 Ameen Eetemadi1,2
Ameen Eetemadi1,2 Navneet Rai2
Navneet Rai2 Beatriz Merchel Piovesan Pereira2,3
Beatriz Merchel Piovesan Pereira2,3 Minseung Kim1,2,4Harold Schmitz5
Minseung Kim1,2,4Harold Schmitz5 Ilias Tagkopoulos1,2,4*
Ilias Tagkopoulos1,2,4*- 1Department of Computer Science, University of California, Davis, Davis, CA, United States
- 2Genome Center, University of California, Davis, Davis, CA, United States
- 3Department of Microbiology, University of California, Davis, Davis, CA, United States
- 4Process Integration and Predictive Analytics (PIPA LLC), Davis, CA, United States
- 5Graduate School of Management, University of California, Davis, Davis, CA, United States
Food and human health are inextricably linked. As such, revolutionary impacts on health have been derived from advances in the production and distribution of food relating to food safety and fortification with micronutrients. During the past two decades, it has become apparent that the human microbiome has the potential to modulate health, including in ways that may be related to diet and the composition of specific foods. Despite the excitement and potential surrounding this area, the complexity of the gut microbiome, the chemical composition of food, and their interplay in situ remains a daunting task to fully understand. However, recent advances in high-throughput sequencing, metabolomics profiling, compositional analysis of food, and the emergence of electronic health records provide new sources of data that can contribute to addressing this challenge. Computational science will play an essential role in this effort as it will provide the foundation to integrate these data layers and derive insights capable of revealing and understanding the complex interactions between diet, gut microbiome, and health. Here, we review the current knowledge on diet-health-gut microbiota, relevant data sources, bioinformatics tools, machine learning capabilities, as well as the intellectual property and legislative regulatory landscape. We provide guidance on employing machine learning and data analytics, identify gaps in current methods, and describe new scenarios to be unlocked in the next few years in the context of current knowledge.
Introduction
During the past two decades, the human microbiome has emerged as a biological system with the potential to significantly influence health and disease (Shreiner et al., 2015). Despite our limited understanding regarding its intricate relationship with the host and its environment (Foster et al., 2017), recent discoveries related to the human microbiome have opened new horizons in food science (Barratt et al., 2017), precision medicine (Wishart, 2016), and biotechnology (Taroncher-Oldenburg et al., 2018) among other fields. In parallel, advances in genomics and bioinformatics have provided inexpensive tools to acquire biological and clinical data, as well as the tools to translate the data into knowledge (Shoaie et al., 2015; Zeevi et al., 2015; Thaiss et al., 2016a; Korem et al., 2017; Baldini et al., 2018; Bauer and Thiele, 2018; Gilbert et al., 2018; Greenhalgh et al., 2018; Knight et al., 2018). Given these advances, the integration of diet, gut microbiome, and human health (DGMH) data has the potential to drive a paradigm shift in the way wellness states are measured, diseases are treated, products are designed, and health interventions are administered. To realize this potential, advances in knowledge are required in order to optimize the composition and metabolic dynamics of microbial communities in relation to desired health and performance outcomes—from dietary interventions and bioengineered products to lifestyle changes and the living environment (Figure 1).
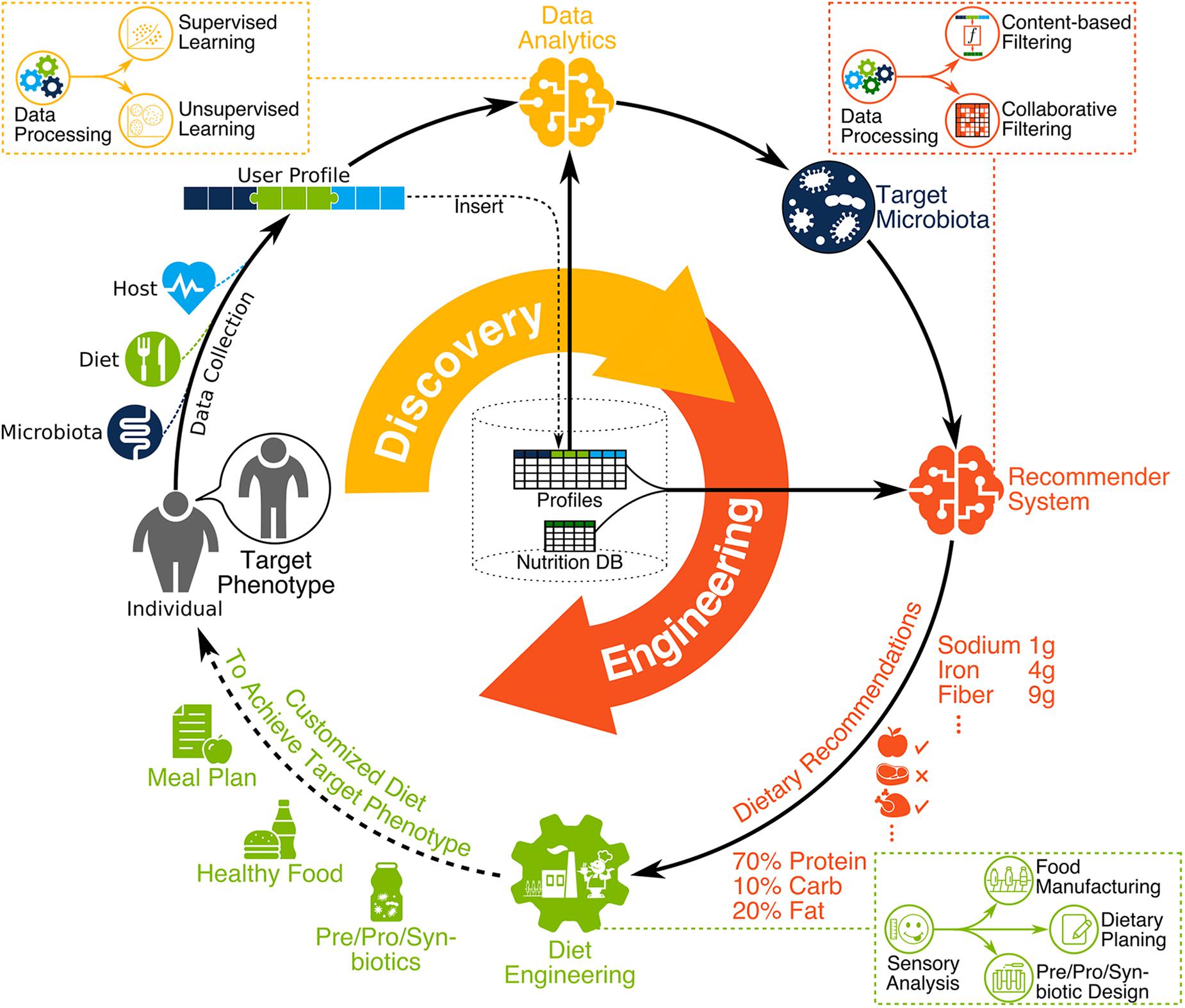
Figure 1. The vision for the next nutrition revolution involves microbiome-aware dietary planning and manufacturing. First, DGMH data is collected, homogenized, and stored, with any new user data integrated as part of a cohesive compendium. Then, DGMH data are analyzed (data analytics) to identify the functional characteristics and target microbiota, personalized to the individual and the desired phenotype. This includes data processing followed by supervised and unsupervised learning using a user profile compendium. Bioinformatic tools are used during data processing to extract meaningful information from raw high-throughput data such as metagenomic sequence reads. Then, the recommendation system provides dietary recommendations to help achieve target microbiota. This includes the integration of user profiles in a compendium along with nutrition DB proceeded by data processing then content-based and collaborative filtering. Finally, diet engineering is performed to create dietary products for the user. This includes the design of prebiotics, probiotics, synbiotics, manufactured food, and detailed dietary planning. In practice, taste and flavor of dietary products is very important to help users commit to any given diet, therefore sensory analysis should inform all dietary engineering efforts.
In this article, we summarize the research that has been done related to DGMH, with a focus on DGMH data and computational methods. We begin with a brief overview of key areas of current knowledge regarding the interaction between diet, health, and the gut microbiome. We then proceed to review the available data sources and the computational methods currently used, investigate the role that machine learning and artificial intelligence (AI) can play in this area, and summarize the intellectual property (IP) and legislative regulatory landscape. We conclude with recommendations to accelerate research and development efforts through better integration of research resources and tools, especially in the context of computational science and data analytics. A glossary of terms is provided in Table 6.
In general, the most recent articles reviewing the computational tools for microbiome data focusing on metagenomic data processing methods provide limited guidance on employing machine learning and data analytics and do not furnish recommendations in the context of DGMH data. The purpose of this manuscript is to help fill this gap by considering relevant literature, describing key challenges and potential solutions, and proposing a framework to improve the potential for research initiatives to accelerate progress in this exciting and potentially revolutionary field.
Current Knowledge: Gut Microbiota and Human Health
Emerging evidence suggests that the intestinal microbiota plays a significant role in modulating human health and behavior [see comprehensive reviews (Sherwin et al., 2018; Pereira et al., 2019; Zmora et al., 2019)]. Several studies have demonstrated that the human intestinal microbiota is seeded before birth (Stinson et al., 2019), and the mode of delivery influences the composition of the gut microbiota (Ferretti et al., 2018; Shao et al., 2019). The gut of a vaginally born newborn is enriched primarily with the vaginal microbiota from the mother, while a cesarean procedure results in the newborn’s gut microbiota being dominated by the microbiota of the mother’s skin as well as points of contact at the hospital (Dominguez-Bello et al., 2010). Microbial diversity is very dynamic during infancy and increases and converges to an adult-type microbiota by 3–5 years of age (Rodríguez et al., 2015). Evidence is also building to suggest that diet plays a key role in shaping the composition of microbial communities in the infant’s gut. For example, species of beneficial bacteria such as Lactobacillus and Bifidobacterium have been found to be dominant in breastfed infants while species of harmful bacteria such as Clostridium, Granulicatella, Citrobacter, Enterobacter, and Bilophila have been found to be dominant in formula-fed infants (Bäckhed et al., 2015). In addition, breastfed babies have higher gut microbial diversity compared to formula-fed babies, and several studies indicate that the diversity of bacteria is directly connected to health (Wang et al., 2008; Bäckhed et al., 2015). An unbalanced composition of the infant’s gut microbiota has been linked to several childhood diseases, including atopic dermatitis (AD) (Abrahamsson et al., 2012; Zheng et al., 2016) obesity (Yuan et al., 2016), and asthma (Thavagnanam et al., 2008).
The composition of the gut microbiota of an adult human is relatively stable (Shreiner et al., 2015), but several factors can influence it, including antibiotic treatment, long-term change in diet, microbial infections, and lifestyle (Willing et al., 2011; Conlon and Bird, 2015; Mathew et al., 2019; Zmora et al., 2019). Several health conditions are linked to changes in a stable and established gut microbiota such as Crohn’s disease (Manichanh et al., 2006), psoriatic arthritis (Scher et al., 2015), type 1 diabetes (de Goffau et al., 2013), atopic eczema (Wang et al., 2008), celiac disease (Schippa et al., 2010), obesity (Castaner et al., 2018), type 2 diabetes (Qin et al., 2012), and arterial stiffness (Menni et al., 2018). However, further research is required to establish direct links between these conditions and the composition of microbial communities in the gut. Interventions, such as oral administration of probiotics/prebiotics and fecal transplants, have shown efficacy on reducing the severity of some conditions, such as diarrhea, acute upper respiratory tract infections, eczema, Crohn’s disease, and ulcerative colitis (Anderson et al., 2012; Mansfield et al., 2014; Hao et al., 2015; Saez-Lara et al., 2015; Goldenberg et al., 2017; Delzenne et al., 2019). See Figure 2 for illustration of factors affecting the gut microbiota.
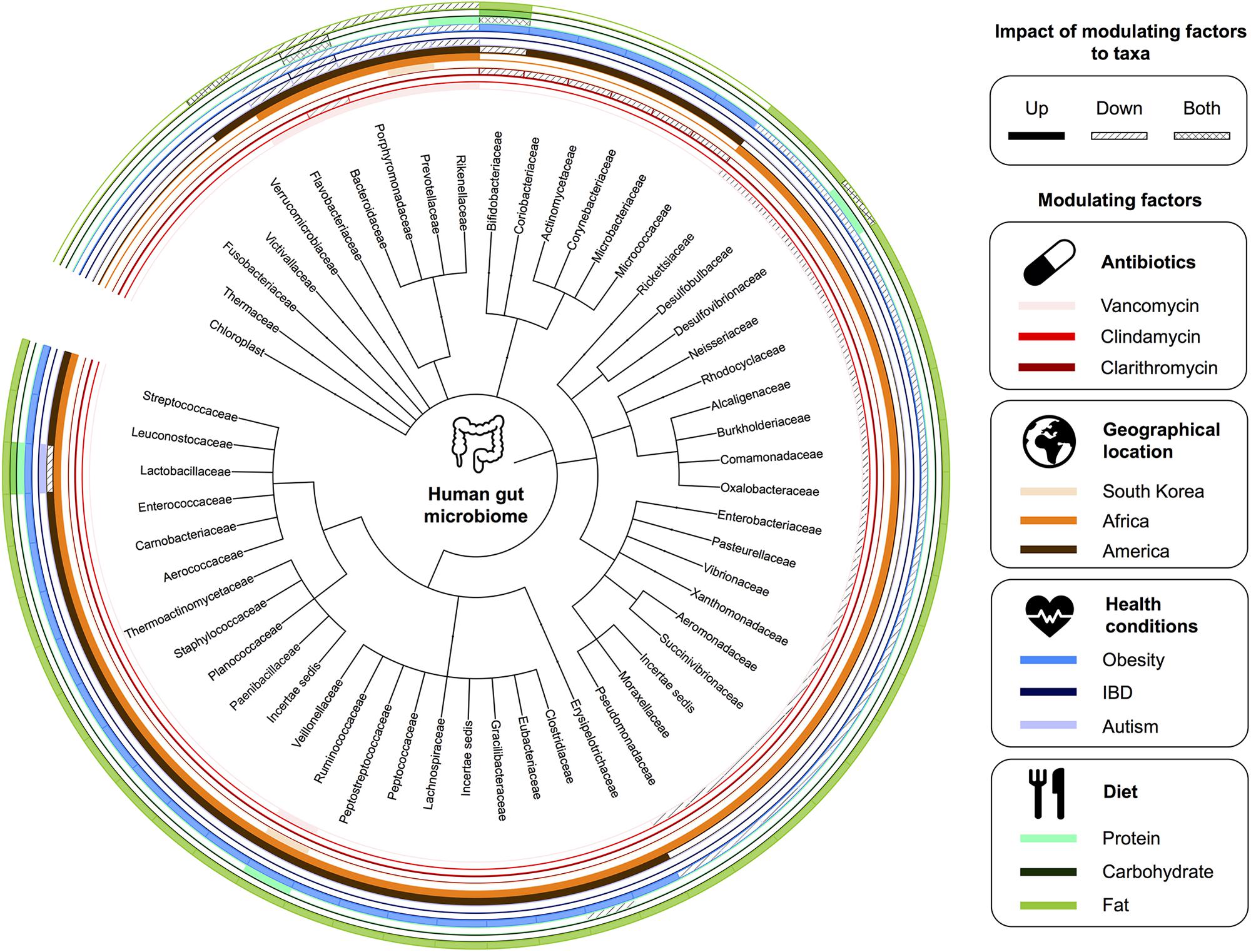
Figure 2. Factors affecting the gut microbiota. A summary of human gut microbiome taxonomy at the family level and the corresponding modulating factors.
Data
The increase in size and heterogeneity of information gathered by microbiome studies present great opportunities and serious data analysis challenges (Wooley et al., 2010), with many tools developed to address them (Breitwieser et al., 2017; Quince et al., 2017). These bioinformatics tools quantify low dimensional biological variables, such as the relative abundance of microbial species and metabolites, by using high dimensional data such as DNA sequence reads and mass spectrometry (MS) signatures as illustrated in Figure 3. Depending on data quality, sample size, and research hypothesis, different information dimensionalities are used, such as gene-level (Vatanen et al., 2018) or functional gene ontology terms (Brown et al., 2011).
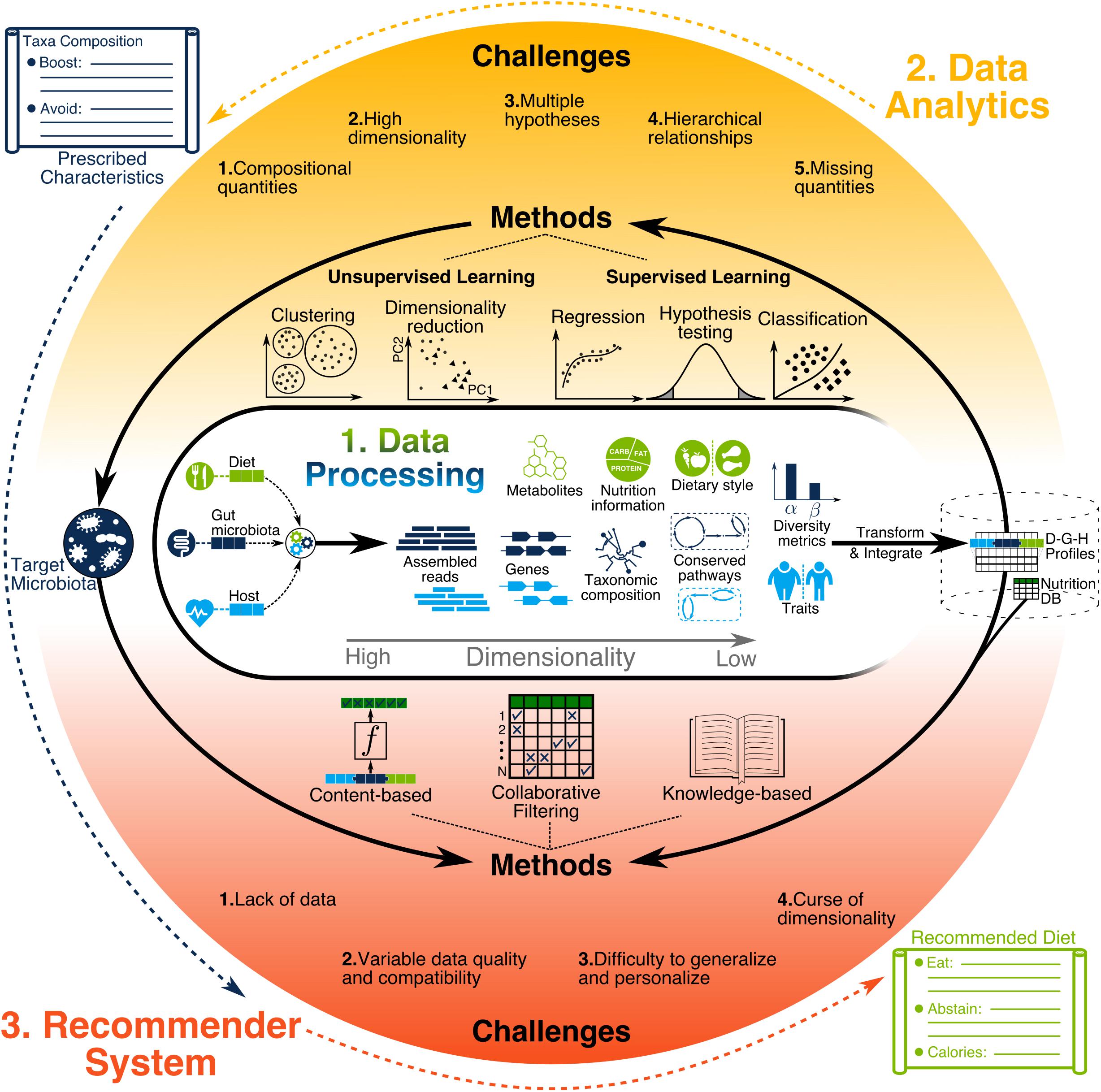
Figure 3. Illustration of data processing, data analytics, and recommendation systems. Data processing generates diverse types of information with different levels of resolution and dimensionality. Such information needs to be transformed and integrated across all users for building a compendium. Next, data analytics methods are used to discover the characteristics of target microbiota prescribed for individuals to achieve their health objectives. Finally, recommendation system methods use the compendium to find the dietary recommendations for helping individuals achieve the target microbiota.
Gut Microbiota Data
Functional characteristics of microbial communities can be revealed using high-throughput metametabolomics (Walker et al., 2014) and metaproteomics (Verberkmoes et al., 2009; Zhang et al., 2018) using MS technologies. Metagenomic and metatranscriptomic content of gut microbiota (which give rise to the functional characteristics) can be quantified using DNA sequencing. The most widely used approach for gut microbiota profiling is marker gene sequencing, which relies on sequencing counts of the hypervariable 16S genes to calculate Operational Taxonomic Units (OTUs) (Amann et al., 1995). Searching OTUs against reference databases such as Greengenes (McDonald et al., 2012) and SILVA (Quast et al., 2012) allows inferring relative taxa abundances in a microbiome sample (Langille et al., 2013). Whole-genome or shotgun metagenomics (Quince et al., 2017) is a recent technique that not only reveals the microbial community structure, but it can also quantify relative abundances of genes, taxa, conserved functional groups, or over-represented pathways. Within-sample (alpha) and cross-sample (beta) diversity of microbiome can be calculated with respect to genetic, taxonomic, functional, or metabolic pathway profiles of samples (Turnbaugh et al., 2009; Martiny et al., 2011; Huttenhower et al., 2012; Lozupone et al., 2012; Heintz-Buschart and Wilmes, 2017; Ranjan et al., 2018). The Shannon index, Chao1, and Abundance-based Coverage Estimator (ACE) are used to measure alpha diversity while UniFrac, weighted UniFrac, and Bray–Curtis calculate beta diversity. In longitudinal studies, the same measures of diversity, or more sophisticated eigenvalue-based analyses, can quantify the microbiota stability across timepoints (Lozupone et al., 2012; Relman, 2012; Coyte et al., 2015; Mehta et al., 2018). Jackknifing and bootstrapping are used to estimate the bias in diversity estimates, particularly when estimating the number of species (i.e., species richness) in samples (Smith and van Belle, 1984). Some of the most significant publicly available microbiome datasets are listed in Table 1.
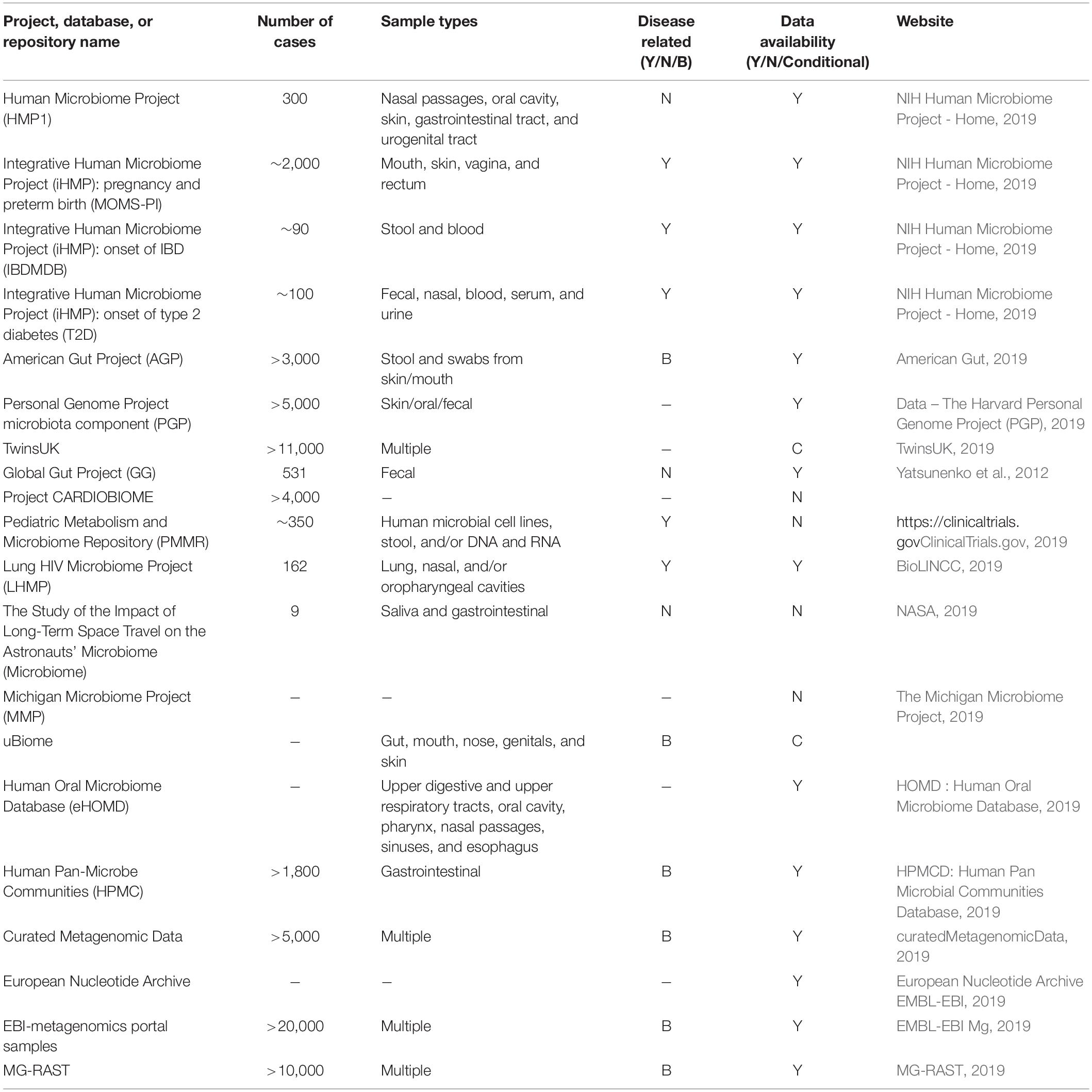
Table 1. Publicly available data from gut microbiota studies.
Diet Data
Various types of dietary information are collected in gut microbiome studies. This includes fine-grain information such as mass spectrometry (MS) signatures and metagenomic reads (Quinn et al., 2016) or coarse grain information such as dietary style [e.g., Western vs. Mediterranean diet (De Filippis et al., 2016)] from study participants. Diet data collection is often questionnaire-based, either through self-reporting or by a trained interviewer. For self-reporting, a food frequency questionnaire (FFQ) and 24-h dietary recall (24HR) can be used where participants report their dietary intake either every 24 h or over a longer period through a checklist of food items (Shim et al., 2014). A dietary record (DR) can also be used where data collection is done when food is consumed (e.g., using smartphones),which minimalizes reliance on participant’s memory. After data collection, the intake amount of macronutrients (fat, carbohydrates, and protein), micronutrients (vitamins and minerals), and food metabolites can be estimated by querying the food items against food composition databases such as the USDA food composition database (US Department of Agriculture and Agricultural Research Service, 2010) and the Canadian nutrient file (Canada, 2010). Note that microbiota of dietary intake can be characterized using metagenomic sequencing as reviewed previously, if not already defined [e.g., probiotics with predefined strains (Sánchez et al., 2017)]. Some studies perform metabolic characterization of dietary intake directly (Quinn et al., 2016), while others rely on pre-characterized metabolic profiles (Zhao et al., 2018). A significant limitation of any analysis is that food composition databases characterize only 0.5% of the known nutritional compounds (Barabási et al., 2019).
Host Data
Profiled host information types can be very high dimensional [e.g., high-throughput genome sequences (Hall et al., 2017)] or low dimensional [e.g., obese vs. non-obese (Thaiss et al., 2014; Cox and Blaser, 2015)]. Host genotype data can come from whole-exome sequencing (WES) (Gopalakrishnan et al., 2018) or a genome-wide association study (GWAS) (Bonder et al., 2016; Turpin et al., 2016). It can also be extended by predicting the whole-genome sequence for each individual through genotype imputation software (Howie et al., 2009), as done in several studies (Bonder et al., 2016; Goodrich et al., 2016; Rothschild et al., 2018). Host transcriptomic profiles can be assessed directly using microarrays (Schwartz et al., 2012; de Steenhuijsen Piters et al., 2016) and RNA-Seq (Thaiss et al., 2016b; Pan et al., 2018) or imputed using tools such as PrediXcan (Gamazon et al., 2015) with GWAS data. The genetic and transcriptomic profiles can be summarized into informative lower-dimensional features through gene ontology categories and metabolic pathways using databases such as MetaCyc (Caspi et al., 2017), KEGG (Kanehisa et al., 2011), Reactome (Fabregat et al., 2017), or GO (Antonazzo et al., 2017). Today, limited microbiome studies perform such analysis (Blekhman et al., 2015; Davenport et al., 2015; Dobson et al., 2015). Other important information such as age, gender, ethnicity, body weight, blood pressure, dietary restrictions, and diseases of a host organism can be extracted from medical records, surveys, and interviews.
Computational Analysis
There have been various reviews concerning microbiome data processing and analysis (Tyler et al., 2014; Tsilimigras and Fodor, 2016; Breitwieser et al., 2017; Quince et al., 2017; Knight et al., 2018). Here we focus on data analytics, machine learning, and AI-based recommendation system methods that enable microbiome-aware systems involving diet and wellness. We provide readers insight into important methods, challenges that arise, suggested solutions as well as blueprints of example scenarios to be used in their research. See Qu et al. (2019), Topçuoglu et al. (2019), and Zhou and Gallins (2019) for further explanation and examples of the machine learning methods discussed here.
Microbiome Data Processing Tools
There are a substantial number of publicly available microbiome data processing methods and pipelines that can generate the various types of data discussed. Table 2 provides a representative summary of such methods and pipelines. For 16S data, QIIME (Caporaso et al., 2010) and MOTHUR (Schloss et al., 2009) provide a wider range of options for the user compared to UPARSE (Edgar, 2013), but all are popular pipelines. QIIME 2 (Bolyen et al., 2019) is now emerging as a powerful replacement to its predecessors, partly due to its extensibility and support. For whole metagenomic sequencing, methods such as Kraken (Wood and Salzberg, 2014), MEGAN (Huson et al., 2016), MetaPhlAn2 (Truong et al., 2015), and HUMAnN (Abubucker et al., 2012) are used.

Table 2. A summary of highlighted methods and pipelines for microbiome data processing.
Challenges in Microbiome Data Processing
Growth in the variety and complexity of data processing tools presents opportunities but also significant challenges for new investigators. First, although best practices have been suggested (Knight et al., 2018), tools are still far from a fully automated user experience that would lead to reliable results. Second, microbial genomes with different abundances are sequenced together, making metagenomic assembly more challenging compared to single genome assembly where the sequence coverage is approximately uniform. Third, the number of uncharacterized microbes (known as microbial dark matter) exacerbates problems associated with unaligned and misaligned sequence reads. Fourth, evaluation of methodology and findings from different studies is difficult since each study may use a different method or a different implementation of the same method in their data processing pipeline. Fifth, data collection and integration of microbiome data from different studies are difficult because of many factors including differences in wet-lab library preparation (e.g., primers used), differences in sequencing devices and their settings (e.g., coverage), and non-uniform methods of formatting and storage for microbiome data and metadata. See Quince et al. (2017) for further discussion concerning microbiome data processing challenges.
Data Analytics and Machine Learning
Data processing is considered to be the step necessary for converting the raw data, such as metagenomics sequence reads, into biologically meaningful representations, such as OTU counts using bioinformatics tools, some of which are done in the sequencing device itself. Data analytics, start after the integration of processed sample data from various information sources (i.e., microbiota, diet, and host), as illustrated in Figure 3. In most cases, all samples are from a single study, which helps ensure consistency with respect to the experimental settings and data processing protocols used. Furthermore, limited resources force the researchers to narrow their data collection to particular information types in order to have sufficient statistical power for hypothesis testing. A recent increase in the number of microbiome studies with publicly available data has enabled cross-study data integration (Pasolli et al., 2016, 2017; Duvallet et al., 2017; Wang et al., 2018; Thomas et al., 2019; Wirbel et al., 2019). In such cases, extra precautions are necessary to minimize biases introduced by inconsistencies among datasets during data collection, sample preparation, sequencing, and data processing.
Challenges in Microbiome Data Analysis
A number of challenges arise when analyzing microbiome data, as summarized in Table 3. The first challenge is due to compositional quantities in microbiome data. Quantities such as the number of reads assigned to a given species, which can only be interpreted as proportions, are called compositional. These quantities cannot be compared directly across multiple samples. Conclusions should not be made based on the number of reads assigned to individual sample features (e.g., OTUs, genes, and functional groups) since they do not represent absolute abundances due to instrumental limitations (Gloor et al., 2017). Instead, the assigned number of reads should be converted to relative abundances and analyzed with that in mind. Some studies perform rarefaction to adjust for differences in library size due to unexhaustive metagenomic sampling. Although several pipelines provide this functionality, it has been found inadmissible for metagenomics microbiome studies as it discards many reads leading to decreased sensitivity in differential abundance testing (McMurdie and Holmes, 2014) and biased estimates for alpha diversity (Willis, 2019). The second challenge is due to the high dimensionality associated with OMICS data. Datasets in which items are characterized by a high number of features while the number of items is limited are called high dimensional. In microbiome studies, a limited number of individuals are characterized using many host, diet, and microbiome features leading to high dimensional datasets (Li, 2015). Dimensionality can be reduced by grouping OTUs into phylogenetic variables, regularization, or unsupervised dimensionality reduction (DR) (explained below). The third challenge is about testing multiple hypotheses in an exploratory analysis. It relates to the fact that, as the number of hypotheses increases, the chance of false discoveries also increases. This can be addressed by increasing sample size and p-value adjustment (explained below). The fourth challenge relates to hierarchical relationships amongst bacterial species due to their shared ancestors. Assumptions such as independence among samples may not hold, leading to wrong estimations of correlation (Felsenstein, 1985) and phylogeny-aware methods to address the issue. The fifth challenge is about missing quantities in sampled data. For example, when marker gene sequencing is used, quantities relating to the amounts of functional genes in the microbiome are not directly available (i.e., missing). Identifying functions of microbial organisms is important for understanding the gut microbiota. Such information can be estimated using metatranscriptomics data, which is often not available. Data imputation tools, such as PICRUSt (Langille et al., 2013), help to mitigate this through gene imputation based on annotated databases.
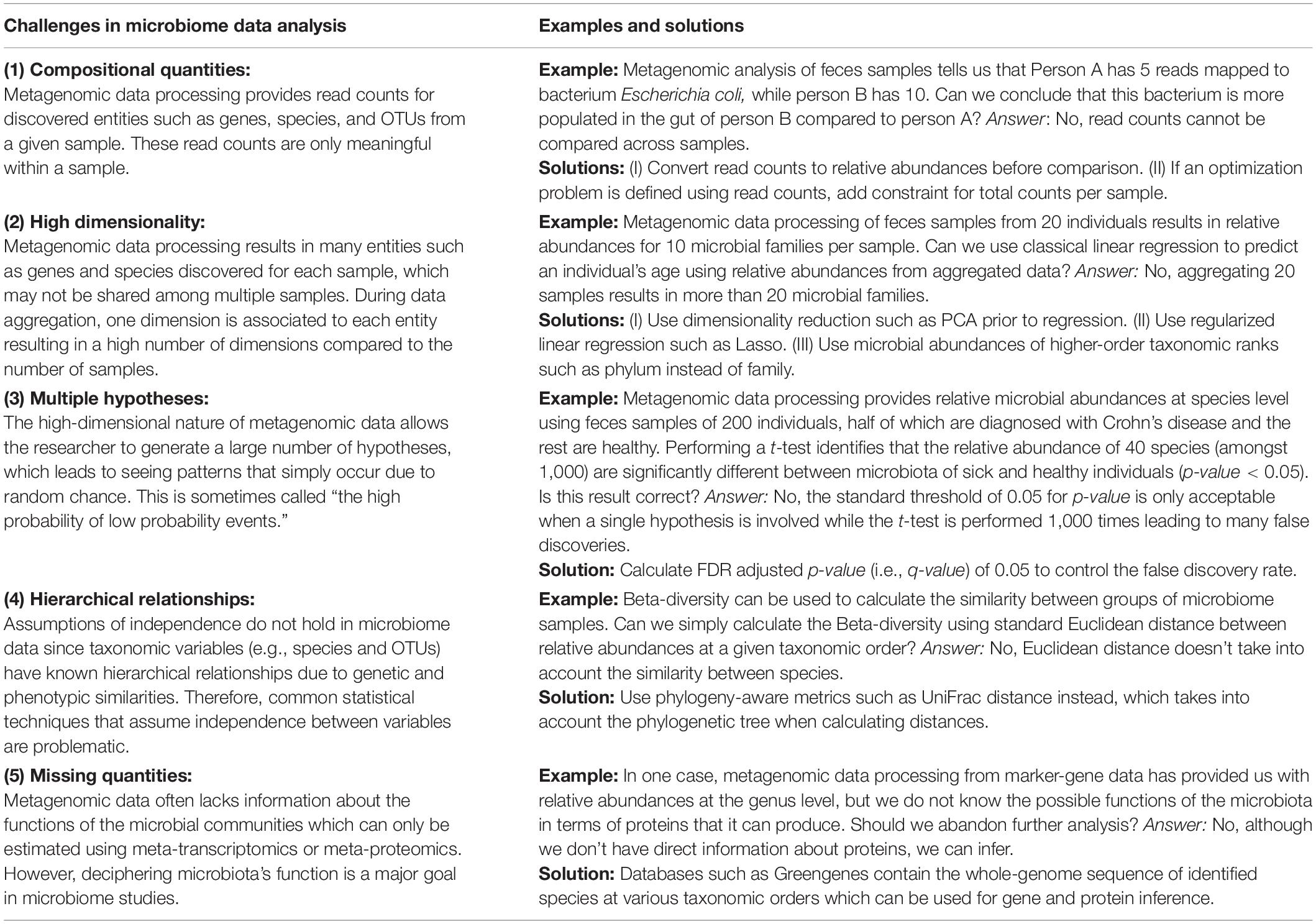
Table 3. Key challenges that arise in microbiome data analysis with examples and suggested solutions.
The methods for identifying microbiota characteristics associated with host phenotypes of interest can be categorized into two main groups, based on whether they use supervised or unsupervised learning. Supervised learning methods require labeled data, while unsupervised learning methods can be used when records are not labeled. More advanced methods include semi-supervised learning (Zhu, 2005), which takes advantage of both labeled and unlabeled data, and transfer learning (Pan and Yang, 2010), which transfers knowledge learned from one task to another, are not discussed here.
Supervised Learning Methods
Hypothesis testing and variation analysis
Analysis of variation may involve single or multiple variables. For a single variable hypothesis, the student’s t-test or non-parametric tests, such as Wilcoxon rank-sum or Kruskal–Wallis, can be used. For example, the t-test has been used to show that patients with ADHD have a lower alpha-diversity index of gut microbiota compared to healthy controls (Prehn-Kristensen et al., 2018). Non-parametric tests are good alternatives when the assumptions regarding the data being normally distributed do not hold. For example, the Wilcoxon rank-sum test is used on predicted pathway data, suggesting that enzymes in the “Glycan Biosynthesis and Degradation” pathway increase in summer when compared to winter (Davenport et al., 2014). In cases where a statistical test is repeated with different hypotheses (i.e., multiple hypothesis testing), the statistical significance should be adjusted by methods such as an FDR adjustment (i.e., q-value) (Benjamini and Hochberg, 1995) or Holm’s procedure (Rice, 1989).
When the hypothesis that is investigated contains multiple variables, MANOVA (Smith et al., 1962) or non-parametric alternatives such as PERMANOVA (Anderson, 2001) or ANOSIM (Clarke, 1993) can be used. The samples are first assigned to multiple groups (e.g., based on some feature values). The goal is to quantify how much this grouping can explain the distribution of values in any given sample feature (response variable). The simplest case is the popular method called analysis of variance (ANOVA), which considers a single response variable with a normal distribution. For instance, in a recent study, two bacterial phyla (Bacteroidetes and Firmicutes) were identified using ANOVA with different relative abundance in the microbiota of children living in a rural African village compared to European children (De Filippo et al., 2010). ANOVA can be generalized to multivariate analysis of variance (MANOVA), which can have multiple response variables. For example, it is used to investigate the overall difference in composition between the microbiota of children with Prader–Willi syndrome and children with simple obesity, before and after treatment (Zhang et al., 2015). In many cases, normal distribution assumptions do not hold; hence, non-parametric methods are used. In one study, PERMANOVA is used to detect taxonomic differences in the microbiota of patients with Crohn’s disease when compared to healthy controls (Pascal et al., 2017).
Regression and correlation analysis
A general understanding of the extent of association among pairs of variables can be achieved using correlation analysis. Correlation metrics measure different types of relationships. For example, the Bray–Curtis measures abundance similarities (Bray and Curtis, 1957), the Pearson correlation coefficient quantifies linear relationships, and the Spearman correlation coefficient quantifies rank relationships (Spearman, 1904). In (Weiss et al., 2016), the authors perform a simulation-based comparison on a range of correlation metrics for microbiome data. Metrics such as SparCC (Friedman and Alm, 2012) and LSA (Ruan et al., 2006) perform particularly better as they are designed to capture complex relationships in compositional microbiome data. For example, SparCC is used for analyzing the TwinUK dataset to identify bacterial taxa whose abundances are influenced by host genetics (Goodrich et al., 2014). This was done by creating a correlation network between microbial families based on their intraclass correlation. More recently, the phylogenetic isometric log-ratio (PhILR) transform has been introduced (Silverman et al., 2017) to transform compositional data into non-compositional space where standard data analytic techniques are applicable. Usage of such transformations should be limited to features that are compositional and phylogenetic in nature.
Regression methods aim to predict the change in one continuous variable using other variables. Correlation analysis can be considered a special case of regression with a single input variable. Standard linear regression can be used for various DGMH predictive tasks. However, when variables relate to OTU abundances, the typical assumptions of a linear relationship, normal distribution, and dependence do not hold. For example, when the goal is to predict the composition of OTUs [normalized for summing up to one (Tyler et al., 2014)], zero-inflated continuous distributions are used. Often a two-part regression model is used where part I is a logistical model to calculate the probability that the given OTU is present. Part II is a generalized linear regression using beta distribution to predict relative abundance assuming the presence of OTU in the sample (Ospina and Ferrari, 2012; Chen and Li, 2016; Peng et al., 2016). Phylogenetic comparative methods (PCMs) such as phylogenetic generalized least squares (PGLS) are used to control for dependence among observations given the phylogenetic hierarchies (Washburne et al., 2018). Ignoring the phylogenetic ancestry of microbial species can increase the chance of false discovery in regression models (Felsenstein, 1985). PCMs are not widely used in microbiome studies today, which may be one reason for a high number of false positives that can be alleviated by using them (Bradley et al., 2018).
Canonical correlation analysis (CCA) can be used (Hotelling, 1992) to investigate the correlation between two groups of variables (e.g., abundances of microbiome OTUs and metabolites). CCA finds linear transformation pairs that are maximally correlated when applied to data while ensuring orthogonality for different transformation pairs. The original CCA, however, fails for high dimensional microbiome data when the number of variables exceeds the number of samples. This can be addressed using regularization, giving rise to sparse CCA methods (Witten et al., 2009). For example, a sparse CCA is applied to investigate correlations between the gut microbiome and metabolome features in type 1 diabetes (Kostic et al., 2015).
Classification
In supervised classification, the goal is to build a predictive model (classifier) using labeled training data. The labels can have binary or categorical values (in contrast to regression where labels are continuous and numerical). In one study, a classifier was built to predict the geographical origin of sample donors using relative OTU abundances estimated from 16s rRNA gut samples (Yatsunenko et al., 2012). This was done using the method called Random Forests (RF), which constructs an ensemble of decision trees (Breiman, 2001). In a different study, the classification task was to identify healthy vs. unhealthy donors given relative OTU abundance data (including species level) coming from shotgun metagenomics sequencing of the gut (as well as other body sites) (Pasolli et al., 2016). In addition to RF, they used the support vector machine (SVM) classifier, which is a powerful method for building generalizable and interpretable models and is mathematically well understood (Suykens and Vandewalle, 1999). In their study, RF classifiers performed better than SVM except in a few datasets. Both RF and SVM have built-in capability to deal with overfitting issues that arise in high-dimensional datasets. RF achieves this using an ensemble-based technique where the prediction is made based on predictions from many trained classifiers. In SVM, parameters of the predictive model are constrained based on a priori defined criteria. Note that constraining the model parameters is often mathematically equivalent to regularization (Scholkopf and Smola, 2001). In both cases, the objective is to minimize the value of a loss function that calculates the overall error in model predictions. When regularization is used, the loss function not only depends on prediction errors but also on the magnitude of model parameters. For example, in L1 regularization, the absolute values of model parameters are scaled and added to the loss function. Therefore, when two models have a similar error, the model with smaller parameter values will be selected during training. L1 regularization is commonly used for feature selection by picking only the non-zero features of the trained model because such a model achieves a low prediction error while using a subset of features.
Artificial neural networks (ANN) can also be used for classification and are shown to outperform other techniques in many areas of biology (Kim et al., 2016, 2017; Singh et al., 2016; Eetemadi and Tagkopoulos, 2018) as well as computer vision and natural language processing, to name a few (LeCun et al., 2015). Recently, a new ANN-based method called Regularization of Learning Networks (RLN) was designed and evaluated microbiome data. RLN provides an efficient way for tuning regularization parameters of a neural network when a different regularization coefficient is assigned for each parameter (Shavitt and Segal, 2018). They use RLN to predict human traits (e.g., BMI, cholesterol) from estimated relative OTU abundances and gene abundances. We expect the development of new classification methods that can deal with the aforementioned challenges arising in DGMH data by considering the biological phenomenon, properties of measurement instruments, and upstream data processing pipelines.
Unsupervised Learning Methods
Dimensionality reduction
High-dimensional datasets can provide a high resolution and multifaceted view of a phenomenon such as gut microbiota. Predictive performance in data analytics can increase significantly given such data. Many data analytics methods, however, fall short when presented with high-dimensional data that necessitates using DR. Once dimensionality is reduced, data visualization and analytics become more accessible. Principal component analysis (PCA) is one of the most widely used DR methods. It replaces the original features with a few uncorrelated features called principal components (PCs), which are linear combinations of the original features and preserve most of the variance within the data. In one study, PCA was applied to predicted abundances of about 10 million genes from the gut microbiota of donors (Li et al., 2014). Reducing dimensionality from 10 million to two dimensions only enabled visualization of data on a standard two-dimensional scatter-plot (i.e., PCA plot) showing a clear distinction between the microbiota of Danish and Chinese donors. In another study, the top five PCs of individual bacteria’s genome (sequenced from infant fecal samples) were used to create a classifier for predicting antibiotic resistance (Rahman et al., 2018).
The relationships among features in a microbiome study can be used in DR, giving rise to various factor analysis (FA) methods we review here briefly. Multiple factor analysis (MFA) is an extension of PCA that considers predefined grouping of features during DR to ensure equal representation for all groups of features in derived PCs (Abdi et al., 2013). In one study (Robertson et al., 2018), MFA is used for simultaneous 2D visualization of host and microbiome features (see Morgan et al., 2012; Raymond et al., 2016 for other examples). Exploratory factor analysis (EFA) is used to identify unobserved latent features called factors to explain the correlations among observed features (Yong and Pearce, 2013). Factors that are identified by EFA are uncorrelated to each other similar to PCs in PCA; however, PCs are used to explain overall variance instead of correlations. EFA has been used in a recent study to extract four factors explaining the correlations among 25 top taxa for studying the association of microbiome with early childhood neurodevelopmental outcomes in 309 infants (Sordillo et al., 2019). Confirmatory factor analysis (CFA) and structural equation modeling (SEM) can be used to examine the extent to which a hypothesized model of latent features and their relationships with observed variables are supported by the data (Schreiber et al., 2006). In a recent study, a theoretical framework is proposed and examined using CFA to model the influence of maternal and infant factors on the milk microbiota (Moossavi et al., 2019). The R packages lavaan (Rosseel, 2012) and FactoMineR (Lê et al., 2008), as well as the IBM SPSS software (IBM Corp, 2013), are widely used for factor analysis.
Another related method is principal coordinate analysis (PCoA), also called multidimensional scaling (MDS) (Kruskal, 1964), which is commonly employed for 2- and 3-dimensional visualization of beta diversity. It can deal with situations where distances between individual feature vectors from samples cannot be used directly (e.g., due to significant sparsity and phylogenetic relationships). PCoA takes a matrix of distances among samples (e.g., UniFrac distance between OTU abundances of a pair of sample donors) as input. It then assigns new coordinates such as PC1 and PC2 to each sample such that the Euclidean distances in the new coordinate are similar to the ones in the matrix. For example, PCoA was applied given UniFrac distances between OTU abundances (from 16S rRNA samples) from the gut microbiota of donors (Yatsunenko et al., 2012). Two-dimensional visualization using PC1 and PC2 showed that the gut microbiota of donors who lived in the United States is distinct from the gut microbiota in donors living in Amerindian and Malawian villages.
Linear discriminant analysis (LDA) is also a DR technique, although supervised and closely related to regression and ANOVA. Unlike PCA and PCoA, it requires class labels. It generates new features that are linear combinations of the original ones while separating samples with respect to their class labels. In one study, LDA was used to distinguish gut microbiota samples based on diet but not for DR (Paulson et al., 2013). Successful usage of LDA for high dimensional microbiome data may require regularization to account for overfitting as similarly used for high-dimensional microarray (Guo et al., 2006).
The optimal amount of reduction in dimensionality (e.g., the number of principal components) varies given the data and the task downstream. For data visualization tasks, it is largely constrained by the limitations of human visual perception (three dimensional). For downstream supervised learning tasks, we are often interested in the maximum amount of DR without a significant decrease in predictive power. This is showcased in Bartenhagen et al. (2010), where the impact of the amount of DR on classification performance is evaluated for gene expression data.
Cluster analysis
Similar microbial communities are expected to exhibit analogous effects on the host organism (Gould et al., 2018). Once a similarity measure is defined, various cluster analysis methods can be used to find groups of samples with similar microbiota. In one study, three robust microbiota clusters (called enterotypes) were identified using cluster analysis from 16s rRNA data of fecal samples (Arumugam et al., 2011). It was later shown that such clustering results are not only sensitive to data but also to choices made during analysis (Koren et al., 2013). We enumerate four important choices impacting cluster analysis results (other than upstream data processing). First is the distance measure. Standard distance metrics such as the Euclidean and Manhattan distance are simple, well understood, and supported in many clustering libraries. Applicability of such metrics depends on prior compositionality aware transformations such as ILR. Beta-diversity metrics such as weighted and unweighted UniFrac distances are designed for microbiome analysis considering compositionality and phylogenetic dependencies of microbiome data. Researchers should pay attention to the properties of the distance metric used in order to better understand the clustering results. Second is the clustering algorithm. Algorithms such as Partition Around Medoids (Kaufman and Rousseeuw, 1987) and Hierarchical Clustering (Murtagh and Contreras, 2012) can employ various distance metrics. Others, such as k-means, are tied to a single distance measure but computationally less demanding. Third is the number of clusters. Clustering algorithms often require the number of clusters to be provided as input. When unknown, the number that provides higher cluster scoring is picked. Prediction strength (Tibshirani and Walther, 2005), silhouette index (Rousseeuw, 1987), and Calinski–Harabasz (Caliñski and Harabasz, 1974) are popular cluster scoring metrics. Fourth is the method used to identify the robustness of clustering results. Often a cluster scoring metric that is not used to identify the number of clusters is used as a robustness measure. Recent studies consider the effect of the above choices during cluster analysis to better understand how results can be generalized (Hildebrand et al., 2013; Costea et al., 2018).
The integration of data from disparate omics data types (also called integrative omics) and other heterogeneous metadata enables a more comprehensive look into the underlying biology (Karczewski and Snyder, 2018). Integrative omics data analysis methods have been categorized into three types (Kim and Tagkopoulos, 2018). First is data-to-data, where disparate data types are analyzed together. For example, CCA can be used to investigate the correlations between metagenomics and metabolomics data, as discussed before. Second is data-to-knowledge, where the knowledge gained from analyzing some data types are used to inform analysis of other data types. For example, a metagenomics analysis of colon cancer patients can lead to further investigation of candidate genes using targeted proteomics analysis. Third is knowledge-to-knowledge, where the data types are initially analyzed separately, but the acquired knowledge is integrated together afterward to either identify hypotheses that are supported by multiple data types or create a more complete view of a given phenomenon. For example, differentially expressed genes and differentially abundant metabolites in the digestive tract of patients with Crohn’s disease can be used together for narrowing down pathways involved in disease etiology. See Huang et al. (2017), Karczewski and Snyder (2018), Kim and Tagkopoulos (2018), and Jiang et al. (2019) for comprehensive reviews.
Recommendation Systems and Artificial Intelligence
The human microbiome is referred to as “our second genome” and has a major influence on our health (Grice and Segre, 2012). Although it is known for its resilience (Lozupone et al., 2012; Relman, 2012), unlike the human genome, it has considerable plasticity hence providing ample opportunities in the design of new types of food, medical interventions, and dietary recommendations (Gentile and Weir, 2018). Despite recent progress in microbiome research, switching from population-wide dietary recommendations to microbiome-aware recommendations is not yet realized. See Table 4, for a representative summary of recent microbiome-aware diet recommendation studies. Once a personalized healthy target microbiome is identified using data analytics methods, a recommendation system (RS) can utilize this information to suggest the path toward establishing it in the host and ensuring the health benefits. One approach is to use a knowledge-based RS where recommendations are made using a limited number of approved drugs and dietary prescriptions. Although this would be a good starting point, such a system would be limited in its ability to provide precise and personalized recommendations that usually need a platform that can create new products or processes on a case-by-case basis. Recent studies simulate a virtual gut microbiome by integrating known metabolic pathways of microbial species with the individual’s microbiome and diet (Shoaie et al., 2015; Baldini et al., 2018; Bauer and Thiele, 2018; Greenhalgh et al., 2018). Such mechanistic modeling is very promising, however, it is currently hindered by numerous challenges, such as incomplete characterization of an individual’s gut and metabolic pathways of their microbiome. There is considerable research on AI-based RS related to food, drug design, and health (Tran et al., 2017; Suphavilai et al., 2018), but its application with microbiome data is in its early stages (Zeevi et al., 2015; Thaiss et al., 2016a; Korem et al., 2017). Commercial investments in this area have already started, with companies such as UBiome and DayTwo using 16S rRNA technology to provide insights into our personal microbiota and suggest dietary recommendations.

Table 4. Highlighted microbiome-aware diet recommendation studies.
Recommendation system is defined as “any system that guides a user in a personalized way to interesting or useful objects in a large space of possible options or that produces such objects as output” (Burke, 2002). Microbiome-aware diet recommendations can be generated from knowledge-based, content-based, or collaborative filtering, as described next.
Knowledge-Based Recommendation Systems
An ideal knowledge-based RS would be based on in silico models that can correctly simulate an individual’s gut. It requires proper characterization of the gut microbiome, human intestinal cells, intestinal and dietary metabolite concentrations, their interactions through metabolic pathways, and realistic objective functions for modeling such complex dynamics. Such a knowledge-based RS was devised in a recent study involving 28 patients with Crohn’s disease and 26 healthy individuals (Bauer and Thiele, 2018). Researchers integrated genome-scale metabolomic reconstructions (GENREs) of 818 microbes from http://vmh.life (Noronha et al., 2018) with the individual’s microbiome abundances after metagenomic data processing in the R package BacArena (Bauer et al., 2017). Their in silico simulations provide personalized metabolic supplements for improving patient’s SCFA levels. Earlier studies have created a metabolic model of the gut microbiome on a smaller scale (Shoaie et al., 2015). See Magnúsdóttir and Thiele (2018) for a comprehensive review. Despite their promise, there are several challenges for the application of such knowledge-based RSs. The first challenge is the limited availability and accuracy of GENREs for gut microbes. A recent study has identified 1,520 unique microbes in the human gut (Zou et al., 2019), while the number of microbes that have GENREs is only 818 (Noronha et al., 2018). In one study (Tramontano et al., 2018), 75% of the GENREs required updates [from previously constructed GENREs (Magnúsdóttir et al., 2017)] so that in silico simulations could recapitulate growth on new media. This suggests that in silico GENREs of the gut microbiome are far from complete, however, progress is being made toward closing this gap. The second challenge is the metabolic characterization of the media inside the intestine on which gut microbes grow. This includes identifying the dietary metabolites available to microbes at different sites in the gut, which necessitates meticulous dietary data processing. The third challenge relates to the computational complexity of in silico simulations, which increases as host and microbial GENREs become more comprehensive. Although more challenges can be enumerated, their inclusion here would go beyond the scope of this article.
Content-Based Recommendation Systems
In content-based RSs, the recommendations are made based on the item’s content (often characterized using item features). This is in contrast to collaborative filtering RSs where recommendations are based on preferences of other users for each item. In one landmark study (Zeevi et al., 2015), authors use a content-based RS for meal recommendations with the goal of improving post-meal glucose levels. Each meal is first characterized based on its nutritional profile (macronutrients and micronutrients). Then a regression model is trained to predict post-meal glucose level based on the meal’s nutritional profile, the individual’s microbiome features, and other personal information. For each new user and meal, post-meal glucose levels are predicted by the model, and the meal with the minimum post-meal glucose level is recommended to the user. The same methodology is used in a later study using only microbiome features of individuals to predict post-meal glucose levels in a bread-type recommendation system (Korem et al., 2017). Several challenges arise when building content-based RSs. The first challenge is variable data quality and compatibility. When a group of users (or items) are overrepresented in the data, the predictive model tends to be biased toward their favorite items. As a result, the quality of recommendations will be highly variable. Stratified sampling can be used to alleviate this issue. The second challenge is difficulty in generalizing and personalizing recommendations, particularly when feature vectors are not informative for predictions (also relevant to the “missing quantities” challenge mentioned in Table 3). This is in contrast to collaborative filtering RSs, where latent features are learned instead of being defined a priori. Hybrid RS methods are designed to take advantage of collaborative filtering RSs to address such inherent challenges in context-based RSs (and vice versa) (Burke, 2002). For an extensive review of context-based RSs, methods see (Lops et al., 2011).
Collaborative Filtering Recommendation Systems
In collaborative filtering RSs, each user is characterized by the items (foods or ingredients here) they have previously rated, bought, or generally acted upon. Recommendations are given based on the idea that users who assign the same rating to existing items are expected to have a similar rating profile for all items. Matrix completion is one of the most popular collaborative filtering methods (Su and Khoshgoftaar, 2009; Ekstrand et al., 2011). User-assigned scores are first organized in a sparse matrix where columns correspond to different items and rows to various users. In cases where most users only have evaluated a few items, most of the matrix remains empty. Matrix completion fills the rest of the matrix through the similarities discovered amongst users and items. See Su and Khoshgoftaar (2009) and Ekstrand et al. (2011) for a comprehensive review. Collaborative filtering RSs have not been used for microbiome-aware food recommendations. We describe an example here to showcase how it can be used. Consider a matrix where each column corresponds to a dietary plan and each row to a person—a specific value can represent gut microbiome alpha diversity during the time which the user followed a particular dietary plan. Assuming that each person has only tried a few dietary plans, most of the matrix will be empty. Here we can use matrix completion to fill the matrix with predicted alpha diversities to create a complete matrix. This can be used to recommend dietary plans for a person with the goal of maximizing gut microbiota diversity. Several challenges arise in collaborative filtering RS. The first challenge is the lack of data for new users (“cold-start”). Note that the recommendations rely on similarities among users, while new users have not tried any of the items available in the database. The second challenge is the curse of dimensionality. As the number of items increases, the chance of having user scores for the same item combinations decreases, hence items and users become equally dissimilar (also relevant to the “high dimensionality” challenges in Table 3). In such cases, hybrid RS can be used. Next, we bring up a few example scenarios.
Example Scenarios
We discussed various data analytics and recommendation system methods for microbiome discovery and diet engineering, as illustrated in Figures 1, 3. Applicability of each method depends on research objectives and data availability. Here we explain particular scenarios illustrated in Figure 4 as blueprints for integrating relevant techniques in a single pipeline. In scenario A, the goal is to identify metabolic pathways that are enriched in the gut microbiome of healthy adults using 16S rRNA data (see Duvallet et al., 2017; Thomas et al., 2019; Wirbel et al., 2019 for similar works). In scenario B, the goal is to provide recommended probiotic intake for supporting a healthy gut microbiome. First, the study participants would be profiled based on the probiotic products they consume (each containing specific OTUs) as well as their gut microbiome. Next, microbiome scores will be calculated for each participant based on the distance between enriched pathways of their microbiome and the target healthy microbiome. Then a regression model is trained to predict microbiome scores based on OTU intakes. Finally, the OTU intake concentration that is predicted to have an optimal microbiome score would be used as the recommended probiotic intake. In scenario C, the goal is to identify optimal diets for health, performance, and disease. A compendium needs to be built following a consistent data collection and processing pipeline for study participants. The compendium serves the training data necessary for building machine learning models to predict health metrics such as post-meal glucose level (Zeevi et al., 2015; Korem et al., 2017) or post-dieting weight regain (Thaiss et al., 2016a). The predictive models can then be used as the key part of a recommendation system by identifying the expected impact of a given diet on health for new individuals. In scenario D, the goal is to recommend metabolic supplements needed by an individual’s microbiota to secrete vital compounds. First, OTU abundances of each individual are identified using a metagenomic data processing pipeline. Then, individual gut metabolic pathways are reconstructed using online resources such as the Virtual Metabolic Human database (Noronha et al., 2018). Finally, constraint-based reconstruction and analysis (COBRA) tools (Bauer et al., 2017; Baldini et al., 2018) are used to perform in silico simulations of GENREs to identify metabolic intake requirements to secrete vital compounds of interest. This mechanistically sound approach has been used in a few recent studies (Shoaie et al., 2015; Bauer and Thiele, 2018).
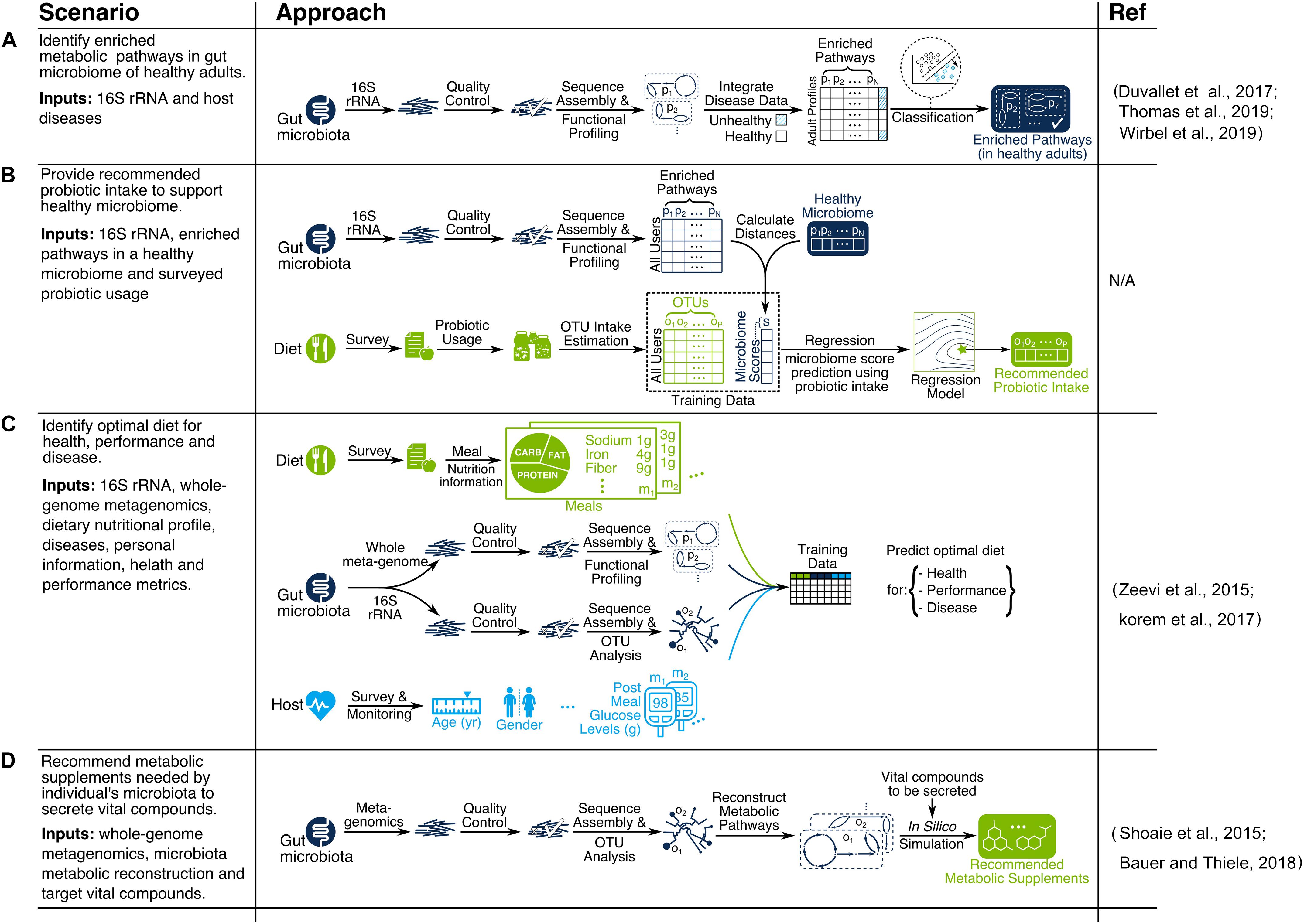
Figure 4. Examples of microbiome-aware diet recommendation pipelines for scenarios (A–D).
Intellectual Property Development
The potential application impact generated by research on the relationship between the gut microbiome and diet can be visualized by the abundant number of patent applications on the topic, as well as more generally in the field of microbiome and health research. A search for “gut microbiome” and “diet” returns over 2,500 patents on Google, deposited by universities, institutes, and companies such as MicroBiome, Microbiome Therapeutics, Gutguide, Whole Biome Inc., UBiome, and others, from as early as 2004. However, it is important to note that most of these hits are less than a decade old, demonstrating the relatively early stages in which this area still resides. The exponential growth in patent applications related to the microbiome since 2007 correlates to a similar curve for the academic publications in the same period (Fankhauser et al., 2018).
One of the earliest available patent applications (US20050239706A1) related to the topic of the microbiome and nutrition describes methods to regulate weight by manipulating the gut microbiome. Additional patents also aim to use the gut microbiome as a therapeutic target, monitoring and altering the composition with the goal of manipulating the host phenotype such as weight gain/loss and obesity. In general, weight management with the manipulation of the gut microbiome (US20110123501A1 and US20100172874A1) appears as a favored theme for early patent applications in the area of microbiome and diet. Several patents describe novel probiotics and their uses (WO2007136553A2), often relating them to specific target phenotypes such as weight loss (EP2178543B1, US9371510B2, US9113641B2, EP2216036A1, EP2296489A1, and WO2010091991A1). Multiple applications for probiotics focused on weight loss were deposited by Nestec SA, which offers research and consulting services to the food company Nestlé S.A.
With the development of computational techniques to analyze larger datasets, and more research on the relationship of the microbiome and the host homeostasis and disease, patent applications related to gut microbiome and diet have subsequently extended to other health conditions beyond obesity and weight control. Among the newest patent applications related to the gut microbiome and diet is a patent describing the characterization, diagnostics, and treatment of a locomotor system condition based on microbiome data (US20170372027A1). Other applications include metagenomic methods specific for the comparison of healthy individuals and those with gut dysbiosis (WO2017216820A1), diagnostic tools for Crohn’s disease, inflammatory bowel disease, irritable bowel syndrome, ulcerative colitis, and celiac disease using microbiome and other types of data (US20170286620A1), and devices such as capsules to acquire and monitor microbiome and metabolites in the gut (US20170281091A1). Research on the gut–brain axis relationship also resulted in several applications aimed at monitoring and manipulating the gut microbiome to enhance cognition or treat mental-health conditions (WO2017171563A1 and WO2017160711A1). A recent and thorough review of patents related to the microbiome identified cancer diagnosis and treatment and CRISPR technology as recent trends in the field (Fankhauser et al., 2018). Table 5 shows a summary of highlighted patents relating to DGMH.
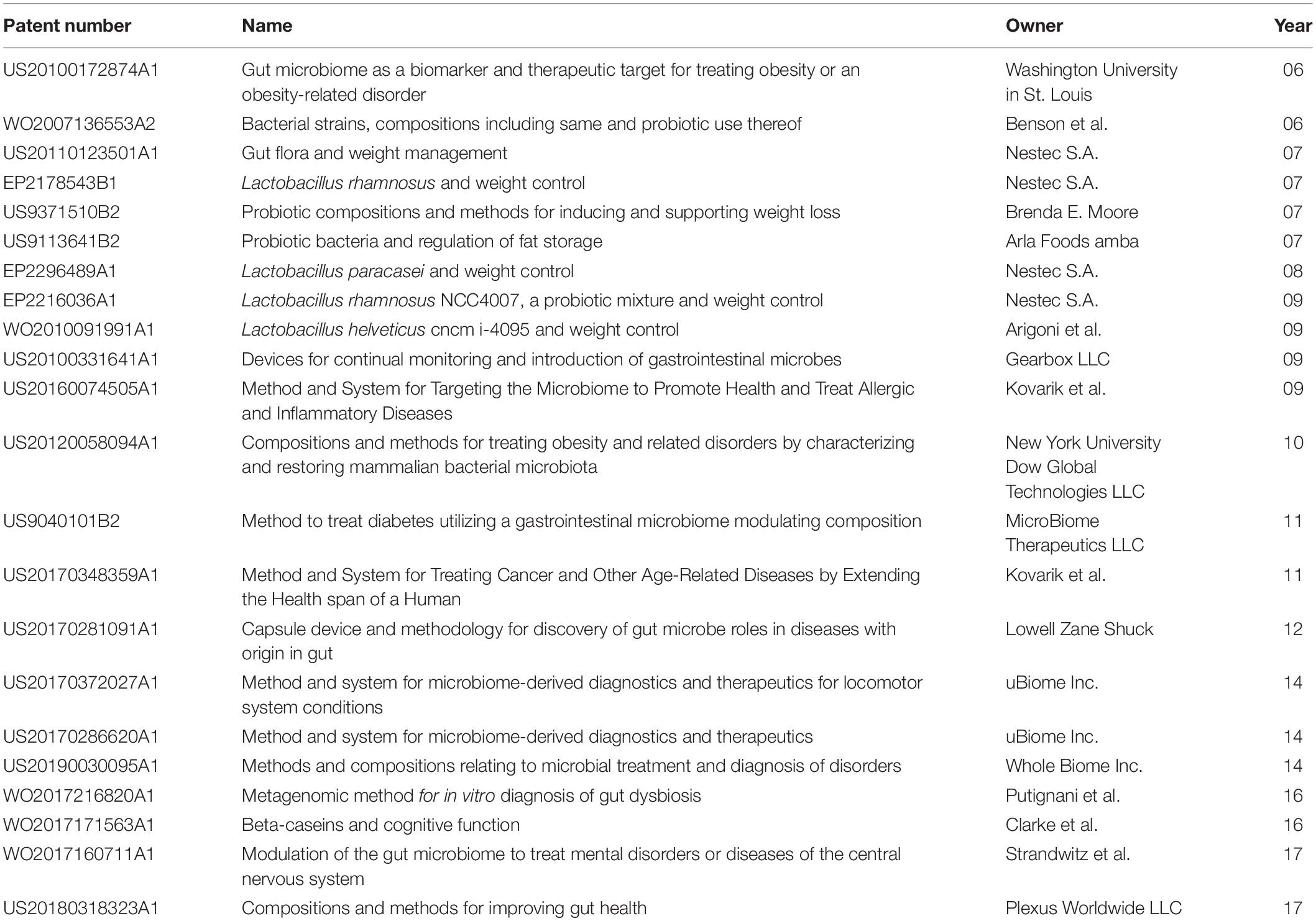
Table 5. Highlighted patents relating to diet, gut microbiome, and human health.

Table 6. Glossary of terms.
Even though there is already a considerable number of patent applications for technologies aiming to manipulate the gut microbiome for multiple health conditions, regulatory legislation has not yet become specific to deal with the new scientific advances in the field. In Europe, the European Food Safety Authority (EFSA) is responsible for regulating and approving food products with health claims, including probiotics, while in the United States, the Food and Drug Administration (FDA) assumes a similar role. Legislation and regulatory aspects are changing in an attempt to keep up with the ever-evolving field. Recently, the FDA has released a statement (Food and Drug Administration, 2018) clarifying existing regulations and announcing the intention to work closely with the United States National Institutes of Health to ensure public safety. Currently, there is no probiotic approved to be marketed in the United States as a live biotherapeutic product, defined by the agency as a “biological product other than a vaccine that contains live organisms used to prevent or treat a disease or condition in humans” (Food and Drug Administration, 2016,2018). This means that, even though probiotics are legally available as dietary supplements or food ingredients, they cannot yet have claims to cure, treat, or prevent any diseases per current regulation (Food and Drug Administration, 2018), since those claims are reserved for drugs. Classification of food ingredients targeting the microbiome, but not composed of living organisms, microbiota-directed foods or MDFs, prebiotics, and dietary fiber, is also challenging based on the available legislation. Depending on the health claims, such products can fall under the categories of drugs or dietary supplements, which have different requirements for approval (Green et al., 2017).
Conclusion
Significant advances in microbiology, genomics, analytical chemistry, computational science, bioinformatics, and other critical disciplines have begun to converge such that it is possible to foresee a new era of health and nutrition research enabling the design of food products capable of optimizing health via predictable interactions with the gut microbiome. Despite the exciting potential in this context demonstrated by pioneering research efforts of many investigators, including those cited in this brief review, the complexity of the microbiome, the chemical composition of food, and their interplay in situ remains a daunting challenge in the context of achieving necessary breakthroughs. However, recent advances in high-throughput sequencing and metabolomics profiling, compositional analysis of food, and the emergence of electronic health records as an opportunity to integrate health information provide new sources of data that can contribute to addressing this challenge. Indeed, it is now clear that computational science will play an essential role in this effort as it will provide the foundation to integrate these data layers and derive insights capable of revealing and understanding the complex interactions between diet, microbiome, and health.
The human microbiome is exceptionally plastic, which presents both challenges and opportunities (Gentile and Weir, 2018). Due to its temporal and inter-individual variability, it is difficult to discover statistically significant signatures that unambiguously constitute a healthy versus non-healthy microbiota. At the same time, its potential for adaptation to diet and other environmental factors makes the gut microbiome an excellent target for diet-related interventions to improve health. In this article, we presented a brief overview of the current state of knowledge and potential avenues for research at the interface of diet, gut microbiome, and human health, with particular emphasis on the role that computational science and data analytics can play in accelerating this research. Using these tools, we envision a future in which diets, as well as food and dietary supplement products, can be better designed for specific populations, and, in some cases, for individuals, in order to optimize gut microbiota and health via a platform integrating two distinct systems. The first system will be responsible for identifying the optimal target microbiota (discovery) given the desired target, individual, and environment, while the second will provide recommendations for achieving that target microbiota (engineering). Recognizing this distinction and the requirement for seamless interaction between the two can reinforce collaborative research in this evolving field where some teams focus on microbiota discovery and others on diet engineering.
Microbiome research has attracted much interest in the past few years and given rise to various software tools and pipelines for metagenomic data processing and analysis. Many of these tools address similar problems and researchers may choose a variety of tools depending on the context. Interestingly, recent research has shown that synthetic datasets can be used to assess the performance of competing tools given a project’s assumptions and hence provide useful benchmarks (Ounit and Lonardi, 2016; Hitch and Creevey, 2018). We further believe that progress in simulation-based studies can give rise to new data processing and analytics pipelines customized for each project based on factors such as sequencing technology, data availability, dimensionality, and variability. This can help to build standard protocols for addressing challenges like the ones mentioned in Tables 3, 4.
Our current knowledge about the relationship between diet, gut microbiome, and human health is evolving fast. Many data analysis methods exist for discovering characteristics that can define a healthy microbiota and the factors influencing it. We believe that proper integration of recommendation systems with existing research developments will have an unprecedented impact on our way of life. Given the accelerated pace of advances in sequencing and computational tools, we expect the next decade to be the era of computational nutrition that will revolutionize our relationship with food and diet.
Author Contributions
AE, NR, BP, MK, HS, and IT wrote the manuscript. AE and MK created figures with input from all authors. IT supervised all aspects of the work. All authors reviewed, revised, and approved the manuscript.
Funding
The authors acknowledge an unrestricted gift from Mars Inc. to IT, and an NSF SBIR award to PIPA LLC. The funders were not involved in the study design, collection, analysis, and interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
MK and IT are employed or have a financial interest in PIPA LLC. HS has a financial interest in T.O.P. LLC and March Capital US LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Polina Hadjipanagiotou and the Tagkopoulos lab for their comments on the article.
References
Abdi, H., Williams, L. J., and Valentin, D. (2013). Multiple factor analysis: principal component analysis for multitable and multiblock data sets. Wiley Interdiscipl. Rev. Comput. Statist. 5, 149–179. doi: 10.1002/wics.1246
Abrahamsson, T. R., Jakobsson, H. E., Andersson, A. F., Björkstén, B., Engstrand, L., and Jenmalm, M. C. (2012). Low diversity of the gut microbiota in infants with atopic eczema. J. Allergy Clin. Immunol. 129, 434–440.e1-2.
Abubucker, S., Segata, N., Goll, J., Schubert, A. M., Izard, J., Cantarel, B. L., et al. (2012). Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 8:e1002358. doi: 10.1371/journal.pcbi.1002358
Agarwala, R., and Morgulis, A. (2011). BMTagger: Best Match Tagger for Removing Human Reads from Metagenomics Datasets. Available at: https://ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger/ (accessed March 14, 2020).
Amann, R. I., Ludwig, W., and Schleifer, K.-H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Mol. Biol. Rev. 59, 143–169. doi: 10.1128/mmbr.59.1.143-169.1995
American Gut (2019). What’s in Your Gut? Available at: http://americangut.org (accessed February 11, 2019).
Amir, A., McDonald, D., Navas-Molina, J. A., Kopylova, E., Morton, J. T., Xu, Z. Z., et al. (2017). Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2:e0191-16. doi: 10.1128/mSystems.00191-16
Anderson, J., Edney, R., and Whelan, K. (2012). Systematic review: faecal microbiota transplantation in the management of inflammatory bowel disease. Aliment. Pharmacol. Therap. 36, 503–516. doi: 10.1111/j.1365-2036.2012.05220.x
Anderson, M. J. A. (2001). new method for non-parametric multivariate analysis of variance. Austr. Ecol. 26, 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x
Antonazzo, G., Attrill, H., Brown, N., Marygold, S. J., McQuilton, P., Ponting, L., et al. (2017). Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–D338.
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473:174.
Bäckhed, F., Roswall, J., Peng, Y., Feng, Q., Jia, H., Kovatcheva-Datchary, P., et al. (2015). Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microb. 17, 690–703. doi: 10.1016/j.chom.2015.04.004
Baldini, F., Heinken, A. K., Heirendt, L., Magnusdottir, S., Fleming, R., and Thiele, I. (2018). The Microbiome Modeling Toolbox: from microbial interactions to personalized microbial communities. Bioinformatics 35, 2332–2334. doi: 10.1093/bioinformatics/bty941
Barabási, A.-L., Menichetti, G., and Loscalzo, J. (2019). The unmapped chemical complexity of our diet. Nat. Food 1, 33–37. doi: 10.1038/s43016-019-0005-1
Barratt, M. J., Lebrilla, C., Shapiro, H.-Y., and Gordon, J. I. (2017). The gut microbiota, food science, and human nutrition: a timely marriage. Cell Host Microb 22, 134–141. doi: 10.1016/j.chom.2017.07.006
Bartenhagen, C., Klein, H.-U., Ruckert, C., Jiang, X., and Dugas, M. (2010). Comparative study of unsupervised dimension reduction techniques for the visualization of microarray gene expression data. BMC Bioinform. 11:567. doi: 10.1186/1471-2105-11-567
Bauer, E., and Thiele, I. (2018). From metagenomic data to personalized in silico microbiotas: predicting dietary supplements for Crohn’s disease. NPJ Syst. Biol. Appl. 4:27.
Bauer, E., Zimmermann, J., Baldini, F., Thiele, I., and Kaleta, C. (2017). BacArena: individual-based metabolic modeling of heterogeneous microbes in complex communities. PLoS Comput. Biol. 13:e1005544. doi: 10.1371/journal.pcbi.1005544
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. Ser. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
BioLINCC (2019). The Lung HIV Microbiome Project (LHMP). Available at: https://biolincc.nhlbi.nih.gov/studies/lhmp/ (accessed February 11, 2019).
Blekhman, R., Goodrich, J. K., Huang, K., Sun, Q., Bukowski, R., Bell, J. T., et al. (2015). Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 16:191. doi: 10.1186/s13059-015-0759-1
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857.
Bonder, M. J., Kurilshikov, A., Tigchelaar, E. F., Mujagic, Z., Imhann, F., Vila, A. V., et al. (2016). The effect of host genetics on the gut microbiome. Nat. Genet. 48:1407. doi: 10.1038/ng.3663
Bradley, P. H., Nayfach, S., and Pollard, K. S. (2018). Phylogeny-corrected identification of microbial gene families relevant to human gut colonization. PLoS Computat. Biol. 14:e1006242. doi: 10.1371/journal.pcbi.1006242
Bragg, L., Stone, G., Imelfort, M., Hugenholtz, P., and Tyson, G. W. (2012). Fast, accurate error-correction of amplicon pyrosequences using Acacia. Nat. Methods 9:425. doi: 10.1038/nmeth.1990
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349. doi: 10.2307/1942268
Breitwieser, F. P., Lu, J., and Salzberg, S. L. A. (2017). review of methods and databases for metagenomic classification and assembly. Briefings Bioinform. 20, 1125–1136. doi: 10.1093/bib/bbx120
Brown, C. T., Davis-Richardson, A. G., Giongo, A., Gano, K. A., Crabb, D. B., Mukherjee, N., et al. (2011). Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One 6:e25792. doi: 10.1371/journal.pone.0025792
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12:59. doi: 10.1038/nmeth.3176
Burke, R. (2002). Hybrid recommender systems: survey and experiments. User Model. User Adapt. Interact. 12, 331–370.
Caliñski, T., and Harabasz, J. A. (1974). dendrite method for cluster analysis. Commun. Statist. Theor. Methods 3, 1–27. doi: 10.1080/03610927408827101
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13:581. doi: 10.1038/nmeth.3869
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7:335.
Caspi, R., Billington, R., Fulcher, C. A., Keseler, I. M., Kothari, A., Krummenacker, M., et al. (2017). The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 46, D633–D639.
Castaner, O., Goday, A., Park, Y.-M., Lee, S.-H., Magkos, F., Shiow, S.-A. T. E., et al. (2018). The gut microbiome profile in obesity: a systematic review. Int. J. Endocrinol. 2018:9.
Chen, E. Z., and Li, H. A. (2016). two-part mixed-effects model for analyzing longitudinal microbiome compositional data. Bioinformatics 32, 2611–2617. doi: 10.1093/bioinformatics/btw308
Clarke, K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Austr. J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
ClinicalTrials.gov (2019). Pediatric Metabolism and Microbiome Repository - Full Text View. Available at: https://clinicaltrials.gov/ct2/show/NCT02959034 (accessed February 11, 2019).
Conlon, M., and Bird, A. (2015). The impact of diet and lifestyle on gut microbiota and human health. Nutrients 7, 17–44. doi: 10.3390/nu7010017
Costea, P. I., Hildebrand, F., Manimozhiyan, A., Bäckhed, F., Blaser, M. J., Bushman, F. D., et al. (2018). Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 3:8. doi: 10.1038/s41564-017-0072-8
Cox, L. M., and Blaser, M. J. (2015). Antibiotics in early life and obesity. Nat. Rev. Endocrinol. 11:182. doi: 10.1038/nrendo.2014.210
Coyte, K. Z., Schluter, J., and Foster, K. R. (2015). The ecology of the microbiome: networks, competition, and stability. Science 350, 663–666. doi: 10.1126/science.aad2602
curatedMetagenomicData (2019). curatedMetagenomicData. Available at: http://waldronlab.io/curatedMetagenomicData (accessed February 11, 2019).
Data – The Harvard Personal Genome Project (PGP) (2019). Available at: https://pgp.med.harvard.edu/data (accessed February 11, 2019).
Davenport, E. R., Cusanovich, D. A., Michelini, K., Barreiro, L. B., Ober, C., and Gilad, Y. (2015). Genome-wide association studies of the human gut microbiota. PLoS One 10:e0140301. doi: 10.1371/journal.pone.0140301
Davenport, E. R., Mizrahi-Man, O., Michelini, K., Barreiro, L. B., Ober, C., and Gilad, Y. (2014). Seasonal variation in human gut microbiome composition. PLoS One 9:e90731. doi: 10.1371/journal.pone.0090731
De Filippis, F., Pellegrini, N., Vannini, L., Jeffery, I. B., La Storia, A., Laghi, L., et al. (2016). High-level adherence to a mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 65, 1812–1821. doi: 10.1136/gutjnl-2015-309957
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 107, 14691–14696. doi: 10.1073/pnas.1005963107
de Goffau, M. C., Luopajärvi, K., Knip, M., Ilonen, J., Ruohtula, T., Härkönen, T., et al. (2013). Fecal microbiota composition differs between children With β-cell autoimmunity and those without. Diabetes 62, 1238–1244. doi: 10.2337/db12-0526
de Steenhuijsen Piters, W. A., Heinonen, S., Hasrat, R., Bunsow, E., Smith, B., Suarez-Arrabal, M.-C., et al. (2016). Nasopharyngeal microbiota, host transcriptome, and disease severity in children with respiratory syncytial virus infection. Am. J. Respir. Crit. Care Med. 194, 1104–1115. doi: 10.1164/rccm.201602-0220oc
Delzenne, N. M., Olivares, M., Neyrinck, A. M., Beaumont, M., Kjølbæk, L., Larsen, T. M., et al. (2019). Nutritional interest of dietary fiber and prebiotics in obesity: Lessons from the MyNewGut consortium. Clin. Nutr. 39, 414–424. doi: 10.1016/j.clnu.2019.03.002
DeSantis, T., Hugenholtz, P., Keller, K., Brodie, E., Larsen, N., Piceno, Y., et al. (2006). NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 2006, W394–W399.
Dobson, A. J., Chaston, J. M., Newell, P. D., Donahue, L., Hermann, S. L., Sannino, D. R., et al. (2015). Host genetic determinants of microbiota-dependent nutrition revealed by genome-wide analysis of Drosophila melanogaster. Nat. Commun. 6:6312. doi: 10.1038/ncomms7312
Dominguez-Bello, M. G., Costello, E. K., Contreras, M., Magris, M., Hidalgo, G., Fierer, N., et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U.S.A. 107:11971. doi: 10.1073/pnas.1002601107
Duvallet, C., Gibbons, S. M., Gurry, T., Irizarry, R. A., and Alm, E. J. (2017). Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 8:1784. doi: 10.1038/s41467-017-01973-8
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2013). highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10:996. doi: 10.1038/nmeth.2604
Edgar, R. C. (2016). UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. BioRxiv. 2016:081257.
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Eetemadi, A., and Tagkopoulos, I. (2018). Genetic Neural Networks: An artificial neural network architecture for capturing gene expression relationships. Bioinformatics 19:bty945. doi: 10.1093/bioinformatics/bty945
Ekstrand, M. D., Riedl, J. T., and Konstan, J. A. (2011). Collaborative filtering recommender systems. Found. Trends Hum. Comput. Interact. 4, 81–173.
EMBL-EBI Mg (2019). MGnify home page > EMBL-EBI [Internet]. MGnify. Available at: https://www.ebi.ac.uk/metagenomics/ (accessed February 11, 2019)Google Scholar
European Nucleotide Archive EMBL-EBI (2019). Available from: https://www.ebi.ac.uk/ena (accessed February 11, 2019).
Fabregat, A., Jupe, S., Matthews, L., Sidiropoulos, K., Gillespie, M., Garapati, P., et al. (2017). The reactome pathway knowledgebase. Nucleic Acids Res. 46, D649–D655.
Fankhauser, M., Moser, C., and Nyfeler, T. (2018). Patents as early indicators of technology and investment trends: analyzing the microbiome space as a case study. Front. Bioeng. Biotechnol. 6:84. doi: 10.3389/fbioe.2018.00084
Felsenstein, J. (1985). Phylogenies and the comparative method. Am. Naturalist. 125, 1–15. doi: 10.1086/284325
Ferretti, P., Pasolli, E., Tett, A., Asnicar, F., Gorfer, V., Fedi, S., et al. (2018). Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microb. 24, 133–145. doi: 10.1016/j.chom.2018.06.005
Food and Drug Administration (2016). Early Clinical Trials with Live Biotherapeutic Products: Chemistry, Manufacturing, and Control Information. Available at: https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/General/UCM292704.pdf
Food and Drug Administration (2018). Statement from FDA Commissioner Scott Gottlieb, M.D., on Advancing The Science and Regulation of Live Microbiome-Based Products Used to Prevent, Treat, or Cure Diseases in Humans. Available at: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm617168.htm
Foster, K. R., Schluter, J., Coyte, K. Z., and Rakoff-Nahoum, S. (2017). The evolution of the host microbiome as an ecosystem on a leash. Nature 548:43. doi: 10.1038/nature23292
Friedman, J., and Alm, E. J. (2012). Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8:e1002687. doi: 10.1371/journal.pcbi.1002687
Gamazon, E. R., Wheeler, H. E., Shah, K. P., Mozaffari, S. V., Aquino-Michaels, K., Carroll, R. J., et al. (2015). A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47:1091. doi: 10.1038/ng.3367
Gentile, C. L., and Weir, T. L. (2018). The gut microbiota at the intersection of diet and human health. Science 362, 776–780. doi: 10.1126/science.aau5812
Gilbert, J. A., Blaser, M. J., Caporaso, J. G., Jansson, J. K., Lynch, S. V., and Knight, R. (2018). Current understanding of the human microbiome. Nat. Med. 24:392. doi: 10.1038/nm.4517
Gloor, G. B., Macklaim, J. M., Pawlowsky-Glahn, V., and Egozcue, J. J. (2017). Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 8:2224. doi: 10.3389/fmicb.2017.02224
Goldenberg, J. Z., Yap, C., Lytvyn, L., Lo, C. K.-F., Beardsley, J., Mertz, D., et al. (2017). Probiotics for the prevention of clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst. Rev. 112:CD006095.
Goodrich, J. K., Davenport, E. R., Beaumont, M., Jackson, M. A., Knight, R., Ober, C., et al. (2016). Genetic determinants of the gut microbiome in UK twins. Cell Host Microb. 19, 731–743. doi: 10.1016/j.chom.2016.04.017
Goodrich, J. K., Waters, J. L., Poole, A. C., Sutter, J. L., Koren, O., Blekhman, R., et al. (2014). Human genetics shape the gut microbiome. Cell 159, 789–799. doi: 10.1016/j.cell.2014.09.053
Gopalakrishnan, V., Spencer, C., Nezi, L., Reuben, A., Andrews, M., Karpinets, T., et al. (2018). Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 359, 97–103.
Gould, A. L., Zhang, V., Lamberti, L., Jones, E. W., Obadia, B., Korasidis, N., et al. (2018). Microbiome interactions shape host fitness. Proc. Natl. Acad. Sci. U.S.A. 115, E11951–E11960.
Green, J. M., Barratt, M. J., Kinch, M., and Gordon, J. I. (2017). Food and microbiota in the FDA regulatory framework. Science 357, 39–40. doi: 10.1126/science.aan0836
Greenhalgh, K., Ramiro-Garcia, J., Heinken, A., Ullmann, P., Bintener, T., Pacheco, M. P., et al. (2018). Integrated in vitro and in silico modelling delineates the molecular effects of a symbiotic regimen on colorectal cancer-derived cells. Cell Rep. 27, 1621–1632.e9. doi: 10.1016/j.celrep.2019.04.001
Grice, E. A., and Segre, J. A. (2012). The human microbiome: our second genome. Annu. Rev. Genom. Hum. Genet. 13, 151–170. doi: 10.1146/annurev-genom-090711-163814
Guo, Y., Hastie, T., and Tibshirani, R. (2006). Regularized linear discriminant analysis and its application in microarrays. Biostatistics 8, 86–100. doi: 10.1093/biostatistics/kxj035
Hall, A. B., Tolonen, A. C., and Xavier, R. J. (2017). Human genetic variation and the gut microbiome in disease. Nat. Rev. Genet. 18:690. doi: 10.1038/nrg.2017.63
Hao, Q., Dong, B. R., and Wu, T. (2015). Probiotics for preventing acute upper respiratory tract infections. Cochrane Database Syst. Rev. 2:CD006895.
Heintz-Buschart, A., and Wilmes, P. (2017). Human gut microbiome: function matters. Trends Microbiol. 26, 563–574. doi: 10.1016/j.tim.2017.11.002
Hildebrand, F., Nguyen, T. L. A., Brinkman, B., Yunta, R. G., Cauwe, B., Vandenabeele, P., et al. (2013). Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 14:R4. doi: 10.1186/gb-2013-14-1-r4
Hitch, T. C., and Creevey, C. J. (2018). Spherical: an iterative workflow for assembling metagenomic datasets. BMC Bioinformatics 19:20. doi: 10.1186/s12859-018-2028-2
HOMD : Human Oral Microbiome Database (2019). Available at: http://www.homd.org/index.php (accessed February 11, 2019).
Hotelling, H. (1992). “Relations between two sets of variates,” in Breakthroughs in statistics, eds S. Kotz, and N. L. Johnson, (New York, NY: Springer), 162–190. doi: 10.1007/978-1-4612-4380-9_14
Howie, B. N., Donnelly, P., and Marchini, J. A. (2009). flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5:e1000529. doi: 10.1371/journal.pgen.1000529
HPMCD: Human Pan Microbial Communities Database (2019). Available at: http://www.hpmcd.org (accessed February 11, 2019).
Huang, S., Chaudhary, K., and Garmire, L. X. (2017). More is better: recent progress in multi-omics data integration methods. Front. Genet. 8:84. doi: 10.3389/fgene.2017.00084
Huson, D. H., Beier, S., Flade, I., Górska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12:e1004957. doi: 10.1371/journal.pcbi.1004957
Huttenhower, C., Gevers, D., Knight, R., Abubucker, S., Badger, J. H., Chinwalla, A. T., et al. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486:207. doi: 10.1038/nature11234
Jiang, D., Armour, C. R., Hu, C., Mei, M., Tian, C., Sharpton, T. J., et al. (2019). Microbiome multi-omics network analysis: statistical considerations, limitations, and opportunities. Front. Genet. 10:995.
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., and Tanabe, M. (2011). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114.
Karczewski, K. J., and Snyder, M. P. (2018). Integrative omics for health and disease. Nat. Rev. Genet. 19:299. doi: 10.1038/nrg.2018.4
Katoh, K., and Standley, D. M. M. A. F. F. T. (2013). multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evolut. 30, 772–780. doi: 10.1093/molbev/mst010
Kaufman, L., and Rousseeuw, P. (1987). Clustering by Means Of Medoids. North-Holland: Delft university.
Kim, M., and Tagkopoulos, I. (2018). Data integration and predictive modeling methods for multi-omics datasets. Mol. Omics. 14, 8–25. doi: 10.1039/c7mo00051k
Kim, M., Eetemadi, A., and Tagkopoulos, I. (2017). DeepPep: deep proteome inference from peptide profiles. PLoS Comput. Biol. 13:e1005661. doi: 10.1371/journal.pcbi.1005661
Kim, M., Rai, N., Zorraquino, V., and Tagkopoulos, I. (2016). Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli. Nat. Commun. 7:13090. doi: 10.1038/ncomms13090
Knight, R., Vrbanac, A., Taylor, B. C., Aksenov, A., Callewaert, C., Debelius, J., et al. (2018). Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16, 410–422.
Korem, T., Zeevi, D., Zmora, N., Weissbrod, O., Bar, N., Lotan-Pompan, M., et al. (2017). Bread affects clinical parameters and induces gut microbiome-associated personal glycemic responses. Cell Metab. 25, 1243–1253. doi: 10.1016/j.cmet.2017.05.002
Koren, O., Knights, D., Gonzalez, A., Waldron, L., Segata, N., Knight, R., et al. (2013). A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput. Biol. 9:e1002863. doi: 10.1371/journal.pcbi.1002863
Kostic, A. D., Gevers, D., Siljander, H., Vatanen, T., Hyötyläinen, T., Hämäläinen, A.-M., et al. (2015). The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microb. 17, 260–273. doi: 10.1016/j.chom.2015.01.001
Kruskal, J. B. (1964). Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 29, 1–27. doi: 10.1007/bf02289565
Kultima, J. R., Coelho, L. P., Forslund, K., Huerta-Cepas, J., Li, S. S., Driessen, M., et al. (2016). MOCAT2: a metagenomic assembly, annotation and profiling framework. Bioinformatics 32, 2520–2523. doi: 10.1093/bioinformatics/btw183
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31:814. doi: 10.1038/nbt.2676
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357. doi: 10.1038/nmeth.1923
Lê, S., Josse, J., and Husson, F. (2008). FactoMineR: an R package for multivariate analysis. J. Statist. Softw. 25, 1–18.
Li, D., Liu, C.-M., Luo, R., Sadakane, K., and Lam, T.-W. M. E. G. A. H. I. T. (2015). an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, H. (2015). Microbiome, metagenomics, and high-dimensional compositional data analysis. Ann. Rev. Statist. Appl. 2, 73–94. doi: 10.1146/annurev-statistics-010814-020351
Li, J., Jia, H., Cai, X., Zhong, H., Feng, Q., Sunagawa, S., et al. (2014). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 32:834. doi: 10.1038/nbt.2942
Lops, P., De Gemmis, M., and Semeraro, G. (2011). Content-Based Recommender Systems: State Of The Art And Trends. In: Recommender Systems Handbook. Berlin: Springer, 73–105.
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K., and Knight, R. (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489:220. doi: 10.1038/nature11550
Magnúsdóttir, S., and Thiele, I. (2018). Modeling metabolism of the human gut microbiome. Curr. Biotechnol. 51, 90–96. doi: 10.1016/j.copbio.2017.12.005
Magnúsdóttir, S., Heinken, A., Kutt, L., Ravcheev, D. A., Bauer, E., Noronha, A., et al. (2017). Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35:81. doi: 10.1038/nbt.3703
Manichanh, C., Rigottier-Gois, L., Bonnaud, E., Gloux, K., Pelletier, E., Frangeul, L., et al. (2006). Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 55, 205–211. doi: 10.1136/gut.2005.073817
Mansfield, J. A., Bergin, S. W., Cooper, J. R., and Olsen, C. H. (2014). Comparative probiotic strain efficacy in the prevention of eczema in infants and children: a systematic review and meta-analysis. Mil. Med. 179, 580–592. doi: 10.7205/MILMED-D-13-00546
Martiny, A. C., Martiny, J. B. H., Weihe, C., Field, A., and Ellis, J. (2011). Functional metagenomics reveals previously unrecognized diversity of antibiotic resistance genes in gulls. Front. Microbiol. 2:238. doi: 10.3389/fmicb.2011.00238
Mathew, S., Smatti, M. K., Al Ansari, K., Nasrallah, G. K., Al Thani, A. A., and Yassine, H. M. (2019). Mixed viral-bacterial infections and their effects on gut microbiota and clinical illnesses in children. Sci. Rep. 9:865. doi: 10.1038/s41598-018-37162-w
McDonald, D., Price, M. N., Goodrich, J., Nawrocki, E. P., DeSantis, T. Z., Probst, A., et al. (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6:610. doi: 10.1038/ismej.2011.139
McMurdie, P. J., and Holmes, S. (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 10:e1003531. doi: 10.1371/journal.pcbi.1003531
Mehta, R. S., Abu-Ali, G. S., Drew, D. A., Lloyd-Price, J., Subramanian, A., Lochhead, P., et al. (2018). Stability of the human faecal microbiome in a cohort of adult men. Nat. Microbiol. 3:347. doi: 10.1038/s41564-017-0096-0
Menni, C., Lin, C., Cecelja, M., Mangino, M., Matey-Hernandez, M. L., Keehn, L., et al. (2018). Gut microbial diversity is associated with lower arterial stiffness in women. Eur. Heart J. 39, 2390–2397. doi: 10.1093/eurheartj/ehy226
MG-RAST (2019). Available at: http://www.mg-rast.org (accessed February 11, 2019).
Moossavi, S., Sepehri, S., Robertson, B., Bode, L., Goruk, S., Field, C. J., et al. (2019). Composition and variation of the human milk microbiota are influenced by maternal and early-life factors. Cell Host Microb. 25, 324–335. doi: 10.1016/j.chom.2019.01.011
Morgan, X. C., Tickle, T. L., Sokol, H., Gevers, D., Devaney, K. L., Ward, D. V., et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13:R79.
Murtagh, F., and Contreras, P. (2012). Algorithms for hierarchical clustering: an overview. Wiley Interdiscipl. Rev. Data Min. Knowl. Discov. 2, 86–97.
NASA (2019). Study of the Impact of Long-Term Space Travel on the Astronauts’ Microbiome. Available at: https://www.nasa.gov/mission_pages/station/research/experiments/explorer/Investigation.html?#id=982 (accessed February 11, 2019).
NIH Human Microbiome Project - Home (2019). Available at: https://hmpdacc.org/hmp (accessed February 11, 2019).
Noronha, A., Modamio, J., Jarosz, Y., Guerard, E., Sompairac, N., Preciat, G., et al. (2018). The virtual metabolic human database: integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 47, D614–D624.
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Ospina, R., and Ferrari, S. L. A. (2012). general class of zero-or-one inflated beta regression models. Comput. Statist. Data Analy. 56, 1609–1623. doi: 10.1016/j.csda.2011.10.005
Ounit, R., and Lonardi, S. (2016). Higher classification sensitivity of short metagenomic reads with CLARK-S. Bioinformatics 32, 3823–3825. doi: 10.1093/bioinformatics/btw542
Pan, S. J., and Yang, Q. A. (2010). Survey on transfer learning. IEEE Trans. Knowl. Data Eng. 10, 1345–1359.
Pan, W.-H., Sommer, F., Falk-Paulsen, M., Ulas, T., Best, P., Fazio, A., et al. (2018). Exposure to the gut microbiota drives distinct methylome and transcriptome changes in intestinal epithelial cells during postnatal development. Genome Med. 10:27. doi: 10.1186/s13073-018-0534-5
Pascal, V., Pozuelo, M., Borruel, N., Casellas, F., Campos, D., Santiago, A., et al. (2017). A microbial signature for Crohn’s disease. Gut 66, 813–822.
Pasolli, E., Schiffer, L., Manghi, P., Renson, A., Obenchain, V., Truong, D. T., et al. (2017). Accessible, curated metagenomic data through ExperimentHub. Nat. Methods 14:1023. doi: 10.1038/nmeth.4468
Pasolli, E., Truong, D. T., Malik, F., Waldron, L., and Segata, N. (2016). Machine learning meta-analysis of large metagenomic datasets: tools and biological insights. PLoS Comput. Biol. 12:e1004977. doi: 10.1371/journal.pcbi.1004977
Paulson, J. N., Stine, O. C., Bravo, H. C., and Pop, M. (2013). Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10:1200. doi: 10.1038/nmeth.2658
Peng, X., Li, G., and Liu, Z. (2016). Zero-inflated beta regression for differential abundance analysis with metagenomics data. J. Comput. Biol. 23, 102–110. doi: 10.1089/cmb.2015.0157
Pereira, J., Rea, K., Nolan, Y., O’Leary, O., Dinan, T., and Cryan, J. (2019). Depression’s unholy trinity: dysregulated stress, immunity, and the microbiome. Ann. Rev. Psychol. 71, 49–78. doi: 10.1146/annurev-psych-122216-011613
Prehn-Kristensen, A., Zimmermann, A., Tittmann, L., Lieb, W., Schreiber, S., Baving, L., et al. (2018). Reduced microbiome alpha diversity in young patients with ADHD. PLoS One 13:e0200728. doi: 10.1371/journal.pone.0200728
Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F., et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490:55. doi: 10.1038/nature11450
Qu, K., Guo, F., Liu, X., Lin, Y., and Zou, Q. (2019). Application of machine learning in Microbiology. Front. Microbiol. 10:827. doi: 10.3389/fmicb.2019.00827
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596.
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J., and Segata, N. (2017). Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35:833. doi: 10.1038/nbt.3935
Quinn, R. A., Navas-Molina, J. A., Hyde, E. R., Song, S. J., Vázquez-Baeza, Y., Humphrey, G., et al. (2016). From sample to multi-omics conclusions in under 48 hours. mSystems 1:e0038-6.
Rahman, S. F., Olm, M. R., Morowitz, M. J., and Banfield, J. F. (2018). Machine learning leveraging genomes from metagenomes identifies influential antibiotic resistance genes in the infant gut microbiome. mSystems 3:e00123-17. doi: 10.1128/mSystems.00123-17
Ranjan, R., Rani, A., Finn, P. W., and Perkins, D. L. (2018). Evaluating bacterial and functional diversity of human gut microbiota by complementary metagenomics and metatranscriptomics. bioRxiv [Preprint]. Available at: https://www.biorxiv.org/content/10.1101/363200v1
Raymond, F., Ouameur, A. A., Déraspe, M., Iqbal, N., Gingras, H., Dridi, B., et al. (2016). The initial state of the human gut microbiome determines its reshaping by antibiotics. ISME J. 10:707. doi: 10.1038/ismej.2015.148
Relman, D. A. (2012). The human microbiome: ecosystem resilience and health. Nutr. Rev. 70(Suppl._1), S2–S9.
Rice, W. R. (1989). Analyzing tables of statistical tests. Evolution 43, 223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x
Robertson, R. C., Kaliannan, K., Strain, C. R., Ross, R. P., Stanton, C., and Kang, J. X. (2018). Maternal omega-3 fatty acids regulate offspring obesity through persistent modulation of gut microbiota. Microbiome 6:95. doi: 10.1186/s40168-018-0476-6
Rodríguez, J. M., Murphy, K., Stanton, C., Ross, R. P., Kober, O. I., Juge, N., et al. (2015). The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 26:10.3402/mehd.v26.26050. doi: 10.3402/mehd.v26.26050
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. V. (2016). a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Rosseel, Y. (2012). Lavaan: an R package for structural equation modeling and more. Version 0.5–12 (BETA). J. Statist. Softw. 48, 1–36.
Rothschild, D., Weissbrod, O., Barkan, E., Kurilshikov, A., Korem, T., Zeevi, D., et al. (2018). Environment dominates over host genetics in shaping human gut microbiota. Nature 555:210. doi: 10.1038/nature25973
Rousseeuw, P. J. (1987). Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65. doi: 10.1016/0377-0427(87)90125-7
Ruan, Q., Dutta, D., Schwalbach, M. S., Steele, J. A., Fuhrman, J. A., and Sun, F. (2006). Local similarity analysis reveals unique associations among marine bacterioplankton species and environmental factors. Bioinformatics 22, 2532–2538. doi: 10.1093/bioinformatics/btl417
Saez-Lara, M. J., Gomez-Llorente, C., Plaza-Diaz, J., and Gil, A. (2015). The role of probiotic lactic acid bacteria and bifidobacteria in the prevention and treatment of inflammatory bowel disease and other related diseases: a systematic review of randomized human clinical trials. Biomed. Res. Int. 2015:505878. doi: 10.1155/2015/505878
Sánchez, B., Delgado, S., Blanco-Míguez, A., Lourenço, A., Gueimonde, M., and Margolles, A. (2017). Probiotics, gut microbiota, and their influence on host health and disease. Mol. Nutr. Food Res. 61:1600240. doi: 10.1002/mnfr.201600240
Scher, J. U., Ubeda, C., Artacho, A., Attur, M., Isaac, S., Reddy, S. M., et al. (2015). Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthrit. Rheumatol. 67, 128–139. doi: 10.1002/art.38892
Schippa, S., Iebba, V., Barbato, M., Di Nardo, G., Totino, V., Checchi, M. P., et al. (2010). A distinctive “microbial signature” in celiac pediatric patients. BMC Microbiol. 10, 1471–2180.
Schloss, P. D., and Handelsman, J. (2005). Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 71, 1501–1506. doi: 10.1128/aem.71.3.1501-1506.2005
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schmieder, R., and Edwards, R. (2011). Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6:e17288. doi: 10.1371/journal.pone.0017288
Scholkopf, B., and Smola, A. J. (2001). Learning with Kernels: Support Vector Machines, Regularization, Optimization, And Beyond. Cambridge, MA: MIT press.
Schreiber, J. B., Nora, A., Stage, F. K., Barlow, E. A., and King, J. (2006). Reporting structural equation modeling and confirmatory factor analysis results: a review. J. Educ. Res. 99, 323–338.
Schwartz, S., Friedberg, I., Ivanov, I. V., Davidson, L. A., Goldsby, J. S., Dahl, D. B., et al. (2012). A metagenomic study of diet-dependent interaction between gut microbiota and host in infants reveals differences in immune response. Genom. Biol. 13:r32. doi: 10.1186/gb-2012-13-4-r32
Shao, Y., Forster, S. C., Tsaliki, E., Vervier, K., Strang, A., Simpson, N., et al. (2019). Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 574, 117–121. doi: 10.1038/s41586-019-1560-1
Shavitt, I., and Segal, E. (2018). Regularization learning networks: deep learning for tabular datasets. Adv. Neural Inform. Process. Syst. 1384–1394. Available at : http://papers.nips.cc/paper/7412-regularization-learning-networks-deep-learning-for-tabular-datasets
Sherwin, E., Dinan, T. G., and Cryan, J. F. (2018). Recent developments in understanding the role of the gut microbiota in brain health and disease. Ann. N. Y. Acad. Sci. 1420, 5–25. doi: 10.1111/nyas.13416
Shim, J.-S., Oh, K., and Kim, H. C. (2014). Dietary assessment methods in epidemiologic studies. Epidemiol. Health 36:e2014009. doi: 10.4178/epih/e2014009
Shoaie, S., Ghaffari, P., Kovatcheva-Datchary, P., Mardinoglu, A., Sen, P., Pujos-Guillot, E., et al. (2015). Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 22, 320–331. doi: 10.1016/j.cmet.2015.07.001
Shreiner, A. B., Kao, J. Y., and Young, V. B. (2015). The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 31, 69–75. doi: 10.1097/MOG.0000000000000139
Silverman, J. D., Washburne, A. D., Mukherjee, S., and David, L. A. A. (2017). phylogenetic transform enhances analysis of compositional microbiota data. eLife 6:e21887. doi: 10.7554/eLife.21887
Singh, R., Lanchantin, J., Robins, G., and Qi, Y. (2016). Deepchrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics 32, i639–i648. doi: 10.1093/bioinformatics/btw427
Smith, E. P., and van Belle, G. (1984). Nonparametric estimation of species richness. Biometrics 40, 119–129.
Smith, H., Gnanadesikan, R., and Hughes, J. B. (1962). Multivariate analysis of variance (MANOVA). Biometrics 18, 22–41.
Sordillo, J. E., Korrick, S., Laranjo, N., Carey, V., Weinstock, G. M., Gold, D. R., et al. (2019). Association of the infant gut microbiome with early childhood neurodevelopmental outcomes: an ancillary study to the VDAART randomized clinical trial. JAMA Netw. Open 2:e190905. doi: 10.1001/jamanetworkopen.2019.0905
Spearman, C. (1904). The proof and measurement of association between two things. Am. J. Psychol. 15, 72–101.
Stinson, L. F., Boyce, M. C., Payne, M. S., and Keelan, J. A. (2019). The not-so-sterile womb: Evidence that the human fetus is exposed to bacteria prior to birth. Front. Microbiol. 10:1124. doi: 10.3389/fmicb.2019.01124
Su, X., and Khoshgoftaar, T. M. (2009). A survey of collaborative filtering techniques. Adv. Artif. Intellig. 2009:421425.
Suphavilai, C., Bertrand, D., and Nagarajan, N. (2018). Predicting cancer drug response using a recommender system. Bioinformatics 34, 3907–3914. doi: 10.1093/bioinformatics/bty452
Suykens, J. A., and Vandewalle, J. (1999). Least squares support vector machine classifiers. Neural Process. Lett. 9, 293–300.
Taroncher-Oldenburg, G., Jones, S., Blaser, M., Bonneau, R., Christey, P., Clemente, J. C., et al. (2018). Translating Microbiome Futures. London: Nxature Publishing Group.
Thaiss, C. A., Itav, S., Rothschild, D., Meijer, M. T., Levy, M., Moresi, C., et al. (2016a). Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature 540:544. doi: 10.1038/nature20796
Thaiss, C. A., Levy, M., Korem, T., Dohnalová, L., Shapiro, H., Jaitin, D. A., et al. (2016b). Microbiota diurnal rhythmicity programs host transcriptome oscillations. Cell 167, 1495–1510. doi: 10.1016/j.cell.2016.11.003
Thaiss, C. A., Zeevi, D., Levy, M., Zilberman-Schapira, G., Suez, J., Tengeler, A. C., et al. (2014). Transkingdom control of microbiota diurnal oscillations promotes metabolic homeostasis. Cell 159, 514–529. doi: 10.1016/j.cell.2014.09.048
Thavagnanam, S., Fleming, J., Bromley, A., Shields, M. D., and Cardwell, C. R. A. (2008). meta-analysis of the association between Caesarean section and childhood asthma. Clin. Exp. Allergy 38, 629–633. doi: 10.1111/j.1365-2222.2007.02780.x
The Michigan Microbiome Project (2019). University of Michigan | Center for Microbial Systems. Available at: https://microbe.med.umich.edu/research/michigan-microbiome-project (accessed February 11, 2019).
Thomas, A. M., Manghi, P., Asnicar, F., Pasolli, E., Armanini, F., Zolfo, M., et al. (2019). Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 25, 667–678. doi: 10.1038/s41591-019-0405-7
Tibshirani, R., and Walther, G. (2005). Cluster validation by prediction strength. J. Comput. Graph. Statist. 14, 511–528. doi: 10.1198/106186005x59243
Topçuoğlu, B. D., Lesniak, N. A., Ruffin, M., Wiens, J., and Schloss, P. D. (2019). Effective application of machine learning to microbiome-based classification problems. BioRxiv [Preprint]. Available at: https://www.biorxiv.org/content/10.1101/816090v1 (accessed March 14, 2020).
Tramontano, M., Andrejev, S., Pruteanu, M., Klünemann, M., Kuhn, M., Galardini, M., et al. (2018). Nutritional preferences of human gut bacteria reveal their metabolic idiosyncrasies. Nat. Microbiol. 3:514. doi: 10.1038/s41564-018-0123-9
Tran, T. N. T., Atas, M., Felfernig, A., and Stettinger, M. (2017). An overview of recommender systems in the healthy food domain. J. Intellig. Inform. Syst. 50, 501–526. doi: 10.1007/s10844-017-0469-0
Truong, D. T., Franzosa, E. A., Tickle, T. L., Scholz, M., Weingart, G., Pasolli, E., et al. (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12:902. doi: 10.1038/nmeth.3589
Tsilimigras, M. C., and Fodor, A. A. (2016). Compositional data analysis of the microbiome: fundamentals, tools, and challenges. Ann. Epidemiol. 26, 330–335. doi: 10.1016/j.annepidem.2016.03.002
Turnbaugh, P. J., Hamady, M., Yatsunenko, T., Cantarel, B. L., Duncan, A., Ley, R. E., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457:480. doi: 10.1038/nature07540
Turpin, W., Espin-Garcia, O., Xu, W., Silverberg, M. S., Kevans, D., Smith, M. I., et al. (2016). Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 48:1413. doi: 10.1038/ng.3693
TwinsUK (2019). The Biggest twin Registry In The Uk for The Study Of Ageing Related Diseases. Available at: http://twinsuk.ac.uk (accessed February 11, 2019)Google Scholar
Tyler, A. D., Smith, M. I., and Silverberg, M. S. (2014). Analyzing the human microbiome: a “how to” guide for physicians. Am. J. Gastroenterol. 109:983. doi: 10.1038/ajg.2014.73
US Department of Agriculture and Agricultural Research Service (2010). USDA National Nutrient Database for Standard Reference, Release 28. Agricultural Research Service. Washington, D.C: USDA.
Vatanen, T., Franzosa, E. A., Schwager, R., Tripathi, S., Arthur, T. D., Vehik, K., et al. (2018). The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature 562:589. doi: 10.1038/s41586-018-0620-2
Verberkmoes, N. C., Russell, A. L., Shah, M., Godzik, A., Rosenquist, M., Halfvarson, J., et al. (2009). Shotgun metaproteomics of the human distal gut microbiota. ISME J. 3:179. doi: 10.1038/ismej.2008.108
Walker, A., Pfitzner, B., Neschen, S., Kahle, M., Harir, M., Lucio, M., et al. (2014). Distinct signatures of host–microbial meta-metabolome and gut microbiome in two C57BL/6 strains under high-fat diet. ISME J. 8:2380. doi: 10.1038/ismej.2014.79
Wang, J., Kurilshikov, A., Radjabzadeh, D., Turpin, W., Croitoru, K., Bonder, M. J., et al. (2018). Meta-analysis of human genome-microbiome association studies: the MiBioGen consortium initiative. Biomed. Central 6:101.
Wang, M., Karlsson, C., Olsson, C., Adlerberth, I., Wold, A. E., Strachan, D. P., et al. (2008). Reduced diversity in the early fecal microbiota of infants with atopic eczema. J. Allergy Clin. Immunol. 121, 129–134. doi: 10.1016/j.jaci.2007.09.011
Washburne, A. D., Morton, J. T., Sanders, J., McDonald, D., Zhu, Q., Oliverio, A. M., et al. (2018). Methods for phylogenetic analysis of microbiome data. Nat. Microbiol. 3:652. doi: 10.1038/s41564-018-0156-0
Weiss, S., Van Treuren, W., Lozupone, C., Faust, K., Friedman, J., Deng, Y., et al. (2016). Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 10:1669. doi: 10.1038/ismej.2015.235
Willing, B. P., Russell, S. L., and Finlay, B. B. (2011). Shifting the balance: antibiotic effects on host–microbiota mutualism. Nat. Rev. Microbiol. 9:233. doi: 10.1038/nrmicro2536
Willis, A. D. (2019). Rarefaction, alpha diversity, and statistics. Front. Microbiol. 10:2407. doi: 10.3389/fmicb.2019.02407
Wirbel, J., Pyl, P. T., Kartal, E., Zych, K., Kashani, A., Milanese, A., et al. (2019). Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 25, 679–689. doi: 10.1038/s41591-019-0406-6
Wishart, D. S. (2016). Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 15:473. doi: 10.1038/nrd.2016.32
Witten, D. M., Tibshirani, R., and Hastie, T. A. (2009). penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics 10, 515–534. doi: 10.1093/biostatistics/kxp008
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genom. Biol. 15:R46. doi: 10.1186/gb-2014-15-3-r46
Wooley, J. C., Godzik, A., and Friedberg, I. A. (2010). primer on metagenomics. PLoS Comput. Biol. 6:e1000667. doi: 10.1371/journal.pcbi.1000667
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486:222. doi: 10.1038/nature11053
Yong, A. G., and Pearce, S. A. (2013). beginner’s guide to factor analysis: focusing on exploratory factor analysis. Tutor. Quant. Methods Psychol. 9, 79–94. doi: 10.20982/tqmp.09.2.p079
Yuan, C., Gaskins, A. J., Blaine, A. I., Zhang, C., Gillman, M. W., Missmer, S. A., et al. (2016). Cesarean birth and risk of offspring obesity in childhood, adolescence and early adulthood. JAMA Pediatr. 170:e162385. doi: 10.1001/jamapediatrics.2016.2385
Zeevi, D., Korem, T., Zmora, N., Israeli, D., Rothschild, D., Weinberger, A., et al. (2015). Personalized nutrition by prediction of glycemic responses. Cell 163, 1079–1094. doi: 10.1016/j.cell.2015.11.001
Zhang, C., Yin, A., Li, H., Wang, R., Wu, G., Shen, J., et al. (2015). Dietary modulation of gut microbiota contributes to alleviation of both genetic and simple obesity in children. eBio Med. 2, 968–984. doi: 10.1016/j.ebiom.2015.07.007
Zhang, X., Deeke, S. A., Ning, Z., Starr, A. E., Butcher, J., Li, J., et al. (2018). Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat. Commun. 9:2873. doi: 10.1038/s41467-018-05357-4
Zhao, L., Zhang, F., Ding, X., Wu, G., Lam, Y. Y., Wang, X., et al. (2018). Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 359, 1151–1156. doi: 10.1126/science.aao5774
Zheng, H., Liang, H., Wang, Y., Miao, M., Shi, T., Yang, F., et al. (2016). Altered gut microbiota composition associated with eczema in infants. PLoS One 11:e0166026. doi: 10.1371/journal.pone.0166026
Zhou, Y.-H., and Gallins, P. A. (2019). review and tutorial of machine learning methods for microbiome host trait prediction. Front. Genet. 10:579. doi: 10.3389/fgene.2019.00579
Zhu, X. (2005). Semi-Supervised Learning Literature Survey. Wisconsin: University of Wisconsin-Madison.
Zmora, N., Suez, J., and Elinav, E. (2019). You are what you eat: diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 16, 35–56. doi: 10.1038/s41575-018-0061-2
Keywords: microbiota, gut microbiome, machine learning, artificial intelligence, data analytics, nutrition
Citation: Eetemadi A, Rai N, Pereira BMP, Kim M, Schmitz H and Tagkopoulos I (2020) The Computational Diet: A Review of Computational Methods Across Diet, Microbiome, and Health. Front. Microbiol. 11:393. doi: 10.3389/fmicb.2020.00393
Received: 23 October 2019; Accepted: 26 February 2020;
Published: 03 April 2020.
Edited by:
Hyun-Seob Song, University of Nebraska–Lincoln, United StatesReviewed by:
Babak Momeni, Boston College, United StatesMatthew John Wade, Newcastle University, United Kingdom
Copyright © 2020 Eetemadi, Rai, Pereira, Kim, Schmitz and Tagkopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilias Tagkopoulos, aWxpYXN0QHVjZGF2aXMuZWR1