 Peidong Xu1,2†
Peidong Xu1,2† Huanwei Wang
Huanwei Wang Wenbo Liu
Wenbo Liu- 1Key Laboratory of Green Prevention and Control of Tropical Plant Diseases and Pests, Ministry of Education, College of Plant Protection, Hainan University, Haikou, China
- 2School of Life Sciences, Hainan University, Haikou, China
Soft rot pectobacteria are devastating plant pathogens with a global distribution and a broad host range. Pectobacterium aroidearum L6, previously isolated from leaves of Syngonium podophyllum, is a pectolytic bacterial pathogen that causes typical soft rot on S. podophyllum. There is a shortage for genome data of P. aroidearum, which seriously hinders research on classification and pathogenesis of Pectobacterium. We present here the complete genome sequence of P. aroidearum L6. The L6 strain carries a single 4,995,896-bp chromosome with 53.10% G + C content and harbors 4,306 predicted protein-coding genes. We estimated in silico DNA–DNA hybridization and average nucleotide identity values in combination with the whole-genome-based phylogeny from 19 Pectobacterium strains including P. aroidearum L6. The results showed that L6 and PC1 formed a population distinct from other populations of the Pectobacterium genus. Phylogenetic analysis based on 16S rRNA and genome sequences showed a close evolutionary relationship among Pectobacterium species. Overall, evolutionary analysis showed that L6 was in the same branch with PC1. In comparison with 18 Pectobacterium spp. reference pathogens, strain L6 had 2,712 gene families, among which 1,632 gene families were identified as orthologous to those strains, as well as 1 putative unique gene family. We discovered 478 genes, 10.4% of the total of predicted genes, that were potentially related to pathogenesis using the Virulence Factors of Pathogenic Bacteria database. A total of 25 genes were related to toxins, 35 encoded plant cell-wall degrading enzymes, and 122 were involved in secretion systems. This study provides a foundation for a better understanding of the genomic structure of P. aroidearum and particularly offers information for the discovery of potential pathogenic factors and the development of more effective strategies against this pathogen.
Introduction
Soft rot Pectobacteriaceae are considered to be one of the top ten most important agricultural phytopathogens (Mansfield et al., 2012). The family Pectobacteriaceae consisted of Pectobacterium spp. and Dickeya spp., formerly characterized as pectinolytic Erwinia spp. (Pérombelon, 2002; Charkowski et al., 2012; Adeolu et al., 2016). They cause destructive soft rot on a variety of field crops, fruits, ornamentals, and vegetables, including the staple food crop potato (Toth et al., 2003, 2011; Ma et al., 2007). Pectobacterium was first established in 1945 (Waldee, 1945; Ma et al., 2007). However, the classification of Pectobacterium spp. is not clear. Hauben et al. (1998) established the genus Pectobacterium that included three species and five subspecies: P. cacticidum, P. chrysanthemi, P. cypripedii, P. carotovorum subsp. atrosepticum, P. carotovorum subsp. betavasculorum, P. carotovorum subsp. carotovorum, P. carotovorum subsp. odoriferum, and P. carotovorum subsp. wasabiae. Subsequently, three subspecies of P. carotovorum were elevated into the species level, namely P. atrosepticum, P. odoriferum, and P. wasabiae (Gardan et al., 2003; Duarte et al., 2004; Nykyri et al., 2012). The classification of the genus Pectobacterium has been subjected to wide revision over the last decade, and it is likely that some of the genome-sequenced strains have been incorrectly assigned to P. carotovorum (Gardan et al., 2003; Khayi et al., 2016; Pritchard et al., 2016; Zhang et al., 2016). For instance, P. carotovorum subsp. carotovorum tends to serve as a catchall for pectobacteria isolates differing from the specific descriptions of the other pectobacteria taxa, and P. aroidearum was classified as a novel species in 2013 (Nabhan et al., 2013). The genome-sequenced strain PC1 (formerly classified as P. carotovorum subsp. carotovorum) is actually P. aroidearum under the new classification (Nabhan et al., 2013).
In the age of genomics, the Pectobacterium genus has been subjected to revision based on the development of bioinformatics. Several pectolytic bacterial strains were thought to belong to a novel Pectobacterium species after several taxonomic analyses including 16S rRNA gene sequence, DNA–DNA hybridization (DDH), genomics, and comparative genomics. These include P. actinidiae (Koh et al., 2012), P. polaris (Dees et al., 2017), P. peruviense (Waleron et al., 2018), P. zantedeschiae (Waleron et al., 2019), P. punjabense (Sarfraz et al., 2018, 2020), and P. aroidearum (Nabhan et al., 2013). The species of the genus Pectobacterium has 18 of child taxa with a validly published with correct name and four proposed species not yet validated based on The List of Prokaryotic names with Standing in Nomenclature1 (Table 1; Adeolu et al., 2016; Parte et al., 2020). Eleven species had complete genome data, and 7 had not complete assembly based on the National Center for Biotechnology Information (NCBI) genome database2 (Table 1). Only strain PC1 of P. aroidearum has its whole genome sequenced. Thus, there is a shortage of whole-genome data for P. aroidearum.
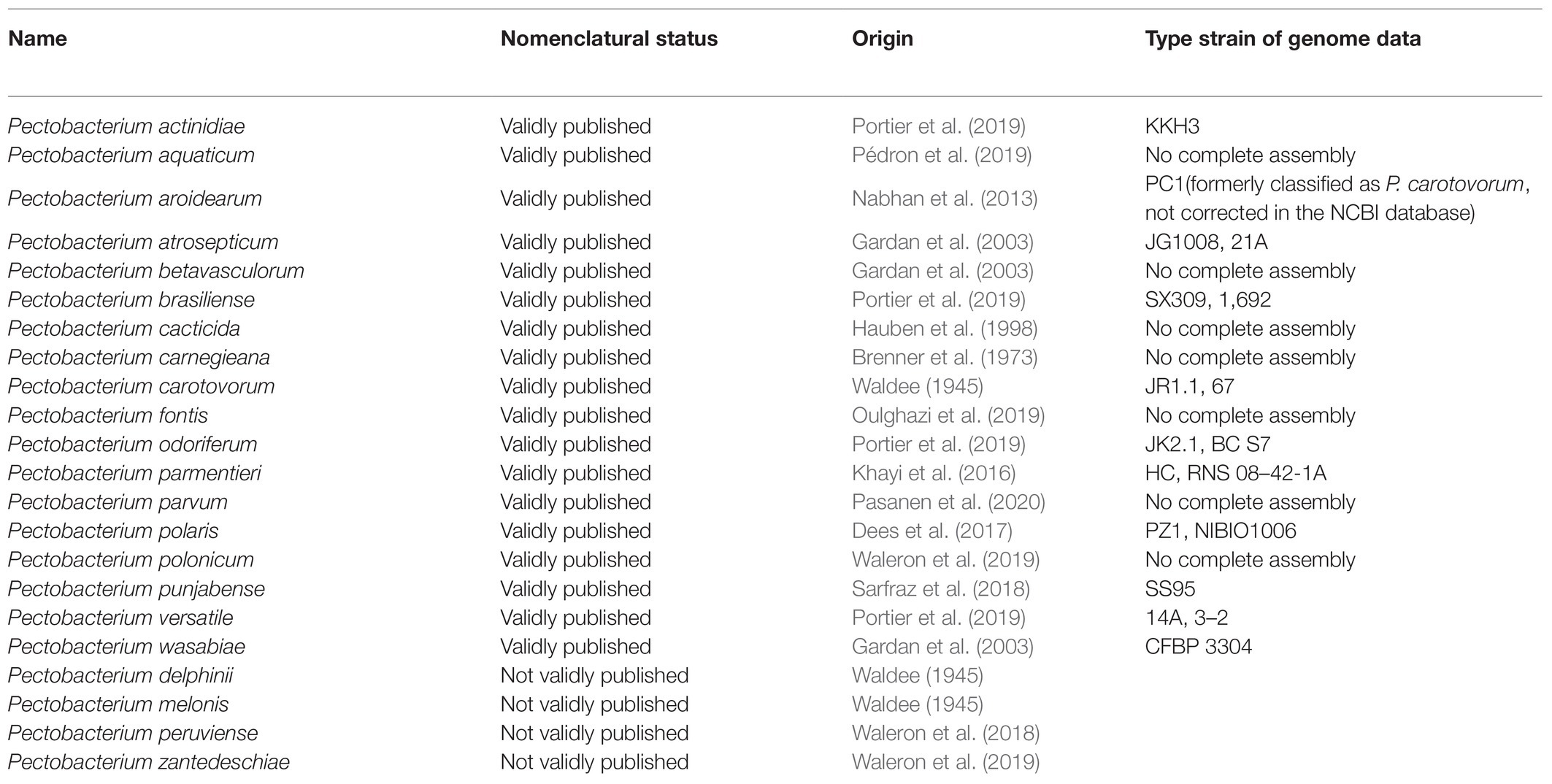
Table 1. Nomenclatural status and type strain of genome data of the Pectobacterium genus.
Currently, no methods and chemicals are effective in controlling Pectobacterium disease or preventing the spread of these pathogens (Zhang et al., 2017). In addition, planting patterns and storage conditions are not applicable for control of the disease (Yaganza et al., 2014). Bacterial strain L6, isolated from Syngonium podophyllum soft rot samples in Hainan Province, was recognized as P. aroidearum (Xu et al., 2020). There is a shortage for whole-genome data of P. aroidearum, which seriously hinders research on classification and pathogenesis of Pectobacterium. Genome comparison revealed that most virulence genes are highly conserved in the Pectobacterium strains, especially for the key virulence determinants involved in the biosynthesis of extracellular enzymes and secretion systems (Li et al., 2019). The functional genomics methods are the effective ways to elucidate that this pathogen interacts with plants and causes disease (Toth et al., 2015). In this study, we sequenced the whole genome of P. aroidearum L6. Then, we compared it with genome analyses of 18 Pectobacterium reference strains. Furthermore, the genome annotation and comparative genomics analysis provided a foundation for a better understanding of the genomic structure of P. aroidearum and particularly offered information for the discovery of potential pathogenic factors and the development of more effective strategies against this pathogen.
Materials and Methods
Strain and Type Strain Genome Sequences
Strain L6 was previously isolated from S. podophyllum soft rot samples in a plant nursery in the Haidian campus of Hainan University, Haikou, Hainan Province, China, in July 2019. Samples were collected from symptomatic S. podophyllum for bacterial isolation. Internal fragments containing symptomatic tissues were transferred to 1 mL of sterile distilled water, after 20 min, rinsed with 70% alcohol and sterile distilled water, and cultured onto Luria-Bertani (LB) medium for 48 h at 28°C to differentiate and characterize the bacterial pathogen. A total of 10 bacterial colonies were isolated from infected tissues. The isolated colonies were subcultured until the pure culture of the suspected bacterium was obtained. Two representative isolates (L5 and L6) were selected for further tests and one isolate preserved in Key Laboratory of Green Prevention and Control of Tropical Plant Diseases and Pests (Hainan University), Ministry of Education as P. aroidearum L6.
The pathogenicity of P. aroidearum L6 was previously reported (Xu et al., 2020). The putative pathogen was re-inoculated to confirm its pathogenicity in the incubator and field on the leaves of S. podophyllum. Typical symptoms of soft rot were observed 12–24 h after inoculation. P. aroidearum was re-isolated from a diseased leaf, fulfilling Koch’s postulates. L6 was grown on LB broth for 12–24 h at 28°C. And genomic DNA of L6 was extracted by the Bacteria Genomic DNA Extraction Kit (Tiangen Biotech Co. Ltd., Beijing, China). Successful extraction of genomic DNA was confirmed by 0.8% agarose gels and quantified by Nanodrop ND-2000 (Thermo Fisher Scientific, United States). All complete genome sequences of Pectobacterium were retrieved from NCBI. Type strain genome sequences of 11 species which had complete assembly genome data were used, including P. aroidearum PC1 (GCF_000023605.1), P. carotovorum subsp. carotovorum JR1.1 (NZ_CP034237.1), P. carotovorum subsp. carotovorum 67 (NZ_CP034211.1), P. atrosepticum JG10-08 (NZ_CP007744.1), P. atrosepticum 21A (NZ_CP009125.1), P. brasiliense SX309 (NZ_CP020350.1), P. brasiliense 1,692 (NZ_CP047495.1), P. odoriferum JK2.1 (NZ_CP034938.1), P. odoriferum BC S7 (NZ_CP009678.1), P. polaris PZ1 (NZ_CP046377.1), P. polaris NIBIO1006 (NZ_CP017481.1), P. actinidiae KKH3 (NZ_JRMH01000001.1), P. wasabiae CFBP 3304 (NZ_CP015750.1), P. parmentieri HC (NZ_CP046376.1), P. parmentieri RNS 08–42-1A (NZ_CP015749.1), P. versatile 14A (NZ_CP034276.1), P. versatile 3–2 (NZ_CP024842.1), and P. punjabense SS95 (NZ_CP038498.1).
Genome Sequencing and Assembly
The P. aroidearum L6 genome was sequenced using a PacBio RS II platform and Illumina HiSeq 4,000 platform. For Illumina HiSeq sequencing, the fragments of 470 bp (with the approximate insert size of 350 bp) from adaptor-ligated DNA were recovered according to standard protocols. The libraries with different indices were multiplexed and loaded on an Illumina HiSeq instrument. Cutadapt (v1.9.1; Martin, 2011) was employed for quality control, and reads whose base groups have quality value below 20 at both ends, sequences containing more than 10% N base, or less than 75 bp in length were removed. The Illumina data were used for estimate and correction. Four SMRT cells zero-mode waveguide (a nano-optical device used to confine light to a small observation volume) arrays of sequencing were used in the PacBio platform to generate the subreads set. PacBio subreads (length < 1 kb) were removed. The program pbdagcon3 was used for self-correction. Draft genomic unitigs, which are uncontested groups of fragments, were assembled using the Celera Assembler (Myers et al., 2000) against a high-quality corrected circular consensus sequence subreads set. To improve accuracy of the genome sequences, the Genome Analysis Toolkit4 (Mckenna et al., 2010) and SOAP tool packages (SOAP2, SOAPsnp, SOAPindel; Li et al., 2013) were used to make single-base corrections. To trace the presence of any plasmid, the filtered Illumina reads were mapped using SOAP to the bacterial plasmid database.5
Genome Component Prediction
Gene prediction was performed on the L6 genome assembly using NCBI Prokaryotic Genome Annotation Pipeline6 (Tatusova et al., 2016; Haft et al., 2018; Li et al., 2021). The tRNA, rRNA, and sRNA recognition made use of tRNAscan-SE (Lowe and Eddy, 1997), RNAmmer (Lagesen et al., 2007), and the Rfam database (Gardner et al., 2009). Tandem repeats annotation was obtained using the Tandem Repeat Finder7 (Wirawan et al., 2010), and the minisatellite DNA and microsatellite DNA were selected based on the number and length of repeat units. The Genomic Island Suite of Tools GIST v1.0 was used for genomic island analysis with Island Path-DIOMB, SIGI-HMM, and Island Picker method (Hasan et al., 2012). Prophage regions were predicted using the PHAge Search Tool web server9 (Zhou et al., 2011) and CRISPR identification using CRISPR Finder (Grissa et al., 2007).
Phylogenetic Analysis
The maximum-likelihood (ML) phylogenetic analysis was inferred with FastME 2.1.6.1 including SPR postprocessing (Lefort et al., 2015) from the Genome BLAST Distance Phylogeny approach (GBDP) distances calculated using default settings from 16S rRNA gene sequences and genome sequences. Branch support was inferred from 1,000 pseudo-bootstrap replicates each. The trees were rooted at the midpoint (Farris, 1972) and visualized with PhyD3 (Kreft et al., 2017).
Average Nucleotide Identity and in silico DNA–DNA Hybridization Analysis
Pairwise comparison of their Average Nucleotide Identity (ANI) was based on BLAST+ (ANIb) from JSpeciesWS10 (Richter et al., 2015), while in silico DNA–DNA Hybridization (isDDH) was conducted using GBDP by the Type (Strain) Genome Server11 (Meier-Kolthoff and Göker, 2019). One-hundred distance replicates were calculated each. The DDH values and confidence intervals were calculated using the recommended settings of the Genome-to-Genome Distance Calculator (GGDC 2.1) for GGDC formula 2 (Meier-Kolthoff et al., 2013).
Comparative Genomics
Pectobacterium aroidearum L6 was compared with reference 18 Pectobacterium strains by using their genome sequence and gene sequence. BLAST core/pan genes of the six strains were clustered using CD-HIT (Fu et al., 2012) rapid clustering of similar proteins software with a threshold of 50% pairwise identity and 0.7 length difference cutoff in amino acids. Gene families were constructed using genes of L6 and reference strains. We carried out gene family TreeFam clustering treatment for the alignment results by Hcluster_sg software (Maqbool and Babri, 2007) and multiple sequence alignment with the clustered gene family using Muscle software (Edgar, 2004a,b). The phylogenetic tree was constructed using multiple sequence alignment results based on Muscle by the TreeBeST (Nandi et al., 2010) using the ML method.
Gene Annotation and Protein Classification
The function annotation is accomplished by analysis of protein sequences. We align genes with databases to obtain the highest quality corresponding annotations. Seven databases were used for general function annotation: Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa et al., 2016), Clusters of Orthologous Groups (COG; Galperin et al., 2015; Makarova et al., 2015), Non-Redundant Protein Database (NR), Swiss-Prot (UniProt Consortium, 2015), Gene Ontology (GO; Ashburner et al., 2000; Philip et al., 2014), TrEMBL (Apweiler et al., 2004), and Evolutionary Genealogy of Genes (EggNOG: Non-supervised Orthologous Groups; Huerta-Cepas et al., 2016) databases. Four databases were used for pathogenicity and drug resistance analysis. Virulence factors and resistance genes were identified based on the core dataset in Virulence Factors of Pathogenic Bacteria (VFDB; Chen et al., 2016) and the Antibiotic Resistance Genes Database (Liu and Pop, 2009); the other two databases were Pathogen Host Interactions (PHI; Winnenburg et al., 2006) and Carbohydrate-Active enZYmes Database (CAZy; Levasseur et al., 2013). Type III secretion system (T3SS) effector proteins were detected using EffectiveT3 (Vargas et al., 2012).
Results
Genomic Features Among P. aroidearum L6 and Reference Strains
The P. aroidearum L6 genome was sequenced using a PacBio RS II platform and Illumina HiSeq 4,000 platform. The raw data were filtered, and this generated 1, 274 Mb of clean data. The reads were assembled into a genome and then assessed. We obtained a genome size of 4,995,896 bp, which consisted of a circular chromosome with no plasmid (Figure 1). GC-Depth analysis was performed on the assembly results to show the assembly length (4,995,896 bp), coverage length (4,995,089), coverage (99.98%), reads usage percent (95.3%), and depth (260) of P. aroidearum L6. The average G + C content of the genome was 53.10%. A total of 4,306 putative protein-coding sequences, with total length of 4,298,622 bp (86.04% of total genome length; Table 2) and average length of 998.29 bp, were annotated on the P. aroidearum L6 genome. The genome encoded 77 tRNA operons and 40 sRNA genes. In addition, a total of 22 rRNA operons were present on the chromosome: eight 5S rRNAs, seven 16S rRNAs, and seven 23S rRNAs.
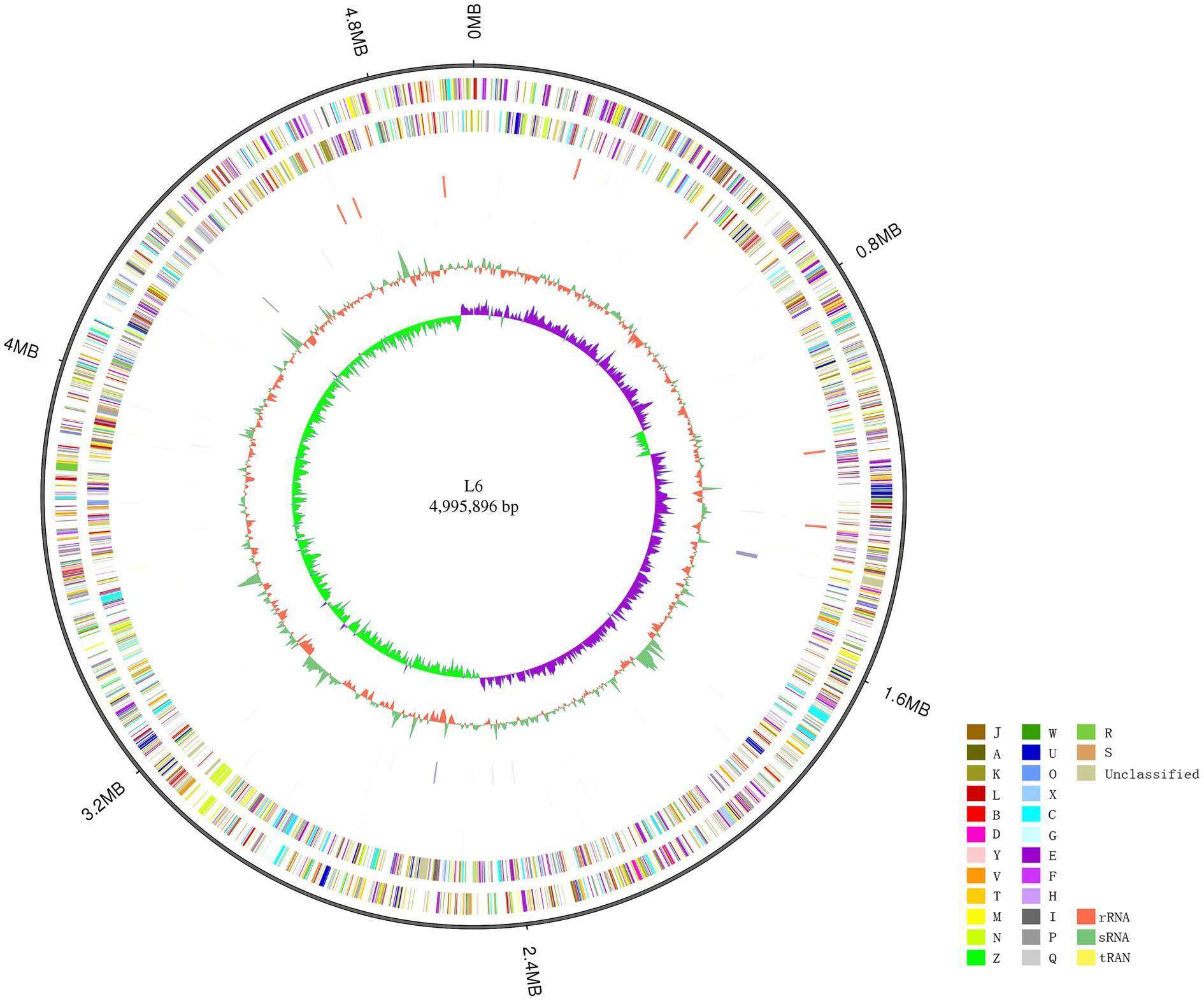
Figure 1. Circular representation of the Pectobacterium aroidearum L6 genome. From outer to inner: first circle is genome size; second and third circles are forward and reverse strand gene, respectively, colored according to cluster of COG classification (A, RNA processing and modification; B, chromatin structure and dynamics; C, energy production and conversion; D, cell cycle control, cell division, and chromosome partitioning; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure, and biogenesis; K, transcription; L, replication, recombination, and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, and chaperones; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport, and catabolism; R, general function prediction only; S, function unknown; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; V, defense mechanisms; W, extracellular structures; X, mobilome: prophages, transposons; Y, nuclear structure; Z, cytoskeleton); fourth and fifth circles are ncRNA (yellow indicates tRNA, orange indicates rRNA, and green indicates sRNA) and repeat, respectively; seventh circle is GC content (green indicates greater than average value, and orange indicates less than average value); and eighth circle is GC-SKEW (GC-SKEW = (G − C)/(G + C), purple indicates >0, and green indicates <0).
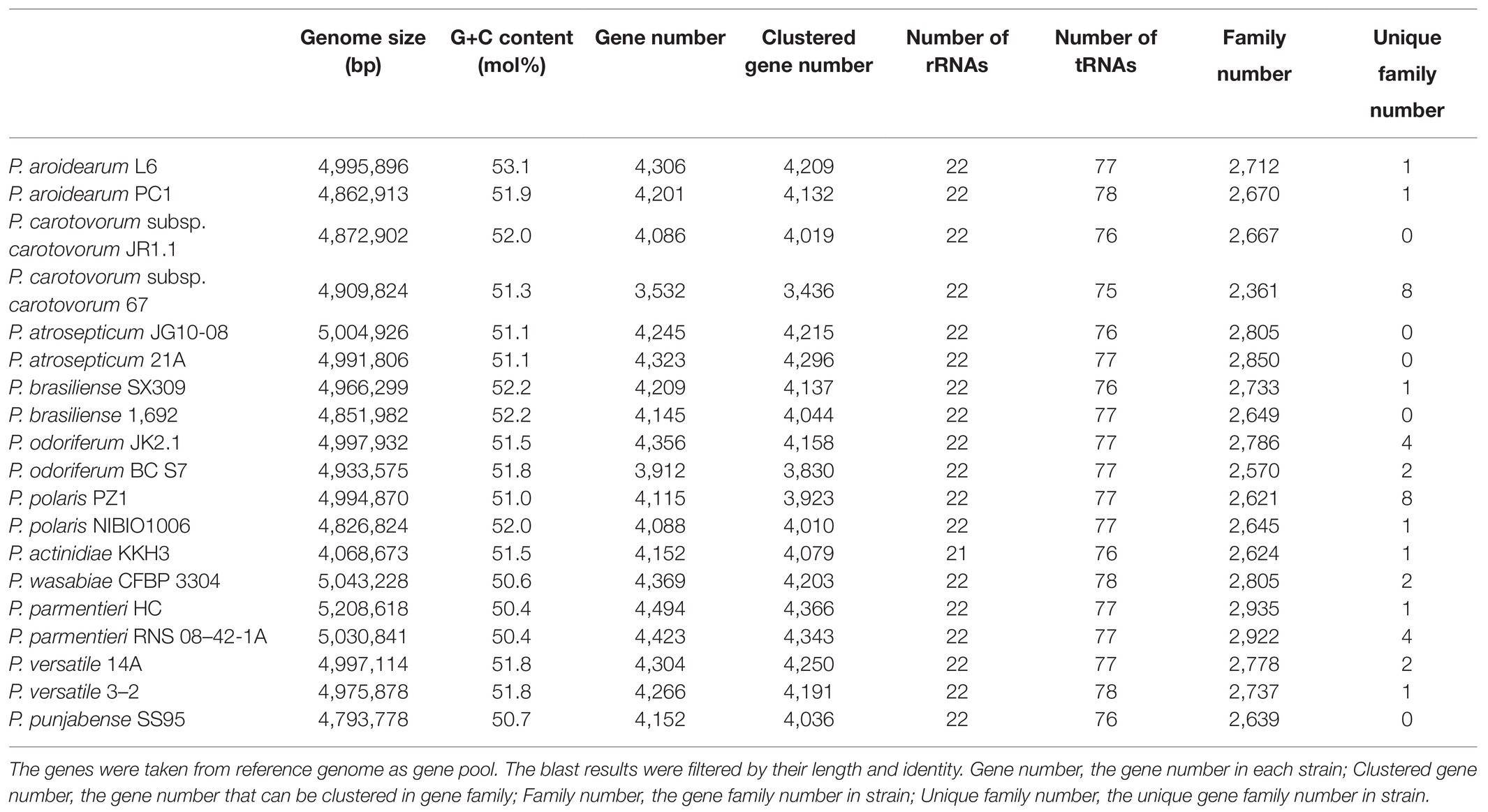
Table 2. Genomic features of the P. aroidearum L6 genome and comparison with genomes of reference strains.
Phylogenetic Analysis of P. aroidearum L6
The 16S rRNA is the most useful and is commonly used as a molecular clock in the systematic classification of bacteria. Its evolution has good clock properties, being highly conserved in structure and function, and can well reflect the differences between different bacteria (Coenye and Vandamme, 2010). Thus, the accessibility of a large quantity of completely sequenced bacterial genomes allows the speedy and reliable determination of intragenomic sequence heterogeneity of 16S rRNA genes. The phylogenetic tree was inferred with FastME 2.1.6.1 from GBDP distances calculated from Pectobacterium species 16S rRNA and genome sequences. The branch lengths were scaled in terms of GBDP distance formula d5. The numbers on branches are GBDP pseudo-bootstrap support values >60% from 1,000 replications, with an average branch support of 91.1%. The delta value was 0.315 based on 16S rRNA and 0.156 based on genome sequences. A phylogram based on computing of the 16S rRNA suggested a close relationship between both P. aroidearum L6 and PC1 genomes (Figure 2A). The phylogenetic relationships among P. aroidearum L6 and reference strains were determined based on genome sequence results (Figure 2B). The whole-genome-based phylogenetic tree showed that L6 was most closely related to P. aroidearum PC1.
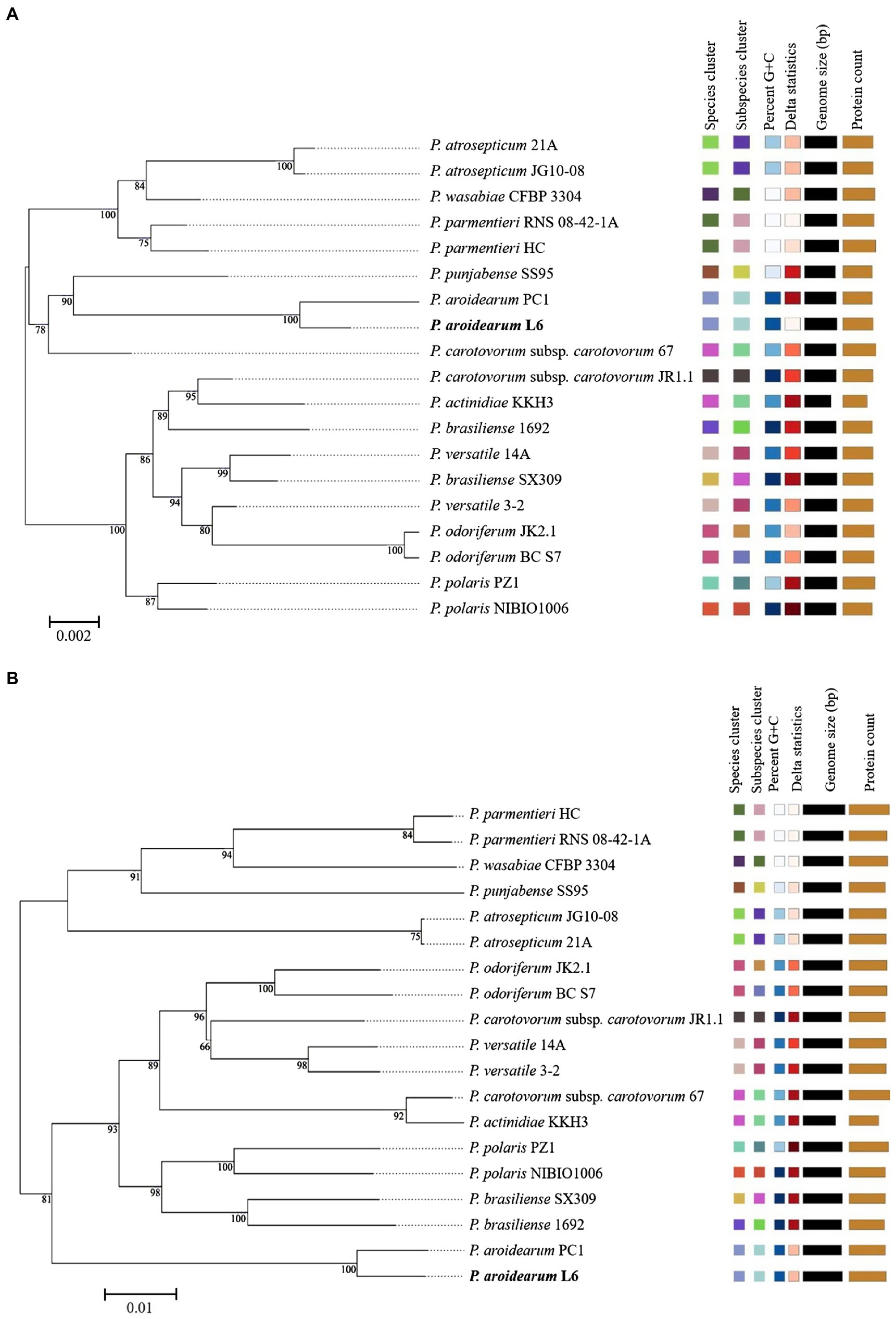
Figure 2. ML phylogenetic analysis for P. aroidearum L6 and reference strains. (A) Phylogenetic tree drawn using the 16S rRNA of Pectobacterium spp. (B) Phylogenetic tree drawn using the genomes of Pectobacterium spp. Bootstrap values are indicated in % of repetitions.
Assessment of Taxonomy of Pectobacterium Species Using isDDH and ANI
We used isDDH and ANI to determine species delineation of Pectobacterium spp. Using empirical evidence based on classified species and their comparisons with DDH and ANI values, the same species were set at ≥70% identity in isDDH and ≥95% identity in ANI. The isDDH and ANI values (Figure 3) were consistent with their phylogenetic relationships (Figure 2), and members of the same phylogenetic clade also showed high isDDH and ANI values. The ranges of isDDH and ANI values for 18 Pectobacterium strains were 88–100 and 36–100. The P. aroidearum L6 and PC1 were evaluated to species level with isDDH = 83 and ANI = 98; furthermore, P. carotovorum subsp. carotovorum JR1.1 and 67 were not the same species with isDDH = 51 and ANI = 93, indicating that these two strains were misidentified.
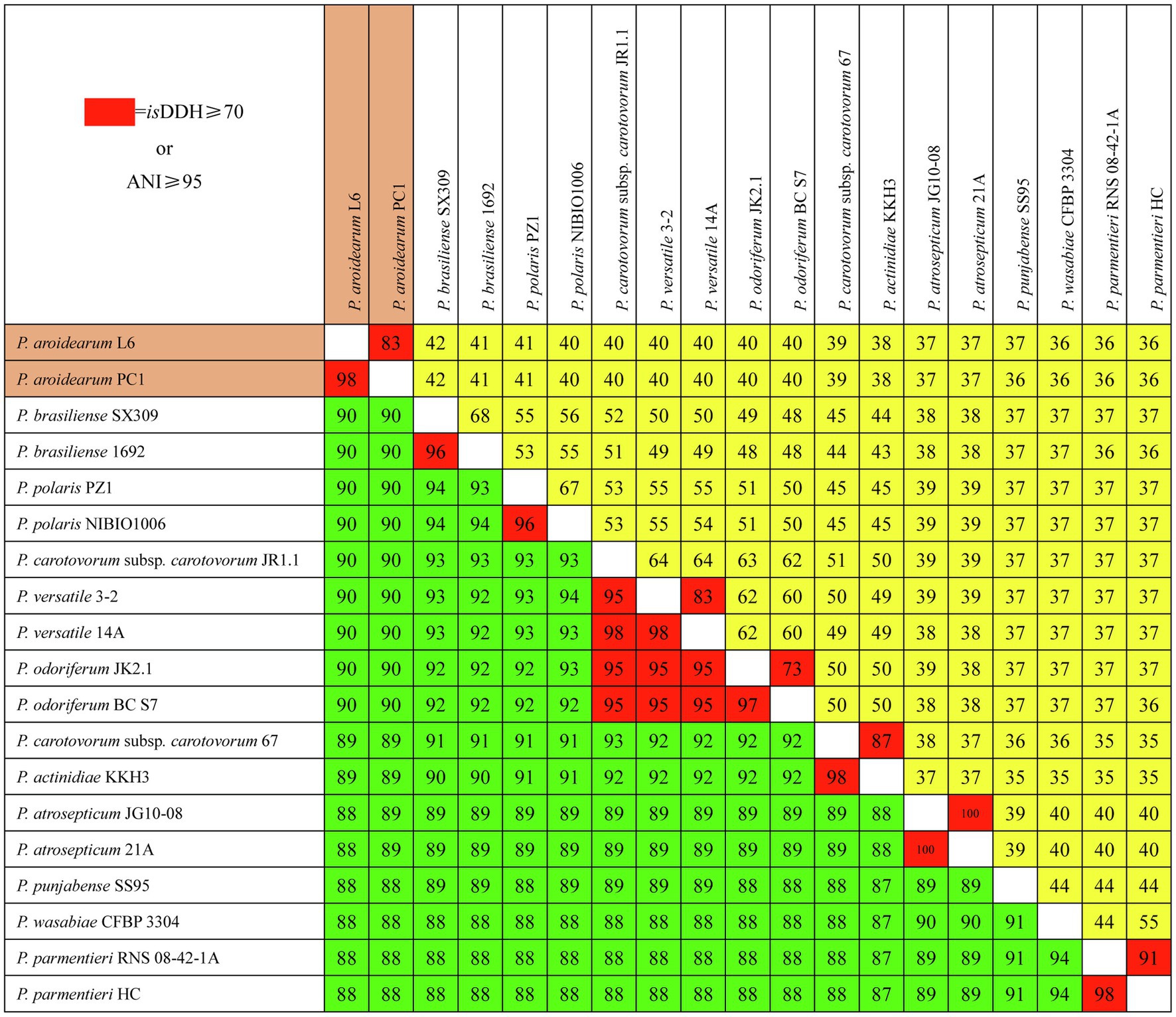
Figure 3. Pairwise comparisons of isDDH and ANI values of Pectobacterium species. The upper triangle (yellow portion) displays isDDH values (%), and the lower triangle (green portion) displays ANI values (%). Boxes with isDDH ≥ 70% or ANI ≥ 95% are colored red.
Analysis of the Core Genome Among Pectobacterium Species and Reference Strains
The genomes of 18 Pectobacterium strains with P. aroidearum L6 were compared. The dispensable gene heatmap showed percentage of dispensable genes among strains. The identity matrix was calculated based on BLASTP. The strains L6 and PC1 had the highest genetic similarity. Otherwise, P. punjabense SS95 also grouped together with P. aroidearum L6 and PC1 based on the dispensable gene heat map (Figure 4A). Analysis of pan genes among L6 strain and reference strains was carried out. There were 1944 genes shared by all of the bacteria. Among them, 132 genes were unique to L6 and 100 genes were unique to PC1 (Figure 4B). Research on special genes and core genes is important for the detection of functional differences and similarities between samples and provides molecular evidence for phenotype differences and similarities. A gene family is a group of genes that have the same ancestor and comprises more than two gene copies. The members of a gene family have similarity in structure and function, and the proteins produced are also similar. Gene families can be used to detect evolutionary history and gene differentiation. The gene family statistics showed that the final core genome was 1,632 gene families. One gene family was unique to L6, and one gene family was unique to PC1 (Figures 4C,D).
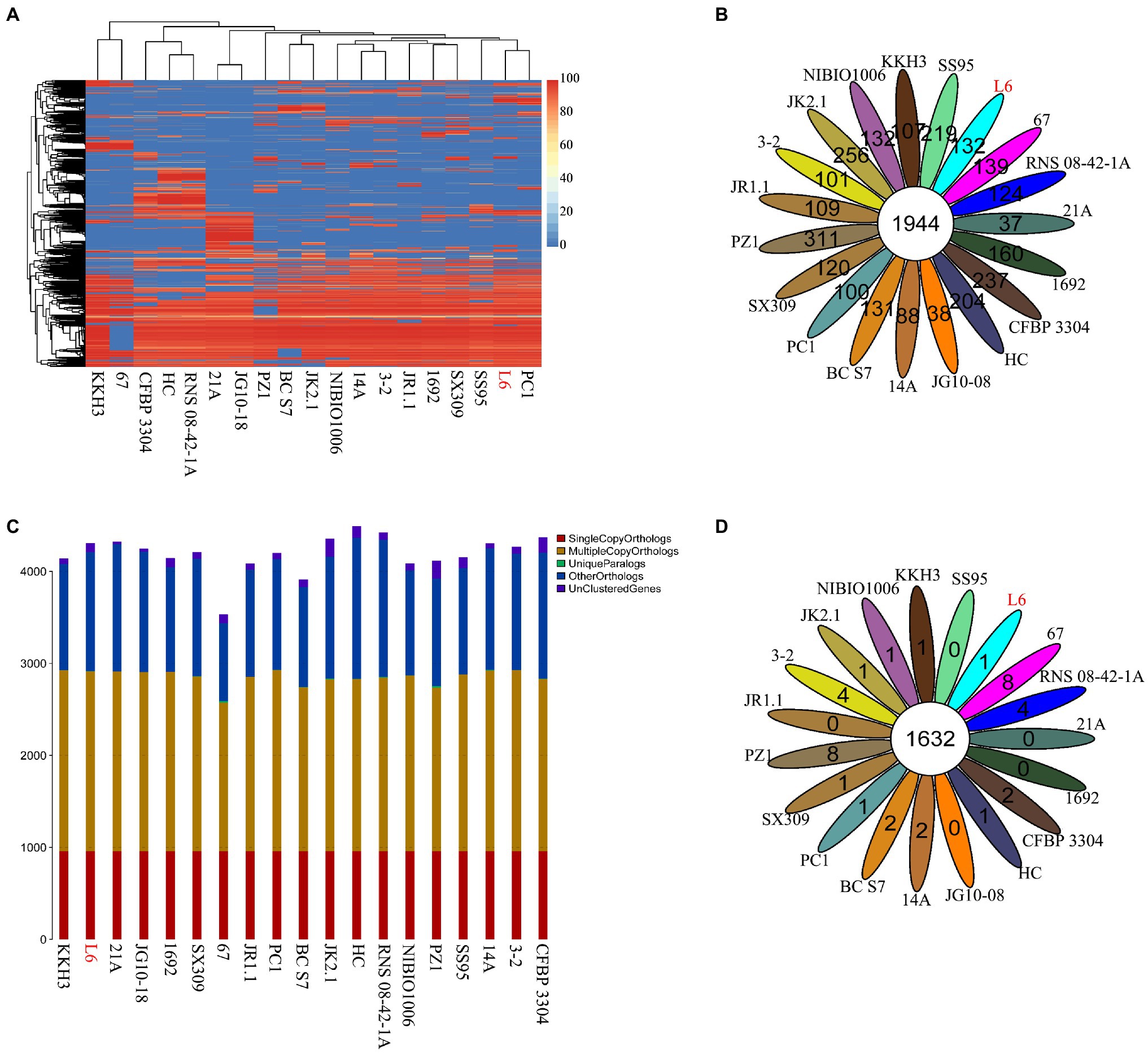
Figure 4. Core genes and dispensable genes and orthologs in the P. aroidearum L6 genome and reference strain genomes. (A) Dispensable gene heat map (left, dispensable gene cluster; top, strain cluster; red gradient bar represents the scale of similarity percentage); (B) Venn diagram of pan genes (each ellipse represents one strain, and the number in the ellipse is the cluster number. One cluster has genes of >50% identity and <0.3 length diversity); (C) Ortholog number (Single Copy Orthologs, the number of single-copy homologous genes in the species common gene families; Multiple Copy Orthologs, the number of multiple-copy homologous genes in the species common gene families; Unique Paralogs, genes in specific gene families; Other Orthologs, other genes; and Unclustered Genes, genes that have not been clustered into any families); (D) Venn diagram of orthologs in gene family (each ellipse represents one strain, and the number in the ellipse is the family number).
Analysis of Gene Function Annotation of P. aroidearum L6
To further determine the difference in functions encoded by 4,306 genes of P. aroidearum L6, we analyzed the data using GO, COG, and KEGG. A total of 3,013 (65.59%) genes could be annotated to one or more of the GO definitions. In our study, 6,763 genes were annotated to biological processes, 2,441 to cellular components, and 3,691 to molecular functions in GO analysis (Figure 5A). Most gene functions focused on cellular process (1,640), metabolic process (1,678), single-organism process (1,411), membrane (772), binding (1,231), and catalytic activity (1,601). There were 3,625 (78.92%) predicted genes assigned to COG categories (Figure 5B): 43.78% (1,871) of the genes were related to metabolism, 26.11% (1,116) to cellular processes, 17.92% (766) to information, and 12.19% (521) to poorly. A total of 3,081 genes were annotated using the KEGG database (Figure 5C). Among the categories, metabolism was the largest group, containing metabolic pathway (690 genes, 22.40%), biosynthesis of secondary metabolites (321 genes, 10.42%), biosynthesis of antibiotics (234 genes, 7.59%), microbial metabolism in diverse environments (229 genes, 7.43%), and others. The cluster of environmental information processing primarily consisted of ABC transporters (279 genes, 9.06%) and two-component system (173 genes, 5.62%).
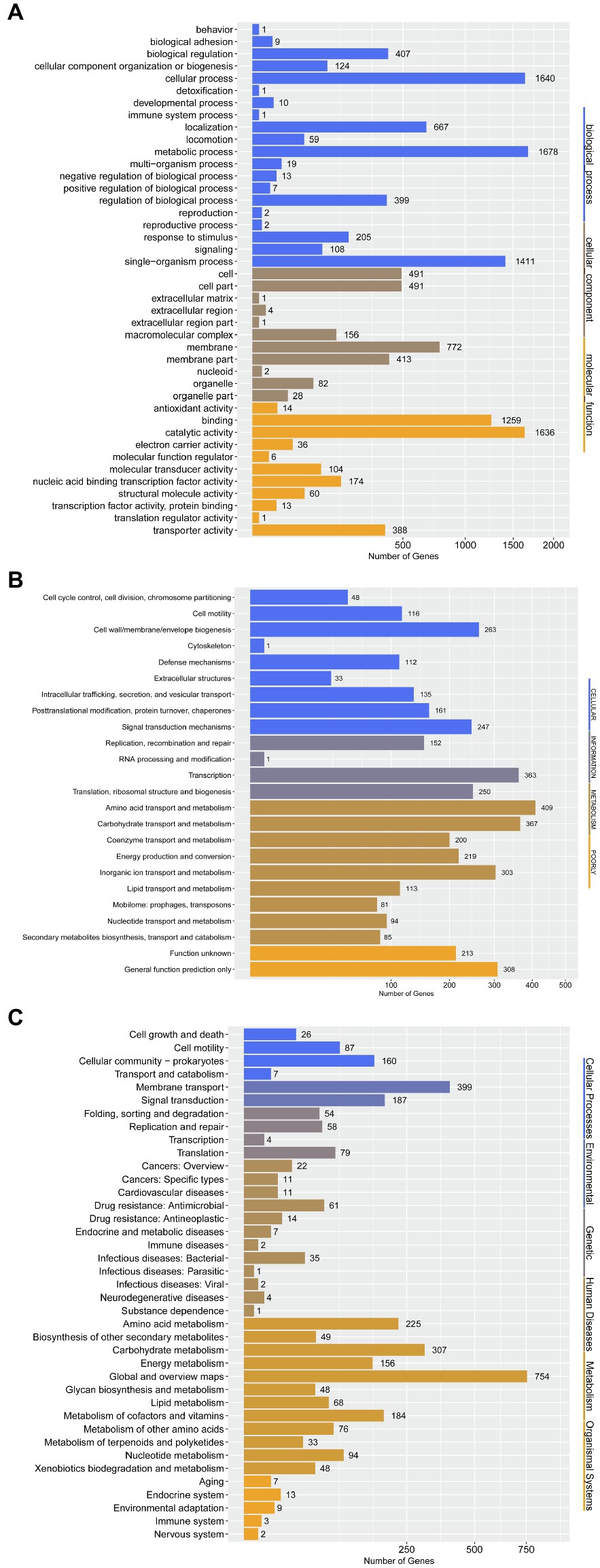
Figure 5. Gene annotation by GO, COG, and KEGG for P. aroidearum L6. (A) GO function classification of genes in L6. GO analysis was performed for three main categories: cellular components, molecular function, and biological processes. (B) COG function classification of genes in L6, grouped into four main parts: metabolism, cellular processes, information, and poorly. (C) The KEGG pathway classification of genes in L6 contains six groups: cellular processes, environmental, genetic, human diseases, metabolism, and organismal systems.
Pathogenic Candidate Genes Obtained Through Gene Annotation Screening
We predicted genes associated with pathogenicity using GO, COG, KEGG, and especially VFDB, PHI, and T3SS, which are important databases for predicting bacterial pathogenicity. The VFDB database mainly focuses on the infectious agents among bacteria, mycoplasma, and chlamydia. A total of 478 (10.40%) genes were annotated to the VFDB definitions. The PHI database contains relationships between pathogens and hosts, and it predicted 432 (9.40%) genes. The T3SS has close relationship with Gram-negative pathogens and aids in determining infection mechanisms and toxicity at the molecular level. There were 723 (15.74%) predicted genes assigned to T3SS categories. Moreover, we screened the relevant genes encoding plant cell-wall degrading enzymes (PCWDEs), toxins, and secretion systems for P. aroidearum L6 (Table 3). There were 25 genes related to toxins, and 35 genes encoded PCWDEs, including 28 encoding pectinases, three encoding cellulases, and four encoding proteinases. In addition, 122 genes were involved in six types of secretion systems: 14 genes in Type I (T1SS), 23 in Type II (T2SS), 29 in Type III (T3SS), 25 in Type IV (T4SS), none in Type V (T5SS), and 31 in Type VI (T6SS) secretion systems.

Table 3. Pathogenic candidate genes of P. aroidearum L6 identified through gene annotation screening.
Discussion
Plant bacterial soft rot is one of the destructive diseases of cabbage, tomato, and potato (Meng et al., 2017; Wang et al., 2017; Cui et al., 2019). And it always can cause more serious losses than any other bacterial disease (Qi et al., 2021). Most of the soft rot disease of vegetables is caused by Pectobacterium spp., one of the top ten bacterial plant pathogens. Pectobacterium spp. has attracted more attention about its wide distribution and diversity (He et al., 2021). Pectobacterium usually exists in soils with a broad range of hosts; thus, Pectobacterium species cause soft rot disease in plants of at least 16 dicotyledonous and 11 monocotyledonous angiosperm families (Ma et al., 2007; Nabhan et al., 2013). Pectobacterium aroidearum was classified as a novel species in 2013 (Nabhan et al., 2013). In previous studies, bacterial soft rot disease was caused by P. aroidearum in calla (Zantedeschia aethiopica; Nabhan et al., 2013), potato (Solanum tuberosum; Moretti et al., 2016), Chinese cabbage (Brassica rapa; Xie et al., 2017), zucchini (Cucurbita pepo; Moraes et al., 2017), konjac (Amorphophallus konjac; Sun et al., 2019), pepper (Capsicum annuum; Moraes et al., 2020), and carrot (Daucus carota; Tang et al., 2021). In our previous research, P. aroidearum as a pathogen on S. podophyllum was found in China (Xu et al., 2020). It is important to prevent the spread of this pathogen because many ornamental and edible plant species are susceptible to P. aroidearum.
The classification of the genus Pectobacterium has been subject to wide revision over the last decade. Pectobacterium spp. are highly phenotypically, genetically, and pathogenically particularly heterogeneous, indicating a need for re-evaluation and a better classification of these species (Dees et al., 2017). Three subspecies of P. carotovorum were reclassified as one subspecies (P. carotovorum subsp. carotovorum), and P. carotovorum subsp. odoriferum and P. carotovorum subsp. brasiliense were reclassified as P. odoriferum and P. brasiliense, respectively, based on genomics (Liu and Filiatrault, 2020; Liu et al., 2020). A lot of genomes of Pectobacterium have been sequenced, annotated, and analyzed previously (Oulghazi et al., 2020; Pedersen et al., 2020; Jonkheer et al., 2021). But there was no information about the whole genome of P. aroidearum-type strain SCRI 109. At present, only strain PC1 of P. aroidearum has sequenced its whole genome (PC1 formerly classified as P. carotovorum subsp. carotovorum, the classification has not been corrected in the NCBI database; He et al., 2021). To understand the molecular mechanisms of taxonomy and pathogenic factors in P. aroidearum, the whole genome of L6 was successively sequenced in this study. And it will be the first public report on the genome of P. roidearum.
Comparison of genomes is an efficient method for classification and detection of bacterial and fungal pathogens (Pritchard et al., 2012; Bühlmann et al., 2013; Malapi-Wight et al., 2016; Tang et al., 2017; Van Dam et al., 2018). Zoledowska et al. (2018) researched the comparative genomic of 15 P. parmentieri strains and found the high genomic variation among P. parmentieri strains. Pédron et al. (2019) established the taxonomic status of six Pectobacterium strains based on phylogenetic data, ANI values, and isDDH results by comparative genomics and identified a novel species of the genus Pectobacterium named Pectobacterium aquaticum. He et al. (2021) analyzed comparative genomics of four Pectobacterium strains and obtained three kinds of highly conserved key pathogenic genes related to cell-wall degrading enzymes in Pectobacterium strain PC1, including 19 pectinase genes, 25 cellulase genes, and 22 protease genes. Zhang et al. (2016) compared 85 genomes of the genera Dickeya and Pectobacterium and found that at least ten tested genomes from these genera were misnamed in GenBank based on ANI, isDDH, and whole genome. In our study, L6 and PC1 were grouped in one population distinct from other populations of the Pectobacterium genus and we also found some strains were misnamed in GenBank. (P. carotovorum subsp. carotovorum JR1.1 and 67 were not the same species.) It is effective for re-evaluating current prokaryotic species definition and establishing a unified prokaryotic species definition frame by using whole-genome sequences for taxonomically challenging genera (Zhang et al., 2016; Qi et al., 2021).
Currently, once plants are infected, there is no effective method to control bacterial soft rot (Sun et al., 2019). It is also very possible to develop new control methods. By screening pathogenic genes based on whole-genome sequences of Pectobacterium species and analyzing the pathogenic mechanism at the molecular level, Zhang et al. (2017) discovered a total of 168 genes related to pathogenesis including nine specific genes encoding toxins on the genome of P. atroseptica JG10-08. Huang et al. (2019) selected five putative effectors from the genome of P. carotovorum subsp. brasiliense BZA12 and discovered that candidate effector A12GL002483 was localized in the cell nucleus and induced cell death. We discovered 478 genes, 10.4% of total predicted genes, that were potentially related to pathogenesis according to the VFDB database. Previous research has shown that soft rot pathogenesis basically relies on toxins, PCWDEs, and the secretion system. Toxins play a key role in the pathogenicity of Pectobacterium species. We discovered 29 genes related to toxins in P. aroidearum L6. Moreover, PCWDEs are crucial in three distinct pathogenic functions: degradation, nutrition, and feedback regulation (Franza et al., 2002; Yang et al., 2007). The pathogens benefit from the nutrients produced after degradation; these degradation products accumulate in the host and can induce bacterium to generate more enzymes (Zhang et al., 2017). Therefore, the production of PCWDEs is characteristic of infection by Pectobacterium species. The PCWDEs consist of pectinases, cellulases, and proteinases. In this study, we identified a total of 36 genes encoding PCWDEs: 29 encoding pectinases, three encoding cellulases, and four encoding proteinases.
In addition to toxins and PCWDEs, secretion systems play a critical role in plant bacterial disease development. There are six types of secretion systems to export extracellular enzymes and effector proteins in bacteria (Lory, 1998). Through secretion systems, effectors can be transported inside the plant cell and promote rapid infection of the host plant (Holst et al., 1996). Among them, the Hrp gene cluster encoding the T3SS is particular important in many Gram-negative pathogens; this is a multi-protein complex bacterial structure to deliver virulence effector proteins directly into plant cells (Tampakaki et al., 2004; Tam et al., 2007; Xie et al., 2019). In our study, there were 723 predicted genes assigned to T3SS categories. Furthermore, 122 genes were involved in the six types of secretion system in P. aroidearum L6 based on GO, COG, and KEGG: 14 in T1SS, 23 in T2SS, 29 in T3SS, 25 in T4SS, and 31 in T6SS. Pasanen et al. (2020) identified a novel species Pectobacterium parvum and found it contained SPI-1-type Type III secretion island by comparing between the genomes of Pectobacterium species. In the genome of Pseudomonas syringae, Kang et al. (2014) researched the role of T3SS effectors in the disruption of actin cytoskeleton and inhibition of endocytosis. In the genome of Shewanella sp., Alex and Antunes (2019) detected the genes encoding for T3SS core components and four copies of homologs of T3SS effector. Currently, the effectors of P. aroidearum pathogenesis have not been studied. All these genes in P. aroidearum L6 have potential virulence functions. Thus, further research on the pathogenic factors in L6 may reveal the mechanism of Pectobacterium species infection of plants.
Conclusion
The classification of the genus Pectobacterium has long been unclear. Pectobacterium spp. are highly phenotypically, genetically, and pathogenically particularly heterogeneous, can cause severe soft rot in plant hosts, and have a wide host range. Our results suggest that P. aroidearum L6 synthesizes and transports virulence factors. Moreover, 182 genes were involved in toxins, PCWDEs, and the secretion system. The results of this research will serve as a foundation for a better understanding of the genomic structure of P. aroidearum. The discovery of potential pathogenic factors can help in preventing spread and outbreak of this pathogen and providing effective biological measures against it.
Data Availability Statement
The datasets used and/or analyzed in the current study are available from the corresponding author on reasonable request. The genome sequence of P. aroidearum L6, including all assemblies and annotations, generated for this study is available at NCBI GenBank with accession number CP065044. The 16S ribosomal RNA sequence is available with accession code MT120309.
Author Contributions
WL and PX designed the experiments and wrote the manuscript. PX, HW, CQ, and ZL performed collection and bioinformatics analysis. CL and WM revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was financially supported by the Hainan Major Research Fund of Science and Technology (grant no. ZDKJ201817), the National Natural Science Foundation of China (grant nos. 31560495, 31760499, and 31660033), the National Natural Rubber Industry Technical System (grant no. CARS-34-GW8), the Opening Project Fund of Key Laboratory of Biology and Genetic Resources of Rubber Tree, Ministry of Agriculture and Rural Affairs, P. R. China (grant no. RRI-KLOF201903), and the Hainan Province Graduate Student Innovation Research Project (grant nos. Hyb2018-07 and Hyb2019-02).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1. ^https://www.bacterio.net/genus/pectobacterium
2. ^https://www.ncbi.nlm.nih.gov/genome/?term=Pectobacterium
3. ^https://github.com/PacificBiosciences/pbdagcon
4. ^https://www.broadinstitute.org/gatk/
5. ^http://www.ebi.ac.uk/genomes/plasmid.html; last accessed July 8, 2016
6. ^https://www.ncbi.nlm.nih.gov/genome/annotation_prok/
7. ^http://tandem.bu.edu/trf/trf.html
9. ^http://phast.wishartlab.com/
References
Adeolu, M., Alnajar, S., Naushad, S., and Gupta, R. S. (2016). Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceaefam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 66, 5575–5599. doi: 10.1099/ijsem.0.001485
Alex, A., and Antunes, A. (2019). Whole-genome comparisons among the genus Shewanella reveal the enrichment of genes encoding ankyrin-repeats containing proteins in sponge-associated bacteria. Front. Microbiol. 10:5. doi: 10.3389/fmicb.2019.00005
Apweiler, R., Bairoch, A., Wu, C. H., Barker, W. C., Boeckmann, B., Ferro, S., et al. (2004). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32, D115–D119. doi: 10.1093/nar/gkh131
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Brenner, D. J., Steigerwalt, A. G., Miklos, G. V., and Fanning, G. R. (1973). Deoxyribonucleic acid relatedness among Erwiniae and other Enterobacteriaceae: the soft-rot organisms (genus Pectobacterium Waldee). Int. J. Syst. Bacteriol. 23, 205–216. doi: 10.1099/00207713-23-3-205
Bühlmann, A., Pothier, J. F., Tomlinson, J. A., Frey, J. E., Boonham, N., Smits, T. H. M., et al. (2013). Genomics-informed design of loop-mediated isothermal amplification for detection of phytopathogenic Xanthomonas arboricola pv. pruni at the intraspecific level. Plant Pathol. 62, 475–484. doi: 10.1111/j.1365-3059.2012.02654.x
Charkowski, A., Blanco, C., Condemine, G., Expert, D., Franza, T., Hayes, C., et al. (2012). The role of secretion systems and small molecules in soft-rot Enterobacteriaceae pathogenicity. Annu. Rev. Phytopathol. 50, 425–449. doi: 10.1146/annurev-phyto-081211-173013
Chen, L. H., Zheng, D. D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis-10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Coenye, T., and Vandamme, P. (2010). Intragenomic heterogeneity between multiple 16S ribosomal RNA operons in sequenced bacterial genomes. FEMS Microbiol. Lett. 228, 45–49. doi: 10.1016/S0378-1097(03)00717-1
Cui, W., He, P., Munir, S., He, P., He, Y., Li, X., et al. (2019). Biocontrol of soft rot of chinese cabbage using an endophytic bacterial strain. Front. Microbiol. 10:1471. doi: 10.3389/fmicb.2019.01471
Dees, M. W., Lysøe, E., Rossmann, S., Perminow, J., and Brurberg, M. B. (2017). Pectobacterium polaris sp. nov., isolated from potato (Solanum tuberosum). Int. J. Syst. Evol. Microbiol. 67, 5222–5229. doi: 10.1099/ijsem.0.002448
Duarte, V., de Boer, S. H., Ward, L. J., and de Oliveira, A. M. R. (2004). Characterization of atypical Erwinia carotovora strains causing blackleg of potato in Brazil. J. Appl. Microbiol. 96, 535–545. doi: 10.1111/j.1365-2672.2004.02173.x
Edgar, R. C. (2004a). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar, R. C. (2004b). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 5:113. doi: 10.1186/1471-2105-5-113
Farris, J. S. (1972). Estimating phylogenetic trees from distance matrices. Am. Nat. 106, 645–668. doi: 10.2307/2459725
Franza, T., Michaud-Soret, I., Piquerel, P., and Expert, D. (2002). Coupling of iron assimilation and pectinolysis in Erwinia chrysanthemi 3937. Mol. Plant-Microbe Interact. 15, 1181–1191. doi: 10.1094/MPMI.2002.15.11.1181
Fu, L., Niu, B., Zhu, Z., Zhu, Z. W., Wu, S., and Li, W. Z. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Eugene, V. K. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Gardan, L., Gouy, C., Christen, R., and Samson, R. (2003). Elevation of three subspecies of Pectobacterium carotovorum to species level: Pectobacterium atrosepticum sp. nov., Pectobacterium betavasculorum sp. nov. and Pectobacterium wasabiae sp. nov. Int. J. Syst. Evol. Microbiol. 53, 381–391. doi: 10.1099/ijs.0.02423-0
Gardner, P. P., Daub, J., Tate, J. G., Nawrocki, E. P., Kolbe, D. L., Lindgreen, S., et al. (2009). Rfam: updates to the RNA families database. Nucleic Acids Res. 37, D136–D140. doi: 10.1093/nar/gkn766
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Haft, D. H., DiCuccio, M., Badretdin, A., Brover, V., Chetvernin, V., O'Neill, K., et al. (2018). RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 46, D851–D860. doi: 10.1093/nar/gkx1068
Hasan, M. S., Liu, Q., Wang, H., Fazekas, J., Chen, B., and Che, D. S. (2012). GIST: genomic island suite of tools for predicting genomic islands in genomic sequences. Bioinformation 8, 203–205. doi: 10.6026/97320630008203
Hauben, L., Moore, E. R., Vauterin, L., Steenackers, M., Mergaert, J., Verdonck, L., et al. (1998). Phylogenetic position of phytopathogens within the Enterobacteriaceae. Syst. Appl. Microbiol. 21, 384–397. doi: 10.1016/S0723-2020(98)80048-9
He, X., Lu, T., and Zhou, X. (2021). Whole genome sequencing and comparative genomics analysis of Pectobacterium carotovorum identifies key pathogenic genes, molecular phylogenetics and evolution. Mol. Phylogenet. Evol. 17:107114. doi: 10.1016/j.ympev.2021.107114
Holst, O., Ulmer, A. J., Brade, H., Flad, H. D., and Rietschel, E. T. (1996). Biochemistry and cell biology of bacterial endotoxins. FEMS Immunol. Med. Microbiol. 16, 83–104. doi: 10.1111/j.1574-695X.1996.tb00126.x
Huang, Y. F., Liu, C. Y., Wang, H., Guan, T. S., Liu, L., and Yu, S. Y. (2019). Bioinformatic analysis of the complete genome sequence of Pectobacterium carotovorum subsp. brasiliense BZA12 and candidate effector screening. J. Plant Path. 101, 39–49. doi: 10.1007/s42161-018-0126-7
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. D1, D286–D293. doi: 10.1093/nar/gkv1248
Jonkheer, E. M., Balázs Br ankovics, Houwers, I. M., Wolf, J. M. V. D., and Lee, T. A. J. V. D. (2021). The pectobacterium pangenome, with a focus on pectobacterium brasiliense, shows a robust core and extensive exchange of genes from a shared gene pool. BMC Genomics 22:265. doi: 10.1186/s12864-021-07583-5
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kang, Y., Jelenska, J., Cecchini, N. M., Li, Y., Lee, M. W., Kovar, D. R., et al. (2014). HopW1 from Pseudomonas syringae disrupts the actin cytoskeleton to promote virulence in Arabidopsis. PLoS Pathog. 10:e1004232. doi: 10.1371/journal.ppat.1004232
Khayi, S., Cigna, J., Chong, T. M., Quêtu-Laurent, A., Chan, K., Hélias, V., et al. (2016). Transfer of the potato plant isolates of Pectobacterium wasabiae to Pectobacterium parmentieri sp. nov. Int. J. Syst. Evol. Microbiol. 66, 5379–5383. doi: 10.1099/ijsem.0.001524
Koh, Y. J., Kim, G. H., Lee, Y. S., Sohn, S. H., Koh, H. S., Kwon, S., et al. (2012). Pectobacterium carotovorum subsp. actinidiae subsp. nov. a new bacterial pathogen causing canker-like symptoms in yellow kiwifruit, Actinidia chinensis. N. Z. J. Crop Hortic. Sci. 40, 269–279. doi: 10.1080/01140671.2012.707129
Kreft, L., Botzki, A., Coppens, F., Vandepoele, K., and Van, B. M. (2017). PhyD3: a phylogenetic tree viewer with extended phyloXML support for functional genomics data visualization. Bioinformatics 33, 2946–2947. doi: 10.1093/bioinformatics/btx324
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lefort, V., Desper, R., and Gascuel, O. (2015). FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32, 2798–2800. doi: 10.1093/molbev/msv150
Levasseur, A., Drula, E., Lombard, V., Coutinho, P. M., and Henrissat, B. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels. 6:41. doi: 10.1186/1754-6834-6-41
Li, S. T., Li, R. Q., Li, H., Lu, J. L., Li, Y. R., Bolund, L., et al. (2013). SOAPindel: efficient identification of indels from short paired reads. Genome Res. 23, 195–200. doi: 10.1101/gr.132480.111
Li, W. J., O’Neill, K. R., Haft, D. H., DiCuccio, M., Chetvernin, V., Badretdin, A., et al. (2021). RefSeq: expanding the prokaryotic genome annotation pipeline reach with protein family model curation. Nucleic Acids Res. 49, D1020–D1028. doi: 10.1093/nar/gkaa1105
Li, L., Yuan, L. F., Shi, Y. X., Xie, X. W., Chai, A., Wang, Q., et al. (2019). Comparative genomic analysis of Pectobacterium carotovorum subsp. brasiliense SX309 provides novel insights into its genetic and phenotypic features. BMC Genomics 20:486. doi: 10.1186/s12864-019-5831-x
Liu, Y. Y., and Filiatrault, M. J. (2020). Complete genome sequence of the necrotrophic plant-pathogenic bacterium Pectobacterium brasiliense 1692. Microbiol. Resour. Announc. 9:e00037-20. doi: 10.1128/MRA.00037-20
Liu, Y. Y., Helmann, T., Stodghill, P., and Filiatrault, M. (2020). Complete genome sequence resource for the necrotrophic plant-pathogenic bacterium Pectobacterium carotovorum WPP14. Plant Dis. 105, 196–198. doi: 10.1094/PDIS-05-20-1059-A
Liu, B., and Pop, M. (2009). ARDB-antibiotic resistance genes database. Nucleic Acids Res. 37, D443–D447. doi: 10.1093/nar/gkn656
Lory, S. (1998). Secretion of proteins and assembly of bacterial surface organelles: shared pathways of extracellular protein targeting. Curr. Opin. Microbiol. 1, 27–35. doi: 10.1016/s1369-5274(98)80139-2
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Ma, B., Hibbing, M. E., Kim, H. S., Reedy, R. M., Yedidia, I., and Breuer, J. (2007). Host range and molecular phylogenies of the soft rot Enterobacterial genera Pectobacterium and Dickeya. Phytopathology 97, 1150–1163. doi: 10.1094/PHYTO-97-9-1150
Makarova, K., Wolf, Y., and Koonin, E. (2015). Archaeal clusters of orthologous genes (arCOGs): an update and application for analysis of shared features between Thermococcales, Methanococcales, and Methanobacteriales. Life 5, 818–840. doi: 10.3390/life5010818
Malapi-Wight, M., Demers, J. E., Veltri, D., Marra, R. E., and Crouch, J. A. (2016). LAMP detection assays for boxwood blight pathogens: a comparative genomics approach. Sci. Rep. 6:26140. doi: 10.1038/srep26140
Mansfield, J., Genin, S., Magori, S., Citovsky, V., Sriariyanum, M., Ronald, M., et al. (2012). Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 13, 614–629. doi: 10.1111/j.1364-3703.2012.00804.x
Maqbool, O., and Babri, H. (2007). Hierarchical clustering for software architecture recovery. IEEE Trans. Softw. Eng. 33, 759–780. doi: 10.1109/TSE.2007.70732
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi: 10.14806/ej.17.1.200
Mckenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., and Göker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10:2182. doi: 10.1038/s41467-019-10210-3
Meng, X. L., Chai, A., Shi, Y. X., Xie, X. W., Ma, Z. H., and Li, B. J. (2017). Emergence of bacterial soft rot in cucumber caused by Pectobacterium carotovorum subsp. brasiliense in China. Plant Dis. 101, 279–287. doi: 10.1094/PDIS-05-16-0763-RE
Moraes, A. J. G., Baia, A. D. B., Souza, E. B., Peixoto, A. R., and Gama, M. A. S. (2020). First report of Pectobacterium aroidearum causing soft rot of pepper (Capsicum annuum) fruits in Brazil. Plant Dis. 104:3054. doi: 10.1094/PDIS-02-20-0403-PDN
Moraes, A. J. G., Souza, E. B., Mariano, R. L. R., Silva, A. M. F., Lima, N. B., Peixoto, A. R., et al. (2017). First report of Pectobacterium aroidearum and Pectobacterium carotovorum subsp. brasiliensis causing soft rot of Cucurbita pepo in Brazil. Plant Dis. 101, 379–380. doi: 10.1094/PDIS-08-16-1168-PDN
Moretti, C., Fakhr, R., Cortese, C., De, V. P., Cerri, M., Geagea, L., et al. (2016). Pectobacterium aroidearum and Pectobacterium carotovorum subsp. carotovorumas causal agents of potato soft rot in Lebanon. Eur. J. Plant Pathol. 144, 205–211. doi: 10.1007/s10658-015-0743-3
Myers, E. W., Sutton, G. G., Delcher, A. L., Dew, I. M., Fasulo, D. P., Flanigan, M. J., et al. (2000). A whole-genome assembly of Drosophila. Science 287, 2196–2204. doi: 10.1126/science.287.5461.2196
Nabhan, S., De Boer, S. H., Maiss, E., and Wydra, K. (2013). Pectobacterium aroidearum sp. nov. a soft rot pathogen with preference for monocotyledonous plants. Int. J. Syst. Evol. Microbiol. 63, 2520–2525. doi: 10.1099/ijs.0.046011-0
Nandi, T., Ong, C., Singh, A. P., Justin, B., Timothy, A., Mitali, S. T., et al. (2010). A genomic survey of positive selection in Burkholderia pseudomallei provides insights into the evolution of accidental virulence. PLoS Pathog. 6:e1000845. doi: 10.1371/journal.ppat.1000845
Nykyri, J., Niemi, O., Koskinen, P., Nokso-Koivisto, J., Pasanen, M., Broberg, M., et al. (2012). Revised phylogeny and novel horizontally acquired virulence determinants of the model soft rot phytopathogen pectobacterium wasabiae scc3193. PLoS Pathog. 8:e1003013. doi: 10.1371/journal.ppat.1003013
Oulghazi, S., Cigna, J., Lau, Y. Y., Moumni, M., Chan, K. G., and Faure, D. (2019). Transfer of the waterfall source isolate Pectobacterium carotovorum M022 to Pectobacterium fontis sp. nov., a deep-branching species within the genus Pectobacterium. Int. J. Syst. Evol. Microbiol. 69, 470–475. doi: 10.1099/ijsem.0.003180
Oulghazi, S., Moumni, M., Khayi, S., Robic, K., and Faure, D. (2020). Diversity of Pectobacteriaceae species in potato growing regions in northern Morocco. Microorganisms 8:895. doi: 10.3390/microorganisms8060895
Parte, A. C., Sardà Carbasse, J., Meier-Kolthoff, J. P., Reimer, L. C., and Göker, M. (2020). List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Pasanen, M., Waleron, M., Schott, T., Cleenwerck, I., Misztak, A., Waleron, K., et al. (2020). Pectobacterium parvum sp. nov., having a Salmonella SPI-1-like type III secretion system and low virulence. Int. J. Syst. Evol. Microbiol. 70, 2440–2448. doi: 10.1099/ijsem.0.004057
Pedersen, J. S., Carstens, A. B., Djurhuus, A. M., Kot, W., and Hansen, L. H. (2020). Isolation and complete genome sequencing of Pectobacterium phage Jarilo, representing a novel genus of bacteriophages within the subfamily Autographivirinae. bioRxiv [Preprint]. doi: 10.1101/2020.01.16.909176
Pédron, J., Claire, B., Taghouti, G., Portier, P., and Barny, M.-A. (2019). Pectobacterium aquaticum sp. nov., isolated from waterways. Int. J. Syst. Evol. Microbiol. 69, 745–751. doi: 10.1099/ijsem.0.003229
Pérombelon, M. C. M. (2002). Potato diseases caused by soft rot erwinias: an overview of pathogenesis. Plant Pathol. 51, 1–12. doi: 10.1046/j.0032-0862.2001.Shorttitle.doc.x
Philip, J., David, B., Chang, H. Y., Fraser, M., Li, W. Z., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Portier, P., Pedron, J., Taghouti, G., Fischer-Le Saux, M., Caullireau, E., Bertrand, C., et al. (2019). Elevation of Pectobacterium carotovorum subsp. odoriferum to species level as Pectobacterium odoriferum sp. nov., proposal of Pectobacterium brasiliense sp. nov. and Pectobacterium actinidiae sp. nov., emended description of Pectobacterium carotovorum and description of Pectobacterium versatile sp. nov., isolated from streams and symptoms on diverse plants. Genomics Taxon. 69, 3214–3223. doi: 10.1099/ijsem.0.003611
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods. 8, 12–24. doi: 10.1039/c5ay02550h
Pritchard, L., Humphris, S., Saddler, G. S., Parkinson, N. M., Bertrand, V., Elphinstone, J. G., et al. (2012). Detection of phytopathogens of the genus Dickeya using a PCR primer prediction pipeline for draft bacterial genome sequences. Plant Pathol. 62, 587–596. doi: 10.1111/j.1365-3059.2012.02678.x
Qi, T., Wang, S., Deng, L. L., Yi, L. H., and Zeng, K. F. (2021). Controlling pepper soft rot by Lactobacillus paracasei WX322 and identification of multiple bacteriocins by complete genome sequencing. Food Control. 121:107629. doi: 10.1016/j.foodcont.2020.107629
Richter, M., Rosselló-Móra, R., Glöckner, F. O., and Peplies, J. (2015). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Sarfraz, S., Oulghazi, S., Cigna, J., Sahi, S. T., Riaz, K., Tufail, M. R., et al. (2020). Complete genome sequence of the type strain Pectobacterium punjabense SS95, isolated from a potato plant with blackleg symptoms. Microbiol. Res. Announc. 9:e00420. doi: 10.1128/MRA.00420-20
Sarfraz, S., Riaz, K., Oulghazi, S., Cigna, J., Sahi, S. T., Khan, S. H., et al. (2018). Pectobacterium punjabense sp. nov., isolated from blackleg symptoms of potato plants in Pakistan. Int. J. Syst. Evol. Microbiol. 68, 3551–3556. doi: 10.1099/ijsem.0.003029
Sun, M. M., Liu, H., Huang, J. B., Peng, J. B., Fei, F. H., Zhang, Y., et al. (2019). A loop-mediated isothermal amplification assay for rapid detection of Pectobacterium aroidearum that causes soft rot in konjac. Int. J. Mol. Sci. 20:1937. doi: 10.3390/ijms20081937
Tam, V. C., Serruto, D., Dziejman, M., Brieher, W., and Mekalanos, J. J. (2007). A type III secretion system in Vibrio cholerae translocates a formin/spire hybrid-like actin nucleator to promote intestinal colonization. Cell Host Microbe 1, 95–107. doi: 10.1016/j.chom.2007.03.005
Tampakaki, A. P., Fadouloglou, V. E., Gazi, A. D., Panopoulos, N. J., and Kokkinidis, M. (2004). Conserved features of type III secretion. Cell. Microbiol. 6, 805–816. doi: 10.1111/j.1462-5822.2004.00432.x
Tang, W. Q., Chang, C. Y., Lee, Y. J., and Chu, C. C. (2021). First report of Pectobacterium aroidearum causing bacterial soft rot of carrot in Taiwan. Plant Dis. 105:695. doi: 10.1094/PDIS-08-20-1824-PDN
Tang, J. T., Zheng, L., Jia, Q., Liu, H., Hsiang, T., and Huang, J. B. (2017). PCR markers derived from comparative genomics for detection and identification of the rice pathogen Ustilaginoidea virens in plant tissues. Plant Dis. 101, 1515–1521. doi: 10.1094/PDIS-08-16-1088-RE
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Toth, I. K., Bell, K. S., Holeva, M. C., and Birch, P. R. J. (2003). Soft rot erwiniae: from genes to genomes. Mol. Plant Pathol. 4, 17–30. doi: 10.1046/j.1364-3703.2003.00149.x
Toth, I., Humphris, S., Campbell, E., and Pritchard, L. (2015). Why genomics research on Pectobacterium and Dickeya makes a difference. Am. J. Potato Res. 92, 218–222. doi: 10.1007/s12230-015-9446-8
Toth, I. K., van der Wolf, J. M., Saddler, G., Lojkowska, E., Helias, V., Pirhonen, M., et al. (2011). Dickeya species: an emerging problem for potato production in Europe. Plant Pathol. 60, 385–399. doi: 10.1111/j.1365-3059.2011.02427.x
UniProt Consortium (2015). UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. doi: 10.1093/nar/gku989
Van Dam, P., de Sain, M., ter Horst, A., van der Gragt, M., and Rep, M. (2018). Use of comparative genomics-based markers for discrimination of host specificity in Fusarium oxysporum. Appl. Environ. Microbiol. 84:e01868–17. doi: 10.1128/AEM.01868-17
Vargas, W. A., Martin, J. M. S., Rech, G. E., Rivera, L. P., Benito, E. P., Diaz-Minguez, J. M., et al. (2012). Plant defense mechanisms are activated during biotrophic and necrotrophic development of Colletotricum graminicola in maize. Plant Physiol. 158, 1342–1358. doi: 10.1104/pp.111.190397
Waldee, E. L. (1945). Comparative studies of some peritrichous phytopathogenic bacteria. Iowa State J. Sci. 19, 435–484.
Waleron, M., Misztak, A., Waleron, M., Franczuk, M., Jonca, J., Wielgomas, B., et al. (2019). Pectobacterium zantedeschiae sp. nov. a new species of a soft rot pathogen isolated from Calla lily (Zantedeschia spp.). Syst. Appl. Microbiol. 42:153. doi: 10.1016/j.syapm.2018.08.004
Waleron, M., Misztak, A., Waleron, M., Franczuk, M., Wielgomas, B., and Waleron, K. (2018). Transfer of Pectobacterium carotovorum subsp. carotovorum strains isolated from potatoes grown at high altitudes to Pectobacterium peruviense sp. nov. Syst. Appl. Microbiol. 41, 85–93. doi: 10.1016/j.syapm.2017.11.005
Wang, L., L, X. B., Suo, H. C., An, K., Luo, H. M., and Liu, X. J. (2017). Soft rot of potatoes caused by Bacillus amyloliquefaciens in Guangdong province China. Can. J. Plant pathol. 39, 533–539. doi: 10.1080/07060661.2017.1381994
Winnenburg, R., Baldwin, T. K., Urban, M., Rawlings, C., Köhler, J., Hammond-Kosack, K. E., et al. (2006). PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 34, D459–D464. doi: 10.1093/nar/gkj047
Wirawan, A., Kwoh, C. K., Hsu, L. Y., and Koh, T. H. (2010). “INVERTER: integrated variable number tandem repeat finder,” in Computational Systems-Biology and Bioinformatics. eds. J. H. Chan, Y.-S. Ong, and S.-B. Cho (Berlin: Springer), 151–164.
Xie, H., Li, X. Y., Ma, Y. L., and Tian, Y. (2017). First report of Pectobacterium aroidearum causing soft rot of Chinese cabbage in China. Plant Dis. 102:674. doi: 10.1094/PDIS-07-17-1059-PDN
Xie, Y. P., Shao, X. L., and Deng, X. (2019). Regulation of type III secretion system in Pseudomonas syringae. Environ. Microbiol. 21, 4465–4477. doi: 10.1111/1462-2920.14779
Xu, P. D., Wei, D. D., Li, Z. P., Qin, C. X., Li, X., Lin, C. H., et al. (2020). First report of bacterial soft rot on Syngonium podophyllum caused by Pectobacterium aroidearum in China. Plant Dis. 104:2720. doi: 10.1094/PDIS-03-20-0552-PDN
Yaganza, E. S., Tweddell, R. J., and Arul, J. (2014). Postharvest application of organic and inorganic salts to control potato (Solanum tuberosum L.) storage soft rot: plant tissue-salt physicochemical interactions. J. Agric. Food Chem. 62, 9223–9231. doi: 10.1021/jf5017863
Yang, J. K., Tian, B. Y., Liang, L. L. M., and Zhang, K. Q. (2007). Extracellular enzymes and the pathogenesis of nematophagous fungi. Appl. Microbiol. Biotechnol. 75, 21–31. doi: 10.1007/s00253-007-0881-4
Zhang, Y., Fan, Q., and Loria, R. (2016). A re-evaluation of the taxonomy of phytopathogenic genera Dickeya and Pectobacterium using whole genome sequencing data. Syst. Appl. Microbiol. 39, 252–259. doi: 10.1016/j.syapm.2016.04.001
Zhang, D., Zhou, Y., Zhao, D. M., Zhou, J. M., Yang, Z. H., and Zhu, M. M. (2017). Complete genome sequence and pathogenic genes analysis of Pectobacterium atroseptica JG10-08. Genes Genom. 39, 945–955. doi: 10.1007/s13258-017-0559-y
Zhou, Y., Liang, Y. J., Lynch, K. H., Dennis, J. J., and Wishart, D. S. (2011). PHAST: a fast phage search tool. Nucleic Acids Res. 39(Suppl. 2), W347–W352. doi: 10.1093/nar/gkr485
Keywords: genome sequence, taxonomy, pathogenic gene, Pectobacterium aroidearum, comparative genomics
Citation: Xu P, Wang H, Qin C, Li Z, Lin C, Liu W and Miao W (2021) Analysis of the Taxonomy and Pathogenic Factors of Pectobacterium aroidearum L6 Using Whole-Genome Sequencing and Comparative Genomics. Front. Microbiol. 12:679102. doi: 10.3389/fmicb.2021.679102
Edited by:
Ralf Koebnik, Plant Health Institute, FranceReviewed by:
Mohammad Arif, University of Hawaii at Manoa, United StatesMelanie J. Filiatrault, United States Department of Agriculture, United States
Ewa Lojkowska, University of Gdansk, Poland
Copyright © 2021 Xu, Wang, Qin, Li, Lin, Liu and Miao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenbo Liu, c2F1Y2hlckBoYWluYW51LmVkdS5jbg==; Weiguo Miao, bWlhb0BoYWluYW51LmVkdS5jbg==
†These authors have contributed equally to this work