 Gilda Varliero
Gilda Varliero Christina Bienhold
Christina Bienhold Florian Schmid
Florian Schmid Antje Boetius
Antje Boetius Massimiliano Molari
Massimiliano Molari- 1Max Planck Institute for Marine Microbiology, Bremen, Germany
- 2School of Biological Sciences, University of Bristol, Bristol, United Kingdom
- 3HGF-MPG Joint Research Group on Deep Sea Ecology and Technology, Alfred Wegener Institute for Polar and Marine Research, Bremerhaven, Germany
- 4Helmholtz Centre for Ocean Research Kiel, GEOMAR, Kiel, Germany
- 5MARUM Center for Marine Environmental Sciences, University of Bremen, Bremen, Germany
Ultraslow spreading ridges account for one-third of the global mid-ocean ridges. Their impact on the diversity and connectivity of benthic deep-sea microbial assemblages is poorly understood, especially for hydrothermally inactive, magma-starved ridges. We investigated bacterial and archaeal diversity in sediments collected from an amagmatic segment (10∘–17∘E) of the Southwest Indian Ridge (SWIR) and in the adjacent northern and southern abyssal zones of similar water depths within one biogeochemical province of the Indian Ocean. Microbial diversity was determined by 16S ribosomal RNA (rRNA) gene sequencing. Our results show significant differences in microbial communities between stations outside and inside the SWIR, which were mostly explained by environmental selection. Community similarity correlated significantly with differences in chlorophyll a content and with the presence of upward porewater fluxes carrying reduced compounds (e.g., ammonia and sulfide), suggesting that trophic resource availability is a main driver for changes in microbial community composition. At the stations in the SWIR axial valley (3,655–4,448 m water depth), microbial communities were enriched in bacterial and archaeal taxa common in organic matter-rich subsurface sediments (e.g., SEEP-SRB1, Dehalococcoida, Atribacteria, and Woesearchaeota) and chemosynthetic environments (mainly Helicobacteraceae). The abyssal stations outside the SWIR communities (3,760–4,869 m water depth) were dominated by OM1 clade, JTB255, Planctomycetaceae, and Rhodospirillaceae. We conclude that ultraslow spreading ridges create a unique environmental setting in sedimented segments without distinct hydrothermal activity, and play an important role in shaping microbial communities and promoting diversity, but also in connectivity among deep-sea habitats.
Introduction
The deep seafloor beyond the shelf break comprises about 67% of the Earth’s lithosphere (Jørgensen and Boetius, 2007), making it the largest ecological realm worldwide. The increasing anthropogenic impact such as climate change, littering and industrial exploitation of deep-sea resources raises global concerns about the future of deep-sea biodiversity and the lack of marine conservation approaches for seabed habitats (Danovaro et al., 2017). In deep sea sediments, the largest fraction of taxonomic richness and biomass is contributed by members of the Bacteria and Archaea, which represent around 90% of the total benthic biomass and have a key role in organic matter remineralization and nutrient cycles (Jørgensen and Boetius, 2007; Wei et al., 2010). Thus, the investigation of spatial patterns of microbial diversity is crucial to better understand the mechanisms controlling the diversity and connectivity between deep-sea habitats. Deciphering factors that influence spatial turnover is relevant to the assessment of ecological function dynamics in the deep-sea. Advances in high-throughput 16S rRNA gene sequencing techniques have enabled the comparative analysis of microbial biogeographic patterns across marine environments (e.g., Sogin et al., 2006; Zinger et al., 2014), including the deep seafloor (Schauer et al., 2010; Durbin and Teske, 2011; Sylvan et al., 2012; Jacob et al., 2013; Anderson et al., 2015; Ristova et al., 2015; Ruff et al., 2015; Bienhold et al., 2016). Distance-decay relationships (i.e., a decrease in taxonomic similarity with increasing geographic distance) and a relatively high degree of endemism, investigated at various taxonomic resolution, have been reported both at local and regional (tens to hundreds of kilometers; Schauer et al., 2010; Jacob et al., 2013; Ishibashi et al., 2015; Shulse et al., 2016; Walsh et al., 2016) and at global scales (Ruff et al., 2015; Mino et al., 2017). Selection, drift, dispersal and mutation are the four evolutionary and ecological interplay processes that shape the microbial biogeography (Hanson et al., 2012). The presence of substantial endemism and distance-decay relationships has been interpreted as a rather rapid diversification (selection and drift) and limited dispersal across ocean basins (Hanson et al., 2012; Bienhold et al., 2016). Whilst environmental selection has been shown to play an important role in shaping deep-sea benthic microbial communities (e.g., Bienhold et al., 2012, 2016), the physical mechanisms responsible for dispersal limitation (e.g., currents and seafloor geomorphology) are poorly understood (Zinger et al., 2014). Patterns of deep-sea bacterial biogeography observed at the global scale suggest that seafloor geomorphology (i.e., mid-ocean ridges and oceanic trenches), deep-water masses and landmasses may represent barriers to dispersal (Nunoura et al., 2015; Bienhold et al., 2016; Salazar et al., 2016; Wenzhöfer et al., 2016).
Mid-ocean ridges (MOR) are undersea mountain ranges forming the largest continuous topographic feature on Earth, a global network almost 85,000 km long (Kennett, 1982). At active MOR oceanic lithosphere formation coincides with substantial fluxes of heat. Seafloor hydrothermal circulation is generated by downward percolation of seawater, through the fractured ocean crust, that is heated at depth. When the fluid becomes buoyant it rises rapidly back to the seafloor where it is expelled into the overlying water column. This fluid generates strong geochemical and physical gradients and provides a chemical energy source for microbial growth, which supports chemosynthesis-based food chains and promotes high physiological diversity (German et al., 2011; Sievert and Vetriani, 2012; Gollner et al., 2015; Goffredi et al., 2017). Despite the substantial passive dispersal of microbes in the ocean (De Rezende et al., 2013), there is increasing evidence that geochemistry and geographical isolation play a role in structuring vent microbial communities (e.g., Flores et al., 2012; Akerman et al., 2013; Campbell et al., 2013; Mino et al., 2017), as has been observed for vent macrofauna (e.g., Rogers et al., 2012). MOR vent fields have been estimated to occur every 25–90 km, but only a small fraction thereof have been mapped and investigated, indicating that microbial diversity and biogeography patterns are largely unknown for most of the ridge segments (Beaulieu et al., 2015). Even less it is known about the microbial diversity at inactive segments where surface expressions of hydrothermalism are absent. It has been proposed that ridges can be stepping stones and pathways for the dispersal of slope fauna into the open ocean and/or may act as barriers to the dispersal of abyssal seafloor fauna (Wilson and Kaufmann, 1987; Vinogradova, 1997; Dinter, 2001; Mironov, 2006; Gebruk et al., 2010), but no studies have yet investigated the role that mid-ocean ridges may play for the connectivity of deep-sea benthic microbial communities.
The Southwest Indian Ridge (SWIR) is a major plate boundary of the world oceans, separating the African and Antarctic plates and extending from the Bouvet triple junction (BTJ) in the South Atlantic Ocean to the east Rodrigues triple junction (RTJ) in the Indian Ocean (Sauter and Cannat, 2013). The SWIR segment in the southern Atlantic Ocean separates the Agulhas basin to the north and the Weddell/Enderby plains to the south, which belong to the same biogeochemical deep-sea floor province as defined by sedimentary organic carbon content, bottom hydrography (i.e., temperature and salinity) and organic matter flux (Seiter et al., 2004; Watling et al., 2013). Due to the presence of Antarctic cold dense bottom water masses flowing eastward, SWIR forms a barrier to the northward and southward flow of bottom water (Larqué et al., 1997; Haine et al., 1998; Orsi et al., 1999; Rutgers van der Loeff et al., 2016). Furthermore, the SWIR is an ultraslow-spreading ridge, which is characterized by low magma input and scant hydrothermal circulation, and by a deep ridge valley that is on average 4,000 m deep, with ridge flanks that rise up to 1,000 m depth (Dick et al., 2003). These features make the SWIR an interesting place to test whether ridges can limit the dispersal of deep-sea benthic microbial communities in the absence of hydrothermalism. Specifically, we assessed bacterial and archaeal community structure and diversity based on the 16S rRNA gene to investigate whether the western section of the SWIR (i) acts as a physical barrier between communities to the North and South, limiting microbial dispersal, and (ii) promotes isolation of microbial communities inside the ridge.
Materials and Methods
Sample Collection
The sediment samples were collected in the segment 10°–17°E of the SWIR during the expedition ANTXXIX/8 with the research vessel Polarstern (PS81) in 2013 (Figure 1A). The SWIR segment studied here is an amagmatic accretionary ridge segment with a spreading rate of less than 15 mm yr-1 and the major percentage of the axial seafloor is constituted by mantle rocks (Dick et al., 2003). In the earlier investigation of this SWIR segment the presence of a hydrothermal plume has been suggested based on turbidity maxima in the water column (Bach et al., 2002). During the PS81 expedition we investigated the turbidity plumes but found them not linked to hydrothermal emissions (Schlindwein, 2014). Clear signals of white or black smoker type venting were lacking, suggesting that this system is a quiescent ridge segment. However, the ridge flanks and trough were heavily sedimented, indicating a productive surface ocean and a relatively substantial input of plankton debris, foremost diatom ooze (Figure 1B). Sediment samples have been taken from the seabed at a depth range of 3,655 and 4,869 m. Surface sediment samples, 0–40 cm below the seafloor (bsf), were collected with a TV-guided multi-corer device (TV-MUC) and subsurface samples, from ca. 50 to 600 cm bsf, with a gravity core (GC). For surface sediments two replicate TV-MUC cores were collected from each site, one used for porewater and one for sediment sampling. For subsurface sediments one GC was collected at A1, A2, and A3, and sediment and porewater were collected from the same GC (Table 1). Cores were sliced on board (0–1, 1–5, and 5–10 cm for MUC cores, and every 40 cm for GC cores) and sediment samples for DNA analysis were stored at -80°C. Porewater was collected at intervals of 1 cm, starting from the water overlying the sediment to the bottom of the core in MUC cores, and every 40 cm in GC cores. Sediments were sampled at 2 reference stations (S0 and N0), located to the south and north of the SWIR, respectively, and at 5 stations inside the SWIR valley (A1, A2, A2m, A3, and A3m). The stations inside the ridge were selected based on environmental data and visual observations: Area 1 (A1) had highest heat flow values (up to 1,000 mW m-2; Schlindwein, 2014); at Area 2 a signature for a hydrothermal plume had previously been reported by Bach et al. (2002), but only based on turbidity maxima in the water column, with stronger turbidity plumes at station A2m than at station A2 (Figure 1B); Area 3 represents the axial valley of the ridge, with station A3 sampled in the central part and station A3m located close to the site where a vesicomyid clam, a typical inhabitant of reduced chemosynthetic habitats, was discovered (Supplementary Figure S6). Additionally, in order to better investigate the effect of the geographical distance and the SWIR on benthic microbial diversity and connectivity in the South Atlantic Polar Front, the microbial communities were also investigated in sediments from two stations (N1 and N2) sampled during the Polarstern cruise PS79 in 2012 (Ruff et al., 2014), and located northwest of the SWIR segment investigated here (Figure 1). Major changes along porewater profiles occurred in the first top 5 cm of sediments (Figure 2 and Supplementary Figure S1), hence microbial communities were described for this top sediment layers (0–5 cm bsf) and for two subsurface layers (110 and 410 cm), the latters as representative of subsurface microbial community.
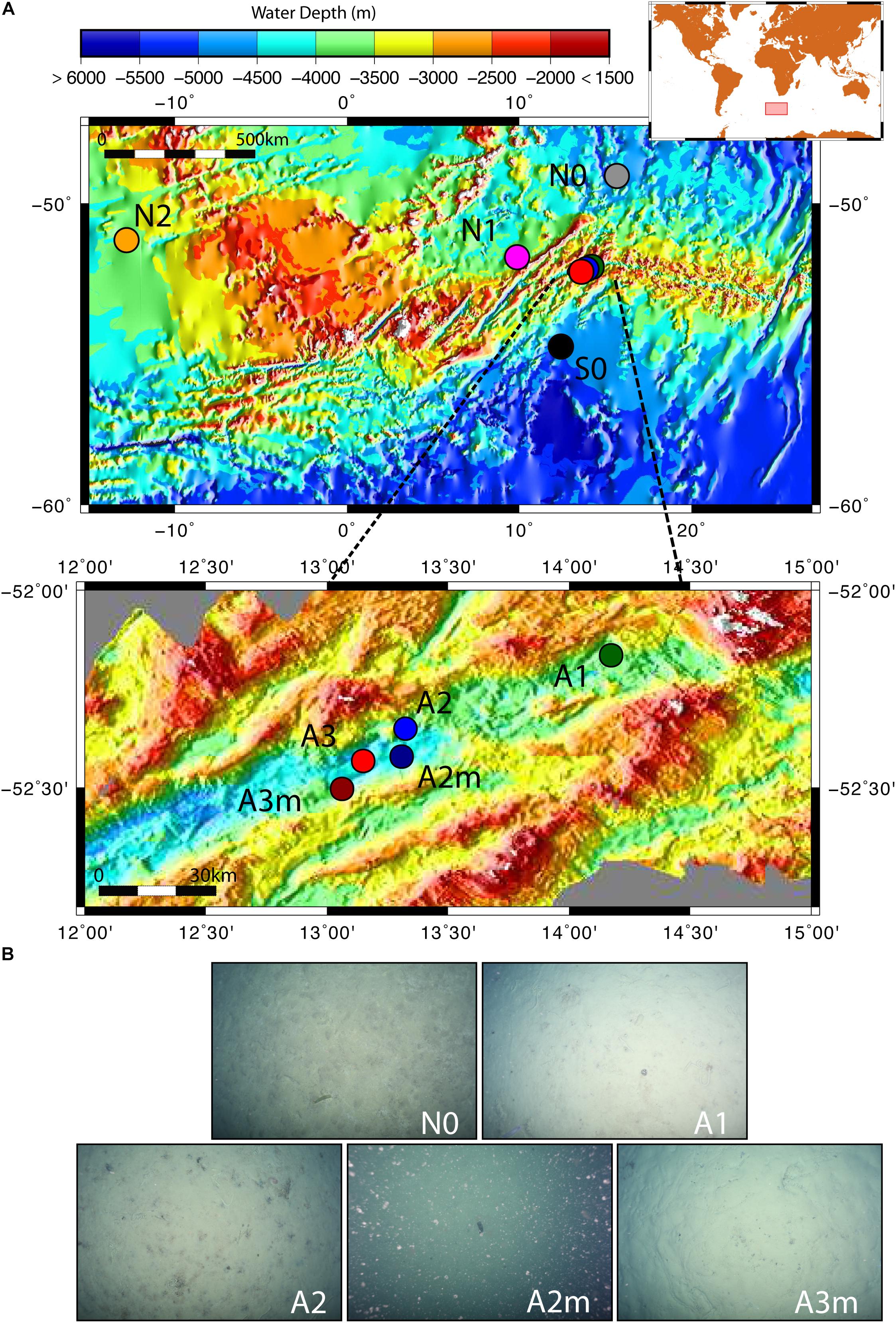
Figure 1. Map of the investigated sites and seafloor pictures. (A) The zoom-in shows the stations inside the Southwest Indian Ridge valley; (B) seafloor photos for station to the north of the SWIR (N0) and for stations inside the SWIR showing diatom ooze sediments; seafloor picture at A2m also shows resuspended sediment particles, which were likely responsible of turbidity signals measured at south ridge flank (Schlindwein, 2014). Green, blue and red dots represent stations inside SWIR, and orange, magenta, gray and black dots represent stations outside SWIR.
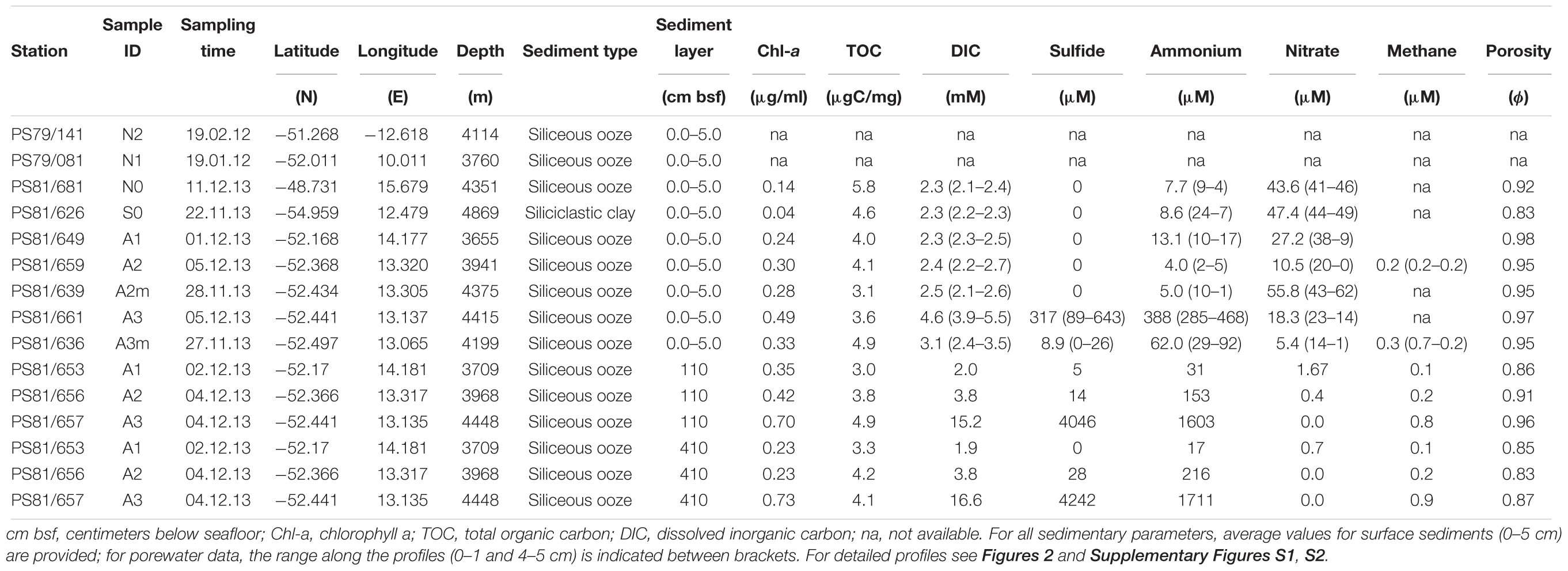
Table 1. Description of investigated sites.
Porewater and Sediment Biogeochemistry
For surface sediments, two replicate cores were collected from each site, one used for porewater and one for sediment sampling. For subsurface sediments, sediment and porewater were collected from the same gravity core. The subsampling of cores was performed immediately after recovery in a temperature controlled lab at 2°C. Profiles of dissolved components like inorganic carbon (DIC), nutrients [NH4+, PO43-, NO2-, NO3- + NO2-, Si(OH)4], sulfate, sulfide and manganese (Mn) in the sediments were assessed by extracting porewater with Rhizons (SMS type MOM 19.21.21F, mean pore size 0.15 μm; Rhizosphere Research Products). For DIC 2 mL porewater were filled headspace-free into glass vials leaving no headspace and stored at 4°C. DIC was assessed via flow injection analysis (Grasshoff et al., 2007). Nutrients were measured with a continuous Flow Nutrient Analyzer “QuAAtro39” (Seal Analytical) according to Grasshoff et al. (2007). Sulfide samples were fixed in plastic vials pre-filled with 0.5 mL 2% ZnAc before being stored at 4°C, and analysis was performed according to procedures described by Cline (1969). Porewater samples for Mn analysis were fixed in plastic vials pre-filled with 0.2 mL 1 M HCl before being stored at 4°C. Mn concentrations were assessed by atomic absorption spectrometry. Solid phase sediment samples were collected by slicing the core in 0–1, 1–5, and 5–10 cm layers, and were preserved for analyses of porosity, chloroplastic pigment equivalents (CPEs), total organic carbon (TOC) and total organic nitrogen (TON). Samples were prepared and analyzed as described in Böer et al. (2009). At the investigated water depths (3,655–4,869 m) no photosynthesis occurs, thus the sedimentary amount of chlorophyll pigments is used as a measure of the amount and freshness of phytodetritus sinking from the productive photic water layers.
For CH4 gas analysis 5 mL of sediment was collected with cut-off 5 mL syringes and added to 10 mL 2.5% NaOH in glass crimp vials, mixed, stored upside down at 4°C and then analyzed by gas chromatography (Focus GC, Thermo Fisher Scientific) as described in Thang et al. (2012). The entire biogeochemical dataset has been deposited in the PANGAEA database1.
We used Fick’s law to estimate the vertical diffusive flux J of geochemical constituents in the sediment cores,
Here ∅ is porosity, Ds = is the sediment diffusion coefficient, calculated from the sediment deviated tortuosity 𝜃 = 1.1 for a porosity of 0.9 (Matyka et al., 2008) and D0 is the tracer diffusion coefficient in seawater. We used values of D0 = 4.64 × 10-6 [cm2 s-1] for sulfate, D0 = 9.17 × 10-6 for sulfide, D0 = 9.03 × 10-6 [cm2 s-1] for ammonium, D0 = 3.02 × 10-6 [cm2 s-1] for manganese, D0 = 9.03 × 10-6 [cm2 s-1] for nitrate. Coefficients are taken from Schulz (2000). δC is the difference in concentration [mmol L-1] and δz is the difference in depth [m].
Microbial Abundance and Activity
For microbial cell count, the top 1 cm of sediment in MUC cores was fixed in 2% buffered formaldehyde/water and stored at 4°C until subsequent analysis. Microbial abundance was estimated by epifluorescence microscopy after staining with Acridine Orange following the procedure described by Hobbie et al. (1977) and modified by Böer et al. (2009). Catalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH) was applied to enumerate the active fraction of bacterial and archaeal assemblages (Amann and Fuchs, 2008). Samples were stored and processed according to the procedure of Ishii et al. (2004) and for archaeal cell-wall permeabilization according to Molari and Manini (2012). Hybridization conditions were applied as previously described for EUB338I-III, targeting members of the Bacteria (Amann et al., 1990; Daims et al., 1999), ARCH915 targeting most members of the Archaea (Stahl and Amann, 1991), and NON338 as negative control (Amann et al., 1995). For checking the reliability of the FISH signal the CARD-FISH filter was counter-stained with DAPI, and up to 700 EUB-FISH-stained cells and 100 ARCH-FISH-stained cells were counted per sample. The relative abundances of Bacteria and Archaea were based on total AODC counts, as the latter gives more reliable counts than DAPI in sediment composed by small particle size (<62.5 μm) (e.g., Schippers et al., 2005). The CARD-FISH efficiency (i.e., sum of bacterial and archaeal relative abundances) does not reach 100% of AODC counts, as not all cells are captured by FISH potentially due to incomplete coverage of probes, low ribosome content and lack of proper cell-wall permeabilization (Amann and Fuchs, 2008).
Total microbial activity was estimated by uptake of 14C-labeled inorganic carbon. Dark CO2 fixation (DCF) rates were estimated following the procedures described by Molari et al. (2013) including some modifications. DCF rates were measured incubating 1 mL of sediment slurry (∼1:1 mixture of sediment and filtrated 0.22-μm bottom seawater) in triplicate with 12 μL 14C-labeled sodium bicarbonate (0.25 mCi mL-1, final activity 3 μCi mL-1) in the dark at in situ temperature (2–4°C). The incubations were terminated by the addition of 1 mL formaldehyde in seawater (final concentration 2%) after 12 h. Two controls per sample were killed with 1 mL formaldehyde in seawater (final concentration 2%) before addition of the tracer. Samples were stored at 4°C until further processing. At MPI laboratory, the samples were centrifuged at 12,000 rpm for 5 min, the supernatant was discarded, and remaining sediment pellets were washed three times with 1× PBS. The sediment pellets were resuspended with 1 mL 3 M HCl, transferred into a new 50 mL vial and mixed constantly by bubbling with pressurized air for 4 h. The samples where mixed with 8 mL of the Scintillation cocktail Ultima GoldTM and centrifuged at 3,500 rpm for 30 min. The supernatants were transferred into a 20 mL scintillation vials and the pellets were resuspended in 8 mL Ultima GoldTM and centrifuged a second time. The supernatants were combined and measured with a liquid scintillation counter up to 10 min. The DPM were converted in moles of inorganic carbon incorporated per unit of sediment volume and time using the formula described by Molari et al. (2013).
DNA Extraction and Sequencing
DNA was extracted from 1 g of homogenized sediment from 0–1, 1–5, 110, and 410 cm sediment layers using the FastDNATM SPIN Kit for Soil (Q-BIOgene, Heidelberg, Germany) following the manual protocol. Then, an isopropanol precipitation was performed on the extracted DNA, and DNA samples were stored at -20°C. DNA extracts from 0–1 to 1–5 cm were pooled at equal volumes and DNA amount prior to sequencing.
Amplicon sequencing was done at the CeBiTec laboratory (Centrum für Biotechnologie, Universität Bielefeld) on an Illumina MiSeq machine. For the 16S rRNA gene amplicon library preparation we used the bacterial primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 785R (5′-GACTACHVGGGTATCTAATCC-3′) and the archaeal primers Arch349F (5′-GYGCASCAGKCGMGAAW-3′) and Arch915R (5′-GTGCTCCCCCGCCAATTCCT-3′) (Wang and Qian, 2009; Klindworth et al., 2013) which amplify the 16S rRNA gene hypervariable region V3–V4 in Bacteria (400–425 bp fragment length) and the V3–V5 region in Archaea (510 bp fragment length). The amplicon library was sequenced with the MiSeq v3 chemistry, in a 2 × 300 bp paired run with >50,000 reads per sample, following the standard instructions of the 16S Metagenomic Sequencing Library Preparation protocol (Illumina, Inc., San Diego, CA, United States).
The quality cleaning of the sequences was performed with several software tools. Primer clipping was performed with cutadapt (Martin, 2011). TRIMMOMATIC (Bolger et al., 2014) was used to remove the sequences of low quality (for Bacteria SLIDINGWINDOW:4:10 MINLEN:300; for Archaea SLIDINGWINDOW:6:13 MINLEN:450); this step was performed before the merging of reverse and forward reads for the bacterial dataset and after the merging for the archaeal dataset in order to enhance the number of retained reads for long archaeal 16S fragments. The merging of forward and reverse reads was performed with PEAR (Zhang et al., 2014). Clustering of sequences into OTUs (operational taxonomic units) was done using the SWARM algorithm, based on one nucleotide difference between amplicons (parameter settings: -b 3 -d 1 -f; Mahé et al., 2014). The taxonomic classification was based on the SILVA 128 database (Quast et al., 2013). During this step, sequences with less than 90% of similarity with SILVA sequences were removed.
The total number of sequences obtained in this study is reported in Supplementary Table S1. Sequences were deposited at the European Nucleotide Archive (ENA) under accession number PRJEB23821; the sequences were archived using the service of the German Federation for Biological Data (GFBio; Diepenbroek et al., 2014). Absolute singletons (SSOabs), i.e., OTUs consisting of sequences occurring only once in the full dataset (Gobet et al., 2013), accounted for 92–98% of all OTUs (20–56% of all sequences) for bacterial and archaeal datasets, respectively, and they were not included in diversity and community analyses.
Data Analysis
Alpha-diversity was assessed as species richness, exponential of Shannon index and inverse of Simpson index, corresponding to Hill’s numbers of order q = 0 (H0), q = 1 (H1) and q = 2 (H2), respectively (Hill, 1973; Chao et al., 2014). Estimated richness (Chao1), shared and unique OTUs (i.e., OTUs that are only present at one station) were calculated by rarifying the sequences to the smallest dataset (25,167 sequences for the domain Bacteria and 1,190 sequences for Archaea) 100 times and taking the average values, to account for differences in sequencing depth between samples. Non-parametric Mantel tests based on the Spearman correlation coefficient with significance assessed based on 1000 Monte Carlo permutations were used to determine correlations between genetic, spatial, and environmental distance matrices (Legendre and Legendre, 1998). To determine the strength of the relationship between similarity in community composition, geographic distance, and environmental settings linear models were fitted. Differences in microbial community composition were visualized with non-metric multidimensional scaling plots, and analysis of similarity (ANOSIM; Clarke, 1993) was used to assess significant differences between groups of samples from outside and within the ridge, and from surface and subsurface sediments. Redundancy analysis (RDA) in combination with variation partitioning (VP) was conducted to test the effect of environment variables on variations in microbial community composition. The analysis of microbial community composition was carried out on dominant bacterial and archaeal taxa, here defined as those composed by OTUs that represent more than 0.1% of the total number of sequences in each sample. Principal component analysis (PCA) was performed at bacterial class and family levels to identify which taxa were responsible for differences between stations. Prior to the analyses: (i) the environmental variables were standardized (i.e., z-scored) and filtered by collinearity based on the variance inflation factor (VIF) of less than five, which retained chlorophyll a, TOC and DIC; specifically DIC, ammonia, sulfide, and porosity were highly correlated (Pearson r > 0.95, p < 0.001), thus we selected DIC as a proxy for the presence of upward porewater fluxes carrying reduced compounds (e.g., ammonia and sulfide); (ii) the OTU dataset was standardized using the Hellinger transformation (Legendre and Gallagher, 2001). All analyses were carried out in the R statistical environment (R Development Core Team, 2013) with the packages vegan (Oksanen et al., 2016), ggplot2 (Wickham, 2009), devtools (Wickham and Chang, 2015), factoextra (Kassambara, 2015), ade4 (Dray and Dufour, 2007), plyr (Wickham, 2011), reshape (Wickham, 2007), and usdm (Naimi et al., 2014), as well as with custom R scripts. The rarefaction curves and the diversity analyses were performed with the iNEXT package (Hsieh et al., 2016) using default parameters (i.e., endpoint = double of the sample size; knots = 40).
The phylogenetic trees were constructed with the RAxML software (Stamatakis, 2014) and the R environment (R Development Core Team, 2013) using the ape (Paradis et al., 2004), phyloseq (McMurdie and Holmes, 2015), and ggplot2 (Wickham, 2009) packages. The sequences for the tree backbone were retrieved from the SILVA SSU Ref database (v128) and it was built with the maximum likelihood method (1,000 bootstraps). Sequences obtained in this study with Illumina tag sequencing were added to the tree backbone using the parsimony method.
Results
Sediment Biogeochemistry
At all sampled stations, the seafloor consisted of diatom ooze, with exception of the southernmost station (S0), where the sediment was siliciclastic clay (Table 1). The porewater profiles of surface sediments showed different patterns between the stations inside and outside the SWIR (Figure 2). Specifically, anomalies in DIC, ammonia, phosphate, and sulfide concentrations were observed at the SWIR western stations (Table 1), with steepest gradients at A3–A3m (Figure 2 and Supplementary Figure S1A). A depletion of nitrate below 0.1 m depth was observed in all cores from inside the SWIR valley, but not at the reference sites outside the valley (N0 and S0). The shallow nitrate depletion inside the axial valley suggests a lower oxygen penetration compared to the reference sites outside the axial valley. The concentration and C:N ratio of organic matter in surface sediments did not show remarkable differences between stations (Supplementary Figure S2). Values increased somewhat in subsurface SWIR sediments in the western part of the segment (Supplementary Figure S1B). In the top 5 cm of sediments the amount of chlorophyll-a (Chl-a) and its contribution to total CPE increased in the stations located inside the SWIR, with highest values at A3 (Table 1). In subsurface SWIR sediments the CPEs increased westward, whereas the Chl-a contribution to CPEs decreased (Supplementary Figure S1B).
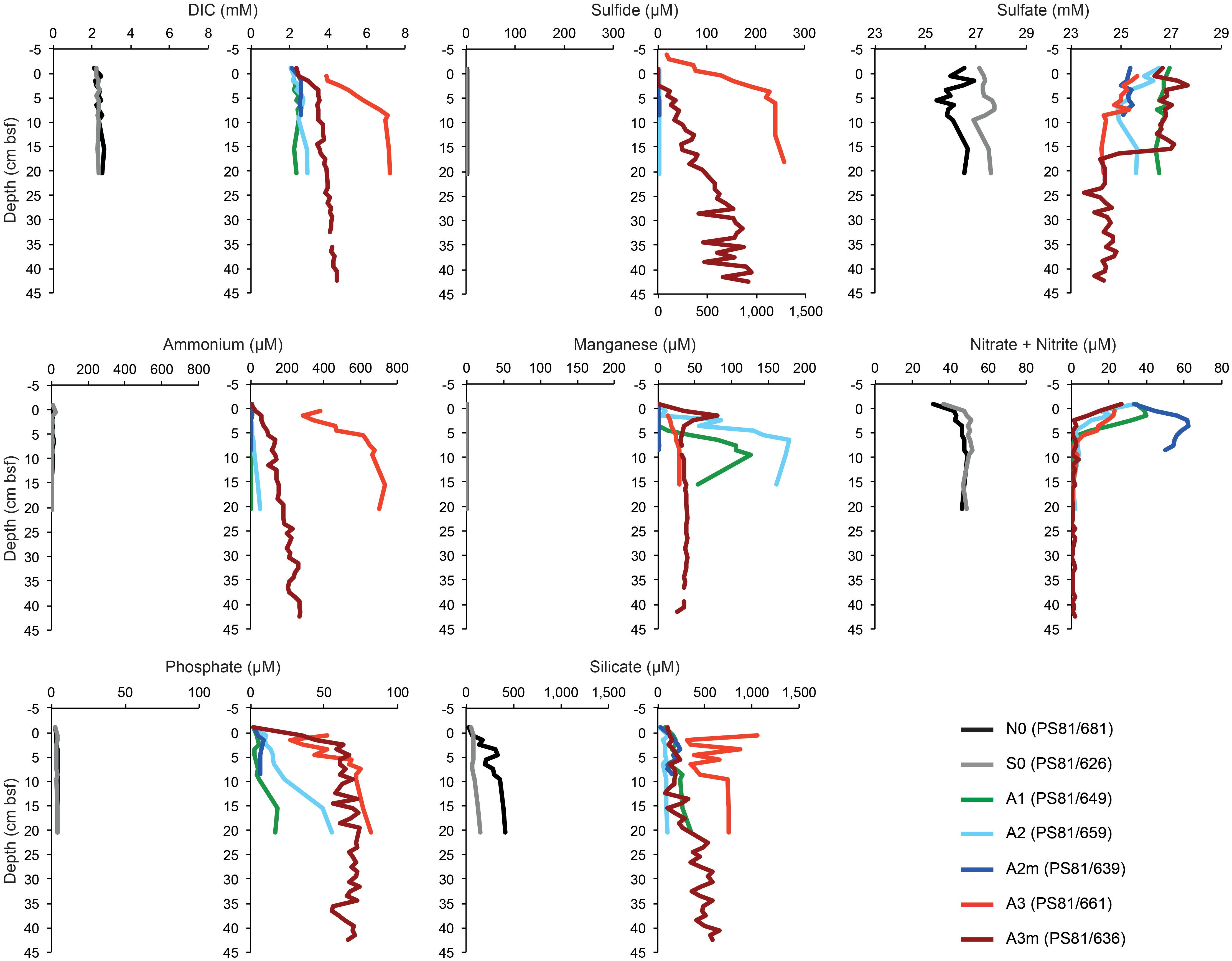
Figure 2. The porewater profiles of surface sediments (0–5 cm) outside and inside the SWIR. The gray (southern reference) and black lines (northern reference) represent outside SWIR stations, the colored lines inside samples. The sulfide plot values for station A3 (PS81/661) refer to the bottom x-axis, for all other stations the values refer to the top x-axis. DIC, dissolved inorganic carbon; cm bsf, centimeters below seafloor.
Diffusive porewater flux rates are listed in Table 2. Fluxes of sulfide, DIC, ammonium and nitrate were observed at A3 and A3m, with the former showing the strongest fluxes. A weak upward flux of ammonium was also observed in the subsurface sediments at A2. Nitrate fluxes into the sediment were observed at the majority of sites inside the axial valley (A3m, A3, and A2), but not at the reference sites (N0, S0).
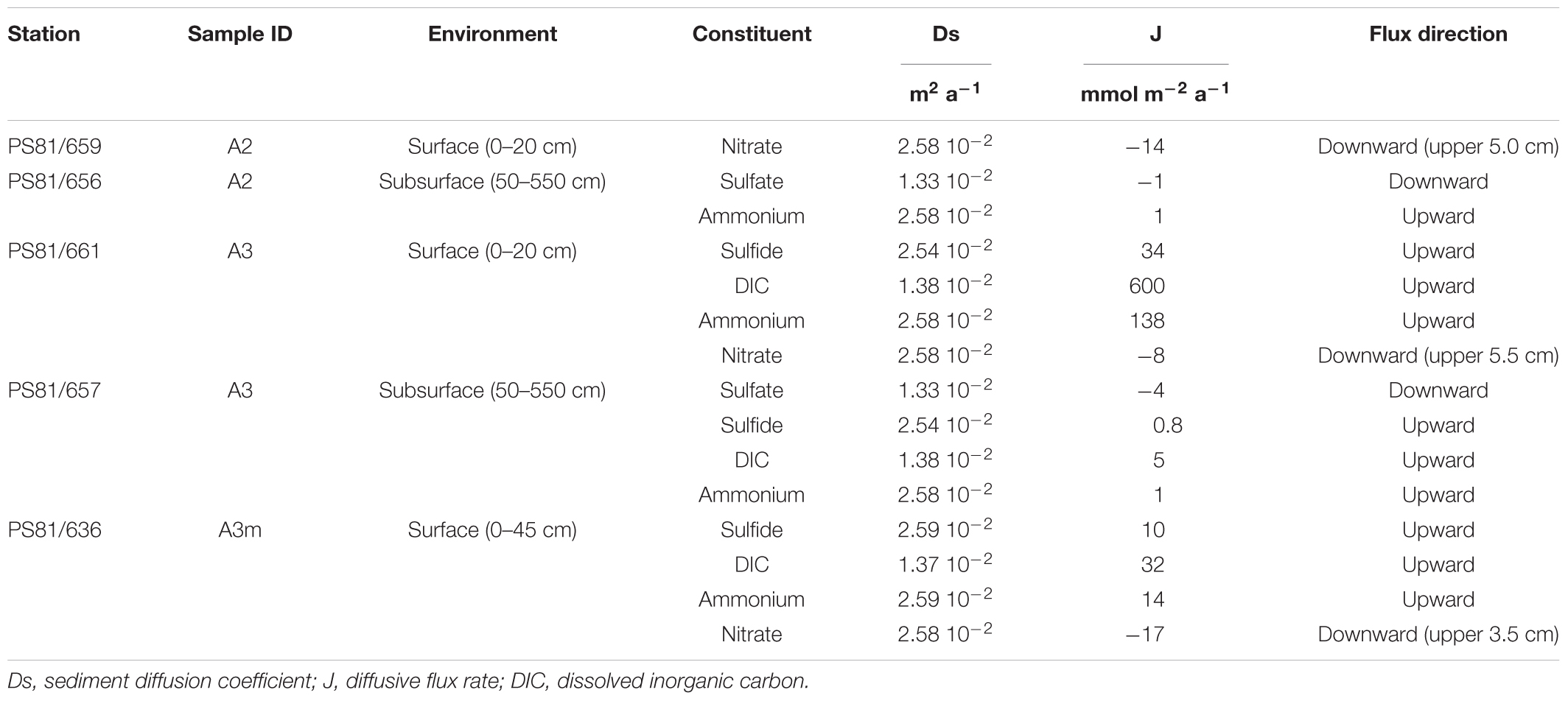
Table 2. Diffusive flux rates (J) of various geochemical constituents in the SWIR ridge valley areas A2 and A3.
Microbial Abundances and Activity
Total cell numbers in the sediments, as determined by AODC (acridine orange direct counts), ranged between 0.25 cells × 109 cells ml-1 wet sediment and 1.4 × 109 cells ml-1 wet sediment (Table 3). Highest numbers were detected at the northern reference site (N0) and slightly lower in the axial valley. The lowest cell numbers were found at the station A3m. The CARD-FISH efficiency ranged from 43 to 67% (the sum of bacterial and archaeal cells counts relative to the total cell counts determined by AODC) and the number of active cells showed the same pattern as the AODC results. Bacteria with a median relative abundance of 48 ± 14% dominated over Archaea (3 ± 1%) at all stations (Table 3).
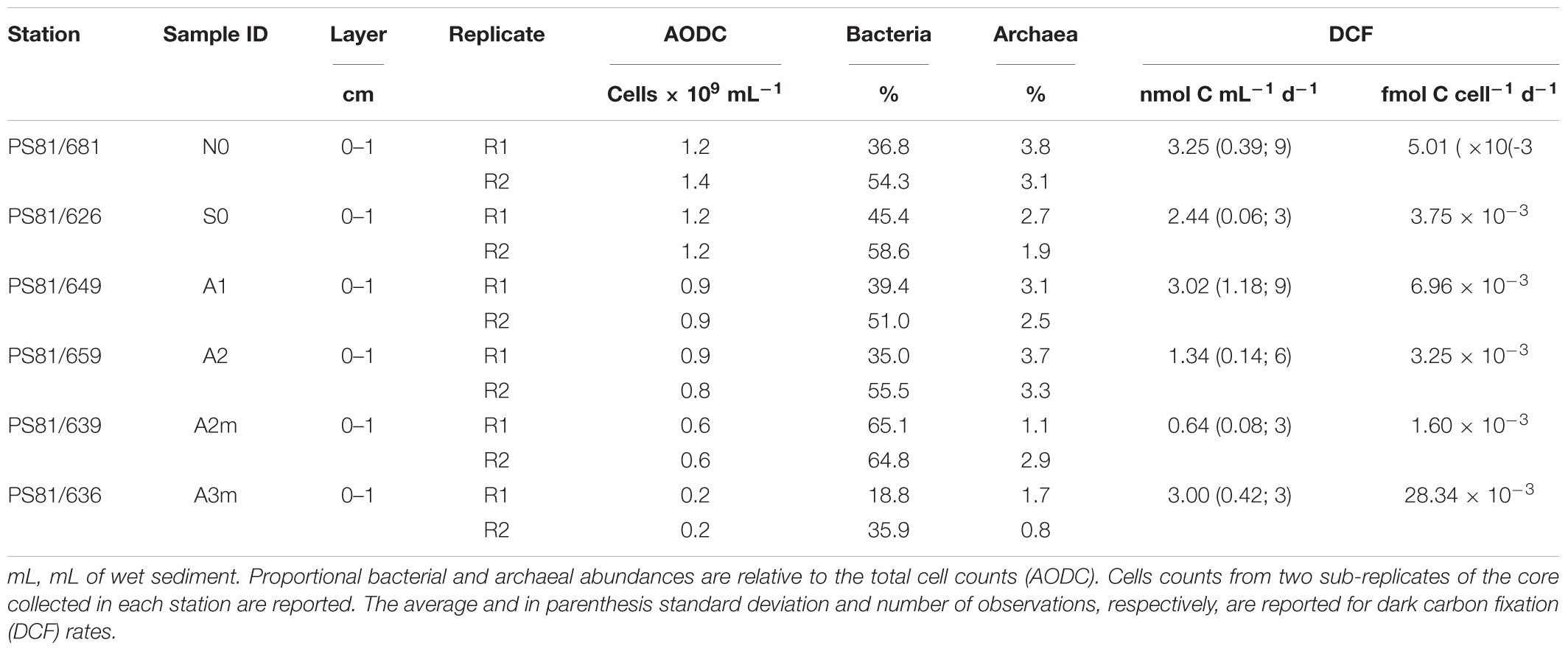
Table 3. Benthic microbial abundances and activity.
Rates of dark carbon fixation (DCF) were highest at N0 and lowest at A2m (Table 3). However, we did not identify any clear pattern between stations outside and inside the ridge. Similar trends were observed for microbial activity per cell, with exception of A3m that showed a value up to 15 times higher than the other stations (0.03 fmol C cell-1 d-1).
Microbial Alpha-Diversity
Rarefaction analysis showed that we captured more than 90% of bacterial and archaeal diversity of non-singletons at the stations investigated, as the rarefaction curves reached a plateau for Hill diversity indices H1 and H2, both in surface and subsurface samples (Supplementary Figure S3). Bacterial diversity indices did not show any apparent patterns between stations both in surface and subsurface layers (Supplementary Table S1A); H1 and H2 were higher in surface than in subsurface sediments (Supplementary Table S1A). Archaeal communities showed a substantially lower diversity than bacterial communities, which could be due to lower intragenomic heterogeneity of the amplified 16S rRNA gene region compared to region V3–V4 of Bacteria (Sun et al., 2013; Oton et al., 2016). Archaeal diversity indices in surface sediments were more than two times higher at A3 with the reduced sediments than at all the other stations, and H1 and H2 decreased westward in subsurface sediments (Supplementary Table S1B).
Differences in Microbial Community Composition
The most important bacterial classes in the surface layer (0–5 cm) of the areas outside the rift valley (S0, N0, N1, and N2), which alone constituted 23–36% of the total sequence abundance in each sample, were Gammaproteobacteria, Acidimicrobiia and Alphaproteobacteria (Figure 3). Stations A1, A2, and A2m on the SWIR were dominated by these taxa as well (14–18%), but also by JTB23, Betaproteobacteria, Deltaproteobacteria, Flavobacteria, and Verrucomicrobiae, which all together represented between 31 and 36% of the total microbial community. The SWIR stations A3 and A3m exhibited a higher diversity of the most abundant taxa, i.e., about 37% of the bacterial community were represented by Atribacteria, Bacteroidetes BD2-2, “Ca. Marinimicrobia” (SAR406), Omnitrophica, Thermoflexia, Aminicenantes, Cytophagia, Sphingobacteriia, Acetothermia, Anaerolineae, BD2-11 terrestrial group, Ignavibacteria, JG30-KF-CM66, LCP-89 and Subgroup 21, in addition to the taxa already mentioned above. Dissimilarities in bacterial community composition between stations in area A3 and other stations were mainly explained by Bacteroidetes BD2.2, Thermoflexia, Dehalococcoidia, “Ca. Marinimicrobia” (SAR406), Anaerolineae, Atribacteria, Parcubacteria, Pla3 Lineage, Spirochaetes, and Deltaproteobacteria (Supplementary Figure S4). Specifically, surface sediments of Area 3 were enriched in Desulfobacteraceae (mostly SEEP-SRB1), Desulfarculaceae (mostly Desulfatiglans), Thermoflexaceae (Thermoflexus), Spirochaetaceae, and mostly at A3m, also by Helicobacteraceae (Sulfurimonas and Sulfurovum) (Figure 4 and Supplementary Table S2). In all subsurface samples the dominant bacterial taxa were Dehalococcoida, Atribacteria, and Aminicenantes (Figure 3). Archaea were dominated by Marine Group I in all surface samples (63–98% of the archaeal sequences), except for station A3, where the dominant taxon was Woesearchaeota (DHVEG-6) with a relative sequence proportion of 39% (Figure 3). Furthermore, in Area 3 and in subsurface sediments the archaeal communities were also composed of Diapherotrites, Altiarchaeales, Lokiarchaeata, Thermoplasmata, and Group C3.
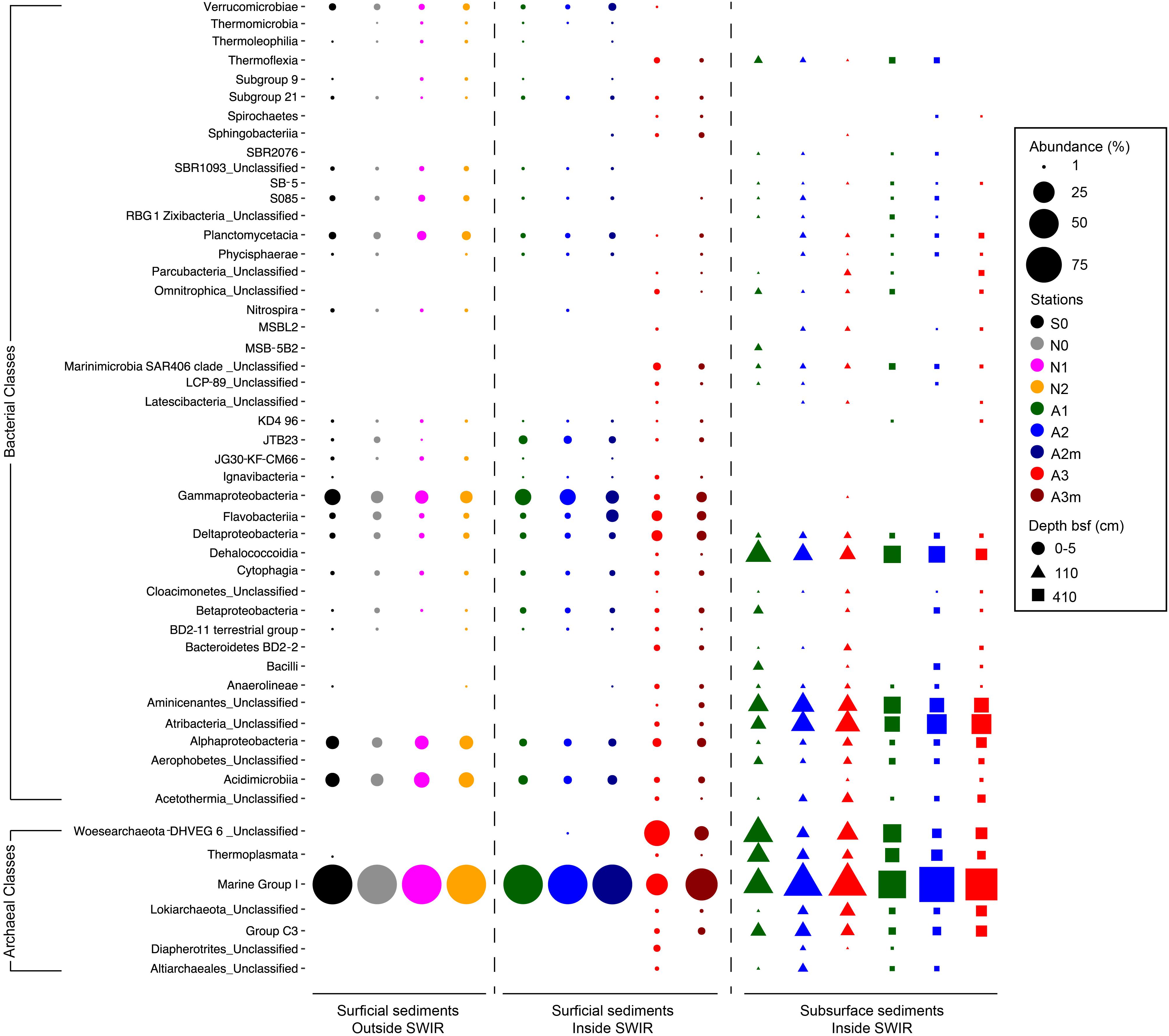
Figure 3. Bacterial and archaeal community composition in surface and subsurface sediments. Relative sequence abundance of (A) bacterial and (B) archaeal 16S rRNA at Class level resolution. Dominant community is here showed (i.e., community composed by those taxa present in the dataset with a relative abundance higher than 1%). For those taxa unclassified at Class level the Phylum is reported.

Figure 4. Bacterial community composition in surficial and subsurface sediments at Family level. Here we reported Families responsible for differences between bacterial communities inside and outside the SWIR and between surficial and subsurface bacterial communities. Families were selected after PCA analysis carried out on Hellinger transformed dominant bacterial and archaeal taxa (i.e., OTU > 0.1%) comparing (i) 0–5 cm layer of A3–A3m versus all stations, (ii) 0–5 cm layer of stations inside SWIR (A1 and A2–A2m) versus outside SWIR stations (N0, N1, N2, and S0), and (iii) 0–5 cm layer of all stations versus subsurface layer of SWIR stations.
Microbial community composition in surface sediments differed significantly between stations outside and inside the SWIR (ANOSIM, r > 0.40, p < 0.05); these differences were more pronounced when Area 3 (A3–A3m) was considered as a discrete group (ANOSIM, r > 0.90, p < 0.01; Figures 5B,C). At station A3 both bacterial and archaeal communities shared the lowest number of OTUs with all other stations (13 ± 2% and 12 ± 3%, respectively; Figures 5D-G and Supplementary Table S3). Bacterial and archaeal communities in surface sediments of SWIR stations (A1, A2, and A2m) shared a higher number of OTUs with each other (36 ± 2% and 57 ± 3%, respectively) than with communities outside the SWIR (27 ± 2% and 40 ± 5%, respectively; Figures 5D–G and Supplementary Table S3). Surface and subsurface bacterial and archaeal communities were significantly different (ANOSIM, r = 0.98 and r = 0.61, respectively, p = 0.001; Figures 5B,C), with the highest number of shared OTUs between 110 and 410 cm layers (Figures 5D,E and Supplementary Table S3). Shared bacterial OTUs between 110 cm layer and the top 0–5 cm layer increased remarkably westward (from 0.3 to 16%), whereas shared archaeal OTUs did not show any clear pattern between stations (11–14%). In surface sediments of Area 3, 46 ± 8, 89 ± 1, and 4 ± 1% of OTUs affiliated with the taxa Dehalococcoida, Atribacteria, and Woesearchaeota were shared with subsurface sediments; this corresponds to 46 ± 12, 93 ± 2, and 12 ± 4% of total sequences assigned to them in surficial sediments, respectively. In surface sediments the number of both bacterial and archaeal unique OTUs increased at the stations A3 and A3m compared to the stations outside the SWIR (Supplementary Table S1). About 3–8% of all bacterial OTUs were unique to one station, with highest values at A3m (Supplementary Table S1A). Unique archaeal OTUs represented 16–34% and 1–3% of total OTUs in surface sediments of Area 3 (A3 and A3m) and other stations, respectively (Supplementary Table S1B). In subsurface samples the contribution of bacterial and archaeal unique OTUs ranged between 3 and 6% and between 2 and 7%, respectively.
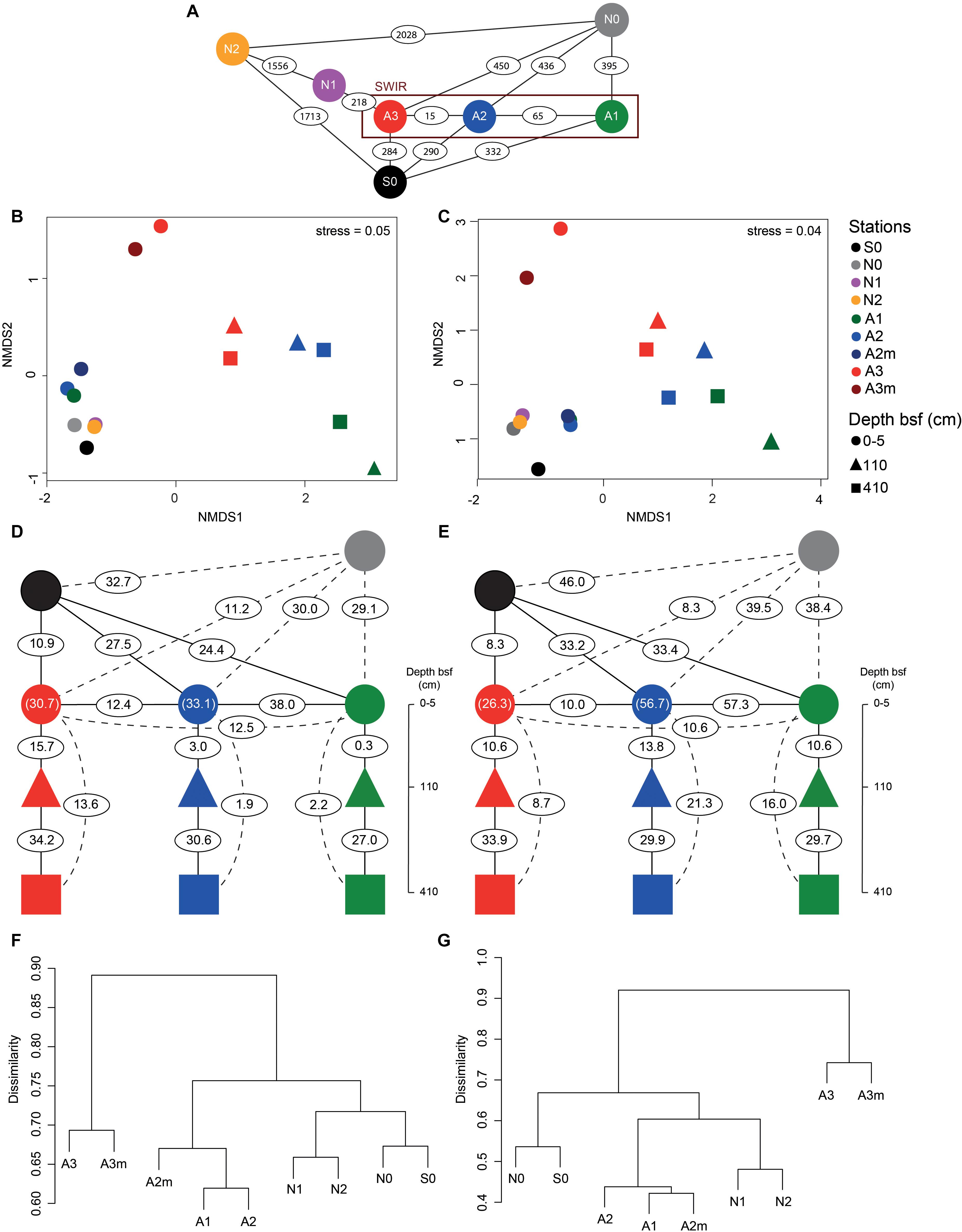
Figure 5. Beta-diversity of bacterial and archaeal communities in surface and subsurface sediments. (A) Geographic distance in kilometers between the investigated stations. Non-metric multidimensional scaling (nMDS) using Bray–Curtis distance on Hellinger transformed (B) bacterial and (C) archaeal community structure at OTU level. Percentage of (D–F) bacterial and (E–G) archaeal shared OTU between stations as defined by Jaccard dissimilarity matrix based on presence/absence OTU table (with 100 sequence re-samplings per sample on the smallest dataset); the numbers in parentheses represent the shared OTU between A2/A2m (green blue) and A3/A3m (red circle). For full matrices see Supplementary Table S3. bsf, below the seafloor.
Factors Controlling Microbial Community Structure
The variables chlorophyll a (Chl-a), total organic carbon (TOC), and dissolved inorganic carbon (DIC) did not show collinearity (VIF < 4) and were therefore used as descriptors of changes in the environmental setting at different stations. DIC, ammonia and sulfide were significantly positively correlated (Pearson r > 0.95, p < 0.001), thus we used DIC as a proxy for the presence of porewater flux and the availability of reduced compounds.
Bacterial and archaeal community similarity (i.e., proportion of shared OTUs) between samples did not show significant relationships with geographic distance, even when stations at Area 3 were excluded or only stations outside the SWIR (N0, N1, N2, and S0) were considered (Figures 6A,B). In contrast, the proportion of bacterial and archaeal OTUs shared between stations (not including N1 and N2) were significantly related with differences in the environmental setting (Spearman ρ = 0.63 and p < 0.01, Spearman ρ = 0.66 and p < 0.05), and correlated negatively with differences in Chl-a content and DIC concentration (Supplementary Table S4 and Figures 6C–F). The extent of the relationship between Chl-a and OTU variations was different between stations inside and outside the ridge, whereas the variations related to DIC concentration were mostly related with larger differences between stations in Area 3 and other stations (similarity < 20%). Accordingly, a combination of Chl-a and DIC concentrations explained 46 and 61% of the variance in bacterial and archaeal community structure, respectively (Supplementary Table S4). Both bacterial and archaeal community structures were mainly explained by DIC (23 and 29%, respectively) rather than Chl-a (10 and 20%, respectively; Supplementary Figure S5).
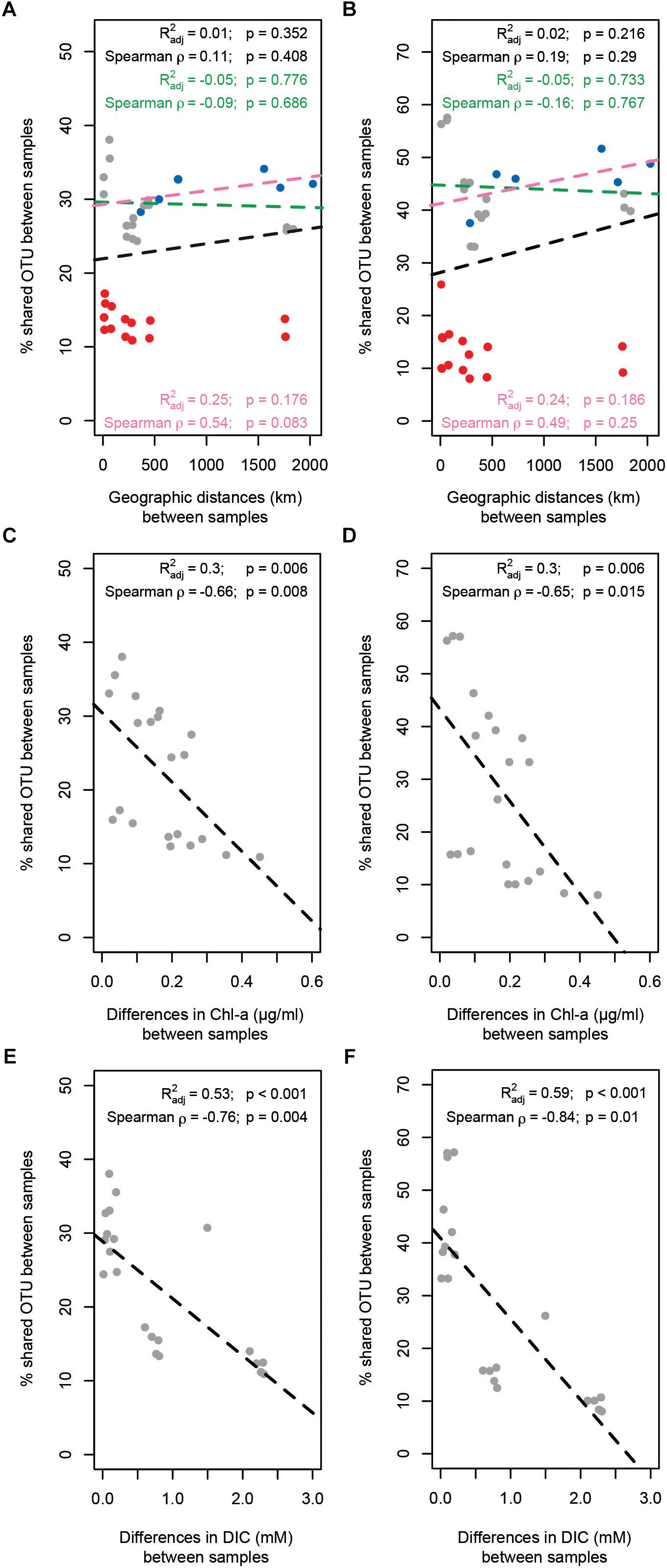
Figure 6. Relationship between geographic distance, environmental patterns and similarity in surface microbial community composition. The proportion of (A) bacterial and (B) archaeal shared OTUs between samples did not show any significant relationship with geographic distance; three different scenarios have been evaluated: full data-set including all stations (all dots; black text and dotted line); excluding stations in Area 3 (red dots; green text and dotted line); considering only the stations outside SWIR (blue dots; pink text and line). The proportion of (C–E) bacterial and (D–F) archaeal shared OTUs between samples decreased significantly with differences in chlorophyll a content (Chl-a; C,D) and dissolved inorganic carbon concentration (DIC; E,F). Dotted lines are linear model fits. Linear models’ R2, Spearman’s rho correlations, and their significance (Mantel tests with 1000 permutations) are reported in each panel.
Discussion
Ocean ridges are the largest continuous topographic feature on Earth, representing diverse geobiological habitats including hydrothermal vents, different types of hard and soft bottom including typical pelagic sediments (Orcutt et al., 2011). Where active venting occurs, substantial energy may be delivered to specific chemosynthetic groups of bacteria that provide the basis of a food web independent of photosynthetically produced matter, including symbiotic interactions with animals. MOR rocks host their own microbial communities that add substantial diversity to this deep-sea realm (Santelli et al., 2008). However, the specific role of non-hydrothermal MOR sediments in shaping benthic microbial diversity and connectivity in the deep sea is largely unknown. Furthermore, the increasing interest in mining seafloor massive sulfide (SMS) deposits at oceanic ridges calls for a better baseline knowledge about the diversity, variability and connectivity of benthic biological communities, in order to assess and forecast significant ecological impacts (Boetius and Haeckel, 2018; Miller et al., 2018). In this study, the diversity of bacterial and archaeal 16S rRNA genes at the sedimented SWIR segment 10°–17°E and in the adjacent sedimentary seafloor north and south of the ridge (Figure 1) within a water depth range of 3,655–4,869 m were investigated to test whether the SWIR may act as a physical barrier (i) reducing the benthic north-south microbial genetic flux in the South Atlantic Polar Front and (ii) promoting isolation of microbial communities inside the ridge. The topography (high ridge flanks and deep valley) and axial orientation (parallel to the main eastward bottom water flow) of SWIR limit the northward and southward flow of bottom water. The SWIR segment studied here is located in a region with relatively high sedimentation rates for an open ocean system (ca. 1–30 cm kyr-1 in the last 200,000 years; Nürnberg et al., 1997; Mackensen et al., 2001). This, together with the V-shape topography at this site, produced a high sediment thickness (ca. 80 m) at the bottom of the axial ridge valley (Supplementary Figure S6). The area is within a relatively productive biogeochemical province (ca. 51.7 × 106 km2) represented by an annual productivity of 8.4 mol C m-2 yr-1 and an estimated carbon flux of 85.8 mmol C m-2 yr-1 to the seafloor at 4,487 m depth (Seiter et al., 2004; Watling et al., 2013). The higher accumulation of Chl-a inside the SWIR compared to outside (up to 10 times; Table 1), and the increase of Chl-a toward the SWIR deepest axial stations (Supplementary Figure S1) is likely an effect produced by the V-shaped topographyat this site.
The detection of significant differences in bacterial and archaeal community structure at stations located outside and inside the SWIR supports at first sight the idea of the MOR acting as a physical barrier limiting microbial dispersal (Figure 5). Nevertheless, stations to the north of SWIR were not more similar to each other than with the station to the south (e.g., N1–N2 vs. N0–S0; Figures 5F,G). As described above, sedimentary matter composition and porewater chemistry indicated substantial differences in biogeochemistry, especially in the stations A3 and A3m. The environmental setting explained a large fraction of variance in bacterial and archaeal communities (45 and 61%, respectively; Supplementary Figure S5 and Supplementary Table S4), indicating environmental selection rather than isolation-by-distance as a main controlling factor.
The presence of the negative relationship between community similarity and differences in sedimentary chlorophyll a (Figures 6C,D) suggests food availability as a major driver of differences in bacterial and archaeal community composition between stations. This type of relationship has been shown before for deep-sea sediments in the Arctic Ocean (Bienhold et al., 2012; Jacob et al., 2013) and the primary role of trophic resource availability in shaping deep-sea benthic microbial community structure globally has been substantiated in more recent studies (Bienhold et al., 2016; Danovaro et al., 2016). This is reflected by the increase in the relative abundance of specific taxa related to phytoplankton/complex organic matter degradation at the SWIR stations in comparison to adjacent northern and southern sites (Figure 4). This includes the class Flavobacteriia, which has also shown positive correlations with chlorophyll pigments in an Arctic region (Bienhold et al., 2012). Flavobacteria have been associated with the ability to hydrolyze complex plant polymers (Humphry et al., 2001; Knoll et al., 2001; Williams et al., 2013). More specifically, the relative sequence abundance of the genus Flavobacterium was considerably higher at SWIR stations than in the adjacent seafloor. Pelagic and deep-sea benthic members of this taxon were previously shown to respond positively to phytoplankton blooms (Teeling et al., 2012, 2016; Ruff et al., 2014; Vigneron et al., 2017), indicating their potential role in complex phytodetritus matter degradation. Another class that seems to be selected for by higher organic matter availability at SWIR stations is Verrucomicrobiae; members of this class are known to play a role in polysaccharide degradation (Cardman et al., 2014; Li et al., 2016).
The different porewater biogeochemistry in Area 3 explained the majority of the observed differences in community composition between stations A3 and A3m with other stations (Figures 6E,F). Area 3 is located in the deepest axial section of the ridge valley where the large sink of organic matter can enhance early diagenesis processes (D’Hondt et al., 2002) and where the underlying lithosphere (i.e., peridotite) can promote serpentinization processes (Schmid and Schlindwein, 2016). These features produce an unusual geological setting in the deep sea, which is likely responsible for the observed upward efflux of anoxic porewater enriched in reduced compounds (Table 2), such as ammonia and sulfide in the surface sediments and potentially methane and hydrogen in deepest subsurface sediments. Hence, the highest per-cell microbial activity measured at A3m (Table 3) may be a consequence of the availability of multiple energy sources. During the PS81 expedition, no evidence for recent hydrothermal activity was found, neither at the seafloor nor in the water column of the amagmatic accretionary SWIR segment studied here (Schlindwein, 2014). Only one large veneroid bivalve of the family Vesicomyidae (genus Christineconcha, identified by Sergei Galkin, IORAS) was discovered, that had crawled onto an ocean bottom seismometer, which was deployed close to stations A3m 1 year before (Schlindwein, 2014; Supplementary Figure S6). Such Vesicomyids (i.e., genus Christineconcha) are known to inhabit reduced sediments with sulfide fluxes (Krylova and Von Cosel, 2011; Decker et al., 2012).
The most remarkable difference to other sites was the higher relative sequence proportion of potential sulfate-reducing bacteria (i.e., Desulfobacteraceae and Desulfarculaceae), which represented about 7% of all bacterial sequences at station A3 compared to less than 0.01% at stations outside the SWIR (Figure 3). This indicates that the high amount of organic carbon and the anaerobic conditions at Area 3 may support the development of sulfate-reducing communities, which are typically found in subsurface OM rich continental margin and cold-seep sediments (Teske, 2010). Interestingly about 35% of sequences related to potential sulfate reducers belonged to the genus SEEP-SRB1 (Supplementary Table S2), which is a sulfate-reducing partner of anaerobic methanotrophic archaea ANME-2 (Schreiber et al., 2010) and dominates methane-rich sediments (Ruff et al., 2015); however, we did not detect ANME-2 sequences in any of our samples. Most of the SEEP-SRB1 sequences found in Area 3 were closely related to sequences found in other chemosynthetic habitats and in SWIR vent fields (Supplementary Figure S7). Hydrogen sulfide, the catabolic product of sulfate reducers, probably favored sulfur-oxidizing bacteria in surface sediments of Area 3 (i.e., Helicobacteraceae), where their relative sequence proportion (ca. 0.2–1%; Figure 4) increased up to 500 times compared to outside the ridge. Sulfur oxidizers are a fundamental component of hydrothermal chemosynthetic communities and Helicobacteraceae dominate benthic chemolithotrophic communities at rocky hydrothermal vents of SWIR (Ding et al., 2017) and worldwide MOR (e.g., Flores et al., 2011; Sievert and Vetriani, 2012; Meier et al., 2017).
In surface sediments of Area 3, Dehalococcoida and Atribacteria were another important component of bacterial assemblages. These two taxa also dominated subsurface bacterial communities (Figure 3) and they are typically reported from subsurface environments associated with methane hydrates, hydrocarbon seeps, and petroleum reservoirs (Webster et al., 2004; Inagaki et al., 2006; Pham et al., 2009; Orcutt et al., 2011; Kobayashi et al., 2012; Parkes et al., 2014). There was no evidence for the presence of hydrocarbons in our sediments, and methane was only measured at nanomolar concentrations (Table 1), thus the presence of Dehalococcoida and Atribacteria in surface sediments could be due to an upward transport of porewater flux from deeper sediment layers. The high permeability and porosity of sediment in the ridge axial valley would allow for porewater circulation that could be responsible for an increased connectivity between subsurface and surface bacterial communities in this area compared to the more consolidated sediments outside the ridge (Figure 5D).
Area 3 also differed in archaeal diversity (Supplementary Figure S3 and Supplementary Table S1B) and community structure (Figures 3, 5C) from the other stations. The increased diversity and endemism of archaeal populations of Area 3 (Supplementary Table S1B) was associated with a decrease in their relative abundance, which was less than 2% of total microbial cells (Table 3). The dominance of Marine group I (Thaumarchaeota) suggests that archaea could have a primary role in nitrification (Könneke et al., 2005; Molari et al., 2013). However, the contribution of this taxon to total archaeal sequences did not increase in Area 3 where we found the highest concentrations of ammonia. Marine group I members use oxygen to oxidize ammonia, thus they may be limited by oxygen availability under the anoxic conditions in Area 3. Nevertheless, Marine group I was also a dominant group in the investigated anoxic subsurface layers, as reported for other deep-sea subsurface sediments (e.g., Durbin and Teske, 2010), which may mean that the metabolic diversity and ecological niche of this archaeal taxon is not fully elucidated yet (Offre et al., 2013). A large proportion of archaeal sequences found in surface sediments of Area 3 and subsurface layers of other SWIR stations belonged to Woesearchaeota (Figure 3), an enigmatic archaeal group of the monophyletic DPANN superphylum (Rinke et al., 2013; Castelle et al., 2015). Woesearchaeota have been detected in various environments including marine hydrothermal habitats (Takai and Horikoshi, 1999; Li et al., 2015), subsurface marine sediments, acid mines, groundwater (Baker et al., 2010; Castelle et al., 2015; Shcherbakova et al., 2016), high-altitude lakes (Ortiz-Alvarez and Casamayor, 2016), and recently in SWIR hydrothermal chimneys (Ding et al., 2017). Recent genome reconstructions support an anaerobic heterotrophic lifestyle (Liu et al., 2018), and their small genome size and the fact that most of the core biosynthetic pathways were partial or absent also suggest that Woesearchaeota have a host-associated/syntrophic or parasitic lifestyle, maybe even with bacteria (Baker et al., 2010; Castelle et al., 2015) or methanogenic Archaea (Liu et al., 2018). Despite the limited information about the ecological role of Woesearchaeota, the presence of this archaeal taxon in surface sediments of Area 3 supports connectivity between surface and anaerobic subsurface environments.
Conclusion
Our study suggests that the amagmatic SWIR fragment investigated here substantially enhanced microbial diversity by providing additional biogeochemical niches, foremost via sediment accumulation in the axial valley. Accordingly, variations in microbial community composition were driven by changes in trophic resource availability and the biogeochemical setting at this site. In the axial valley, porewater circulation also promoted connectivity between subsurface and surface communities, and favored microbial taxa typically associated with reduced sediments, including such found at hydrothermal vent fields on the SWIR and in other deep-sea regions. This indicates a potential role of ultraslow spreading ridges in connecting spatially isolated chemosynthetic communities in the deep sea, instead of inducing isolation. In this regard the findings reported here expand our knowledge about microbial community dynamics in these systems, and stimulate future research to better elucidate the role of magma-starved ridges in deep-sea biodiversity and connectivity.
Author Contributions
AB and MM designed the study and performed field observation and sampling activities. MM carried out cell counts and analyzed biogeochemical data. GV analyzed Illumina sequencing data and calculated the phylogenetic tree. FS calculated porewater fluxes. MM and GV made statistical analysis. MM, GV, CB, FS, and AB interpreted the results and wrote the manuscript.
Funding
The authors acknowledge the financial support of the ERASMUS+ programme to GV, the ERC Adv. Grant Abyss (#294757) to AB, and the Max Planck Society (MPG).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the captain, crew, and chief scientist of RV Polarstern expedition PS81 and Dr. Katrin Knittel (Molecular Ecology, MM-MPI, Bremen) who provided sediment samples collected during RV Polarstern expedition PS79. We are also grateful for the bioinformatics support by Dr. Christiane Hassenrück (Biogeochemistry and Geology, ZMT, Bremen), and technical support by M. Meiners, E. Weiz-Bersch, M. Alisch, W. Stiens, and R. Stiens (HGF MPG Joint Research Group for Deep-Sea Ecology and Technology).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00665/full#supplementary-material
Footnotes
References
Akerman, N. H., Butterfield, D. A., and Huber, J. A. (2013). Phylogenetic diversity and functional gene patterns of sulfur-oxidizing subseafloor Epsilonproteobacteria in diffuse hydrothermal vent fluids. Front. Microbiol. 4:185. doi: 10.3389/fmicb.2013.00185
Amann, R., and Fuchs, B. M. (2008). Single-cell identification in microbial communities by improved fluorescence in situ hybridization techniques. Nat. Rev. Microbiol. 6, 339–348. doi: 10.1038/nrmicro1888
Amann, R. I., Binder, B. J., Olson, R. J., Chisholm, S. W., Devereux, R., and Stahl, D. A. (1990). Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56, 1919–1925. doi: 10.1111/j.1469-8137.2004.01066.x
Amann, R. I., Ludwig, W., Schleifer, K. H., Amann, R. I., and Ludwig, W. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. These include: phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiology 59, 143–169.
Anderson, R. E., Sogin, M. L., and Baross, J. A. (2015). Biogeography and ecology of the rare and abundant microbial lineages in deep-sea hydrothermal vents. FEMS Microbiol. Ecol. 91, 1–11. doi: 10.1093/femsec/fiu016
Bach, W., Banerjee, N. R., Dick, H. J. B., and Baker, E. T. (2002). Discovery of ancient and active hydrothermal systems along the ultra-slow spreading Southwest Indian Ridge 10°-16°E. Geochem. Geophys. Geosyst. 3, 1–14. doi: 10.1029/2001GC000279
Baker, B. J., Comolli, L. R., Dick, G. J., Hauser, L. J., Hyatt, D., Dill, B. D., et al. (2010). Enigmatic, ultrasmall, uncultivated Archaea. Proc. Natl. Acad. Sci. U.S.A. 107, 8806–8811. doi: 10.1073/pnas.0914470107
Beaulieu, S. E., Baker, E. T., and German, C. R. (2015). Where are the undiscovered hydrothermal vents on oceanic spreading ridges? Deep Sea Res. Part II Top. Stud. Oceanogr. 121, 202–212. doi: 10.1016/j.dsr2.2015.05.001
Bienhold, C., Boetius, A., and Ramette, A. (2012). The energy-diversity relationship of complex bacterial communities in Arctic deep-sea sediments. ISME J. 6, 724–732. doi: 10.1038/ismej.2011.140
Bienhold, C., Zinger, L., Boetius, A., and Ramette, A. (2016). Diversity and biogeography of bathyal and abyssal seafloor bacteria. PLoS One 11:e0148016. doi: 10.1371/journal.pone.0148016
Böer, S. I., Arnosti, C., Van Beusekom, J. E. E., and Boetius, A. (2009). Temporal variations in microbial activities and carbon turnover in subtidal sandy sediments. Biogeosciences 6, 1149–1165. doi: 10.5194/bg-6-1149-2009
Boetius, A., and Haeckel, M. (2018). Mind the seafloor. Science 359, 34–36. doi: 10.1126/science.aap7301
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Campbell, B. J., Polson, S. W., Allen, L. Z., Williamson, S. J., Lee, C. K., Wommack, K. E., et al. (2013). Diffuse flow environments within basalt- and sediment-based hydrothermal vent ecosystems harbor specialized microbial communities. Front. Microbiol. 4:182. doi: 10.3389/fmicb.2013.00182
Cardman, Z., Arnosti, C., Durbin, A., Ziervogel, K., Cox, C., Steen, A. D., et al. (2014). Verrucomicrobia are candidates for polysaccharide-degrading bacterioplankton in an Arctic fjord of Svalbard. Appl. Environ. Microbiol. 80, 3749–3756. doi: 10.1128/AEM.00899-14
Castelle, C. J., Wrighton, K. C., Thomas, B. C., Hug, L. A., Brown, C. T., Wilkins, M. J., et al. (2015). Genomic expansion of domain Archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr. Biol. 25, 690–701. doi: 10.1016/j.cub.2015.01.014
Chao, A., Gotelli, N. J., Hsieh, T. C., Sander, E. L., Ma, K. H., Colwell, R. K., et al. (2014). Rarefaction and extrapolation with Hill numbers: a framework for sampling and estimation in species diversity studies. Ecol. Monogr. 84, 45–67. doi: 10.1890/13-0133.1
Clarke, K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
Cline, J. (1969). Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14, 454–458. doi: 10.4319/lo.1969.14.3.0454
Daims, H., Brühl, A., Amann, R., Schleifer, K. H., and Wagner, M. (1999). The domain-specific probe EUB338 is insufficient for the detection of all bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444. doi: 10.1016/S0723-2020(99)80053-8
Danovaro, R., Aguzzi, J., Fanelli, E., Billett, D., Gjerde, K., Jamieson, A., et al. (2017). An ecosystem-based deep-ocean strategy. Science 355, 452–454. doi: 10.1126/science.aah7178
Danovaro, R., Molari, M., Corinaldesi, C., and Dell’Anno, A. (2016). Macroecological drivers of Archaea and bacteria in benthic deep-sea ecosystems. Sci. Adv. 2:e1500961. doi: 10.1126/sciadv.1500961
De Rezende, J. R., Kjeldsen, K. U., Hubert, C. R. J., Finster, K., Loy, A., and Jørgensen, B. B. (2013). Dispersal of thermophilic Desulfotomaculum endospores into Baltic Sea sediments over thousands of years. ISME J. 7, 72–84. doi: 10.1038/ismej.2012.83
Decker, C., Olu, K., Cunha, R. L., and Arnaud-Haond, S. (2012). Phylogeny and diversification patterns among vesicomyid bivalves. PLoS One 7:e33359. doi: 10.1371/journal.pone.0033359
D’Hondt, S., Rutherford, S., and Spivack, A. J. (2002). Metabolic activity of subsurface life in deep-sea sediments. Science 295, 2067–2070. doi: 10.1126/science.1064878
Dick, H. J. B., Lin, J., and Schouten, H. (2003). An ultraslow-spreading class of ocean ridge. Nature 426, 405–412. doi: 10.1038/nature02128
Diepenbroek, M., Glöckner, F. O., Grobe, P., Güntsch, A., Huber, R., König-Ries, B., et al. (2014). “Towards an integrated biodiversity and ecological research data management and archiving platform: the German federation for the curation of biological data (GFBio),” in Informatik 2014, eds E. Plödereder, L. Grunske, E. Schneider, and D. Ull (Bonn: Gesellschaft für Informatik e.V), 1711–1724.
Ding, J., Zhang, Y., Wang, H., Jian, H., Leng, H., and Xiao, X. (2017). Microbial community structure of deep-sea hydrothermal vents on the ultraslow spreading southwest Indian ridge. Front. Microbiol. 8:1012. doi: 10.3389/fmicb.2017.01012
Dinter, W. P. (2001). Biogeography of the OSPAR Maritime Area. Bonn: Federal Agency for Nature Conservation.
Dray, S., and Dufour, A. B. (2007). The ade4 Package: implementing the duality diagram for ecologists. J. Stat. Softw. 22, 1–20. doi: 10.18637/jss.v022.i04
Durbin, A. M., and Teske, A. (2010). Sediment-associated microdiversity within the Marine Group I Crenarchaeota. Environ. Microbiol. Rep. 2, 693–703. doi: 10.1111/j.1758-2229.2010.00163.x
Durbin, A. M., and Teske, A. (2011). Microbial diversity and stratification of South Pacific abyssal marine sediments. Environ. Microbiol. 13, 3219–3234. doi: 10.1111/j.1462-2920.2011.02544.x
Flores, G. E., Campbell, J. H., Kirshtein, J. D., Meneghin, J., Podar, M., Steinberg, J. I., et al. (2011). Microbial community structure of hydrothermal deposits from geochemically different vent fields along the Mid-Atlantic Ridge. Environ. Microbiol. 13, 2158–2171. doi: 10.1111/j.1462-2920.2011.02463.x
Flores, G. E., Wagner, I. D., Liu, Y., and Reysenbach, A. L. (2012). Distribution, abundance, and diversity patterns of the thermoacidophilic “deep-sea hydrothermal vent euryarchaeota 2.” Front. Microbiol. 3:47. doi: 10.3389/fmicb.2012.00047
Gebruk, A. V., Budaeva, N. E., and King, N. J. (2010). Bathyal benthic fauna of the mid-Atlantic ridge between the azores and the reykjanes ridge. J. Mar. Biol. Assoc. U.K. 90, 1–14. doi: 10.1017/S0025315409991111
German, C. R., Ramirez-Llodra, E., Baker, M. C., Tyler, P. A., Baco-Taylor, A., Boetius, A., et al. (2011). Deep-water chemosynthetic ecosystem research during the census of marine life decade and beyond: a proposed deep-ocean road map. PLoS One 6:e23259. doi: 10.1371/journal.pone.0023259
Gobet, A., Boetius, A., and Ramette, A. (2013). Ecological coherence of diversity patterns derived from classical fingerprinting and Next Generation Sequencing techniques. Environ. Microbiol. 16, 2672–2681. doi: 10.1111/1462-2920.12308
Goffredi, S. K., Johnson, S., Tunnicliffe, V., Caress, D., Clague, D., Escobar, E., et al. (2017). Hydrothermal vent fields discovered in the southern Gulf of California clarify role of habitat in augmenting regional diversity. Proc. R. Soc. B Biol. Sci. 284:20170817. doi: 10.1098/rspb.2017.0817
Gollner, S., Govenar, B., Fisher, C. R., and Bright, M. (2015). Size matters at deep-sea hydrothermal vents: different diversity and habitat fidelity patterns of meio- and macrofauna. Mar. Ecol. Prog. Ser. 520, 57–66. doi: 10.3354/meps11078
Grasshoff, K., Kremling, K., and Ehrhardt, M. (2007). Methods of Seawater Analysis, 3rd Edn. Hoboken, NJ: Wiley. doi: 10.1002/9783527613984
Haine, T. W. N., Watson, A. J., Liddicoat, M. I., and Dickson, R. R. (1998). The flow of Antarctic bottom water to the southwest Indian Ocean estimated using CFCs. J. Geophys. Res. Ocean. 103, 27637–27653. doi: 10.1029/98JC02476
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C., and Martiny, J. B. H. (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 497–506. doi: 10.1038/nrmicro2795
Hill, M. O. (1973). Diversity and evenness: a unifying notation and its consequences. Ecology 54, 427–432. doi: 10.2307/1934352
Hobbie, J. E., Daley, R. J., and Jasper, S. (1977). Use of nuclepore filters for counting bacteria by fluorescence microscopy. Appl. Environ. Microbiol. 33, 1225–1228.
Hsieh, T. C., Ma, K. H., and Chao, A. (2016). iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol. 7, 1451–1456. doi: 10.1111/2041-210X.12613
Humphry, D. R., George, A., Black, G. W., and Cummings, S. P. (2001). Flavobacterium frigidarium sp. nov., an aerobic, psychrophilic, xylanolytic and laminarinolytic bacterium from Antarctica. Int. J. Syst. Evol. Microbiol. 51, 1235–1243. doi: 10.1099/00207713-51-4-1235
Inagaki, F., Nunoura, T., Nakagawa, S., Teske, A., Lever, M., Lauer, A., et al. (2006). Biogeographical distribution and diversity of microbes in methane hydrate-bearing deep marine sediments on the Pacific Ocean Margin. Proc. Natl. Acad. Sci. U.S.A. 103, 2815–2820. doi: 10.1073/pnas.0511033103
Ishibashi, J. I., Okino, K., and Sunamura, M. (eds) (2015). “Intra-field variation of prokaryotic communities on and below the seafloor in the back-arc hydrothermal system of the southern Mariana trough,” in Subseafloor Biosphere Linked to Hydrothermal Systems: TAIGA Concept, (Tokyo: Springer), 301–311. doi: 10.1007/978-4-431-54865-2
Ishii, K., Mußmann, M., MacGregor, B. J., and Amann, R. I. (2004). An improved fluorescence in situ hybridization protocol for the identification of bacteria and Archaea in marine sediments. FEMS Microbiol. Lett. 50, 203–212. doi: 10.1016/j.femsec.2004.06.015
Jacob, M., Soltwedel, T., Boetius, A., and Ramette, A. (2013). Biogeography of deep-sea benthic bacteria at regional scale (LTER HAUSGARTEN, Fram Strait, Arctic). PLoS One 8:e72779. doi: 10.1371/journal.pone.0072779
Jørgensen, B. B., and Boetius, A. (2007). Feast and famine - Microbial life in the deep-sea bed. Nat. Rev. Microbiol. 5, 770–781. doi: 10.1038/nrmicro1745
Kassambara, A. (2015). factoextra: Visualization of the Outputs of a Multivariate Analysis. R Package version.
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Knoll, S., Zwisler, W., and Simon, M. (2001). Bacterial colonization of early stages of limnetic diatom microaggregates. Aquat. Microb. Ecol. 25, 141–150. doi: 10.3354/ame025141
Kobayashi, H., Endo, K., Sakata, S., Mayumi, D., Kawaguchi, H., Ikarashi, M., et al. (2012). Phylogenetic diversity of microbial communities associated with the crude-oil, large-insoluble-particle and formation-water components of the reservoir fluid from a non-flooded high-temperature petroleum reservoir. J. Biosci. Bioeng. 113, 204–210. doi: 10.1016/j.jbiosc.2011.09.015
Könneke, M., Bernhard, A. E., De La Torre, J. R., Walker, C. B., Waterbury, J. B., and Stahl, D. A. (2005). Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437, 543–546. doi: 10.1038/nature03911
Krylova, M. E., and Von Cosel, R. (2011). A new genus of large Vesicomyidae (Mollusca, Bivalvia, Vesicomyidae, Pliocardiinae) from the Congo margin, with the first record of the subfamily Pliocardiinae in the Bay of Biscay (northeastern Atlantic). Zoosystema 33, 83–99. doi: 10.5252/z2011n1a4
Larqué, L., Maamaatuaiahutapu, K., and Garçon, V. (1997). On the intermediate and deep water flows in the South Atlantic Ocean. J. Geophys. Res. Ocean. 102, 12425–12440. doi: 10.1029/97JC00629
Legendre, P., and Gallagher, E. D. (2001). Ecologically meaningful transformations for ordination of species data. Oecologia 129, 271–280. doi: 10.1007/s004420100716
Legendre, P., and Legendre, L. (1998). Numerical Ecology, 2nd Edn. Cambridge: Cambridge University Press. doi: 10.1017/CBO9781107415324.004
Li, M., Baker, B. J., Anantharaman, K., Jain, S., Breier, J. A., and Dick, G. J. (2015). Genomic and transcriptomic evidence for scavenging of diverse organic compounds by widespread deep-sea Archaea. Nat. Commun. 6:8933. doi: 10.1038/ncomms9933
Li, M., Jain, S., and Dick, G. J. (2016). Genomic and transcriptomic resolution of organic matter utilization among deep-sea bacteria in Guaymas basin hydrothermal plumes. Front. Microbiol. 7:1125. doi: 10.3389/fmicb.2016.01125
Liu, X., Li, M., Castelle, C. J., Probst, A. J., Zhou, Z., Pan, J., et al. (2018). Insights into the ecology, evolution, and metabolism of the widespread Woesearchaeotal lineages. Microbiome 6:102. doi: 10.1186/s40168-018-0488-2
Mackensen, A., Rudolph, M., and Kuhn, G. (2001). Late pleistocene deep-water circulation in the subantarctic eastern Atlantic. Glob. Planet. Change 30, 197–229. doi: 10.1016/S0921-8181(01)00102-3
Mahé, F., Rognes, T., Quince, C., de Vargas, C., and Dunthorn, M. (2014). Swarm: robust and fast clustering method for amplicon-based studies. PeerJ 2:e593. doi: 10.7717/peerj.593
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Matyka, M., Khalili, A., and Koza, Z. (2008). Tortuosity-porosity relation in porous media flow. Phys. Rev. E 78(2 Pt 2):026306. doi: 10.1103/PhysRevE.78.026306
McMurdie, P. J., and Holmes, S. (2015). Shiny-phyloseq: web application for interactive microbiome analysis with provenance tracking. Bioinformatics 31, 282–283. doi: 10.1093/bioinformatics/btu616
Meier, D. V., Pjevac, P., Bach, W., Hourdez, S., Girguis, P. R., Vidoudez, C., et al. (2017). Niche partitioning of diverse sulfur-oxidizing bacteria at hydrothermal vents. ISME J. 11, 1545–1558. doi: 10.1038/ismej.2017.37
Miller, K. A., Thompson, K. F., Johnston, P., and Santillo, D. (2018). An overview of seabed mining including the current state of development, environmental impacts, and knowledge gaps. Front. Mar. Sci. 4:418. doi: 10.3389/fmars.2017.00418
Mino, S., Nakagawa, S., Makita, H., Toki, T., Miyazaki, J., Sievert, S. M., et al. (2017). Endemicity of the cosmopolitan mesophilic chemolithoautotroph Sulfurimonas at deep-sea hydrothermal vents. ISME J. 11, 909–919. doi: 10.1038/ismej.2016.178
Mironov, A. N. (2006). Centers of marine fauna redistribution. Entomol. Rev. 86, S32–S44. doi: 10.1134/S0013873806100034
Molari, M., and Manini, E. (2012). Reliability of CARD-FISH procedure for enumeration of Archaea in deep-sea surficial sediments. Curr. Microbiol. 64, 242–250. doi: 10.1007/s00284-011-0056-5
Molari, M., Manini, E., and Dell’Anno, A. (2013). Dark inorganic carbon fixation sustains the functioning of benthic deep-sea ecosystems. Glob. Biogeochem. Cycles 27, 212–221. doi: 10.1002/gbc.20030
Naimi, B., Hamm, N. A. S., Groen, T. A., Skidmore, A. K., and Toxopeus, A. G. (2014). Where is positional uncertainty a problem for species distribution modelling? Ecography 37, 191–203. doi: 10.1111/j.1600-0587.2013.00205.x
Nunoura, T., Takaki, Y., Hirai, M., Shimamura, S., Makabe, A., Koide, O., et al. (2015). Hadal biosphere: insight into the microbial ecosystem in the deepest ocean on Earth. Proc. Natl. Acad. Sci. U.S.A. 112, E1230–E1236. doi: 10.1073/pnas.1421816112
Nürnberg, C. C., Bohrmann, G., Schlüter, M., and Frank, M. (1997). Barium accumulation in the Atlantic sector of the Southern Ocean: results from 190,000-year records. Paleoceanography 12, 594–603. doi: 10.1029/97PA01130
Offre, P., Spang, A., and Schleper, C. (2013). Archaea in biogeochemical cycles. Annu. Rev. Microbiol. 67, 437–457. doi: 10.1146/annurev-micro-092412-155614
Oksanen, J., Blanchet, F., Kindt, R., Legendre, P., and O’Hara, R. (2016). Vegan: Community Ecology Package. R Package 2.3-3. Available at: https://cran.r-project.org/web/packages/vegan/vegan.pdf
Orcutt, B. N., Sylvan, J. B., Knab, N. J., and Edwards, K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 75, 361–422. doi: 10.1128/MMBR.00039-10
Orsi, A. H., Johnson, G. C., and Bullister, J. L. (1999). Circulation, mixing, and production of Antarctic Bottom Water. Prog. Oceanogr. 43, 55–109. doi: 10.1016/S0079-6611(99)00004-X
Ortiz-Alvarez, R., and Casamayor, E. O. (2016). High occurrence of Pacearchaeota and Woesearchaeota (Archaea superphylum DPANN) in the surface waters of oligotrophic high-altitude lakes. Environ. Microbiol. Rep. 8, 210–217. doi: 10.1111/1758-2229.12370
Oton, E. V., Quince, C., Nicol, G. W., Prosser, J. I., and Gubry-Rangin, C. (2016). Phylogenetic congruence and ecological coherence in terrestrial Thaumarchaeota. ISME J. 10, 85–96. doi: 10.1038/ismej.2015.101
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Parkes, R. J., Cragg, B., Roussel, E., Webster, G., Weightman, A., and Sass, H. (2014). A review of prokaryotic populations and processes in sub-seafloor sediments, including biosphere: geosphere interactions. Mar. Geol. 352, 409–425. doi: 10.1016/j.margeo.2014.02.009
Pham, V. D., Hnatow, L. L., Zhang, S., Fallon, R. D., Jackson, S. C., Tomb, J. F., et al. (2009). Characterizing microbial diversity in production water from an Alaskan mesothermic petroleum reservoir with two independent molecular methods. Environ. Microbiol. 11, 176–187. doi: 10.1111/j.1462-2920.2008.01751.x
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Development Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J. F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
Ristova, P. P., Wenzhöfer, F., Ramette, A., Felden, J., and Boetius, A. (2015). Spatial scales of bacterial community diversity at cold seeps (Eastern Mediterranean Sea). ISME J. 9, 1306–1318. doi: 10.1038/ismej.2014.217
Rogers, A. D., Tyler, P. A., Connelly, D. P., Copley, J. T., James, R., Larter, R. D., et al. (2012). The discovery of new deep-sea hydrothermal vent communities in the Southern ocean and implications for biogeography. PLoS Biol. 10:e1001234. doi: 10.1371/journal.pbio.1001234
Ruff, S. E., Biddle, J. F., Teske, A. P., Knittel, K., Boetius, A., and Ramette, A. (2015). Global dispersion and local diversification of the methane seep microbiome. Proc. Natl. Acad. Sci. U.S.A. 112, 4015–4020. doi: 10.1073/pnas.1421865112
Ruff, S. E., Probandt, D., Zinkann, A. C., Iversen, M. H., Klaas, C., Würzberg, L., et al. (2014). Indications for algae-degrading benthic microbial communities in deep-sea sediments along the Antarctic Polar Front. Deep Sea Res. Part II Top. Stud. Oceanogr. 108, 6–16. doi: 10.1016/j.dsr2.2014.05.011
Rutgers van der Loeff, M., Venchiarutti, C., Stimac, I., van Ooijen, J., Huhn, O., Rohardt, G., et al. (2016). Meridional circulation across the Antarctic Circumpolar Current serves as a double 231Pa and 230Th trap. Earth Planet. Sci. Lett. 455, 73–84. doi: 10.1016/j.epsl.2016.07.027
Salazar, G., Cornejo-Castillo, F. M., Benítez-Barrios, V., Fraile-Nuez, E., Álvarez-Salgado, X. A., Duarte, C. M., et al. (2016). Global diversity and biogeography of deep-sea pelagic prokaryotes. ISME J. 10, 596–608. doi: 10.1038/ismej.2015.137
Santelli, C. M., Orcutt, B. N., Banning, E., Bach, W., Moyer, C. L., Sogin, M. L., et al. (2008). Abundance and diversity of microbial life in ocean crust. Nature 453, 653–656. doi: 10.1038/nature06899
Sauter, D., and Cannat, M. (2013). “The ultraslow spreading southwest Indian ridge,” in Diversity of Hydrothermal Systems on Slow Spreading Ocean Ridges, eds P. A. Rona, C. W. Devey, J. Dyment, and B. J. Murton (Washington, DC: American Geophysical Union), 153–173. doi: 10.1029/2008GM000843
Schauer, R., Bienhold, C., Ramette, A., and Harder, J. (2010). Bacterial diversity and biogeography in deep-sea surface sediments of the South Atlantic Ocean. ISME J. 4, 159–170. doi: 10.1038/ismej.2009.106
Schippers, A., Neretin, L. N., Kallmeyer, J., Ferdelman, T. G., Cragg, B. A., Parkes, R. J., et al. (2005). Prokaryotic cells of the deep sub-seafloor biosphere identified as living bacteria. Nature 433, 861–864. doi: 10.1038/nature03302
Schlindwein, V. (2014). “The expedition of the research vessel “Polarstern” to the Antarctic in 2013 (ANT-XXIX/8),” in Berichte zur Polar- und Meeresforschung = Reports on Polar and Marine Research, Vol. 672, ed. Vera Schlindwein with Contributions of the Participants (Bremerhaven: Alfred Wegener Institute for Polar and Marine Research), 111. doi: 10.2312/BzPM_0672_2014
Schmid, F., and Schlindwein, V. (2016). Microearthquake activity, lithospheric structure, and deformation modes at an amagmatic ultraslow spreading Southwest Indian Ridge segment. Geochem. Geophys. Geosyst. 17, 2905–2921. doi: 10.1002/2016GC006271
Schreiber, L., Holler, T., Knittel, K., Meyerdierks, A., and Amann, R. (2010). Identification of the dominant sulfate-reducing bacterial partner of anaerobic methanotrophs of the ANME-2 clade. Environ. Microbiol. 12, 2327–2340. doi: 10.1111/j.1462-2920.2010.02275.x
Schulz, H. D. (2000). “Quantification of early diagenesis: dissolved constituents in marine pore water,” in Marine Geochemistry, eds H. D. Schulz and M. Zabel (Berlin: Springer-Verlag), 85–128.
Seiter, K., Hensen, C., Schröter, J., and Zabel, M. (2004). Organic carbon content in surface sediments - Defining regional provinces. Deep Sea Res. Part I Oceanogr. Res. Pap. 51, 2001–2026. doi: 10.1016/j.dsr.2004.06.014
Shcherbakova, V., Yoshimura, Y., Ryzhmanova, Y., Taguchi, Y., Segawa, T., Oshurkova, V., et al. (2016). Archaeal communities of Arctic methane-containing permafrost. FEMS Microbiol. Ecol. 92:fiw135. doi: 10.1093/femsec/fiw135
Shulse, C. N., Maillot, B., Smith, C. R., and Church, M. J. (2016). Polymetallic nodules, sediments, and deep waters in the equatorial North Pacific exhibit highly diverse and distinct bacterial, Archaeal, and microeukaryotic communities. Microbiologyopen 6:e00428. doi: 10.1002/mbo3.428
Sievert, S. M., and Vetriani, C. (2012). Chemoautotrophy at deep-sea vents: past, present, and future. Oceanography 25, 218–233. doi: 10.5670/oceanog.2012.21
Sogin, M. L., Morrison, H. G., Morrison, H. G., Huber, J. A., Huber, J. A., Mark Welch, D., et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Stahl, D. A., and Amann, R. (1991). “Development and application of nucleic acid probes,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (Hoboken, NJ: John Wiley & Sons),205–242.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun, D. L., Jiang, X., Wu, Q. L., and Zhou, N. Y. (2013). Intragenomic heterogeneity of 16S rRNA genes causes overestimation of prokaryotic diversity. Appl. Environ. Microbiol. 79, 5962–5969. doi: 10.1128/AEM.01282-13
Sylvan, J. B., Toner, B. M., and Edwards, K. J. (2012). Life and death of deep-sea vents: bacterial diversity and ecosystem succession on inactive hydrothermal sulfides. mBio 3:e00279-11. doi: 10.1128/mBio.00279-11
Takai, K., and Horikoshi, K. (1999). Genetic diversity of Archaea in deep-sea hydrothermal vent environments. Genetics 152, 1285–1297. doi: 10.1016/s0723-2020(87)80053-x
Teeling, H., Fuchs, B. M., Becher, D., Klockow, C., Gardebrecht, A., Bennke, C. M., et al. (2012). Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336, 608–611. doi: 10.1126/science.1218344
Teeling, H., Fuchs, B. M., Bennke, C. M., Krüger, K., Chafee, M., Kappelmann, L., et al. (2016). Recurring patterns in bacterioplankton dynamics during coastal spring algae blooms. eLife 5:e11888. doi: 10.7554/eLife.11888
Teske, A. (2010). “Sulfate-reducing and methanogenic hydrocarbon-oxidizing microbial communities in the marine environment A,” in Handbook of Hydrocarbon and Lipid Microbiology, Vol. 1, eds K. N. Timmis, T. McGenity, J. R. van der Meer, and V. de Lorenzo (Berlin: Springer-Verlag), 1–6. doi: 10.1007/978-3-540-77587-4
Thang, N. M., Brüchert, V., Formolo, M., Wegener, G., Ginters, L., Jørgensen, B. B., et al. (2012). The impact of sediment and carbon fluxes on the biogeochemistry of methane and sulfur in littoral Baltic sea sediments (Himmerfjärden, Sweden). Estuaries Coast. 36, 98–115. doi: 10.1007/s12237-012-9557-0
Vigneron, A., Alsop, E. B., Cruaud, P., Philibert, G., King, B., Baksmaty, L., et al. (2017). Comparative metagenomics of hydrocarbon and methane seeps of the Gulf of Mexico. Sci. Rep. 7:16015. doi: 10.1038/s41598-017-16375-5
Vinogradova, N. G. (1997). Zoogeography of the abyssal and Hadal zones. Adv. Mar. Biol. 32, 325–387. doi: 10.1016/S0065-2881(08)60019-X
Walsh, E. A., Kirkpatrick, J. B., Rutherford, S. D., Smith, D. C., Sogin, M., and D’Hondt, S. (2016). Bacterial diversity and community composition from seasurface to subseafloor. ISME J. 10, 979–989. doi: 10.1038/ismej.2015.175
Wang, Y., and Qian, P. Y. (2009). Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One 4:e7401. doi: 10.1371/journal.pone.0007401
Watling, L., Guinotte, J., Clark, M. R., and Smith, C. R. (2013). A proposed biogeography of the deep ocean floor. Prog. Oceanogr. 111, 91–112. doi: 10.1016/j.pocean.2012.11.003
Webster, G., Parkes, R. J., Fry, J. C., and Weightman, A. J. (2004). Widespread occurrence of a novel division of bacteria identified by 16S rRNA gene sequences originally found in deep marine sediments. Appl. Environ. Microbiol. 70, 5708–5713. doi: 10.1128/AEM.70.9.5708-5713.2004
Wei, C. L., Rowe, G. T., Briones, E. E., Boetius, A., Soltwedel, T., Caley, M. J., et al. (2010). Global patterns and predictions of seafloor biomass using random forests. PLoS One 5:e15323. doi: 10.1371/journal.pone.0015323
Wenzhöfer, F., Oguri, K., Middelboe, M., Turnewitsch, R., Toyofuku, T., Kitazato, H., et al. (2016). Benthic carbon mineralization in hadal trenches: assessment by in situ O2 microprofile measurements. Deep Sea Res. Part 1 Oceanogr. Res. Pap. 116, 276–286. doi: 10.1016/j.dsr.2016.08.013
Wickham, H. (2007). Reshaping data with the reshape package. J. Stat. Softw. 21, 1–20. doi: 10.1016/S0142-1123(99)00007-9
Wickham, H. (2009). ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer. doi: 10.1007/978-0-387-98141-3
Wickham, H. (2011). The split-apply-combine strategy for data analysis. J. Stat. Softw. 40, 1–29. doi: 10.18637/jss.v040.i01
Wickham, H., and Chang, W. (2015). Devtools: Tools to Make Developing R Code Easier. R Package version.
Williams, T. J., Wilkins, D., Long, E., Evans, F., Demaere, M. Z., Raftery, M. J., et al. (2013). The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics. Environ. Microbiol. 15, 1302–1317. doi: 10.1111/1462-2920.12017
Wilson, R. R. R. Jr., and Kaufmann, R. R. S. (1987). Seamount biota and biogeography. Geophys. Monogr. Ser. 43, 355–377. doi: 10.1029/GM043p0355
Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2014). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. doi: 10.1093/bioinformatics/btt593
Keywords: Southwest Indian Ridge, seamounts, deep-sea, connectivity, diversity, bacteria, archaea
Citation: Varliero G, Bienhold C, Schmid F, Boetius A and Molari M (2019) Microbial Diversity and Connectivity in Deep-Sea Sediments of the South Atlantic Polar Front. Front. Microbiol. 10:665. doi: 10.3389/fmicb.2019.00665
Received: 29 December 2018; Accepted: 18 March 2019;
Published: 09 April 2019.
Edited by:
Jennifer F. Biddle, University of Delaware, United StatesReviewed by:
Rika Anderson, Carleton College, United StatesAdrien Vigneron, Laval University, Canada
Copyright © 2019 Varliero, Bienhold, Schmid, Boetius and Molari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimiliano Molari, bWFtb2xhcmlAbXBpLWJyZW1lbi5kZQ==