 Jun Chen1,2
Jun Chen1,2 Haoxin Lv3Zhixia Zhang2Hua Zhang1,4Bei Zhang5
Haoxin Lv3Zhixia Zhang2Hua Zhang1,4Bei Zhang5 Xing Wang1
Xing Wang1 Yuan Liu1Miao Zhang1Huili Pang1Guangyong Qin1Lei Wang6*
Yuan Liu1Miao Zhang1Huili Pang1Guangyong Qin1Lei Wang6* Zhongfang Tan1*
Zhongfang Tan1*- 1School of Agricultural Sciences, Zhengzhou University, Zhengzhou, China
- 2Henan Key Laboratory of Ion-Beam Bioengineering, School of Physics, Zhengzhou University, Zhengzhou, China
- 3School of Food Science and Technology, Henan University of Technology, Zhengzhou, China
- 4School of Food and Biological Engineering, Henan University of Animal Husbandry and Economy, Zhengzhou, China
- 5Department of Chemical and Environmental Engineering, Jiaozuo University, Jiaozuo, China
- 6Academy of Animal Science and Veterinary Medicine, Qinghai University, Xining, China
Leuconostoc mesenteroides strains were a type of epiphytic bacterium widely used in fermented foods and products in the biochemical and pharmaceutical industries but data on its presence in foods from Qinghai-Tibet Plateau in China was scarce. In this study, molecular analysis based on multilocus sequence typing (MLST) with eight housekeeping genes (pyrG, groeL, rpoB, recA, uvrC, murC, carB, and pheS) was carried out on 45 L. mesenteroides strains isolated from different plants and dairy products from Qinghai-Tibet Plateau in China. The objective of this study was to perform genetic diversity analysis and explore the relationship between strains and isolate samples or separate regions. A total of 25 sequence types (STs) were identified with a diversity of up to 55.6%, which were grouped into one clonal complexes (CCs), 3 doublets and 17 singletons by eBURST. The results of minimum spanning tree and clustering analysis indicated these L. mesenteroides strains from the Qinghai-Tibet Plateau were relatively weakly related to the isolated region. However, there was a close relationship between the genotypes of L. mesenteroides strains and the type of the isolated sample, which was consistent with the results of API 50CH. The MLST scheme presented in this study provides a shareable and comparable sequence database and enhances our knowledge of the population diversity of L. mesenteroides strains which will be further used for the selection of industrial strains.
Introduction
Leuconostoc mesenteroides, a species of Leuconostoc, is usually found to be associated with plant, meat, dairy, and other food products. L. mesenteroides is a kind of gram-positive, facultative anaerobic lactic acid bacterium usually used as starter cultures or as natural inoculum in many fermented foods such as cheese, sauerkraut, butter milk, wine, and kefir (Campedelli et al., 2015). Some studies have shown that L. mesenteroides plays significant roles in fermentation of foods, such as sauerkraut from Europe and kimchi from East Asia (Beganovic et al., 2011), and it contributes to the improvement of the nutritional and organoleptic properties of fermented foods. In addition, L. mesenteroides is also widely used in the field of in the biochemical and pharmaceutical industries (Choi et al., 2017). The demand for producing high quality dextran and their derivatives continues increasing all over the world (Naessens et al., 2005). Compared to the immense applications in pharmaceutical, food and biotechnology industries, that in public health and food safety should be paid more attention and further investigations need to cover the origin of L. mesenteroides because of its frequent detection in different foods and its interaction to some gastrointestinal problem in humans (Franco-Cendejas et al., 2017; Menegueti et al., 2018). In our study, Qula (a kind of fermented dairy food) is very popular in the Qinghai-Tibet Plateau region, but there are few reports about the microorganisms and food safety in Qula which poses some potential threats to diet health. Compared to other dairy production, the main component of Qula, has higher lipid, protein, lactose, and mineral levels (Zhang B. et al., 2015). In view of these excellent traits, the development of L. mesenteroides is of great potential in the application, so it is necessary to carry out diversity characterization analysis. L. mesenteroides species contain four subspecies: L. mesenteroides subsp. mesenteroides, L. mesenteroides subsp. cremoris, L. mesenteroides subsp. dextranicum and L. mesenteroides subsp. Suionicum (Jeon et al., 2017). The identity between the four subspecies was as high as 99%, which meant traditional molecular typing approaches such as 16s rRNA for characterization of the species at the isolate level were either unavailable or unreliable at the molecular level, and a standard protocol for their detection and characterization was not available. Only a higher resolution molecular typing technique can be used to explore its genetic evolution. Considering the important role in the fermented food industry and the production of dextrans and bacteriocins with promising developing prospect, the exploration of molecular character of L. mesenteroides was in great need to categorize candidate microorganisms for further safe utilization in production.
Generally, typing methods of bacteria contained phenotyping and genotyping. Phenotypic methods were traditional including serotypes, phage-types, biotypes, and antibiograms, which often brought about uncertain identification (Dan et al., 2014). Therefore, genotypic methods were playing important roles in phylogenetic classification and identification of bacterial species. Till now, many molecular methods had been used for the typing of Leuconostoc genus like pulsed-field gel electrophoresis (PFGE), restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), random amplified polymorphic DNA (RAPD)-PCR, repetitive element palindromic PCR (rep-PCR), and multilocus sequence typing (MLST) (Vihavainen and Bjorkroth, 2009; Alegria et al., 2013; Sharma et al., 2018, 2020). MLST was a technique for distinguishing accurately between different isolates within a species. MLST was first proposed and applied in Maiden et al. (1998), which was a new molecular analysis method developed on the basis of Multilocus Enzyme Electrophoresis (MLEE) methods. This typing methods for intraspecies identification of pathogens were essential epidemiological tools in infection prevention and control. Shortly thereafter, MLST was applied to analyze non-pathogenic food production bacteria including LAB (Sabat et al., 2013). For example, Tanigawa and Watanabe used MLST to compare 7 housekeeping genes in 41 isolates of Lactobacillus delbrueckii and demonstrated MLST was efficient for identificating isolates to subspecies level (Tanigawa and Watanabe, 2011). MLST was a technique for distinguishing accurately between different isolates within a species. Compared to other methods, MLST scheme had clear results and the sequence data could be transferred worldwide with a global internet database which can realize the transmission of genetic evolution information between a wide range of strains (Olsen et al., 2014). The research field of MLST had a qualitative breakthrough until Mora promoted multilocus technology from pathogenic bacteria to non-pathogenic bacteria in Mora et al. (2000). He explored the genetic diversity of Pediococcus acidilactic by using MLST, finding that AcH/PA-1 pediocin produced by Pediococcus acidilactici had a certain relationship with its genotype (Mora et al., 2000). In Ruiz-Garbajosa et al. (2006) used MLST to amplify 110 strains of Enterococcus faecalis. The results showed that the genetic relationship was related with the isolated source (Ruiz-Garbajosa et al., 2006). Although the population biology of some LAB species had been characterized by MLST methods, to date, there was little MLST protocol available for Leuconostoc species.
In this study, based on the effective method of MLST, 41 L. mesenteroides strains isolated from some parts of the Qinghai-Tibet Plateau and 4 standard strains were used to explore the population structure and the relationship between the genetic relationship and the factors including the isolate region and the isolated samples. The aim of the present study was to utilize the genomic sequence to construct a multilocus sequence typing (MLST) approach for L. mesenteroides, which would provide a tool for characterizing isolates from Qinghai-Tibet Plateau to study globally. In addition, establishment of a sequence-based MLST library would enable global surveillance of L. mesenteroides.
Materials and Methods
Obtain of the Experimental Strains
We obtained 41 strains of L. mesenteroides isolated from crops and dairy products from some parts of the Qinghai-Tibet Plateau from 2008 to 2011 (Table 1), and 4 standard strains purchased from Japan and China Culture Collection Center (Table 2). All of the strains were initially identified by physiological and biochemical characteristics, including experiments of Gram reactions, catalase activity and gas production in the presence of glucose as described (Kozaki et al., 1992). Furthermore, the species were identified based on 16S rRNA gene sequence analysis with the prokaryotic 16S rDNA universal primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTA CCTTGTTACGACTT-3′) (Zhang et al., 2016). The PCR products were sequenced by the Huada Biotech Company (Zhengzhou, China) and subjected to BLAST analysis on the NCBI website1. Among these 41 strains of L. mesenteroides isolated from the Tibetan plateau, the series of qz strains represented the isolated strains naturally attached to the surface of common crops in some places of the Qinghai-Tibet Plateau, including the potato (n = 16), oat (n = 7) and wheat (n = 2). The series of cw strains (n = 16) represented strains isolated from a kind of dairy product called Qula. Stock cultures were stored at −80°C in 20% (v/v) glycerol.
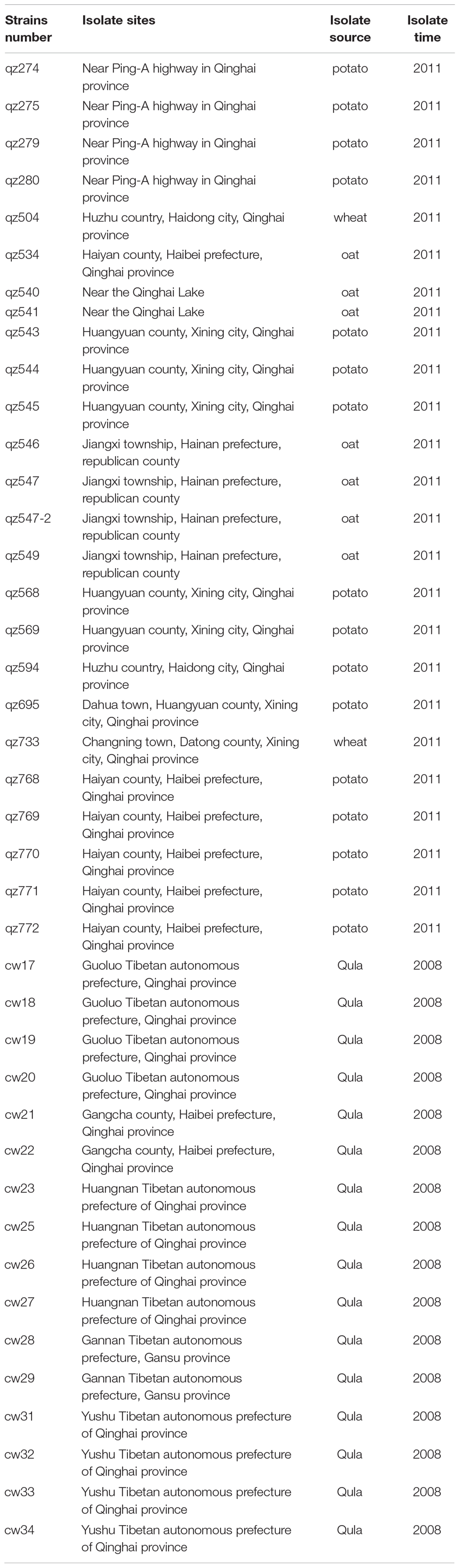
Table 1. Basic information of Leuconostoc mesenteroides.

Table 2. Standard strains of Leuconostoc mesenteroides.
Extraction and Detection of Total Genomic DNA
All of the L. mesenteroides strains were incubated for two consecutive generations of 24 h each in de Man, Rogosa and Sharpe (MRS) broth medium at 37°C. Total genomic DNA was extracted according to the manufacturer’s instructions in Whole Genome DNA Kit (NEP062, Beijing Dingguo, China) and the method described by Pitcher et al. (1989), and the extracted DNA was measured by a microscopic UV spectrophotometer (Thermo Fisher Scientific, Wilmington, United States) at a uniform concentration of 100 ng/μL before use. The total DNA extraction process mainly included the following steps and respective systems. lysozyme: 20 μg/ml, fully dissolved and divided into 1 ml per tube for storage in the refrigerator. Proteinase K: 20 mg/ml, stored at −20°C. RNase A: 10 mg/ml, 10 mg RNase A added to a concentration of 1 M Tris (pH 7.5) 10 μl, 2.5 M NaCl 6 μl, sterile water 984 μl, heated at 100°C for 15 min cooled and stored at −20°C. 5 × TBE Electrophoresis buffer reservoir: 54 g of Tris, 27.5 g of boric acid, 20 ml of 0.5 M/L EDTA (pH 8.0), 1000 ml of fixed volume of ultrapure water. The phylogenic tree was constructed using the neighbor-joining method with MEGA 7 software.
Amplification and Sequencing of Housekeeping Genes
The selected 8 housekeeping genes were relatively evenly distributed: pyrG, groeL, rpoB, recA, uvrC, murC, carB, pheS. The distance distribution of these 8 housekeeping genes and the location of the whole genome in the reference strain L. mesenteroides ATCC8293 were shown in Figure 1, and the specific details were shown in Table 3.
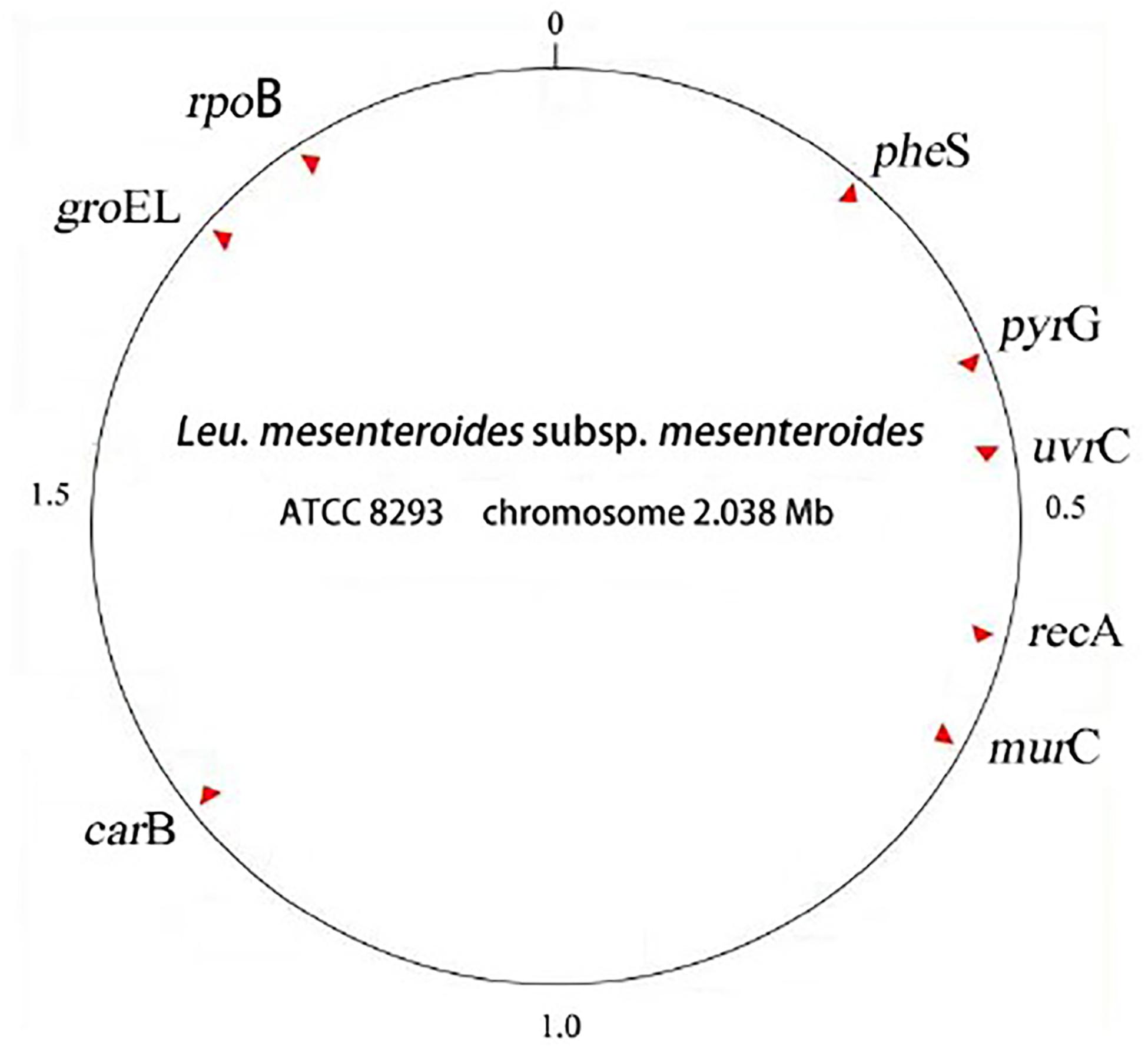
Figure 1. Locations of 8 MLST loci in the genome of Leuconostoc mesenteroides subsp. Mesenteroides ATCC 8293.
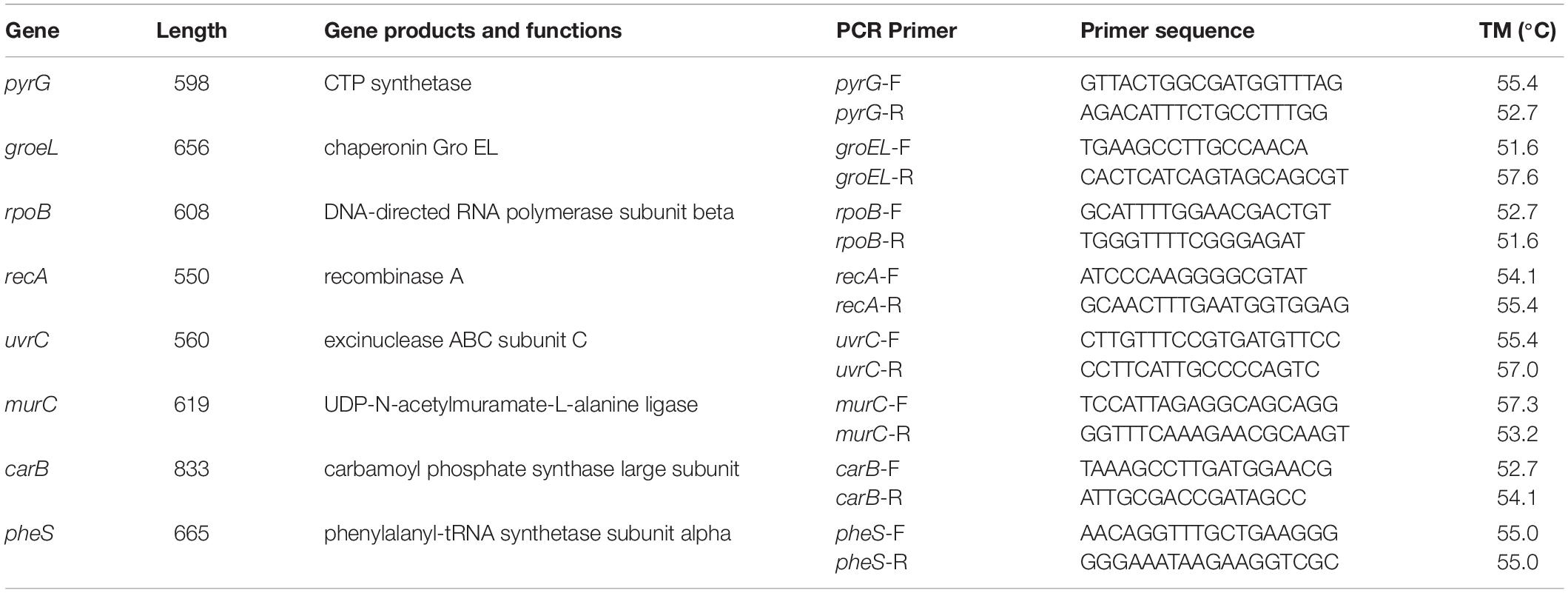
Table 3. Genes and primers used for MLST.
In the MLST scheme, the number of 7 housekeeping gene loci was the most common, and the length of the amplified fragments was concentrated around 450bp. In this experiment, in order to increase the discrimination between strains, 8 housekeeping genes were selected. The location of housekeeping genes was shown in Figure 1. It was clear that the 8 selected housekeeping genes (pyrG, grooveL, rpoB, recA, uvrC, murC, carB, pheS) were unlinked and evenly distributed along the genome of L. mesenteroides ATCC 8293, and there was a certain distance between each loci which can reflect the genetic information of L. mesenteroides more comprehensively (Makarova et al., 2006). The whole genome sequence size of the standard strain ATCC 8293 was 2.038 Mbp. Among them, the pheS, pyrG, uvrC genes were distributed at 0-0.5 Mbp, the recA and murC genes were distributed in the 0.5-1.0 Mbp interval, the carB gene was at 1.0-1.5 Mbp, within the range of chromosome 1.5-2.038 Mbp, groEL and rpoB genes were distributed.
The primers for the eight housekeeping genes pyrG, groEL, rpoB, recA, uvrC, murC, carB, and pheS were designed via the software Primer5.0, and then the correctness of the primers was determined by sequence alignment check. The primers used in this experiment (Table 3) were synthesized by Biotechnology (Shanghai) Co., Ltd. PCR amplifications were performed with the methods described by Lhomme et al. (2015); Sharma et al. (2018). The PCR reaction program included several steps, pre-denaturation at 94°C for 5 min, denaturation at 94°C for 1 min, annealing at 50°C for 1 min, extension at 72°C for 1 min, extension at 72°C for 10 min, in which the second step to the fourth step were repeated 35 times. The annealing temperature of rpoB was 48°C, and the annealing temperature of UvrC and MurC was 52°C. The PCR product was verified in 1% agarose, and after being qualified, it was sent to Shanghai Meiji Biological Company for sequencing.
Gene Sequence Database Alignment and Analysis of MLST
The bidirectional sequence bases that had been successfully sequenced were spliced together without error, and then imported into BioNumerics V6.0 (Applied-Maths, Sint Maartens-Latem, Belgium) software to determine the sequence typing. Used the online eBURST v3.0 software2 to analyze the resulting STs (Francisco et al., 2009), and determined the clonal complex (Urwin and Maiden, 2003). Finally, the minimum spanning tree analysis of all STs was performed by using Prims’s algorithm in the BioNumerics software to explore the relationship between STs and different isolate regions and samples.
The Simpson Index was used to evaluate the discriminatory power of the MLST typing method established in this study for different L. mesenteroides strains. Simpson’s diversity index (D) was calculated according to the results of MLST (Hunter and Gaston, 1988). DNA sequence analyses were performed, including allelic variation of selected 8 housekeeping genes. The number of polymorphic sites, Tajima’s D value, nucleotide diversity (π), G + C content and the dN/dS ratio were calculated with the software DnaSp v5.1. The linkage equilibrium between alleles at the 8 housekeeping genes was detected with the program LIAN and the index of association (IA) was calculated (Murray, 2005).
STs Clustering Analysis
The study of genetic diversity of species was based on genetic distance, which described the genetic structure and interspecies differences of a population by reflecting the phylogenetic evolutionary relationships of that population. It was generally believed that ST in populations with longer genetic distances had existed in nature for longer periods of time and have evolved in different regions. Distance-based clustering construction methods mainly included Unweighted pair-group Method Using an Arithmetic Average (UPGMA), Neighbor-joining method (NJ), and Maximum-likelihood method (ML). In this study, the UPGMA method was used to construct clustering map. An UPGMA dendrogram was constructed by START 2.0 software via UPGMA method described by Dan et al. (2014). The most prominent advantage of this method was that the results were clear, less distortion, and better representation of the affinity between different strains.
Carbon Source Fermentation Clustering Analysis
The test was carried out using an Analytical Profile Index (API 50CH, bioMerieux, France) test strip containing 49 carbon sources and a control. The experimental method was operated according to the instructions and strains were cultivated in a constant temperature incubator at 37°C. The yellow color represents positive result, yellow-green means weak positive, and blue indicates negative. The experimental results were recorded at 24 and 48 h, respectively. Analyzed the results of the carbon source fermentation experiment, and the clustering analysis of API 50CH was performed. Furthermore, the results of the cluster analysis of carbon source fermentation experiments were compared with those of STs cluster analysis, and the relationship between the characteristics of carbon source fermentation and the STs of L. mesenteroides strains from different isolated regions and sample were discussed. It also provided a reference for the research of nutritional physiology, phylogenetic relationship and interspecies relationship of different isolated strains.
Results and Discussion
Sequence Alignment and Analysis of Typing Results
All 45 L. mesenteroides isolates were identified by 16S rRNA sequencing and a phylogenetic tree was constructed using the neighbor-joining method (Figure 2). They were all L. mesenteroides, which belong to different subspecies. In order to identify and distinguish these strains more accurately, the method of MLST was adopted. At present, there is no relevant data of L. mesenteroides on the PubMLST website. The eight genes (pyrG, groeL, rpoB, recA, uvrC, murC, carB and pheS) were successfully amplified for all 45 L. mesenteroides isolates. The sequences obtained for the eight housekeeping genes were imported into BioNumerics V6.0 software to determine the allele number. The allele numbers of each strain were linked in series in a certain order as Allelic Profiles. This set of values corresponds to a number representing the ST of the strain. The results of the typing of 45 strains were shown in Table 4. Finally, 45 L. mesenteroides were divided into 25 sequence types (STs) with a diversity of up to 55.6%. Among them, ST8, ST17, and ST19 were the most frequent, each containing 4 strains, accounting for 8.9% of all strains respectively. Next were ST9, ST10, ST13, and ST14, containing three isolated strains each, accounting for 6.7% of the total number of strains, respectively. What’s more, all of the isolated strains contained in these 4 STs were derived from crops. The remaining STs such as ST5, ST6, ST16, ST20, ST21 only had one member. The results indicated that the MLST profiles of L. mesenteroides isolates from Qinghai-Tibet Plateau in China were highly diverse. The genetic variations of the L. mesenteroides isolates for the different loci also have been shown in Table 4. Furthermore, it can be observed that the allelic frequency was dominated for alleles 1 and 2 compared to other alleles among the 8 selected housekeeping gene loci which was uniform with the results of Sharma et al. (2018). The apparent low levels of biodiversity in groEL, recA, pheS suggested that these gene sequences of the loci were somewhat conserved which have the lower discriminatory ability than the other housekeeping loci, and the remaining 5 loci had more polymorphic sites. Our work led to the identification of new alleles and new STs, confirming that the L. mesenteroides population had a great genetic diversity. Similar work on Leuconostoc citreum strains were performed, which also revealed the diversity of STs of 13 bacterial strains isolated from South Korea (Sharma et al., 2018). Besides, the genomic variation is associated with ecological and social interactions, and local selection pressures and population of bacteria could cause micro-evolutionary changes (Cordero and Polz, 2014). The isolated strains were examined for their genetic diversity using 7 selected seven housekeeping genes with the lower discriminatory ability rather than 8 housekeeping loci used in our study. Initially, 10 housekeeping genes were selected for MLST from the complete genome of L. gelidum. subsp. gasicomitatum LMG 18811T in the study of Johansson et al. (2011). however three of the 10 genes including grpoA, dnaA and atpA were deleted because they were either located too close to another gene or bring with too little variation (Rahkila et al., 2015). MLST was considered to be the most discriminating method for studying molecular epidemiology and population structure of bacteria (Baldo et al., 2006; Liang et al., 2013). Although this approach has been developed for several LAB, such as Oenococcus oeni (de las Rivas et al., 2004) and Lactobacillus casei (Diancourt et al., 2007), until this study there had been no MLST protocol used for L. mesenteroides, and the genetic diversity of this species in Chinese traditional fermented dairy food Qula had never been investigated. In this study, we used MLST with 8 housekeeping genes on 45 L. mesenteroides isolates from a large region in Qinghai-Tibet Plateau in China, making up for the blank of related research in this area.
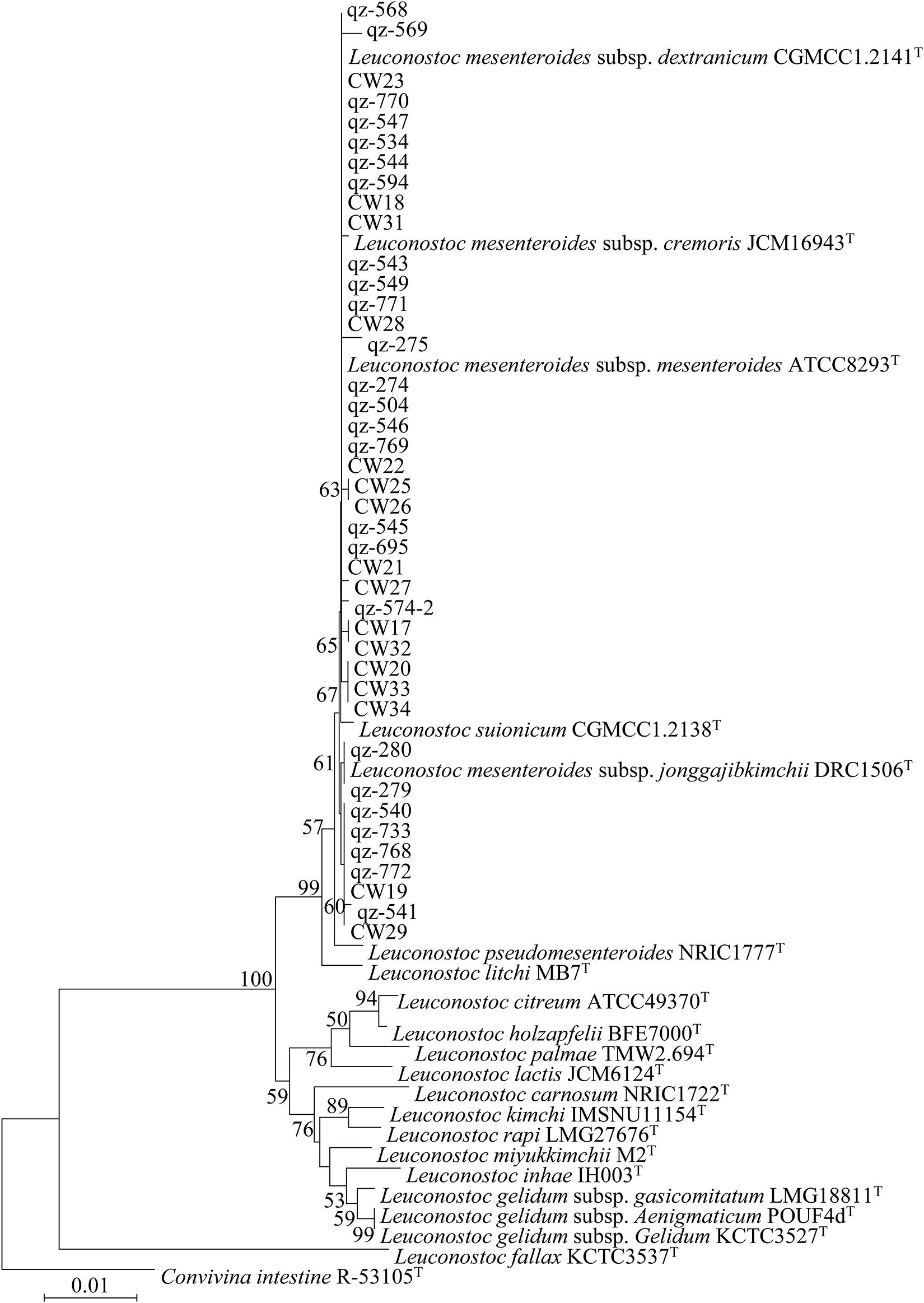
Figure 2. Phylogenetic trees of 45 Leuconostoc mesenteroides strains based on 16S rRNA gene sequences.
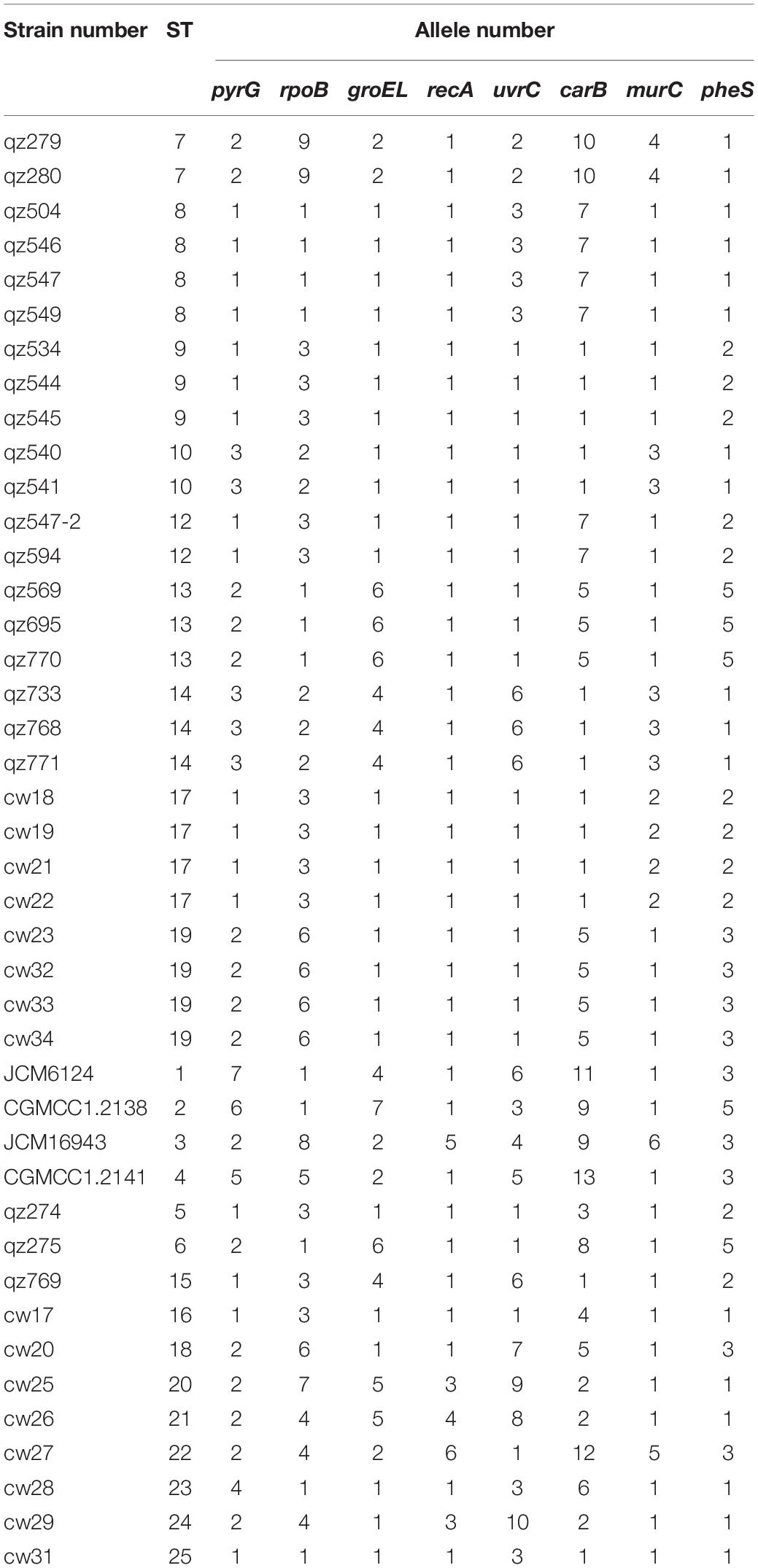
Table 4. STs and Allelic profiles of 45 Leuconostoc mesenteroides.
The D value of 0.939 calculated from Simpson’s equation indicates that the method of MLST had a high discriminatory power. Allelic variation and detection for recombination were also detected, described in Table 5. The number of alleles for each locus ranged from 5 to 13, and the number of polymorphic sites ranged from 4 to 14. The G + C content was from 37.71 to 42.58%. The average pairwise nucleotide diversity per site among the 8 genes ranged from 0.00081 to 0.00428. The dN/dS ratios of 8 loci ranged from 0.05 to 1.39. The ratio of dN/dS was less than 1 except for the gene pheS, indicating that external selection pressure did not interfere too much with these genes to accelerate their evolution. A similar result of dN/dS was found for the allelic variation at the MLST loci, that was, most dN/dS ratios were less than 1 except for that of murC (Zhang W. et al., 2015). The values from Tajima’s D test, which measures deviation from the standard neutral model of evolution, ranged from −1.72542 to 0.46268. The value of IA in this study was 0.6246, indicating that the 8 selected housekeeping genes were evenly distributed throughout the genome with complete linkage equilibrium and that there was no high frequency recombination in these strains. These results above suggested balancing selection for the genes, which is typical for housekeeping genes.
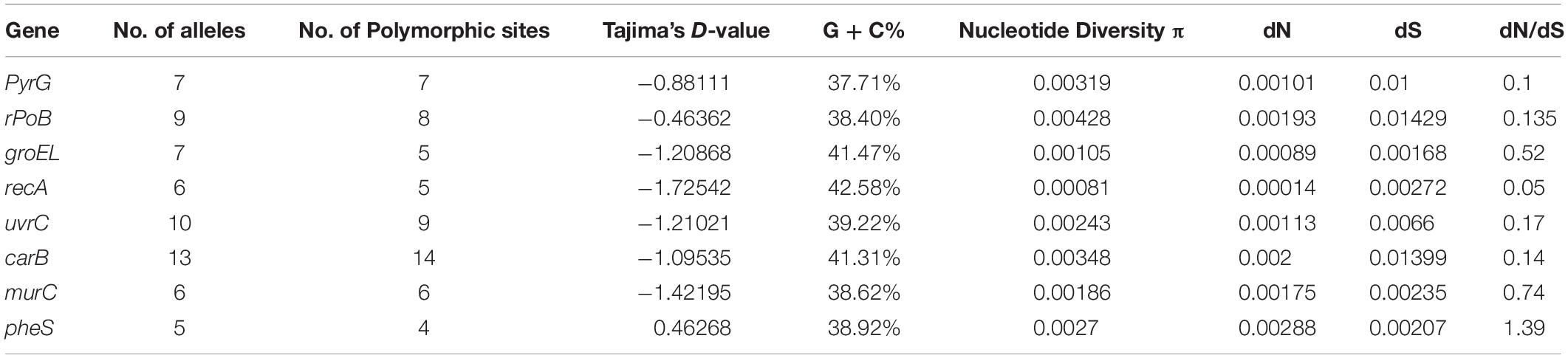
Table 5. Allelic variation and nucleotide diversity of the seven MLST loci.
Analysis of Different STs Clonal Complexes
The online software eBURST was used to classify the MLST data of isolated representative strains and obtain non-overlapping ST groups or clonal complexes. A connection that included three and more ST types was commonly referred to as a Clonal Complex (CC) (Francisco et al., 2009). Generally speaking, the core ST of the clonal complex was the original sequence type. Although eBURST analysis was only a hypothesis of the origin and evolution relationship model of the clonal complex, it can be used to explore and analyze the diversity of bacterial clones. The eBURST aimed to identify closely related strains based on allelic profiles and is less affected by recombination than many sequence-based methods (Turner et al., 2007).
According to Figure 3, 25 STs were analyzed by eBURST to eventually form a clonal complex, three doublets and 15 singletons. The clonal complex contained 4 STs. The general clonal complex was numbered by the core ST, so the core ST9 was named CC9. According to Table 4, CC9 contained a total of 10 isolates, accounting for 22.2% of the total strains. Among them, three ST types were from plants, including ST5, ST9, and ST12, while the four strains in ST17 were isolated from dairy product Qula. The Singletons contained a total of 15 ST types, containing 21 strains, accounting for 46.7% of the total and the distance between these strains is scattered, indicating that the genetic differences among the strains were relatively greater than those of other strains, possibly due to the higher conservativeness of L. mesenteroides and the lower rate of mutation of the housekeeping genes. It can also be drawn from the chart the genetic polymorphism of L. mesenteroides was high, and different strains have different evolutionary processes. The STs of strains with different isolation regions but the same kind of samples were also different, possibly due to regional environmental variation, so the strains were subjected to different selection pressures resulting in slight changes in nucleotide sequence. Similar works have been reported for Leuconostoc lactis isolates from traditional dairy products (Dan et al., 2014). Analysis of their MLST by eBURST produced two CCs and six singletons. The majority of L. lactis isolates from dairy products were found in these two CCs, while the remaining isolates from various samples including milk, pickle and yogurt were scattered into unique STs. Comparatively, 46 STs were grouped into three CCs and 17 singletons by goeBURST, and the STs in CC1 consisted mostly of strains from modified-atmosphere-packaged poultry, beef, pork, minced pork and beef (Rahkila et al., 2015). It has been reported in the previous study that usually in the genus Leuconostoc recombination could occur because of various mobile elements including transposable elements, bacteriophages and genomic islands (Meslier et al., 2012).
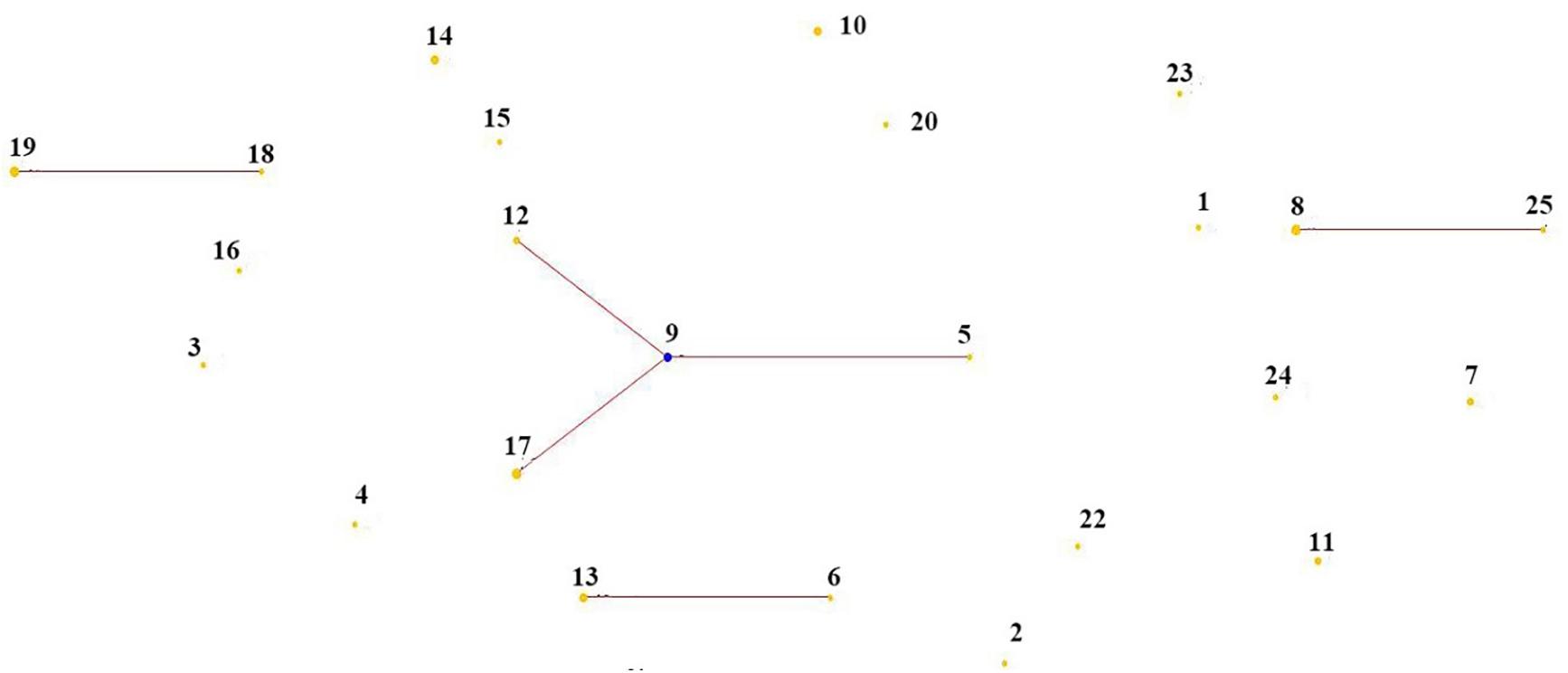
Figure 3. Population structure of 45 Leuconostoc mesenteroides strains by eBURST analysis.
Analysis of Genetic Evolution of ST in Different Isolation Region and Different Isolated Samples
A minimum-spanning tree of 25 ST phenotypes was constructed using the software BioNumerics V6.0 to investigate the relationship between STs and different regions or isolated samples. The minimum spanning tree of ST of L. mesenteroides isolated from different region was shown in Figure 4. It can be seen from the figure that the ST9 core group had 4 STs, except for ST17 which contained four isolated strains from dairy products, all of which were from plants. The isolated regions of the four nearest groups of affinities were all relatively close, and their isolated strain affinities were correspondingly close. However, the different isolated strains from the same region were not all clustered together when viewed as a whole, for example, a total of eight isolated strains were isolated from Haibei State, ST9 (qz534), ST10 (qz772), ST13 (qz770), ST14 (qz768, qz771), ST15 (qz769), and ST17 (cw21, cw22), which were relatively distant from each other on the minimal generative tree, indicating their distant kinship. One of the great strengths of MLST was that it promoted the comparison of the results between different laboratories (Xu et al., 2015). In Dan’s study, clustering by region amongst the isolates was evident in the minimum-spanning tree (Dan et al., 2014). Comparable results had been reported in Leuconostoc lactis, where no significant associations between STs and the sources of the isolates could be found (Picozzi et al., 2010). Conclusions about the correlation between microbial diversity and geography still remain conflicting (Foschino et al., 2001; Lhomme et al., 2016; Yang et al., 2017). In the study of Yang, the genotypic diversity of Lactobacillus sanfranciscensis strains typed by MLST indicated geographical origin had no relation to ST of strains. In other study, the cheese isolates did not cluster together based on geographical origins, which suggesting that environmental selective pressures for strains from cheese had weaker association with geographical origin (Cai et al., 2007). The low association in L. mesenteroides strains in this study probably because of the genetic diversity of individual L. mesenteroides isolates.
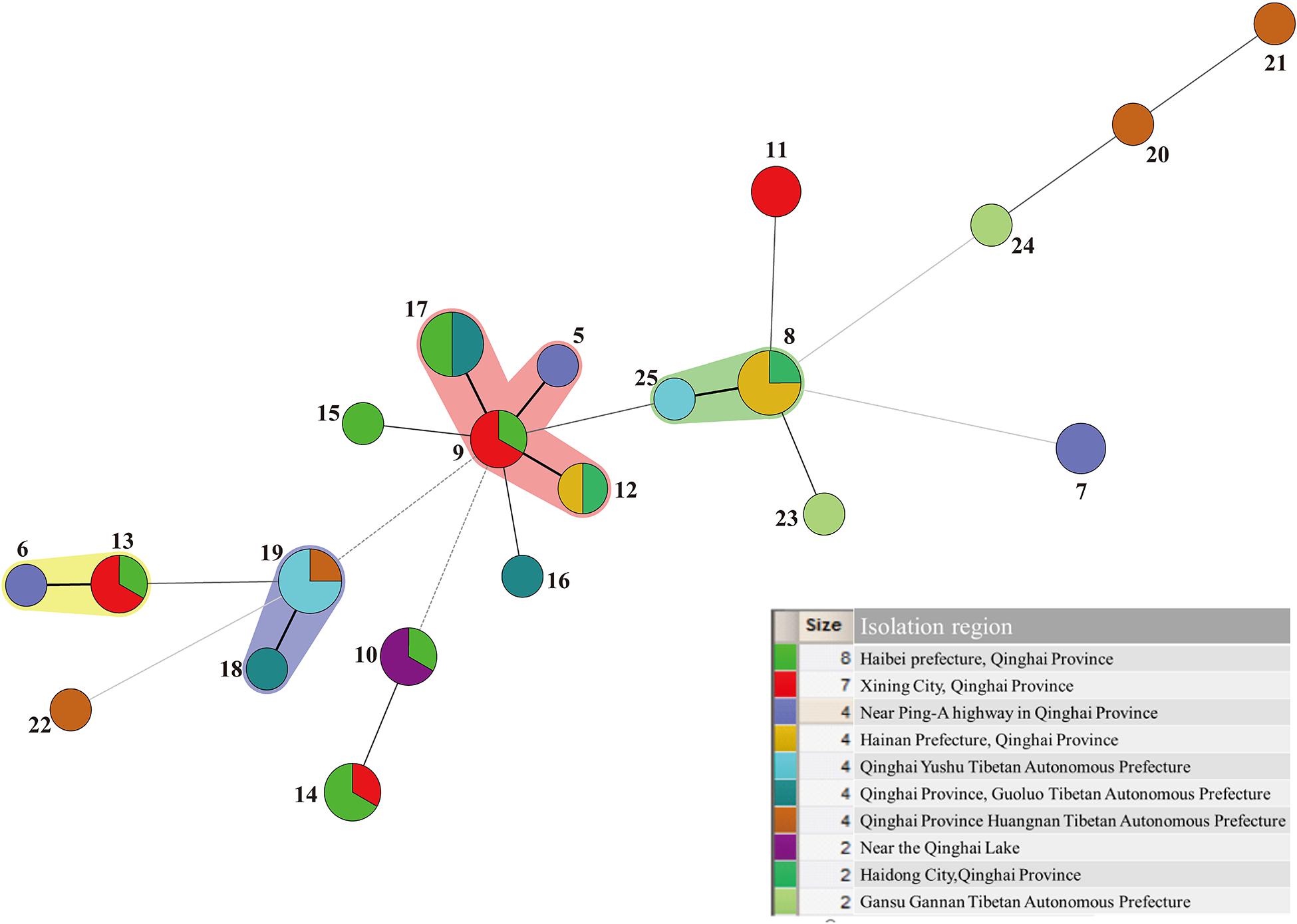
Figure 4. Minimum-spanning tree analysis of 45 Leuconostoc mesenteroides strains based on MLST date according to region. Each circle indicates a sequence type (ST), the size of the circle is proportional to the number of strains and the type of line between isolates indicates the strength of the genetic relationship between these isolates (black line = strong relationship, gray line = intermediate relationship and dotted line = weak relationship). The different colors represent the strains isolated from different region.
The minimum spanning Tree of STs for the different isolated samples was shown in Figure 5. Red color indicated isolated samples from oats, wheat, potatoes, while green color indicated isolated samples from dairy Qula and 4 blue circles were standard strains. Besides, Figure 5 also had four shaded areas, and the STs grouping with the closest kinship was consistent. However, the population structure of the isolated sample group had a more obvious correlation than the population structure of the isolated region. Interestingly, the strains contained in pink shaded area were all derived from plants except ST17 which came from Qula. It was found that most of the solid lines were connected to the same isolated sample except for the green shaded area, ST25 from dairy products and ST8 from plants. In addition, the strains contained in the other shaded areas were all the same source of isolated samples. The standard strain had a far-reaching relationship with the experimental strain. The whole picture showed that the relationship between the strains was more closely related to the isolated samples. Previous studies on the population genetics of food-associated bacterial species indicated that the division into subpopulations reflects adaptations to different niches rather than geographical isolation (Bridier et al., 2010). However, different results have been reported that the evolution of different Leuconostoc strains was not related to respective food sources (Sharma et al., 2018).
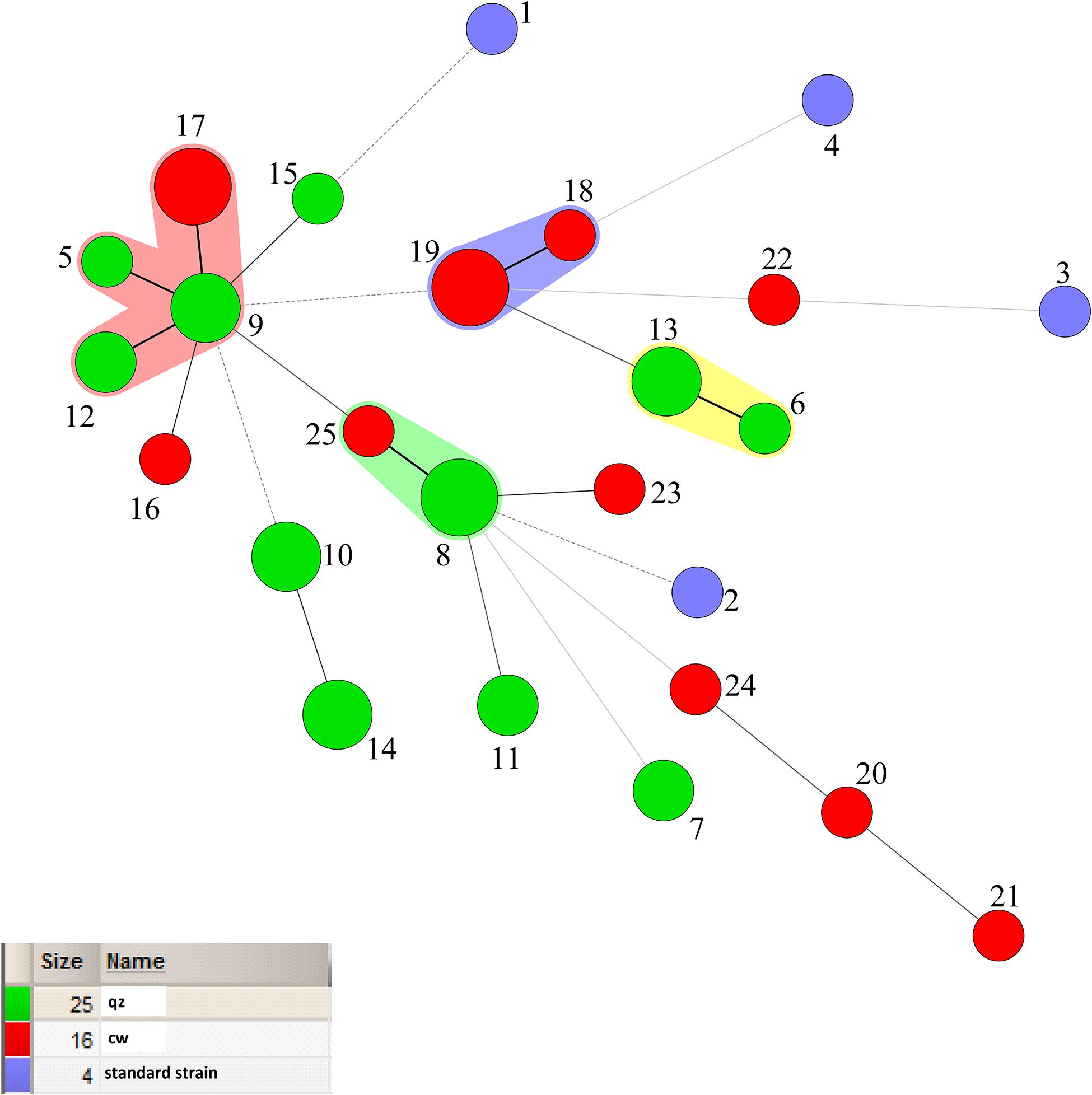
Figure 5. Minimum-spanning tree analysis of 45 Leuconostoc mesenteroides strains based on MLST date according to samples. Each circle indicates a sequence type (ST), the size of the circle is proportional to the number of strains sharing the same ST, and the type of line between isolates indicates the strength of the genetic relationship between these isolates (black line = strong relationship, gray line = intermediate relationship and dotted line = weak relationship). The different colors represent the strains isolated from different samples.
Clustering Analysis of STs
In this study, the similarity coefficient was calculated by BioNumerics V6.0 software using the categorical algorithm, and a cluster diagram of 45 strains (25 STs) was constructed using UPGMA method, as shown in Figure 6. The characterization of genetic diversity was also reflected in the UPGMA dendrogram. The four standard strains had further genetic distance with other strains, which is consistent with the theory that genetic distances exist in nature for a long time. The branches of ST1 and ST2 were relatively close, suggesting the information of the constructed cluster graph was reliable. Most of the STs were showing some degree of relationship. There were 4 clusters in the branches with confidence above 80%, which were the first group ST6 and ST13 each with 4 strains respectively; ST18 and ST19 in second group were all from Qula; ST8 and ST25 in the third group contained 9 strains and in the fourth group consisted of 10 strains. These four groups were all on the same branch, and the genetic distance was close, indicating that the group differentiation time was short and the relationship was close, which was consistent with the results of the four shaded areas of the minimum spanning tree. Interestingly, the most of Qula-derived L. mesenteroides were clustered together, suggesting that they may have a common recent ancestor, despite their different geographical locations. The other genetic distances were relatively far, indicating these strains may have undergone some variation during the evolution process. Similar work had been reported for the UPGMA tree based on the MLST scheme. No clear link existed between ST and the original source of each isolate (Dan et al., 2014). There was no significant relationship between STs and the various sources of the strains in L. sanfranciscensis (Picozzi et al., 2010). The absence of association in L. lactis may due to the genetic diversity of strains (Passerini et al., 2010). It was found that the evolution of different Leuconostoc strains was not related to respective food sources (Sharma et al., 2018). A systematic and detailed study on the MLST scheme of L. mesenteroides has been reported back in Zhang W. et al. (2015). In comparison, our strains come from a wider variety of sources, not only from dairy products, but also from agricultural crops. The differences in selected housekeeping genes and the strain source, including the geographical location of the strains and the source of the isolated samples may have contributed to the different MLST results.
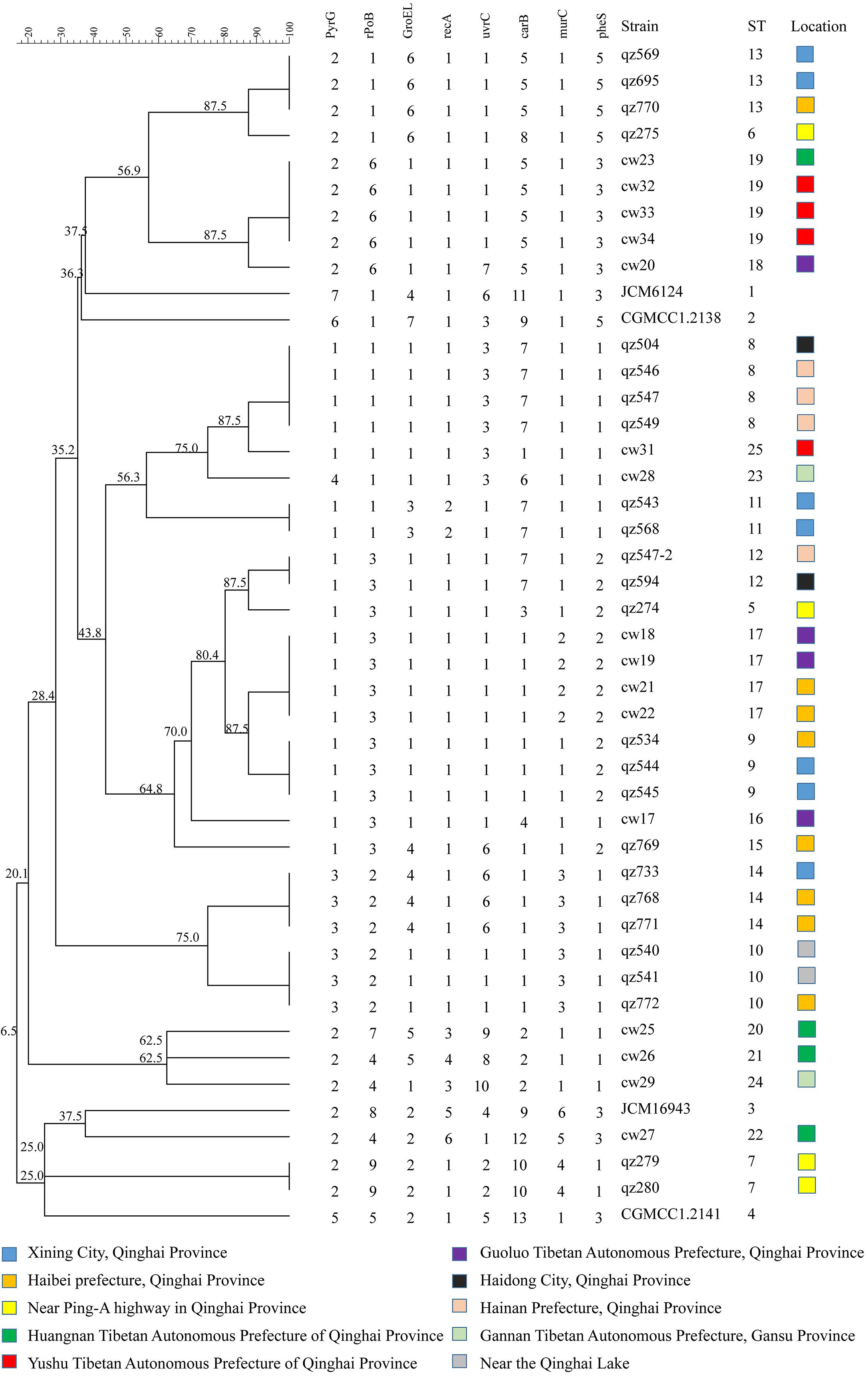
Figure 6. An UPGMA dendrogram of the ST of 45 Leuconostoc mesenteroides strains. The different colors represent the strains isolated from different location. Each row shows in turn the number of alleles, ST and isolated location for each strain.
Clustering Analysis of Carbon Source Fermentation
The results of carbon source fermentation characteristics of L. mesenteroides isolated from oat, wheat, potato and Qula showed most of the strains from the same kinds of samples had a certain similarity in carbon source utilization capacity. Clustering analysis can classify bacteria with similar carbon source metabolism characteristics, thus effectively reflecting the metabolic relationship of different strains. Taking the utilization of 49 carbon sources as clustering variables, the clustering analysis was performed using the system clustering function in SPSS software. The results, reported in Figure 7, showed that the sorts of samples clustering was more obvious, indicating the correlation was greater, while the region clustering was not obvious, which was consistent with the results of the ST clustering analysis. The carbon source fermentation results of 16 strains of L. mesenteroides from Qula were relatively close, so the clustering distance is closer, as shown, cw17, cw32, cw25, cw26, cw19, cw21, cw33, cw34, cw18, cw22, cw29, cw27 were clustered together, and they were all derived from the sample of Qula, and this group of similar carbon fermentation characteristics also included qz541 from oats and qz545 from potatoes. It was found that in ST clustering analysis in Figure 6 cw17, cw18, cw19, cw21, cw22, qz545 were also clustered together, which suggested that for strains from the same isolated samples, not only STs, but also carbon source fermentation characteristics may cluster together. In addition, we found that some strains with the same ST were similar in the carbon source fermentation clustering, for example, the strains of qz768, qz733, qz771 were all belong to ST14, and their carbon source fermentation results were clustered together, similar groups were ST8, ST11, ST17, and ST19. In a word, there may be a relationship between the ST and carbon source fermentation characteristics of L. mesenteroides. Other surveys had similarly found, Lactose utilization was less prevalent in plant isolates than in those from cheese and human GI tracts, probably due to relatively recent acquisitions of lactose metabolic genes (Cai et al., 2007). However, there was no significant relationship between the location of the isolated strains and the carbon source fermentation characteristics compared to the sorts of isolated sample of strains, which was similar with the results of ST clustering analysis. Moreover, it was reported the metabolic properties were slightly different because of microbial evolutionary adaption to the environment (Lee and Chang, 2016). The metabolic fingerprinting of Arcobacter butzleri resulted less discriminatory than the genome-based approach (Fanelli et al., 2020). Different results was reported that the metabolic properties of strains were not correlated with the two different source of isolation from shellfish (Fanelli et al., 2019). Furthermore, compared to previous studies on MLST scheme of L. mesenteroides (Zhang W. et al., 2015), phenotypic studies of carbon source fermentation and genotypic studies of MLST were combined for the first time, revealing the existence of a link between them. Many L. mesenteroides strains isolated from the same sample, their carbon source fermentation results clustered together, and some strains with the same ST also clustered together (Figure 7). The results tentatively confirmed that there was a link between genotype and phenotype, which deserved further in-depth study.

Figure 7. Clustering analysis of carbon source fermentation of Leuconostoc mesenteroides. The different colors represent the strains isolated from different location.
Conclusion
The presented MLST scheme was found to be a robust tool for the investigating the population structure of L. mesenteroides from Qinghai-Tibet Plateau. A total of 25 STs were generated from 45 strains of L. mesenteroides with a diversity of 55.6%. 25 STs were analyzed by eBURST to form a clonal complex CC9, three doublets and 15 singletons. The methods of minimum spanning tree and ST clustering analysis were used to explore the relationship between strains of L. mesenteroides from Qinghai-Tibet Plateau. It was found that STs of strains from a region did not show obvious aggregation, indicating the correlation between the strains and the isolated region was relatively weak. Specifically, the STs did not cluster significantly according to the isolated regions of the strains in either the minimum spanning tree analysis or in the STs clustering analysis. However, there was a more obvious relevance between the relationship and the types of the isolated samples, that is, the representative strains of the same type of samples were more closely related, and their STs had a tendency to cluster together. The results of clustering analysis of STs and carbon source fermentation showed that there was a certain correlation between the ST and types of isolated sample and the carbon source fermentation characteristics of these strains. This is because we found that the carbon source fermentation results of strains from the same isolated samples had a tendency to cluster together, especially for strains isolated from Qula, and there were some strains with the same ST whose carbon source fermentation results also clustered together. This study revealed the diversity of L. mesenteroides from the Qinghai-Tibet Plateau and supplemented the relevant information about L. mesenteroides in the MLST online database, and provided certain contribution and clues to the molecular evolution and prevalence of this species. Further work will be focused on the biodiversity, clonal population structure and genetic recombination to obtain a better understanding of the evolution and population genetics of L. mesenteroides.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
JC, ZZ, and ZT designed the study and wrote the manuscript. JC, HL, and ZZ performed the experiments. JC and ZZ conducted the statistical and bioinformatics analysis. LW, HZ, BZ, and YL performed the analysis of some of the data. All the authors reviewed and approved the final version of the manuscript.
Funding
This work was supported by the Qinghai Province Key R&D and Transformation Plan of China (No. 2020-NK-127).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Alegria, A., Delgado, S., Florez, A. B., and Mayo, B. (2013). Identification, typing, and functional characterization of Leuconostoc spp. strains from traditional, starter-free cheeses. Dairy Sci. Technol. 93, 657–673. doi: 10.1007/s13594-013-0128-3
Baldo, L., Hotopp, J. C. D., Jolley, K. A., Bordenstein, S. R., Biber, S. A., Choudhury, R. R., et al. (2006). Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl. Environ. Microbiol. 72, 7098–7110. doi: 10.1128/Aem.00731-06
Beganovic, J., Pavunc, A. L., Gjuracic, K., Spoljarec, M., Suskovic, J., and Kos, B. (2011). Improved sauerkraut production with probiotic strain Lactobacillus plantarum L4 and Leuconostoc mesenteroides LMG 7954. J. Food Sci. 76, M124–M129. doi: 10.1111/j.1750-3841.2010.02030.x
Bridier, J., Claisse, O., Coton, M., Coton, E., and Lonvaud-Funel, A. (2010). Evidence of distinct populations and specific subpopulations within the species Oenococcus oeni. Appl. Environ. Microbiol. 76, 7754–7764. doi: 10.1128/Aem.01544-10
Cai, H., Rodriguez, B. T., Zhang, W., Broadbent, J. R., and Steele, J. L. (2007). Genotypic and phenotypic characterization of Lactobacillus casei strains isolated from different ecological niches suggests frequent recombination and niche specificity. Microbiology Sgm 153(Pt 8), 2655–2665. doi: 10.1099/mic.0.2007/006452-0
Campedelli, I., Florez, A. B., Salvetti, E., Delgado, S., Orru, L., Cattivelli, L., et al. (2015). Draft genome sequence of three antibiotic-resistant Leuconostoc mesenteroides strains of dairy origin. Genome Announc. 3:e01018-15. doi: 10.1128/genomeA.01018-15
Choi, S. Y., Ryu, S. H., Park, J. I., Jeong, E. S., Park, J. H., Ham, S. H., et al. (2017). Anti-obesity effect of robusta fermented with Leuconostoc mesenteroides in high-fat diet-induced obese mice. Exp. Ther. Med. 14, 3761–3767. doi: 10.3892/etm.2017.4990
Cordero, O. X., and Polz, M. F. (2014). Explaining microbial genomic diversity in light of evolutionary ecology. Nat. Rev. Microbiol. 12, 263–273. doi: 10.1038/nrmicro3218
Dan, T., Liu, W. J., Sun, Z. H., Lv, Q., Xu, H. Y., Song, Y. Q., et al. (2014). A novel multi-locus sequence typing (MLST) protocol for Leuconostoc lactis isolates from traditional dairy products in China and Mongolia. BMC Microbiol. 14:150. doi: 10.1186/1471-2180-14-150
de las Rivas, B., Marcobal, A., and Munoz, R. (2004). Allelic diversity and population structure in Oenococcus oeni as determined from sequence analysis of housekeeping genes. Appl. Environ. Microbiol. 70, 7210–7219. doi: 10.1128/Aem.70.12.7210-7219.2004
Diancourt, L., Passet, V., Chervaux, C., Garault, P., Smokvina, T., and Brisse, S. (2007). Multilocus sequence typing of Lactobacillus casei reveals a clonal population structure with low levels of homologous recombination. Appl. Environ. Microbiol. 73, 6601–6611. doi: 10.1128/Aem.01095-07
Fanelli, F., Chieffi, D., Di Pinto, A., Mottola, A., Baruzzi, F., and Fusco, V. (2020). Phenotype and genomic background of Arcobacter butzleri strains and taxogenomic assessment of the species. Food Microbiol. 89:103416. doi: 10.1016/j.fm.2020.103416
Fanelli, F., Di Pinto, A., Mottola, A., Mule, G., Chieffi, D., Baruzzi, F., et al. (2019). Genomic characterization of Arcobacter butzleri isolated from shellfish: novel insight into antibiotic resistance and virulence determinants. Front. Microbiol. 10:670. doi: 10.3389/fmicb.2019.00670
Foschino, R., Arrigoni, C., Picozzi, C., Mora, D., and Galli, A. (2001). Phenotypic and genotypic aspects of Lactobacillus sanfranciscensis strains isolated from sourdoughs in Italy. Food Microbiol. 18, 277–285. doi: 10.1006/fmic.2000.0399
Francisco, A. P., Bugalho, M., Ramirez, M., and Carrico, J. A. (2009). Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics 10:152. doi: 10.1186/1471-2105-10-152
Franco-Cendejas, R., Colin-Castro, C. A., Hernandez-Duran, M., Lopez-Jacome, L. E., Ortega-Pena, S., Ceron-Gonzalez, G., et al. (2017). Leuconostoc mesenteroides periprosthetic knee infection, an unusual fastidious Gram-positive bacteria: a case report. BMC Infect. Dis. 17:227. doi: 10.1186/s12879-017-2315-y
Hunter, P. R., and Gaston, M. A. (1988). Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J. Clin. Microbiol. 26, 2465–2466. doi: 10.1128/JCM.26.11.2465-2466.1988
Jeon, H. H., Kim, K. H., Chun, B. H., Ryu, B. H., Han, N. S., and Jeon, C. O. (2017). A proposal of Leuconostoc mesenteroides subsp jonggajibkimchii subsp nov and reclassification of Leuconostoc mesenteroides subsp suionicum (Gu et al., 2012) as Leuconostoc suionicum sp nov based on complete genome sequences. Int. J. Syst. Evol. Microbiol. 67, 2225–2230. doi: 10.1099/ijsem.0.001930
Johansson, P., Paulin, L., Sade, E., Salovuori, N., Alatalo, E. R., Bjorkroth, K. J., et al. (2011). Genome sequence of a food spoilage lactic acid bacterium, Leuconostoc gasicomitatum LMG 18811(T), in association with specific spoilage reactions. Appl. Environ. Microbiol. 77, 4344–4351. doi: 10.1128/Aem.00102-11
Kozaki, M., Uchimura, T., and Okada, S. (1992). Experimental Manual for Lactic Acid Bacteria. Tokyo: Asakurasyoten, 29–72.
Lee, S. H., and Chang, H. C. (2016). Isolation of antifungal activity of Leuconostoc mesenteroides TA from kimchi and characterization of its antifungal compounds. Food Sci. Biotechnol. 25, 213–219. doi: 10.1007/s10068-016-0032-8
Lhomme, E., Lattanzi, A., Dousset, X., Minervini, F., De Angelis, M., Lacaze, G., et al. (2015). Lactic acid bacterium and yeast microbiotas of sixteen French traditional sourdoughs. Int. J. Food Microbiol. 215, 161–170. doi: 10.1016/j.ijfoodmicro.2015.09.015
Lhomme, E., Onno, B., Chuat, V., Durand, K., Orain, S., Valence, F., et al. (2016). Genotypic diversity of Lactobacillus sanfranciscensis strains isolated from French organic sourdoughs. Int. J. Food Microbiol. 226, 13–19. doi: 10.1016/j.ijfoodmicro.2016.03.008
Liang, J. G., Ducatelle, R., Pasmans, F., Smet, A., Haesebrouck, F., and Flahou, B. (2013). Multilocus sequence typing of the porcine and human gastric pathogen Helicobacter suis. J. Clin. Microbiol. 51, 920–926. doi: 10.1128/Jcm.02399-12
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Makarova, K., Slesarev, A., Wolf, Y., Sorokin, A., Mirkin, B., Koonin, E., et al. (2006). Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. U.S.A. 103, 15611–15616. doi: 10.1073/pnas.0607117103
Menegueti, M. G., Gaspar, G. G., Laus, A. M., Basile-Filho, A., Bellissimo-Rodrigues, F., and Auxiliadora-Martins, M. (2018). Bacteremia by Leuconostoc mesenteroides in an immunocompetent patient with chronic Chagas disease: a case report. BMC Infect. Dis. 18:547. doi: 10.1186/s12879-018-3452-7
Meslier, V., Loux, V., and Renault, P. (2012). Genome sequence of Leuconostoc pseudomesenteroides strain 4882, isolated from a dairy starter culture. J. Bacteriol. 194, 6637–6637. doi: 10.1128/Jb.01696-12
Mora, D., Fortina, M. G., Parini, C., Daffonchio, D., and Manachini, P. L. (2000). Genomic subpopulations within the species Pediococcus acidilactici detected by multilocus typing analysis: relationships between pediocin AcH/PA-1 producing and non-producing strains. Microbiology (Reading, England) 146(Pt 8), 2027–2038. doi: 10.1099/00221287-146-8-2027
Murray, S. S. (2005). Evaluation of linkage disequilibrium and its effect on non-parametric multipoint linkage analysis using two high density single-nucleotide polymorphism mapping panels. BMC Genet. 6(Supp. l):S85. doi: 10.1186/1471-2156-6-S1-S85
Naessens, M., Cerdobbel, A., Soetaert, W., and Vandamme, E. J. (2005). Leuconostoc dextransucrase and dextran: production, properties and applications. J. Chem. Technol. Biotechnol. 80, 845–860. doi: 10.1002/jctb.1322
Olsen, J. S., Scholz, H., Fillo, S., Ramisse, V., Lista, F., Tromborg, A. K., et al. (2014). Analysis of the genetic distribution among members of Clostridium botulinum group I using a novel multilocus sequence typing (MLST) assay. J. Microbiol. Methods 96, 84–91. doi: 10.1016/j.mimet.2013.11.003
Passerini, D., Beltramo, C., Coddeville, M., Quentin, Y., Ritzenthaler, P., Daveran-Mingot, M. L., et al. (2010). Genes but not genomes reveal bacterial domestication of Lactococcus Lactis. PLoS One 5:12. doi: 10.1371/journal.pone.0015306
Picozzi, C., Bonacina, G., Vigentini, I., and Foschino, R. (2010). Genetic diversity in Italian Lactobacillus sanfranciscensis strains assessed by multilocus sequence typing and pulsed-field gel electrophoresis analyses. Microbiology Sgm 156, 2035–2045. doi: 10.1099/mic.0.037341-0
Pitcher, D. G., Saunders, N. A., and Owen, R. J. (1989). Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett. Appl. Microbiol. 8, 151–156. doi: 10.1111/j.1472-765X.1989.tb00262.x
Rahkila, R., Johansson, P., Sade, E., Paulin, L., Auvinen, P., and Bjorkroth, J. (2015). Multilocus sequence typing of Leuconostoc gelidum subsp gasicomitatum, a psychrotrophic lactic acid bacterium causing spoilage of packaged perishable foods. Appl. Environ. Microbiol. 81, 2474–2480. doi: 10.1128/Aem.04013-14
Ruiz-Garbajosa, P., Bonten, M. J. M., Robinson, D. A., Top, J., Nallapareddy, S. R., Torres, C., et al. (2006). Multilocus sequence typing scheme for Enterococcus faecalis reveals hospital-adapted genetic complexes in a background of high rates of recombination. J. Clin. Microbiol. 44, 2220–2228. doi: 10.1128/Jcm.02596-05
Sabat, A. J., Budimir, A., Nashev, D., Sa-Leao, R., Van Dijl, J. M., Laurent, F., et al. (2013). Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Eurosurveillance 18, 17–30.
Sharma, A., Kaur, J., Lee, S., and Park, Y. S. (2018). Analysis of Leuconostoc citreum strains using multilocus sequence typing. Food Sci. Biotechnol. 27, 1755–1760. doi: 10.1007/s10068-018-0417-y
Sharma, A., Lee, S., and Park, Y. S. (2020). Molecular typing tools for identifying and characterizing lactic acid bacteria: a review. Food Sci. Biotechnol. 29, 1301–1318. doi: 10.1007/s10068-020-00802-x
Tanigawa, K., and Watanabe, K. (2011). Multilocus sequence typing reveals a novel subspeciation of Lactobacillus delbrueckii. Microbiology Sgm 157, 727–738. doi: 10.1099/mic.0.043240-0
Turner, K. M. E., Hanage, W. P., Fraser, C., Connor, T. R., and Spratt, B. G. (2007). Assessing the reliability of eBURST using simulated populations with known ancestry. BMC Microbiol. 7:30. doi: 10.1186/1471-2180-7-30
Urwin, R., and Maiden, M. C. J. (2003). Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 11, 479–487. doi: 10.1016/j.tim.2003.08.006
Vihavainen, E. J., and Bjorkroth, K. J. (2009). Diversity of Leuconostoc gasicomitatum associated with meat spoilage. Int. J. Food Microbiol. 136, 32–36. doi: 10.1016/j.ijfoodmicro.2009.09.010
Xu, H. Y., Liu, W. J., Zhang, W. Y., Yu, J., Song, Y. Q., Menhe, B., et al. (2015). Use of multilocus sequence typing to infer genetic diversity and population structure of Lactobacillus plantarum isolates from different sources. BMC Microbiol. 15:241. doi: 10.1186/s12866-015-0584-4
Yang, H. Y., Liu, T. J., Zhang, G. H., Chen, J. C., Gu, J. S., Yuan, L., et al. (2017). Genotyping of Lactobacillus sanfranciscensis isolates from Chinese traditional sourdoughs by multilocus sequence typing and multiplex RAPD-PCR. Int. J. Food Microbiol. 258, 50–57. doi: 10.1016/j.ijfoodmicro.2017.07.013
Zhang, B., Tan, Z. F., Wang, Y. P., Li, Z. W., Jiao, Z., and Huang, Q. C. (2015). Dynamic changes of the microbial communities during the preparation of traditional Tibetan Qula cheese. Dairy Sci. Technol. 95, 167–180. doi: 10.1007/s13594-014-0194-1
Zhang, B., Wang, Y. P., Tan, Z. F., Li, Z. W., Jiao, Z., and Huang, Q. C. (2016). Screening of probiotic activities of Lactobacilli strains isolated from traditional Tibetan Qula, a raw Yak milk cheese. Asian Australas. J. Anim. Sci. 29, 1490–1499. doi: 10.5713/ajas.15.0849
Keywords: Leuconostoc mesenteroides, multilocus sequence typing, genomes, phylogenetics, housekeeping genes
Citation: Chen J, Lv H, Zhang Z, Zhang H, Zhang B, Wang X, Liu Y, Zhang M, Pang H, Qin G, Wang L and Tan Z (2021) Multilocus Sequence Typing of Leuconostoc mesenteroides Strains From the Qinghai-Tibet Plateau. Front. Microbiol. 12:614286. doi: 10.3389/fmicb.2021.614286
Received: 05 October 2020; Accepted: 05 January 2021;
Published: 25 January 2021.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Beatrix Stessl, University of Veterinary Medicine Vienna, AustriaAnusak Kerdsin, Kasetsart University, Thailand
Copyright © 2021 Chen, Lv, Zhang, Zhang, Zhang, Wang, Liu, Zhang, Pang, Qin, Wang and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Wang, d2FuZ2xlaTM4MjM5NkAxNjMuY29t; Zhongfang Tan, dHpob25nZmFuZ0B6enUuZWR1LmNu