 Tetyana Milojevic
Tetyana Milojevic Wolfram Weckwerth
Wolfram Weckwerth- 1Extremophiles/Space Biochemistry Group, Department of Biophysical Chemistry, University of Vienna, Vienna, Austria
- 2Department of Ecogenomics and Systems Biology, University of Vienna, Vienna, Austria
- 3Vienna Metabolomics Center, University of Vienna, Vienna, Austria
Since the dawn of space exploration, the survivability of terrestrial life in outer space conditions has attracted enormous attention. Space technology has enabled the development of advanced space exposure facilities to investigate in situ responses of microbial life to the stress conditions of space during interplanetary transfer. Significant progress has been made toward the understanding of the effects of space environmental factors, e.g., microgravity, vacuum and radiation, on microorganisms exposed to real and simulated space conditions. Of extreme importance is not only knowledge of survival potential of space-exposed microorganisms, but also the determination of mechanisms of survival and adaptation of predominant species to the extreme space environment, i.e., revealing the molecular machinery, which elicit microbial survivability and adaptation. Advanced technologies in –omics research have permitted genome-scale studies of molecular alterations of space-exposed microorganisms. A variety of reports show that microorganisms grown in the space environment exhibited global alterations in metabolic functions and gene expression at the transcriptional and translational levels. Proteomic, metabolomic and especially metabolic modeling approaches as essential instruments of space microbiology, synthetic biology and metabolic engineering are rather underrepresented. Here we summarized the molecular space-induced alterations of exposed microorganisms in terms of understanding the molecular mechanisms of microbial survival and adaptation to drastic outer space environment.
Introduction
The outer space environment, which is characterized by a high vacuum and an intense radiation, provides hostile conditions to any form of life. With the upcoming long-term space explorations, it is becoming increasingly important to understand the molecular mechanisms of survival in outer space. Remarkably, a few extremophilic microbial species have been shown to withstand the drastic influence of the outer space factors (Saffary et al., 2002; Sancho et al., 2007; Cockell et al., 2011; Wassmann et al., 2012; Kawaguchi et al., 2013; Selbmann et al., 2015), and numerous studies have significantly proved the possibility of microbial life transfer through space (Horneck et al., 2008; Nicholson, 2009). Considerable progress has been made toward the understanding of the effects of space environmental factors, e.g., microgravity, vacuum and radiation, on microorganisms exposed to real and simulated space conditions. However, we still have been missing an explicit knowledge of molecular mechanisms permitting survival and adaptation in the outer space environment. Space parameters affect microorganisms by altering a variety of physiological features, including cell metabolism, proliferation rate, cell division, cell motility, virulence, and biofilm production (Figure 1). These physiological perturbations of space-exposed microorganisms are very poorly understood at the molecular level. In this context, omics-based analyses combined with classical phenotyping and physiological measurements provide the integrative suite of tools to functionally decipher the mechanisms of microbial survivability in outer space. Exploring the relevant mechanisms underlying metabolic and physiological changes which microorganisms experience during exposure to outer space, the omics-based approach integrates the different fragments of biological information to understand the flow of information from genomes, mRNA, proteins to metabolites (Weckwerth, 2011a; Ott et al., 2017, 2019b,a), and explains how the microorganisms adapt to this extreme environment.
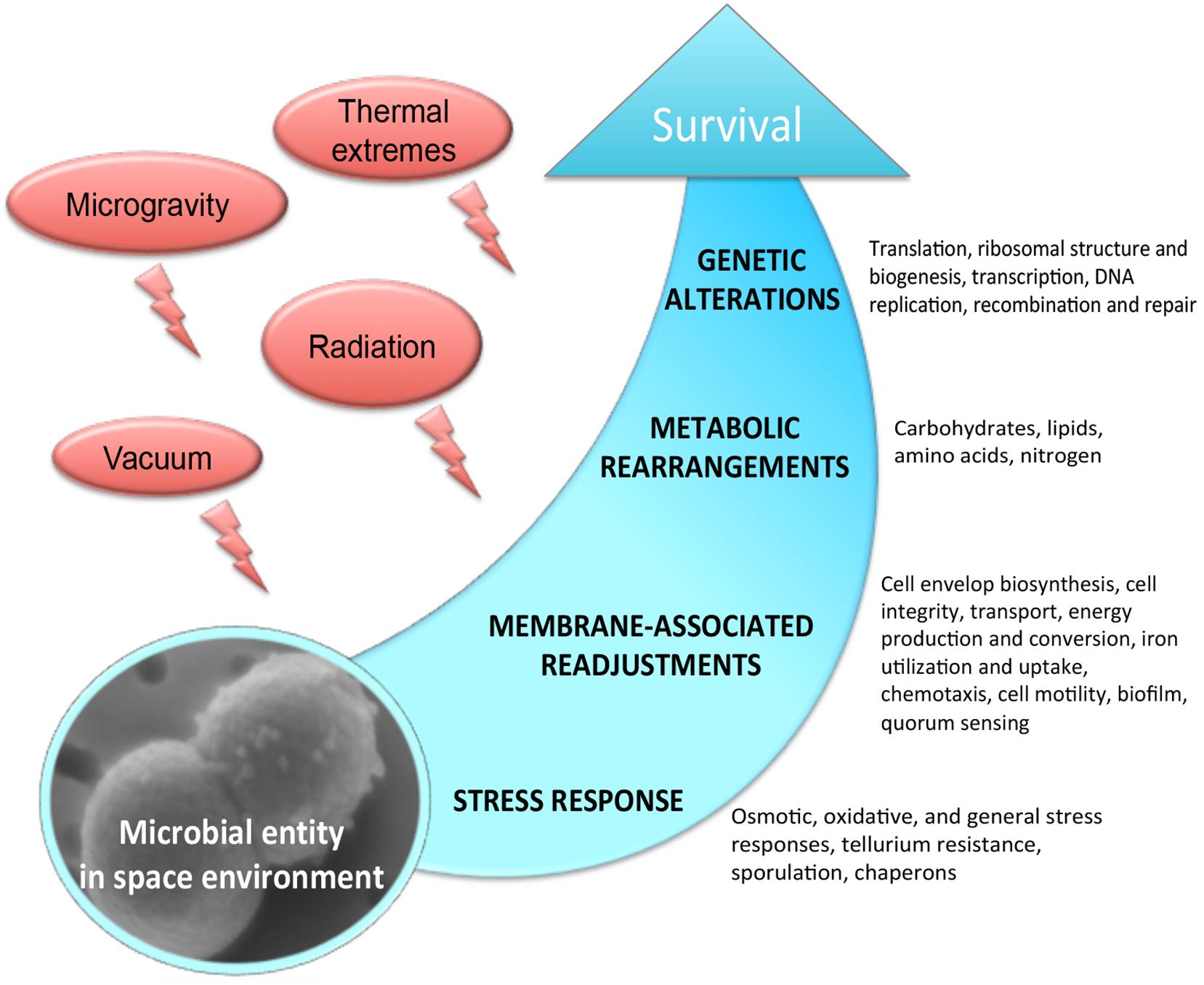
Figure 1. Molecular response experienced by microorganisms in the outer space environment.
Systems biology is an interdisciplinary field that incorporates the results of genome-scale molecular analysis–omics techniques–such as genomics, transcriptomics, proteomics, and metabolomics and genome-scale metabolic and regulatory biomathematical models to reveal complex molecular interactions underlying molecular evolution, functional and phenotypical diversity and molecular adaptation (Kitano, 2000; Ideker et al., 2001; Weckwerth, 2003, 2011a,b). The roots of systems biology go back to the development of the General System Theory by Ludwig von Bertalanffy in the early 30’s of the 20th century. He published one of the first explicit articles about open systems, feedback regulation and self-organization in living systems and proposed the basic mathematical framework for systems biology (Bertalanffy, 1940, 1969; Weckwerth, 2016, 2019).
In this review we focus on molecular mechanisms of microbial survivability in the outer space environment revealed with the help of global and integrative –omics approaches of systems biology that have been recently used to study microorganisms exposed to real and simulated space conditions. The use of –omics in space life sciences potentially has a pivotal role in the understanding of the molecular machineries implicated by microorganisms to tolerate the harsh conditions of space.
Metabolic Changes
Exposure to space environment strongly modifies the expression of genes and the abundance of proteins related to metabolism (Table 1). Space parameters primarily affect the genes involved in metabolism, raising further alterations of the microbial growth. However, energy and regenerative power are needed for the microbial cell to counteract the drastic effects of space exposure by launching a number of regenerative activities and repair mechanisms.
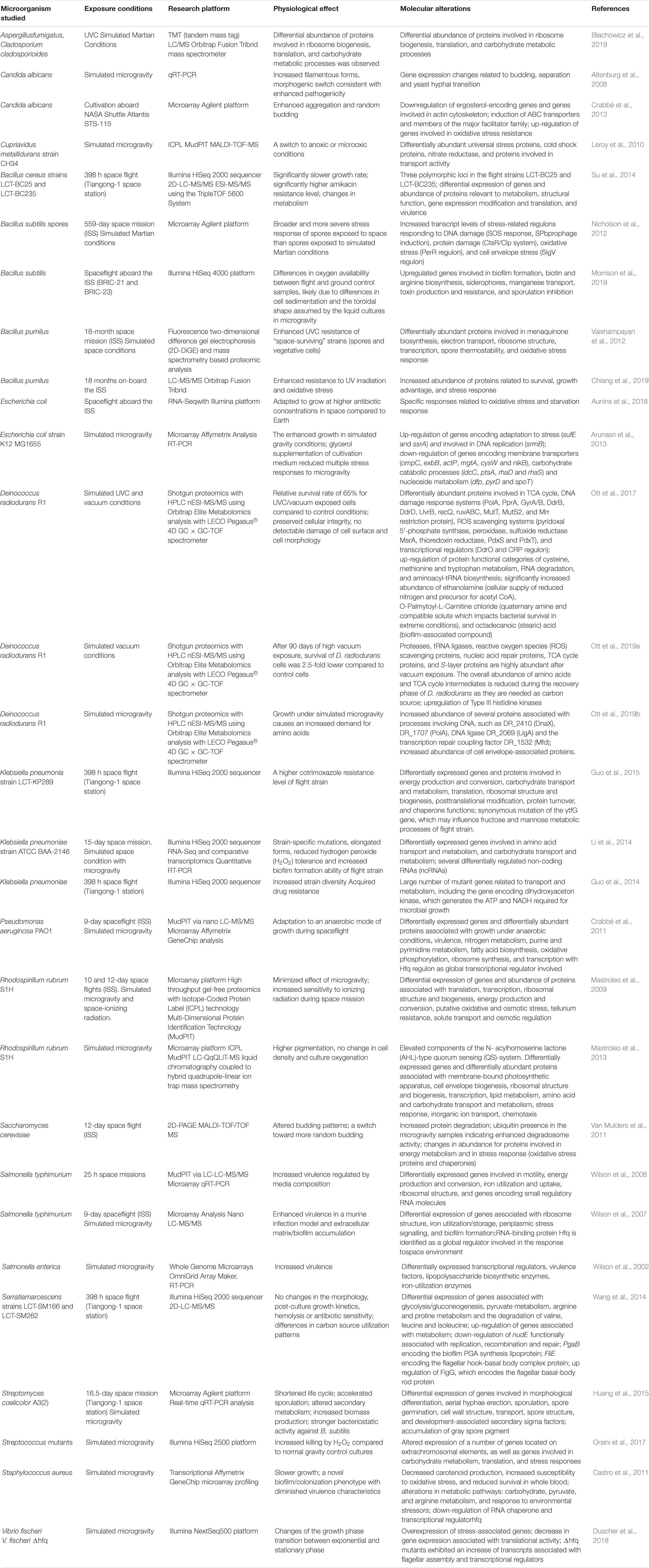
Table 1. Omics studies of the microbial physiology in real and simulated outer space environment.
Carbohydrate Metabolism
The molecular mechanisms of adaptive reactions under stress conditions require additional energy and carbohydrate metabolism is crucial in generating that energy (Minic, 2015). Carbon source utilization by various microorganisms has been primarily affected during short- and long-term exposure to space conditions. In response to space environmental stress, microorganisms with great adaptability to survive successfully exhibit certain flexibility in carbon source utilization. Strains of Serratia marcescens, Escherichia coli, and Klebsiella pneumoniae after a long-term flight on-board of the SHENZHOU-8 spacecraft exhibited a difference in carbon source utilization (as suggested by multi-omics analysis), while their morphology or growth patterns were not affected (Li et al., 2014, 2015; Wang et al., 2014; Guo et al., 2015; Zhang et al., 2015). Strains of S. marcescens after spaceflight displayed a positive reaction in the sole-carbon-source utilization of D-Mannitol, D-Raffinose, and N-Acetyl neuraminic acid (Wang et al., 2014). Correspondingly, proteomic analysis showed up-regulation of the proteins of the glycolysis/gluconeogenesis pathway of S. marcescens grown during spaceflight, which was suggested to reflect adaptive changes of the strain aboard the spacecraft (Wang et al., 2014). The strain of E. coli after spaceflight showed increased carbon source utilization and 2,58-fold up-regulation of maltose regulon periplasmic protein malmM (shown in proteomic analysis), which can be explained by the increased demand for additional carbon substrates during stress adaptability to the space environment (Zhang et al., 2015). Klebsiella pneumoniae gained the ability to use D-Mannose after spaceflight, possibly reflecting an overall slowed down metabolism of this microorganism to better adapt to the space environment (Guo et al., 2015). Numerous K. pneumoniae genes differentially expressed after spaceflight were involved in carbohydrate transport and metabolism, as suggested by genomic and transcriptomic analyses (Li et al., 2014). Integrative proteotranscriptomic analysis of Bacillus cereus strain flown on-board of the SHENZHOU-8 spacecraft has shown that genes of glucose metabolism (glpX gene product, trehalose-6-phosphate hydrolase) were differentially expressed during spaceflight (transcriptomic analysis reported in Su et al., 2014). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is one of the key enzymes in glycolytic pathway that serves to break down glucose for carbon molecules and energy. GAPDH provides an important source of NADH during glycolysis and contributes to the various regulatory functions (Sirover, 2012, 2014). Proteomic analysis showed that GAPDH was significantly up-regulated in Bacillus pumilus spores exposed to outer space (Vaishampayan et al., 2012). Apart from the differential expression of GAPDH under a variety of stress conditions (Nicholls et al., 2012; Hildebrandt et al., 2015), a microgravity induced up-regulation of this housekeeping protein together with pyruvate kinase and a subunit from pyruvate decarboxylase was observed in proteomic analysis of Saccharomyces cerevisiae after the 2-day trip on-board of the Soyuz TMA-9 vehicle (Van Mulders et al., 2011). RNAseq-based analysis of Streptococcus mutants displayed an alteration in early stationary-phase metabolism under the influence of simulated microgravity, and expression of phosphortransferase system genes associated with transport of carbohydrates (trehalose, mannose, glucose, mannitol, and cellobiose) was significantly increased (Orsini et al., 2017). Proteomic profiling of several fungal strains exposed to simulated Martian conditions revealed that carbohydrate metabolic functional category was among significantly over-represented biological processes (Blachowicz et al., 2019). A set of up-regulated proteins involved in carbohydrate metabolism included enzymes (e.g., isocitrate lyase AcuD, cellobiohydrolases, exo-polygalacturonase, and chitin deacetylases), which enable exposed fungal strains using an alternative carbon source and permit morphogenetic alterations in the course of growth and differentiation. The authors reported on adjustments in carbohydrate metabolism as an adaptive response to simulated Martian conditions (Blachowicz et al., 2019).
The analyzed –omics-assisted investigations indicate characteristic changes in carbohydrate metabolism of microorganisms cultivated during spaceflight and exposed to outer space. These changes are directed to restore energy balance in stress conditions and frequently serve to satisfy the increased demand in carbon source during adaptive reactions to the space environment.
Amino Acid Metabolism
Apart from their main contribution as substrates for protein synthesis, amino acids act as signaling molecules exerting regulatory functions (Sturme et al., 2002; Rinaldo et al., 2018), and serve as indirect carbon sources through citrate cycle. Frequently observed down-regulation of enzymes involved in amino acid transport and metabolism of space-exposed microorganisms is usually associated with their arrest in growth. Among all the genes of K. pneumonia, E. coli and B. cereus affected during spaceflight on-board of space vehicle, functional category of amino acid transport and metabolism was the most represented (Li et al., 2014, 2015; Su et al., 2014; Guo et al., 2015; Zhang et al., 2015). The strain of K. pneumoniae after spaceflight characterized by increased biofilm formation was suggested to use amino acids as an indirect carbon source through TCA cycle in stress-related space conditions (Li et al., 2014). Proteomic analysis showed that most key enzymes of the TCA cycle were more abundantly represented in Deinococcus radiodurans cells after the exposure to UVC/vacuum conditions (Ott et al., 2017). Proteomic analysis of S. marcescens and K. pneumonia revealed that proteins involved in arginine and proline metabolism, and degradation pathways of valine, leucine, and isoleucine were down regulated after spaceflight (Wang et al., 2014; Guo et al., 2015). However, another study by Morrison et al., 2019 reported that genes of arginine biosynthesis were up-regulated during spaceflight of B. subtilis aboard the ISS (based on the RNA-seq analysis). Such discrepancies between different reports can be very well stated by the experimental set up of the studies. One group of the studies reported on molecular profiles during spaceflight, i.e., avoiding re-cultivation of returned samples (e.g., Morrison et al., 2019), while another group of investigations analyzed space-returned strains upon their recovery in liquid medium (e.g., Li et al., 2014). Serine hydroxymethyltransferase responsible for the enzymatic catalysis of the reversible conversion of L-serine to L-glycine was up-regulated in proteomic analysis of space-returned spores of B. pumilus (Vaishampayan et al., 2012). Proteomic analysis of spaceflight grown cells of P. aeruginosa indicated the down-regulation of ArcA, an enzyme associated with the fermentation of arginine (Crabbé et al., 2011). Proline and arginine metabolism implement in microbial mechanisms of stress survival (Zhao and Houry, 2010; Goh et al., 2011; Liang et al., 2013). Various studies have shown evidence that proline metabolism leads to increased production of endogenous reactive oxygen species (ROS) (Liang et al., 2013).
Experimental data obtained in the post-flight/post-exposure analysis suggest that amino acids metabolism is majorly down regulated in order to favor the suppressed microbial growth and proliferation. The observed suppression of proline metabolism in a number of space exposed bacterial strains can be considered as one of the microbial strategies to minimize the generation of endogenous metabolically produced ROS in order to cope efficiently with radiation-induced damage. The functional categories of cysteine and methionine metabolism, however, can be up-regulated in response to radiation component of the outer space environment (Ott et al., 2017). Methionine and cysteine are sulfur-containing amino acids, which significantly contribute to the antioxidant defense system of exposed microorganisms. Cysteine chemistry, i.e., cysteine-mediated redox signaling is important biochemical response against ROS damage (Paulsen and Carroll, 2013). Methionines located on the surface of protein structures act as effective endogenous antioxidants to defend functionally essential molecules against oxidative damage (Levine et al., 2000). In case with outer space-associated radiation exposure, activation of cysteine and methionine metabolism is one of the most obvious microbial responses to oxidative damage.
Lipid Metabolism
Microorganisms adapt their membrane lipid composition to stressful environmental conditions by adjusting the relative amounts of different types of lipids and the degree of unsaturation of fatty acyl residues. Strains of Bacillus horneckiae sp. isolated from the Phoenix spacecraft were characterized by altered lipid profiles based on the results of traditional biochemical lipid analyses (Vaishampayan et al., 2010). Radiation-induced elevated generation of ROS causes multiple disintegrative disorders, including oxidative changes in lipid metabolism. The genes involved in lipid biosynthesis (enoyl-CoA hydratase/isomerase with fatty acid synthase activity) and in fatty acid metabolism were down-regulated in RNAseq-based analysis of B. cereus and K. pneumoniae strains after spaceflight (Li et al., 2014; Su et al., 2014). The authors connect it with the detected reduced growth of B. cereus after spaceflight as an adaptive strategy that enable the survival and maintenance of the energy status. Transcriptomic analysis of space exposed B. subtilis spores and Rhodospirillum rubrum exposed to modeled microgravity in frames of the Micro-Ecological Life Support System Alternative (MELiSSA) project showed the down-regulation of genes encoding lipid biosynthetic enzymes (Nicholson et al., 2012; Mastroleo et al., 2013). Multi-omic analyses showed that metabolic pathways associated with fatty acid metabolism, phospholipid biosynthetic process, and cellular lipid biosynthetic processes were affected in E. coli and Enterococcus faecium strains after spaceflight (Chang et al., 2013a; Li et al., 2015).
Studies of microbial biochemistry in microgravity conditions generally suggest the up-regulation of genes encoding lipoproteins and lipopolysaccharide biosynthetic enzymes (Wilson et al., 2007; Leroy et al., 2010). Several of them are associated with bacterial biofilm formation, virulence, and pathogenicity. The gene encoding rhamnosyltransferase (rhlA) involved in surfactant biosynthesis was found among the major virulence-associated genes of P. aeruginosa stimulated in spaceflight along with the accumulation of rhamnolipids under simulated microgravity conditions, which might be connected to low shear liquid sensing (transcriptomic analysis in Crabbé et al., 2011).
In summary, many of changes in lipid metabolism observed under the space environmental conditions aim to adjust the energy status toward the slowed down growth of exposed microorganisms. However, the mobilization of certain microbial lipoproteins and lipopolysaccharides is activated in spaceflight and under simulated microgravity conditions to adapt cell-cell contacts and communication toward the low-shear growth environment.
Membrane-Associated Processes
The destructive effect of space vacuum (10−7 to 10−4 Pa) triggers cellular integrity of space-exposed microorganisms, influencing numerous processes in membrane apparatus of microbial cell (Horneck et al., 2010). Among them are rearrangements of lipid bilayers, changes in membrane fluidity and permeability, and alteration of membrane bound enzymatic activities. Cell membrane that carries a function of physical barrier and protect the cell from extracellular environment can be affected in conditions of microgravity, causing the altered uptake or excretion rates.
Cell Envelope Biosynthesis and Maintenance
Microbiological and biochemical analysis of the survival and behavior of C. metallidurans in the MESSAGE-1 flight experiment by means of flow cytometry-assisted analysis of cell physiology and proteomic profiling revealed a minor damage of cell membrane; the remaining viable cells acquired a higher membrane potential than the ground control cells (Leys et al., 2009). The genes involved in the cell wall/membrane/envelope biogenesis of E. faecium were among the differentially expressed genes with the greatest change in expression after flight on the SHENZHOU-8 spacecraft, as suggested by comparative transcriptomic and proteomic analyses. Comparative genomic analysis of returned after spaceflight E. faecium strain revealed mutation of the arpU gene associated with cell wall growth, which in turn may affect the expression of molecular players responsible for cell wall and membrane biogenesis of E. faecium (Chang et al., 2013a). The post-flight transcriptomic and proteomic studies of E. coli revealed the up-regulation of the envelope stress induced periplasmic protein Spy and the yfbE gene encoding predicted pyridoxal phosphate-dependent enzyme with regulatory functions in cell wall biogenesis (Li et al., 2015; Zhang et al., 2015). A number of differentially expressed genes involved in cell wall and spore structure were described microarray-based analysis of Streptomyces coelicolor during spaceflight and simulated microgravity (Huang et al., 2015). Modeled microgravity affected gene groups of Salmonella involved in type III secretion, lipopolysaccharides, and cell wall synthesis (microarray-based analysis in Wilson et al., 2002). Proteomic analysis showed that D. radiodurans grown in simulated microgravity showed an increased abundance of cell envelope-associated proteins (Ott et al., 2019a). Cell envelope biosynthesis and maintenance was triggered in multi-omics analysis of R. rubrum by spaceflight and simulated space environment (the top 20 of the most induced genes) (Mastroleo et al., 2009, 2013). Cell envelope of B. subtilis exposed to 1.5 years of simulated Martian and space conditions was massively affected as suggested by a comprehensive transcriptomic analysis with a number of up-regulated membrane and cell envelope stress proteins (SigV regulon) (Nicholson et al., 2012).
A number of spaceflight induced alterations associated with cell envelope, e.g., a thickened cell envelope and intensive vesiculation (Zea et al., 2017), are quick stress responses to enhance microbial adaptation rates and regulate the level of protein accumulation in the cell envelope. These autonomous stress responses are mediated by means of altered cell envelope protein machinery, allowing exposed cells to export stress products (e.g., damage or misfolded proteins) and to achieve alleviated stress response.
Transport
The effects of simulated and real microgravity on microbial behavior and metabolism in liquid cultures aboard spacecraft are most likely mediated by alterations of extracellular environment. The response of the cell to changes in the extracellular environment includes a cascade of cellular transport events that operate nutrient uptake, cellular waste disposal, solute transport and quorum-sensing signaling. The Suf along with other ABC membrane transporters were identified as differentially abundant in S. typhimurium in proteomic response to cultivation aboard spacecraft (Wilson et al., 2008). Spaceflight significantly affected the transport machinery of E. coli, up-regulating a number of transporters, as shown in multi-omics analysis (Li et al., 2015). The multi-omics based comparison of the strains after spaceflight and the control strains of E. coli, K. pneumonia, and R. rubrum showed that ontological categories “transmembrane transporter activity” were overrepresented among all differently expressed genes and differently abundant proteins during spaceflight (Mastroleo et al., 2009; Li et al., 2014, 2015). The genes encoding for multidrug efflux and arsenite membrane-bound transporters were 3 to 4 fold overexpressed in transcriptomic analysis of spores of B. subtilus exposed to simulated Mars conditions or/and real space (Nicholson et al., 2012). The genes encoding for probable metal-transporting P-type ATPase and dctA C4-dicarboxylate transport protein were overexpressed as suggested by post-flight transcriptomic analysis of P. aeruginosa (Crabbé et al., 2011). Microarray analysis identified that the group of membrane transport genes belongs to low-shear modeled microgravity (LSMMG) regulon of Salmonella and the gene sbmA encoding ABC superfamily transporter was up-regulated during spaceflight (Wilson et al., 2007). Post-flight proteotranscriptomic analysis of R. rubrum revealed up-regulated genes that are involved in solute transport and osmotic regulation, and are probably related to oxidative stress (Mastroleo et al., 2009). Antibiotic-producing S. coelicolor in conditions of simulated and real space microgravity displayed a significant up-regulation of several transporters which contribute to its enhanced bioactivity and BldK ABC transporter complex which is essential for aerial mycelium formation (microarray-based analysis in Huang et al., 2015).
Due to the lack of gravity-driven convective flows, extracellular mass transport becomes essentially limited to diffusion under the conditions of spaceflight and simulated microgravity. Cells cultured in liquid medium during spaceflight may experience a deficiency in oxygen and nutrients availability (Zea et al., 2016), which reflects altered transport functions. The elevated transport machinery of microgravity-affected microorganisms aims to facilitate not only nutrient uptake, but also cellular waste removal, distribution of solute and trafficking of quorum-sensing signaling molecules.
Chemotaxis, Cell Motility
Microgravity in conditions of spaceflight affects extracellular fluid properties, this way altering relationship between microbial cell and extracellular environment (Horneck et al., 2010). The involvement of flagellar apparatus and chemotaxis machinery after spaceflight and real space exposure has been detected applying –omics assisted analyses of several bacterial strains. 5.1-fold up-regulation of the fliL gene required for flagellar formation was indicated in the transcriptomic response of space-exposed spores of B. subtilis (Nicholson et al., 2012). The flgG gene encoding the flagellar basal-body rod protein was up-regulated in transcriptomic response of S. marcescens to spaceflight conditions, while down-regulation was shown in the same study for the FliE gene that encodes the flagellar hook-basal body complex protein (Wang et al., 2014). In addition, many K. pneumoniae genes affecting cell motility were differentially expressed in the strain exposed to simulated space condition (Li et al., 2014). The expression of flagellar assembly genes in LSMMG conditions was also increased in the transcriptome of the mutualistic bacterium Vibrio fischeri (Duscher et al., 2018). Flagellar assembly and bacterial chemotaxis were the most significantly enriched functional categories among all affected genes differently expressed by E. coli during spaceflight, e.g., genes encoding bacterial flagellin and methyl-accepting chemotaxis protein II (Li et al., 2015). Transcriptomic response of R. rubrum to spaceflight and simulated microgravity indicated the induced expression (upregulated > 2.6-fold) of the gene encoding for Flagellar hook-associated protein 2 (FliD filament cap protein) (Mastroleo et al., 2009). Genes of S. typhimurium involved in motility and chemotaxis response were identified as differentially expressed in response to spaceflight cultivation (Wilson et al., 2007). Transcriptomic and proteomic analyses of P. aeruginosa exposed to spaceflight conditions revealed that several chemotaxis transducers were up-regulated in response to spaceflight (Crabbé et al., 2011).
The reported effects of simulated microgravity and spaceflight on the various physiological properties of microorganisms are associated with the potential disruption of the quiescent extracellular environment. The molecular components of bacterial motility and chemotaxis response are activated in microgravity-exposed microorganisms in order to gain a balanced connection between the cell and its environment. The induced flagellar action results in mixing of the local fluid surrounding of microbial cell and removal of the cell from its quiescent location. The induction of molecular machinery responsible for motility and chemotaxis can locally influence the correct redistribution and availability of nutrients, substrates, solutes and other biologically active molecules at the extracellular environment level.
Energy Production and Conversion
In line with the increased demand in energy needed to cope with stress in space environment, there are several post-flight –omics assisted observations of altered membrane bioenergetics and electron transport chains with elevated abundances of electron transfer proteins and ATP synthase. Comparative proteomic analysis of B. pumilus spores long-termly exposed to a variety of real space conditions at the ISS in the EXPOSE facility revealed the up-regulation of the alpha-ketoglutarate decarboxylase enzyme which is associated with menaquinone biosynthesis and involved in the membrane-associated electron transport system (Palaniappan et al., 1994; Vaishampayan et al., 2012). Further proteomic studies with space-exposed B. pumilus showed that the altered protein abundances in the category of energy metabolism might be a microbial strategy to better cope with stressful environments (Chiang et al., 2019). Post-flight proteotranscriptomic analysis of R. rubrum identified significant up-regulation of multiple clusters of genes relevant to energy production and conversion (succinate dehydrogenase, ubiquinone oxidoreductase) (Mastroleo et al., 2009). The hydN gene encoding Fe-S center-bearing protein responsible for electron transport from formate to hydrogen together with a set of S. typhimurium genes involved in the formation of the Hyc hydrogenase (respiratory enzyme of H2-uptake) were differentially expressed in response to spaceflight cultivation (Wilson et al., 2007). The gene hppA encoding membrane-bound proton-translocating pyrophosphatase and five F0F1 ATP synthase subunits were up-regulated after spaceflight of R. rubrum (Mastroleo et al., 2009). The subunits of ATP synthase of microgravity-exposed S. cerevisiae have been differentially regulated during short-term spaceflight (Van Mulders et al., 2011). The down-regulation of CcoP2, a cytochrome with high affinity for oxygen, has been observed in proteomic analysis of the P. aeruginosa cells grown in spaceflight (Crabbé et al., 2011). This cytochrome is not active in the anaerobic lifestyle of P. aeruginosa, but is induced under microaerophilic conditions (Crabbé et al., 2011). The authors reported that P. aeruginosa adopted an anaerobic mode of growth during spaceflight, and switched to anaerobic metabolism, which was accompanied by the down-regulation of CcoP2 under oxygen-limiting conditions. Spaceflight induced mainly genes involved in anaerobic metabolism of this pathogen and anaerobic respiration occurred through denitrification, i.e., in the presence of the alternative electron acceptor nitrate or nitrite (Crabbé et al., 2011).
Microbial adaptation to spaceflight conditions may lead to alterations associated with energy sources utilization. Hereby, a corresponding molecular pull of energy-converting enzymes responsible for a switch from one energy source to another is affected in conditions of spaceflight and simulated microgravity. Energy management also facilitates the survival of outer space exposed microorganisms by modulating electron transfer proteins.
Iron Utilization and Uptake
Iron as the most abundant transition metal in biological systems is incorporated into protein cofactors and plays important regulatory, redox and catalytic roles in microbial world. Prokaryotes have evolved powerful iron assimilation and storage systems, which supply sufficient iron for growth and metabolism. Microorganisms show an enormous diversity and abundance of iron-dependent redox proteins, which majorly harbor iron within hemes and in the form of iron–sulfur (Fe–S) clusters. Bacteria and Archaea enormously depend on these two classes of cofactors for their energy metabolism. Iron availability influences the expression of bacterial genes encoding high-affinity iron uptake pathways and, in pathogenic microorganisms, virulence determinants (Butt and Thomas, 2017). The metal-specific repressor Fur (ferric uptake regulator) (Lee and Helmann, 2007) appears to be a very global and well-conserved regulator, which participates in regulation of iron uptake and homeostasis in bacteria. Fur coordinates the expression of various genes and acts as an iron-responsive, DNA-binding repressor protein (Hantke, 1987; Lee and Helmann, 2007). The Fur protein employs Fe(II) as a cofactor and binds to a “Fur box” with the palindromic consensus sequence GATAATGATAATCATTATC in the promoters of iron-regulated genes, resulting in repression of these genes, while under low-iron conditions, the Fur protein is released from this operator site and transcription takes place. The Fur repressor is involved in the transcriptional control of the operons encoding the pathways for the production of the siderophores (responsible for delivering the iron into the cells), virulence-associated genes, the manganese- and iron-containing superoxide dismutase genes and in the fur gene autoregulation (Hall and Foster, 1996; Lee and Helmann, 2007).
The Fur protein and iron utilization/storage system were shown to play a role in the Salmonella ground-based LSMMG (Wilson et al., 2002) and the spaceflight-induced multi-omics molecular responses (Wilson et al., 2002, 2008). LSMMG induces acid resistance in Salmonella and Fur was shown to be essential in this process. The fur mutant strain of Salmonella did not demonstrate any detectable acid resistance to be induced by LSMMG (Wilson et al., 2002). Furthermore, several genes involved in iron metabolism were induced (fepD encoding for ferric enterobactin transporter, STM1537 encoding for Ni/Fe-hydrogenase1 b-type cytochrome subunit, hscB responsible for assembly of Fe-S clusters) and down regulated (feoB encoding for ferrous iron transport protein, yliG putative Fe-S oxidoreductase, sufC, sufS) by LSMMG, as indicated by whole genome microarray analysis (Wilson et al., 2002). Additionally, the authors mention a number of potential Fur-binding sites that were located upstream of several different LSMMG regulated genes. The Fur-binding site-associated genes regulated by LSMMG included those identified to be regulated by Fur (fepD, sufC, sufS, and feoB) and those not earlier described as Fur regulated (Wilson et al., 2002). Wilson et al. (2007, 2008) reported that several molecular components of iron utilization/storage system were altered during spaceflight. Proteomic analysis showed that the abundances of Fur, iron-dependent alcohol dehydrogenase, bactoferrin, and electron transport protein with Fe-S center were increased after spaceflight, while a number of other proteins involved in iron utilization and uptake (e.g., cytoplasmic ferritin, siderophore receptor TonB, Fe-S cluster formation protein, and ferric enterobactin receptor) were underrepresented during growth of S. typhimurium in spaceflight (Wilson et al., 2007, 2008). These findings indicate the involvement of Fur in transmitting the LSMMG and spaceflight induced signals.
Proteotranscriptomic analysis of P. aeruginosa showed that the gene bfrB encoding bacterioferritin was down-regulated in cells grown on-board (Crabbé et al., 2011). Apart from the primarily function of iron storage, bacterioferritins participate in defense against oxidative stress and radical damage (Velayudhan et al., 2007). The down-regulation of bfrB in this case may be linked to the anerobiosis switch of P. aeruginosa during spaceflight (Crabbé et al., 2011). 4 to 6-fold down-regulation of several genes encoding putative iron (III) dicitrate transporting proteins was shown in transcriptomic analysis of spores of B. subtilus exposed to 1.5 year to real space conditions (Nicholson et al., 2012). The analysis of differential gene expression of R. rubrum in MESSAGE 2 spaceflight experiment (Mastroleo et al., 2009) revealed the up-regulation of the Fur repressor, which connects cellular iron status to oxidative stress by scavenging iron (Lee and Helmann, 2007), and the down-regulation of hfq, which negatively controls fur expression in E. coli (Vecerek et al., 2003). During MESSAGE 2 experiment the genes involved in the iron acquisition and redox balance, e.g., ferredoxin (Rru_A0077), Fe-S cluster-related gene (Rru_A1069), dps-related gene (Rru_A1499), superoxide dismutase (Rru_A1760) and bacterioferritin bfr (Rru_A2195) were induced (Mastroleo et al., 2009).
The analyzed –omics based investigations indicate that modeled microgravity and spaceflight conditions operate molecular iron-binding elements for their regulatory and oxidative stress pathways, encompassing a novel environmental signal.
Genetic Machinery
The potential effects of space radiation target genetic stability of space-traveling microorganisms. Both by direct damage to DNA and indirect consequences due to ROS generation, radiation component of space environment affects genetic machinery of exposed microorganisms (Figure 2), causing a wide variety of changes starting from gene expression patterns (Table 1) and frequently leading to space-induced DNA mutagenesis (Horneck and Rabbow, 2007; Moeller et al., 2012). Global alterations in gene expression at the translational and transcriptional levels induced by adaptation of exposed microorganisms to radiation- and microgravity-filled space environment have been confirmed by proteomic and transcriptomic analyses (Figure 2), while genomics techniques revealed a number of mutant microbial strains after spaceflight (Vaishampayan et al., 2010; Chang et al., 2013a, b; Li et al., 2014; Zhang et al., 2015).
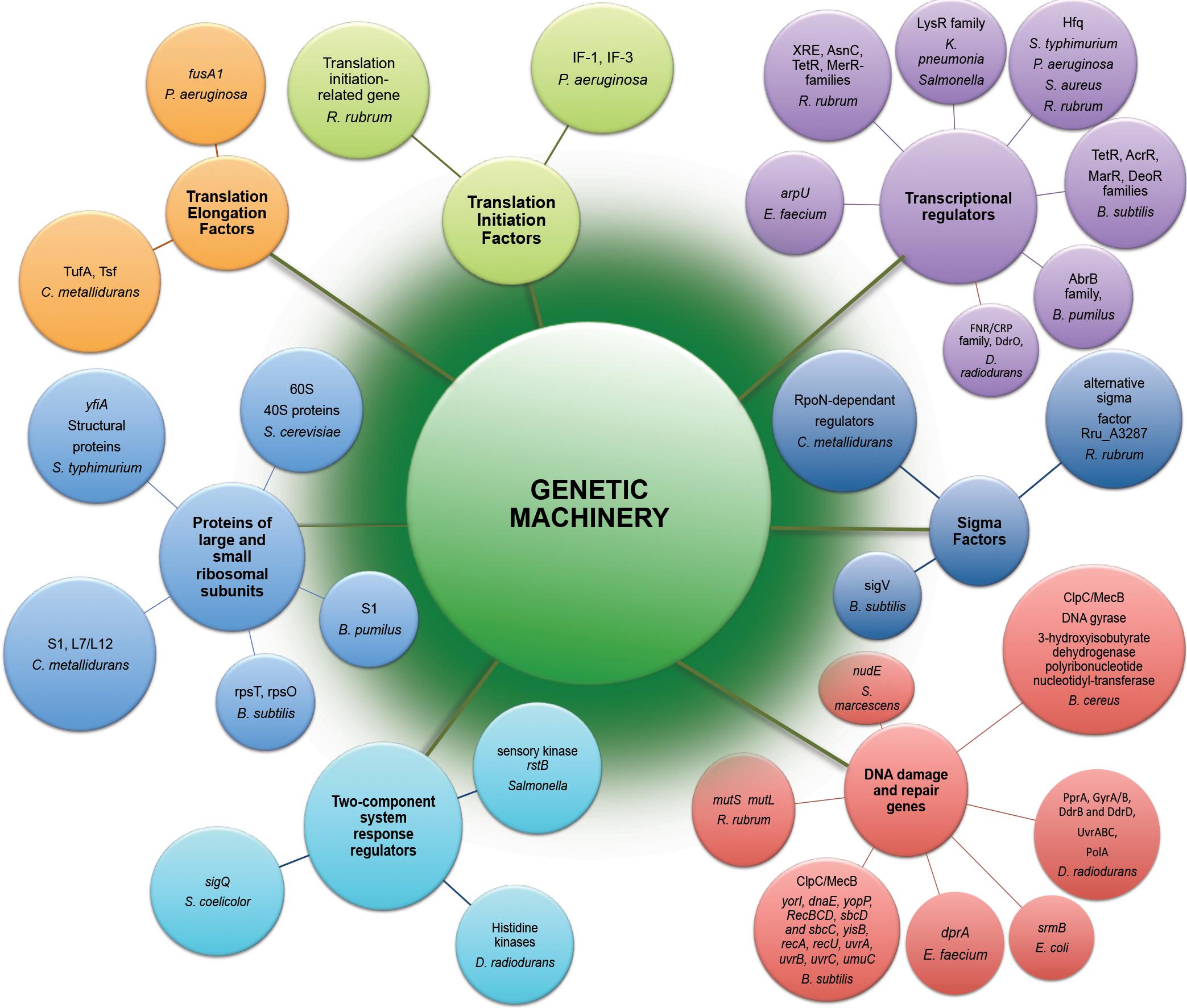
Figure 2. Genetic machinery implicated in molecular response to exposure of microorganisms to the outer space environment. Represented are differently expressed genes and differently abundant proteins of microorganisms exposed to real and simulated space conditions (Table 1) revealed by means of –omics assisted methodological approaches.
Translation, Ribosomal Structure, and Biogenesis
The multi-omics post-flight observations of K. pneumonia, E. faecium and R. rubrum cultivated on-board spacecraft revealed that the functional category “translation, ribosomal and biogenesis” was one of most highly represented category among all the up-regulated genes (Mastroleo et al., 2009; Chang et al., 2013a; Guo et al., 2015). More than 20 ribosomal protein-encoding genes of R. rubrum, 1 translation initiation-related gene, and 2 translation elongation-related genes have been found up-regulated in transcriptomic response to spaceflight (Mastroleo et al., 2009). Ribosomal apparatus of R. rubrum, P. aeruginosa, S. typhimurium, B. pumilus, B. subtilis, B. cereus, S. cerevisiae and C. metallidurans has been also massively affected in conditions of modeled microgravity (Wilson et al., 2002; Leroy et al., 2010; Mastroleo et al., 2013), spaceflight (Leys et al., 2009; Su et al., 2014; Zhang et al., 2015) and in real space conditions (Nicholson et al., 2012; Vaishampayan et al., 2012) with a number of down-regulated ribosomal genes and proteins as suggested by the multi-omics based analysis. Of 115 genes of P. aeruginosa that were down-regulated during spaceflight, 40 were involved in the synthesis of ribosomes (Crabbé et al., 2011). The P. aeruginosa genes encoding translational elongation factor (fusA1) and translation initiation factors IF-1 and IF-3 were down-regulated in transcriptomic response to spaceflight cultivation (Crabbé et al., 2011). Several enzymes involved in ribosomal protein translation (translation elongation factors TufA, Tsf) were more abundantly represented in proteomic analysis of cells of C. metallidurans after the exposure to spaceflight within the MESSAGE experiments (Leys et al., 2009).
The elevated level of translation-related genes and proteins (Figure 2) can support the higher abundances of proteins related to the metabolic and stress responses in microorganisms exposed to space conditions.
Transcription
One of the most noteworthy and challenging matters in the understanding of the effects of space environmental factors is the identification of global master regulators of space-induced response (Figure 2), that serve to globally reprogram microbial physiology in order to permit the adaptation of microorganisms to the space environment.
Transcriptomic analysis of R. rubrum (Mastroleo et al., 2009) cultivated aboard spacecraft, B. subtilis (Nicholson et al., 2012) and B. pumilus (Vaishampayan et al., 2012) spores exposed to outer space conditions and Salmonella in LSMMG conditions (Wilson et al., 2002) revealed a wide repertoire of transcriptional regulators involved in response to a long-term exposure to outer space. Interestingly, genomic analysis of E. faecium (Chang et al., 2013a) and K. pneumonia (Guo et al., 2015) mutant strains after spaceflight identified point mutations in genes encoding transcriptional regulators, including arpU gene that plays a role in cell wall growth and division by controlling the muramidase-2 export (Chang et al., 2013a). Remarkably high in B. subtilis spores exposed to simulated and real space conditions was 24- to 29-fold up-regulation of fruR transcriptional repressor of fru operon (DeoR family), which is implicated in metabolism of carbohydrates triggered in majority of space exposed microorganisms (Nicholson et al., 2012). Transcriptional regulator of AbrB family responsible for the repression of various starvation-induced differentiation processes was up-regulated in proteomic analysis of space-exposed spores of B. pumilus (Vaishampayan et al., 2012). The elevated transcript levels of several of B. subtilis master regulators were revealed in transcriptomic analysis; the stimulation of these master stress-responsive genes was higher in space-exposed spores of B. subtilis, than in spores exposed to simulated Mars environment (Nicholson et al., 2012).
Apart from the transcription factors, the rstB gene encoding a putative membrane sensory kinase was identified in microarray analysis of Salmonella LSMMG response (Wilson et al., 2002), acting as part of a two-component system to accomplish signal transduction and reprogram the cell physiology in conditions of LSMMG. Multi-omics analyses of B. subtilis, C. metallidurans, R. rubrum, and S. coelicolor identified several sigma factors involved in response to the space conditions (Leys et al., 2009; Mastroleo et al., 2009; Nicholson et al., 2012; Huang et al., 2015). Proteometabolomic analysis of D. radiodurans exposed to simulated space conditions showed the involvement of the transcriptional regulator of FNR/CRP family and DdrO, the transcriptional regulator of HTH_3 family in response to UVC/vacuum combined stress (Ott et al., 2017). The RNA chaperone and global regulator Hfq (Sauter et al., 2003; Vogel and Luisi, 2011; Sauer, 2013) has been directly involved in mechanisms of spaceflight and LSMMG microbial responses (Wilson et al., 2007, 2008; Mastroleo et al., 2009; Castro et al., 2011; Crabbé et al., 2011) (Figure 2). Apart from the highly abundant transcriptional regulator of FNR/CRP family, specific histidine kinases might be also involved in the regulation of vacuum stress response in D. radiodurans as suggested by proteomic analysis (Ott et al., 2019b).
The adaptation of microorganisms to the spaceflight conditions and microbial survivability in the outer space environment is realized in the complex of responses, which are controlled under the regulation of specific genetic elements – master regulators of transcriptional response. The –omics based analyses revealed a number of upstream transcriptional regulators which affect downstream gene expression activity in response to space environmental parameters.
DNA Replication, Recombination, and Repair
Extremophilic microorganisms in the outer space environment tolerate radiation stress and cope with radiation-induced DNA damage by means of their DNA repair molecular machinery (Figure 2). A comparative multi-omic analysis of B. cereus and E. faecium strains after spaceflight (Chang et al., 2013a; Su et al., 2014) and transcriptomic analysis of B. subtilis long-termly exposed to simulated Martian and real outer space conditions (Nicholson et al., 2012) showed that the functional category of DNA replication, recombination, and repair was among the most abundantly represented categories of proteins and genes differently transcribed in comparison with a control ground strains. The DNA mismatch repair genes mutS and mutL were up-regulated in transcriptomic response to space flight of R. rubrum (Mastroleo et al., 2009). Putative replicative DNA helicase (yorI) and DNA polymerase III alpha-subunit 3 (dnaE) were 3- to 14-fold up-regulated in space-exposed and simulated Mars-exposed B. subtilis spores (Nicholson et al., 2012). The srmB gene encoding DNA helicase was up-regulated in E. coli under simulated microgravity conditions (Arunasri et al., 2013). A number of other up-regulated genes associated with DNA repair and recombination were identified in transcriptional response of B. subtilis to real and simulated space conditions (Figure 2) (Nicholson et al., 2012). Especially remarkable was 44-fold up-regulation of the gene umuC (yqjW) encoding Y-family DNA polymerase, responsible for translesion bypass DNA repair in space-exposed B. subtilis spores (Nicholson et al., 2012).
A recent proteomic analysis of D. radiodurans exposed to simulated space conditions indicated the up-regulation of DNA damage response proteins PprA, GyrA/B, DdrB and DdrD in UVC/vacuum-affected cells, along with high constitutive RecA levels (Ott et al., 2017). Moreover, UVC/vacuum stress conditions stimulated a number of proteins involved in detoxification process and aimed to remove damaged nucleotides from D. radiodurans cells (e.g., UvrB, a helixase subunit of the DNA excision repair endonuclease complex, MutT/nudix, and MutS2 families proteins involved in mismatch excision repair). The abundances of the proteins (recQ and ruvABC) responsible for recombinational DNA repair and the Mrr restriction system protein were also significantly increased (Ott et al., 2017). Furthermore, an increase in proteins of the UvrABC nucleotide excision repair machinery and polymerase PolA was observed during the 1st hours of recovery D. radiodurans after 90 days of exposure to simulated vacuum conditions of Low Earth Orbit (proteomic-based analyses in Ott et al., 2019b).
Obviously, “space travelers” utilize a striking up-regulation of DNA damage response genes and proteins as an important strategy to cope with space radiation induced DNA damage. Various DNA damage response systems react in order to handle with the stressful situation caused by spaceflight, outer space exposure and simulated space conditions.
General Cellular Responses
Among other characteristics of the influence of the space environment on microbial cell behavior and physiology are numerous stress responses, altered tellurium resistance, biofilm formation, sporulation and virulence of several opportunistic and obligate pathogenic microorganisms.
General Stress Response
Proteins of general stress response function to protect cells, restore damage to cellular and molecular structures (e.g., DNA, the cell envelope, and proteins), and to provide microorganisms the ability to recover from the stress they experience. Overexpression of stress response genes was observed by real-time-PCR in E. coli under modeled reduced gravity conditions (Vukanti and Leff, 2012), in proteomic response of C. metallidurans grown aboard spacecraft (Leys et al., 2009) and in multi-omics analysis of R. rubrum exposed to spaceflight and simulated microgravity (Mastroleo et al., 2009, 2013) (Figure 3). Induction of stress-responsive proteins with the function of stress/protein folding and oxidative stress has been shown in proteomic analysis of S. cerevisiae aboard Soyuz TMA-9 (Van Mulders et al., 2011). Systems for monitoring protein quality within the cell devoted to control protein damage and misfolding implement in transcriptomic-assisted stress response of B. subtilis to real space and simulated Martian conditions (Nicholson et al., 2012) and proteomic response of S. cerevisiae to microgravity conditions during short-term spaceflight (Van Mulders et al., 2011) (Figure 3). The increased abundances of a number of chaperons and proteases were observed in proteomic response of D. radiodurans to UVC/vacuum (Ott et al., 2017), implying the involvement of proteolytic regulation and quality monitoring in response to simulated space conditions. An increased abundance of S. cerevisiae ubiquitin in microgravity conditions was detected in proteomic analysis, indicating an increased degradosome activity and suggesting that cells of exposed microorganisms are prone to experience protein damage and/or misfolding as a consequence of exposure to space parameters (Van Mulders et al., 2011).
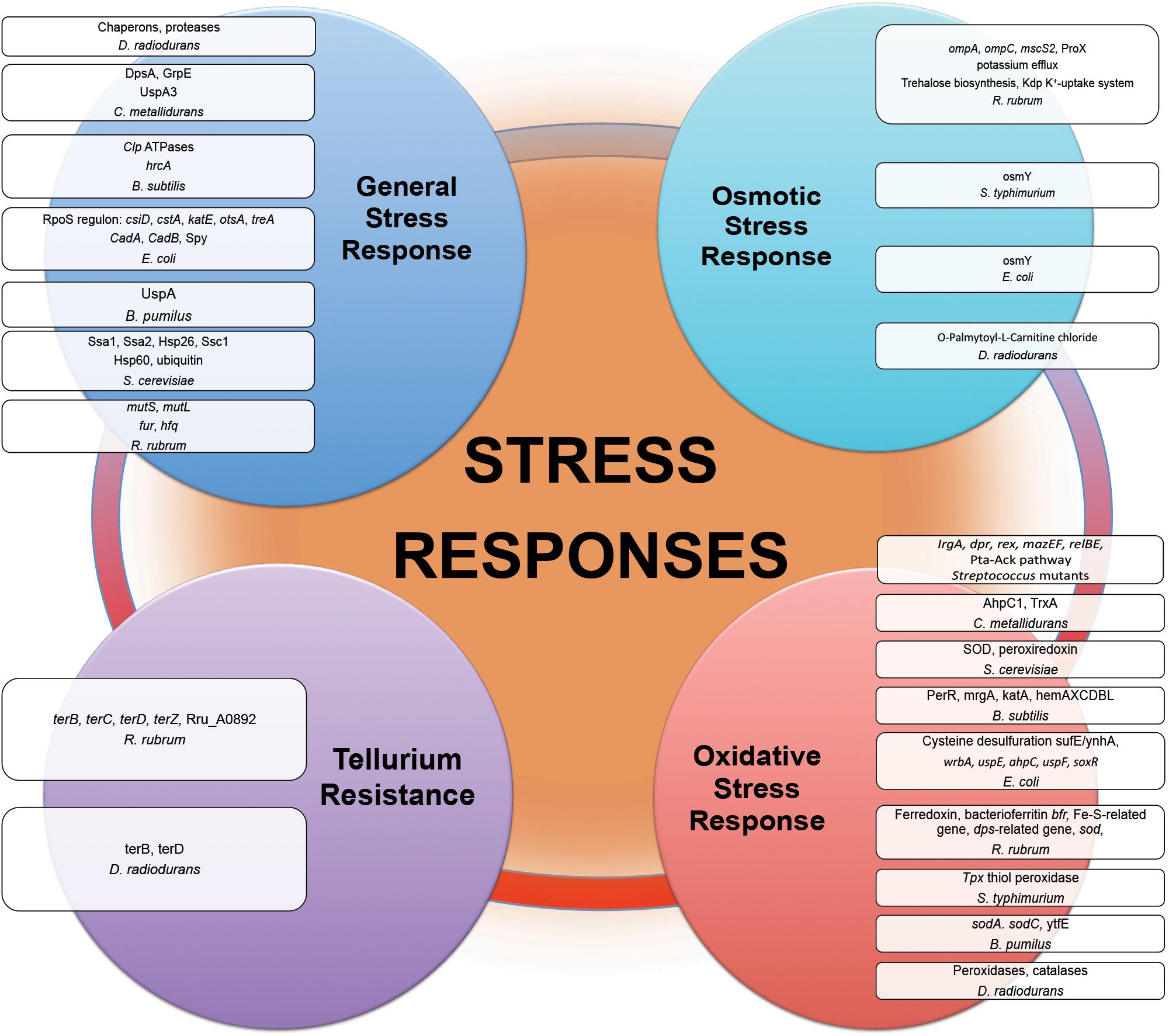
Figure 3. Stress responses experienced by microorganisms in outer space real and simulated conditions. Proteins and genes of numerous stress responses with altered abundance and expression after exposure of microorganisms (Table 1) to the outer space real and simulated environment.
Various heat shock proteins and numerous chaperone proteins are frequently more abundantly represented in exposed microorganisms. Being involved in diverse metabolic processes and responsible for protein folding, these proteins can prevent or reverse protein misfolding. By binding to proteins, which are misfolded and damaged in response to the outer space environmental stresses, these molecular chaperones can direct the misfolded proteins to the associated proteases for degradation. The elevated level of different types of proteases under the influence of space-associated environmental factors indicates the involvement of quality monitoring and proteolytic regulation in response to outer space environmental stress.
Oxidative Stress Response
Comparative -omics assisted investigations revealed various universal ROS scavengers, e.g., superoxide dismutase (SOD), and redox active proteins (peroxiredoxin, thiol peroxidase, thioredoxin, catalase, sulfoxide reductase MsrA) induced in “real” space-exposed spores of B. pumilus and B. subtilis; E. coli, R. rubrum, and S. typhimurium on-board of spacecraft, and D. radiodurans exposed to space simulating conditions, manifesting the up-regulation of antioxidant defense mechanisms during long-term spaceflight (Figure 3) (Wilson et al., 2008; Mastroleo et al., 2009; Nicholson et al., 2012; Vaishampayan et al., 2012; Ott et al., 2017). The PerR regulon consisting of a cluster of genes, which are stimulated in response to oxidative stress, was activated in transcriptomic analysis of B. subtilis spores long-termly exposed to ionizing radiation, vacuum, and extreme desiccation in outer space (Nicholson et al., 2012). Being essential for the resistance to oxidative stress, SOD (Fukai and Ushio-Fukai, 2011) by scavenging the superoxide (O2–) radical, provides an efficient antioxidant defense in conditions of outer space. In opposite to its bacterial counterpart under the long-term influence of real space factors, SOD from spaceflight microgravity-exposed yeast S. cerevisiae along with the other oxidative stress protein peroxiredoxin was down-regulated in proteomic analysis after short-time space flight (Van Mulders et al., 2011). Together with the suppression of enzymes involved in oxidative metabolism, SOD down-regulation in microgravity conditions may indicate the shift of S. cerevisiae to anaerobiosis during spaceflight. P. aeruginosa has also shown an adaptation to anaerobic mode of growth during spaceflight with a number of up-regulated genes involved in anaerobic metabolism (Crabbé et al., 2011). The increased abundances of ROS scavenging proteins, e.g., peroxidases and catalases, were observed in proteomic analysis of D. radiodurans exposed to simulated Low Earth Orbit vacuum conditions (Ott et al., 2019b). The stress response genes contribute to the increased antibiotic tolerance of E. coli in microgravity during spaceflight aboard the ISS, as suggested by RNA-Seq assisted analysis (Aunins et al., 2018).
Mining data from –omics-assisted studies clearly shows that antioxidant defense mechanisms are important part of microbial responses to cope with space-induced oxidative damage.
Osmotic Stress Response
Results of post-flight proteotranscriptomic analysis of E. coli and R. rubrum indicated the altered expression of a number of genes involved in solute transport and osmotic regulation (Figure 3) (Mastroleo et al., 2009; Li et al., 2015; Zhang et al., 2015). The osmoprotectant glycine betaine transporter ProX of R. rubrum has been indicated as one of the very few proteins up-regulated in proteomic response under the conditions of simulation of ISS-ionizing radiation (Mastroleo et al., 2009). Osmotically inducible proteins were among up-regulated proteins in proteomic analyses of S. typhimurium and E. coli after spaceflight (Wilson et al., 2007; Zhang et al., 2015). S. typhimurium grown in conditions of modeled microgravity displayed increased resistance to multiple environmental stresses, including resistance to osmotic stress (Wilson et al., 2002). Notably, metabolomics analysis of cells of D. radiodurans exposed to simulated space conditions revealed the elevated level of a palmitoyl-derivative of carnitine, a quaternary amine compound with various physiological effects (Ott et al., 2017). Carnitine is a compatible solute and important osmoprotectant, which can augment thermotolerance, cryotolerance and barotolerance, thus influencing bacterial survival in extreme conditions (Meadows and Wargo, 2015). This compatible solute can help to cope with osmotic stress as a damaging desiccation effect of vacuum (Horneck et al., 2010; Frosler et al., 2017) by binding additional water molecules, stabilizing proteins and cell membranes, and thus blocking complete desiccation of the cell. The up-regulation of O-Palmitoyl-L-Carnitine chloride was suggested to play a role in the defense of D. radiodurans against combined stress conditions of UVC and vacuum (Ott et al., 2017).
Space environmental parameters inflict a cellular stress state that has the characteristics similar to an osmotic stress. Along with an activation of cell wall-associated machinery and integrity pathways, the production of osmoprotective compounds (e.g., compatible solutes) that increase the osmotolerance serve as a microbial strategy to cope with outer space environmental stressors.
Tellurium Resistance
Tellurium resistance does not necessarily constitute a distinct resistance determinant in microorganisms, but it may represent a resulting effect of a specific metabolic function, such as oxidative stress response (Taylor, 1999; Chasteen et al., 2009). Post-flight analysis of tellurium resistance of R. rubrum sent to the ISS in frames of MELiSSA project and in conditions of modeled microgravity indicated an enhanced expression of genes involved in tellurium resistance in transcriptomic analysis (Figure 3) (Mastroleo et al., 2009). The tellurium resistance proteins TerB and TerZ are associated with the resistance of E. coli against various damaging agents (e.g., heavy metal ions and UV radiation), and contribute to the preservation of the intracellular reducing environment, probably by directly reversing disulfide bonds (Slade and Radman, 2011). Oxidative stress-responsive proteins within tellurium resistance operon in D. radiodurans were found to be up-regulated immediately after gamma-irradiation (Slade and Radman, 2011). Moreover, TerB and TerD were up-regulated in proteomic response of D. radiodurans exposed to UVC/vacuum simulated space conditions (Ott et al., 2017). The enhanced tellurium resistance of exposed microorganisms can be either linked to another metabolic function or a part of metal sensing stress response system.
Sporulation
Frequently, space microbiology concentrates on spore-forming bacteria such as Bacillus due to the remarkable resistance of their spores to harsh conditions (Nicholson et al., 2000). Antibiotic-producing and spore-forming Streptomyces also represent additional interest due to the modulation of secondary metabolites production in space environment. The sporulation process of microgravity exposed S. coelicolor was intensified during spaceflight, and increased accumulation of the gray spore pigment and the fastened transition from aerial hyphae to mature spores were observed on-board the SHENZHOU-8 spacecraft (Huang et al., 2015). Global transcriptional analysis revealed the differential expression of genes involved in morphological differentiation and development of streptomycetes, which are mainly linked to aerial hyphae erection, sporulation, spore germination, cell wall structure, spore structure, and development-associated secondary sigma factors (Huang et al., 2015).
Proteotranscriptomic analysis of B. cereus after short-term spaceflight showed the down-regulation protein MreB, which determines rod-shape of B. cereus (Su et al., 2014). A number of genes involved in sporulation were induced in transcriptional analysis during germination of B. subtilis spores after the exposure to simulated Martian and real space conditions (Nicholson et al., 2012). Significant up-regulation of the cgeAB operon involved in maturation of the outermost spore layer together with increased transcript levels of genes encoding minor and major acid-soluble spore proteins has been observed in this study. The genes cotG, cotT, and cotVWXY encoding protein components of the spore coat, the safAcoxA operon responsible for spore morphogenesis and spore cortex formation, and rapAphrA operon which controls initiation of sporulation were also significantly up-regulated in B. subtilis (Nicholson et al., 2012). Comparative proteomics analysis indicated that the outer spore coat protein A was modulated in spores of B. pumilus exposed to the UV- space–and UV-Mars–conditions (Vaishampayan et al., 2012).
Sporulation is a widely used strategy that helps to various spore-forming microorganisms to adapt and survive in harsh conditions of outer space. –Omics-assisted investigations helped to reveal the various molecular components involved in sporulation across different microbial species after exposure to the space environment.
Pathogenicity, Virulence, and Biofilm Formation
The molecular mechanisms underlying alterations of microbial virulence in space conditions have been successfully resolved with using state-of-the-art –omics technologies (Figure 4). A number of studies report that potentially pathogenic microorganisms display altered virulence and pathogenicity in real space or simulated microgravity conditions (Leys et al., 2004; Chopra et al., 2006; Taylor, 2015). Microbial morphogenic alterations have been described coherent with the enhanced pathogenicity of exposed microbes. Human opportunistic pathogen Candida albicans exhibited a morphogenic switch consistent with enhanced pathogenicity in simulated microgravity conditions (Altenburg et al., 2008). Microgravity induced the increased frequency of filamentous forms that contributes to the virulence and budding abnormalities (aberrant budding and cell clumping phenotype) of C. albicans and S. cerevisiae (Altenburg et al., 2008; Van Mulders et al., 2011). Spaceflight-cultured C. albicans exhibited random budding phenotype in accordance with the gene expression data (Crabbé et al., 2013). Genomic response analysis indicated that the expression of a set of genes involved in cell polarity and budding was significantly altered in these yeasts in simulated microgravity conditions. The conserved genes responsible for cell budding and separation were similarly down-regulated in microgravity exposed C. albicans and S. cerevisiae, thus suggesting that conservation of the genetic response takes place between the two distant yeast species (Altenburg et al., 2008; Van Mulders et al., 2011). Significant reduction in the expression of these genes appears to be consistent with the aberrant budding and cell-clumping phenotype of microgravity exposed cells. Increased virulence of S. typhimurium cultivated in spaceflight was accompanied by bacteria cellular aggregation, clumping and extracellular matrix formation, which is coherent with biofilm production (Wilson et al., 2007). Concomitantly, expression of Salmonella genes involved in biofilm formation was altered in microarray analysis during spaceflight (Wilson et al., 2007). A number of genes related to biofilm formation were up-regulated in transcriptomic profiling of B. subtilis during spaceflight aboard the ISS, which can be connected to the oxygen availability the in liquid cultures under the microgravity influence (Morrison et al., 2019).
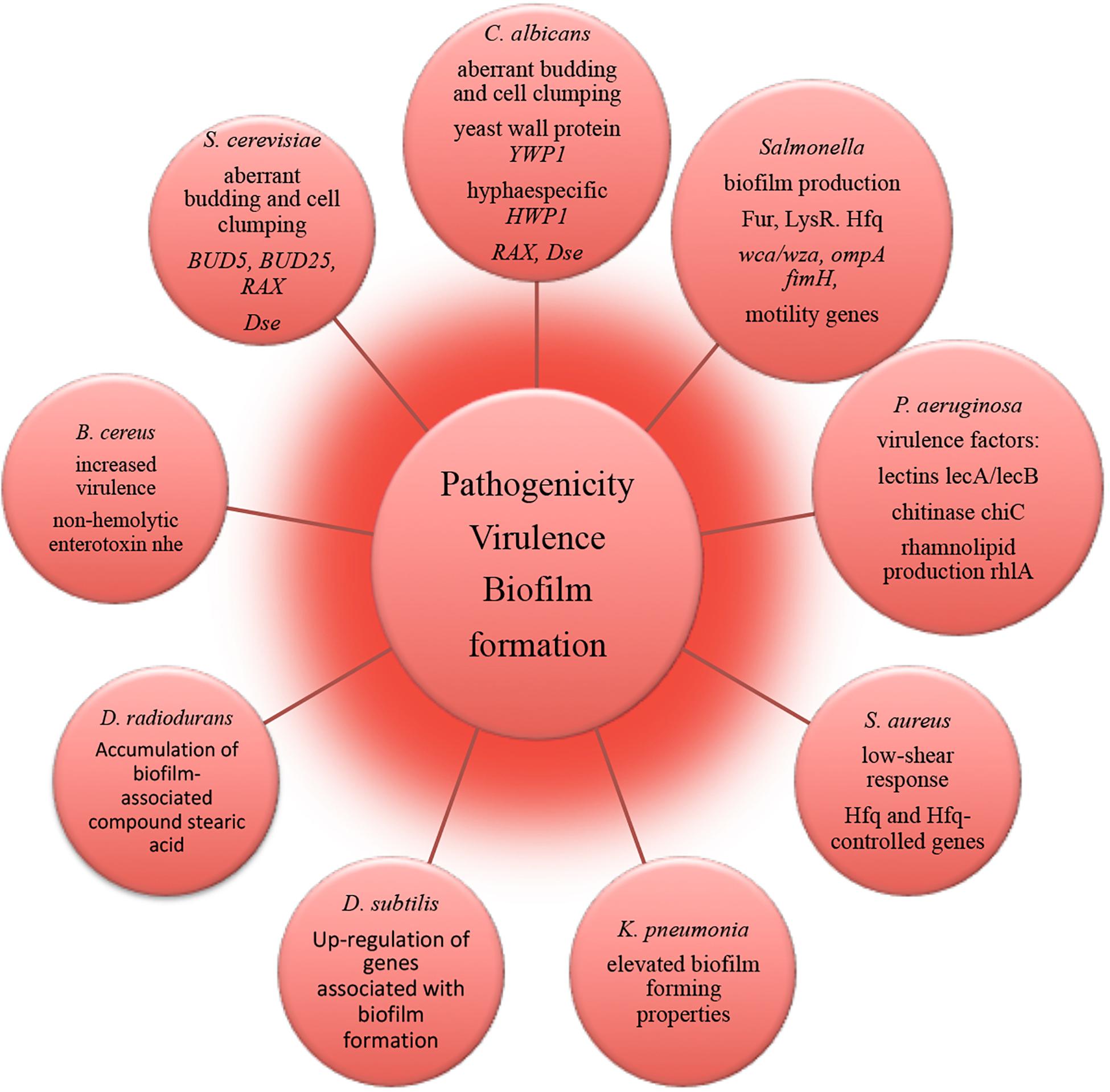
Figure 4. Molecular alterations underlying microbial pathogenicity, virulence and biofilm formation in outer space environment, resolved with –omics assisted investigations.
The strain of human pathogen K. pneumonia returned from a spaceflight exhibited elevated biofilm forming properties as an important virulence characteristic (Li et al., 2014). The amount of biofilm-associated compound stearic acid (Saravanakumari and Mani, 2010; Vecino et al., 2015; Ge et al., 2016) was significantly elevated in cells of D. radiodurans exposed to vacuum and UVC-radiation (Ott et al., 2017). Although the cells of D. radiodurans do not naturally produce stearic acid in big quantities under non-stressed conditions (Melin et al., 1998), the observed accumulation of this biofilm-associated compound may lead to the high survival of D. radiodurans in dry multilayers under UVC/vacuum combined stress by preserving the structural integrity of cell membranes in conditions of vacuum-induced dehydration.
RNA-binding global regulatory protein Hfq has been shown to coordinate a regulatory response in bacterial reprogramming during spaceflight, altering bacterial gene expression and virulence under the influence of spaceflight conditions (multi-omics analyses in Wilson et al., 2007, 2008; Mastroleo et al., 2009; Crabbé et al., 2011). P. aeruginosa and human pathogen B. cereus cultured in the microgravity environment of spacecraft responded with the induction of several genes encoding virulence factors (Crabbé et al., 2011; Su et al., 2014). The obtained results of –omics studies of biofilm formation, microbial virulence and pathogenicity in space conditions and future follow-up investigations should continue to deliver novel molecular candidates for pharmacological intervention to prevent and control infectious diseases and to identify novel targets for vaccines and therapeutic development in order to keep crewmembers safe and healthy. This will ultimately facilitate long-term interplanetary transfer and productive space exploration.
Space Systems Biology – a Framework for Integration of Outer Space Parameters With Omics Technology and Joined Mathematical Modeling
The –omics based approach has recently opened a window for a deep insight into molecular machinery implicated in survivability of space exposed microorganisms by revealing expression, metabolic functioning, and regulation of the genes and proteins encoded by the genomes of “space travelers.” Metabolic alterations mediated by genetic regulations affect the diverse biological activities of space-exposed microorganisms (Figure 1). Space induced metabolic rearrangements trigger the restoration of energy status of exposed microbial cell (Mastroleo et al., 2009; Wang et al., 2014; Zhang et al., 2015). Frequently, the exposed microbial entity is posed in “energy saving mode” by the regulatory molecular network in order to reduce the need for synthesis of cellular material in non-growing cells and complement the increased energy demands for the maintenance of genetic stability and cellular integrity in the space environment. Several global regulatory molecules have been identified which orchestrate the molecular response of few space-exposed microorganisms (Figure 2). Of especial attention is a group of hypothetical proteins of “unknown function” which are often numerically abundantly represented in space responses (over 50% of space exposed B. subtilis genes) (Nicholson et al., 2012). One of the strategies to assign novel functions for these hypothetical proteins might be the exploration of new space-related environmental and stress conditions.
Experimental parameters during space exposure affect microbial survival rates and may lead to certain discrepancies in –omics assisted analysis of returned/exposed microorganisms. The composition of cultivation medium influences the microbial space response (Benoit and Klaus, 2007; Baker and Leff, 2006; Wilson et al., 2008), for instance, by providing specific antioxidants presented in rich medium, which may protect microbial cell against ionizing radiation. The majority of space experiments have been performed on-board of spaceflights, where microorganisms are cultivated in protected environment of spacecraft (Table 1). Only a few microbial species were exposed unprotected to real space conditions outside the ISS and then subsequently investigated with –omics techniques (Nicholson et al., 2012; Vaishampayan et al., 2012). There is an increasing demand in new space experiments to broad our knowledge of molecular mechanisms of microbial survivability under the conditions of real outer space or its selected parameters. Concomitantly, the design of space exposure experiments has to be critically assessed to accommodate sufficient number of independent biological repetitions enabling a comprehensive statistical analysis of obtained –omics data. This is an extremely necessary prerequisite to avoid artifacts during the evaluation of the multitude of effects of outer space environment on microorganisms. In many instances, a multi-omics post-flight analysis faces the problem of limited amount of the microbiological samples exposed to the space environment. In this connection, the development of valid technical approaches enabling simultaneous efficient extraction of DNA, RNA, proteins, and metabolites from a minimal amount of microbial cells is highly desirable to overcome this limiting step (Weckwerth et al., 2004; Valledor et al., 2014; Ott et al., 2017). Another important issue, which requires critical reassessment, is the frequent absence of detailed reports on the environmental conditions during space exposure of microorganisms and corresponding ground control experiments. Due to the high sensitivity of the –omics techniques used in post-flight analysis and the partial occurrence of uncontrolled conditions of the space experiments, the appearance of stress-related artifacts cannot be ruled out. Providing of a record of controlled parameters (e.g., temperature, humidity, pressure profiles) during flight, simulated, and control experiments is highly anticipated to achieve a comprehensive and artifacts-free analysis of the effects of the space environment on physiology and molecular machinery of microorganisms.
In addition to the proteotranscriptomic profiling, we propose that new space experiments should include a detailed metabolomic analysis of exposed microorganisms. This novel approach provides a rich source of new findings of fine molecular network regulating the space response (Ott et al., 2017, 2019a,b). Another aspect which is not yet addressed is the combination of molecular data with a genome-scale metabolic reconstruction of the respective species which is rather routine standard nowadays for the analysis of organisms (Weckwerth, 2011b). Summarizing the comprehensive review of metabolic alterations of microorganisms in space conditions reveals a multifactorial response trajectory which is not intuitively leading to a causal understanding. Thus, it is of utmost importance to integrate isolated biochemical parts into a global statistical and mathematical model. Here, we propose hybrid modeling approaches integrating statistical regression models and genome-scale metabolic reconstruction (Weckwerth, 2019). In Figure 5 the proposed workflow for this hybrid modeling approach is shown: (i) for microorganisms with the available genome sequences genome-scale metabolic reconstruction is a straightforward approach (Thiele and Palsson, 2010; Weckwerth, 2011b); (ii) in a next step, –omics technology is applied as well as classical physiological and morphological parameter are measured to build a comprehensive statistical regression model of the response trajectory to environmental space conditions. Decisive for this regression model is the experimental design and the comprehensive monitoring of the environmental parameters. (iii) Finally, the statistical and the genome-scale models are joined by biomathematical approaches to represent a global model of metabolic regulation in outer space conditions (Weckwerth, 2019; Wilson et al., 2020). Based on a thorough hybrid modeling approach of the microbial systems in outer space as described above also metabolic engineering strategies can be developed upon revealing the elements of adaptation: from intrinsic protective mechanisms to elevated repair abilities, and a merging of these adaptation strategies.
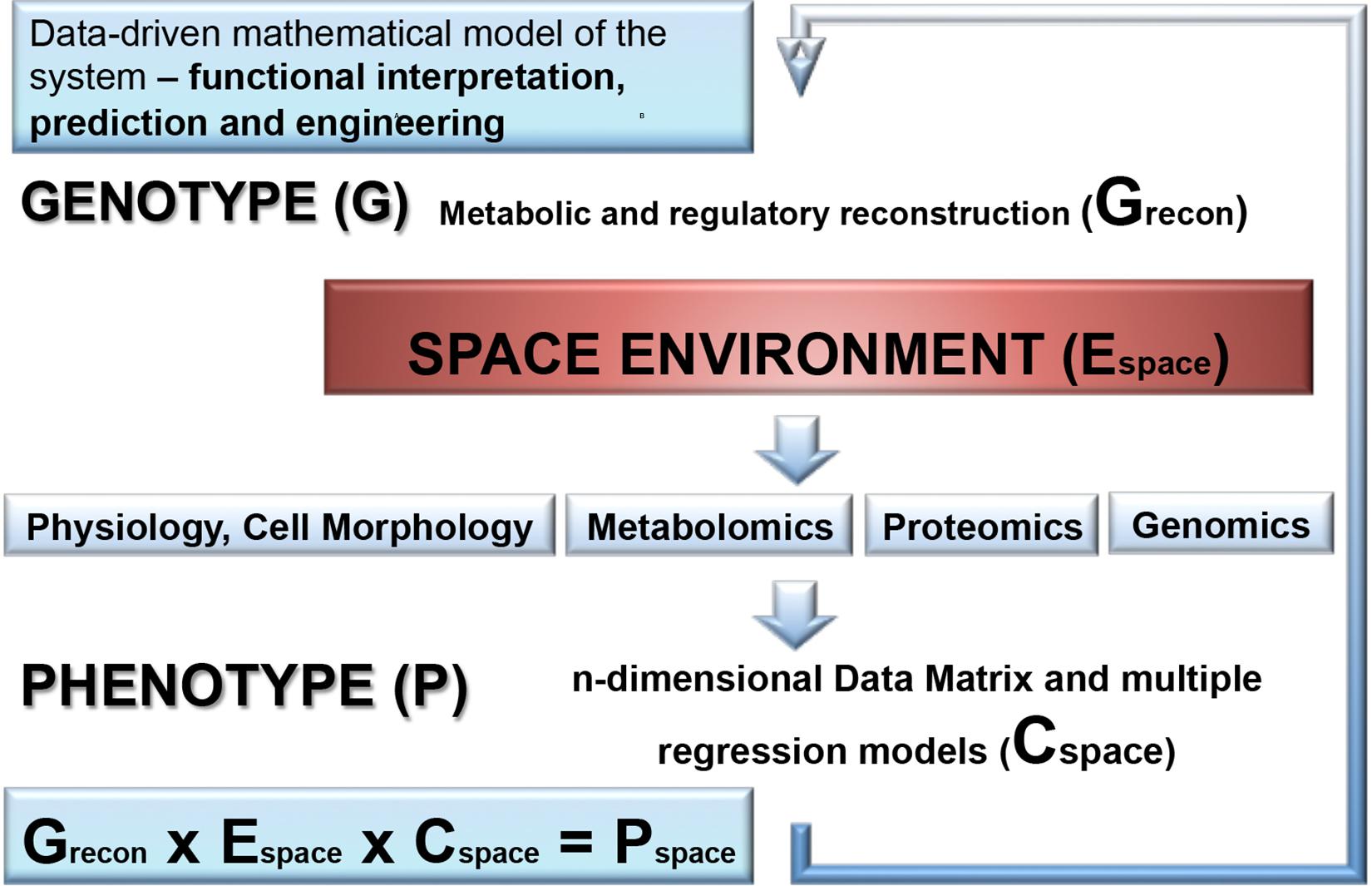
Figure 5. A combination of molecular data with a genome-scale metabolic reconstruction of the microbial species exposed to the space environment. First, genome-scale metabolic and regulatory reconstruction of the respective microorganism is performed based on the available genome sequence and gene annotation information and results into a genotype matrix (Grecon). This provides the basic predictive metabolic model to eventually integrate statistical models of molecular data generated by OMICS technology, physiological and morphological data as well as environmental parameter which are accurately monitored in outer space conditions. The molecular and physio-morphometric data are correlated with environmental parameter by multiple regression methods and generate a comprehensive data covariance matrix times environmental data matrix (Cspace × Espace). The resulting data covariance model is eventually connected with the genotype matrix Grecon to generate a biomathematical data-driven regulatory and predictive model for the response trajectory of the microorganism in outer space conditions (Pspace). Overall, this is an iterative process improving step by step the predictive models (for further information see Weckwerth et al., 2004; Weckwerth and Morgenthal, 2005; Weckwerth, 2011a, 2016, 2019; Sun and Weckwerth, 2012; Doerfler et al., 2013; Nägele et al., 2014, 2016; Sun et al., 2015).
Author Contributions
Both authors conceived the review, conducted the literature search, created the figures, wrote and revised the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The study was conducted within the MOMEDOS (Molecular Mechanisms of Deinococcus radiodurans survivability in Outer Space) project 854013, funded by the FFG (Österrei- chische Forschungsförderungsgesellschaft—https://www.ffg.at/, Austrian Space Applications Programme ASAP) to TM.
Acknowledgments
The authors would like to acknowledge 18th EANA Conference European Astrobiology Network Association (Freie Universität Berlin, Germany, 24–28 September 2018) for giving a possibility to present partial data from this review article.
References
Altenburg, S. D., Nielsen-Preiss, S. M., and Hyman, L. E. (2008). Increased filamentous growth of Candida albicans in simulated microgravity. Genom. Proteom. Bioinf. 6, 42–50. doi: 10.1016/S1672-0229(08)60019-4
Arunasri, K., Adil, M., Venu Charan, K., Suvro, C., Himabindu, R. S., and Shivaji, S. (2013). Effect of simulated microgravity on E. coli K12 MG1655 growth and gene expression. PLoS One 8:e57860. doi: 10.1371/journal.pone.0057860
Aunins, T. R., Erickson, K. E., Prasad, N., Levy, S. E., Jones, A., Shrestha, S., et al. (2018). Spaceflight Modifies Escherichia coli gene expression in response to antibiotic exposure and reveals role of oxidative stress response. Front. Microbiol. 9:310. doi: 10.3389/fmicb.2018.00310
Baker, P. W., and Leff, L. (2006). Mir space station bacteria responses to modeled reduced gravity under starvation conditions. Adv Space Res. 38, 1152–1158. doi: 10.1111/j.1365-2672.2005.02593.x
Benoit, M. R., and Klaus, D. M. (2007). Microgravity, bacteria and the influence of motility. Adv. Space Res. 39, 1225–1232. doi: 10.1089/ast.2010.0536
Bertalanffy, L. V. (1940). Der organismus als physikalisches system betrachtet. Naturwissenschaften 33, 522–531.
Blachowicz, A., Chiang, A. J., Elsaesser, A., Kalkum, M., Ehrenfreund, P., Stajich, J. E., et al. (2019). Proteomic and metabolomic characteristics of extremophilic fungi under simulated mars conditions. Front. Microbiol. 10:1013. doi: 10.3389/fmicb.2019.01013
Butt, A. T., and Thomas, M. S. (2017). Iron acquisition mechanisms and their role in the virulence of burkholderia species. Front. Cell Infect. Microbiol. 7:460. doi: 10.3389/fcimb.2018.00305
Castro, S. L., Nelman-Gonzalez, M., Nickerson, C. A., and Ott, C. M. (2011). Induction of attachment-independent biofilm formation and repression of Hfq expression by low-fluid-shear culture of Staphylococcus aureus. Appl. Environ. Microbiol. 77, 6368–6378. doi: 10.1128/AEM.00175-11
Chang, D., Zhu, Y., An, L., Liu, J., Su, L., Guo, Y., et al. (2013a). A multi-omic analysis of an Enterococcus faecium mutant reveals specific genetic mutations and dramatic changes in mRNA and protein expression. BMC Microbiol. 28:304. doi: 10.1186/1471-2180-13-304
Chang, D., Zhu, Y., Fang, X., Li, T., Wang, J., Guo, Y., et al. (2013b). Draft Genome Sequences of the Enterococcus faecium Strain LCT-EF258. Genome Announc. 1, e147–e112. doi: 10.1128/genomeA.00147-12
Chasteen, T. G., Fuentes, D. E., Tantaleán, J. C., and Vásquez, C. C. (2009). Tellurite: history, oxidative stress, and molecular mechanisms of resistance. FEMS Microbiol. Rev. 33, 820–832. doi: 10.1111/j.1574-6976.2009.00177.x
Chiang, A. J., Malli Mohan, G. B., Singh, N. K., Vaishampayan, P. A., Kalkum, M., and Venkateswaran, K. (2019). Alteration of proteomes in first- generation cultures of Bacillus pumilus spores exposed to outer space. mSystems 4:e00195-19. doi: 10.1128/mSystems.00195-19
Chopra, V., Fadl, A. A., Sha, J., Chopra, S., Galindo, C. L., and Chopra, A. K. (2006). Alterations in the virulence potential of enteric pathogens and bacterial-host cell interactions under simulated microgravity conditions. J. Toxicol. Environ. Health 69, 1345–1370. doi: 10.1080/15287390500361792
Cockell, C. S., Rettberg, P., Rabbow, E., and Olsson-Francis, K. (2011). Exposure of phototrophs to 548 days in low Earth orbit: microbial selection pressures in outer space and on early earth. ISME J. 5, 1671–1682. doi: 10.1038/ismej.2011.46
Crabbé, A., Nielsen-Preiss, S. M., Woolley, C. M., Barrila, J., Buchanan, K., McCracken, J., et al. (2013). Spaceflight enhances cell aggregation and random budding in Candida albicans. PLoS One 8:e80677. doi: 10.1371/journal.pone.0080677
Crabbé, A., Schurr, M. J., Monsieurs, P., Morici, L., Schurr, J., Wilson, J. W., et al. (2011). Transcriptional and proteomic responses of Pseudomonas aeruginosa PAO1 to spaceflight conditions involve Hfq regulation and reveal a role for oxygen. Appl. Environ. Microbiol. 77, 1221–1230. doi: 10.1128/AEM.01582-10
Doerfler, H., Lyon, D., Nägele, T., Sun, X., Fragner, L., Hadacek, F., et al. (2013). Granger causality in integrated GC-MS and LC-MS metabolomics data reveals the interface of primary and secondary metabolism. Metabolomics 9, 564–574. doi: 10.1007/s11306-012-0470-0
Duscher, A. A., Conesa, A., Bishop, M., Vroom, M. M., Zubizarreta, S. D., and Foster, J. S. (2018). Transcriptional profiling of the mutualistic bacterium Vibrio fischeri and an hfq mutant under modeled microgravity. NPJ Microgravity 4:25. doi: 10.1038/s41526-018-0060-1
Frosler, J., Panitz, C., Wingender, J., Flemming, H. C., and Rettberg, P. (2017). Survival of deinococcus geothermalis in biofilms under desiccation and simulated space and martian conditions. Astrobiology 17, 431–447. doi: 10.1089/ast.2015.1431
Fukai, T., and Ushio-Fukai, M. (2011). Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal 15, 1583–1606. doi: 10.1089/ars.2011.3999
Ge, X., Shi, X., Shi, L., Liu, J., Stone, V., Kong, F., et al. (2016). Involvement of NADH oxidase in biofilm formation in Streptococcus sanguinis. PLoS One 11:e0151142. doi: 10.1371/journal.pone.0151142
Goh, K., Chua, D., Beck, B., McKee, M. L., and Bhagwat, A. A. (2011). Arginine-dependentacid-resistance pathway in Shigella boydii. Arch. Microbiol. 193, 179–185. doi: 10.1007/s00203-010-0656-7
Guo, Y., Li, J., Liu, J., Wang, T., Li, Y., Yuan, Y., et al. (2015). Effects of Space Environment on Genome, Transcriptome, and Proteome of Klebsiella pneumoniae. Arch. Med. Res. 46, 609–618. doi: 10.1016/j.arcmed.2015.11.001
Guo, Y., Li, Y., Su, L., Chang, D., Liu, W., Wang, T., et al. (2014). Comparative genomic analysis of Klebsiella pneumonia (LCT-KP214) and a mutant strain (LCT-KP289) obtained after spaceflight. BMC Genomics 12:589. doi: 10.1186/1471-2164-15-589
Hall, H. K., and Foster, J. W. (1996). The role of fur in the acid tolerance response of Salmonella typhimurium is physiologically and genetically separable from its role in iron acquisition. J. Bacteriol. 178, 5683–5691. doi: 10.1128/jb.178.19.5683-5691.1996
Hantke, K. (1987). Selection procedure for deregulated iron transport mutants (fur) in Escherichia coli K 12: fur not only affects iron metabolism. Mol. Gen. Genet. 210, 135–139. doi: 10.1007/bf00337769
Hildebrandt, T., Knuesting, J., Berndt, C., Morgan, B., and Scheibe, R. (2015). Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 396, 523–537. doi: 10.1515/hsz-2014-0295
Horneck, G., Klaus, D. M., and Mancinelli, R. L. (2010). Space microbiology. Microbiol. Mol. Biol. Rev. 74, 121–156. doi: 10.1128/MMBR.00016-09
Horneck, G., and Rabbow, E. (2007). Mutagenesis by outer space parameters other than cosmic rays. Adv. Space Res. 40, 445–454. doi: 10.1016/j.asr.2006.12.049
Horneck, G., Stöffler, D., Ott, S., Hornemann, U., Cockell, C. S., Moeller, R., et al. (2008). Microbial rock inhabitants survive hypervelocity impacts on Mars-like host planets: first phase of lithopanspermia experimentally tested. Astrobiology 1, 17–44. doi: 10.1089/ast.2007.0134
Huang, B., Liu, N., Rong, X., Ruan, J., and Huang, Y. (2015). Effects of simulated microgravity and spaceflight on morphological differentiation and secondary metabolism of Streptomyces coelicolor A3(2). Appl. Microbiol. Biotechnol. 99, 4409–4422. doi: 10.1007/s00253-015-6386-7
Ideker, T., Galitski, T., and Hood, L. (2001). A new approach to decoding life: systems biology. Annu. Rev. Genom. Hum. G. 2, 343–372. doi: 10.1146/annurev.genom.2.1.343
Kawaguchi, Y., Yang, Y., Kawashiri, N., Shiraishi, K., Takasu, M., Narumi, I., et al. (2013). The possible interplanetary transfer of microbes: assessing the viability of Deinococcus spp. under the ISS Environmental conditions for performing exposure experiments of microbes in the Tanpopo mission. Orig. Life Evol. Biosph. 43, 411–428. doi: 10.1007/s11084-013-9346-1
Lee, J. W., and Helmann, J. D. (2007). Functional specialization within the Fur family of metalloregulators. Biometals 20, 485–499. doi: 10.1007/s10534-006-9070-7
Leroy, B., Rosier, C., Erculisse, V., Leys, N., Mergeay, M., and Wattiez, R. (2010). Differential proteomic analysis using isotope-coded protein-labeling strategies: comparison, improvements and application to simulated microgravity effect on Cupriavidus metallidurans CH34. Proteomics 10, 2281–2291. doi: 10.1002/pmic.200900286
Levine, R. L., Moskovitz, J., and Stadtman, E. R. (2000). Oxidation of methionine in proteins: roles in antioxidant defense and cellular regulation. IUBMB Life 50, 301–307. doi: 10.1080/713803735
Leys, N., Baatout, S., Rosier, C., Dams, A., Heeren, C., Wattiez, R., et al. (2009). The response of Cupriavidus metallidurans CH34 to spaceflight in the international space station. Antonie Van Leeuwenhoek 96, 227–245. doi: 10.1007/s10482-009-9360-5
Leys, N. M., Hendrickx, L., De Boever, P., Baatout, S., and Mergeay, M. (2004). Space flight effects on bacterial physiology. J. Biol. Regul. Homeost. Agents 18, 193–199.
Li, J., Liu, F., Wang, Q., Ge, P., Woo, P. C., Yan, J., et al. (2014). Genomic and transcriptomic analysis of NDM-1 Klebsiella pneumoniae in spaceflight reveal mechanisms underlying environmental adaptability. Sci. Rep. 4:6216. doi: 10.1038/srep06216
Li, T., Chang, D., Xu, H., Chen, J., Su, L., Guo, Y., et al. (2015). Impact of a short-term exposure to spaceflight on the phenotype, genome, transcriptome and proteome of Escherichia coli. Int. J. Astrobiology 14, 435–444. doi: 10.1017/S1473550415000038
Liang, X., Zhang, L., Natarajan, S. K., and Becker, D. F. (2013). Proline mechanisms of stress survival. Antioxid. Redox Signal 20, 998–1011. doi: 10.1089/ars.2012.5074
Mastroleo, F., Van Houdt, R., Atkinson, S., Mergeay, M., Hendrickx, L., Wattiez, R., et al. (2013). Modelled microgravity cultivation modulates N-acylhomoserine lactone production in Rhodospirillum rubrum S1H independently of cell density. Microbiology 159, 2456–2466. doi: 10.1099/mic.0.066415-0
Mastroleo, F., Van Houdt, R., Leroy, B., Benotmane, M. A., Janssen, A., Mergeay, M., et al. (2009). Experimental design and environmental parameters affect Rhodospirillum rubrum S1H response to space flight. ISME J. 3, 1402–1419. doi: 10.1038/ismej.2009.74
Meadows, J. A., and Wargo, M. J. (2015). Carnitine in bacterial physiology and metabolism. Microbiology 161, 1161–1174. doi: 10.1099/mic.0.000080
Melin, A. M., Peuchant, E., Perromat, A., and Clerc, M. (1998). Sensitivity to oxidative damage of two Deinococcus radiodurans strains. J. Appl. Microbiol. 84, 531–537. doi: 10.1046/j.1365-2672.1998.00376.x
Minic, Z. (2015). Proteomic studies of the effects of different stress conditions on central carbon metabolism in microorganisms. J. Proteomics Bioinform. 8, 80–90. doi: 10.4172/jpb.1000355
Moeller, R., Reitz, G., The Protect Team Nicholson, W. L., and Horneck, G. (2012). Mutagenesis in bacterial spores exposed to space and simulated martian conditions: data from the EXPOSE-E spaceflight experiment PROTECT. Astrobiology 12, 457–468. doi: 10.1089/ast.2011.0739
Morgenthal, K., Wienkoop, S., Scholz, M., Selbig, J., and Weckwerth, W. (2005). Correlative GC-TOF-MS-based metabolite profiling and LC-MS-based protein profiling reveal time-related systemic regulation of metabolite-protein networks and improve pattern recognition for multiple biomarker selection. Metabolomics 1, 109–121.
Morrison, M. D., Fajardo-Cavazos, P., and Nicholson, W. L. (2019). Comparison of Bacillus subtilis transcriptome profiles from two separate missions to the international space station. NPJ Microgravity 5:1. doi: 10.1038/s41526-018-0061-0
Nägele, T., Furtauer, L., Nagler, M., Weiszmann, J., and Weckwerth, W. (2016). A strategy for functional interpretation of metabolomic time series data in context of metabolic network information. Front. Mol. Biosci. 3:6. doi: 10.3389/fmolb.2016.00006
Nägele, T., Mair, A., Sun, X., Fragner, L., Teige, M., and Weckwerth, W. (2014). Solving the differential biochemical Jacobian from metabolomics covariance data. PLoS One 9:e92299. doi: 10.1371/journal.pone.0092299
Nicholls, C., Li, H., and Liu, J. P. (2012). GAPDH: a common enzyme with uncommon functions. Clin. Exp. Pharmacol. Physiol. 39, 674–679. doi: 10.1111/j.1440-1681.2011.05599.x
Nicholson, W. L. (2009). Ancient micronauts: interplanetary transport of microbes by cosmic impacts. Trends Microbiol. 17, 243–250. doi: 10.1016/j.tim.2009.03.004
Nicholson, W. L., Moeller, R., Protect Team, and Horneck, G. (2012). Transcriptomic responses of germinating Bacillus subtilis spores exposed to 1.5 years of space and simulated martian conditions on the EXPOSE-E experiment PROTECT. Astrobiology 12, 469–486. doi: 10.1089/ast.2011.0748
Nicholson, W. L., Munakata, N., Horneck, G., Melosh, H. J., and Setlow, P. (2000). Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol. Mol. Biol. Rev. 64, 548–572. doi: 10.1128/mmbr.64.3.548-572.2000
Orsini, S. S., Lewis, A. M., and Rice, K. C. (2017). Investigation of simulated microgravity effects on Streptococcus mutans physiology and global gene expression. NPJ Microgravity 3:4. doi: 10.1038/s41526-016-0006-4
Ott, E., Kawaguchi, Y., Kölbl, D., Chaturvedi, P., Yamagishi, N. K., et al. (2017). Proteometabolomic response of Deinococcus radiodurans exposed to UVC and vacuum conditions: initial studies prior to the Tanpopo space mission. PLoS One 12:e0189381. doi: 10.1371/journal.pone.0189381
Ott, E., Fuchs, F., Moeller, R., Hemmersbach, R., Kawaguchi, Y., Yamagishi, A., et al. (2019a). Molecular response of Deinococcus radiodurans to simulated microgravity explored by proteometabolomic approach. Sci. Rep. 9:18462. doi: 10.1038/s41598-019-54742-6
Ott, E., Kawaguchi, Y., Özgen, N., Yamagishi, A., Rabbow, E., Rettberg, P., et al. (2019b). Proteomic and metabolomic profiling of Deinococcus radiodurans recovering after exposure to simulated low earth orbit vacuum conditions. Front. Microbiol. 10:909. doi: 10.3389/fmicb.2019.00909
Palaniappan, C., Taber, H., and Meganathan, R. (1994). Biosynthesis of o-succinylbenzoic acid in Bacillus subtilis: identification of menD mutants and evidence against the involvement of the alpha-ketoglutarate dehydrogenase complex. J. Bacteriol. 176, 2648–2653. doi: 10.1128/jb.176.9.2648-2653.1994
Paulsen, C. E., and Carroll, K. S. (2013). Cysteine-mediated redox signaling: chemistry, biology and tools for discovery. Chem. Rev. 113, 4633–4679. doi: 10.1021/cr300163e
Rinaldo, S., Giardina, G., Mantoni, F., Paone, A., and Cutruzzolà, F. (2018). Beyond nitrogen metabolism: nitric oxide, cyclic-di-GMP and bacterial biofilms. FEMS Microbiol. Lett. 365:6. doi: 10.1093/femsle/fny029
Saffary, R., Nandakumar, R., Spencer, D., Robb, F. T., Davila, J. M., Swartz, M., et al. (2002). Microbial survival of space vacuum and extreme ultraviolet irradiation: strain isolation and analysis during a rocket flight. FEMS Microbiol. Lett. 215, 163–168. doi: 10.1111/j.1574-6968.2002.tb11386.x
Sancho, L. G., de la Torre, R., Horneck, G., Ascaso, C., de Los Rios, A., Pintado, A., et al. (2007). Lichens survive in space: results from the 2005 LICHENS experiment. Astrobiology 3, 443–454. doi: 10.1089/ast.2006.0046
Saravanakumari, P., and Mani, K. (2010). Structural characterization of a novel xylolipid biosurfactant from Lactococcus lactis and analysis of antibacterial activity against multi-drug resistant pathogens. Biores. Technol. 101, 8851–8854. doi: 10.1016/j.biortech.2010.06.104
Sauer, E. (2013). Structure and RNA-binding properties of the bacterial LSm protein Hfq. RNA Biol. 10, 610–618. doi: 10.4161/rna.24201
Sauter, C., Basquin, J., and Suck, D. (2003). Sm-like proteins in Eubacteria: the crystal structure of the Hfq protein from Escherichia coli. Nucleic Acids Res. 31, 4091–4098. doi: 10.1093/nar/gkg480
Selbmann, L., Zucconi, L., Isola, D., and Onofri, S. (2015). Rock black fungi: excellence in the extremes, from the Antarctic to space. Curr. Genet. 3, 335–345. doi: 10.1007/s00294-014-0457-7
Sirover, M. A. (2012). Subcellular dynamics of multifunctional protein regulation: mechanisms of GAPDH intracellular translocation. J. Cell Biochem. 113, 2193–2200. doi: 10.1002/jcb.24113
Sirover, M. A. (2014). Structural analysis of glyceraldehyde-3-phosphate dehydrogenase functional diversity. Int. J. Biochem Cell Biol. 57, 20–26. doi: 10.1016/j.biocel.2014.09.026
Slade, D., and Radman, M. (2011). Oxidative stress resistance in Deinococcus radiodurans. Microbiol. Mol. Biol. Rev. 75, 133–191. doi: 10.1128/MMBR.00015-10
Sturme, M. H., Kleerebezem, M., Nakayama, J., Akkermans, A. D., Vaugha, E. E., and de Vos, W. M. (2002). Cell to cell communication by autoinducing peptides in gram-positive bacteria. Antonie Van Leeuwenhoek 81, 233–243. doi: 10.1023/a:1020522919555
Su, L., Zhou, L., Liu, J., Cen, Z., Wu, C., Wang, T., et al. (2014). Phenotypic, genomic, transcriptomic and proteomic changes in Bacillus cereus after a short-term space flight. Adv. Space Res. 53, 18–29. doi: 10.1016/j.asr.2013.08.001
Sun, X., Langer, B., and Weckwerth, W. (2015). Challenges of inversely estimating jacobian from metabolomics data. Front. Bioeng. Biotechnol. 3:188. doi: 10.3389/fbioe.2015.00188
Sun, X., and Weckwerth, W. (2012). COVAIN: a toolbox for uni- and multivariate statistics, time-series and correlation network analysis and inverse estimation of the differential Jacobian from metabolomics covariance data. Metabolomics 8, 81–93. doi: 10.1007/s11306-012-0399-3
Taylor, D. E. (1999). Bacterial tellurite resistance. Trends Microbiol. 7, 111–115. doi: 10.1016/s0966-842x(99)01454-7
Taylor, P. W. (2015). Impact of space flight on bacterial virulence and antibiotic susceptibility. Infect. Drug Resist. 30, 249–262. doi: 10.2147/IDR.S67275
Thiele, I., and Palsson, B. O. (2010). A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121. doi: 10.1038/nprot.2009.203
Vaishampayan, P., Probst, A., Krishnamurthi, S., Ghosh, S., Osman, S., McDowall, A., et al. (2010). Bacillus horneckiae sp. nov., isolated from a spacecraft-assembly clean room. Int. J. Syst. Evol. Microbiol. 60, 1031–1037. doi: 10.1099/ijs.0.008979-0
Vaishampayan, P. A., Rabbow, E., Horneck, G., and Venkateswaran, K. J. (2012). Survival of Bacillus pumilus spores for a prolonged period of time in real space conditions. Astrobiology 12, 487–497. doi: 10.1089/ast.2011.0738
Valledor, L., Escandón, M., Meijón, M., Nukarinen, E., Cañal, M. J., and Weckwerth, W. (2014). A universal protocol for the combined isolation of metabolites, DNA, long RNAs, small RNAs, and proteins from plants and microorganisms. Plant J. 79, 173–180. doi: 10.1111/tpj.12546
Van Mulders, S. E., Stassen, C., Daenen, L., Devreese, B., and van Eijsden Siewers, V. (2011). The influence of microgravity on invasive growth in Saccharomyces cerevisiae. Astrobiology 11, 45–55. doi: 10.1089/ast.2010.0518
Vecerek, B., Moll, I., Afonyushkin, T., Kaberdin, V., and Bläsi, U. (2003). Interaction of the RNA chaperone Hfq with mRNAs: direct and indirect roles of Hfq in iron metabolism of Escherichia coli. Mol. Microbiol. 50, 897–909. doi: 10.1046/j.1365-2958.2003.03727.x
Vecino, X., Barbosa-Pereira, L., Devesa-Rey, R., Cruz, J. M., and Moldes, A. B. (2015). Optimization of extraction conditions and fatty acid characterization of Lactobacillus pentosus cell-bound biosurfactant/bioemulsifier. J. Sci. Food Agric. 95, 313–320. doi: 10.1002/jsfa.6720
Velayudhan, J., Castor, M., Richardson, A., Main-Hester, K. L., and Fang, F. C. (2007). The role of ferritins in the physiology of Salmonella enterica sv. Typhimurium: a unique role for ferritin B in iron-sulphur cluster repair and virulence. Mol. Microbiol. 63, 1495–1507. doi: 10.1111/j.1365-2958.2007.05600.x
Vogel, J., and Luisi, B. F. (2011). Hfq and its constellation of RNA. Nat. Rev. Microbiol. 15, 578–589. doi: 10.1038/nrmicro2615
Vukanti, R., and Leff, L. G. (2012). Expression of multiple stress response genes by Escherichia coli under modeled reduced gravity. Microgravit. Sci. Tecnol. 24, 267–279. doi: 10.1007/s12217-012-9310-0
Wang, Y., Yuan, Y., Liu, J., Su, L., Chang, D., Guo, Y., et al. (2014). Transcriptomic and proteomic responses of Serratia marcescens to spaceflight conditions involve large-scale changes in metabolic pathways. Adv. Space Res. 53, 1108–1117. doi: 10.1016/j.asr.2014.01.018
Wassmann, M., Moeller, R., Rabbow, E., Panitz, C., Horneck, G., Reitz, G., et al. (2012). Survival of spores of the UV-resistant Bacillus subtilis strain MW01 after exposure to low-earth orbit and simulated martian conditions: data from the space experiment ADAPT on EXPOSE-E. Astrobiology 5, 498–507. doi: 10.1089/ast.2011.0772
Weckwerth, W. (2003). Metabolomics in systems biology. Annu. Rev. Plant. Biol. 54, 669–689. doi: 10.1146/annurev.arplant.54.031902.135014
Weckwerth, W. (2011a). Green systems biology - From single genomes, proteomes and metabolomes to ecosystems research and biotechnology. J. Proteomics. 75, 284–305. doi: 10.1016/j.jprot.2011.07.010
Weckwerth, W. (2011b). Unpredictability of metabolism – the key role of metabolomics science in combination with next-generation genome sequencing. Anal. Bioanal. Chem. 400, 1967–1978. doi: 10.1007/s00216-011-4948-9
Weckwerth, W. (2016). Systemtheoretische Konzepte der Genomweiten Molekularen Analyse und Datenintegration in der Biologie. Aus der Schriftenreihe der Hülsenberger Gespräche. Hamburg: Heigener Europrint GmbH, 68–71.
Weckwerth, W. (2019). Toward a unification of system-theoretical principles in biology and ecology—the stochastic lyapunov matrix equation and its inverse application. Front. Appl. Math. Stat. 5:29. doi: 10.3389/fams.2019.00029
Weckwerth, W., and Morgenthal, K. (2005). Metabolomics: from pattern recognition to biological interpretation. Drug Discov. Today 10, 1551–1558. doi: 10.1016/S1359-6446(05)03609-3
Weckwerth, W., Wenzel, K., and Fiehn, O. (2004). Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co-regulation in biochemical networks. Proteomics 4, 78–83. doi: 10.1002/pmic.200200500
Wilson, J. L., Nägele, T., Linke, M., Demel, F., Fritsch, S. D., Mayr, H. K., et al. (2020). Inverse data-driven modeling and multiomics analysis reveals phgdh as a metabolic checkpoint of macrophage polarization and proliferation. Cell Rep. 30, 1542–1552.e7. doi: 10.1016/j.celrep.2020.01.011
Wilson, J. W., Ott, C. M., Höner zu Bentrup, K., Ramamurthy, R., Quick, L., Porwollik, S., et al. (2007). Space flight alters bacterial gene expression and virulence and reveals a role for global regulator Hfq. Proc. Natl. Acad. Sci. U.S.A 104, 16299–163304. doi: 10.1073/pnas.0707155104
Wilson, J. W., Ott, C. M., Quick, L., Davis, R., Höner zu Bentrup, K., Crabbé, A., et al. (2008). Media ion composition controls regulatory and virulence response of Salmonella in spaceflight. PLoS One 3:e3923. doi: 10.1371/journal.pone.0003923
Wilson, J. W., Ramamurthy, R., Porwollik, S., McClelland, M., Hammond, T., Allen, P., et al. (2002). Microarray analysis identifies Salmonella genes belonging to the low-shear modeled microgravity regulon. Proc. Natl. Acad. Sci. U.S.A. 15, 13807–13812. doi: 10.1073/pnas.212387899
Zea, L., Larsen, M., Estante, F., Qvortrup, K., Moeller, R., and Dias, et al. (2017). Phenotypic changes exhibited by E. coli cultured in space. Front. Microbiol. 8:1598. doi: 10.3389/fmicb.2017.01598
Zea, L., Prasad, N., Levy, S. E., Stodieck, L., Jones, A., Shrestha, S., et al. (2016). A molecular genetic basis explaining altered bacterial behavior in space. PLoS One 11:e0164359. doi: 10.1371/journal.pone.0164359
Zhang, X., Fang, X., and Liu, C. (2015). Genomic and proteomic analysis of Escherichia coli after spaceflight reveals changes involving metabolic pathways. Arch. Med. Res. 46, 181–185. doi: 10.1016/j.arcmed.2015.03.007
Keywords: microbes in space, space missions, –omics technology, extremophiles, outer space
Citation: Milojevic T and Weckwerth W (2020) Molecular Mechanisms of Microbial Survivability in Outer Space: A Systems Biology Approach. Front. Microbiol. 11:923. doi: 10.3389/fmicb.2020.00923
Received: 03 December 2019; Accepted: 20 April 2020;
Published: 15 May 2020.
Edited by:
André Antunes, Macau University of Science and Technology, ChinaReviewed by:
Felipe Gómez, Centro de Astrobiología (CSIC-INTA), SpainAlexander Josef Probst, University of Duisburg-Essen, Germany
Copyright © 2020 Milojevic and Weckwerth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tetyana Milojevic, dGV0eWFuYS5taWxvamV2aWNAdW5pdmllLmFjLmF0