 Ekaterina Samatova
Ekaterina Samatova Marija Liutkute
Marija Liutkute Marina V. Rodnina
Marina V. Rodnina- Department of Physical Biochemistry, Max Planck Institute for Biophysical Chemistry, Göttingen, Germany
Protein homeostasis of bacterial cells is maintained by coordinated processes of protein production, folding, and degradation. Translational efficiency of a given mRNA depends on how often the ribosomes initiate synthesis of a new polypeptide and how quickly they read the coding sequence to produce a full-length protein. The pace of ribosomes along the mRNA is not uniform: periods of rapid synthesis are separated by pauses. Here, we summarize recent evidence on how ribosome pausing affects translational efficiency and protein folding. We discuss the factors that slow down translation elongation and affect the quality of the newly synthesized protein. Ribosome pausing emerges as important factor contributing to the regulatory programs that ensure the quality of the proteome and integrate the cellular and environmental cues into regulatory circuits of the cell.
Translation Regulation in Bacteria
Translation is an essential step in the expression of protein-coding genes, which defines the composition of the cellular proteome. Timely production of functional proteins is of central importance in maintaining cell viability, as the proteins – which comprise 60–80% of cellular biomass in bacteria – play key roles in every process in living cells. Translation is the most conserved and energy-demanding process in the cell, which consumes two-thirds of the total cellular energy during rapid growth (Russell and Cook, 1995; Klumpp et al., 2013; Basan et al., 2015). More than 40% of the protein synthesis capacity is dedicated to maintaining the translational machinery itself and in particular the ribosomes (Li et al., 2014). In bacteria, translational control ensures rapid response to changes in environmental cues, which is then followed by global changes in cell physiology, including adjustments in transcriptional profiles, alterations in ribosome biogenesis, and switching to ribosome hibernation programs. However, even under constant environmental conditions, some mRNAs are translated more often than others, resulting in a characteristic copy number of proteins synthesized from their respective mRNA, which is defined as translational efficiency (TE).
In principle, translation can be regulated at any of its steps, i.e., initiation, elongation, termination, or ribosome recycling. The initiation step, at which the ribosome selects the mRNA and finds the open reading frame (ORF), to a large extent, controls the frequency at which a given mRNA is translated (Milon and Rodnina, 2012; Gualerzi and Pon, 2015; Tollerson and Ibba, 2020; Figure 1). The basal translation level is determined by the accessibility of the ribosome binding site on the mRNA, the nature of the start codon, the position of the Shine-Dalgarno (SD) sequence relative to the start codon and its complementarity to the anti-SD sequence in the 16S rRNA, and the presence of A/U rich sequences that may be specifically recognized by ribosomal protein bS1 (Gualerzi and Pon, 2015; Cifuentes-Goches et al., 2019; Saito et al., 2020). The accessibility of the ribosome binding site can change depending on the environment conditions due to ligand- or temperature-induced re-folding of the mRNA or its interactions with proteins, which regulates the protein synthesis levels of selected mRNAs and is generally referred to as riboswitches (Geissmann et al., 2009; Kortmann and Narberhaus, 2012; Breaker, 2018).
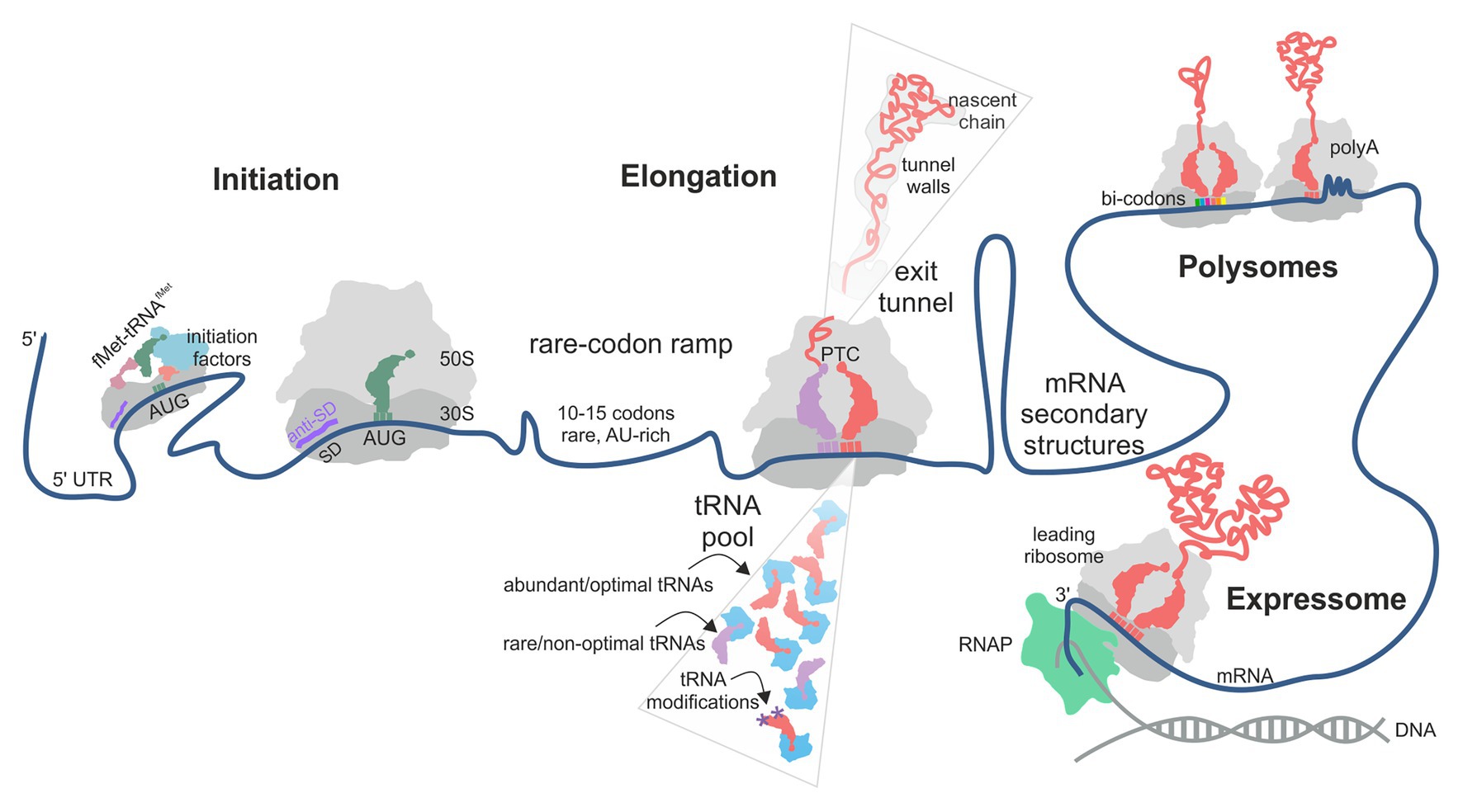
Figure 1. Factors contributing to translational efficiency (TE) and protein folding in bacterial translation. The schematic follows the mRNA direction from the 5' (left) to the 3' (right) end. The TE of an mRNA is largely determined at the translation initiation step when the 30S ribosomal subunit is recruited to the start codon on the mRNA. In some cases, next 30S subunit can be recruited to a stand-by site upstream of the initiation site. A rare-codon ramp of 10–15 A/U-rich codons at the beginning of the coding region can increase TE by disfavoring mRNA secondary structures at the start codon. During translation, rare codons, low-abundance aa-tRNAs, lack of tRNA modifications and the interactions of the nascent chain with the polypeptide exit tunnel of the ribosome may cause ribosome pausing. mRNA secondary structures can regulate ribosome occupancy at the upstream sequences. Some mRNA contexts, such as particular bi-codons, poly(Pro) and poly(Lys) sequences, cause rearrangements in the peptidyl transferase center (PTC) and promote formation of unusual structures in the A-site, thereby promoting ribosome stalling. Interactions between ribosomes in a polysome and of a leading ribosome with the RNA polymerase (RNAP) may provide yet another source of pausing.
Growing evidence suggests that also the elongation step can affect TE and the quality of the corresponding protein (Rodnina, 2018; Tollerson and Ibba, 2020). At each step of translation elongation, a new amino acid is added to the growing nascent chain, and the ribosome moves along the mRNA by one codon. The rate of elongation is not uniform along the mRNA, with periods of rapid movement separated by pauses. Translation termination can contribute to the diversity of the cellular proteome by producing C-terminally extended protein isoforms due to translational readthrough of a stop codon. Finally, ribosome recycling releases the ribosomal subunits from the mRNA that has been translated, thereby maintaining the pool of ribosomes available for initiation on new mRNAs. While mechanisms of translation regulation at the initiation step are well understood, the contribution of the kinetic tuning of elongation to TE and protein quality is a complex question. Although the idea that the speed and the quality of translation are related is not new, it remained controversial due to the complexity and pleiotropic origins of the effects. In this review, we will summarize new results that shed light on ribosome pausing during elongation in bacteria and discuss the role of pauses in the control of translation efficiency and protein folding.
Non-Uniform Rate of Translation and Translational Efficiency
Variation of translation rates may be caused by several factors (Figure 1). The mRNA may contain sequences that – under given environmental conditions – can cause pausing. These include codon choice and distribution along the mRNA (Mohammad et al., 2019), codon context (Gamble et al., 2016), specific sequences, such as poly(A) tracts (Koutmou et al., 2015), and the mRNA structure (Wen et al., 2008). The composition of the aa-tRNA pool is important as well (Rudorf et al., 2014; Vieira et al., 2016; Dykeman, 2020). The translatome of the cell, i.e., the pool of mRNA that are translated at a given moment, the tRNA charging levels, and tRNA modification efficiency may change under conditions of nutritional stress, thereby affecting the efficiency of decoding of some codons (Elf et al., 2003; Ranjan and Leidel, 2019; Dykeman, 2020). Finally, the sequence of the nascent peptide may regulate translation by, e.g., stalling translation at specific sites (Ito and Chiba, 2013; Wilson et al., 2016) or modulating translation via interactions of charged residues in the nascent peptide with the polypeptide exit tunnel wall of the ribosome. Ribosome pausing prolongs the time required to complete translation of a given mRNA, which – in the simplest model – could result in fewer copies of protein per mRNA per unit of time and thus a lower TE. Thus, a simple model would predict an effect of ribosome pausing on the TE, which we will discuss in detail below.
Codon Usage
It is long known that the same protein sequence can be encoded in the mRNA in various ways using synonymous codons, which specify the insertion of the same amino acid but differ in their nucleotide composition. Synonymous codons can be read by a single tRNA, such as tRNALys or tRNAPhe, which in Escherichia coli decode their codons AAA/AAG and UUU/UUC, respectively. However, for most four- and six-codon families, there are several tRNAs that deliver the same amino acid but differ in their decoding preferences; such tRNAs are called isoacceptor tRNAs. Isoacceptor tRNAs can usually recognize several synonymous codons through non-Watson-Crick “wobble” base pairing at the third codon position. How many and which codons can be decoded by one particular tRNA is controlled by post-transcriptional tRNA modification (Grosjean and Westhof, 2016). The rate of decoding a particular codon depends not only on the complementarity of the codon-anticodon complex but also on the concentration and properties of the respective cognate aa-tRNA in the total tRNA pool.
The preference for particular codons (codon usage bias) and their distribution along the mRNA are non-random and specific for each organism (Grantham et al., 1980; Ikemura, 1985; Moura et al., 2005). Analysis of synonymous codon frequencies in different genes has shown a bias toward usage of optimal codons in highly expressed mRNAs (Ikemura, 1981; Quax et al., 2013), which led to the suggestion that codon usage bias defines the TE, and that the presence of slowly translated codons results in lower TE (Ikemura, 1981; Sharp and Li, 1987; Dong et al., 1996; Tuller et al., 2010; Quax et al., 2013). However, later bioinformatics analyses did not support this assertion (Kudla et al., 2009; Allert et al., 2010; Subramaniam et al., 2014; Cambray et al., 2018). Expression studies found little or no correlation between codon bias and gene expression in E. coli using a synthetic library of 154 variants of green fluorescent protein (GFP) with random synonymous substitutions (Kudla et al., 2009), as well as a synthetic library of 285 genes (Allert et al., 2010) and 244,000 genes fused with GFP (Cambray et al., 2018) under normal conditions. In contrast, the enrichment in rapidly decoded codons in highly expressed genes becomes important under conditions of amino acid starvation, where the difference in the rate of aminoacylation of tRNA isoacceptors has a significant impact on the TE (Elf et al., 2003; Subramaniam et al., 2013, 2014; Wohlgemuth et al., 2013).
Interestingly, translation of the first 10–15 codons of ORFs appears to be special, as these stretches are enriched with non-optimal codons which are mostly A/U-rich, at least in E. coli (Goodman et al., 2013; Cambray et al., 2018; Verma et al., 2019; Osterman et al., 2020; Figure 1). Translation of these stretches is relatively slow (Oh et al., 2011), which would predict a lower TE, but in fact the high A/U content correlates with high TE (Moreira et al., 2019; Verma et al., 2019; Osterman et al., 2020). One possible explanation is that the A/U-rich sequences at the beginning of the ORF act as a translation enhancer (Qing et al., 2003) by reducing the stability of mRNA secondary structures around the ribosome binding site, thereby facilitating initiation and increasing TE (Gu et al., 2010; Goodman et al., 2013; Bhattacharyya et al., 2018). Alternatively, A/U-rich sequences may be preferentially recruited by the ribosomal protein bS1, which is the largest ribosomal protein and an RNA chaperone (Duval et al., 2013; Byrgazov et al., 2015). bS1 is composed of six contiguous domains known to bind to such sequences in the 5' UTR (Boni et al., 1991; Duval et al., 2013). Whether it also recruits mRNAs through the A/U sequences at the beginning of the coding region has not been tested so far.
One important question is whether rare codons are decoded more slowly than the abundant codons, thereby locally slowing down the progression of ribosomes along the mRNA, which is called ribosome pausing. This question should be readily answered by ribosome profiling, a method that enables the detailed measurements of translation in vivo at single codon resolution (Ingolia et al., 2009). While in mammalian and yeast cells ribosome profiling demonstrated a robust anti-correlation between codon optimality and ribosome occupancy (Weinberg et al., 2016; Wu et al., 2019), for bacteria, and E. coli in particular, the method has been more problematic due to technical caveats of stopping translation and alignment of ribosome-protected mRNA fragments (Oh et al., 2011; Subramaniam et al., 2014). Recent improvement of sample preparation for the ribosome profiling in E. coli, including rapid cell harvesting and methods to arrest translation, confirmed that also in bacteria, the ribosome density correlates with the codon-adaptation index, consistent with the expectation that rare codons are decoded by lower-abundance tRNAs more slowly than more abundant codons by their respective tRNAs (Mohammad et al., 2019), although the correlation is not as strong as in yeast cells (Weinberg et al., 2016). As a complementary method, ribosome pausing sites can be identified by the so-called integrated Nascent chain Profiling, iNP (Ito et al., 2011; Chadani et al., 2016). The approach is to isolate peptidyl-tRNAs accumulating when the ribosome pauses at a certain codon for a long time, which is defined as ribosome stalling, and identifying the sequence of the nascent peptide and its respective tRNA. Such analysis could potentially provide very accurate information on the duration of ribosome pausing but lacks so far the high-throughput character of the ribosome profiling technique.
Some aspects of the codon usage bias go beyond the simple distribution of rare and abundant codons. Bioinformatics analysis of E. coli genome suggests that certain codon pairs are overrepresented (i.e., codon pairs observed more frequently than predicted) in the genes coding for non-abundant proteins and underrepresented in highly expressed genes (Boycheva et al., 2003; Moura et al., 2005; Tats et al., 2008; Guo et al., 2012). The order of codons in the codon pairs is crucial; for example, the codon pair AAG-UUA is overrepresented in highly expressed genes and UUA-AAG in poorly expressed genes (Boycheva et al., 2003). Although no experimental data are available on how codon combinations may regulate the elongation rate in bacteria, translation inhibition by specific codon pairs has been demonstrated in yeast (Gamble et al., 2016). In yeast, translation of such inhibitory pairs is slower than expected for the sum of translation times for each codon individually; inhibition is abolished by replacing the sequence with synonymous codons or by reversing the codon order (Gamble et al., 2016; Tesina et al., 2020). The structure of the ribosome stalled on CGA-CCG and CGA-CGA inhibitory codon pairs showed that in both cases, the mRNA conformation in the A-site disturbs the aa-tRNA binding or accommodation, which causes the ribosome stalling (Figure 2; Tesina et al., 2020).
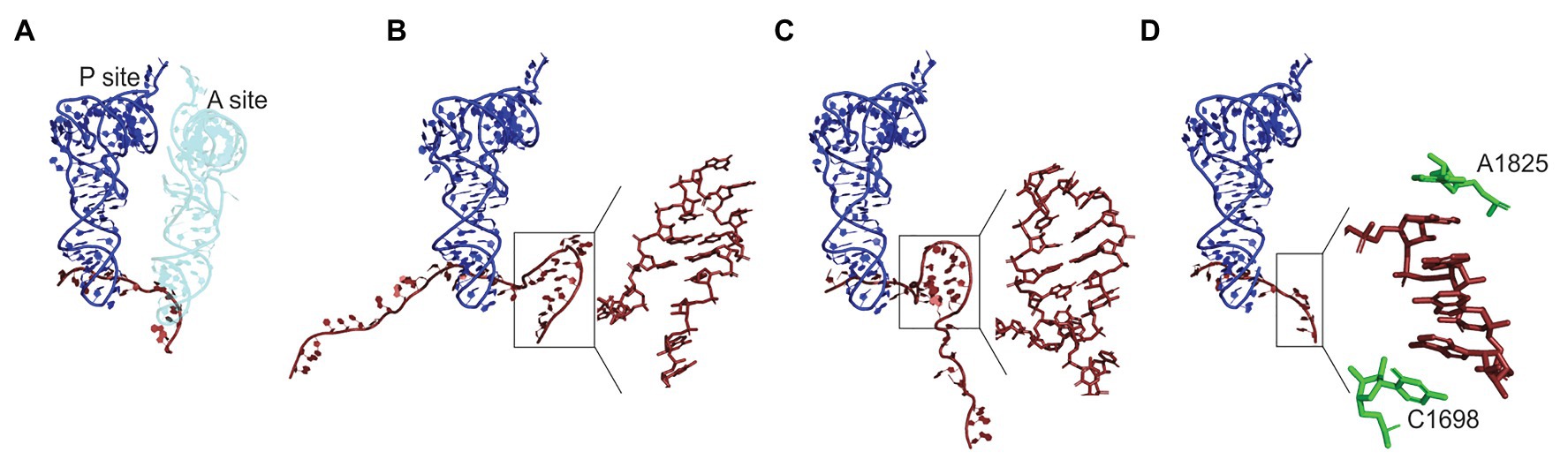
Figure 2. Regulation of the A-site accessibility by unconventional mRNA secondary structures formed upon ribosome pausing. (A) Positions of the P- (dark blue) and A-site (light blue) tRNAs on the mRNA (dark red) in Escherichia coli ribosome (PDB: 7K00; Watson et al., 2020). The density of the ribosome is omitted for clarity. The A-site is occupied by tRNA. In panels (B–D), the A-site tRNA is absent and the P-site tRNA is used for alignment. (B) An mRNA element in the A-site of yeast ribosome stalled on CGA-CCG inhibitory bicodon (PDD: 6T4Q; Tesina et al., 2020). Inset: a close up of the secondary structure element. (C) A hairpin formed in the A-site of E. coli ribosome stalled on a take-off site of gene 60 mRNA of bacteriophage T4 prior to bypassing (PDB: 5NP6; Agirrezabala et al., 2017). Inset: a close up of the secondary structure element. (D) Mammalian ribosome stalled on a poly(A) sequence (PDB: 6SGC; Chandrasekaran et al., 2019). Inset: a close-up of the single stranded helix in the A-site. Residues A1825 and C1698 of 18S rRNA (green) stabilize helix formation by stacking.
Another interesting phenomenon is “codon clustering,” which is found in both pro- and eukaryotes. Rare codons often occur in clusters, rather than being randomly distributed along the mRNA (Makhoul and Trifonov, 2002; Clarke and Clark, 2008; Chartier et al., 2012). In bacteria, including E. coli, codon pairs formed by rare codons are overused, whereas pairs formed by common codons are underused, which is not the case in eukaryotes, where this tendency is reversed (Buchan et al., 2006). Finally, some reports suggest “co-occurrence” of synonymous codons that use the same tRNA in close proximity on the mRNA coding sequence (Cannarrozzi et al., 2010; Shao et al., 2012), although the functional significance of such clustering is not understood. The complexity and strong species-specificity of overall codon distribution suggests that cells adapt their genetic programs to the needs for the protein production beyond amino acid selection.
One of the major factors that can affect protein production in the cell is the stability of the mRNA transcript. In fact, codon usage appears to be a major determinant of mRNA stability in yeast and mammals (Hanson and Coller, 2018). In E. coli, codon content can modulate mRNA stability (Boel et al., 2016; Cambray et al., 2018), but the mechanisms that link rare codons with the mRNA stability are likely to be different in bacteria and eukaryotes, because bacteria lack the specific Ccr4-Not complex that in eukaryotes senses the unoccupied A-site and initiates mRNA degradation by deadenylation and decapping (Buschauer et al., 2020). The precise effect of rare codons on mRNA stability in bacteria likely depends on a delicate balance between their propensity to form secondary structures and to stall ribosomes along the ORF. Both can shield the mRNA from ribonucleases, thereby extending the mRNA lifetime and increasing TE but can also reduce the initiation frequency and increase the translation time, respectively, which decreases TE (Boel et al., 2016; Cambray et al., 2018). Furthermore, slow translation elongation due to poor codon usage may deplete the pool of free ribosomes, which in turn can reduce the initiation efficiency and thereby tends to decrease TE (Cambray et al., 2018). Thus, the relationship between mRNA stability, TE, and availability of translational components may be very complex and affected not only by the codon usage in a given mRNA but also in the pool of cellular mRNAs as a whole.
Mechanistically, codon-dependent variations in the elongation rates can arise at each of the three elongation phases: during (i) aa-tRNA selection according to the mRNA codon in the A-site (decoding); (ii) peptide bond formation between the incoming amino acid and the nascent polypeptide in the peptidyl transferase center; and (iii) tRNA-mRNA translocation, which exposes the next codon in the A-site. These reactions, alone or in combination, establish the rate of elongation at a particular codon. In the following, we will summarize potential mechanisms of pausing at each of these phases.
Aa-tRNA Abundance
Aa-tRNA is delivered to the A-site of the ribosome in a ternary complex with EF-Tu and GTP. The rate of decoding is determined by the rate of selection of an aa-tRNA cognate to a given codon from the total pool of aa-tRNAs, followed by GTP hydrolysis by EF-Tu and accommodation of aa-tRNA in the peptidyl transferase center of the ribosome (Rodnina, 2018). For every single codon, there is a different distribution of cognate, near-cognate, and non-cognate aa-tRNAs, with their different decoding properties, such as the number and geometry of mismatches in the codon-anticodon complex, and aa-tRNA concentrations. The competition between the aa-tRNAs results in variations in decoding times for different codon-aa-tRNA pairs (Rudorf et al., 2014; Vieira et al., 2016; Dykeman, 2020), even though the rates of reactions on the decoding pathway, such as binding, GTP hydrolysis, and tRNA accommodation, are similar for different cognate aa-tRNAs (Rodnina et al., 2005; Ledoux and Uhlenbeck, 2008). Although in bacteria, global codon usage matches the tRNA abundance, transient changes in the transcriptome composition due to transcriptional responses may shift mRNA codon bias relative to tRNA concentrations, which is predicted to have strong effects on decoding and may lead to additional, unexpected ribosome pauses (Rudorf, 2019; Dykeman, 2020). Under conditions of rapid growth, tRNAs are almost fully charged (Dittmar et al., 2005). Depletion of amino acids, e.g., during starvation, results in selective aa-tRNA charging levels for some tRNA isoacceptors, with some becoming low and others remaining high (Elf et al., 2003). Codons read by isoacceptors that retain high charging can be used for efficient translation of genes that are essential during amino-acid starvation (Dittmar et al., 2005; Figure 1).
In this context, modifications at the tRNA anticodon region play an important role, and their absence can lead to ribosome stalling at the respective codons (Ranjan and Leidel, 2019). In particular, the positions 34 and 37 in the tRNA anticodon stem-loop (ASL) affect several elongation steps, including aa-tRNA selection and translocation. Modifications at both positions affect the structure and conformational dynamics of the ASL and provide chemical groups for the non-canonical interactions with the mRNA (Agris, 2008). The modifications at position 37 are mainly associated with reading frame maintenance (Agris et al., 2018; Hou et al., 2019). The N6-threonylcarbamoyladenosine-(t6A) modification, which is commonly found at position 37 of Arg-, Asn-, Ile-, Lys-, Met-, and Ser-tRNAs, is essential for the viability of E. coli (Juhling et al., 2009; Thiaville et al., 2015); t6A-deficient yeast strains are viable but have increased protein aggregation due to global mistranslation (Pollo-Oliveira et al., 2020). The modifications at position 34 are particularly important as modulators of wobble-position decoding. For example, the 5-oxyacetic acid (cmo5) modification at U34 expands the decoding capacity of tRNA allowing to decode all four synonymous codons for Ala, Ser, and Val (Agris et al., 2018) and expands the codon recognition by wobble interaction toward unusual base pairing (Lim and Curran, 2001; Agris et al., 2007; Grosjean and Westhof, 2016). Ser-tRNA(UGA), carrying the cmo5 modification at U34, can form stable complexes with all four codons, adopting specific conformations to stabilize each type of interaction (Weixlbaumer et al., 2007). Another cmo5-modified tRNA, Ala-tRNA(UGC), can read the non-Watson Crick synonymous codon GCC correctly, using a U:C base pairing at the third codon position, albeit slower than codons with a conventional wobble base pairing (Kothe and Rodnina, 2007). The lack of the 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U34) modification in tRNALys, tRNAGlu, and tRNAGln in yeast results in ribosome queuing at the respective codons, affects expression of a subset of genes enriched for AAA, CAA, and GAA codons and leads to protein aggregation (Rezgui et al., 2013; Zinshteyn and Gilbert, 2013; Nedialkova and Leidel, 2015; Ranjan and Rodnina, 2016). Although some of these specific examples are derived from yeast, it is likely that in E. coli, the effect of these modifications is similar, because the basic mechanisms of codon-anticodon recognition are universally conserved in pro- and eukaryotes. In summary, the decoding properties, concentrations, and chemical modifications of aa-tRNA play an important role during decoding and may contribute to translational pausing and the overall TE of the gene.
Amino Acid Composition of the Nascent Peptide
Experiments with model substrates show that the rate of peptide bond formation depends on the nature of the amino acids acting as peptide donors and acceptors (Wohlgemuth et al., 2008; and earlier papers cited therein). However, for many aa-tRNAs, the chemistry step is rate-limited by the preceding step of aa-tRNA accommodation in the peptidyl transferase center on the large ribosomal subunit, which is similar for various aa-tRNAs (Wohlgemuth et al., 2008). One exception is the peptide bond formation involving consecutive Pro residues, which is much slower than with other amino acids. The rate of Pro codon translation is context-dependent with the time of pausing modulated by the preceding or following amino acid (Peil et al., 2013; Woolstenhulme et al., 2015; Mohammad et al., 2019). In bacteria, peptide bond formation between consecutive Pro residues is accelerated by EF-P (Doerfel et al., 2013; Ude et al., 2013), which helps to orient and stabilize the reactants in the peptidyl-transferase center (Doerfel et al., 2015; Huter et al., 2017). Nevertheless, even in the presence of EF-P, ribosomes pause on sequences presenting a combination of Pro, Gly, and Asp codons (Mohammad et al., 2019). Pausing on poly-Pro stretches may regulate the amount of the respective proteins in the cell, as suggested for the Cad module, a lysine-dependent acid-resistance stress-response system. The membrane-integrated sensor CadC regulates induction of the cadBA operon encoding the lysine/cadaverine antiporter CadB and the lysine decarboxylase CadA. Activation of wild-type CadC requires two stimuli, low pH and exogenous lysine, to induce cadBA expression; in the absence of exogenous lysine, CadC is inhibited by its co-sensor, lysine permease (LysP). CadC gene contains a stretch of five Pro codons, which are normally translated with the help of EF-P. In the absence of functional EF-P, the ribosome is stalled at the poly-Pro track, which reduces the copy number of CadC and abolishes the expression of the downstream cadAB operon, resulting in an inactivation of the module. In contrast, introducing mutations into the poly-Pro track results in a higher TE of all proteins encoded by the operon and makes the system less dependent on both external stimuli due to a change in the ratio between CadC and LysP. This example illustrates the potential regulatory role of EF-P and the poly-Pro stretches in fine-tuning protein expression (Ude et al., 2013).
Ribosome pauses can also be caused by the interactions of the newly synthesized nascent peptide with the walls of the polypeptide exit tunnel via distortion of the optimal geometry at the peptidyl transferase center (PTC; Ito and Chiba, 2013; Wilson et al., 2016). This phenomenon is best illustrated by the so-called “arrest peptides” such as SecM, MifM, VemP, and ErmCL (Ito and Chiba, 2013). These sequences are about 20 amino acids long. They cause PTC distortion through the interaction with the polypeptide exit tunnel of the ribosome, which results in the reduced rate of the peptide bond formation and ribosome stalling when slowly reacting amino acids are in the A and P sites of the PTC (such as proline and glycine; Woolstenhulme et al., 2013; Wilson et al., 2016; Seip et al., 2018; Mohammad et al., 2019).
Peptidyl transferase center also becomes distorted during translation of poly(A) sequences into stretches of poly-Lys both in E. coli and eukaryotes (Koutmou et al., 2015; Chandrasekaran et al., 2019). The ribosomes tend to stall and shift the reading frame when they encounter poly(A) sequences longer than 9 nucleotides (nt). The inhibition of translation, as shown for mammalian ribosomes, is due to conformational changes in both PTC and the decoding center (Chandrasekaran et al., 2019). Poly-Lys in the polypeptide exit tunnel stabilizes the PTC in a conformation that is inhibitory to peptide bond formation. In parallel, the poly(A) stretch of the mRNA adopts a single-stranded helix conformation in the decoding center, which is stabilized by the interactions with the rRNA (Figure 2). The reconfigured decoding center disfavors aminoacyl-tRNA binding to the A-site, thereby hindering the elongation even further (Chandrasekaran et al., 2019; Tesina et al., 2020). Given the high degree of evolutionary conservation of the functional centers of the ribosome and the consistent stalling of E. coli ribosomes by poly-Lys nascent peptide, same mechanism is likely to operate in bacteria as well.
In addition to the specialized stalling sequences, also shorter patches of amino acids may have an impact on the TE. Statistical analysis of ~6,000 genes from different organisms expressed in E. coli showed that the amino acid identity has a significant impact on protein expression. Bioinformatics analysis of E. coli proteome revealed that several amino acid triplets (for example, CMY, MWC, GPP, and WMC) and thousands of quadruplets (for example, CMYW) are either completely absent or are several-fold less abundant than expected for random sequences (Navon et al., 2016). Single-molecule FRET experiments suggested that the presence of such sequences in the mRNA increases the elongation time (Navon et al., 2016). This indicates that ribosomes tend to stall while translating these codon combinations, but the exact reason for stalling is not yet known.
The amino acid charge distribution along the nascent chain may affect translation elongation as well (Lu and Deutsch, 2008; Tuller et al., 2011). Positively charged amino acids upstream of the A-site codon slow down the ribosome irrespective of codon identity or codon distribution (Lu and Deutsch, 2008; Charneski and Hurst, 2013; Sabi and Tuller, 2015), and there is a linear correlation between the extent of ribosome pausing and the length of positively charged segments (Charneski and Hurst, 2013). The effect of charged amino acids on translation also depends on its location in the nascent peptide. Codons encoding for positively and negatively charged amino acids are non-randomly distributed along the mRNA with higher frequency of positive charges at the beginning and negative charges at the end (Duc and Song, 2018). This observation led to the suggestion that the incorporation of positively charged amino acids at the beginning of elongation helps the N-terminus of the nascent polypeptide to move toward and enter the negatively charged part of the exit tunnel (Duc and Song, 2018). Once the N-terminus passed the exit tunnel, the mean elongation rate increases with the hydrophobicity of the nascent chain (Duc and Song, 2018). While some of these studies have been conducted using yeast as a model organism (Charneski and Hurst, 2013; Sabi and Tuller, 2015; Duc and Song, 2018), the tendencies observed in yeast may be common for bacteria as well, because residues lining the exit tunnel are highly conserved in the zone proximal to the PTC and diverge only around the vestibule zone (Duc et al., 2019). Mechanistically, the effects of interactions of the nascent peptide with the exit tunnel are still poorly understood. Nascent peptide interactions with the ribosome may provide a new regulatory mechanism for sequence-specific modulation of translation speed.
mRNA Structure and tRNA-mRNA Translocation
The nucleotide sequence of the mRNA determines the potential secondary structure, which may create obstacles when the ribosome moves along the mRNA during the translocation phase (Figure 1). The coding regions of mRNA generally appear more structured than non-coding ones, but the structures are dynamic (Del Campo et al., 2015). Ribosome pausing at mRNA structures is likely transient due to the intrinsic helicase activity of the ribosome that can unwind thermodynamically stable mRNA structures (Takyar et al., 2005; Qu et al., 2011). The TE of ORFs within a polycistronic mRNAs negatively correlates with increasing levels of mRNA structure (Burkhardt et al., 2017). However, probing of RNA secondary structure with the so-called selective 2' hydroxyl acylation analyzed by primer extension (SHAPE) method in conjunction with parallel analysis of RNA structure (PARS) shows that only the mRNA structures at the initiation site are important and there is no significant correlation between mRNA structure in the coding region and TE (Del Campo et al., 2015; Mustoe et al., 2018). Stable in vivo mRNA structures mostly represent evolutionarily conserved functional elements, which serve as signals for programmed recoding events, such as frameshifting and bypassing (Rouskin et al., 2014), or for membrane insertion of inner membrane proteins (Del Campo et al., 2015). For example, mRNA structures that stimulate frameshifting can reduce the rate of translation by as much as 10-fold (Wen et al., 2008; Chen et al., 2013, 2014; Caliskan et al., 2014, 2017; Kim et al., 2014; Bock et al., 2019) by inhibiting the dissociation of deacylated tRNA from the E-site (Chen et al., 2013; Caliskan et al., 2014). On the other hand, the overall effect of mRNA structure on pausing may be counterbalanced by the selection of highly abundant codons participating in structure formation (Gorochowski et al., 2015). Vice versa, in bacteria, unstructured mRNA regions are often comprised of codons decoded by low abundance tRNAs. This, in general, can smoothen out the variation in global translation rates (Gorochowski et al., 2015). Of note, also mRNA interactions with the rRNA were suggested to slow down translation, in particular through the interactions of SD-like sequences in the mRNA coding region with the anti-SD sequence in the 16S rRNA (Li et al., 2012). Early ribosome profiling experiments seemed to support this notion (Li et al., 2012). However, more recent work using optimized ribosome profiling protocols has shown that the respective accumulation of mRNA reads was caused by technical problems and that there is no indication for pervasive ribosome pausing caused by potential SD-like sequences (Martens et al., 2015; Chadani et al., 2016; Mohammad et al., 2016, 2019). This result is consistent with the lack of peptidyl-tRNA accumulation which would be expected if the ribosome were stalled at SD-like sequences (Chadani et al., 2016).
In addition to large mRNA secondary structure elements that can modulate stalling, recent data suggest that local mRNA structures may have an unexpected effect that would be difficult to predict based on the thermodynamic stabilities of the respective structures alone (Figure 2). Pausing at these unusual structures is regulated by the interactions of the nascent peptide with the exit tunnel. As mentioned above, poly(A) stretches can form a single-stranded helix in the A-site, which is stabilized by the rRNA in the decoding region and coincides with the formation of the catalytically inactive conformation of the PTC (Figure 2D; Chandrasekaran et al., 2019; Tesina et al., 2020). Similarly, CGA-CCG and CGA-CGA sequences form secondary structures in the A-site that inhibit aa-tRNA binding (Figure 2B; Tesina et al., 2020). The gene 60 mRNA of the bacteriophage T4 codes for a sequence that can stall its own translation at a very specific codon due to interactions of the nascent peptide with the exit tunnel. The interaction induces an inactive PTC conformation and the formation of a small mRNA hairpin in the A-site (Figure 2C; Agirrezabala et al., 2017). Thus, nascent-peptide-mediated formation of local mRNA structures is an emerging new mechanism for regulation translational pauses both in bacteria and eukaryotes.
Coupling of Transcription and Translation, Polysomes, and Ribosome Collisions
In many bacteria, including E. coli, transcription and translation are coupled. The mRNA emerging from the RNA polymerase (RNAP) is recruited by the leading ribosome that resides in close proximity or is even physically linked to RNAP (Landick et al., 1985; Burmann et al., 2010; Proshkin et al., 2010; Kohler et al., 2017; O’Reilly et al., 2020; Wang et al., 2020; Webster et al., 2020), forming the so-called expressome (Kohler et al., 2017; Figure 1). The rates of transcription and translation in the expressome are coordinated and change together in response to growth conditions (Vogel and Jensen, 1994). The coordination between RNAP can be either direct, i.e., via the bridging proteins (Burmann et al., 2010; Proshkin et al., 2010), or indirect, e.g., through second messengers such as ppGpp (Chen and Fredrick, 2020). Regardless of the coupling mechanism, the leading ribosome cannot move faster than the RNAP does, so that the pausing of RNAP may dictate the behavior of the leading ribosome (Vogel and Jensen, 1994; Proshkin et al., 2010). Surprisingly, however, it is rather the ribosome that appears to modulate the pace of RNAP. For example, acceleration or deceleration of translation by antibiotics or mutations in ribosomal proteins result in corresponding changes in the speed of RNAP (Proshkin et al., 2010). Even more remarkable, the rate of transcription correlates with the number of rare codons in a gene, presumably by slowing down the progression of the ribosome along the mRNA (Proshkin et al., 2010). How exactly the coordination between the ribosome and RNAP affects the TE for a given protein is not clear, but it clearly provides a mechanism by which bacteria can globally react to changes in growth rate and environmental conditions. One surprising aspect of the tight coupling between the RNAP and the leading ribosome is that such hand-over of the emerging mRNA transcript might prevent formation of the potential regulatory secondary structures. To be operational, such structures would have either to delay the progression of the leading ribosome relative to the RNAP or to form behind the leading ribosome. Interestingly, a recent paper suggests that coupled RNAP-ribosome movement is not a general hallmark of bacterial gene expression; rather, in some bacteria transcription is much faster than translation (Johnson et al., 2020). This finding would predict a much larger contribution of mRNA elements, such as riboswitches and secondary structure elements, in translation regulation of bacteria with “runaway” transcription, because a delay in ribosome movement would allow the emerging mRNA structures to fold. Similar regulatory mechanisms may also become more pervasive in bacteria with the coupled transcription-translation once the first round of translation is completed.
Another interesting, but yet poorly studied aspect of translation regulation is polysome formation. In exponentially growing E. coli cells, 70% of the ribosomes are found in polysomes (Jacobson and Baldassare, 1976). Ribosomes can load onto the mRNA in intervals of 1–3 s (Kennell and Riezman, 1977; Mitarai et al., 2008). The average polysome packing density is 1.3 ribosomes per 100 nt of mRNA, which yields an average ribosome spacing of 77 nt (Siwiak and Zielenkiewicz, 2013), with examples ranging from one ribosome every 72 nt on luc mRNA (Brandt et al., 2009) to one in every 100 nt on lacZ mRNA (Kierzek et al., 2001; Mitarai et al., 2008). Polysome formation may increase TE by protecting mRNA from degradation and thereby increasing its lifetime for producing more protein from the same mRNA or by affecting the accessibility of the ribosome binding site at the initiation step of translation (Andreeva et al., 2018). Obviously, increasing the number of ribosomes translating simultaneously the same mRNA should also increase TE. However, too high ribosome loading in a polysome, in combination with stalling of the leading ribosome, may lead to ribosome queuing which tends to reduce TE by slowing down translation after the stalling ribosome (Sorensen and Pedersen, 1991; Mitarai et al., 2008). One extreme form of such queueing are ribosome collisions, which in eukaryotes, elicit cotranslational degradation of both mRNA and nascent peptide, thereby affecting TE through a degradation pathway (Simms et al., 2017; Schuller and Green, 2018; Joazeiro, 2019; Collart and Weiss, 2020). While less is known about the ribosome collisions in bacteria, one key component of the sensing machinery that identifies stalled ribosomes, Rqc2, has a bacterial homolog, RqcH, which recognizes obstructed 50S subunits and promotes nascent chain proteolysis (Lytvynenko et al., 2019). In contrast to eukaryotic Rqc2, RqcH directly marks nascent chains for degradation by appending C-terminal poly-Ala tails that act as degrons recognized by the ClpXP protease (Lytvynenko et al., 2019). We note that it is not clear whether ribosome collisions are the only trigger of the ribosome quality control mechanism in bacteria or it is yet another general mechanism to cope with incomplete translation, overlapping with the functions of tmRNA, ArfA, and ArfB systems that sense and discard ribosomes stalled on truncated mRNAs. Thus, translation can be modulated not only by mRNA elements but also by RNAP or ribosomes bound to a given mRNA transcript, which can also attenuate the strength of the regulatory signals encoded by the mRNA.
Non-Uniform Elongation as a Timer for Protein Folding
Ribosome pausing may have evolved not only to regulate the production of a given protein but also to ensure the quality of synthesized proteins (Komar, 2009; Chaney and Clark, 2015). There is growing evidence that synonymous substitutions of natural rare codons or alterations in rare tRNA abundance changes the kinetics of translation and increases protein aggregation and susceptibility to protease digestion (Komar et al., 1999; Cortazzo et al., 2002; Zhang et al., 2009; Buhr et al., 2016; Konczal et al., 2019; Walsh et al., 2020; Figure 3). A study of a gamma-B crystallin, a mammalian eye-lens protein, demonstrated that synonymous codon substitutions change the kinetics of translation and the lifetime of translation intermediates (Figure 3A; Buhr et al., 2016). NMR and proteolysis assays showed that this change in translation kinetics and cotranslational folding trajectory alters protein stability, solubility, and conformation (Figure 3A; Buhr et al., 2016). Similar changes in the cotranslational folding trajectory, protein function, and stability were observed for a number of other proteins, such as Sufl (Figure 3B; Zhang et al., 2009), chloramphenicol acetyltransferase (CAT; Komar et al., 1999; Walsh et al., 2020; Shuf1 in Figure 3C), or the Echinococcus granulosus fatty acid binding protein1 (EgFABP1; Cortazzo et al., 2002). It seems that cotranslational protein folding may be one of the main driving forces that preserve the non-uniform rate of translation through maintaining the codon usage bias.
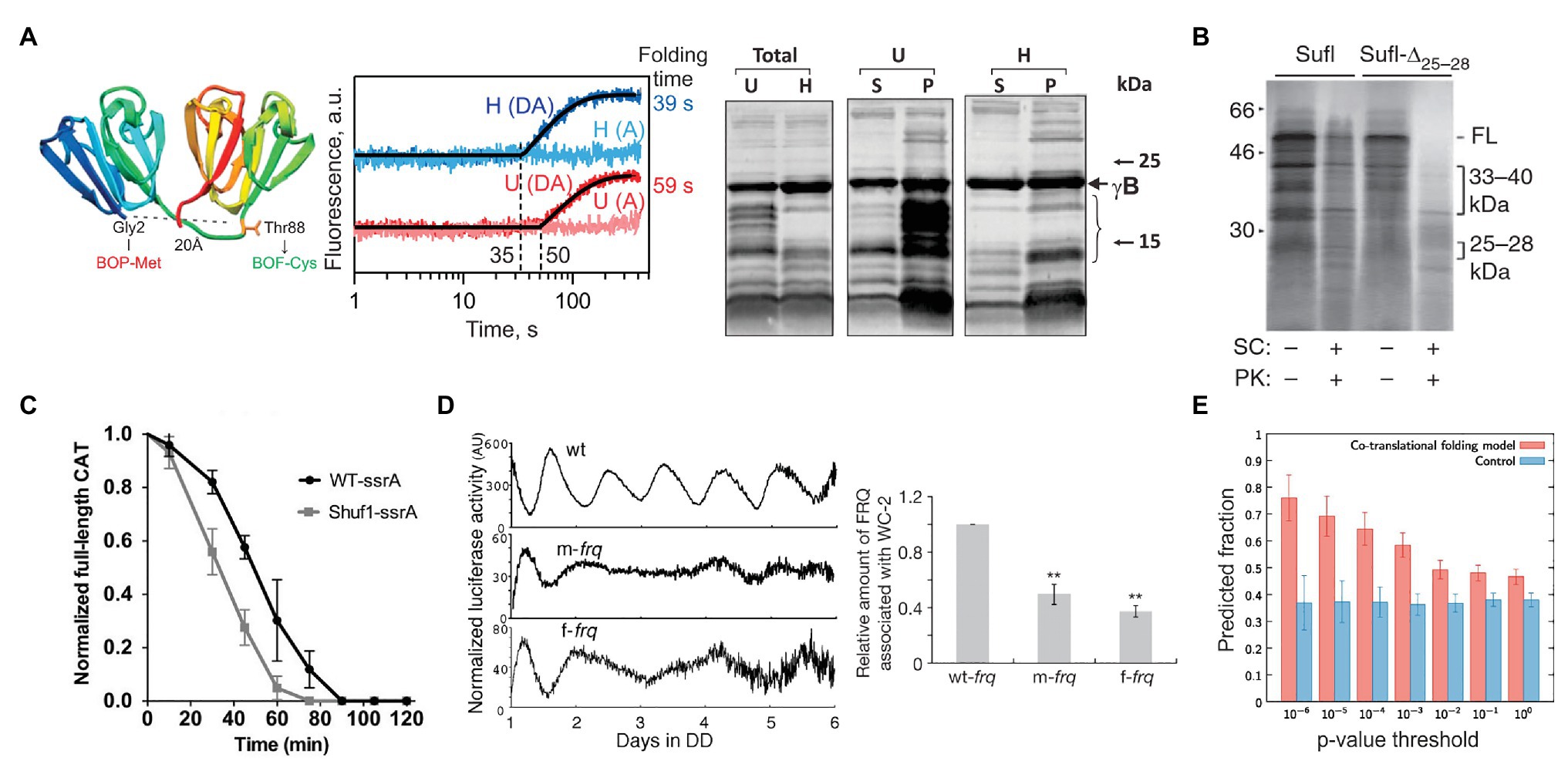
Figure 3. Examples of how rare codons can affect protein folding and function. (A) Gamma-B crystallin (Buhr et al., 2016). Left panel: Crystal structure of bovine gamma-B crystallin (PDB: 4GCR). Indicated are the protein positions used to introduce FRET labels. Middle panel: Cotranslational folding of un-optimized (U) and harmonized (H) gamma-B crystallin monitored in real time. The translation times, indicated by the delay in the time courses, are 35 s for the H and 50 s for the U construct; the delay coincides with the emergence of the N-terminal domain of gamma-B crystallin from the exit tunnel of the ribosome. The folding times, derived from exponential fitting, are 39 s for the H and 59 s for the U construct, demonstrating that folding kinetics is affected by synonymous codon replacements. Right panel: Expression of U and H proteins in E. coli. Shown are the total protein (Total) for the U and H variants as well as soluble (S) and pellet (P) fractions for each construct. (B) Stability of an E. coli protein Sufl (Zhang et al., 2009). Comparison of the proteinase K sensitivity of the wild-type wt Sufl and SufI-Δ25–28, in which two rare Leu codons at positons 244 and 252 were substituted by abundant codons. (C) Stability of the E. coli chloramphenicol acetyltransferase (CAT; Walsh et al., 2020). The CAT activity was measured in a ClpXP degradation assay of ssrA-tagged wt CAT and a ssrA-tagged Shuf1-CAT variant. In Shuf1-CAT, the pattern of synonymous codons is changed locally, but the global codon usage was largely unchanged. (D) Activity of the circadian clock protein FRQ (Zhou et al., 2013) tracked via Luciferase (Luc) reporter assay. Right panel: Effect of codon optimization of the circadian rhythm. m-frq, rare codons were optimized; f-frq, all codons were optimized. Left panel: Comparing periodicity and expression levels of wt and optimized FRQ variants. (E) Analysis of 500 E. coli proteins determining the fraction of conserved, slowly translated, rare codon-enriched regions that account for the predicted intermediates in a cotranslational folding model (orange) vs. changing threshold value of p (Jacobs and Shakhnovich, 2017). Randomized control sequences are shown in blue. **p < 0.01.
A number of well-documented cases in eukaryotic organisms comprehensively demonstrate how synonymous mutations result in specific phenotypes (Kimchi-Sarfaty et al., 2007; Xu et al., 2013; Kim et al., 2015). A silent mutation of Ile codon AUC to a rare AUU in the coding sequence of the human MDR1 protein alters translation speed and affects the timing of cotranslational folding, resulting in a protein that has an altered conformation and a changed affinity to its substrates (Kimchi-Sarfaty et al., 2007). The change in protein function alters the pharmacokinetics of individuals carrying this silent mutation and is associated with adverse therapeutic outcomes in breast cancer patients (Cizmarikova et al., 2010). Another example is a protein from Neurospora crassa that functions as part of the circuit establishing the molecular circadian rhythm (Zhou et al., 2013). The gene coding for protein FREQUENCY (FRQ) has multiple regions with non-optimal codons, read by low abundance tRNAs. When codon usage is optimized, the total amount of translated protein is high, but the circadian rhythm is changed (Figure 3D; Cheng et al., 2001), indicating that the codon-optimized protein is unable to interact with its partners due to its altered structure (Zhou et al., 2013). In cyanobacterium Synechococcus elongatus, the molecular circadian clock proteins KaiBC also have clusters of non-optimal codons. When the coding sequences are optimized, the periodicity of expression remains unchanged compared to the wild-type protein under optimal growth conditions (at 30°C; Xu et al., 2013). However, when the temperature is lowered to 18–25°C, the codon-optimized KaiBC strains grow significantly slower than the wild-type cells (Xu et al., 2013). It is tempting to speculate that the difference may originate from changes in translation kinetics and cotranslational folding at lower temperatures. While at higher temperatures, KaiBC might be able to refold to its functional form post-translationally, at lower temperatures, this is no longer possible, perhaps due to an overload of the chaperone networks in the cold.
Membrane proteins were shown to have multiple pauses during translation in vivo (Chadani et al., 2016). They have long clusters of rare codons that may play a role in defining the time window for correct targeting and membrane insertion of these proteins (Zalucki and Jennings, 2007; Chartier et al., 2012; Mercier et al., 2020). The topology of transmembrane helices generally follows the “positive-inside” rule, according to which the distribution of positively charged residues at the N-terminus of the bacterial inner membrane proteins determines the orientation of the first transmembrane helix with its N-terminus in the cytoplasm or the periplasm (von Heijne, 1989). However, computer simulations suggested that variations in the translation rate may alter transmembrane helix topology (Zhang and Miller, 2012; Niesen et al., 2017), indicating that the “positive-inside” rule is not the only driver of correct membrane insertion. In fact, a recent biochemical study which monitored cotranslational insertion of transmembrane segments of E. coli EmrD protein, showed that a prolonged ribosome stalling at a critical position can alter the topology of transmembrane segments (Mercier et al., 2020), underscoring the important link between the rate of translation and protein biogenesis and folding.
The primary effect of ribosome pauses on protein folding is likely cotranslational, arising as the nascent peptide moves through the exit tunnel and emerges from the ribosome. Many types of folding events occur in the micro- to millisecond time scale, whereas the rate of translation in bacteria is about 10–20 amino acids/s. Thus, rapid folding events that are much faster than amino acid addition to the nascent chain occur at quasi-equilibrium conditions (O’Brien et al., 2014; Liutkute et al., 2020). So why should further slowing down translation at particular positions affect protein folding? It is known that the distribution of rare codons with respect to the protein structure is non-random. Amino acids that are buried within the protein structure and are key to the overall stability of a protein are generally encoded by optimal codons (Zhou et al., 2009). In large proteins, clusters of rare codons are found every 125–155 amino acids and mark domain boundaries (Makhoul and Trifonov, 2002; Zhang et al., 2009). Slowing down translation at these critical junctions would provide the domains with enough time for correct folding and prevent inter-domain aggregation (Makhoul and Trifonov, 2002; Zhang et al., 2009; Vasquez et al., 2016). Notably, it is the rare-codon cluster positions, rather than the codon identity that are conserved within homologous coding sequences across eukaryotic, bacterial, and archaeal species (Chaney et al., 2017), which provides a basis for rational design of genes for heterologous expression and increasing protein solubility (for review, see Komar, 2016). Molecular dynamics simulations indicate that conserved rare codons are associated with cotranslational folding intermediates that may be even smaller than protein domains, and that this mechanism is conserved across multiple prokaryotic species (Figure 3E; Jacobs and Shakhnovich, 2017). Computer simulations that estimate the cotranslational folding as a function of translation rate suggest that the extent of domain folding is defined by the states populated early during synthesis and that this entire process can be derailed by single synonymous codon substitutions (O’Brien et al., 2012). Taking into account that ribosome pausing can be caused not only by rare codons, but also by a variety of different factors described above, or even emerge due to stochastic variations in elongation transit times, the link between translation and ribosome pausing may be an important factor for defining cotranslational protein folding.
Finally, alterations in translation rate can also affect the concerted binding of cofactors and proteins involved in nascent-chain processing (Nissley and O’Brien, 2014). Disturbing the recruitment of chaperones (TF, DnaK, and GroEL) and other ribosome-associated factors has been shown to hinder protein biogenesis in eukaryotes (Zhang et al., 2010), and SRP-mediated targeting in prokaryotes (Zalucki and Jennings, 2007) and eukaryotes (Pechmann et al., 2014). In vivo the specific kinetics of translation and the associated trajectories of cotranslational folding is aided by the entire proteostasis network, including chaperones and the protein degradation machinery, to produce functional proteins (Mogk et al., 2011; Collart and Weiss, 2020). Any changes in the kinetics of translation that affect either the overall protein fold or the way how the protein quality control machinery interacts with nascent proteins may cause visible phenotypic outcomes (Walsh et al., 2020).
High translation speed might not always be preferable if it comes at the expense of the protein’s quality in terms of solubility, stability, and function. Rare codons and other sources for ribosome pausing at specific points of the coding sequence may be under selective pressure to keep translation slower and less efficient but generate proteins that are functionally active and structurally stable throughout their lifetime. Thus, ribosome pausing emerges as a mechanism that allows cells to balance translational efficiency with correct protein folding and thus achieve optimal fitness under given environmental conditions.
Concluding Remarks
In recent years, it became increasingly clear that beyond the sequence information mRNA contains regulatory signals that attenuate translation and affect TE and protein folding. Although the importance of ribosome pausing is clearly established, many mechanistic questions remain open. For example, it is not clear whether only a fraction of ribosomes pause while others continue translation; what is the structure of pausing ribosomes; and what causes them to resume translation. Among the most interesting novel findings are sequences that stall translation through combined effects on the mRNA structure and on the conformation of the PTC. As the mRNA structures that fold in the A-site appear specific for each case studied, it would be interesting to see more of such examples to understand the underlying principles of regulation. Another emerging theme is how interactions between the nascent peptide and the exit tunnel of the ribosome can stall ribosomes by inactivating the PTC. Also here more examples of stalling structures would help to understand the prevalence of this phenomenon and its contribution to regulatory programs. Finally, the mechanism by which ribosome pausing affects nascent protein folding is poorly understood and would require development of new techniques to monitor protein folding in real time in atomic detail. The improvement of ribosome profiling techniques in conjunction with nascentome analysis and mass spectrometry, the increasing availability of high-resolution and high-throughput structural analysis of the stalling complexes, and development of biophysical methods that monitor the dynamics of ribosomes and nascent chains at single codon resolution will answer these questions and reveal yet another level of translation regulation acting as a “code-in-the-code” in cellular transcriptome.
Author Contributions
All authors contributed to the writing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Max Planck Society and by the European Research Council (ERC) Advanced Investigator Grant RIBOFOLD (proposal number no 787926).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Wolfgang Wintermeyer for critical reading of the manuscript.
Abbreviations
TE, Translation efficiency – the number of protein molecules produced from one mRNA; ASL, tRNA anticodon stem-loop; PTC, Peptidyl transferase center; RNAP, RNA polymerase; SD, Shine-Dalgarno.
References
Agirrezabala, X., Samatova, E., Klimova, M., Zamora, M., Gil-Carton, D., Rodnina, M. V., et al. (2017). Ribosome rearrangements at the onset of translational bypassing. Sci. Adv. 3:e1700147. doi: 10.1126/sciadv.1700147
Agris, P. F. (2008). Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 9, 629–635. doi: 10.1038/embor.2008.104
Agris, P. F., Eruysal, E. R., Narendran, A., Vare, V. Y. P., Vangaveti, S., and Ranganathan, S. V. (2018). Celebrating wobble decoding: half a century and still much is new. RNA Biol. 15, 537–553. doi: 10.1080/15476286.2017.1356562
Agris, P. F., Vendeix, F. A., and Graham, W. D. (2007). tRNA’s wobble decoding of the genome: 40 years of modification. J. Mol. Biol. 366, 1–13. doi: 10.1016/j.jmb.2006.11.046
Allert, M., Cox, J. C., and Hellinga, H. W. (2010). Multifactorial determinants of protein expression in prokaryotic open reading frames. J. Mol. Biol. 402, 905–918. doi: 10.1016/j.jmb.2010.08.010
Andreeva, I., Belardinelli, R., and Rodnina, M. V. (2018). Translation initiation in bacterial polysomes through ribosome loading on a standby site on a highly translated mRNA. Proc. Natl. Acad. Sci. U. S. A. 115, 4411–4416. doi: 10.1073/pnas.1718029115
Basan, M., Zhu, M., Dai, X., Warren, M., Sevin, D., Wang, Y. P., et al. (2015). Inflating bacterial cells by increased protein synthesis. Mol. Syst. Biol. 11:836. doi: 10.15252/msb.20156178
Bhattacharyya, S., Jacobs, W. M., Adkar, B. V., Yan, J., Zhang, W. L., and Shakhnovich, E. I. (2018). Accessibility of the Shine-Dalgarno sequence dictates N-terminal codon bias in E. coli. Mol. Cell 70, 894.e5–905.e5. doi: 10.1016/j.molcel.2018.05.008
Bock, L. V., Caliskan, N., Korniy, N., Peske, F., Rodnina, M. V., and Grubmuller, H. (2019). Thermodynamic control of-1 programmed ribosomal frameshifting. Nat. Commun. 10:4598. doi: 10.1038/s41467-019-12648-x
Boel, G., Letso, R., Neely, H., Price, W. N., Wong, K. H., Su, M., et al. (2016). Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 529, 358–363. doi: 10.1038/nature16509
Boni, I. V., Isaeva, D. M., Musychenko, M. L., and Tzareva, N. V. (1991). Ribosome-messenger recognition: mRNA target sites for ribosomal protein S1. Nucleic Acids Res. 19, 155–162. doi: 10.1093/nar/19.1.155
Boycheva, S., Chkodrov, G., and Ivanov, I. (2003). Codon pairs in the genome of Escherichia coli. Bioinformatics 19, 987–998. doi: 10.1093/bioinformatics/btg082
Brandt, F., Etchells, S. A., Ortiz, J. O., Elcock, A. H., Hartl, F. U., and Baumeister, W. (2009). The native 3D organization of bacterial polysomes. Cell 136, 261–271. doi: 10.1016/j.cell.2008.11.016
Breaker, R. R. (2018). Riboswitches and translation control. Cold Spring Harb. Perspect. Biol. 10:a032797. doi: 10.1101/cshperspect.a032797
Buchan, J. R., Aucott, L. S., and Stansfield, I. (2006). tRNA properties help shape codon pair preferences in open reading frames. Nucleic Acids Res. 34, 1015–1027. doi: 10.1093/nar/gkj488
Buhr, F., Jha, S., Thommen, M., Mittelstaet, J., Kutz, F., Schwalbe, H., et al. (2016). Synonymous codons direct cotranslational folding toward different protein conformations. Mol. Cell 61, 341–351. doi: 10.1016/j.molcel.2016.01.008
Burkhardt, D. H., Rouskin, S., Zhang, Y., Li, G. W., Weissman, J. S., and Gross, C. A. (2017). Operon mRNAs are organized into ORF-centric structures that predict translation efficiency. eLife 6:e22037. doi: 10.7554/eLife.22037
Burmann, B. M., Schweimer, K., Luo, X., Wahl, M. C., Stitt, B. L., Gottesman, M. E., et al. (2010). A NusE:NusG complex links transcription and translation. Science 328, 501–504. doi: 10.1126/science.1184953
Buschauer, R., Matsuo, Y., Sugiyama, T., Chen, Y. H., Alhusaini, N., Sweet, T., et al. (2020). The Ccr4-Not complex monitors the translating ribosome for codon optimality. Science 368:eaay6912. doi: 10.1126/science.aay6912
Byrgazov, K., Grishkovskaya, I., Arenz, S., Coudevylle, N., Temmel, H., Wilson, D. N., et al. (2015). Structural basis for the interaction of protein S1 with the Escherichia coli ribosome. Nucleic Acids Res. 43, 661–673. doi: 10.1093/nar/gku1314
Caliskan, N., Katunin, V. I., Belardinelli, R., Peske, F., and Rodnina, M. V. (2014). Programmed-1 frameshifting by kinetic partitioning during impeded translocation. Cell 157, 1619–1631. doi: 10.1016/j.cell.2014.04.041
Caliskan, N., Wohlgemuth, I., Korniy, N., Pearson, M., Peske, F., and Rodnina, M. V. (2017). Conditional switch between frameshifting regimes upon translation of dnaX mRNA. Mol. Cell 66, 558.e554–567.e554. doi: 10.1016/j.molcel.2017.04.023
Cambray, G., Guimaraes, J. C., and Arkin, A. P. (2018). Evaluation of 244,000 synthetic sequences reveals design principles to optimize translation in Escherichia coli. Nat. Biotechnol. 36, 1005–1015. doi: 10.1038/nbt.4238
Cannarrozzi, G., Schraudolph, N. N., Faty, M., von Rohr, P., Friberg, M. T., Roth, A. C., et al. (2010). A role for codon order in translation dynamics. Cell 141, 355–367. doi: 10.1016/j.cell.2010.02.036
Chadani, Y., Niwa, T., Chiba, S., Taguchi, H., and Ito, K. (2016). Integrated in vivo and in vitro nascent chain profiling reveals widespread translational pausing. Proc. Natl. Acad. Sci. U. S. A. 113, E829–E838. doi: 10.1073/pnas.1520560113
Chandrasekaran, V., Juszkiewicz, S., Choi, J., Puglisi, J. D., Brown, A., Shao, S., et al. (2019). Mechanism of ribosome stalling during translation of a poly(A) tail. Nat. Struct. Mol. Biol. 26, 1132–1140. doi: 10.1038/s41594-019-0331-x
Chaney, J. L., and Clark, P. L. (2015). Roles for synonymous codon usage in protein biogenesis. Annu. Rev. Biophys. 44, 143–166. doi: 10.1146/annurev-biophys-060414-034333
Chaney, J. L., Steele, A., Carmichael, R., Rodriguez, A., Specht, A. T., Ngo, K., et al. (2017). Widespread position-specific conservation of synonymous rare codons within coding sequences. PLoS Comput. Biol. 13:e1005531. doi: 10.1371/journal.pcbi.1005531
Charneski, C. A., and Hurst, L. D. (2013). Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol. 11:e1001508. doi: 10.1371/journal.pbio.1001508
Chartier, M., Gaudreault, F., and Najmanovich, R. (2012). Large-scale analysis of conserved rare codon clusters suggests an involvement in co-translational molecular recognition events. Bioinformatics 28, 1438–1445. doi: 10.1093/bioinformatics/bts149
Chen, M., and Fredrick, K. (2020). RNA polymerase’s relationship with the ribosome: not so physical, most of the time. J. Mol. Biol. 432, 3981–3986. doi: 10.1016/j.jmb.2020.03.018
Chen, J., Petrov, A., Johansson, M., Tsai, A., O’Leary, S. E., and Puglisi, J. D. (2014). Dynamic pathways of-1 translational frameshifting. Nature 512, 328–332. doi: 10.1038/nature13428
Chen, C. L., Zhang, H. B., Broitman, S. L., Reiche, M., Farrell, I., Cooperman, B. S., et al. (2013). Dynamics of translation by single ribosomes through mRNA secondary structures. Nat. Struct. Mol. Biol. 20, 582–588. doi: 10.1038/nsmb.2544
Cheng, P., Yang, Y., and Liu, Y. (2001). Interlocked feedback loops contribute to the robustness of the Neurospora circadian clock. Proc. Natl. Acad. Sci. U. S. A. 98, 7408–7413. doi: 10.1073/pnas.121170298
Cifuentes-Goches, J. C., Hernandez-Ancheyta, L., Guarneros, G., Oviedo, N., and Hernandez-Sanchez, J. (2019). Domains two and three of Escherichia coli ribosomal S1 protein confers 30S subunits a high affinity for downstream A/U-rich mRNAs. J. Biochem. 166, 29–40. doi: 10.1093/jb/mvz006
Cizmarikova, M., Wagnerova, M., Schonova, L., Habalova, V., Kohut, A., Linkova, A., et al. (2010). MDR1 (C3435T) polymorphism: relation to the risk of breast cancer and therapeutic outcome. Pharm. J. 10, 62–69. doi: 10.1038/tpj.2009.41
Clarke, T. F. T., and Clark, P. L. (2008). Rare codons cluster. PLoS One 3:e3412. doi: 10.1371/journal.pone.0003412
Collart, M. A., and Weiss, B. (2020). Ribosome pausing, a dangerous necessity for co-translational events. Nucleic Acids Res. 48, 1043–1055. doi: 10.1093/nar/gkz763
Cortazzo, P., Cervenansky, C., Marin, M., Reiss, C., Ehrlich, R., and Deana, A. (2002). Silent mutations affect in vivo protein folding in Escherichia coli. Biochem. Biophys. Res. Commun. 293, 537–541. doi: 10.1016/S0006-291x(02)00226-7
Del Campo, C., Bartholomaus, A., Fedyunin, I., and Ignatova, Z. (2015). Secondary structure across the bacterial transcriptome reveals versatile roles in mRNA regulation and function. PLoS Genet. 11:e1005613. doi: 10.1371/journal.pgen.1005613
Dittmar, K. A., Sorensen, M. A., Elf, J., Ehrenberg, M., and Pan, T. (2005). Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Rep. 6, 151–157. doi: 10.1038/sj.embor.7400341
Doerfel, L. K., Wohlgemuth, I., Kothe, C., Peske, F., Urlaub, H., and Rodnina, M. V. (2013). EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science 339, 85–88. doi: 10.1126/science.1229017
Doerfel, L. K., Wohlgemuth, I., Kubyshkin, V., Starosta, A. L., Wilson, D. N., Budisa, N., et al. (2015). Entropic contribution of elongation factor P to proline positioning at the catalytic center of the ribosome. J. Am. Chem. Soc. 137, 12997–13006. doi: 10.1021/jacs.5b07427
Dong, H. J., Nilsson, L., and Kurland, C. G. (1996). Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 260, 649–663. doi: 10.1006/jmbi.1996.0428
Duc, K. D., Batra, S. S., Bhattacharya, N., Cate, J. H. D., and Song, Y. S. (2019). Differences in the path to exit the ribosome across the three domains of life. Nucleic Acids Res. 47, 4198–4210. doi: 10.1093/nar/gkz106
Duc, K. D., and Song, Y. S. (2018). The impact of ribosomal interference, codon usage, and exit tunnel interactions on translation elongation rate variation. PLoS Genet. 14:e1007166. doi: 10.1371/journal.pgen.1007166
Duval, M., Korepanov, A., Fuchsbauer, O., Fechter, P., Haller, A., Fabbretti, A., et al. (2013). Escherichia coli ribosomal protein S1 unfolds structured mRNAs onto the ribosome for active translation initiation. PLoS Biol. 11:e1001731. doi: 10.1371/journal.pbio.1001731
Dykeman, E. C. (2020). A stochastic model for simulating ribosome kinetics in vivo. PLoS Comput. Biol. 16:e1007618. doi: 10.1371/journal.pcbi.1007618
Elf, J., Nilsson, D., Tenson, T., and Ehrenberg, M. (2003). Selective charging of tRNA isoacceptors explains patterns of codon usage. Science 300, 1718–1722. doi: 10.1126/science.1083811
Gamble, C. E., Brule, C. E., Dean, K. M., Fields, S., and Grayhack, E. J. (2016). Adjacent codons act in concert to modulate translation efficiency in yeast. Cell 166, 679–690. doi: 10.1016/j.cell.2016.05.070
Geissmann, T., Marzi, S., and Romby, P. (2009). The role of mRNA structure in translational control in bacteria. RNA Biol. 6, 153–160. doi: 10.4161/rna.6.2.8047
Goodman, D. B., Church, G. M., and Kosuri, S. (2013). Causes and effects of N-terminal codon bias in bacterial genes. Science 342, 475–479. doi: 10.1126/science.1241934
Gorochowski, T. E., Ignatova, Z., Bovenberg, R. A. L., and Roubos, J. A. (2015). Trade-offs between tRNA abundance and mRNA secondary structure support smoothing of translation elongation rate. Nucleic Acids Res. 43, 3022–3032. doi: 10.1093/nar/gkv199
Grantham, R., Gautier, C., and Gouy, M. (1980). Codon frequencies in 119 individual genes confirm consistent choices of degenerate bases according to genome type. Nucleic Acids Res. 8, 1893–1912. doi: 10.1093/nar/8.9.1893
Grosjean, H., and Westhof, E. (2016). An integrated, structure- and energy-based view of the genetic code. Nucleic Acids Res. 44, 8020–8040. doi: 10.1093/nar/gkw608
Gu, W., Zhou, T., and Wilke, C. O. (2010). A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput. Biol. 6:e1000664. doi: 10.1371/journal.pcbi.1000664
Gualerzi, C. O., and Pon, C. L. (2015). Initiation of mRNA translation in bacteria: structural and dynamic aspects. Cell. Mol. Life Sci. 72, 4341–4367. doi: 10.1007/s00018-015-2010-3
Guo, F. B., Ye, Y. N., Zhao, H. L., Lin, D., and Wei, W. (2012). Universal pattern and diverse strengths of successive synonymous codon bias in three domains of life, particularly among prokaryotic genomes. DNA Res. 19, 477–485. doi: 10.1093/dnares/dss027
Hanson, G., and Coller, J. (2018). Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 19, 20–30. doi: 10.1038/nrm.2017.91
Hou, Y. M., Masuda, I., and Gamper, H. (2019). Codon-specific translation by m(1)G37 methylation of tRNA. Front. Genet. 9:713. doi: 10.3389/fgene.2018.00713
Huter, P., Arenz, S., Bock, L. V., Graf, M., Frister, J. O., Heuer, A., et al. (2017). Structural basis for polyproline-mediated ribosome stalling and rescue by the translation elongation factor EF-P. Mol. Cell 68, 515.e6–527.e6. doi: 10.1016/j.molcel.2017.10.014
Ikemura, T. (1981). Correlation between the abundance of Escherichia coli transfer-Rnas and the occurrence of the respective codons in its protein genes. J. Mol. Biol. 146, 1–21. doi: 10.1016/0022-2836(81)90363-6
Ikemura, T. (1985). Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 2, 13–34. doi: 10.1093/oxfordjournals.molbev.a040335
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R., and Weissman, J. S. (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. doi: 10.1126/science.1168978
Ito, K., Chadani, Y., Nakamori, K., Chiba, S., Akiyama, Y., and Abo, T. (2011). Nascentome analysis uncovers futile protein synthesis in Escherichia coli. PLoS One 6:e28413. doi: 10.1371/journal.pone.0028413
Ito, K., and Chiba, S. (2013). Arrest peptides: cis-acting modulators of translation. Annu. Rev. Biochem. 82, 171–202. doi: 10.1146/annurev-biochem-080211-105026
Jacobs, W. M., and Shakhnovich, E. I. (2017). Evidence of evolutionary selection for cotranslational folding. Proc. Natl. Acad. Sci. U. S. A. 114, 11434–11439. doi: 10.1073/pnas.1705772114
Jacobson, L. A., and Baldassare, J. C. (1976). Association of messenger ribonucleic acid with 70S monosomes from down-shifted Escherichia coli. J. Bacteriol. 127, 637–643. doi: 10.1128/JB.127.1.637-643.1976
Joazeiro, C. A. P. (2019). Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 20, 368–383. doi: 10.1038/s41580-019-0118-2
Johnson, G. E., Lalanne, J. B., Peters, M. L., and Li, G. W. (2020). Functionally uncoupled transcription-translation in Bacillus subtilis. Nature 585, 124–128. doi: 10.1038/s41586-020-2638-5
Juhling, F., Morl, M., Hartmann, R. K., Sprinzl, M., Stadler, P. F., and Putz, J. (2009). tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 37, D159–D162. doi: 10.1093/nar/gkn772
Kennell, D., and Riezman, H. (1977). Transcription and translation initiation frequencies of the Escherichia coli lac operon. J. Mol. Biol. 114, 1–21. doi: 10.1016/0022-2836(77)90279-0
Kierzek, A. M., Zaim, J., and Zielenkiewicz, P. (2001). The effect of transcription and translation initiation frequencies on the stochastic fluctuations in prokaryotic gene expression. J. Biol. Chem. 276, 8165–8172. doi: 10.1074/jbc.M006264200
Kim, H. K., Liu, F., Fei, J. Y., Bustamante, C., Gonzalez, R. L., and Tinoco, I. (2014). A frameshifting stimulatory stem loop destabilizes the hybrid state and impedes ribosomal translocation. Proc. Natl. Acad. Sci. U. S. A. 111, 5538–5543. doi: 10.1073/pnas.1403457111
Kim, S. J., Yoon, J. S., Shishido, H., Yang, Z., Rooney, L. A., Barral, J. M., et al. (2015). Protein folding. Translational tuning optimizes nascent protein folding in cells. Science 348, 444–448. doi: 10.1126/science.aaa3974
Kimchi-Sarfaty, C., Oh, J. M., Kim, I. W., Sauna, Z. E., Calcagno, A. M., Ambudkar, S. V., et al. (2007). A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315, 525–528. doi: 10.1126/science.1135308
Klumpp, S., Scott, M., Pedersen, S., and Hwa, T. (2013). Molecular crowding limits translation and cell growth. Proc. Natl. Acad. Sci. U. S. A. 110, 16754–16759. doi: 10.1073/pnas.1310377110
Kohler, R., Mooney, R. A., Mills, D. J., Landick, R., and Cramer, P. (2017). Architecture of a transcribing-translating expressome. Science 356, 194–197. doi: 10.1126/science.aal3059
Komar, A. A. (2009). A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 34, 16–24. doi: 10.1016/j.tibs.2008.10.002
Komar, A. A. (2016). “The art of gene redesign and recombinant protein production: approaches and perspectives” in Protein therapeutics. eds. Z. E. Sauna and C. Kimchi-Sarfaty (Cham: Springer International Publishing), 161–177.
Komar, A. A., Lesnik, T., and Reiss, C. (1999). Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett. 462, 387–391. doi: 10.1016/s0014-5793(99)01566-5
Konczal, J., Bower, J., and Gray, C. H. (2019). Re-introducing non-optimal synonymous codons into codon-optimized constructs enhances soluble recovery of recombinant proteins from Escherichia coli. PLoS One 14:e0215892. doi: 10.1371/journal.pone.0215892
Kortmann, J., and Narberhaus, F. (2012). Bacterial RNA thermometers: molecular zippers and switches. Nat. Rev. Microbiol. 10, 255–265. doi: 10.1038/nrmicro2730
Kothe, U., and Rodnina, M. V. (2007). Codon reading by tRNAAla with modified uridine in the wobble position. Mol. Cell 25, 167–174. doi: 10.1016/j.molcel.2006.11.014
Koutmou, K. S., Schuller, A. P., Brunelle, J. L., Radhakrishnan, A., Djuranovic, S., and Green, R. (2015). Ribosomes slide on lysine-encoding homopolymeric A stretches. eLife 4:e05534. doi: 10.7554/eLife.05534
Kudla, G., Murray, A. W., Tollervey, D., and Plotkin, J. B. (2009). Coding-sequence determinants of gene expression in Escherichia coli. Science 324, 255–258. doi: 10.1126/science.1170160
Landick, R., Carey, J., and Yanofsky, C. (1985). Translation activates the paused transcription complex and restores transcription of the trp operon leader region. Proc. Natl. Acad. Sci. U. S. A. 82, 4663–4667. doi: 10.1073/pnas.82.14.4663
Ledoux, S., and Uhlenbeck, O. C. (2008). Different aa-tRNAs are selected uniformly on the ribosome. Mol. Cell 31, 114–123. doi: 10.1016/j.molcel.2008.04.026
Li, G. W., Burkhardt, D., Gross, C., and Weissman, J. S. (2014). Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157, 624–635. doi: 10.1016/j.cell.2014.02.033
Li, G. W., Oh, E., and Weissman, J. S. (2012). The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 484, 538–541. doi: 10.1038/nature10965
Lim, V. I., and Curran, J. F. (2001). Analysis of codon: anticodon interactions within the ribosome provides new insights into codon reading and the genetic code structure. RNA 7, 942–957. doi: 10.1017/S135583820100214x
Liutkute, M., Samatova, E., and Rodnina, M. V. (2020). Cotranslational folding of proteins on the ribosome. Biomolecules 10:97. doi: 10.3390/biom10010097
Lu, J. L., and Deutsch, C. (2008). Electrostatics in the ribosomal tunnel modulate chain elongation rates. J. Mol. Biol. 384, 73–86. doi: 10.1016/j.jmb.2008.08.089
Lytvynenko, I., Paternoga, H., Thrun, A., Balke, A., Muller, T. A., Chiang, C. H., et al. (2019). Alanine tails signal proteolysis in bacterial ribosome-associated quality control. Cell 178, 76.e22–90.e22. doi: 10.1016/j.cell.2019.05.002
Makhoul, C. H., and Trifonov, E. N. (2002). Distribution of rare triplets along mRNA and their relation to protein folding. J. Biomol. Struct. Dyn. 20, 413–420. doi: 10.1080/07391102.2002.10506859
Martens, A. T., Taylor, J., and Hilser, V. J. (2015). Ribosome A and P sites revealed by length analysis of ribosome profiling data. Nucleic Acids Res. 43, 3680–3687. doi: 10.1093/nar/gkv200
Mercier, E., Wintermeyer, W., and Rodnina, M. V. (2020). Co-translational insertion and topogenesis of bacterial membrane proteins monitored in real time. EMBO J. 39:e104054. doi: 10.15252/embj.2019104054
Milon, P., and Rodnina, M. V. (2012). Kinetic control of translation initiation in bacteria. Crit. Rev. Biochem. Mol. 47, 334–348. doi: 10.3109/10409238.2012.678284
Mitarai, N., Sneppen, K., and Pedersen, S. (2008). Ribosome collisions and translation efficiency: optimization by codon usage and mRNA destabilization. J. Mol. Biol. 382, 236–245. doi: 10.1016/j.jmb.2008.06.068
Mogk, A., Huber, D., and Bukau, B. (2011). Integrating protein homeostasis strategies in prokaryotes. Cold Spring Harb. Perspect. Biol. 3:a004366. doi: 10.1101/cshperspect.a004366
Mohammad, F., Green, R., and Buskirk, A. R. (2019). A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. eLife 8:e42591. doi: 10.7554/eLife.42591
Mohammad, F., Woolstenhulme, C. J., Green, R., and Buskirk, A. R. (2016). Clarifying the translational pausing landscape in bacteria by ribosome profiling. Cell Rep. 14, 686–694. doi: 10.1016/j.celrep.2015.12.073
Moreira, M. H., Barros, G. C., Requiao, R. D., Rossetto, S., Domitrovic, T., and Palhano, F. L. (2019). From reporters to endogenous genes: the impact of the first five codons on translation efficiency in Escherichia coli. RNA Biol. 16, 1806–1816. doi: 10.1080/15476286.2019.1661213
Moura, G., Pinheiro, M., Silva, R., Miranda, I., Afreixo, V., Dias, G., et al. (2005). Comparative context analysis of codon pairs on an ORFeome scale. Genome Biol. 6:R28. doi: 10.1186/gb-2005-6-3-r28
Mustoe, A. M., Busan, S., Rice, G. M., Hajdin, C. E., Peterson, B. K., Ruda, V. M., et al. (2018). Pervasive regulatory functions of mRNA structure revealed by high-resolution SHAPE probing. Cell 173, 181.e18–195.e18. doi: 10.1016/j.cell.2018.02.034
Navon, S. P., Kornberg, G., Chen, J., Schwartzman, T., Tsai, A., Puglisi, E. V., et al. (2016). Amino acid sequence repertoire of the bacterial proteome and the occurrence of untranslatable sequences. Proc. Natl. Acad. Sci. U. S. A. 113, 7166–7170. doi: 10.1073/pnas.1606518113
Nedialkova, D. D., and Leidel, S. A. (2015). Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161, 1606–1618. doi: 10.1016/j.cell.2015.05.022
Niesen, M. J., Wang, C. Y., Van Lehn, R. C., and Miller, T. F. 3rd. (2017). Structurally detailed coarse-grained model for Sec-facilitated co-translational protein translocation and membrane integration. PLoS Comput. Biol. 13:e1005427. doi: 10.1371/journal.pcbi.1005427
Nissley, D. A., and O’Brien, E. P. (2014). Timing is everything: unifying codon translation rates and nascent proteome behavior. J. Am. Chem. Soc. 136, 17892–17898. doi: 10.1021/ja510082j
O’Brien, E. P., Ciryam, P., Vendruscolo, M., and Dobson, C. M. (2014). Understanding the influence of codon translation rates on cotranslational protein folding. Acc. Chem. Res. 47, 1536–1544. doi: 10.1021/ar5000117
O’Brien, E. P., Vendruscolo, M., and Dobson, C. M. (2012). Prediction of variable translation rate effects on cotranslational protein folding. Nat. Commun. 3:868. doi: 10.1038/ncomms1850
O’Reilly, F. J., Xue, L., Graziadei, A., Sinn, L., Lenz, S., Tegunov, D., et al. (2020). In-cell architecture of an actively transcribing-translating expressome. Science 369, 554–557. doi: 10.1126/science.abb3758
Oh, E., Becker, A. H., Sandikci, A., Huber, D., Chaba, R., Gloge, F., et al. (2011). Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell 147, 1295–1308. doi: 10.1016/j.cell.2011.10.044
Osterman, I. A., Chervontseva, Z. S., Evfratov, S. A., Sorokina, A. V., Rodin, V. A., Rubtsova, M. P., et al. (2020). Translation at first sight: the influence of leading codons. Nucleic Acids Res. 48, 6931–6942. doi: 10.1093/nar/gkaa430
Pechmann, S., Chartron, J. W., and Frydman, J. (2014). Local slowdown of translation by nonoptimal codons promotes nascent-chain recognition by SRP in vivo. Nat. Struct. Mol. Biol. 21, 1100–1105. doi: 10.1038/nsmb.2919
Peil, L., Starosta, A. L., Lassak, J., Atkinson, G. C., Virumae, K., Spitzer, M., et al. (2013). Distinct XPPX sequence motifs induce ribosome stalling, which is rescued by the translation elongation factor EF-P. Proc. Natl. Acad. Sci. U. S. A. 110, 15265–15270. doi: 10.1073/pnas.1310642110
Pollo-Oliveira, L., Klassen, R., Davis, N., Ciftci, A., Bacusmo, J. M., Martinelli, M., et al. (2020). Loss of elongator- and KEOPS-dependent tRNA modifications leads to severe growth phenotypes and protein aggregation in yeast. Biomolecules 10:322. doi: 10.3390/biom10020322
Proshkin, S., Rahmouni, A. R., Mironov, A., and Nudler, E. (2010). Cooperation between translating ribosomes and RNA polymerase in transcription elongation. Science 328, 504–508. doi: 10.1126/science.1184939
Qing, G., Xia, B., and Inouye, M. (2003). Enhancement of translation initiation by A/T-rich sequences downstream of the initiation codon in Escherichia coli. J. Mol. Microbiol. Biotechnol. 6, 133–144. doi: 10.1159/000077244
Qu, X. H., Wen, J. D., Lancaster, L., Noller, H. F., Bustamante, C., and Tinoco, I. (2011). The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature 475, 118–121. doi: 10.1038/nature10126
Quax, T. E. F., Wolf, Y. I., Koehorst, J. J., Wurtzel, O., van der Oost, R., Ran, W. Q., et al. (2013). Differential translation tunes uneven production of operon-encoded proteins. Cell Rep. 4, 938–944. doi: 10.1016/j.celrep.2013.07.049
Ranjan, N., and Leidel, S. A. (2019). The epitranscriptome in translation regulation: mRNA and tRNA modifications as the two sides of the same coin? FEBS Lett. 593, 1483–1493. doi: 10.1002/1873-3468.13491
Ranjan, N., and Rodnina, M. V. (2016). tRNA wobble modifications and protein homeostasis. Translation 4:e1143076. doi: 10.1080/21690731.2016.1143076
Rezgui, V. A., Tyagi, K., Ranjan, N., Konevega, A. L., Mittelstaet, J., Rodnina, M. V., et al. (2013). tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine-tune protein translation by promoting ribosome A-site binding. Proc. Natl. Acad. Sci. U. S. A. 110, 12289–12294. doi: 10.1073/pnas.1300781110
Rodnina, M. V. (2018). Translation in prokaryotes. Cold Spring Harb. Perspect. Biol. 10:a032664. doi: 10.1101/cshperspect.a032664
Rodnina, M. V., Gromadski, K. B., Kothe, U., and Wieden, H. J. (2005). Recognition and selection of tRNA in translation. FEBS Lett. 579, 938–942. doi: 10.1016/j.febslet.2004.11.048
Rouskin, S., Zubradt, M., Washietl, S., Kellis, M., and Weissman, J. S. (2014). Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505:701. doi: 10.1038/nature12894
Rudorf, S. (2019). Efficiency of protein synthesis inhibition depends on tRNA and codon compositions. PLoS Comput. Biol. 15:e1006979. doi: 10.1371/journal.pcbi.1006979
Rudorf, S., Thommen, M., Rodnina, M. V., and Lipowsky, R. (2014). Deducing the kinetics of protein synthesis in vivo from the transition rates measured in vitro. PLoS Comput. Biol. 10:e1003909. doi: 10.1371/journal.pcbi.1003909
Russell, J. B., and Cook, G. M. (1995). Energetics of bacterial-growth-balance of anabolic and catabolic reactions. Microbiol. Rev. 59, 48–62. doi: 10.1128/Mmbr.59.1.48-62.1995
Sabi, R., and Tuller, T. (2015). A comparative genomics study on the effect of individual amino acids on ribosome stalling. BMC Genomics 16(Suppl. 10):S5. doi: 10.1186/1471-2164-16-S10-S5
Saito, K., Green, R., and Buskirk, A. R. (2020). Translational initiation in E. coli occurs at the correct sites genome-wide in the absence of mRNA-rRNA base-pairing. eLife 9:e55002. doi: 10.7554/eLife.55002
Schuller, A. P., and Green, R. (2018). Roadblocks and resolutions in eukaryotic translation. Nat. Rev. Mol. Cell Biol. 19, 526–541. doi: 10.1038/s41580-018-0011-4
Seip, B., Sacheau, G., Dupuy, D., and Innis, C. A. (2018). Ribosomal stalling landscapes revealed by high-throughput inverse toeprinting of mRNA libraries. Life Sci. Alliance 1:e201800148. doi: 10.26508/lsa.201800148
Shao, Z. Q., Zhang, Y. M., Feng, X. Y., Wang, B., and Chen, J. Q. (2012). Synonymous codon ordering: a subtle but prevalent strategy of bacteria to improve translational efficiency. PLoS One 7:e33547. doi: 10.1371/journal.pone.0033547
Sharp, P. M., and Li, W. H. (1987). The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. doi: 10.1093/nar/15.3.1281
Simms, C. L., Thomas, E. N., and Zaher, H. S. (2017). Ribosome-based quality control of mRNA and nascent peptides. Wiley Interdiscip Rev. RNA 8:e1366. doi: 10.1002/wrna.1366
Siwiak, M., and Zielenkiewicz, P. (2013). Transimulation—protein biosynthesis web service. PLoS One 8:e73943. doi: 10.1371/journal.pone.0073943
Sorensen, M. A., and Pedersen, S. (1991). Absolute in vivo translation rates of individual codons in Escherichia coli. The two glutamic acid codons GAA and GAG are translated with a threefold difference in rate. J. Mol. Biol. 222, 265–280. doi: 10.1016/0022-2836(91)90211-n
Subramaniam, A. R., Pan, T., and Cluzel, P. (2013). Environmental perturbations lift the degeneracy of the genetic code to regulate protein levels in bacteria. Proc. Natl. Acad. Sci. U. S. A. 110, 2419–2424. doi: 10.1073/pnas.1211077110
Subramaniam, A. R., Zid, B. M., and O’Shea, E. K. (2014). An integrated approach reveals regulatory controls on bacterial translation elongation. Cell 159, 1200–1211. doi: 10.1016/j.cell.2014.10.043
Takyar, S., Hickerson, R. P., and Noller, H. F. (2005). mRNA helicase activity of the ribosome. Cell 120, 49–58. doi: 10.1016/j.cell.2004.11.042
Tats, A., Tenson, T., and Remm, M. (2008). Preferred and avoided codon pairs in three domains of life. BMC Genomics 9:463. doi: 10.1186/1471-2164-9-463
Tesina, P., Lessen, L. N., Buschauer, R., Cheng, J., Wu, C. C., Berninghausen, O., et al. (2020). Molecular mechanism of translational stalling by inhibitory codon combinations and poly(A) tracts. EMBO J. 39:e103365. doi: 10.15252/embj.2019103365
Thiaville, P. C., El Yacoubi, B., Kohrer, C., Thiaville, J. J., Deutsch, C., Iwata-Reuyl, D., et al. (2015). Essentiality of threonylcarbamoyladenosine (t(6)A), a universal tRNA modification, in bacteria. Mol. Microbiol. 98, 1199–1221. doi: 10.1111/mmi.13209
Tollerson, R., and Ibba, M. (2020). Translational regulation of environmental adaptation in bacteria. J. Biol. Chem. 295, 10434–10445. doi: 10.1074/jbc.REV120.012742
Tuller, T., Carmi, A., Vestsigian, K., Navon, S., Dorfan, Y., Zaborske, J., et al. (2010). An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141, 344–354. doi: 10.1016/j.cell.2010.03.031
Tuller, T., Veksler-Lublinsky, I., Gazit, N., Kupiec, M., Ruppin, E., and Ziv-Ukelson, M. (2011). Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 12:R110. doi: 10.1186/gb-2011-12-11-r110
Ude, S., Lassak, J., Starosta, A. L., Kraxenberger, T., Wilson, D. N., and Jung, K. (2013). Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science 339, 82–85. doi: 10.1126/science.1228985
Vasquez, K. A., Hatridge, T. A., Curtis, N. C., and Contreras, L. M. (2016). Slowing translation between protein domains by increasing affinity between mRNAs and the ribosomal anti-Shine Dalgarno sequence improves solubility. ACS Synth. Biol. 5, 133–145. doi: 10.1021/acssynbio.5b00193
Verma, M., Choi, J., Cottrell, K. A., Lavagnino, Z., Thomas, E. N., Pavlovic-Djuranovic, S., et al. (2019). A short translational ramp determines the efficiency of protein synthesis. Nat. Commun. 10:5774. doi: 10.1038/s41467-019-13810-1
Vieira, J. P., Racle, J., and Hatzimanikatis, V. (2016). Analysis of translation elongation dynamics in the context of an Escherichia coli cell. Biophys. J. 110, 2120–2131. doi: 10.1016/j.bpj.2016.04.004
Vogel, U., and Jensen, K. F. (1994). The RNA chain elongation rate in Escherichia coli depends on the growth rate. J. Bacteriol. 176, 2807–2813. doi: 10.1128/jb.176.10.2807-2813.1994
von Heijne, G. (1989). Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature 341, 456–458. doi: 10.1038/341456a0
Walsh, I. M., Bowman, M. A., Soto Santarriaga, I. F., Rodriguez, A., and Clark, P. L. (2020). Synonymous codon substitutions perturb cotranslational protein folding in vivo and impair cell fitness. Proc. Natl. Acad. Sci. U. S. A. 117, 3528–3534. doi: 10.1073/pnas.1907126117
Wang, C., Molodtsov, V., Firlar, E., Kaelber, J. T., Blaha, G., Su, M., et al. (2020). Structural basis of transcription-translation coupling. Science 369, 1359–1365. doi: 10.1126/science.abb5317
Watson, Z. L., Ward, F. R., Meheust, R., Ad, O., Schepartz, A., Banfield, J. F., et al. (2020). Structure of the bacterial ribosome at 2 angstrom resolution. eLife 9:e60482. doi: 10.7554/eLife.60482
Webster, M. W., Takacs, M., Zhu, C., Vidmar, V., Eduljee, A., Abdelkareem, M., et al. (2020). Structural basis of transcription-translation coupling and collision in bacteria. Science 369, 1355–1359. doi: 10.1126/science.abb5036
Weinberg, D. E., Shah, P., Eichhorn, S. W., Hussmann, J. A., Plotkin, J. B., and Bartel, D. P. (2016). Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 14, 1787–1799. doi: 10.1016/j.celrep.2016.01.043
Weixlbaumer, A., Murphy, F. V., Dziergowska, A., Malkiewicz, A., Vendeix, F. A. P., Agris, P. F., et al. (2007). Mechanism for expanding the decoding capacity of transfer RNAs by modification of uridines. Nat. Struct. Mol. Biol. 14, 498–502. doi: 10.1038/nsmb1242
Wen, J. D., Lancaster, L., Hodges, C., Zeri, A. C., Yoshimura, S. H., Noller, H. F., et al. (2008). Following translation by single ribosomes one codon at a time. Nature 452, 598–603. doi: 10.1038/nature06716
Wilson, D. N., Arenz, S., and Beckmann, R. (2016). Translation regulation via nascent polypeptide-mediated ribosome stalling. Curr. Opin. Struct. Biol. 37, 123–133. doi: 10.1016/j.sbi.2016.01.008
Wohlgemuth, I., Brenne, S., Beringer, M., and Rodnina, M. V. (2008). Modulation of the rate of peptidyl transfer on the ribosome by the nature of substrates. J. Biol. Chem. 283, 32229–32235. doi: 10.1074/jbc.M805316200
Wohlgemuth, S. E., Gorochowski, T. E., and Roubos, J. A. (2013). Translational sensitivity of the Escherichia coli genome to fluctuating tRNA availability. Nucleic Acids Res. 41, 8021–8033. doi: 10.1093/nar/gkt602
Woolstenhulme, C. J., Guydosh, N. R., Green, R., and Buskirk, A. R. (2015). High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 11, 13–21. doi: 10.1016/j.celrep.2015.03.014
Woolstenhulme, C. J., Parajuli, S., Healey, D. W., Valverde, D. P., Petersen, E. N., Starosta, A. L., et al. (2013). Nascent peptides that block protein synthesis in bacteria. Proc. Natl. Acad. Sci. U. S. A. 110, E878–E887. doi: 10.1073/pnas.1219536110
Wu, C. C. C., Zinshteyn, B., Wehner, K. A., and Green, R. (2019). High-resolution ribosome profiling defines discrete ribosome elongation states and translational regulation during cellular stress. Mol. Cell 73, 959.e5–970.e5. doi: 10.1016/j.molcel.2018.12.009
Xu, Y., Ma, P., Shah, P., Rokas, A., Liu, Y., and Johnson, C. H. (2013). Non-optimal codon usage is a mechanism to achieve circadian clock conditionality. Nature 495, 116–120. doi: 10.1038/nature11942
Zalucki, Y. M., and Jennings, M. P. (2007). Experimental confirmation of a key role for non-optimal codons in protein export. Biochem. Biophys. Res. Commun. 355, 143–148. doi: 10.1016/j.bbrc.2007.01.126
Zhang, G., Hubalewska, M., and Ignatova, Z. (2009). Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 16, 274–280. doi: 10.1038/nsmb.1554
Zhang, B., and Miller, T. F. 3rd. (2012). Long-timescale dynamics and regulation of sec-facilitated protein translocation. Cell Rep. 2, 927–937. doi: 10.1016/j.celrep.2012.08.039
Zhang, F. L., Saha, S., Shabalina, S. A., and Kashina, A. (2010). Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science 329, 1534–1537. doi: 10.1126/science.1191701
Zhou, M., Guo, J., Cha, J., Chae, M., Chen, S., Barral, J. M., et al. (2013). Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature 495, 111–115. doi: 10.1038/nature11833
Zhou, T., Weems, M., and Wilke, C. O. (2009). Translationally optimal codons associate with structurally sensitive sites in proteins. Mol. Biol. Evol. 26, 1571–1580. doi: 10.1093/molbev/msp070
Keywords: translation, ribosome pausing, tRNA, prokaryotes, cotranslational folding, translation efficiency, nascent peptide
Citation: Samatova E, Daberger J, Liutkute M and Rodnina MV (2021) Translational Control by Ribosome Pausing in Bacteria: How a Non-uniform Pace of Translation Affects Protein Production and Folding. Front. Microbiol. 11:619430. doi: 10.3389/fmicb.2020.619430
Edited by:
Anna Maria Giuliodori, University of Camerino, ItalyReviewed by:
Martin Siemann-Herzberg, University of Stuttgart, GermanyDamon Huber, University of Birmingham, United Kingdom
Copyright © 2021 Samatova, Daberger, Liutkute and Rodnina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina V. Rodnina, cm9kbmluYUBtcGlicGMubXBnLmRl