 Vojtěch Tláskal
Vojtěch Tláskal Petr Baldrian
Petr Baldrian- Laboratory of Environmental Microbiology, Institute of Microbiology of the Czech Academy of Sciences, Praha, Czechia
Deadwood decomposition is responsible for a significant amount of carbon (C) turnover in natural forests. While fresh deadwood contains mainly plant compounds and is extremely low in nitrogen (N), fungal biomass and N content increase during decomposition. Here, we examined 18 genome-sequenced bacterial strains representing the dominant deadwood taxa to assess their adaptations to C and N utilization in deadwood. Diverse gene sets for the efficient decomposition of plant and fungal cell wall biopolymers were found in Acidobacteria, Bacteroidetes, and Actinobacteria. In contrast to these groups, Alphaproteobacteria and Gammaproteobacteria contained fewer carbohydrate-active enzymes and depended either on low-molecular-mass C sources or on mycophagy. This group, however, showed rich gene complements for N2 fixation and nitrate/nitrite reduction—key assimilatory and dissimilatory steps in the deadwood N cycle. We show that N2 fixers can obtain C independently from either plant biopolymers or fungal biomass. The succession of bacteria on decomposing deadwood reflects their ability to cope with the changing quality of C-containing compounds and increasing N content.
Introduction
The decomposition of deadwood in forests is a complex process during which plant polymers such as cellulose, hemicellulose, and lignin are exploited by members of the microbial community. Since the deadwood microbiome is dominated by fungi, their cell wall constituents represent another important resource, especially in the late phases of decomposition. Deadwood is estimated to contain 8% of the total global forest carbon (C) stock (Pan et al., 2011; Martin et al., 2021), making it an important source of carbon. However, the degree of the recalcitrance of C in deadwood components varies considerably and changes over time with the progress of decomposition. Fungi and bacteria produce a wide array of enzymes to access complex C sources, typically termed carbohydrate-active enzymes (CAZymes, Berlemont and Martiny, 2015). C-source niche partitioning in the utilization of C compounds is observed among microbial taxa (Gallardo et al., 2021), as well as various levels of opportunism in C acquisition (Lladó et al., 2019), where released degradation products may be utilized by microbes incapable of decomposition without the need to invest in enzyme production (Berlemont and Martiny, 2013). In contrast to C, the initial nitrogen (N) content in deadwood is extremely low (typically <1%), which leads to strong N limitation of microbial growth and substrate colonization (Tláskal et al., 2017). N accumulates in deadwood during decomposition either due to transport via the mycelium of ectomycorrhizal and soil foraging fungi (Philpott et al., 2014) or via the activity of the diazotrophic bacterial community (Rinne et al., 2016; Tláskal et al., 2021a). Such an increase gradually alleviates N limitation, leading to a more favorable C:N ratio of the whole habitat (Weedon et al., 2009).
A previous study analyzing the decomposition of Fagus sylvatica deadwood through metatranscriptome analysis showed the major role of fungi in C release, as demonstrated by their dominance in CAZyme production (Tláskal et al., 2021a). Notably, even the composition of the bacterial community in deadwood is heavily affected by fungi due to their primary access to C resources (Odriozola et al., 2021). The effect of fungi on bacteria might be direct (such as through antibiosis) or indirect through substrate acidification, allowing for the selection of acid-tolerant bacterial taxa (Valášková et al., 2009; Christofides et al., 2019; Johnston et al., 2019). Fungal biomass formation also represents a nutritional basis for several bacteria (Brabcová et al., 2016; López-Mondéjar et al., 2018). Such mycophagous bacteria developed enzymes for chitin degradation to overcome N limitation by feeding on fungal biomass containing N-rich chitin and chitosan (Uroz et al., 2013; Brabcová et al., 2016; Starke et al., 2020). As a result of fungal modulation and changing conditions in the substrate, the composition and abundance of the bacterial community in deadwood change over time showing an increase of bacterial biomass and taxa succession toward community similar to that in soil (Rinta-Kanto et al., 2016; Tláskal et al., 2017). Among the factors influencing bacterial community composition in deadwood, the most often reported are pH, the C:N ratio, and water content (Folman et al., 2008; Hoppe et al., 2015; Tláskal et al., 2017; Moll et al., 2018; Mieszkin et al., 2021), but the biopolymer composition and fungal abundance are also important (Rinta-Kanto et al., 2016; Odriozola et al., 2021). Due to limited knowledge of the functional traits of deadwood bacteria, it is unclear how exactly these factors influence the members of the bacterial community or vice versa and which adaptations make bacterial species successful at various stages of deadwood decomposition.
While culture-independent methods may provide the best picture of bacterial community composition and gene expression, the characterization of isolates of dominant bacteria represents the best approach for the characterization of their phenotypes and traits (Lladó et al., 2016). The genome sequencing of bacterial isolates has helped distinguish the functional guilds of decomposers and opportunists within soil bacteria (Lladó et al., 2019). While metagenome analysis may help predict bacterial traits based on the construction of metagenome-assembled genomes (MAGs, Tláskal et al., 2021a), this approach has multiple limitations. MAGs are mostly incomplete, subject to potential contamination by non-target sequences, and often difficult to link to existing 16S rRNA gene sequences and, as such, exact bacterial identities (Bowers et al., 2017). Furthermore, the ability to recover dominant bacteria as MAGs is also variable (Nayfach et al., 2020). All of these limitations can be overcome by isolate characterization, which also offers the opportunity for phenotype screening in cultures (Madin et al., 2020).
The present study isolated the dominant bacteria from decomposing deadwood to describe their functional traits through genome sequencing and phenotype analysis and used this information to explain the preference of specific bacterial taxa for certain stages of deadwood decomposition. We focused on the bacterial ability to release C from plant and fungal polymers and bacterial assimilatory and dissimilatory N cycling pathways. We expect dominant bacteria to show differing occurrences throughout deadwood decomposition stages, driven by substrate availability and their metabolic ability to be constrained by taxonomic identity.
Materials and Methods
Strain Isolation
This study was conducted in the Žofínský Prales National Nature Reserve, a temperate unmanaged forest in the south of the Czech Republic (48°39′57″N, 14°42′24″E). The core zone of the forest reserve (42 ha) has never been managed, and any human intervention ceased in 1838 when it was declared a reserve. This reserve thus represents a rare fragment of European temperate virgin forest with deadwood left to spontaneously decompose. The reserve is situated at 730–830 m above sea level and the bedrock is almost homogeneous and consists of finely to medium-grainy porphyritic and biotite granite. The annual average rainfall is 866 mm, and the annual average temperature is 6.2°C (Anderson-Teixeira et al., 2015). At present, the reserve is covered by a mixed forest where Fagus sylvatica predominates in all diameter classes (51.5% of the total living wood volume), followed by Picea abies (42.8%) and Abies alba (4.8%). The mean living tree volume is 690 m3 h−1, and the volume of coarse woody debris (logs, represented by tree trunks and their fragments) ranges from 102 to 310 m3 h−1 with an average of 208 m3 h−1 (Král et al., 2010; Šamonil et al., 2013). Logs are repeatedly surveyed, and the approximate age of each log, the cause of death (stem breakage, windthrow, etc.), and status before downing (fresh, decomposed) is known.
Decomposing wood samples were collected as described previously (Tláskal et al., 2017). Briefly, dead tree trunks (coarse woody debris; CWD) of F. sylvatica, P. abies, and A. alba with breast height diameters between 30 and 100 cm at the time of downing were chosen to evenly represent all decay lengths (age classes) of deadwood and the span of the diameter range. Trees decomposing as standing snags were omitted to exclude CWD with unclear decay lengths. The CWD decay length ranged from <5 to >38 years. In October 2013, four samples were obtained by drilling with an electrical drill vertically covering sapwood and heartwood along the whole decomposing stem. Part of the material was used for the description of fungal and bacterial communities and wood chemistry (Baldrian et al., 2016; Tláskal et al., 2017), while another part was used for the isolation of bacteria. For this latter category of subsamples, wood chips from each stem were pooled together and transported to the laboratory, where they were cooled until the next day. Approximately 2 g of wood material were shaken with 15 mL of Ringer solution for 2 h and this suspension was diluted 104–106×. Five hundred microlitres of each dilution were plated on nutrient-limited NB medium (0.26 g L−1 nutrient broth, 15 g L−1 agar, pH 5) and WYA4 medium (0.1 g L−1 yeast extract, 15 g L−1 agar, pH 5, Valášková et al., 2009) with cycloheximide (50 mg L−1). Occurrences of bacterial colonies were recorded on Petri dishes incubated at 24°C for 8 weeks and marked with a marker indicating the week of the first appearance. Colony PCR was used to infer the taxonomy of the selected strains that appeared during the course of incubation. PCR premix and cycling conditions were as follows: 2.5 μL of 10× buffer for DyNAzyme DNA Polymerase; 0.75 μL of DyNAzyme II DNA polymerase (2 u μL−1); 0.75 μL of BSA (20 mg mL−1); 0.5 μL of PCR Nucleotide Mix (10 mM); 1 μL of primer eub530f (10 μM, 5′-GTG CCA GCM GCN GCG G); 1 μL of primer eub1100br (10 μM, 5'-GGG TTN CGN TCG TTG, Lane, 1991) and sterile ddH2O up to 25 μL, with amplification started at 94°C for 5 min followed by 35 cycles of 94°C for 1 min, 62°C for 1 min, 72°C for 1 min and a final setting of 72°C for 10 min. Sanger sequencing was performed from the reverse primer as an external service. The obtained sequences were compared by BLASTn with bacterial 16S rRNA-based community data from the same deadwood objects that were used for isolation (Tláskal et al., 2017). Bacterial strains with high similarity (>97%) and coverage (>90%) relative to the most abundant deadwood OTUs were selected for further cultivation. The strains that were cultivable on laboratory media in subsequent passages and those that exhibited sufficient growth and biomass production were used for genome sequencing and culturing tests. By this approach, we were able to select 18 bacterial strains with high similarity to highly abundant bacterial OTUs from the studied deadwood.
Cultivation Tests
The activity of the cell wall-associated fraction of enzymes was measured in cells after 2 weeks of incubation in triplicate in 50 mL of liquid GY-VL55 medium as described previously (Lasa et al., 2019). To assess the ability to grow on cellulose, carboxymethyl cellulose medium (CMC) was prepared (5 g L−1 CMC, 2 g L−1 yeast extract, 15 g L−1 agar, pH 5). Plates were incubated at 24°C for 2 weeks. After incubation, 12 mL of 1% Congo Red was poured onto each plate, staining was performed for 30 min, the Congo Red was replaced with 12 mL of 1 M NaCl, the NaCl was left for 30 min, the NaCl was replaced with 12 mL of H2O, and the H2O was left overnight. The presence of a halo around the colonies indicated CMC utilization.
DNA Extraction, Sequencing, Genome Assembly, and Annotation
Selected bacterial strains were cultivated in 50 mL of GY-VL55 liquid medium shaken in Erlenmeyer flasks (Lladó et al., 2019) for 2 weeks at 23°C. Cells were collected by centrifugation, and DNA was extracted by an ArchivePure DNA Yeast and Gram-+ Kit (5 Prime, Germany) according to the manufacturer's instructions. DNA was sheared by a Bioruptor Pico sonication device (Diagenode, Belgium) to an average length of 550 bp, and sequencing adapters were ligated by the TruSeq DNA PCR-Free Library Prep Kit (Illumina Inc., USA). Ligated libraries were sequenced in-house on the Illumina MiSeq platform.
Genome assembly of each isolate was performed using a Unicycler 0.4.7 (Wick et al., 2017) in normal mode with the following programs: SPAdes 3.13.0 (Bankevich et al., 2012), BLAST 2.7.1+ (Altschul et al., 1997), Bowtie 2.2.4 (Langmead et al., 2009), SAMtools 1.9 (Li et al., 2009), and Pilon 1.22 (Walker et al., 2014). Prokka 1.13.3 (Seemann, 2014) with RNAmmer (Lagesen et al., 2007) was used for gene calling, annotation, and rRNA gene identification. The taxonomy of the closest complete genome in the NCBI database with >97% similarity of 16S rRNA gene sequences were used to assign strains at the genus level. GToTree v1.5.39 (Lee, 2019) together with Prodigal (Hyatt et al., 2010), HMMER3 (Eddy, 2011), Muscle (Edgar, 2004), trimAI (Capella-Gutiérrez et al., 2009), FastTree2 (Price et al., 2010), and GNU Parallel (Tange, 2018) was used to construct a phylogenetic tree of the genomes based on a set of HMM profiles for 74 bacterial single-copy genes.
For CAZyme annotation, Prokka gene prediction was used to obtain the sequences of genes. Amino acid sequences were compared with the dbCAN database version 07312018 using the run-dbcan.py script (Zhang et al., 2018) and HMMER 3.2.1 (Eddy, 2011). CAZymes with an e-value ≤ 1E−30 were considered for further annotation and their most probable target substrates were identified based on the genes with characterized functions in the CAZy database (http://www.cazy.org; Lombard et al., 2014). Functions of predicted genes were annotated with the hmmsearch function in HMMER 3.2.1, using the FOAM database as a source of HMMs for relevant genes (Prestat et al., 2014). FOAM functions with an e-value ≤ 1E−30 were considered for further annotation. In the case of genes with more than one hit in the dbCAN and the FOAM databases, the function with the lowest e-value was selected.
Metagenome Mapping
The metagenomes of naturally decomposing F. sylvatica deadwood were obtained in a previous study (Tláskal et al., 2021a, Tláskal et al., under review) and used to quantify genome coverage in deadwood of various decay lengths. This metagenome study included 25 CWD samples of F. sylvatica. The CWD samples were classified into five decay length classes of <4, 4–7, 8–19, 20–41, and > 41 years. Metagenomic reads were mapped to genomes as described previously (Lladó et al., 2019) using Bowtie v2.3.4.1 (Langmead and Salzberg, 2013). Metagenomic reads from sample with NCBI accession SRR10968255 were omitted from mapping due to under-sampling. The number of mapped reads was normalized to the sequencing depth of each sample, divided by the genome length of individual strains, and multiplied by the average genome size of sequenced bacteria. Differences among groups were tested by the Kruskal-Wallis test using the agricolae package (de Mendiburu, 2017) and considered to be statistically significant at the level P < 0.05.
16S rRNA Gene-Based Phylogeny of OTUs and Strains
Dereplicated 16S rRNA genes of strains with sequenced genomes were merged with representative OTU sequences (most frequent sequences from each OTU, Tláskal et al., 2017). Only the globally most abundant OTUs represented by an abundance >0.5% in >10 samples were selected (n = 86 OTUs). Sequences were aligned by the MAFFT online tool (Katoh et al., 2018) with default settings. A phylogenetic tree was constructed using FastTree2 (Price et al., 2010) with default settings. The taxonomy of the tree tips was assigned with DECIPHER v2.0 (Wright, 2016) using SILVA SSU database release 138 (Quast et al., 2013). A phylogenetic tree of 16S rRNA sequences from all 959 obtained isolates was constructed by the same approach. The diversity of the whole isolated collection was inferred by clustering using VSEARCH 2.4.3 (Rognes et al., 2016). Tables were processed using R 4.0.2 (R Core Team, 2020) and tidyverse 1.3.0 (Wickham et al., 2019). The resulting images were processed with Inkscape 1.0 (https://inkscape.org/).
Results
Isolation yielded 959 isolates, with Gammaproteobacteria being the most represented class, followed by the class Alphaproteobacteria and the phyla Actinobacteria, Bacteroidetes, Acidobacteria, and Firmicutes (Supplementary Figure 1A). The largest share of isolates was represented by the Burkholderia-Caballeronia-Paraburkholderia complex (17%), followed by the genera Mucilaginibacter (7%) and Pseudomonas (7%, Supplementary Figure 1B). Isolates clustered into 467 OTUs (>99% similarity) and were binned to 4.5% of OTUs from the 16S rRNA amplicon study from the same CWD samples published previously (Tláskal et al., 2017). The isolates represented on average 33.5 ± 1.3% of bacteria in a CWD sample. The youngest age class of deadwood, comprising CWD samples with a decay length <5 years, was most represented by the isolates (38.8 ± 2.6%).
The selected sequenced strains binned to 0.7% of bacterial OTUs, representing 15.3 ± 0.8% of bacteria in CWD samples (Supplementary Figure 2; Table 1). The phylogeny of 86 dominant OTUs in the whole amplicon dataset, i.e., those representing >0.5% sequences in >10 CWD samples, showed that we retrieved members of the top five deadwood bacterial phyla and classes of Proteobacteria. These were Acidobacteria, Alphaproteobacteria, Gammaproteobacteria, Actinobacteria, and Bacteroidetes (Figure 1; Table 1). Members of other phyla that represented only a small minority among the dominant OTUs–Firmicutes (n = 1), class Polyangia (n = 1), Planctomycetes (n = 2), Verrucomicrobia (n = 5), and WPS-2 (n = 1)–either were not isolated or failed to grow in subsequent cultures. Except for the strains of Bradyrhizobium (1.5%), each genome represented on average <1% of bacteria across all CWD samples. Up to 26.7% of bacteria were represented in the best-covered CWD samples (Table 1).
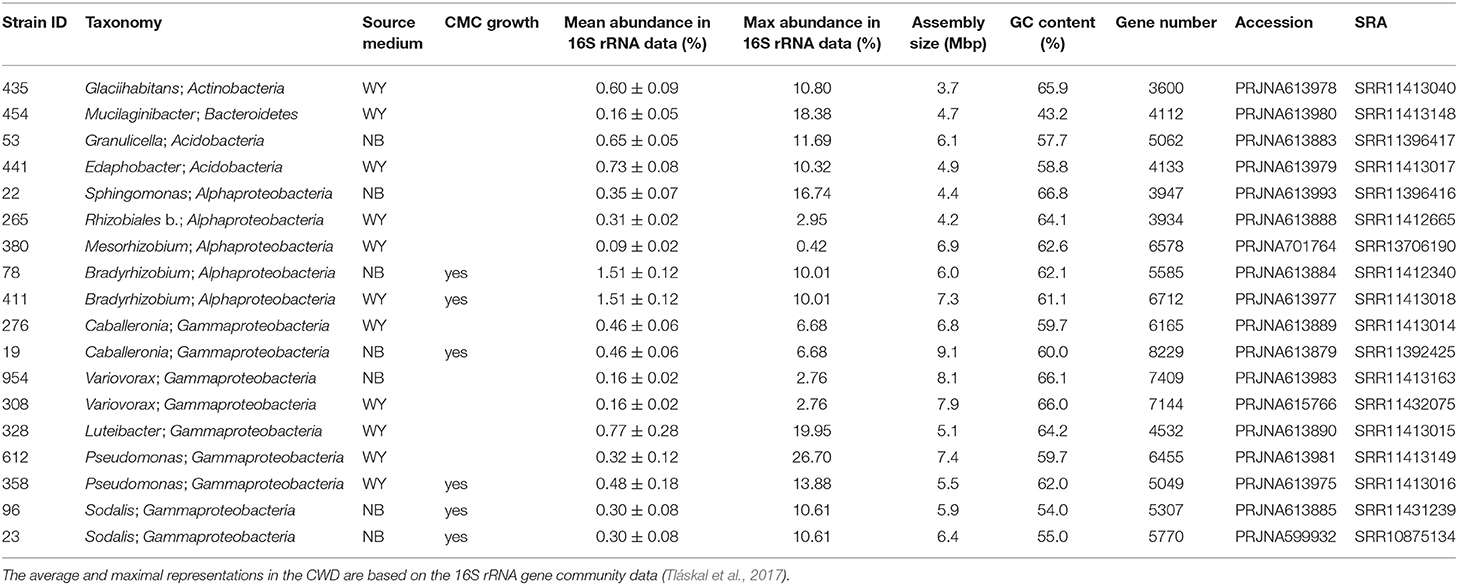
Table 1. Genomic traits of dominant bacteria associated with decomposing deadwood.
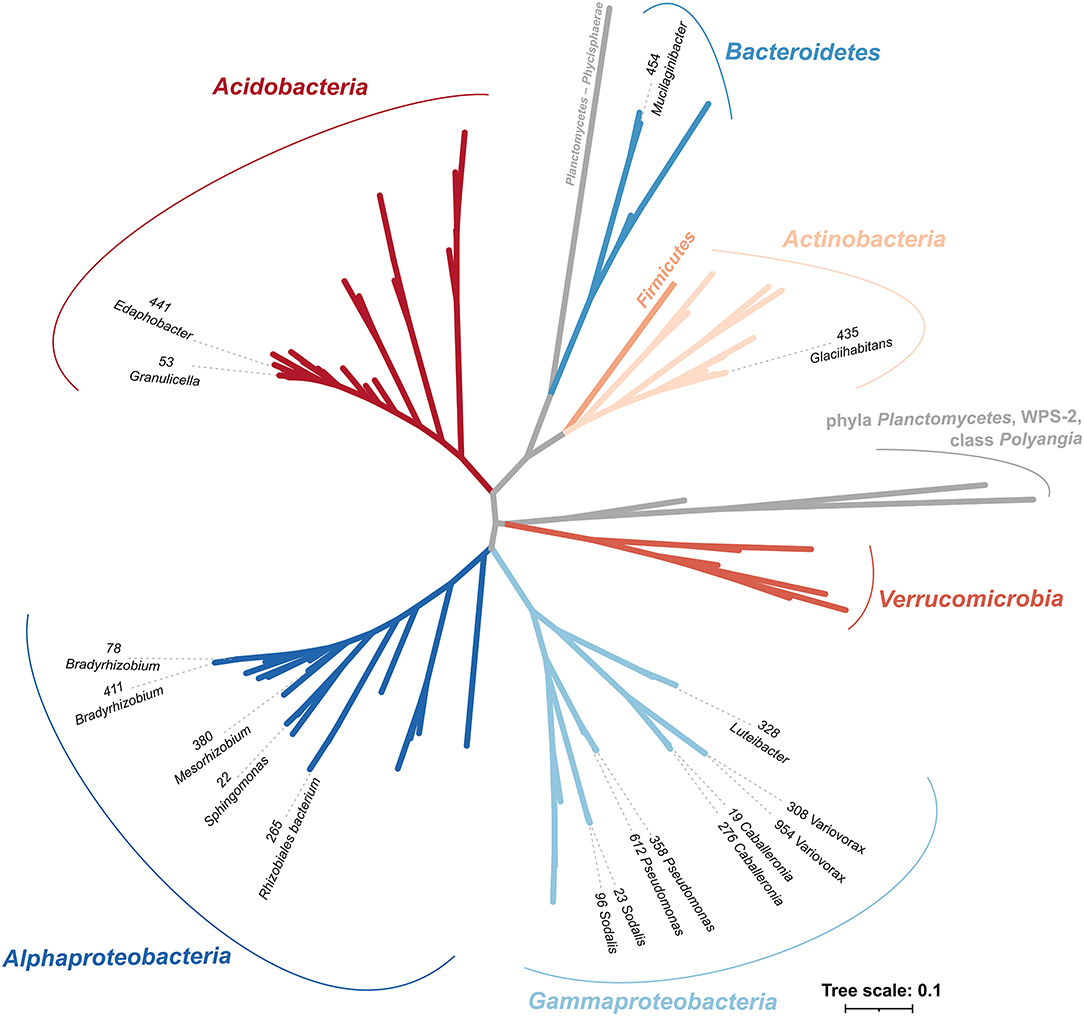
Figure 1. Phylogenetic tree of strains with sequenced genomes in the context of representative sequences of the 87 most abundant OTUs from Fagus sylvatica, Picea abies, and Abies alba deadwood based on the V4 region of 16S rRNA gene. Strains are denoted with strain IDs and genus names and cover the majority of the dominant bacterial phyla in deadwood. Phyla are labeled with different colors, and taxonomy follows the SILVA 138 database. The most abundant bacteria are based on the study of Tláskal et al. (2017).
CAZy analysis showed a considerable level of variation in the carbon utilization traits among strains (Figure 2). Four strains were distinct in terms of high CAZyme counts and diversity: two Acidobacteria (Edaphobacter strain 441 and Granulicella strain 53), Mucilaginibacter strain 454, and Sphingomonas strain 22. The Granulicella strain showed the most CAZymes (n = 187 in 72 CAZy families), representing 3.7% of its total genes. In contrast, six members of the genera Variovorax, Pseudomonas, and Bradyrhizobium contained only 62–75 CAZymes in 28–35 CAZy families, representing 1% of their total genes (Figure 2).
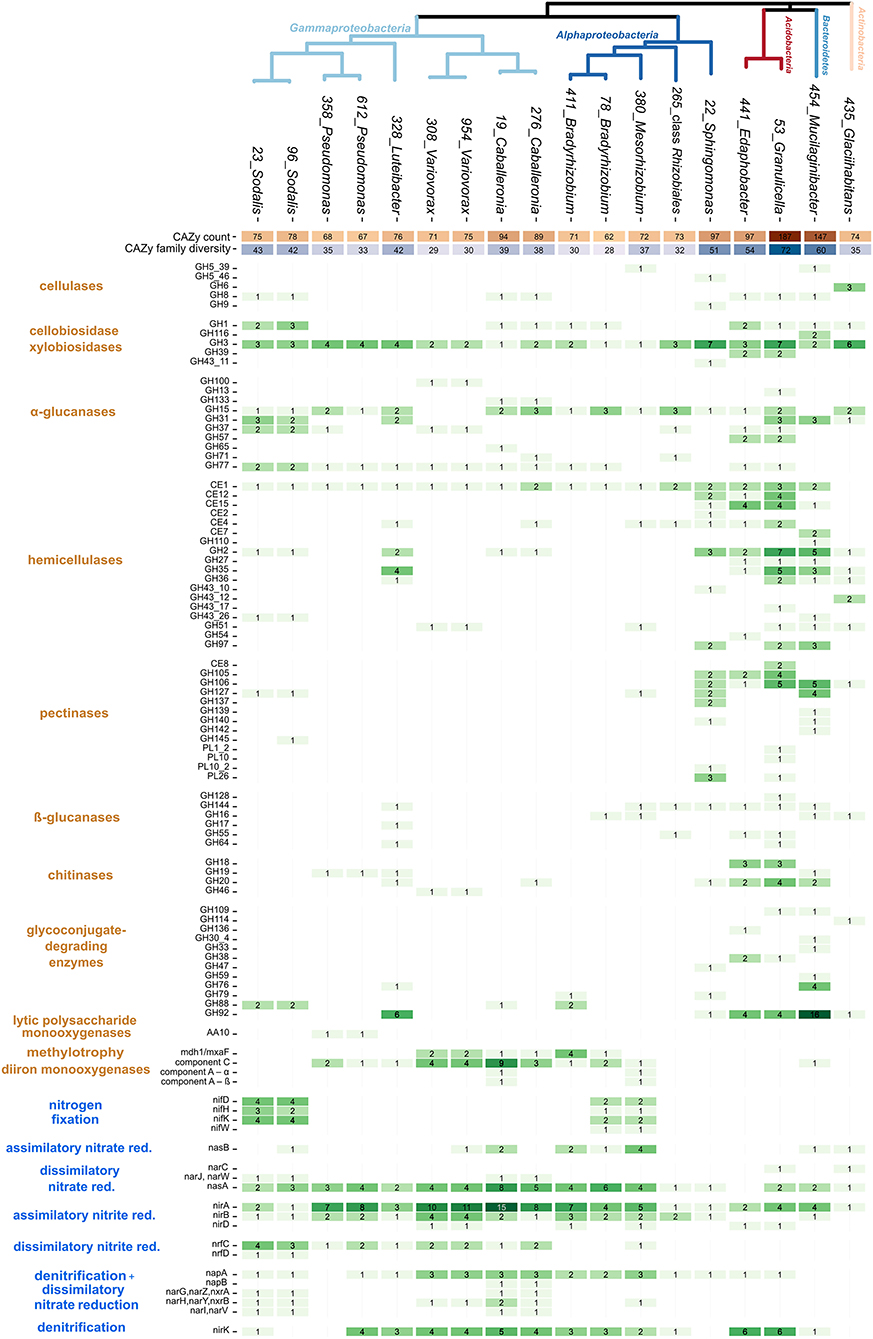
Figure 2. Genomic traits of dominant bacteria associated with decomposing deadwood. Heatmap shows CAZy counts, counts of genes for low-molecular-mass compounds utilization, and counts of N cycle-related genes together with putative functional annotation of each gene. The total number of CAZy genes and counts of CAZy families (CAZy diversity) per genome are depicted as heatmaps in the upper rows. A phylogenetic tree based on single-copy marker genes shows the phylogenetic relatedness of individual strains.
Functional analysis of the identified CAZymes showed the specific C sources targeted by the strains (Figure 2). Genes for cellulose degradation were present in three copies of cellulase GH6 in the genome of Glaciihabitans strain 435. Mucilaginibacter strain 454 and Sphingomonas strain 22 each contained two copies of cellulases. Other strains with a single copy of a cellulase gene included both Acidobacteria strains, both Caballeronia strains, both Sodalis strains, and Mesorhizobium strain 380. An increased number of chitinase genes were present in Granulicella strain 53 and Edaphobacter strain 441 (n = 7 and 5, respectively). Mucilaginibacter strain 454 contained three genes for enzymes involved in chitin degradation, and Luteibacter strain 328 contained two chitinases. Both Pseudomonas strains encoded a single chitinase gene, and genes encoding enzymes targeting chitin were present in both Variovorax strains, Caballeronia strain 276 and Sphingomonas strain 22. An increased number of hemicellulases were present in two Acidobacteria strains, Sphingomonas strain 22, Mucilaginibacter strain 454, and Luteibacter strain 328. CAZymes for the labile substrates cellobiose and xylobiose and α-glucanases were present throughout the isolated strains without evident differences. CAZymes for other labile substrates, pectinases, β-glucanases, and glycoconjugate-degrading enzymes, were preferentially found in high-CAZy-containing Acidobacteria strains, Mucilaginibacter strain 454, Glaciihabitans strain 435, and Sphingomonas strain 22. Two Pseudomonas strains contained lytic polysaccharide monooxygenase genes from CAZy family AA10 for the oxidation of a broad range of polysaccharides. Some of the low-CAZy genomes contained genes for C uptake from alternative sources: the mxaF gene for methanol dehydrogenase, which oxidizes methanol, was present in Variovorax and Caballeronia (both Burkholderiales), and four copies of this gene were present in Bradyrhizobium strain 411. Mesorhizobium together with Caballeronia contained genes for soluble diiron monooxygenases.
Luteibacter strain 328 showed by far the most active cell wall-associated enzymatic fraction. Except for Luteibacter, chitinase activity was highest in Bradyrhizobium strains, Variovorax strains, and Pseudomonas strains. Strains of Pseudomonas, Variovorax, and Luteibacter were not able to grow on CMC medium but displayed cellobiohydrolase and β-glucosidase activities suitable for the degradation of cellulose-derived oligosaccharides (Table 1; Supplementary Figure 3).
Nitrogen cycling-related genes showed a higher occurrence in the genomes of Alphaproteobacteria and Gammaproteobacteria (Figure 2). Bradyrhizobium strain 78, Mesorhizobium strain 380, and the two Sodalis strains contained the nitrogenase operon nifHDK, which enables nitrogen fixation. Genomes of both Sodalis strains and Mesorhizobium strain 380 showed a combination of nitrogenase genes and cellulase genes. The two Caballeronia and the two Sodalis strains contained an increased number of genes for dissimilatory nitrate to nitrite reduction. Assimilatory nitrate reduction genes seem to be limited only to the presence of the nasB gene in a restricted number of taxa. In contrast, the assimilatory nitrite reduction genes nirAB were present in almost all isolated strains. Denitrification steps leading to the production of N2–reduction of nitric oxide and nitrous oxide–were not detected in any genome. However, several genomes (especially those in the genus Sodalis) contained genes for dissimilatory nitrite reduction (nrfC or nrfD), directing N flow into dissimilatory nitrate reduction to the ammonia pathway (DNRA). Similar to denitrification genes, nitrification genes, including those that allow the oxidation of ammonia, were missing in the bacterial genomes examined.
Genome-sequenced bacterial strains differed in their tendency to associate with deadwood of a certain decay length, as demonstrated by the mapping of metagenome reads from decomposing F. sylvatica. Mapping showed that several strains were restricted to very fresh deadwood (<4 years of decomposition), while others tended to increase in abundance at later decomposition stages (Figure 3). The isolates Glaciihabitans strain 435, Pseudomonas strain 358, Sodalis strains 23, and 96 and Sphingomonas strain 22 were significantly more abundant in the fresh CWD, while several other strains also showed high abundances in the fresh deadwood (Edaphobacter strain 441, Caballeronia strain 276 and Mucilaginibacter strain 454) but only inhabited certain CWD samples from the youngest age class. The abundance of other strains, including two Bradyrhizobium strains and Rhizobiales strain 265, peaked later during decomposition. Notably, the aforementioned Alphaproteobacteria represent bacteria with the highest abundance in the examined metagenomes (Figure 3).
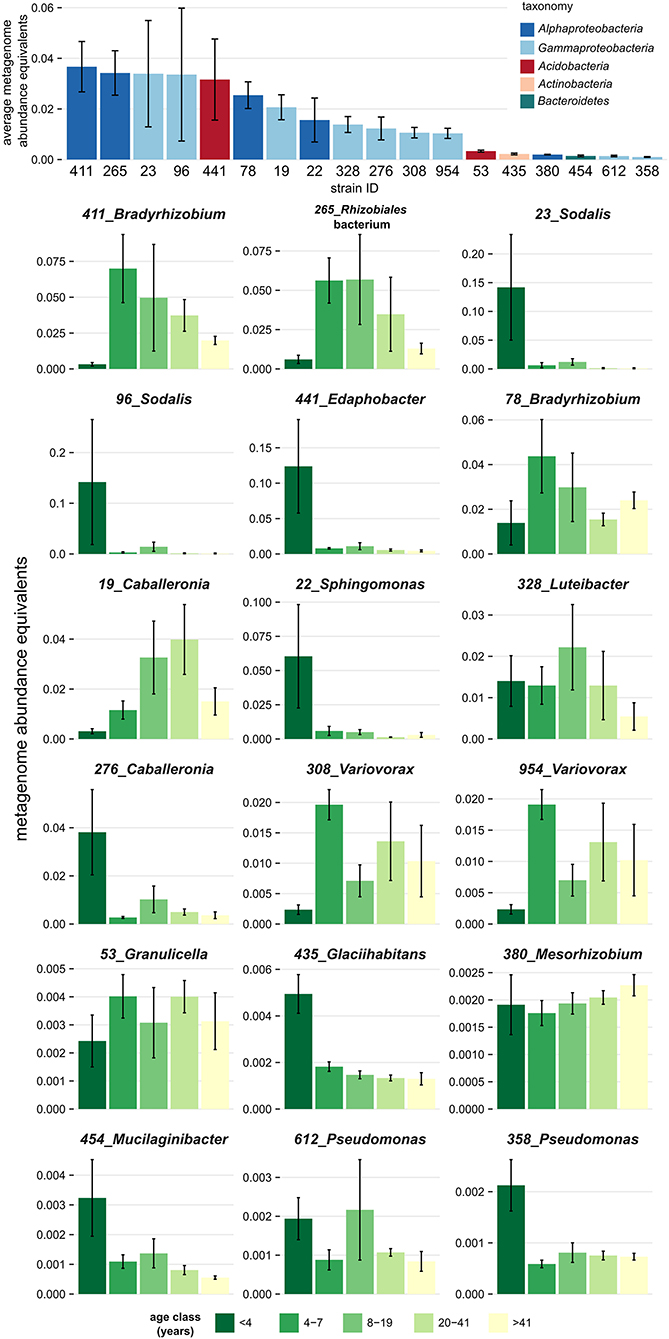
Figure 3. Association of dominant bacteria isolated from decomposing Fagus sylvatica deadwood with a certain age of deadwood. Mapping of the deadwood metagenome reads (Tláskal et al., under review) to genomes of strains shows a distinct overall abundance of these bacteria in metagenomes and distinct occurrence in individual deadwood age classes. Strains are sorted based on the total average metagenome abundance. The height of the bar plots corresponds to abundance equivalents—relative share of mapped reads corrected to the genome size of each strain. Error bars represent standard errors.
Discussion
Using prolonged cultivation, it is possible to capture a wide diversity of bacteria, including slow-growing taxa, with potentially important ecological functions (Sait et al., 2002; Lladó et al., 2016). Our isolation, which resulted in 959 deadwood bacterial isolates, was partly redundant, with many taxa repeatedly captured (Supplementary Figure 1), yet due to the presence of several members of dominant deadwood OTUs, the collection represented a relatively large portion of the bacterial community—on average, approximately one-third of the bacteria present in CWD. In contrast to culture-independent genome resolving, isolation provides complete data about the genome content of a particular strain without the threat of chimeric genomes (Shaiber and Eren, 2019) and represents a suitable approach for fully describing the role of key members of the microbiome (Valášková et al., 2009).
We characterized members of the most abundant deadwood bacterial taxa Acidobacteria, Alphaproteobacteria, Gammaproteobacteria, Actinobacteria, and Bacteroidetes. These dominant phyla appear to be generally occurring in deadwood (Johnston et al., 2016). Although our functional assignment of genes was done as accurately as possible, it should be noted that, in some cases, one CAZy family of enzymes can target different substrates (e.g., GH6; such genes found in Glaciihabitans displayed the highest similarity to known endo-β-1,4-glucanases). Therefore, we show the most probable functions of the CAZymes. Among cultivated bacteria, we identified strains with an increased potential for the utilization of complex carbohydrates, including cellulose, chitin, hemicelluloses, and pectin. These strains belong to the phyla Acidobacteria, Bacteroidetes, Actinobacteria, and the genus Sphingomonas from the Alphaproteobacteria. The strong hydrolytic capabilities of these taxa have been reported previously for some of the soil-inhabiting groups (Uroz et al., 2013; Větrovský et al., 2014; López-Mondéjar et al., 2019). Specifically, Acidobacteria and Bacteroidetes were identified as efficient decomposers of carbohydrate biopolymers (Lladó et al., 2019). This also corresponds to the identified chitinase activity (Lladó et al., 2016) and degradation of fungal mycelium by Acidobacteria including genus Granulicella (López-Mondéjar et al., 2018). This trait was supported by an increased number of chitinase genes in the cultivated Acidobacteria in our study.
In contrast to general degraders, deadwood Gammaproteobacteria and most Alphaproteobacteria harbored lower numbers of CAZymes, indicating a narrow spectrum of potential C sources. The least CAZy-equipped strains were those assigned to the genera Variovorax, Pseudomonas, and Bradyrhizobium. Despite the lack of CAZymes for plant-derived compound degradation, such as cellulose or hemicellulose, these low-CAZy strains have the potential to compensate for this narrow range by decomposition of fungal biomass. Adherence to fungal hyphae or enrichment in the presence of fungi were repeatedly described for Burkholderiales, suggesting a mycophagous lifestyle for these strains (Valášková et al., 2009; Hervé et al., 2016; Brabcová et al., 2018; Starke et al., 2020). Our Variovorax and Caballeronia strains from Burkholderiales indeed contained chitosanase and N-acetylglucosaminidase genes, respectively, which are involved in the degradation of the main fungal structural biopolymers chitin and chitosan. The genus Pseudomonas showed a similar chitinase gene presence to strains of the same genus involved in the decomposition of fungal biomass (Starke et al., 2020). The chitinolytic activity of the membrane-bound enzymes was measured for the Bradyrhizobium strains and the Luteibacter strain. Luteibacter displayed the highest chitinase activity in this study, and its mycolytic and cellulolytic activity was described previously (López-Mondéjar et al., 2016; Mieszkin et al., 2021); Luteibacter additionally showed a generally high activity of membrane-bound enzymes (Lasa et al., 2019).
The bacteria from the classes Alphaproteobacteria and Gammaproteobacteria, which are low in carbohydrate-active enzymes, showed the presence of genes for putative methanol oxidation. Genes for soluble diiron monooxygenases in Caballeronia and Mesorhizobium point to oxidation of other low-molecular-mass C sources rather than performing methanotrophy which was not described for these taxa (Leahy et al., 2003). Altogether, the presence of these genes suggests that Alphaproteobacteria and Gammaproteobacteria utilize simple C compounds generated during lignin decomposition by fungi (Filley et al., 2002; Lenhart et al., 2012). Methylotrophy of Alphaproteobacteria is sometimes accompanied by diazotrophic activity (Vorob'ev et al., 2009). Changes in bacterial deadwood community composition are tightly linked with changes in the pH, water content, and C:N ratio of the substrate (Hoppe et al., 2015; Tláskal et al., 2017). The C:N ratio of fresh deadwood is very high and decreases during decomposition, which is a result of N accumulation partially through fungal translocation and mainly through bacterial N2 fixation (Rayner and Boddy, 1988; Rinne et al., 2016; Tláskal et al., 2021a).
The ability to fix N2 might explain the association of Alphaproteobacteria with low-N conditions and their positive correlation with the C:N ratio (Rinta-Kanto et al., 2016; Mieszkin et al., 2021). Previous studies detected the order Rhizobiales (Alphaproteobacteria) as one of the abundant members in the deadwood community (Hervé et al., 2013) and showed the presence of nitrogenase genes assigned to this group (Hoppe et al., 2015). In line with this, we cultivated two Rhizobiales strains containing the nitrogenase nifHDK operon. The other two nitrogen fixers are enterobacteria from the genus Sodalis, and both are closely related to Sodalis insect endosymbionts that lack nitrogenase genes (Tláskal et al., 2021b). After almost 40 years, non-symbiotic N2-fixing Sodalis and Rhizobiales strains widen a small group of cultivated deadwood diazotrophic bacteria successfully retrieved from naturally decomposing CWD (Seidler et al., 1972; Spano et al., 1982). Sodalis strains were most abundant in early decomposition when the N limitation was most severe, and the abundance of Rhizobiales peaked later. Deadwood seems to be a hotspot of non-symbiotic N2 fixation, and these abundant taxa contribute to the N enrichment of the substrate (Brunner and Kimmins, 2003; Tláskal et al., 2021a). Notably, three out of four nitrogen-fixing strains (the two Sodalis and Mesorhizobium strains) also contained a single cellulase gene, which suggests that these strains might be independent in recalcitrant substrates utilization. Examination of other N cycle pathways shows that genes encoding dissimilatory steps are less common than those encoding assimilatory steps. Specifically, Sodalis strains (and partly Caballeronia strains) contain genes for DNRA, enabling the transformation of oxidized forms of N into ammonium. The balance between DNRA and denitrification is crucial for the soil N budget (Nelson et al., 2016), and a similar mechanism might exist in deadwood. The absence of complete denitrification and the preferred DNRA pathway thus prevents the loss of scarce nitrogen (Tláskal et al., 2021a).
Bacterial community composition was shown to undergo a significant change between the early and late decomposition stages at a deadwood age of ~15 years (Tláskal et al., 2017). Here, several strains showed a preference for very young and almost intact deadwood (though there was a high variation among individual fresh stems), while the same strains were less abundant in the subsequent decomposition stages. Conversely, strains that reached the highest abundance in the late decomposition phase were considerably less abundant in fresh deadwood. Bacteria involved in N2 fixation occur in fresh deadwood as well as in the following decomposition phases, showing the importance of this activity throughout decomposition. As the most readily available plant biopolymers are gradually depleted during decomposition, bacteria with genes for chitin degradation might switch to utilizing C from fungal biomass. Mycophagous bacteria such as Granulicella, Luteibacter, and Burkholderiales indeed tended to be more abundant in the later decomposition stages when fungal biomass in deadwood increases and starts to represent a relatively easily accessible source of both C and N.
Different C utilization strategies were described for soil habitats in which Acidobacteria and Bacteroidetes served as decomposers with a high number of expressed CAZymes, while Proteobacteria were recognized as opportunists relying on C uptake from other labile sources (Lladó et al., 2019). With the high taxonomic and functional overlap in these soil groups, we can identify two distinct strategies of abundant deadwood bacterial taxa: degraders with high decomposition potential and less generalist bacteria filling their C needs by alternative source utilization and mycophagy. Moreover, the latter are active in N cycling, and their presence might be beneficial for the total microbial community due to N2 fixation. Metabolically diverse bacteria thus show specific adaptations to challenging deadwood habitats and complement deadwood decomposition driven by fungi through N enrichment.
Code Availability
The above methods indicate the programs used for analysis within the relevant sections. The code for reproducing the sequence processing is provided at https://github.com/TlaskalV/Deadwood-bacterial-isolates
Data Availability Statement
Data described in this manuscript, including raw sequences from short read sequencing and genome assembly files have been deposited in NCBI under BioProject accession numbers summarized in the Table 1.
Author Contributions
VT and PB conceived the study and wrote the manuscript. VT performed strain isolation, genome processing, and strain growth characterization. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Czech Science Foundation (21-09334J).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.685303/full#supplementary-material
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Anderson-Teixeira, K. J., Davies, S. J., Bennett, A. C., Muller-landau, H. C., and Wright, S. J. (2015). CTFS-ForestGEO: a worldwide network monitoring forests in an era of global change. Glob. Change Biol. 21, 528–549. doi: 10.1111/gcb.12712
Baldrian, P., Zrustová, P., Tláskal, V., Davidová, A., Merhautová, V., and Vrška, T. (2016). Fungi associated with decomposing deadwood in a natural beech-dominated forest. Fungal Ecol. 23, 109–122. doi: 10.1016/j.funeco.2016.07.001
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Alexander, S., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Berlemont, R., and Martiny, A. C. (2013). Phylogenetic distribution of potential cellulases in bacteria. Appl. Environ. Microbiol. 79, 1545–1554. doi: 10.1128/AEM.03305-12
Berlemont, R., and Martiny, A. C. (2015). Genomic potential for polysaccharide deconstruction in Bacteria. Appl. Environ. Microbiol. 81, 1513–1519. doi: 10.1128/AEM.03718-14
Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B. K., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731. doi: 10.1038/nbt.3893
Brabcová, V., Nováková, M., Davidová, A., and Baldrian, P. (2016). Dead fungal mycelium in forest soil represents a decomposition hotspot and a habitat for a specific microbial community. New Phytol. 210, 1369–1381. doi: 10.1111/nph.13849
Brabcová, V., Štursová, M., and Baldrian, P. (2018). Nutrient content affects the turnover of fungal biomass in forest topsoil and the composition of associated microbial communities. Soil Biol. Biochem. 118, 187–198. doi: 10.1016/j.soilbio.2017.12.012
Brunner, A., and Kimmins, J. P. (2003). Nitrogen fixation in coarse woody debris of Thuja plicata and Tsuga heterophylla forests on northern Vancouver Island. Can. J. For. Res. 33, 1670–1682. doi: 10.1139/x03-085
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Christofides, S. R., Hiscox, J., Savoury, M., Boddy, L., and Weightman, A. J. (2019). Fungal control of early-stage bacterial community development in decomposing wood. Fungal Ecol. 42:100868. doi: 10.1016/j.funeco.2019.100868
de Mendiburu, F. (2017). Agricolae: Statistical Procedures for Agricultural Research. R package version 1.2–4.
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Filley, T. R., Cody, G. D., Goodell, B., Jellison, J., Noser, C., and Ostrofsky, A. (2002). Lignin demethylation and polysaccharide decomposition in spruce sapwood degraded by brown rot fungi. Org. Geochem. 33, 111–124. doi: 10.1016/S0146-6380(01)00144-9
Folman, L. B., Gunnewiek, P. J. A. K., Boddy, L., and de Boer, W. (2008). Impact of white-rot fungi on numbers and community composition of bacteria colonizing beech wood from forest soil. FEMS Microbiol. Ecol. 63, 181–191. doi: 10.1111/j.1574-6941.2007.00425.x
Gallardo, C. A., Baldrian, P., and López-mondéjar, R. (2021). Litter-inhabiting fungi show high level of specialization towards biopolymers composing plant and fungal biomass. Biol. Fertil. Soils 57, 77–88. doi: 10.1007/s00374-020-01507-3
Hervé, V., Ketter, E., Pierrat, J.-C., Gelhaye, E., and Frey-Klett, P. (2016). Impact of Phanerochaete chrysosporium on the functional diversity of bacterial communities associated with decaying wood. PLoS ONE 11:e0147100. doi: 10.1371/journal.pone.0147100
Hervé, V., Le Roux, X., Uroz, S., Gelhaye, E., and Frey-Klett, P. (2013). Diversity and structure of bacterial communities associated with Phanerochaete chrysosporium during wood decay. Environ. Microbiol. 16, 2238–2252. doi: 10.1111/1462-2920.12347
Hoppe, B., Krüger, D., Kahl, T., Arnstadt, T., Buscot, F., Bauhus, J., et al. (2015). A pyrosequencing insight into sprawling bacterial diversity and community dynamics in decaying deadwood logs of Fagus sylvatica and Picea abies. Sci. Rep. 5:9456. doi: 10.1038/srep09456
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Johnston, S. R., Boddy, L., and Weightman, A. J. (2016). Bacteria in decomposing wood and their interactions with wood-decay fungi. FEMS Microbiol. Ecol. 92:fiw179. doi: 10.1093/femsec/fiw179
Johnston, S. R., Hiscox, J., Savoury, M., Boddy, L., and Weightman, A. J. (2019). Highly competitive fungi manipulate bacterial communities in decomposing beech wood (Fagus sylvatica). FEMS Microbiol. Ecol. 95:fiy225. doi: 10.1093/femsec/fiy225
Katoh, K., Rozewicki, J., and Yamada, K. D. (2018). MAFFT online service: multiple sequence alignment, interactive sequence choice, and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Král, K., Janík, D., Vrška, T., Adam, D., Hort, L., Unar, P., et al. (2010). Local variability of stand structural features in beech dominated natural forests of Central Europe: Implications for sampling. For. Ecol. Manage. 260, 2196–2203. doi: 10.1016/j.foreco.2010.09.020
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (New York, NY: Wiley), 115–175.
Langmead, B., and Salzberg, S. L. (2013). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Lasa, A. V., Mašínová, T., Baldrian, P., and Fernández-López, M. (2019). Bacteria from the endosphere and rhizosphere of Quercus spp. use mainly cell wall-associated enzymes to decompose organic matter. PLoS ONE 14:e0214422. doi: 10.1371/journal.pone.0214422
Leahy, J. G., Batchelor, P. J., and Morcomb, S. M. (2003). Evolution of the soluble diiron monooxygenases. FEMS Microbiol. Rev. 27, 449–479. doi: 10.1016/S0168-6445(03)00023-8
Lee, M. D. (2019). GToTree: a user-friendly workflow for phylogenomics. Bioinformatics 35, 4162–4164. doi: 10.1093/bioinformatics/btz188
Lenhart, K., Bunge, M., Ratering, S., Neu, T. R., Schüttmann, I., Greule, M., et al. (2012). Evidence for methane production by saprotrophic fungi. Nat. Commun. 3:1046. doi: 10.1038/ncomms2049
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lladó, S. F., Větrovský, T., and Baldrian, P. (2019). Tracking of the activity of individual bacteria in temperate forest soils shows guild-specific responses to seasonality. Soil Biol. Biochem. 135, 275–282. doi: 10.1016/j.soilbio.2019.05.010
Lladó, S. F., Žifčáková, L., Větrovský, T., Eichlerová, I., and Baldrian, P. (2016). Functional screening of abundant bacteria from acidic forest soil indicates the metabolic potential of Acidobacteria subdivision 1 for polysaccharide decomposition. Biol. Fertil. Soils 52, 251–260. doi: 10.1007/s00374-015-1072-6
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
López-Mondéjar, R., Algora, C., and Baldrian, P. (2019). Lignocellulolytic systems of soil bacteria: a vast and diverse toolbox for biotechnological conversion processes. Biotechnol. Adv. 37, 399–404. doi: 10.1016/j.biotechadv.2019.03.013
López-Mondéjar, R., Brabcová, V., Štursová, M., Davidová, A., Jansa, J., Cajthaml, T., et al. (2018). Decomposer food web in a deciduous forest shows high share of generalist microorganisms and importance of microbial biomass recycling. ISME J. 12, 1768–1778. doi: 10.1038/s41396-018-0084-2
López-Mondéjar, R., Zühlke, D., Becher, D., Riedel, K., and Baldrian, P. (2016). Cellulose and hemicellulose decomposition by forest soil bacteria proceeds by the action of structurally variable enzymatic systems. Sci. Rep. 6:25279. doi: 10.1038/srep25279
Madin, J. S., Nielsen, D. A., Brbic, M., Corkrey, R., Danko, D., Edwards, K., et al. (2020). A synthesis of bacterial and archaeal phenotypic trait data. Sci. Data 7:170. doi: 10.1038/s41597-020-0497-4
Martin, A. R., Domke, G. M., Doraisami, M., and Thomas, S. C. (2021). Carbon fractions in the world's dead wood. Nat. Commun. 12:889. doi: 10.1038/s41467-021-21149-9
Mieszkin, S., Richet, P., Bach, C., Lambrot, C., Augusto, L., Buée, M., et al. (2021). Oak decaying wood harbors taxonomically and functionally different bacterial communities in sapwood and heartwood. Soil Biol. Biochem. 155:108160. doi: 10.1016/j.soilbio.2021.108160
Moll, J., Kellner, H., Leonhardt, S., Stengel, E., Dahl, A., Buscot, F., et al. (2018). Bacteria inhabiting deadwood of 13 tree species reveal great heterogeneous distribution between sapwood and heartwood. Environ. Microbiol. 20, 3744–3756. doi: 10.1111/1462-2920.14376
Nayfach, S., Roux, S., Seshadri, R., Udwary, D., Varghese, N., Schulz, F., et al. (2020). A genomic catalog of Earth's microbiomes. Nat. Biotechnol. 39:520. doi: 10.1038/s41587-020-00769-4
Nelson, M. B., Martiny, A. C., and Martiny, J. B. H. (2016). Global biogeography of microbial nitrogen-cycling traits in soil. Proc. Natl. Acad. Sci. U.S.A. 113, 8033–8040. doi: 10.1073/pnas.1601070113
Odriozola, I., Abrego, N., Tláskal, V., Zrustová, P., Morais, D., Větrovský, T., et al. (2021). Fungal communities are important determinants of bacterial community composition in deadwood. mSystems 6, e01017–e01020. doi: 10.1128/mSystems.01017-20
Pan, Y., Birdsey, R. A., Fang, J., Houghton, R., Kauppi, P. E., Kurz, W. A., et al. (2011). A large and persistent carbon sink in the world's forests. Science 333, 988–993. doi: 10.1126/science.1201609
Philpott, T. J., Prescott, C. E., Chapman, W. K., and Grayston, S. J. (2014). Nitrogen translocation and accumulation by a cord-forming fungus (Hypholoma fasciculare) into simulated woody debris. For. Ecol. Manage. 315, 121–128. doi: 10.1016/j.foreco.2013.12.034
Prestat, E., David, M. M., Hultman, J., Taş, N., Lamendella, R., Dvornik, J., et al. (2014). FOAM (Functional Ontology Assignments for Metagenomes): a Hidden Markov Model (HMM) database with environmental focus. Nucleic Acids Res. 42:e145. doi: 10.1093/nar/gku702
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2020). R: A Language and Environment for Statistical Computing. Available online at: http://www.r-project.org/ (accessed June 22, 2020).
Rayner, A. D., and Boddy, L. (1988). Fungal Decomposition of Wood. Its Biology and Ecology. Hoboken, NJ: John Wiley and Sons Ltd.
Rinne, K. T., Rajala, T., Peltoniemi, K., Chen, J., Smolander, A., and Mäkipää, R. (2016). Accumulation rates and sources of external nitrogen in decaying wood in a Norway spruce dominated forest. Funct. Ecol. 31, 530–541. doi: 10.1111/1365-2435.12734
Rinta-Kanto, J. M., Sinkko, H., Rajala, T., Al-Soud, W. A., Sørensen, S. J., Tamminen, M. V., et al. (2016). Natural decay process affects the abundance and community structure of Bacteria and Archaea in Picea abies logs. FEMS Microbiol. Ecol. 92:fiw087. doi: 10.1093/femsec/fiw087
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ. 4:e2584. doi: 10.7717/peerj.2584
Sait, M., Hugenholtz, P., and Janssen, P. H. (2002). Cultivation of globally distributed soil bacteria from phylogenetic lineages previously only detected in cultivation-independent surveys. Environ. Microbiol. 4, 654–666. doi: 10.1046/j.1462-2920.2002.00352.x
Šamonil, P., Schaetzl, R. J., Valtera, M., Goliáš, V., Baldrian, P., Vašíčková, I., et al. (2013). Crossdating of disturbances by tree uprooting: can treethrow microtopography persist for 6000 years? For. Ecol. Manage. 307, 123–135. doi: 10.1016/j.foreco.2013.06.045
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seidler, R. J., Aho, P. E., Raju, P. N., and Evans, H. J. (1972). Nitrogen fixation by bacterial isolates from decay in living white fir trees [Abies concolor (Gord. and Glend.) Lindl.]. J. Gen. Microbiol. 73, 413–416. doi: 10.1099/00221287-73-2-413
Shaiber, A., and Eren, A. M. (2019). Composite metagenome-assembled genomes reduce the quality of public genome repositories. mBio 10, e00725–e00719. doi: 10.1128/mBio.00725-19
Spano, S. D., Jurgensen, M. F., Larsen, M. J., and Harvey, A. E. (1982). Nitrogen-fixing bacteria in Douglas-fir residue decayed by Fomitopsis pinicola. Plant Soil 68, 117–123. doi: 10.1007/BF02374731
Starke, R., Morais, D., Větrovský, T., Mondéjar, R. L., Baldrian, P., and Brabcová, V. (2020). Feeding on fungi: genomic and proteomic analysis of the enzymatic machinery of bacteria decomposing fungal biomass. Environ. Microbiol. 22, 4604–4619. doi: 10.1111/1462-2920.15183
Tláskal, V., Brabcová, V., Větrovský, T., Jomura, M., López-Mondéjar, R., Monteiro, M. O. L., et al. (2021a). Complementary roles of wood-inhabiting fungi and bacteria facilitate deadwood decomposition. mSystems 6, e01078–e01020. doi: 10.1128/mSystems.01078-20
Tláskal, V., Žifčáková, L., Pylro, V. S., and Baldrian, P. (2021b). Ecological divergence within the enterobacterial genus Sodalis: from insect symbionts to inhabitants of decomposing deadwood. Front. Microbiol. 12:668644. doi: 10.3389/fmicb.2021.668644
Tláskal, V., Zrustová, P., Vrška, T., and Baldrian, P. (2017). Bacteria associated with decomposing dead wood in a natural temperate forest. FEMS Microbiol. Ecol. 93:fix157. doi: 10.1093/femsec/fix157
Uroz, S., Courty, P. E., Pierrat, J. C., Peter, M., Buée, M., Turpault, M. P., et al. (2013). Functional profiling and distribution of the forest soil bacterial communities along the soil mycorrhizosphere continuum. Microb. Ecol. 66, 404–415. doi: 10.1007/s00248-013-0199-y
Valášková, V., de Boer, W., Gunnewiek, P. J. A. K., Pospíšek, M., and Baldrian, P. (2009). Phylogenetic composition and properties of bacteria coexisting with the fungus Hypholoma fasciculare in decaying wood. ISME J. 3, 1218–1221. doi: 10.1038/ismej.2009.64
Větrovský, T., Steffen, K. T., and Baldrian, P. (2014). Potential of cometabolic transformation of polysaccharides and lignin in lignocellulose by soil Actinobacteria. PLoS ONE 9:e89108. doi: 10.1371/journal.pone.0089108
Vorob'ev, A. V., de Boer, W., Folman, L. B., Bodelier, P. L. E., Doronina, N. V., Suzina, N. E., et al. (2009). Methylovirgula ligni gen. nov., sp. nov., an obligately acidophilic, facultatively methylotrophic bacterium with a highly divergent mxaF gene. Int. J. Syst. Evol. Microbiol. 59, 2538–2545. doi: 10.1099/ijs.0.010074-0
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly omprovement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Weedon, J. T., Cornwell, W. K., Cornelissen, J. H. C., Zanne, A. E., Wirth, C., and Coomes, D. A. (2009). Global meta-analysis of wood decomposition rates: a role for trait variation among tree species? Ecol. Lett. 12, 45–56. doi: 10.1111/j.1461-0248.2008.01259.x
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wickham, H., Averick, M., Bryan, J., Chang, W., Mcgowan, L. D. A., François, R., et al. (2019). Welcome to the Tidyverse. J. Open Source Softw. 4:1686. doi: 10.21105/joss.01686
Wright, E. S. (2016). Using DECIPHER v2.0 to analyze big biological sequence data in R. R J. 8, 352–359. doi: 10.32614/RJ-2016-025
Keywords: deadwood, bacterial genomes, cellulose, mycophagy, nitrogen fixation
Citation: Tláskal V and Baldrian P (2021) Deadwood-Inhabiting Bacteria Show Adaptations to Changing Carbon and Nitrogen Availability During Decomposition. Front. Microbiol. 12:685303. doi: 10.3389/fmicb.2021.685303
Received: 24 March 2021; Accepted: 04 May 2021;
Published: 17 June 2021.
Edited by:
Svetlana N. Dedysh, Winogradsky Institute of Microbiology, Russian Academy of Sciences (RAS), RussiaReviewed by:
Miha Humar, University of Ljubljana, SloveniaSarah Christofides, Cardiff University, United Kingdom
Copyright © 2021 Tláskal and Baldrian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vojtěch Tláskal, dGxhc2thbEBiaW9tZWQuY2FzLmN6