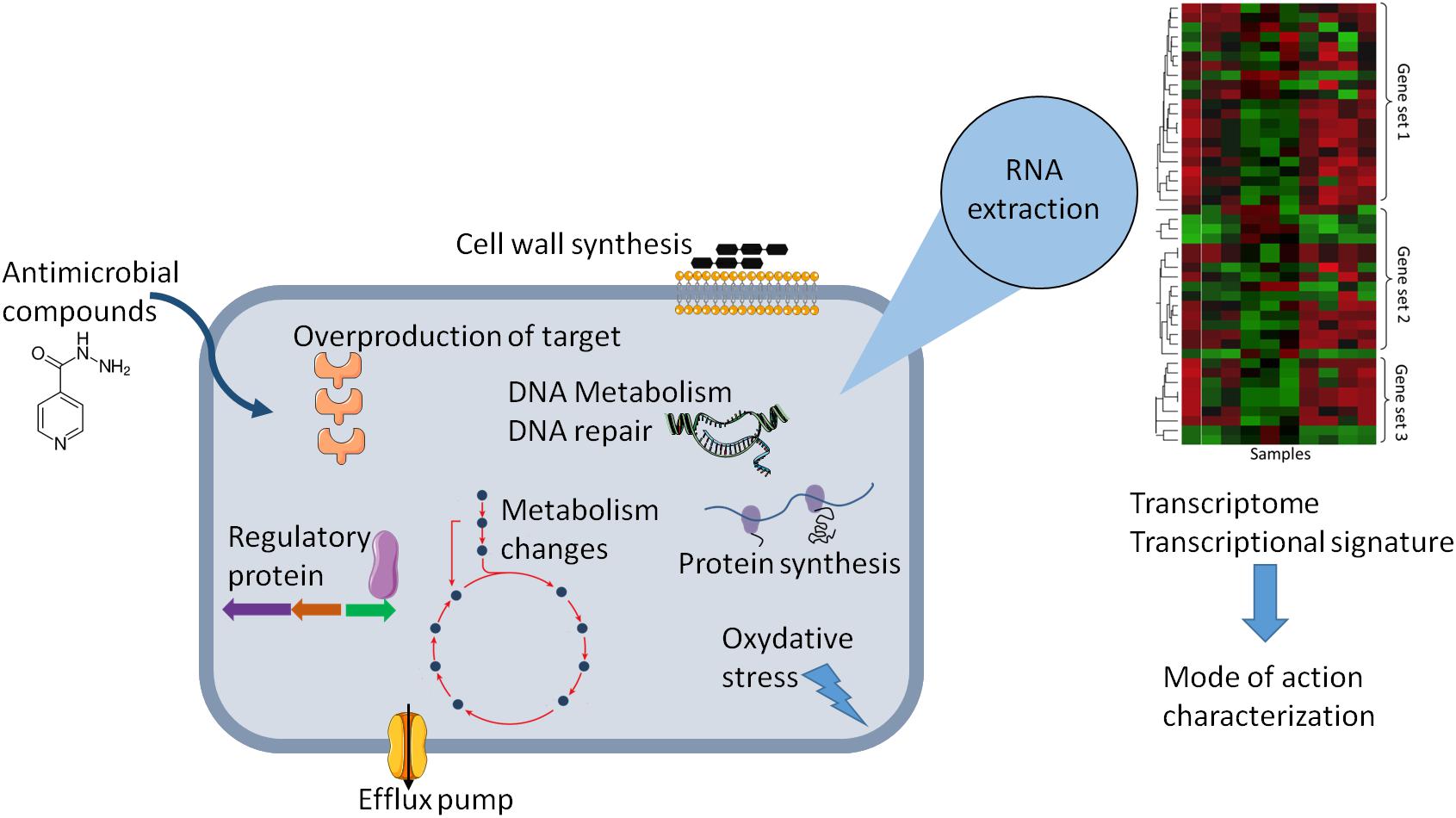
Figure 1. Schematic representation of the effect of antibiotic on the physiology of bacteria that can be explored by change in gene expression. Gene expression dynamic can reflect cellular response in the presence of antibiotic and can lead altered cellular state or adaptive responses and antibiotic tolerance. The transcriptional signature analysis allows to have new insights in the molecular mechanism of antimicrobial compounds or to predict the mode of action of uncharacterized antimicrobial compounds.
The deregulated genes in response to antibiotic treatment in Mycobacterium based on their fold expression, reported in most of the papers in this review, are analyzed and categorized into 10 functional classes: (1) virulence, detoxification, and adaptation; (2) lipid metabolism; (3) information pathways; (4) cell wall and cell processes; (5) insertion sequences and phages; (6) PE and PPE proteins; (7) intermediary metabolism and respiration; (8) proteins with unknown function; (9) regulatory proteins; and (10) conserved hypothetical proteins. From these data, it is possible to propose a role for certain genes in the response and adaptation to a given drug and a transcriptional signature for the drug, possibly highlighting transcriptional regulators and regulatory networks involved in the response.
Isoniazid Induced Changes in Gene Expression
The first study to investigate changes in gene expression after antibiotic treatment of Mycobacterium was published in 1999 (Wilson et al., 1999). In this study, DNA microarrays were used to monitor gene-expression changes in response to isoniazid, one of the most active antibiotics used in TB treatment. Isoniazid is a prodrug and must be activated by a catalase-peroxidase (KatG) of Mycobacterium, to form an isonicotinic acyl anion, which reacts with NAD+ and binds to the active site of an enoyl-acyl carrier protein reductase, InhA (Rawat et al., 2003). This process prevents the synthesis of mycolic acids, components of the mycobacterial cell wall, thus resulting in death of the cells. Among several isoniazid induced genes, the authors highlighted the induction of five genes organized as an operon (fabD-acpM-kasA-kasB-accD6) and encoding type II fatty acid synthase enzymes (FAS-II), involved in the production of membrane phospholipids, as well as the gene encoding FbpC (Ag85C), an enzyme involved in mycolic acid transfer during cell-wall formation. It is possible that these genes are upregulated by a feedback mechanism which responds to the decrease of mature mycolates inside the cell. inhA is not induced in response to isoniazid treatment, nevertheless, by using M. tuberculosis strains with multicopy inhA or kasAB plasmids, it has been observed that the overexpression of inhA, but not kasA, confers resistance to isoniazide (Larsen et al., 2002).
The other genes do not appear to have any link with the biosynthesis of these fatty acids, but may be induced because of the toxic effects of isoniazid. Moreover, upregulation of the expression of the gene efpA, which encodes a major facilitator superfamily (MFS) efflux pump, as well as the upregulation of iniA, which encodes putative pump component, could participate in intrinsic resistance to isoniazid, as has been reported in other studies (Colangeli et al., 2005; Gupta et al., 2010; Li et al., 2015; Narang et al., 2017). iniA is upregulated along with iniB and iniC, these genes are indeed organized in operon under the control of iniBAC promoter and upregulated by various cell envelope inhibitor (Alland et al., 1998, 2000).
Another study investigated gene expression changes in M. tuberculosis following exposure to isoniazid, as well as thiolactomycin and triclosan (Betts et al., 2003). All three drugs are inhibitors of mycolic acid biosynthesis, but have different modes of action. The authors compared the response to these three drugs and proposed a transcriptional profile model that allows discrimination between M. tuberculosis treated with isoniazid, thiolactomycin, or triclosan. The model can then be used to specify the mode of action of uncharacterized mycolic acid biosynthesis inhibitors or characterize new InhA inhibitors during the drug discovery and development process. The gene expression profiles induced by isoniazid treatment were also compared to those induced by thiocarlide (isoxyl), a mycolic acid biosynthesis inhibitor and tetrahydrolipstatin (Waddell et al., 2004).
Isoniazid is less active in slowly or non-replicating mycobacteria. A study investigated isoniazid-regulated genes in drug-treated dormant M. tuberculosis and katG-deficient, isoniazid-resistant M. tuberculosis (Karakousis et al., 2008). Nutrient starvation and progressive oxygen depletion were used as in vitro models of dormancy. The transcriptional profile reflecting the mechanism of action of isoniazid was not found under these conditions nor in the katG mutant, in contrast to wild type M. tuberculosis after isoniazid exposure. The absence of the characteristic isoniazid-response profile correlated with the absent or decreased isoniazid activity in dormant M. tuberculosis. Moreover, this study also showed that the isoniazid-response profile can be found in the early steps of infection in the mouse after isoniazid treatment, but that this response decreases during late-stage infection, correlating with the reduction in isoniazid activity. This may correspond with entry of the bacteria into dormancy at this stage of infection. Isoniazid has no effect in non-replicating bacteria under microaerophilic conditions and the isoniazid-response profile is absent in this condition (Tudo et al., 2010). The authors proposed that the decreased requirement for mycolic acid, modification of the redox state, and cell wall permeability of non-replicating mycobacteria can confer tolerance to isoniazid.
Proteomics approaches provide another means to investigate gene expression and identify changes in protein levels in response to antibiotics. Quantitative proteomics detected increased levels of AcpM, KasA, KasB, and AccD6, proteins involved in the FAS II pathway, after isoniazid treatment, as well as proteins involved in cell division (Hughes et al., 2006). An original study used this approach to compare two isogenic strains of M. tuberculosis obtained from the same patient before and after isoniazid treatment (Nieto et al., 2016). Forty-six proteins for which the abundance was altered were identified after the acquisition of isoniazid resistance. The authors emphasized the low levels of KatG found in the isoniazid-resistant strain. Other proteins for which the abundance was modified belong to several categories, in particular, lipid metabolism, intermediate metabolism, and the respiration pathway, especially decreased levels of enzymes of the tricarboxylic acid (TCA) cycle. The authors suggested that these changes may reflect a compensatory mechanism to the reduced amount of KatG.
These observations may also be related to the fact that low metabolic rate contributes to drug tolerance in the host. Several stresses that limit replication, such as low pH or hypoxia, induce triglyceride synthesis and decrease cellular carbon fluxes from the TCA cycle and the accumulation of triacylglycerol (TAG) is associated with decreased growth and increased antibiotic tolerance, both in vitro and in the mouse infection model (Baek et al., 2011).
In order to be complete, metabolomic (Zampieri et al., 2018) and lipidomic (Pal et al., 2017) are complementary approaches to study the effect of isoniazid on physiology of mycobacteria. The use of high-throughput mass spectrometry-based lipidomic approach revealed differential lipids profile in MDR compared to drug sensitive M. tuberculosis (Pal et al., 2017). Moreover, the composition of different classes of lipids, and not only fatty acyl category, is modified in M. tuberculosis after treatment by isoniazid (Pal et al., 2018).
Transcriptional Signature and Drug Mode of Action
An ambitious study concerning the transcriptional response to antibiotics in Mycobacterium consisted of using whole-genome microarrays to measure the effects of 75 different drugs, drug combinations, and growth conditions (Boshoff et al., 2004). The genes that were down- or upregulated at least three-fold were analyzed and grouped by profile, according to coordinately regulated genes. These profiles corresponded to the groups of drugs classified based on their mechanism of action (cell-wall biosynthesis inhibitors, protein synthesis inhibitors, transcriptional inhibitors, or DNA gyrase inhibitors). They corresponded to the signature of a given drug’s mode of action. For example, isoniazid, ethionamide, thiolactomycin, and cerulenin, which inhibit fatty acid and mycolic acid synthesis, displayed a similar expression profile, with induction of the kas operon (acpM-kasA-kasB-accD6), which encodes an enzyme involved in the FAS II system, and efpA, which encodes an efflux pump protein. These profiles were then used to successfully predict the mechanism of action of two previously uncharacterized compounds, phenothiazine, as a respiratory modulator, and pyridoacridone, as an iron scavenger, as well as the mechanism of action of a purified natural product and the product in the crude extract. This method was later applied to other molecules, such as a lipase inhibitor, tetrahydrolipstatin (Waddell et al., 2004), benzothiazinones (Makarov et al., 2009) targeting arabinose metabolism, or pretomanid (PA-824) (Manjunatha et al., 2009), which has a complex mechanism of action that targets both the bacterial cell wall and respiratory complex. Pretomanid is currently being tested in a phase III clinical trial for the treatment of TB and recently, a study using a metabolomic approach, showed the accumulation of a toxic metabolite, a methylglyoxal induced by pretomanid, which participates in the action of the drug (Baptista et al., 2018).
This transcriptional approach has recently been used to compare the response of M. tuberculosis and M. marinum after treatment with a subinihibitory concentration of ciprofloxacin, ethambutol, isoniazid, streptomycin and rifampicin (Boot et al., 2018) and the authors observe that fingerprint was more distinctive in M. marinum. Then, tree functional stress reporters were constructed on the base of stress fingerprint of M. marinum, and these reporter strains have made it possible to identify the mode of action of three antimycobacterial compounds previously uncharacterized (Boot et al., 2018).
Transcriptome analysis has also been used to study the effect of natural compounds from plants with antimycobacterial activity, such as chelerythrine, on M. tuberculosis (Liang et al., 2011). The authors showed there are many genes that were either up- or downregulated and involved in various pathways, but were unable to propose a mechanism of action of this compound. They noted, however, the up-regulation of genes that encode the 30S and 50S ribosomal proteins, which could be due to an adaptive response or the inhibition of translation by chelerythrine. The genes hsp, hspX, and hspR, involved in the heat shock response, were downregulated. The decrease in the expression of the gene Rv0467, encoding ICL (isocitrate lyase), after chelerythrine treatment is also of interest. Indeed, ICL, a glyoxylate cycle enzyme that converts isocitrate to succinate and glyoxylate, is required for the in vivo growth and virulence of M. tuberculosis and can mediate broad antibiotic tolerance (Nandakumar et al., 2014). It is a potential drug target for M. tuberculosis dormancy (Lee et al., 2015).
On the contrary, ICL has been shown to be induced in Mycobacterium by capreomycin (Fu and Shinnick, 2007), a cyclic peptide produced by Streptomyces capreolus that inhibits protein biosynthesis in Mycobacterium by binding 16S rRNA and 23S rRNA. Capreomycin also induced expression of the genes encoding the 30S and 50S ribosomal proteins, that encode methyltransferase Rv1988, and the gene eis Rv2416c, which enhance intracellular survival of Mycobacterium in macrophages (Fu and Shinnick, 2007). Moreover, most genes specifically induced by capreomycin and PA-824 relative to first-line TB drugs are associated with the stress response and the PE/PPE family expression (Fu and Tai, 2009).
Linezolid is a synthetic antibiotic that belongs to the oxazolidinone class of antimicrobials, which bind to the 50S ribosomal subunits and inhibit protein synthesis. This compound is generally used to treat Gram-positive infections, such as with Staphylococcus or Enterococcus, but has also been recently used for the treatment of patients with drug-resistant TB (Lee et al., 2012). The transcriptional response to subinhibitory linezolid exposure has been studied in M. tuberculosis by microarrays (Liang et al., 2012). The authors reported 729 genes to be differentially regulated by linezolid. They are involved in various pathways, such as protein synthesis, sulfite metabolism, cell-wall permeability, and virulence. Among them, linezolid exposure induced the transcription of chaperone-encoding genes (dnaK and groEL), which may be a sign of a linezolid-induced stress response. Genes involved in the biosynthesis of sulfolipid-I and TAG were downregulated, whereas genes involved in the synthesis of phthiocerol dimycocerosate (DIM), which plays a role in cell-wall permeability and the virulence of M. tuberculosis during macrophage invasion (Astarie-Dequeker et al., 2009), were inhibited. Surprisingly, some of the genes encoding the 50S ribosomal protein were down regulated, whereas others were upregulated in the presence of linezolid. These results are different from those observed for Staphylococcus aureus, for which linezolid treatment increased the expression of ribosomal proteins to adjust protein biosynthesis in response to the reduction in translational efficiency (Bonn et al., 2016). Other genes involved in protein synthesis were also down- or upregulated in M. tuberculosis. These variations in expression level probably affect protein synthesis rates and could participate in the disturbance of homeostasis by linezolid in Mycobacteria.
The most recent study of the transcriptional response to antibiotics in Mycobacterium concerns the response following sublethal kanamycin exposure (Habib et al., 2017). Kanamycin is a second-line drug used for the treatment of patients with MDR-TB. This antibiotic works by binding to the 30S subunit of ribosomes, which interferes with translation and disrupts protein synthesis. Unlike previous studies, in which the authors showed the response to antibiotics to occur relatively early, this study investigated the adaptive response. Indeed, an avirulent strain, M. tuberculosis H37Ra, was subcultured every 3 days with a sublethal concentration of the drug to observe transcriptional changes over time. The authors showed that the growth curve of the bacteria cultured in the presence of kanamycin became the same as that of bacteria cultured in the absence of the drug. This corresponds to an adaptation to the presence of kanamycin over time, with the bacteria changing their physiology to adapt to antibiotic stress. Every 3 days, a portion of both the experimental and control cultures were subjected to RNAseq. The authors identified 98 upregulated and 198 downregulated genes in M. tuberculosis in the presence of kanamycin. Several upregulated genes belonged to different functional categories, such as cell-wall and cellular processes, virulence, and the PE/PPE family, whereas the downregulated genes were involved in lipid metabolism. Members of the PE/PPE family are proteins associated with the membrane or cell wall of mycobacteria and have been shown to interact with mediators of the host immune response and play a role in pathogenesis (Fishbein et al., 2015) and the genotoxic stress response (Namouchi et al., 2016). Several genes with increased or decreased expression encode uncharacterized proteins. In particular, the upregulated gene Ra3160 is one of the most responsive genes to kanamycin. Ra3160 and Ra3161 are members of the dormancy regulon (DosR), known to be involved in the survival of bacteria under conditions of stress and antibacterial tolerance (Sivaramakrishnan and de Montellano, 2013). Ra1750 was strongly induced by kanamycin treatment and was previously shown to be induced during hypoxia through the DosS/DosR system (Sherman et al., 2001; Sivaramakrishnan and de Montellano, 2013). Finally, the authors built gene-interaction clusters which suggest the contribution of these genes to the physiological adaptation of M. tuberculosis in response to sub-lethal kanamycin exposure.
Vancomycin is an antibiotic of the glycopeptide family and inhibits peptidoglycan synthesis of the bacterial cell wall, but is considered to have little effect on Mycobacterium because of the impermeability of the membrane. However, global transcriptomics detected 153 differentially expressed genes in M. tuberculosis after exposure to 10X-MIC vancomycin and 141 differentially expressed genes following exposure to a in subinhibitory concentration of the drug (Provvedi et al., 2009). The high concentration of vancomycin induced the expression of several genes involved in the oxidative-stress response, including the (USP) rv2623; chaperone-encoding genes (hsp and htpX), suggesting that vancomycin causes protein misfolding or aggregation; the toxin-antitoxin (TA) system, which plays a role in the stress response and induction of dormancy; and several genes encoding surface proteins or involved in cell-wall processes. Rv2623 is known to be involved in the regulation of mycobacterial growth and the transition to latency via an ATP-dependent function (Drumm et al., 2009). Thus, induction of the rv2623 gene, by treatment with vancomycin, could contribute to the induction of persistent infection in the host and difficulty in eradicating the disease. Rv1152 is a transcriptional regulator that belongs to the GntR family and negatively regulates the vancomycin-responsive genes, including rv2623 (Zeng et al., 2016). The MIC of vancomycin against M. tuberculosis is too high to allow its use in the clinic, but the discovery of adjuvants to improve its efficacy, such as a Rv1152 inhibitor, should be envisaged. Drug targeting lipid synthesis, such as that of cerulenin, has also been shown to synergistically increase susceptibility to vancomycin (Soetaert et al., 2015). Moreover, vancomycin treatment induces the transcription of the transcriptional regulator whiB6, which is also induced by several types of stress (SDS, ethanol, oxidative stress, heat shock) but slightly downregulated by cycloserine, another inhibitor of cell-wall biosynthesis (Geiman et al., 2006).
Cycloserine, ethambutol, and isoniazid, which all disrupt formation of the cell wall, but by different modes of action, increase the transcription of the transcriptional regulator whiB2, whereas the transcription of whiB7 is increased by protein synthesis inhibitors, such as erythromycin, tetracycline, streptomycin, and kanamycin (Morris et al., 2005; Geiman et al., 2006), and DNA replication inhibitors, such as fluoroquinolones (Burian et al., 2012a). WhiB2 has been reported to act as a chaperone in vitro (Konar et al., 2012) and it also an ortholog of the Streptomyces sporulation regulators WhiB and it is induced in resting cell formation in M. smegmatis (Wu et al., 2016). WhiB7 is a transcriptional regulator induced by exposure to antibiotics and fatty acids of the eukaryotic host (Morris et al., 2005). The whiB7 promoter can be induced by many diverse compounds, independently of the structure or target of such compounds, and many are respiration and redox homeostasis disrupters (Burian et al., 2012b). Thus, WhiB7 likely regulates redox stress and cell metabolism and likely acts as a transcriptional regulator that coordinates intrinsic drug resistance with antibiotic-induced changes of bacterial physiology (Burian et al., 2012a). A better understanding of the genes regulated by WhiB7 and the identification of compounds that inhibit the products of these genes should provide new insights into the treatment of mycobacterial diseases.
β-lactam antibiotics are a class of broad-spectrum antibiotics that inhibit peptidoglycan synthesis and thus cell wall biosynthesis. However, this class is not usually used for the treatment of TB because M. tuberculosis is intrinsically resistant to β-lactam through the production of β-lactamases (Flores et al., 2005). Nonetheless, the association of β-lactam with β-lactamase inhibitors is currently being reconsidered due to the increase in the appearance of MDR-TB (Wivagg et al., 2014; Story-Roller and Lamichhane, 2018).
Meropenem, a member of the carbapenem class of β-lactams, induces the expression of cell-wall biosynthetic genes, such as FbpC (Ag85C), the β-lactamase BlaC Rv2068c, and those of the stress pathway, such as Hsp, and membrane proteins, and the multidrug-transport integral membrane protein Mmr (Rv3065); whereas genes involved in various metabolic process are downregulated (Lun et al., 2014).
An analysis of the transcriptional response of M. tuberculosis treated with amoxicillin and clavulanate has been performed to clarify the mode of action of the combination of a β-lactam with a β-lactamase inhibitor (Mishra et al., 2017). Such treatment resulted in the upregulation of 481 genes and downregulation of 461; the authors suggest that these modifications represent an adaptation profile that allows tolerance to the combination of drugs. The authors further combined these results with in silico network analysis to highlight genes belonging to several functional classes in this transcriptional response. They included cell-wall processes with genes involved in peptidoglycan biogenesis and the induction of genes involved in membrane permeability and the drug efflux pump, as well as those belonging to lipid metabolism, intermediary metabolism, and respiration. Finally, the main finding of this work was a link between tolerance to the combination of the β-lactam and β-lactamase inhibitor with the redox stress mechanism. Indeed, the authors propose that β-lactam affects the integrity of the membrane and thus disturbs the respiratory chain, redox balance, and ATP generation, all which are involved in killing the bacteria. The mycobacteria compensates for these disturbances by modifying its metabolism, which produces reactive oxygen species (ROS). WhiB4, a regulator involved in the oxidative-stress response (Chawla et al., 2012) was also shown to regulate β-lactamase expression associated with redox homeostasis and thus participate in antibiotic tolerance of M. tuberculosis (Mishra et al., 2017).
Bedaquiline is a new drug approved for the treatment of MDR-TB since 2012, with the ability to inhibit mycobacterial F1F0 ATP synthase (Andries et al., 2005). Transcriptional analyses revealed that M. tuberculosis induces the DosR and activates ATP-generating pathways in the beginning of treatment with to bedaquilin (30 and 180 min) (Koul et al., 2014). The DosR is induced by multiple stresses, especially conditions that inhibit aerobic respiration (Boon and Dick, 2012). Upregulation of the ATP synthase operon could be a strategy to compensate for the inhibition of ATP synthase at the beginning of bedaquiline treatment. A time-course analysis of the transcriptome was performed in M. smegmatis to study the bactericidal mode of action of bedaquiline over time and was coupled to the measurement of oxygen consumption and the proton-motive force (PMF) (Hards et al., 2015). Many of the genes upregulated in response to bedaquiline treatment belong to alternative metabolic pathways for energy generation, in particular cytochrome bd oxidase, as well as those involved in central intermediary metabolism and regulatory functions, whereas genes involved in glycolysis, a source of non-respiratory ATP generation, were downregulated. The authors suggested a new mechanism for the bactericidal effects of bedaquiline, at least in M. smegmatis, consisting of the uncoupling of respiration-driven ATP synthesis, leading to collapse of the transmembrane pH gradient and dissipation of the PMF (Hards et al., 2015).
Upregulation of the dosR regulon and ATP synthase operon is transient given that the expression of these genes is returned to the baseline profile 48–96 h after bedaquiline treatment (Peterson et al., 2016). At these times, the ArsR-type transcription factor Rv0324 and the MarR family transcription regulator Rv0880 have an essential role in the regulation of response to bedaquiline. Moreover, the disruption of these two genes by knocking out makes the bacteria hypersensitive to bedaquiline (Peterson et al., 2016). Rv0324 and Rv0880 would regulate and coordinate the response to bedaquiline treatment that allows the bedaquiline-tolerance state. The authors then managed to demonstrate that pretomanid potentiates killing by bedaquiline via inhibition of the Rv0880 bedaquiline-response regulon (Peterson et al., 2016).
The compound AX-35 is an arylvinylpiperazine amide that target QcrB subunit of the respiratory cytochrome bc1 complex (Ballell et al., 2013; Foo et al., 2018). The treatment of M. tuberculosis by AX-35 causes the upregulation of the cydABCD operon, but not the dosR regulon, which is compatible with the signature profile of compounds inhibiting cytochrome c oxydase (Boshoff et al., 2004). cydA and cydB are the structural genes coding the cytochrome bd terminal oxidase while cydCD encodes an ABC transporter implicated in cytochrome bd assembly. The authors also note an upregulation of tgs genes and lipases genes (lipU and lipR) involved in the biosynthesis of TAG which would correspond to a remodeling of the central carbon metabolism and lipid metabolism induced by AX stress in M. tuberculosis (Foo et al., 2018).
DNA Repair Pathway Induced by Subinhibitory Concentrations of Ciprofloxacin
It is known that bacterial stress causes various adaptive and protective responses and modifies gene expression patterns and cell physiology, which can influence susceptibility to antibiotics (Poole, 2012). However, addition of an antibiotic can itself be considered to be a stress. The effect of antibiotics on bacteria is not only target-specific inhibition. Bactericidal antibiotics can induce the production of toxic metabolites that lead to cellular damage (Belenky et al., 2015). Antibiotics, such as β-lactams, quinolones, and aminoglycosides, can induce the production of ROS in bacteria (Kohanski et al., 2007). In addition, several studies involving different antibiotics and different species of bacteria have shown that ROS cause damage to macromolecules, including DNA, which can result in induction of the SOS response and thus SOS-induced mutagenesis, leading to the appearance of resistance (Dwyer et al., 2009; Kohanski et al., 2010a).
Fluoroquinolones have antimycobacterial activity and are used for the treatment of TB as second-line drugs. They prevent cell division by inhibiting DNA gyrase, which belongs to the type II topoisomerase family, thus preventing the replication of bacterial DNA. In addition, inhibition of DNA gyrase results in the appearance of double-stranded DNA breaks, inducing the SOS response. The SOS system is an error-prone repair system which can induce mutations. Gyrase inhibitors also induce the formation of ROS and oxidative damage (Dwyer et al., 2007; Kohanski et al., 2010a), which can also lead to mutagenesis. The contribution of these mutations to adaptation or antibiotic resistance is still a subject of debate (Kohanski et al., 2010b; Torres-Barcelo et al., 2015). A study in Mycobacterium showed that a subinhibitory concentration of ciprofloxacin, which belongs to the fluoroquinolone class, increased mutation rates (Gillespie et al., 2005). It is in this context that a study was performed to investigate the effects of the response of M. tuberculosis to subinhibitory concentrations of ciprofloxacin by genome-wide expression profiling and qRT-PCR (O’Sullivan et al., 2008). The microarray analysis showed upregulation of 16 genes involved in DNA protection, repair, and recombination, including recA, known to be involved in the SOS response and stimulated by single-stranded DNA. Differential expression of lexA and dnaE2, two other components of the SOS response, was not observed by microarray analysis, but qRT-PCR showed an increase in the expression of these two genes. DnaE2 polymerase is known to contribute to the emergence of drug resistance in M. tuberculosis (Boshoff et al., 2003). It is thus important to extend this research to investigate the possibility of inducing resistance by fluoroquinolone in the treatment of TB.
The transcriptional response to ciprofloxacin has also been studied in S. coelicolor (Patkari and Mehra, 2013), a non-pathogenic soil bacterium belonging to the Actinobacteria phylum, the same as Mycobacterium. The exposure of Streptomyces to ciprofloxacin induced the transcription of the gene encoding the target, DNA gyrase, as well as genes involved in DNA repair and various transporters which could function as efflux pumps. The authors also noted the production of ROS, as well as the induction of SOS genes by ciprofloxacin. Thus, bacteria may increase the transcription of DNA repair and SOS genes to repair damaged DNA as a consequence of ciprofloxacin treatment.
The Transcriptomic Response to Antibiotics During Infection
Most data on the mode of action of antibiotics have been obtained using cultures under laboratory growth conditions and the studies presented in this review up to now have focused on the in vitro effects of drugs against Mycobacterium. However, these approaches may not reflect reality in vivo, in humans infected by mycobacteria or animal models of infection. It is thus important to explore the transcriptional response in vivo during the treatment of TB.
Several studies have investigated the transcriptional response of Mycobacterium inside macrophages, providing new insights into the adaptation of the mycobacteria in the cellular environment and changes in their metabolism during progression of the disease (Waddell and Butcher, 2007). The absence of induction of isoniazide-responsive genes after isoniazide treatment in an animal model and patients suggests that the mycobacteria have become tolerant to isoniazid (Karakousis et al., 2008; Walter et al., 2015). Thus, clinically, the analysis of the antibiotic-response by transcriptomic profiling should make it possible to predict the physiological state of the bacteria and the induction of antibiotic tolerance. Such information could contribute to improving treatment strategies.
Gene expression has been investigated in samples from patients with pulmonary drug susceptible TB treated with the standard combination of isoniazid, rifampicin, pyrazinamide, and ethambutol (Walter et al., 2015). A decline in bacterial mRNA abundance showed killing of most of the bacteria 4 days after starting treatment, but the abundance of bacterial mRNA after that indicates drug tolerance. Genes involved in growth, metabolism, and lipid synthesis were downregulated in the drug-tolerant bacteria, whereas those involved in the stress response and drug efflux pumps were upregulated. In particular, the upregulation of regulatory genes, such as those of the toxin-antitoxin (TA) module, sigma factors, and transcription factors were observed, as in previous transcriptomic studies of persister cells (Betts et al., 2002, 2003; Muttucumaru et al., 2004). Moreover, the transcriptional signature of isoniazid stress was induced at the beginning of treatment but disappeared after 4 days. This suggests that the response to antibiotics of drug-tolerant M. tuberculosis in vivo changes, as observed in drug-treated dormant M. tuberculosis (Karakousis et al., 2008).
The Transcriptomic Response in Clinical Strains and Multidrug Resistant Strains
Global expression profiling studies have also been carried out to investigate the response of resistant clinical strains to antibiotics. Some genes appear to be differentially expressed between sensitive H37Rv strain and drug-resistant strains (Penuelas-Urquides et al., 2013; Yu et al., 2015). Thus, esxG, esxH, rpsA, esxI, and rpmI have been shown to be upregulated, whereas lipF, groES, and narG were downregulated in MDR clinical strains (Penuelas-Urquides et al., 2013). However, the expression of these genes among different clinical strains with similar drug-resistance profiles varies widely (Gonzalez-Escalante et al., 2015). EsxG-EsxH are proteins secreted by the ESX-3 type VII secretion system required for optimal growth and may play a role in the virulence of M. tuberculosis (Ilghari et al., 2011; Tufariello et al., 2016). Nine intergenic regions were also upregulated in the MDR strain (Penuelas-Urquides et al., 2013). Whether these intergenic regions code for proteins or are non-coding RNA with a role in post-transcriptional regulation is yet to be determined (Arnvig et al., 2011).
A transcriptional profiling study to assess the accumulation of drug resistance in the Mumbai area showed genes for cell-wall biosynthesis (emb), protein synthesis (rpl), transcription factors (sig, rpoB), and those involved in various metabolic pathways to be downregulated, whereas genes encoding efflux pumps and transcriptional factors involved in the stress response (whiB) where upregulated in MDR clinical isolates relative to those in a drug-susceptible strain (Chatterjee et al., 2013). Such differences could contribute to resistance to anti-TB drugs, without the presence of drug-resistance mutations. The upregulation of efflux pumps allows expulsion of the antibiotics whereas the decrease in cellular metabolism and transcription could be associated with a form of persistence.
Another comparison of the transcriptome of XDR clinical isolates and susceptible M. tuberculosis revealed downregulation of ethA (de Welzen et al., 2017), which encodes the monooxygenase that activates ethionamide. Such downregulation was due to a mutation in the promoter, but not transcriptional repression by ethR, and the strains with this mutation were resistant to ethionamide without other known ethionamide resistance-determining genotypes.
Transcriptional responses to isoniazid, capreomycin, and rifampicin have been investigated in two XDR strains compared to that of H37Rv, a wildtype strain (Yu et al., 2015). The authors highlighted a correlation between genes overexpressed in the XDR strains and previously published known drug resistance genes and proposed 92 as candidate resistance genes. Regulatory network analysis of these genes showed the importance of the transcriptional regulator CRP (Rv3676) and three two-component regulatory systems (TCS) in drug resistance: SenX3-RegX3, MprA-MprB, and MtrA-MtrB. TCS are involved in sensing the response to stress and adaptation to changes in the environment (Bretl et al., 2011; Parish, 2014) and the coordination of global changes in gene expression. It is interesting to note that SenX3 and MtrA share homology with the gene expression modulator BfmRS, which has recently been shown to enhance pathogenicity and antibiotic resistance in Acinetobacter baumannii (Geisinger et al., 2018).
Rifampicin is a first line TB drug that binds to the pocket of the RNA polymerase β subunit (RpoB) and thus inhibits RNA synthesis. In 95% of cases, M. tuberculosis becomes resistant to rifampicin by target alteration after mutation of the gene rpoB. A transcriptional profiling study was performed using microarray technology on wildtype rifampicin-susceptible and rifampicin-resistant strains exposed to several concentrations of rifampicin to obtain new insights into the response to rifampicin exposure (de Knegt et al., 2013). The transcriptional response induced by rifampicin was different between the rifampicin-resistant and rifampicin-susceptible strains. In particular, the authors highlighted changes in the expression of gene clusters associated with efflux, transport, and virulence. Moreover, the authors observed strong upregulation of both genes of operon Rv0559c-Rv0560c, previously shown to be induced following salicylate exposure (Denkin et al., 2005) and recently shown to be upregulated upon isoniazide exposure (Gamngoen et al., 2018). The mechanism and function of Rv0560c and Rv0559c in response to two of the first-line TB drugs, rifampicin and isoniazid, merit further study.
The mutation in rpoB can be associated with a fitness cost for bacteria (Mariam et al., 2004; Gagneux et al., 2006) but it has been proposed that mutation in genes rpoC could compensate for this fitness cost (Comas et al., 2011). Transcriptional approach seems to confirm this proposition by comparing responses between clinical strains that harbor single mutation in rpoB and clinical strains with mutations in rpoB and rpoC (Xu et al., 2018). The energy metabolism and oxidative phosphorylation machinery are disrupted in the rpoB single mutants but the upregulation of the bd-type menaquinol oxydase (CydAB) and of qcrCAB operon, encoding cytochrome c oxydase, in rpoBC mutants might compensate this phenomenon. rpoC mutations were also associated with upregulation of ribosomal protein genes and alteration in amino acid synthesis pathways, these changes in metabolism may participate in the compensatory response (Xu et al., 2018).
It is well known that the multidrug efflux pumps contribute to antibiotic resistance and/or tolerance, as well as bacterial virulence (Alcalde-Rico et al., 2016). Efflux pumps can be expressed constitutively and participate in intrinsic resistance, or their expression can be stimulated by an effector and thus result in phenotypic resistance to an antibiotic. Various efflux pump genes can be up-regulated in the presence of many antibiotics, as already discussed. An exhaustive study underlined the importance of this mechanism of drug resistance by comparing the differential expression of efflux pump genes in the presence of various antimycobacterial drugs in several clinical MDR isolates of M. tuberculosis (Gupta et al., 2010). This study highlighted the association between differential expression of selective efflux pump genes and drugs, such as ethambutol, isoniazid, and streptomycin, as well as strain-to-strain variations. The same phenomena was observed for the overexpression of efflux pump genes by isoniazid and rifampicin in MDR isolates (Li et al., 2015) and the basal expression level of some genes encoding efflux pumps was higher in MDR than sensitive isolates.
Discussion
Antibiotic-induced changes in gene expression profiles have aided our understanding of the effect of antibiotics on the physiology of M. tuberculosis. The investigation of such changes, measured by microarrays or RNA-seq, can help to identify the mode of action of the drug tested, as well as provide a better vision of the mechanisms of drug tolerance or drug resistance. The genes that promote antibiotic resistance have collectively been called the resistome (Wright, 2007).
The gene expression profile induced by a drug can be considered to be the transcriptional signature characteristic of its mode of action and this signature can be used to predict the mode of action of novel compounds with antimycobacterial activity, identified by whole cell-screening approaches. However, the quality of the expression data is essential to predicting the mode of action of new drugs based on comparisons with the profile induced by well-characterized antimycobacterial compounds, as shown in the study of Boshoff et al. (2004).
It may be difficult to directly compare the presented studies because of the different methods used. The modulation of mycobacterial gene expression depends on not only the class of antibiotic but also the concentration tested. Antibiotics can be tested at high concentrations, equivalent to or higher than the MIC that prevents growth of the bacteria, or at a subinhibitory concentration to explore the physiological adaptation of the bacteria. The duration of exposure to the antibiotic should also be taken into consideration.
In addition to having an effect on a specific target, an antibiotic can affect the global physiology of the bacteria. In transcriptomic studies, it can be difficult to discriminate between changes in RNA expression corresponding to compensation for inhibition of the target pathway by the drug and those that are a response to the toxic effect of the compound. Indeed, the changes in gene expression in response to an antibiotic are a combination of the specific effect of the drug and a global metabolic effect that involves the deregulation of many genes (Mitosch and Bollenbach, 2014). For example, the activity of transcriptional regulators and RNA polymerase can be modified upon antibiotic treatment and thus influence the expression of all bacterial genes. Moreover, antibiotics affect the growth rate of bacteria and it has been shown that changes in bacterial growth rates modify gene expression in bacteria in chemostat culture (Hua et al., 2004).
However, the comparative analysis of transcriptome data from Mycobacterium treated with different antimycobacterial drugs can be used to predict new drug combination. From these comparisons, After having identified Rv0324 and Rv0880 as regulators of bedaquiline tolerance in M. tuberculosis, pretomanid has been identified as a modulator of expression of Rv0880 and a potentiator of bedaquiline activity (Peterson et al., 2016).
We have seen that the transcriptomic data can be used to identify new antibacterial compounds. Genes induced specifically by one class of antibacterial can be used as reporter to identify compounds sharing the same mode of action. In order to identify cell envelope inhibitor, a synthetic compounds library was screened for induction of iniBAC promoter, a marker of cell envelope stress response (Shetty et al., 2018). This approach has identified new acetamide that indirectly targets the essential mycolic acid transporter MmpL3 by disruption of the proton motive force (PMF) that MmpL3 use for translocation to translocate trehalose monomycolates (THM) (Shetty et al., 2018).
The measurement of the variation of mRNA expression is only an indication of the phenotype of bacteria. Metabolomics approaches, which measure small molecules produced during the metabolism, directly reflect the biochemical activity of bacteria. Changes in the metabolome profiling after treatment with antimicrobial compounds give new insight in the bacterial physiological response to antibiotic and the drug mechanism (Zampieri et al., 2017) and this analysis have been used in M. smegmatis to predict the mode of action of uncharacterized antimicrobial compounds (Zampieri et al., 2018).
The environmental context of M. tuberculosis affects mRNA expression, as the regulation of gene expression also depends on the host microenvironment. Once inside macrophages, the bacteria must adapt to the intracellular environment and accommodate the host immune response. Upregulated genes of M. tuberculosis inside macrophages include those involved in fatty acid metabolism, mycolic acid modification, the DosR (Schnappinger et al., 2003), and several members of the WhiB family (Rohde et al., 2007). Thus, the transcriptional response of bacteria to antibiotic exposure in vitro, in the laboratory, is likely to be very different from that in the intracellular context.
A study showed the role of host macrophages in the induction of efflux pumps and drug tolerance of Mycobacteria (Adams et al., 2011) using a zebrafish model of TB infection with M. marinum. The study showed that Rv1258c, a member of the MFS of efflux pumps, which is transcriptionally induced after entry into the macrophage (Schnappinger et al., 2003), promotes intracellular growth in macrophages in the absence of drug treatment, as well as intracellular rifampicin tolerance during treatment (Adams et al., 2011).
The metabolism of mycobacterial cells can change during the dynamics of the infection, in particular when the bacilli enter into the dormant state, in which metabolic activity is reduced, leading to antibiotic tolerance or resistance. Persister mycobacteria are phenotypically resistant to antimycobacterial compounds, although genetically susceptible to these drugs (Gengenbacher and Kaufmann, 2012). Thus, the phenotypic plasticity of mycobacteria allows them to survive in the presence of antibiotics (Ehrt et al., 2018).
After to have identified a set of genes induced in M. tuberculosis persister cells surviving 4 days of isoniazide treatment, the promoter of these genes was fused with fluorescent reporters in mycobacteriophages. This tool helped to identify a population of preexisting persister mycobacteria in the sputum of human patients and the number of these persister cells increased after the beginning of antitubercular therapy (Jain et al., 2016).
In addition, factors of variability inherent to the host, such as the genetic background or the efficiency of the host’s immune response, can also influence the bacterial transcriptional response, as recently shown for S. aureus (Thanert et al., 2017). It has been observed that susceptibility to TB varies according to genetic predisposition among populations (Cooke and Hill, 2001; Thye et al., 2010). Moreover, patient responses to treatment are heterogenic; transcriptomic and metabolomic profiles of patients infected by M. tuberculosis were similar before treatment, but different after treatment (Tientcheu et al., 2015). Thus, human polymorphism also influences the behavior of the bacteria. Given these considerations, the next major challenge will be the exploration of gene expression changes induced by antibiotics during in vivo infection.
Mycobacterium tuberculosis, possesses genes that can be considered to be drug-resistance systems through modulation of their expression, but the interaction between the drug, the bacteria, and the host is complex. Bacteria can change their global gene expression to increase their chances of survival. Gene regulatory networks appear to be important for preserving cell homeostasis under conditions of stress, such as antibiotic exposure. Transcriptional profiles and phenotypic responses of bacteria under stress are generally highly correlated. Phenotypic and transcriptional stress responses can involve distinct gene sets (Jensen et al., 2017). It has been shown in Streptococcus pneumonia that nutrient stress causes a highly organized response, whereas the response to antibiotics is uncoordinated (Jensen et al., 2017). The construction of transcriptomic data sets, complemented with proteomic (Wenzel and Bandow, 2011), metabolomic data (Zampieri et al., 2018), and lipidomic (Pal et al., 2017) should bring new insights into the molecular networks involved in the response to antibiotic exposure.
Deregulated genes identified after antibiotic exposure can be considered to be targets for the identification of new drugs and the development of therapy against TB. These drugs, which would target potential resistant pathways, could be used in combinatorial therapies with existing anti-TB drugs. Antibiotic adjuvants or potentiators are compounds that enhance antibiotic activity by inactivating resistance mechanism (Wright, 2016). As a proof of concept, it was shown that the MIC of isoniazid decreased in the presence of various categories of efflux pump inhibitors in clinical isolates of M. tuberculosis (Jaiswal et al., 2017). Efflux pump inhibitors should thus be considered to improve current TB therapy (Pule et al., 2016).
Author Contributions
JB, SL, and BG have decided the content of the manuscript. JB has analyzed the bibliography and written the manuscript. BG and SL has corrected and approved the manuscript.
Funding
This study was supported by the Sanming project of Medicine in Shenzhen (SZSM201603029).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Institut Pasteur of Shanghai, Chinese Academy of Sciences in Shanghai and the Shenzhen Key Laboratory of Microbiology and Antibacterial Resistance Surveillance, at Shenzhen People’s Hospital, the Second Clinical Medical College of Jinan University, for hosting during the completion of this paper. We also thank the Department of Tuberculosis Control and Prevention, Shenzhen Nanshan Center for Chronic Disease Control and the Institut Pasteur in Paris for the support.
References
Adams, K. N., Takaki, K., Connolly, L. E., Wiedenhoft, H., Winglee, K., Humbert, O., et al. (2011). Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145, 39–53. doi: 10.1016/j.cell.2011.02.022
Alcalde-Rico, M., Hernando-Amado, S., Blanco, P., and Martinez, J. L. (2016). Multidrug efflux pumps at the crossroad between antibiotic resistance and bacterial virulence. Front. Microbiol. 7:1483. doi: 10.3389/fmicb.2016.01483
Alland, D., Kramnik, I., Weisbrod, T. R., Otsubo, L., Cerny, R., Miller, L. P., et al. (1998). Identification of differentially expressed mRNA in prokaryotic organisms by customized amplification libraries (DECAL): the effect of isoniazid on gene expression in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 95, 13227–13232. doi: 10.1073/pnas.95.22.13227
Alland, D., Steyn, A. J., Weisbrod, T., Aldrich, K., and Jacobs, W. R. (2000). Characterization of the Mycobacterium tuberculosis iniBAC promoter, a promoter that responds to cell wall biosynthesis inhibition. J. Bacteriol. 182, 1802–1811. doi: 10.1128/JB.182.7.1802-1811.2000
Almeida Da Silva, P. E., and Palomino, J. C. (2011). Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: classical and new drugs. J. Antimicrob. Chemother. 66, 1417–1430. doi: 10.1093/jac/dkr173
Andries, K., Verhasselt, P., Guillemont, J., Gohlmann, H. W., Neefs, J. M., Winkler, H., et al. (2005). A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science (80-.) 307, 223–227. doi: 10.1126/science.1106753
Arnvig, K. B., Comas, I., Thomson, N. R., Houghton, J., Boshoff, H. I., Croucher, N. J., et al. (2011). Sequence-based analysis uncovers an abundance of non-coding RNA in the total transcriptome of Mycobacterium tuberculosis. PLoS Pathog. 7:e1002342. doi: 10.1371/journal.ppat.1002342
Astarie-Dequeker, C., Le Guyader, L., Malaga, W., Seaphanh, F. K., Chalut, C., Lopez, A., et al. (2009). Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS Pathog. 5:e1000289. doi: 10.1371/journal.ppat.1000289
Bacon, J., James, B. W., Wernisch, L., Williams, A., Morley, K. A., Hatch, G. J., et al. (2004). The influence of reduced oxygen availability on pathogenicity and gene expression in Mycobacterium tuberculosis. Tuberc 84, 205–217. doi: 10.1016/j.tube.2003.12.011
Baek, S. H., Li, A. H., and Sassetti, C. M. (2011). Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 9:e1001065. doi: 10.1371/journal.pbio.1001065
Balganesh, M., Dinesh, N., Sharma, S., Kuruppath, S., Nair, A. V., and Sharma, U. (2012). Efflux pumps of Mycobacterium tuberculosis play a significant role in antituberculosis activity of potential drug candidates. Antimicrob. Agents Chemother. 56, 2643–2651. doi: 10.1128/AAC.06003-11
Ballell, L., Bates, R. H., Young, R. J., Alvarez-Gomez, D., Alvarez-Ruiz, E., Barroso, V., et al. (2013). Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem 8, 313–321. doi: 10.1002/cmdc.201200428
Baptista, R., Fazakerley, D. M., Beckmann, M., Baillie, L., and Mur, L. A. J. (2018). Untargeted metabolomics reveals a new mode of action of pretomanid (PA-824). Sci. Rep. 8:5084. doi: 10.1038/s41598-018-23110-1
Belenky, P., Ye, J. D., Porter, C. B., Cohen, N. R., Lobritz, M. A., Ferrante, T., et al. (2015). Bactericidal antibiotics induce toxic metabolic perturbations that lead to cellular damage. Cell Rep. 13, 968–980. doi: 10.1016/j.celrep.2015.09.059
Betts, J. C., Lukey, P. T., Robb, L. C., McAdam, R. A., and Duncan, K. (2002). Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43, 717–731. doi: 10.1046/j.1365-2958.2002.02779.x
Betts, J. C., McLaren, A., Lennon, M. G., Kelly, F. M., Lukey, P. T., Blakemore, S. J., et al. (2003). Signature gene expression profiles discriminate between isoniazid-, thiolactomycin-, and triclosan-treated Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 47, 2903–2913. doi: 10.1128/AAC.47.9.2903-2913.2003
Bonn, F., Pane-Farre, J., Schluter, R., Schaffer, M., Fuchs, S., Bernhardt, J., et al. (2016). Global analysis of the impact of linezolid onto virulence factor production in S. aureus USA300. Int. J. Med. Microbiol. 306, 131–140. doi: 10.1016/j.ijmm.2016.02.004
Boon, C., and Dick, T. (2012). How Mycobacterium tuberculosis goes to sleep: the dormancy survival regulator DosR a decade later. Futur. Microbiol. 7, 513–518. doi: 10.2217/fmb.12.14
Boot, M., Commandeur, S., Subudhi, A. K., Bahira, M., Smith, T. C., Abdallah, A. M., et al. (2018). Accelerating early antituberculosis drug discovery by creating mycobacterial indicator strains that predict mode of action. Antimicrob. Agents Chemother. 62:e00083-18. doi: 10.1128/AAC.00083-18
Boshoff, H. I., Myers, T. G., Copp, B. R., McNeil, M. R., Wilson, M. A., and Barry, C. E. III (2004). The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J. Biol. Chem. 279, 40174–40184. doi: 10.1074/jbc.M406796200
Boshoff, H. I., Reed, M. B., Barry, C. E. III, and Mizrahi, V. (2003). DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 113, 183–193. doi: 10.1016/S0092-8674(03)00270-8
Bretl, D. J., Demetriadou, C., and Zahrt, T. C. (2011). Adaptation to environmental stimuli within the host: two-component signal transduction systems of Mycobacterium tuberculosis. Microbiol. Mol. Biol. Rev. 75, 566–582. doi: 10.1128/MMBR.05004-11
Brooks, A. N., Turkarslan, S., Beer, K. D., Lo, F. Y., and Baliga, N. S. (2011). Adaptation of cells to new environments. Wiley Interdiscip. Rev. Syst. Biol. Med. 3, 544–561. doi: 10.1002/wsbm.136
Burian, J., Ramon-Garcia, S., Howes, C. G., and Thompson, C. J. (2012a). WhiB7, a transcriptional activator that coordinates physiology with intrinsic drug resistance in Mycobacterium tuberculosis. Expert Rev. Anti Infect Ther. 10, 1037–1047. doi: 10.1586/eri.12.90
Burian, J., Ramon-Garcia, S., Sweet, G., Gomez-Velasco, A., Av-Gay, Y., and Thompson, C. J. (2012b). The mycobacterial transcriptional regulator whiB7 gene links redox homeostasis and intrinsic antibiotic resistance. J. Biol. Chem. 287, 299–310. doi: 10.1074/jbc.M111.302588
Cases, I., de Lorenzo, V., and Ouzounis, C. A. (2003). Transcription regulation and environmental adaptation in bacteria. Trends Microbiol. 11, 248–253. doi: 10.1016/S0966-842X(03)00103-3
Chatterjee, A., Saranath, D., Bhatter, P., and Mistry, N. (2013). Global transcriptional profiling of longitudinal clinical isolates of Mycobacterium tuberculosis exhibiting rapid accumulation of drug resistance. PLoS One 8:e54717. doi: 10.1371/journal.pone.0054717
Chawla, M., Parikh, P., Saxena, A., Munshi, M., Mehta, M., Mai, D., et al. (2012). Mycobacterium tuberculosis WhiB4 regulates oxidative stress response to modulate survival and dissemination in vivo. Mol. Microbiol. 85, 1148–1165. doi: 10.1111/j.1365-2958.2012.08165.x
Colangeli, R., Helb, D., Sridharan, S., Sun, J., Varma-Basil, M., Hazbón, M. H., et al. (2005). The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol. Microbiol. 55, 1829–1840. doi: 10.1111/j.1365-2958.2005.04510.x
Comas, I., Borrell, S., Roetzer, A., Rose, G., Malla, B., Kato-Maeda, M., et al. (2011). Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 44, 106–110. doi: 10.1038/ng.1038
Comas, I., Coscolla, M., Luo, T., Borrell, S., Holt, K. E., Kato-Maeda, M., et al. (2013). Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45, 1176–1182. doi: 10.1038/ng.2744
Cooke, G. S., and Hill, A. V. (2001). Genetics of susceptibility to human infectious disease. Nat. Rev. Genet. 2, 967–977. doi: 10.1038/35103577
de Knegt, G. J., Bruning, O., ten Kate, M. T., de Jong, M., van Belkum, A., Endtz, H. P., et al. (2013). Rifampicin-induced transcriptome response in rifampicin-resistant Mycobacterium tuberculosis. Tuberc 93, 96–101. doi: 10.1016/j.tube.2012.10.013
de Welzen, L., Eldholm, V., Maharaj, K., Manson, A. L., Earl, A. M., and Pym, A. S. (2017). Whole-transcriptome and -genome analysis of extensively drug-resistant Mycobacterium tuberculosis clinical isolates identifies downregulation of etha as a mechanism of ethionamide resistance. Antimicrob. Agents Chemother. 61:e01461-17. doi: 10.1128/AAC.01461-17
Denkin, S., Byrne, S., Jie, C., and Zhang, Y. (2005). Gene expression profiling analysis of Mycobacterium tuberculosis genes in response to salicylate. Arch. Microbiol. 184, 152–157. doi: 10.1007/s00203-005-0037-9
Drumm, J. E., Mi, K., Bilder, P., Sun, M., Lim, J., Bielefeldt-Ohmann, H., et al. (2009). Mycobacterium tuberculosis universal stress protein Rv2623 regulates bacillary growth by ATP-Binding: requirement for establishing chronic persistent infection. PLoS Pathog. 5:e1000460. doi: 10.1371/journal.ppat.1000460
Dwyer, D. J., Kohanski, M. A., and Collins, J. J. (2009). Role of reactive oxygen species in antibiotic action and resistance. Curr. Opin. Microbiol. 12, 482–489. doi: 10.1016/j.mib.2009.06.018
Dwyer, D. J., Kohanski, M. A., Hayete, B., and Collins, J. J. (2007). Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol. Syst. Biol. 3:91. doi: 10.1038/msb4100135
Ehrt, S., and Schnappinger, D. (2009). Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. 11, 1170–1178. doi: 10.1111/j.1462-5822.2009.01335.x
Ehrt, S., Schnappinger, D., and Rhee, K. Y. (2018). Metabolic principles of persistence and pathogenicity in Mycobacterium tuberculosis. Nat. Rev. Microbiol. 16, 496–507. doi: 10.1038/s41579-018-0013-4
Fishbein, S., van Wyk, N., Warren, R. M., and Sampson, S. L. (2015). Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Mol. Microbiol. 96, 901–916. doi: 10.1111/mmi.12981
Flores, A. R., Parsons, L. M., and Pavelka, M. S. Jr. (2005). Genetic analysis of the beta-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to beta-lactam antibiotics. Microbiology 151, 521–532. doi: 10.1099/mic.0.27629-0
Foo, C. S., Lupien, A., Kienle, M., Vocat, A., Benjak, A., Sommer, R., et al. (2018). Arylvinylpiperazine amides, a new class of potent inhibitors targeting QcrB of Mycobacterium tuberculosis. MBio 9:e01276-18. doi: 10.1128/mBio.01276-18
Fu, L. M., and Shinnick, T. M. (2007). Genome-wide exploration of the drug action of capreomycin on Mycobacterium tuberculosis using Affymetrix oligonucleotide GeneChips. J. Infect. 54, 277–284. doi: 10.1016/j.jinf.2006.05.012
Fu, L. M., and Tai, S. C. (2009). The differential gene expression pattern of Mycobacterium tuberculosis in response to capreomycin and PA-824 versus first-line TB drugs reveals stress- and PE/PPE-related drug targets. Int. J. Microbiol. 2009:879621. doi: 10.1155/2009/879621
Gagneux, S., Long, C. D., Small, P. M., Van, T., Schoolnik, G. K., and Bohannan, B. J. M. (2006). The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312, 1944–1946. doi: 10.1126/science.1124410
Gamngoen, R., Putim, C., Salee, P., Phunpae, P., and Butr-Indr, B. (2018). A comparison of Rv0559c and Rv0560c expression in drug-resistant Mycobacterium tuberculosis in response to first-line antituberculosis drugs. Tuberc 108, 64–69. doi: 10.1016/j.tube.2017.11.002
Geiman, D. E., Raghunand, T. R., Agarwal, N., and Bishai, W. R. (2006). Differential gene expression in response to exposure to antimycobacterial agents and other stress conditions among seven Mycobacterium tuberculosis whiB-like genes. Antimicrob. Agents Chemother. 50, 2836–2841. doi: 10.1128/AAC.00295-06
Geisinger, E., Mortman, N. J., Vargas-Cuebas, G., Tai, A. K., and Isberg, R. R. (2018). A global regulatory system links virulence and antibiotic resistance to envelope homeostasis in Acinetobacter baumannii. PLoS Pathog. 14:e1007030. doi: 10.1371/journal.ppat.1007030
Gengenbacher, M., and Kaufmann, S. H. (2012). Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 36, 514–532. doi: 10.1111/j.1574-6976.2012.00331.x
Gillespie, S. H., Basu, S., Dickens, A. L., O’Sullivan, D. M., and McHugh, T. D. (2005). Effect of subinhibitory concentrations of ciprofloxacin on Mycobacterium fortuitum mutation rates. J. Antimicrob. Chemother. 56, 344–348. doi: 10.1093/jac/dki191
Gonzalez-Escalante, L., Penuelas-Urquides, K., Said-Fernandez, S., Silva-Ramirez, B., Bermudez, and de Leon, M. (2015). Differential expression of putative drug resistance genes in Mycobacterium tuberculosis clinical isolates. FEMS Microbiol. Lett. 362:fnv194. doi: 10.1093/femsle/fnv194
Gupta, A. K., Katoch, V. M., Chauhan, D. S., Sharma, R., Singh, M., Venkatesan, K., et al. (2010). Microarray analysis of efflux pump genes in multidrug-resistant Mycobacterium tuberculosis during stress induced by common anti-tuberculous drugs. Microb. Drug Resist. 16, 21–28. doi: 10.1089/mdr.2009.0054
Habib, Z., Xu, W., Jamal, M., Rehman, K., Chen, X., Dai, J., et al. (2017). Adaptive gene profiling of Mycobacterium tuberculosis during sub-lethal kanamycin exposure. Microb. Pathog. 112, 243–253. doi: 10.1016/j.micpath.2017.09.055
Hards, K., Robson, J. R., Berney, M., Shaw, L., Bald, D., Koul, A., et al. (2015). Bactericidal mode of action of bedaquiline. J. Antimicrob. Chemother. 70, 2028–2037. doi: 10.1093/jac/dkv054
Hoagland, D., Liu, J., Lee, R. B., and Lee, R. E. (2016). New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv. Drug Deliv. Rev. 102, 55–72. doi: 10.1016/j.addr.2016.04.026
Hua, Q., Yang, C., Oshima, T., Mori, H., and Shimizu, K. (2004). Analysis of gene expression in Escherichia coli in response to changes of growth-limiting nutrient in chemostat cultures. Appl. Environ. Microbiol. 70, 2354–2366. doi: 10.1128/AEM.70.4.2354-2366.2004
Hughes, M. A., Silva, J. C., Geromanos, S. J., and Townsend, C. A. (2006). Quantitative proteomic analysis of drug-induced changes in mycobacteria. J. Proteome Res. 5, 54–63. doi: 10.1021/pr050248t
Ilghari, D., Lightbody, K. L., Veverka, V., Waters, L. C., Muskett, F. W., Renshaw, P. S., et al. (2011). Solution structure of the Mycobacterium tuberculosis EsxG.EsxH complex: functional implications and comparisons with other M. tuberculosis Esx family complexes. J. Biol. Chem. 286, 29993–30002. doi: 10.1074/jbc.M111.248732
Islam, M. M., Hameed, H. M., Mugweru, J., Chhotaray, C., Wang, C., Tan, Y., et al. (2017). Drug resistance mechanisms and novel drug targets for tuberculosis therapy. J. Genet. Genomics 44, 21–37. doi: 10.1016/j.jgg.2016.10.002
Jain, P., Weinrick, B. C., Kalivoda, E. J., Yang, H., Munsamy, V., Vilcheze, C., et al. (2016). Dual-reporter mycobacteriophages (Φ2DRMS) reveal preexisting Mycobacterium tuberculosis persistent cells in human sputum. MBio 7:e01023-16. doi: 10.1128/mBio.01023-16
Jaiswal, I., Jain, A., Verma, S. K., Singh, P., Kant, S., and Singh, M. (2017). Effect of efflux pump inhibitors on the susceptibility of Mycobacterium tuberculosis to isoniazid. Lung India 34, 499–505. doi: 10.4103/0970-2113.217567
Jensen, P. A., Zhu, Z., and van Opijnen, T. (2017). Antibiotics disrupt coordination between transcriptional and phenotypic stress responses in pathogenic bacteria. Cell Rep. 20, 1705–1716. doi: 10.1016/j.celrep.2017.07.062
Karakousis, P. C., Williams, E. P., and Bishai, W. R. (2008). Altered expression of isoniazid-regulated genes in drug-treated dormant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 61, 323–331. doi: 10.1093/jac/dkm485
Kohanski, M. A., DePristo, M. A., and Collins, J. J. (2010a). Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell 37, 311–320. doi: 10.1016/j.molcel.2010.01.003
Kohanski, M. A., Dwyer, D. J., and Collins, J. J. (2010b). How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 8, 423–435. doi: 10.1038/nrmicro2333
Kohanski, M. A., Dwyer, D. J., Hayete, B., Lawrence, C. A., and Collins, J. J. (2007). A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810. doi: 10.1016/j.cell.2007.06.049
Konar, M., Alam, M. S., Arora, C., and Agrawal, P. (2012). WhiB2/Rv3260c, a cell division-associated protein of Mycobacterium tuberculosis H37Rv, has properties of a chaperone. FEBS J. 279, 2781–2792. doi: 10.1111/j.1742-4658.2012.08662.x
Koul, A., Vranckx, L., Dhar, N., Gohlmann, H. W., Ozdemir, E., Neefs, J. M., et al. (2014). Delayed bactericidal response of Mycobacterium tuberculosis to bedaquiline involves remodelling of bacterial metabolism. Nat. Commun. 5:3369. doi: 10.1038/ncomms4369
Larsen, M. H., Vilchèze, C., Kremer, L., Besra, G. S., Parsons, L., Salfinger, M., et al. (2002). Overexpression of inhA, but not kasA, confers resistance to isoniazid and ethionamide in mycobacterium smegmatis, M. bovis BCG and M. tuberculosis. Mol. Microbiol. 46, 453–466. doi: 10.1046/j.1365-2958.2002.03162.x
Lee, M., Lee, J., Carroll, M. W., Choi, H., Min, S., Song, T., et al. (2012). Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N. Engl. J. Med. 367, 1508–1518. doi: 10.1056/NEJMoa1201964
Lee, Y. V., Wahab, H. A., and Choong, Y. S. (2015). Potential inhibitors for isocitrate lyase of Mycobacterium tuberculosis and non-M. tuberculosis: a summary. Biomed. Res. Int. 2015:895453. doi: 10.1155/2015/895453
Li, G., Zhang, J., Guo, Q., Jiang, Y., Wei, J., Zhao, L. L., et al. (2015). Efflux pump gene expression in multidrug-resistant Mycobacterium tuberculosis clinical isolates. PLoS One 10:e0119013. doi: 10.1371/journal.pone.0119013
Liang, J., Tang, X., Guo, N., Zhang, K., Guo, A., Wu, X., et al. (2012). Genome-wide expression profiling of the response to linezolid in Mycobacterium tuberculosis. Curr. Microbiol. 64, 530–538. doi: 10.1007/s00284-012-0104-9
Liang, J., Zeng, F., Guo, A., Liu, L., Guo, N., Li, L., et al. (2011). Microarray analysis of the chelerythrine-induced transcriptome of Mycobacterium tuberculosis. Curr. Microbiol. 62, 1200–1208. doi: 10.1007/s00284-010-9837-5
Lun, S., Miranda, D., Kubler, A., Guo, H., Maiga, M. C., Winglee, K., et al. (2014). Synthetic lethality reveals mechanisms of Mycobacterium tuberculosis resistance to beta-lactams. MBio 5:e1767-14. doi: 10.1128/mBio.01767-14
Makarov, V., Manina, G., Mikusova, K., Mollmann, U., Ryabova, O., Saint-Joanis, B., et al. (2009). Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science (80-.) 324, 801–804. doi: 10.1126/science.1171583
Manjunatha, U., Boshoff, H. I., and Barry, C. E. (2009). The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun. Integr. Biol. 2, 215–218. doi: 10.4161/cib.2.3.7926
Mariam, D. H., Mengistu, Y., Hoffner, S. E., and Andersson, D. I. (2004). Effect of rpoB mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 48, 1289–1294. doi: 10.1128/AAC.48.4.1289-1294.2004
Mishra, S., Shukla, P., Bhaskar, A., Anand, K., Baloni, P., Jha, R. K., et al. (2017). Efficacy of beta-lactam/beta-lactamase inhibitor combination is linked to WhiB4-mediated changes in redox physiology of Mycobacterium tuberculosis. Elife 6:e25624. doi: 10.7554/eLife.25624
Mitosch, K., and Bollenbach, T. (2014). Bacterial responses to antibiotics and their combinations. Environ. Microbiol. Rep. 6, 545–557. doi: 10.1111/1758-2229.12190
Morris, R. P., Nguyen, L., Gatfield, J., Visconti, K., Nguyen, K., Schnappinger, D., et al. (2005). Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 102, 12200–12205. doi: 10.1073/pnas.0505446102
Murray, J. F., Schraufnagel, D. E., and Hopewell, P. C. (2015). Treatment of tuberculosis: a historical perspective. Ann. Am. Thorac. Soc. 12, 1749–1749. doi: 10.1513/AnnalsATS.201509-632PS
Muttucumaru, D. G., Roberts, G., Hinds, J., Stabler, R. A., and Parish, T. (2004). Gene expression profile of Mycobacterium tuberculosis in a non-replicating state. Tuberc 84, 239–246. doi: 10.1016/j.tube.2003.12.006
Mutz, K. O., Heilkenbrinker, A., Lonne, M., Walter, J. G., and Stahl, F. (2013). Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 24, 22–30. doi: 10.1016/j.copbio.2012.09.004
Namouchi, A., Gomez-Munoz, M., Frye, S. A., Moen, L. V, Rognes, T., Tonjum, T., et al. (2016). The Mycobacterium tuberculosis transcriptional landscape under genotoxic stress. BMC Genomics 17:791. doi: 10.1186/s12864-016-3132-1
Nandakumar, M., Nathan, C., and Rhee, K. Y. (2014). Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat. Commun. 5:4306. doi: 10.1038/ncomms5306
Narang, A., Giri, A., Gupta, S., Garima, K., Bose, M., and Varma-Basil, M. (2017). Contribution of putative efflux pump genes to isoniazid resistance in clinical isolates of Mycobacterium tuberculosis. Int. J. Mycobacteriol. 6, 177–183. doi: 10.4103/ijmy.ijmy_26_17
Nasiri, M. J., Haeili, M., Ghazi, M., Goudarzi, H., Pormohammad, A., Imani Fooladi, A. A., et al. (2017). New insights in to the intrinsic and acquired drug resistance mechanisms in mycobacteria. Front. Microbiol. 8:681. doi: 10.3389/fmicb.2017.00681
Nieto, R. L., Mehaffy, C., and Dobos, K. M. (2016). Comparing isogenic strains of Beijing genotype Mycobacterium tuberculosis after acquisition of Isoniazid resistance: a proteomics approach. Proteomics 16, 1376–1380. doi: 10.1002/pmic.201500403
Nonejuie, P., Burkart, M., Pogliano, K., and Pogliano, J. (2013). Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc. Natl. Acad. Sci. U.S.A. 110, 16169–16174. doi: 10.1073/pnas.1311066110
O’Sullivan, D. M., Hinds, J., Butcher, P. D., Gillespie, S. H., and McHugh, T. D. (2008). Mycobacterium tuberculosis DNA repair in response to subinhibitory concentrations of ciprofloxacin. J. Antimicrob. Chemother. 62, 1199–1202. doi: 10.1093/jac/dkn387
Padiadpu, J., Baloni, P., and Chandra, N. (2015). Gene expression profiles of wild-type and isoniazid-resistant strains of Mycobacterium smegmatis. Data Br. 4, 186–189. doi: 10.1016/j.dib.2015.05.006
Pal, R., Hameed, S., Kumar, P., Singh, S., and Fatima, Z. (2017). Comparative lipidomics of drug sensitive and resistant Mycobacterium tuberculosis reveals altered lipid imprints. 3Biotech 7:325. doi: 10.1007/s13205-017-0972-6
Pal, R., Hameed, S., Sabareesh, V., Kumar, P., Singh, S., and Fatima, Z. (2018). Investigations into isoniazid treated Mycobacterium tuberculosis by electrospray mass spectrometry reveals new insights into its lipid composition. J. Pathog. 2018:1454316. doi: 10.1155/2018/1454316
Palomino, J. C., and Martin, A. (2014). Drug resistance mechanisms in Mycobacterium tuberculosis. Antibiot 3, 317–340. doi: 10.3390/antibiotics3030317
Parish, T. (2014). Two-component regulatory systems of mycobacteria. Microbiol. Spectr. 2:MGM2-0010-2013. doi: 10.1128/microbiolspec.MGM2-0010-2013
Patkari, M., and Mehra, S. (2013). Transcriptomic study of ciprofloxacin resistance in Streptomyces coelicolor A3(2). Mol. Biosyst. 9, 3101–3116. doi: 10.1039/c3mb70341j
Penuelas-Urquides, K., Gonzalez-Escalante, L., Villarreal-Trevino, L., Silva-Ramirez, B., Gutierrez-Fuentes, D. J., Mojica-Espinosa, R., et al. (2013). Comparison of gene expression profiles between pansensitive and multidrug-resistant strains of Mycobacterium tuberculosis. Curr. Microbiol. 67, 362–371. doi: 10.1007/s00284-013-0376-8
Perez, J. C., and Groisman, E. A. (2009). Evolution of transcriptional regulatory circuits in bacteria. Cell 138, 233–244. doi: 10.1016/j.cell.2009.07.002
Peterson, E. J. R., Ma, S., Sherman, D. R., and Baliga, N. S. (2016). Network analysis identifies Rv0324 and Rv0880 as regulators of bedaquiline tolerance in Mycobacterium tuberculosis. Nat. Microbiol. 1:16078. doi: 10.1038/nmicrobiol.2016.78
Poole, K. (2007). Efflux pumps as antimicrobial resistance mechanisms. Ann. Med. 39, 162–176. doi: 10.1080/07853890701195262
Poole, K. (2012). Bacterial stress responses as determinants of antimicrobial resistance. J. Antimicrob. Chemother. 67, 2069–2089. doi: 10.1093/jac/dks196
Provvedi, R., Boldrin, F., Falciani, F., Palu, G., and Manganelli, R. (2009). Global transcriptional response to vancomycin in Mycobacterium tuberculosis. Microbiology 155, 1093–1102. doi: 10.1099/mic.0.024802-0
Pule, C. M., Sampson, S. L., Warren, R. M., Black, P. A., van Helden, P. D., Victor, T. C., et al. (2016). Efflux pump inhibitors: targeting mycobacterial efflux systems to enhance TB therapy. J. Antimicrob. Chemother. 71, 17–26. doi: 10.1093/jac/dkv316
Rawat, R., Whitty, A., and Tonge, P. J. (2003). The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc. Natl. Acad. Sci. U.S.A. doi: 10.1073/pnas.2235848100
Rohde, K., Yates, R. M., Purdy, G. E., and Russell, D. G. (2007). Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 219, 37–54. doi: 10.1111/j.1600-065X.2007.00547.x
Schnappinger, D., Ehrt, S., Voskuil, M. I., Liu, Y., Mangan, J. A., Monahan, I. M., et al. (2003). Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198, 693–704. doi: 10.1084/jem.20030846
Sherman, D. R., Voskuil, M., Schnappinger, D., Liao, R., Harrell, M. I., and Schoolnik, G. K. (2001). Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha -crystallin. Proc. Natl. Acad. Sci. U.S.A. 98, 7534–7539. doi: 10.1073/pnas.121172498
Shetty, A., Xu, Z., Lakshmanan, U., Hill, J., Choong, M. L., Chng, S.-S., et al. (2018). Novel acetamide indirectly targets mycobacterial transporter MmpL3 by proton motive force disruption. Front. Microbiol. 9:2960. doi: 10.3389/fmicb.2018.02960
Sivaramakrishnan, S., and de Montellano, P. R. (2013). The DosS-DosT/DosR mycobacterial sensor system. Biosensing 3, 259–282. doi: 10.3390/bios3030259
Soetaert, K., Rens, C., Wang, X. M., De Bruyn, J., Laneelle, M. A., Laval, F., et al. (2015). Increased vancomycin susceptibility in mycobacteria: a new approach to identify synergistic activity against multidrug-resistant mycobacteria. Antimicrob. Agents Chemother. 59, 5057–5060. doi: 10.1128/AAC.04856-14
Story-Roller, E., and Lamichhane, G. (2018). Have we realized the full potential of beta-lactams for treating drug-resistant TB? IUBMB Life 70, 881–888. doi: 10.1002/iub.1875
Takiff, H. E., and Feo, O. (2015). Clinical value of whole-genome sequencing of Mycobacterium tuberculosis. Lancet Infect Dis. 15, 1077–1090. doi: 10.1016/S1473-3099(15)00071-7
Tandon, R., and Nath, M. (2017). Tackling drug-resistant tuberculosis: current trends and approaches. Mini Rev. Med. Chem. 17, 549–570. doi: 10.2174/1389557516666160606204639
Thanert, R., Goldmann, O., Beineke, A., and Medina, E. (2017). Host-inherent variability influences the transcriptional response of Staphylococcus aureus during in vivo infection. Nat. Commun. 8:14268. doi: 10.1038/ncomms14268
Thye, T., Vannberg, F. O., Wong, S. H., Owusu-Dabo, E., Osei, I., Gyapong, J., et al. (2010). Genome-wide association analyses identifies a susceptibility locus for tuberculosis on chromosome 18q11.2. Nat. Genet. 42, 739–741. doi: 10.1038/ng.639
Tientcheu, L. D., Maertzdorf, J., Weiner, J., Adetifa, I. M., Mollenkopf, H. J., Sutherland, J. S., et al. (2015). Differential transcriptomic and metabolic profiles of M. africanum- and M. tuberculosis-infected patients after, but not before, drug treatment. Genes Immun. 16, 347–355. doi: 10.1038/gene.2015.21
Torres-Barcelo, C., Kojadinovic, M., Moxon, R., and MacLean, R. C. (2015). The SOS response increases bacterial fitness, but not evolvability, under a sublethal dose of antibiotic. Proc. Biol. Sci. 282:20150885. doi: 10.1098/rspb.2015.0885
Tsai, S. H., Lai, H. C., and Hu, S. T. (2015). Subinhibitory doses of aminoglycoside antibiotics induce changes in the phenotype of Mycobacterium abscessus. Antimicrob. Agents Chemother. 59, 6161–6169. doi: 10.1128/AAC.01132-15
Tudo, G., Laing, K., Mitchison, D. A., Butcher, P. D., and Waddell, S. J. (2010). Examining the basis of isoniazid tolerance in nonreplicating Mycobacterium tuberculosis using transcriptional profiling. Futur. Med. Chem. 2, 1371–1383. doi: 10.4155/fmc.10.219
Tufariello, J. M., Chapman, J. R., Kerantzas, C. A., Wong, K. W., Vilcheze, C., Jones, C. M., et al. (2016). Separable roles for Mycobacterium tuberculosis ESX-3 effectors in iron acquisition and virulence. Proc. Natl. Acad. Sci. U.S.A. 113, E348–E357. doi: 10.1073/pnas.1523321113
Udwadia, Z. F., Amale, R. A., Ajbani, K. K., and Rodrigues, C. (2012). Totally drug-resistant tuberculosis in India. Clin. Infect Dis. 54, 579–581. doi: 10.1093/cid/cir889
Voskuil, M. I., Visconti, K. C., and Schoolnik, G. K. (2004). Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberclosis 84, 218–227. doi: 10.1016/j.tube.2004.02.003
Waddell, S. J., and Butcher, P. D. (2007). Microarray analysis of whole genome expression of intracellular Mycobacterium tuberculosis. Curr. Mol. Med. 7, 287–296. doi: 10.2174/156652407780598548
Waddell, S. J., Stabler, R. A., Laing, K., Kremer, L., Reynolds, R. C., and Besra, G. S. (2004). The use of microarray analysis to determine the gene expression profiles of Mycobacterium tuberculosis in response to anti-bacterial compounds. Tuberclosis 84, 263–274. doi: 10.1016/j.tube.2003.12.005
Walter, N. D., Dolganov, G. M., Garcia, B. J., Worodria, W., Andama, A., Musisi, E., et al. (2015). Transcriptional adaptation of drug-tolerant Mycobacterium tuberculosis during treatment of human tuberculosis. J. Infect. Dis. 212, 990–998. doi: 10.1093/infdis/jiv149
Wenzel, M., and Bandow, J. E. (2011). Proteomic signatures in antibiotic research. Proteomics 11, 3256–3268. doi: 10.1002/pmic.201100046
WHO (2017). Global Tuberculosis Report 2017. Available at: http://www.who.int/tb/publications/global_report/en/
Wilson, M., DeRisi, J., Kristensen, H. H., Imboden, P., Rane, S., Brown, P. O., et al. (1999). Exploring drug-induced alterations in gene expression in Mycobacterium tuberculosis by microarray hybridization. Proc. Natl. Acad. Sci. U.S.A. 96, 12833–12838. doi: 10.1073/pnas.96.22.12833
Wivagg, C. N., Bhattacharyya, R. P., and Hung, D. T. (2014). Mechanisms of beta-lactam killing and resistance in the context of Mycobacterium tuberculosis. J. Antibiot. 67, 645–654. doi: 10.1038/ja.2014.94
Wright, G. D. (2007). The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 5, 175–186. doi: 10.1038/nrmicro1614
Wright, G. D. (2016). Antibiotic adjuvants: rescuing antibiotics from resistance. Trends Microbiol. 24:928. doi: 10.1016/j.tim.2016.06.009
Wu, M. L., Gengenbacher, M., Chung, J. C. S., Chen, S. L., Mollenkopf, H. J., Kaufmann, S. H. E., et al. (2016). Developmental transcriptome of resting cell formation in Mycobacterium smegmatis. BMC Genomics 17:837. doi: 10.1186/s12864-016-3190-4
Xu, Z., Zhou, A., Wu, J., Zhou, A., Li, J., Zhang, S., et al. (2018). Transcriptional approach for decoding the mechanism of rpoc compensatory mutations for the fitness cost in rifampicin-resistant Mycobacterium tuberculosis. Front. Microbiol. 9:2895. doi: 10.3389/fmicb.2018.02895
Yu, G., Cui, Z., Sun, X., Peng, J., Jiang, J., Wu, W., et al. (2015). Gene expression analysis of two extensively drug-resistant tuberculosis isolates show that two-component response systems enhance drug resistance. Tuberclosis 95, 303–314. doi: 10.1016/j.tube.2015.03.008
Zampieri, M., Enke, T., Chubukov, V., Ricci, V., Piddock, L., and Sauer, U. (2017). Metabolic constraints on the evolution of antibiotic resistance. Mol. Syst. Biol. 13:917. doi: 10.15252/msb.20167028
Zampieri, M., Szappanos, B., Buchieri, M. V, Trauner, A., Piazza, I., Picotti, P., et al. (2018). High-throughput metabolomic analysis predicts mode of action of uncharacterized antimicrobial compounds. Sci. Transl. Med. 10:eaal3973. doi: 10.1126/scitranslmed.aal3973
Keywords: Mycobacterium, antibiotics, microarrays, RNA-seq, transcriptome, tuberculosis
Citation: Briffotaux J, Liu S and Gicquel B (2019) Genome-Wide Transcriptional Responses of Mycobacterium to Antibiotics. Front. Microbiol. 10:249. doi: 10.3389/fmicb.2019.00249
Received: 12 September 2018; Accepted: 30 January 2019;
Published: 20 February 2019.
Edited by:
Henrietta Venter, University of South Australia, AustraliaReviewed by:
Paras Jain, Intellectual Ventures Management, LLC, United StatesSotirios Vasileiadis, University of Thessaly, Greece
Copyright © 2019 Briffotaux, Liu and Gicquel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julien Briffotaux, anVsaWVuLmJyaWZmb3RhdXhAZ21haWwuY29t