 Rowena Chong
Rowena Chong Yuanyuan Cheng
Yuanyuan Cheng Katherine Belov
Katherine Belov- School of Life and Environmental Sciences, The University of Sydney, Sydney, NSW, Australia
The study of the gut microbiome in threatened wildlife species has enormous potential to improve conservation efforts and gain insights into host-microbe coevolution. Threatened species are often housed in captivity, and during this process undergo considerable changes to their gut microbiome. Studying the gut microbiome of captive animals therefore allows identification of dysbiosis and opportunities for improving management practices in captivity and for subsequent translocations. Manipulation of the gut microbiome through methods such as fecal transplant may offer an innovative means of restoring dysbiotic microbiomes in threatened species to provide health benefits. Finally, characterization of the gut microbiome (including the viral components, or virome) provides important baseline health information and may lead to discovery of significant microbial pathogens. Here we summarize our current understanding of microbiomes in Australian marsupial species.
Introduction
The gut microbiome plays an important role in many physiological processes including nutrition (Kau et al., 2011), immunity (Round and Mazmanian, 2009), metabolism (Musso et al., 2011), brain functions and behavior (Rogers G. et al., 2016). In humans, the highly diverse gut bacterial communities have been found to play a wide range of symbiotic functions that are essential for maintaining the health of the host, and disturbances to the gut microbiome structure have been associated with various diseases, such as diabetes, inflammations, metabolic or autoimmune disorders, infections, and cancer (reviewed in Kho and Lal, 2018). Certain attributes of the gut microbiome have been implicated in an increased risk for an individual to develop certain diseases, such as a high Firmicutes:Bacteroidetes ratio in obesity (Ley et al., 2006), and Enterobacterial blooms in inflammatory diseases of the gut (Zeng et al., 2017). Advances in sequencing technologies in recent years have also allowed the development of new methods for studying the gut virome, another important component of the gut microbial ecosystem, revealing a high richness of gut viral community and various potential beneficial functions of viruses (e.g., bacteriophages) in mediating host microbiome adaptation and stability (Ogilvie and Jones, 2015).
Much of what we know about the gut microbiome so far stems from studies in humans or animal model species, but recent studies have increasingly focused on wildlife biology and conservation (Trevelline et al., 2019). These offer a wealth of knowledge about the abundance and diversity of microbes that inhabit wildlife species across diverse taxa, including the diverse lineage of marsupials. Australian marsupials represent a unique evolutionary lineage of mammals that has dominated the Australian continent. A long history of geographical isolation has led to the diversification of marsupial species in terms of their biology, diets and life history traits (Nipperess, 2015). Here we will review our current understanding of the gut microbiome of marsupials and how this knowledge can be applied to further our understanding of marsupial health, host-microbiome coevolution and conservation.
Baseline Characterization of Marsupial Gut Microbiome
Tasmanian Devil
The Tasmanian devil (Sarcophilus harrisii; “devil” hereinafter) is the world’s largest living carnivorous marsupial from the family Dasyuridae. Once widespread throughout Australia, it became extinct on the mainland about 400 to 3,000 years ago (Archer and Baynes, 1972; Brown, 2006) and is now endemic to the island state of Tasmania. Modern devils are facing extinction due to a fatal contagious cancer called devil facial tumour diseases (DFTD) (Pemberton, 2019). Since its discovery in 1996, DFTD has spread over 75% of the state and caused declines of up to 80% of wild devil populations (Lazenby et al., 2018). This has resulted in devils being listed as Endangered by the IUCN (International Union for Conservation of Nature) and under the Environment Protection and Biodiversity Conservation (EPBC) Act (Australia). A large amount of effort has gone into furthering our understanding of devil biology to facilitate conservation efforts, including population genetics (Jones et al., 2004; Miller et al., 2011) and the etiology of DFTD (Pearse and Swift, 2006; Pye et al., 2016). Tasmanian devils are predominantly scavengers, but are also known to hunt, consuming a wide range of prey items from marcopods to insects, birds and fish (Table 1).
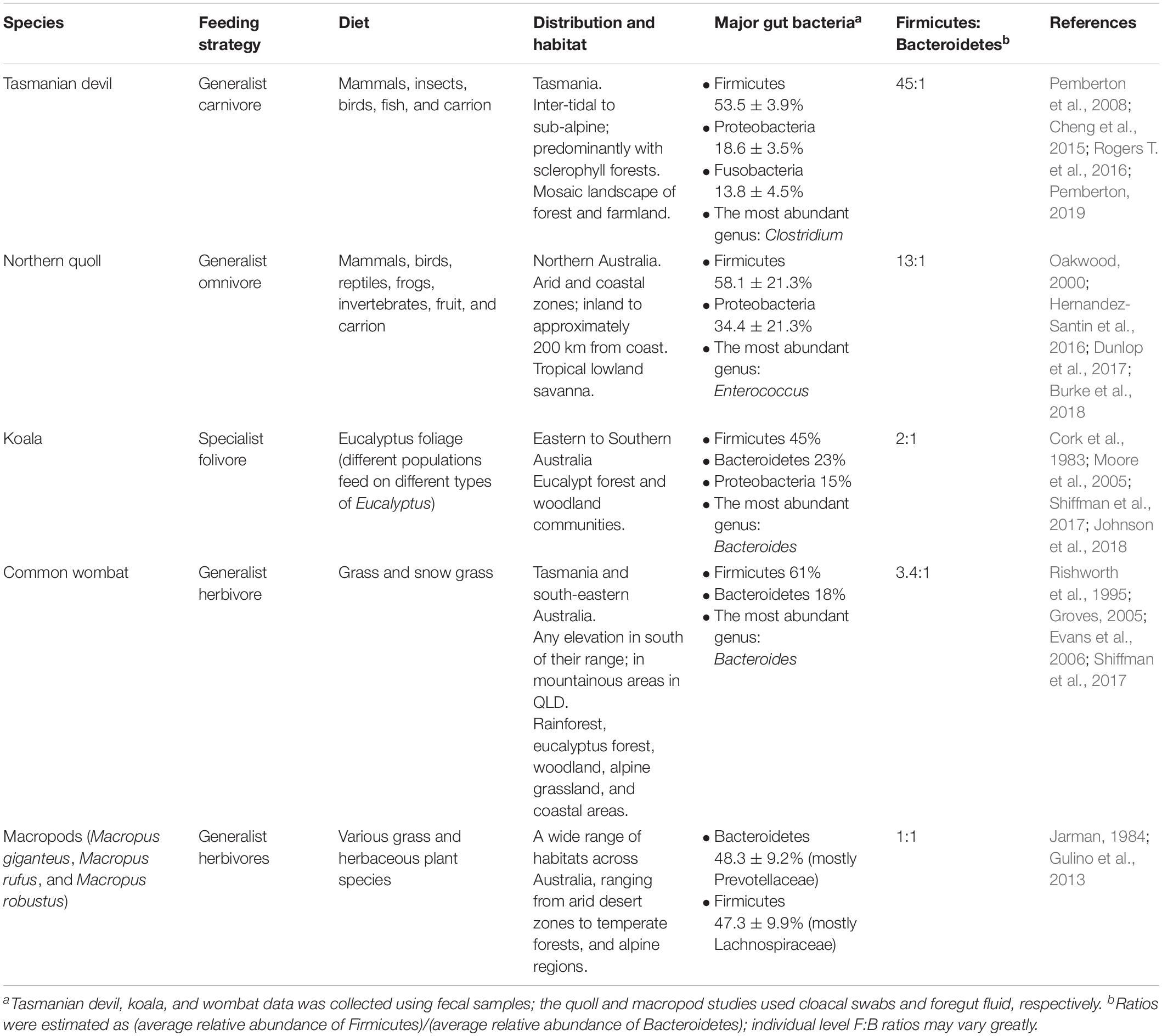
Table 1. Comparison of diet, habitat, and gut microbiome in marsupials.
More recently, the microbiome of the devil also became the focus of research. Initial microbiome characterization on the devils using 16S rRNA gene amplicon sequencing of the V1–V3 region generated baseline information of the bacterial communities in the gut (feces), pouch, skin and oral cavity (Cheng et al., 2015). Across all body sites, bacterial phyla Firmicutes, Proteobacteria, Fusobacteria, Bacteroidetes, and Actinobacteria were the top five constituents. However, compared to the other three microbiome types, the gut microbiome had significantly higher phylotype richness (Cheng et al., 2015). The most abundant bacterial phyla found within the devil gut microbiome was Firmicutes (53.5 ± 3.9%), followed by Proteobacteria (18.6 ± 3.5%), and fusobacteria (13.8 ± 4.5%) (Figure 1 and Table 1; Cheng et al., 2015). Clostridium, a bacterial genus known to contain species with protein decomposition and amino acid degradation activities (Fonknechten et al., 2010), was identified as the most common bacteria in devil gut flora (18.5 ± 2.4%), which speculatively could be an indication of the gut flora having evolved to adapt to the host’s carnivorous feeding strategy. The level of Proteobacteria (primarily Gammaproteobacteria and Alphaproteobacteria) observed in devil gut microbiome is relatively higher than that found in many other mammalian species (on average 8.8% in mammals based on Ley et al., 2008). Particularly Enterobacteriaceae, a family of Gammaproteobacteria, accounts for approximately 9.4% of the devil gut flora. This bacterial family is known to contain many symbionts such as Escherichia coli, Klebsiella spp., and Proteus spp., and in humans, dysbiosis involving Enterobacteriaceae have been associated with various inflammatory gut diseases (Zeng et al., 2017).
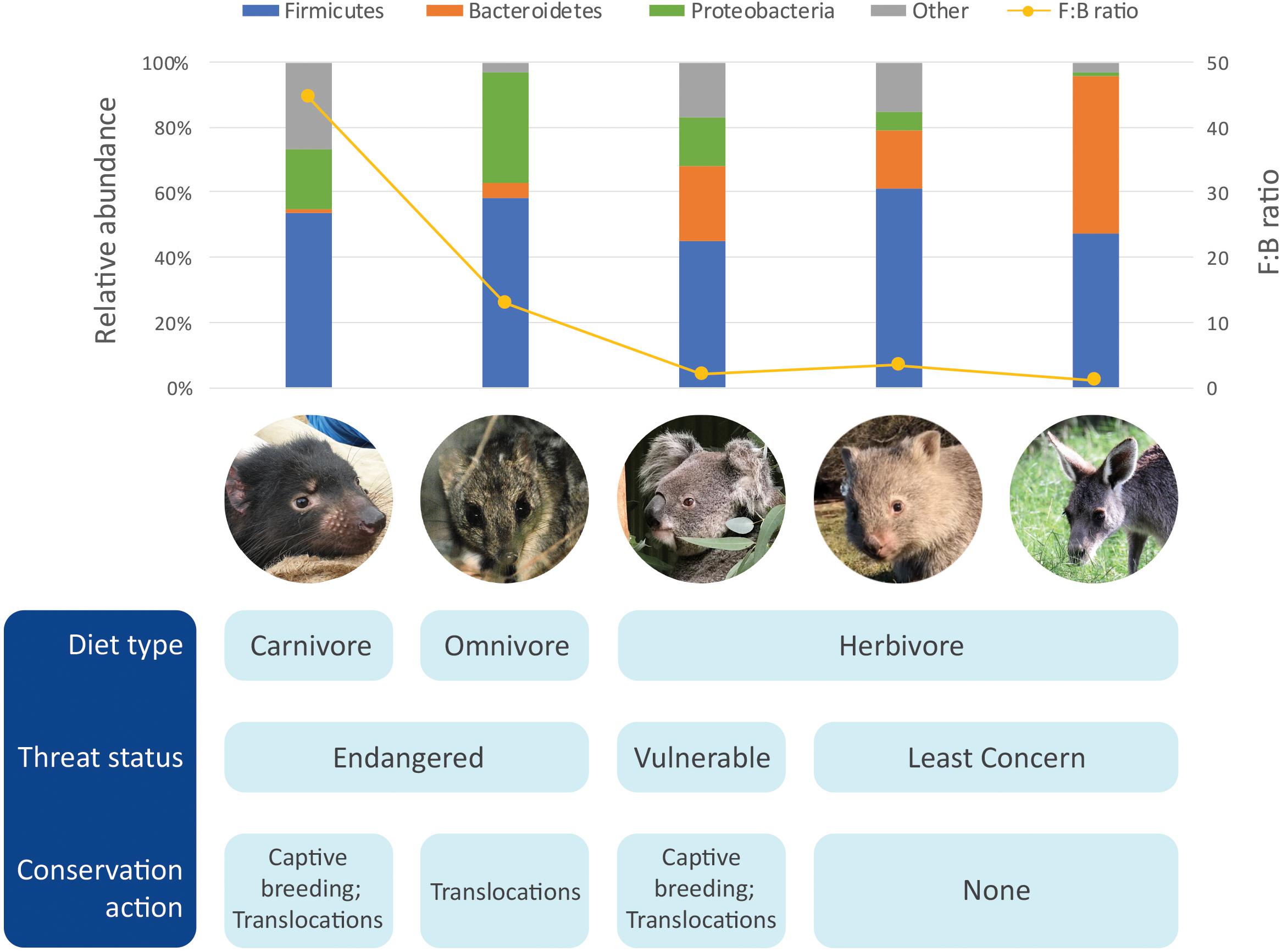
Figure 1. Comparison of gut microbiome of five marsupials [data from Barker et al. (2013), Gulino et al. (2013), Cheng et al. (2015), Shiffman et al. (2017), Burke et al. (2018)].
Another important feature of the devil gut microbiome is the low prevalence of Bacteroidetes (1.2 ± 0.6%), which leads to a high Firmicutes to Bacteroidetes ratio (F:B ratio; approximately 45:1 in devils) (Cheng et al., 2015). It has been found in humans and mice that a high F:B ratio (the “obese microbiome”) is associated with high efficiency in energy harvest from the diet and an increased risk for the host to develop obesity, while the increase of Bacteroidetes and decrease of Firmicutes can lead to weight loss (Ley et al., 2006; Turnbaugh et al., 2006). Interestingly, low levels of Bacteroidetes have also been observed in the gut microbiome of many other carnivorous mammals besides devils, including the cheetah (Acinonyx jubatus) (Menke et al., 2017), spotted hyena (Crocuta crocuta), polar bear (Ursus maritimus) (Ley et al., 2008), and northern quoll (Dasyurus hallucatus; further discussed below). These findings suggest that a high F:B ratio could be a feature of carnivorous species which is possibly related to the need to efficiently harvest and store energy from limited food sources (Cheng et al., 2015). In the devil, this feature is also in line with their feeding habit, whereby they typically gorge up to 40% of their body weight in a single meal, followed by several days of no feeding (Pemberton and Renouf, 1993).
In addition to the gut bacterial microbiome, a recent study reported the characterization of devil fecal virome and the identification of 24 novel marsupial-associated viruses as well as known mammalian pathogens such as rabbit haemorrhagic disease virus (Chong et al., 2019b). Some notable marsupial-associated viruses identified include astroviruses, picobirnaviruses, parvoviruses, papillomaviruses, polyomaviruses and a gammaherpesvirus. Among these, picobirnaviruses have recently been found to possess bacteriophage properties (Krishnamurthy and Wang, 2018) and thus can potentially play a role in the regulation of gut bacterial community and protection against pathogenic bacteria (Mukhopadhya et al., 2019). Prior to this study, only a single gammaherpesvirus affecting both captive and wild devils has been recorded in the literature (Stalder et al., 2015), demonstrating a significant lack of knowledge in this area. Although much is still unknown regarding what roles the viruses identified in devil gut flora may play on host health, the viral sequences isolated through devil gut virome characterization provide a useful resource for future research toward illuminating activities and functions of mammalian gut viruses. Further investigations of gut virome in more marsupial species will be needed to understand the structure and function of viruses in the gut microenvironment of marsupials.
Northern Quoll
The northern quoll (D. hallucatus) is an omnivorous marsupial from the family Dasyuridae. Found predominantly in the northern regions of Australia, they are currently listed as endangered and are found distributed in fragmented areas across northern Australia (Braithwaite and Griffiths, 1994). Northern quolls are generalists consuming a wide prey base including vertebrate and invertebrate prey and fruit (Table 1). Using cloacal swab as a non-invasive proxy for the gut, the gut microbiome of the northern quoll was characterized using 16S rRNA amplicon sequencing of the V3–V4 region (Burke et al., 2018). Similar to its close carnivorous relative, the Tasmanian devil, the northern quoll cloacal microbiome shows high abundance of Firmicutes (58.1 ± 21.3%) and Proteobacteria (34.4 ± 21.3%) and low abundance of Bacteroidetes (4.5 ± 13.85%) (Figure 1; Burke et al., 2018). In addition, the northern quoll gut microbiome was characterized by a high abundance of Enterococcus (27.3 ± 22.4%) compared to other mammalian species (∼1% in humans) (Dubin and Pamer, 2014). The similarities between the northern quoll and devil gut microbiome in the higher taxonomic levels can possibly be attributed to their close phylogenetic relationship, as well as similar carnivorous diets. However, it should be noted that due to different sampling methods that have been used for the two species (feces vs. cloacal swab), the results from the two studies on devils and quolls may not be directly comparable.
Koala
The koala (Phascolarctos cinereus) is an arboreal folivore endemic to Australia and the last surviving member of the family Phascolarctidae. Koalas occur across eastern Australia in a wide range of habitat types (Table 1). Yet they are a dietary specialist, feeding solely on the foliage from species of Eucalyptus (Cork and Sanson, 1991). Various anatomical and physiological adaptations enable the koalas to survive on a diet that is low in proteins and high in lignified fiber and phenolic compounds that would make it toxic to other animals. The hindgut, including the caecum and proximal colon of a koala is significantly enlarged (Cork and Sanson, 1991), making it one of the largest in any known mammals (Krockenberger and Hume, 2007). The mean retention times of solutes and larger particles of digesta in the digestive tract in koalas are both longer than have been reported in most other mammals, including many other eucalypt-specialist marsupial folivores (Krockenberger and Hume, 2007), allowing the potential for relatively extensive microbial degradation and nutrient extraction from the nutritionally poor foliage. In addition, endogenous enzymes produced in the liver have also been found to assist the koala in coping with toxic plant secondary metabolites (PSMs) in their Eucalyptus diets (Ngo et al., 2000). As with other herbivores, the koala relies on microbes in their gut for digestion of plant material through hydrolysis and fermentation.
Characterization of the koala hindgut microbiome revealed a dominance of Firmicutes and Bacteroidetes, consistent with many other species (Figure 1; Barker et al., 2013). The F:B ratio varies significantly across the hindgut, with a low ratio close to 1 (1.3:1) found in the caecum, and significantly higher ratios of 6:1 and 3:1 in the colon and fecal pellet, respectively, suggesting differential microbial fermentation processes taking place at various sites (Barker et al., 2013). Due to its unusual diet, a number of studies have focused on elucidating the gut microbiome’s contribution to the host’s ability to digest and detoxify Eucalyptus. Early investigations using culture-based techniques identified presence of tannin degrading microorganisms across the koala’s gastrointestinal tract, including Streptococcus gallolyticus and Lonepinella koalarum from the Pasteurellaceae family (Osawa, 1990; Osawa et al., 1995). Furthermore, comparative metagenomics analysis of the gut microbiome between koala and its closest living relative, the wombat has enabled identification of other key microbial linages and functional pathways unique to the koala. Several microbial lineages thought to play conserved roles in fiber degradation and urea recycling, both of which are essential metabolic pathways for herbivorous species, were found in both the koala and wombat (Shiffman et al., 2017). For example, fibrolytic bacteria from the genus Bacteroides and Ruminococcus were found consistently across all koala and wombat samples. These fibrolytic bacteria metabolize complex plant compounds into short-chain fatty acids, which can then be easily absorbed by the host (Barboza and Hume, 1992). Urease-containing Succinivibrionaceae bacterium found in both species are thought to assist in urea degradation. In mammals, ammonia, the toxic end-product of protein catabolism, is converted into urea through the urea cycle for elimination; it is estimated that approximately 20% of urea is degraded by urease-expressing gut bacteria through the gastrointestinal tract, with the remaining eliminated through renal excretion (Ramezani et al., 2016). One important distinction between koala and wombat gut microbiome is that members of the family Synergistaceae were detected at relatively high abundance (>4–17%) in the koala but absent in wombat. These bacterial populations are predicted to encode multiple pathways related to the degradation of toxic Eucalyptus plant secondary metabolites (PSMs), therefore playing a key role in the koala’s ability to survive in a specialized dietary niche (Shiffman et al., 2017).
Wombat
The wombat is the koala’s closest living relative, both belonging to the suborder Vombatiformes. The family Vombatidae consists of three species, the common wombat (Vombatus ursinus), the southern-hairy nosed wombat (Lasiorhinus latifrons) and the northern-hairy nosed wombat (Lasiorhinus krefftii). The common wombat is found across a range of habitats in Tasmania and south-eastern Australia (Table 1), with the southern-hairy nosed wombat found in southern Australia, and the northern-hairy nosed wombat isolated to Queensland. Unlike the koala, wombats are a generalist herbivore that primarily grazes on grass (Rishworth et al., 1995).
Characterization of the gut microbiome has been carried out in two species of wombats, the southern hairy-nosed wombat and common wombat. Based on 16S rRNA amplicon sequencing of the V6 to V8 region, there was a dominance of Firmicutes (∼61%) and Bacteroidetes (∼18%) and relatively low F:B ratio (3.4:1) (Figure 1; Shiffman et al., 2017). Compared to the koala, higher levels of xylanases were found (7.6% vs. 1.9% in the koala), which could be attributed to the higher content of hemicellulose in the wombat diet (Rishworth et al., 1995; Hume, 1999). Several distinct microbes were also only detected in the wombat, including unclassified members of the family Christensenellacea, the order Clostridiales, and the genus Ruminococcus (Shiffman et al., 2017).
Macropods
This family of Macropodidae, including species of kangaroos and wallabies, is found in a wide range of habitats across Australia, ranging from arid desert zones to temperate forests and alpine regions. They are grazing generalist herbivores, foraging on a range of grass and herbaceous plant species depending on their environment (Jarman, 1984). All species of macropods are foregut fermenters (Hume, 1999). Consequently, the gastrointestinal tracts of macropods are generally characterized by an enlarged forestomach, sacciform, and tubiform where microbial fermentation of plant material takes place (Hume, 1999). Early studies based on 16S rRNA amplicon sequencing of the V3–V4 region again identified Bacteroidetes and Firmicutes as key constituents of macropod (Macropus giganteus, Macropus rufus, and Macropus robustus) foregut microbiome (48.3 ± 9.19% and 47.3 ± 9.85%, respectively) (Figure 1; Gulino et al., 2013). A number of OTUs identified in the macropod foregut microbiome shared highly percentage of homology to known fibrolytic bacteria such as Ruminococcus flavefaciens and Butyrivibrio fibrisolvens, which were identified as key microbes responsible for fibrolytic digestion (Hespell et al., 1987; Miller et al., 2009). In the tammar wallaby (Macropus eugenii), it has been reported that pouch young (40 and 56 days old) have a gut flora dominated by Firmicutes and Actinobacteria that is distinct to the maternal pouch and oral microbiome, highlighting the possibility of the gut microbiota of marsupial pouch young arising from the maternal milk (Chhour et al., 2010). However, it should be noted that this early study used a low throughput cloning-based method for sequencing 16S rRNA genes, which may not have the power to fully reveal the complexity and comprehensive structure of the microbiomes surveyed.
Implications for Marsupial Biology and Conservation
Dysbiosis in Captivity and Implications for Translocation/Reintroduction
A commonly used tool in conservation management is captive breeding for those species which are suffering significant population declines (Harley et al., 2018). Yet life in captivity can present a range of extreme lifestyle changes, many of which may affect the host microbiome. A growing number of studies have focused on determining the effects of captivity on wildlife microbiomes, with many providing evidence of microbiome perturbations (Amato, 2013; Kohl et al., 2014; McKenzie et al., 2017). Significant differences in the gut microbiome composition of captive animals relative to their wild counterparts have been frequently observed in many species. This is particularly apparent in carnivorous and omnivorous species, where the supply of natural and diverse diets in an artificial setting is often restricted (Nakamura et al., 2011; Guan et al., 2016). In Tasmanian devils, evidence of microbiome dysbiosis has been detected, where captive individuals showed significantly different gut microbiome compositions and lower microbial diversity compared to their wild counterparts (Cheng et al., 2015). Interestingly, the type of captive enclosure influenced gut microbiome composition and diversity in the devils. Of the two types of captive enclosures studied, devils that were housed in more intensive, zoo-based facilities had lower microbial diversity in their gut than those housed in larger, group housing enclosures. Those that are housed in group enclosures also have gut microbiomes that more closely resemble the microbiome of wild devils, suggesting free-range or group enclosures to be a more preferable housing option for managing devil microbiomes in captivity (Cheng et al., 2015). Currently the impact of a depauperate microbiome on devils remains unclear, but it has been suggested that the low diversity of gut microbiome in captive devils may lead to an increased risk of obesity (Le Chatelier et al., 2013), which can consequently cause reduced success rate of captive breeding (Cheng et al., 2015).
In contrast, it has been reported that captivity does not appear to significantly alter the gut microbiome in koalas, as both captive and wild koalas share very similar and consistent microbiome compositions at the phyla and genus level (Alfano et al., 2015). The lack of differentiation between captive and wild microbiomes has mostly been observed in herbivorous species, such as even-toed ungulates (bovids and giraffes) (McKenzie et al., 2017). Alterations to the microbiome in captivity can be driven by many factors, including changes to the natural diets (Clayton et al., 2016), reduced environmental microbial reservoirs (Loudon et al., 2014), cohabitation with other species (Lemieux-Labonté et al., 2016), and antibiotic use (Plummer et al., 2005; Dahlhausen et al., 2018). The strict dietary requirements of koalas mean that captive and wild individuals likely feed on similar Eucalyptus diets, which could have resulted in limited differentiations in their gut microbiome. One limitation though with the study by Alfano et al. (2015) is the small sample size consisting of only two captive koalas (plus data of two wild koalas from an earlier study). Further studies using a larger sample size will be needed to verify the hypothesis that captive and wild koalas have similar gut microbiomes.
An important aspect of many captive breeding programs is the reintroduction of animals back into the wild (Seddon et al., 2007). Microbiome perturbations observed in captivity may underlie poor host health, which may in turn impact the reintroduction success and post-release survival of captive individuals (Redford et al., 2012). For example, increased abundance of pathogenic microbes and disease-associated pathways in the captive cheetah gut microbiome may explain the poor reproductive rates and high prevalence of bacterial infections associated mortality (Menke et al., 2017). Similarly in the grouse (Tetrao urogallus), microbiome disturbances (Wienemann et al., 2011) as well as anatomical changes, such as shorter small intestines and caeca observed in captivity (Liukkonen-Anttila et al., 2000), may compromise digestion likely leading to high mortality of captive birds upon release to the wild (Seiler et al., 2000). With concerns about the consequences of dysbiosis in captivity, and the potential implications for the reintroduction of captive devils back into the wild, the gut microbiomes of translocated devils were monitored for temporal changes over the course of translocation to understand how translocation may influence devil gut microbiome (Chong et al., 2019a). Comparisons between the microbiome of released devils before and after translocation showed significant shifts in composition and diversity, and that released devils began to re-acquire the wild, incumbent microbiome as early as 3–4 weeks post-release (Chong et al., 2019a). This result suggests that microbiome perturbations as a result of captivity in a carnivorous species, such as the devils, are not necessarily permanent. Studies investigating changes in the gut microbiome post-release are scarce but can provide important insights into the impact of translocation on the host-associated microbiome, allowing evaluation and improvement of translocation success.
Gut Microbiome Management in Wildlife Conservation
Bioaugmentation of the microbiome through probiotic therapy or fecal microbiome transplantation is a new and emerging field in microbiome research, especially within the context of wildlife conservation (McKenzie et al., 2018; Trevelline et al., 2019). Augmenting or manipulating the microbiome may provide numerous benefits, such as restoring dysbiotic microbiome for improved physiological functions and animal health (Kueneman et al., 2016), and mitigating disease risk (Bletz et al., 2013). In the wild, koalas often have access to different Eucalyptus spp. but majority feed exclusively on a specific food tree (Brice et al., 2019). The koala gut microbiome has been suggested to play a role in their dietary preferences. For example, the gut microbiome of koalas that preferentially feed on messmate gum (Eucalyptus obliqua) have higher abundances of fibrolytic bacteria and are more adapted to using different complex carbohydrate sources than those feeding on manna gum (Eucalyptus viminalis), one of the main food trees for koalas in many areas (Brice et al., 2019). Microbial richness and diversity were also found to be lower in the microbiome of koalas feeding on manna gum, likely to be linked to a greater energy harvest from this species of Eucalyptus compared to messmate (Brice et al., 2019). Therefore it has been hypothesized that the inability of the majority of koalas to shift diets to messmate is due to a lack appropriate gut microbial assemblage for optimal digestion (Blyton et al., 2019). With the continuous threat of habitat loss and limited resources, the ability to shift diet and utilize different food sources is crucial for the survival and persistence of koalas in the wild. In a study by Blyton et al. (2019), wild koalas that previously fed on manna gum were inoculated with faecally derived microbes from koalas that feed on messmate. Although on average, the treatment koalas did not show a significant increase in their messmate consumption after inoculation, their gut microbiome shifted significantly to resemble the microbiomes of koalas that feed primarily on messmate. Also importantly, a pattern was observed that koalas showing a more prominent shift in the gut flora consumed more messmate. As such, fecal transplant between koalas feeding on different Eucalyptus species may be useful in introducing beneficial microbes to the gut microbiome that will enable koalas to adapt to and utilize more variety of food sources. This may prove to be particularly important when translocating koalas to areas with different Eucalyptus tree species. Meanwhile, it also needs to be emphasized that further research will be needed to evaluate the broader impact and safety (e.g., potential disease transmission) of such treatments in wild species.
Microbiome in Health and Disease
Infectious diseases are major threats to wildlife species. In marsupials, a well-known example is Chlamydia infections in koalas. Infections caused by Chlamydia pecorum and Chlamydia pneumoniae can cause conjunctivitis, blindness, pneumonia, urinary tract and reproductive tract infections, and infertility (Brown et al., 1987). Antibiotic treatments are routinely used in wildlife hospitals to treat infections, but have been suggested to cause disruptions to the normal intestinal microbial communities, resulting in adverse side effects (Polkinghorne et al., 2013; Dahlhausen et al., 2018). Results from a study by Dahlhausen et al. (2018) found that koalas that were treated with antibiotics for chlamydia and subsequently died had lower microbial diversity and abundance of tannin-degrading bacteria, Lonepinella koalarum, in their gut than koalas recovered after treatment. Although the study did not detect a significant difference in the gut bacterial richness between antibiotic-treated koalas and control individuals, possibly at least partly due to the limited number of controls (two koalas), the comparison of microbiome between pre-treatment and post-treatment samples revealed that antibiotic treatments may influence the composition and adaptation of gut microbiome of koalas and affect the abundance of beneficial microbes with functions (such as detoxification of Eucalyptus) essential to the health and survival of the species.
With increasing usage of antibiotics in wildlife medicine, antibiotic resistance is of growing concern for the health and conservation of threatened species (West et al., 2019). Evidence of antibiotic resistance has been detected in a number of wildlife species including Iberian lynx (Lynx pardinus) (Sousa et al., 2014) and the Australian sea lions (Neophoca cinerea) (Delport et al., 2015). In marsupials, bacterial genetic elements associated with antibiotic resistance genes (class 1 integrons) have also been found in the gut microbiome of the endangered brush-tailed rock-wallabies (Petrogale penicillata) living in captivity (Power et al., 2013). This raises concerns about the future effectiveness of antibiotic treatments, as well as the potential spread of resistance into wild populations through the translocation of these captive individuals. Careful use of antibiotic treatment, as well as continuous efforts to develop antibiotic alternatives, are paramount to prevent the rise of antibiotic-resistant diseases in threatened wildlife.
Another emerging field of research in wildlife gut microbiome is the study of the gut virome. So far, the overall knowledge on functions of gut virome is still quite limited even in model species. Most of the current understanding on the potential beneficial effect of gut viruses surrounds bacteriophages, which have been suggested to play a part in regulating and maintaining the balance of bacterial community (Ogilvie and Jones, 2015). Emerging evidence also suggests that gut viruses interact with the host immune system and are likely sources of immune variation (Neil and Cadwell, 2018). The Tasmanian devil was the first marsupial species in which the gut viral communities have been characterized in great depth (Chong et al., 2019b). Identification of viruses, some of which are potentially pathogenic is important for understanding and safeguarding devil health. Further work is required to elucidate the pathogenicity of novel viruses. The use of a metagenomics approach to categorize the viral components of the gut microbiome in marsupial is still in its infancy but has enormous coding potential. Current knowledge on the diversity of viruses found in marsupials is scarce and virome studies will provide important baseline health information, as well as insights into host-microbe interactions and the phylogenetic history of viruses infecting this evolutionary unique group of mammals.
Conclusion
Australia has one of the highest extinction rates of mammals in the world (Woinarski et al., 2015). Conservation biologists are constantly searching for ways to protect threatened wildlife species from extinction. With advances in sequencing technology, our ability to catalog and study the complex host-associated gut microbiome has improved substantially in recent years. Consequently, there has been a paradigm shift focusing on understanding the importance of the gut microbiome in threatened wildlife species and how the knowledge gained can contribute to conservation efforts. In this review, we have provided numerous examples of how studying the gut microbiome has advanced our understanding of marsupial biology (such as the complex microbial digestion of toxic Eucalyptus in koalas), as well as how to facilitate conservation through managing the microbiome in captive populations and during translocations. In addition, the ability to manipulate the gut microbiome through methods such as fecal inoculations proves to be an exciting avenue for future research in wildlife health. For many marsupial species, baseline characterization of their gut microbiome is still required. This will be an essential first step in understanding the overall patterns of microbial composition and diversity, thus providing a springboard for studying dysbiosis, particularly in relation to multiple anthropogenic pressures and environmental changes, such as captive management and habitat disturbances.
Author Contributions
RC wrote the manuscript with input from KB. YC and CH made Figure 1 and Table 1 and carried out major revisions of the manuscript.
Funding
The authors acknowledge the Australian Research Council for funding the projects on the Tasmanian devil microbiome and virome (DP140103260, LP140100508, and LP180100244).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all the Save the Tasmanian Devil Program staff who assisted in collection of samples. Photo credits for photos used in Figure 1: Tasmanian devil, by CH; Northern quoll, by Wildlife Explorer – Picasa Web Albums, CC BY 3.0, https://commons.wikimedia.org/w/index.php?curid=9488385; koala, by Parice Brandies; common wombat, by Elspeth McLennan; eastern gray kangaroo, by YC.
References
Alfano, N., Courtiol, A., Vielgrader, H., Timms, P., Roca, A. L., and Greenwood, A. D. (2015). Variation in koala microbiomes within and between individuals: effect of body region and captivity status. Sci. Rep. 5:10189.
Amato, K. R. (2013). Co-evolution in context: the importance of studying gut microbiomes in wild animals. Microb. Sci. Med. 1, 10–29.
Archer, M., and Baynes, A. (1972). Prehistoric mammal faunas from two small caves in the extreme south- west of Western Australia. J. R. Soc. Western Austral. 55, 80–89.
Barboza, P., and Hume, I. (1992). Hindgut fermentation in the wombats: two marsupial grazers. J. Comp. Physiol. B 162, 561–566.
Barker, C. J., Gillett, A., Polkinghorne, A., and Timms, P. (2013). Investigation of the koala (Phascolarctos cinereus) hindgut microbiome via 16S pyrosequencing. Vet. Microbiol. 167, 554–564. doi: 10.1016/j.vetmic.2013.08.025
Bletz, M. C., Loudon, A. H., Becker, M. H., Bell, S. C., Woodhams, D. C., Minbiole, K. P., et al. (2013). Mitigating amphibian chytridiomycosis with bioaugmentation: characteristics of effective probiotics and strategies for their selection and use. Ecol. Lett. 16, 807–820. doi: 10.1111/ele.12099
Blyton, M. D., Soo, R. M., Whisson, D., Marsh, K. J., Pascoe, J., Le Pla, M., et al. (2019). Faecal inoculations alter the gastrointestinal microbiome and allow dietary expansion in a wild specialist herbivore, the koala. Anim. Microb. 1:6.
Braithwaite, R. W., and Griffiths, A. (1994). Demographic variation and range contraction in the northern quoll, Dasyurus hallucatus (Marsupialia: Dasyuridae). Wildlife Res. 21, 203–217.
Brice, K. L., Trivedi, P., Jeffries, T. C., Blyton, M. D., Mitchell, C., Singh, B. K., et al. (2019). The Koala (Phascolarctos cinereus) faecal microbiome differs with diet in a wild population. PeerJ 7:e6534. doi: 10.7717/peerj.6534
Brown, A., Girjes, A., Lavin, M., Timms, P., and Woolcock, J. (1987). Chlamydial disease in koalas. Aust. Vet. J. 64, 346–350. doi: 10.1111/j.1751-0813.1987.tb06064.x
Brown, O. J. (2006). Tasmanian devil (Sarcophilus harrisii) extinction on the Australian mainland in the mid-Holocene: multicausality and ENSO intensification. Alcheringa Austral. J. Palaeontol. 30, 49–57. doi: 10.1080/03115510608619574
Burke, C., Burnard, D., Polkinghorne, A., Webb, J., and Huston, W. (2018). Cloacal and ocular microbiota of the Endangered Australian Northern Quoll. Microorganisms 6:68. doi: 10.3390/microorganisms6030068
Cheng, Y., Fox, S., Pemberton, D., Hogg, C., Papenfuss, A. T., and Belov, K. (2015). The Tasmanian devil microbiome—implications for conservation and management. Microbiome 3;1.
Chhour, K.-L., Hinds, L. A., Jacques, N. A., and Deane, E. M. (2010). An observational study of the microbiome of the maternal pouch and saliva of the tammar wallaby, Macropus eugenii, and of the gastrointestinal tract of the pouch young. Microbiology 156, 798–808. doi: 10.1099/mic.0.031997-0
Chong, R., Grueber, C. E., Fox, S., Wise, P., Barrs, V. R., Hogg, C. J., et al. (2019a). Looking like the locals-gut microbiome changes post-release in an endangered species. Anim. Microb. 1, 1–10.
Chong, R., Shi, M., Grueber, C. E., Holmes, E. C., Hogg, C. J., Belov, K., et al. (2019b). Fecal viral diversity of captive and wild Tasmanian devils characterized using virion-enriched metagenomics and metatranscriptomics. J. Virol. 93:e00205-19.
Clayton, J. B., Vangay, P., Huang, H., Ward, T., Hillmann, B. M., Al-Ghalith, G. A., et al. (2016). Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. U.S.A. 113, 10376–10381. doi: 10.1073/pnas.1521835113
Cork, S. J., Hume, I. D., and Dawson, T. J. (1983). Digestion and metabolism of a natural foliar diet (Eucalyptus punctata) by an arboreal marsupial, the koala (Phascolarctos cinereus). J. Comp. Physiol. 153, 181–190. doi: 10.1007/bf00689622
Cork, S. J., and Sanson, G. (1991). “Digestion and nutrition in the koala: a review,” in Biology of the Koala, eds A. K. Lee, K. A. Hanasyde, and G. D. Sanson (Sydney: Surrey Beatty), 129–144.
Dahlhausen, K. E., Doroud, L., Firl, A. J., Polkinghorne, A., and Eisen, J. A. (2018). Characterization of shifts of koala (Phascolarctos cinereus) intestinal microbial communities associated with antibiotic treatment. PeerJ 6:e4452. doi: 10.7717/peerj.4452
Delport, T. C., Harcourt, R. G., Beaumont, L. J., Webster, K. N., and Power, M. L. (2015). Molecular detection of antibiotic-resistance determinants in Escherichia coli isolated from the endangered Australian sea lion (Neophoca cinerea). J. Wildlife Dis. 51, 555–563. doi: 10.7589/2014-08-200
Dubin, K., and Pamer, E. G. (2014). Enterococci and their interactions with the intestinal microbiome. Microbiol. Spectr. 5, 15–19.
Dunlop, J. A., Rayner, K., and Doherty, T. S. (2017). Dietary flexibility in small carnivores: a case study on the endangered northern quoll, Dasyurus hallucatus. J. Mammal. 98, 858–866. doi: 10.1093/jmammal/gyx015
Evans, M. C., Macgregor, C., and Jarman, P. J. (2006). Diet and feeding selectivity of common wombats. Wildlife Res. 33, 321–330.
Fonknechten, N., Chaussonnerie, S., Tricot, S., Lajus, A., Andreesen, J. R., Perchat, N., et al. (2010). Clostridium sticklandii, a specialist in amino acid degradation: revisiting its metabolism through its genome sequence. BMC Genomics 11:555. doi: 10.1186/1471-2164-11-555
Groves, C. P. (2005). Mammal Species of the World: A Taxonomic and Geographic Reference, 3rd Edn. Baltimore: Johns Hopkins University Press, 43–44.
Guan, Y., Zhang, H., Gao, X., Shang, S., Wu, X., Chen, J., et al. (2016). Comparison of the bacterial communities in feces from wild versus housed sables (Martes zibellina) by high-throughput sequence analysis of the bacterial 16S rRNA gene. AMB Express 6:98. doi: 10.1186/s13568-016-0254-4
Gulino, L.-M., Ouwerkerk, D., Kang, A. Y., Maguire, A. J., Kienzle, M., and Klieve, A. V. (2013). Shedding light on the microbial community of the macropod foregut using 454-amplicon pyrosequencing. PLoS One 8:e61463. doi: 10.1371/journal.pone.0061463
Harley, D., Mawson, P. R., Olds, L., McFadden, M., and Hogg, C. J. (2018). “The contribution of captive breeding in zoos to the conservation of Australia’s threatened fauna,” in Recovering Australian Threatened Species: A Book of Hope, eds S. Garnett, J. Woinarski, D. Lindenmayer, and P. Latch (Melbourne: CSIRO Publishing), 281–294.
Hernandez-Santin, L., Goldizen, A. W., and Fisher, D. O. (2016). Introduced predators and habitat structure influence range contraction of an endangered native predator, the northern quoll. Biol. Conserv. 203, 160–167. doi: 10.1016/j.biocon.2016.09.023
Hespell, R., Wolf, R., and Bothast, R. (1987). Fermentation of xylans by Butyrivibrio fibrisolvens and other ruminal bacteria. Appl. Environ. Microbiol. 53, 2849–2853. doi: 10.1128/aem.53.12.2849-2853.1987
Jarman, P. J. (1984). The dietary ecology of macropod marsupials. Proc. Nutr. Soc. Austral. 9, 82–87.
Johnson, R. N., O’Meally, D., Chen, Z., Etherington, G. J., Ho, S. Y., Nash, W. J., et al. (2018). Adaptation and conservation insights from the koala genome. Nat. Genet. 50, 1102–1111.
Jones, M. E., Paetkau, D., Geffen, E., and Moritz, C. (2004). Genetic diversity and population structure of Tasmanian devils, the largest marsupial carnivore. Mol. Ecol. 13, 2197–2209. doi: 10.1111/j.1365-294x.2004.02239.x
Kau, A. L., Ahern, P. P., Griffin, N. W., Goodman, A. L., and Gordon, J. I. (2011). Human nutrition, the gut microbiome and the immune system. Nature 474:327. doi: 10.1038/nature10213
Kho, Z. Y., and Lal, S. K. (2018). The human gut microbiome – a potential controller of wellness and disease. Front. Microbiol. 9:1835. doi: 10.3389/fmicb.2018.01835
Kohl, K. D., Skopec, M. M., and Dearing, M. D. (2014). Captivity results in disparate loss of gut microbial diversity in closely related hosts. Conserv. Physiol. 2:cou009. doi: 10.1093/conphys/cou009
Krishnamurthy, S. R., and Wang, D. (2018). Extensive conservation of prokaryotic ribosomal binding sites in known and novel picobirnaviruses. Virology 516, 108–114. doi: 10.1016/j.virol.2018.01.006
Krockenberger, A., and Hume, I. (2007). A flexible digestive strategy accommodates the nutritional demands of reproduction in a free-living folivore, the Koala (Phascolarctos cinereus). Funct. Ecol. 21, 748–756. doi: 10.1111/j.1365-2435.2007.01279.x
Kueneman, J. G., Woodhams, D. C., Harris, R., Archer, H. M., Knight, R., and McKenzie, V. J. (2016). Probiotic treatment restores protection against lethal fungal infection lost during amphibian captivity. Proc. R. Soc. B Biol. Sci. 283:20161553. doi: 10.1098/rspb.2016.1553
Lazenby, B. T., Tobler, M. W., Brown, W. E., Hawkins, C. E., Hocking, G. J., Hume, F., et al. (2018). Density trends and demographic signals uncover the long-term impact of transmissible cancer in Tasmanian devils. J. Appl. Ecol. 55, 1368–1379. doi: 10.1111/1365-2664.13088
Le Chatelier, E., Nielsen, T., Qin, J., Prifti, E., Hildebrand, F., Falony, G., et al. (2013). Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546.
Lemieux-Labonté, V., Tromas, N., Shapiro, B. J., and Lapointe, F.-J. (2016). Environment and host species shape the skin microbiome of captive neotropical bats. PeerJ 4:e2430. doi: 10.7717/peerj.2430
Ley, R., Turnbaugh, P., and Klein, S. (2006). Human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651.
Liukkonen-Anttila, T., Saartoala, R., and Hissa, R. (2000). Impact of hand-rearing on morphology and physiology of the capercaillie (Tetrao urogallus). Comp. Biochem. Physiol. A Mol. Integr. Physiol. 125, 211–221. doi: 10.1016/s1095-6433(99)00174-9
Loudon, A. H., Woodhams, D. C., Parfrey, L. W., Archer, H., Knight, R., McKenzie, V., et al. (2014). Microbial community dynamics and effect of environmental microbial reservoirs on red- backed salamanders (Plethodon cinereus). ISME J. 8:830. doi: 10.1038/ismej.2013.200
McKenzie, V. J., Kueneman, J. G., and Harris, R. N. (2018). Probiotics as a tool for disease mitigation in wildlife: insights from food production and medicine. Ann. N.Y. Acad. Sci. 1429, 18–30. doi: 10.1111/nyas.13617
McKenzie, V. J., Song, S. J., Delsuc, F., Prest, T. L., Oliverio, A. M., Korpita, T. M., et al. (2017). The effects of captivity on the mammalian gut microbiome. Integr. Comp. Biol. 57, 690–704.
Menke, S., Melzheimer, J., Thalwitzer, S., Heinrich, S., Wachter, B., and Sommer, S. (2017). Gut microbiomes of free-ranging and captive Namibian cheetahs: diversity, putative functions, and occurrence of potential pathogens. Mol. Ecol. 26, 5515–5527. doi: 10.1111/mec.14278
Miller, M. E. B., Antonopoulos, D. A., Rincon, M. T., Band, M., Bari, A., Akraiko, T., et al. (2009). Diversity and strain specificity of plant cell wall degrading enzymes revealed by the draft genome of Ruminococcus flavefaciens FD-1. PLoS One 4:e6650. doi: 10.1371/journal.pone.0006650
Miller, W., Hayes, V. M., Ratan, A., Petersen, D. C., Wittekindt, N. E., Miller, J., et al. (2011). Genetic diversity and population structure of the endangered marsupial Sarcophilus harrisii (Tasmanian devil). Proc. Natl. Acad. Sci. U.S.A. 108, 12348–12353.
Moore, B. D., Foley, W. J., Wallis, I. R., Cowling, A., and Handasyde, K. A. (2005). Eucalyptus foliar chemistry explains selective feeding by koalas. Biol. Lett. 1, 64–67. doi: 10.1098/rsbl.2004.0255
Mukhopadhya, I., Segal, J. P., Carding, S. R., Hart, A. L., and Hold, G. L. (2019). The gut virome: the ‘missing link’ between gut bacteria and host immunity? Ther. Adv. Gastroenterol. 12:1756284819836620.
Musso, G., Gambino, R., and Cassader, M. (2011). Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu. Rev. Med. 62, 361–380. doi: 10.1146/annurev-med-012510-175505
Nakamura, N., Amato, K. R., Garber, P., Estrada, A., Mackie, R. I., and Gaskins, H. R. (2011). Analysis of the hydrogenotrophic microbiota of wild and captive black howler monkeys (Alouatta pigra) in palenque national park, Mexico. Am. J. Primatol. 73, 909–919. doi: 10.1002/ajp.20961
Neil, J. A., and Cadwell, K. (2018). The intestinal virome and immunity. J. Immunol. 201, 1615–1624. doi: 10.4049/jimmunol.1800631
Ngo, S., Kong, S., Kirlich, A., McKinnon, R., and Stupans, I. (2000). Cytochrome P450 4A, peroxisomal enzymes and nicotinamide cofactors in koala liver. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 127, 327–334. doi: 10.1016/s0742-8413(00)00160-2
Nipperess, D. A. (2015). “A separate creation: diversity, distinctiveness and conservation of Australian wildlife,” in Austral Ark: the State of Wildlife in Australia and New Zealand, ed. G. I. Holwell (Cambridge, MA: Cambridge University Press), 1–23. doi: 10.1017/cbo9781139519960.003
Oakwood, M. (2000). Reproduction and demography of the northern quoll, Dasyurus hallucatus, in the lowland savanna of northern Australia. Austral. J. Zool. 48, 519–539.
Ogilvie, L. A., and Jones, B. V. (2015). The human gut virome: a multifaceted majority. Front. Microbiol. 6:918. doi: 10.3389/fmicb.2015.00918
Osawa, R. (1990). Formation of a clear zone on tannin-treated brain heart infusion agar by a Streptococcus sp. isolated from feces of koalas. Appl. Environ. Microbiol. 56, 829–831. doi: 10.1128/aem.56.3.829-831.1990
Osawa, R., Rainey, F., Fujisawa, T., Lang, E., Busse, H., Walsh, T., et al. (1995). Lonepinella koalarum gen. nov., sp. nov., a new tannin-protein complex degrading bacterium. Syst. Appl. Microbiol. 18, 368–373. doi: 10.1016/s0723-2020(11)80430-3
Pearse, A., and Swift, K. (2006). Allograft theory: transmission of devil facial-tumour disease. Nature 439, 549–549. doi: 10.1038/439549a
Pemberton, D. (2019). “The tasmanian devil: a uniquely threatened animal,” in Saving the Tasmanian Devil: Recovery through Science-based Management, eds C. Hogg, S. Fox, D. Pemberton, and K. Belov (Melbourne: CSIRO PUBLISHING).
Pemberton, D., Gales, S., Bauer, B., Gales, R., Lazenby, B., and Medlock, K. (2008). The diet of the Tasmanian devil, Sarcophilus harrisii, as determined from analysis of scat and stomach contents. Pap. Proc. R. Soc. Tasmania 142, 13–22. doi: 10.26749/rstpp.142.2.13
Pemberton, D., and Renouf, D. (1993). A field-study of communication and social-behavior of the Tasmanian devil at feeding sites. Austral. J. Zool. 41, 507–526.
Plummer, S. F., Garaiova, I., Sarvotham, T., Cottrell, S. L., Le Scouiller, S., Weaver, M. A., et al. (2005). Effects of probiotics on the composition of the intestinal microbiota following antibiotic therapy. Int. J. Antimicrob. Agents 26, 69–74. doi: 10.1016/j.ijantimicag.2005.04.004
Polkinghorne, A., Hanger, J., and Timms, P. (2013). Recent advances in understanding the biology, epidemiology and control of chlamydial infections in koalas. Vet. Microbiol. 165, 214–223. doi: 10.1016/j.vetmic.2013.02.026
Power, M. L., Emery, S., and Gillings, M. R. (2013). Into the wild: dissemination of antibiotic resistance determinants via a species recovery program. PLoS One 8:e63017. doi: 10.1371/journal.pone.0063017
Pye, R. J., Pemberton, D., Tovar, C., Tubio, J. M., Dun, K. A., Fox, S., et al. (2016). A second transmissible cancer in Tasmanian devils. Proc. Natl. Acad. Sci. U.S.A. 113, 374–379.
Ramezani, A., Massy, Z. A., Meijers, B., Evenepoel, P., Vanholder, R., and Raj, D. S. (2016). Role of the gut microbiome in uremia: a potential therapeutic target. Am. J. Kidney Dis. 67, 483–498. doi: 10.1053/j.ajkd.2015.09.027
Redford, K. H., Segre, J. A., Salafsky, N., del Rio, C. M., and McAloose, D. (2012). Conservation and the microbiome. Conserv. Biol. 26, 195–197. doi: 10.1111/j.1523-1739.2012.01829.x
Rishworth, C., McIlroy, J., and Tanton, M. (1995). Diet of the common wombat, Vombatus ursinus, in plantations of Pinus radiata. Wildlife Res. 22, 333–339.
Rogers, G., Keating, D., Young, R., Wong, M., Licinio, J., and Wesselingh, S. (2016). From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol. Psychiatry 21:738. doi: 10.1038/mp.2016.50
Rogers, T., Fox, S., Pemberton, D., and Wise, P. (2016). Sympathy for the devil: captive-management style did not influence survival, body-mass change or diet of Tasmanian devils 1 year after wild release. Wildlife Res. 43, 544–552.
Round, J. L., and Mazmanian, S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9;313. doi: 10.1038/nri2515
Seddon, P. J., Armstrong, D. P., and Maloney, R. F. (2007). Developing the science of reintroduction biology. Conserv. Biol. 21, 303–312. doi: 10.1111/j.1523-1739.2006.00627.x
Seiler, C., Angelstam, P., and Bergmann, H.-H. (2000). Conservation releases of captive-reared grouse in Europe–What do we know and what do we need. Cahiers d’Ethologie 20, 235–252.
Shiffman, M. E., Soo, R. M., Dennis, P. G., Morrison, M., Tyson, G. W., and Hugenholtz, P. (2017). Gene and genome-centric analyses of koala and wombat fecal microbiomes point to metabolic specialization for Eucalyptus digestion. PeerJ 5:e4075. doi: 10.7717/peerj.4075
Sousa, M., Gonçalves, A., Silva, N., Serra, R., Alcaide, E., Zorrilla, I., et al. (2014). Acquired antibiotic resistance among wild animals: the case of Iberian Lynx (Lynx pardinus). Vet. Q. 34, 105–112. doi: 10.1080/01652176.2014.949391
Stalder, K., Vaz, P. K., Gilkerson, J. R., Baker, R., Whiteley, P., Ficorilli, N., et al. (2015). Prevalence and clinical significance of herpesvirus infection in populations of Australian marsupials. PLoS One 10:e0133807. doi: 10.1371/journal.pone.0133807
Trevelline, B. K., Fontaine, S. S., Hartup, B. K., and Kohl, K. D. (2019). Conservation biology needs a microbial renaissance: a call for the consideration of host-associated microbiota in wildlife management practices. Proc. R. Soc. B 286, 20182448. doi: 10.1098/rspb.2018.2448
Turnbaugh, P. J., Ley, R. E., Mahowald, M. A., Magrini, V., Mardis, E. R., and Gordon, J. I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027.
West, A. G., Waite, D. W., Deines, P., Bourne, D. G., Digby, A., McKenzie, V. J., et al. (2019). The microbiome in threatened species conservation. Biol. Conserv. 229, 85–98. doi: 10.1016/j.biocon.2018.11.016
Wienemann, T., Schmitt-Wagner, D., Meuser, K., Segelbacher, G., Schink, B., Brune, A., et al. (2011). The bacterial microbiota in the ceca of Capercaillie (Tetrao urogallus) differs between wild and captive birds. Syst. Appl. Microbiol. 34, 542–551. doi: 10.1016/j.syapm.2011.06.003
Woinarski, J. C., Burbidge, A. A., and Harrison, P. L. (2015). Ongoing unraveling of a continental fauna: decline and extinction of Australian mammals since European settlement. Proc. Natl. Acad. Sci. U.S.A. 112, 4531–4540. doi: 10.1073/pnas.1417301112
Keywords: gut microbiome, wildlife conservation, marsupial, captivity, translocation, dysbiosis
Citation: Chong R, Cheng Y, Hogg CJ and Belov K (2020) Marsupial Gut Microbiome. Front. Microbiol. 11:1058. doi: 10.3389/fmicb.2020.01058
Received: 16 November 2019; Accepted: 29 April 2020;
Published: 29 May 2020.
Edited by:
Lifeng Zhu, Nanjing Normal University, ChinaReviewed by:
Zhihong Sun, Inner Mongolia Agricultural University, ChinaHua Chen, Mingke Biotechnology, China
Copyright © 2020 Chong, Cheng, Hogg and Belov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine Belov, a2F0aHkuYmVsb3ZAc3lkbmV5LmVkdS5hdQ==