 Adriana A. Pedroso
Adriana A. Pedroso Margie D. Lee
Margie D. Lee John J. Maurer
John J. Maurer- 1Department of Population Health, University of Georgia, Athens, GA, United States
- 2Department of Biomedical Sciences and Pathobiology, Virginia Polytechnic Institute and State University, Blacksburg, VA, United States
- 3Department of Animal and Poultry Sciences, Virginia Polytechnic Institute and State University, Blacksburg, VA, United States
The transfer of the intestinal microbiota from adult to juvenile animals reduces Salmonella prevalence and abundance. The mechanism behind this exclusion is unknown, however, certain member species may exclude or promote pathogen colonization and Salmonella abundance in chickens correlates with intestinal community composition. In this study, newly hatched chicks were colonized with Salmonella Typhimurium and 16S rRNA libraries were generated from the cecal bacterial community at 21, 28, 35, and 42 days of age. Salmonella was quantified by real-time PCR. Operational taxonomic units (OTUs) were assigned, and taxonomic assignments were made, using the Ribosomal Database Project. Bacterial diversity was inversely proportional to the Salmonella abundance in the chicken cecum (p < 0.01). In addition, cecal communities with no detectable Salmonella (exclusive community) displayed an increase in the abundance of OTUs related to specific clostridial families (Ruminococcaceae, Eubacteriaceae, and Oscillospiraceae), genera (Faecalibacterium and Turicibacter) and member species (Ethanoligenens harbinense, Oscillibacter ruminantium, and Faecalibacterium prausnitzii). For cecal communities with high Salmonella abundance (permissive community), there was a positive correlation with the presence of unclassified Lachnospiraceae, clostridial genera Blautia and clostridial species Roseburia hominis, Eubacterium biforme, and Robinsoniella peoriensis. These findings strongly support the link between the intestinal bacterial species diversity and the presence of specific member species with Salmonella abundance in the chicken ceca. Exclusive bacterial species could prove effective as direct-fed microbials for reducing Salmonella in poultry while permissive species could be used to predict which birds will be super-shedders.
Introduction
Salmonella is a γ-proteobacter capable of colonizing the gastrointestinal tract of many animal species (Sanchez et al., 2002). The evolution of the genus Salmonella involved the acquisition of a pathogenicity island encoding a type III secretion system that mediates cell invasion (Baumler et al., 1998). This pathogenicity island (SPI1) is central to Salmonella’s ability to cause disease in many animal species (Galan and Curtiss, 1989; Watson et al., 1998; Lichtensteiger and Vimr, 2003) but is primarily responsible for inducing the inflammation associated with gastroenteritis (Jung et al., 1995; Hapfelmeier et al., 2004). By eliciting this inflammation, Salmonella infection creates a metabolically favorable environment for the pathogen that results in improved growth in the intestine (Winter et al., 2010; Thiennimitr et al., 2011; Rivera-Chavez et al., 2016).
Salmonella infection causes the most severe symptoms in young mammals (Wray and Wray, 2000), and chickens exhibit symptoms with Salmonella if chicks are infected in ovo or shortly after hatch (Wray and Wray, 2000). If challenged at 2 days of age, many chicks fail to exhibit any symptoms even though nearly all of the birds become colonized and shed the organism for 4–6 weeks (Cheng et al., 2015). The microbiota of the hatchling evolves quickly so that by 3 days posthatch, several dozen distinct bacterial species inhabit the chicken gastrointestinal tract, with γ-proteobacter accounting for ∼2% of the total species population (Lu et al., 2003a; Pedroso et al., 2016). Within the next 3 weeks, species diversity in the intestine increases with a succession of bacterial species in the ileum and cecum and γ-proteobacter become a minor component of the intestinal community (Lu et al., 2003a). The community composition of the chicken ileum and cecum are similar within the first week post hatch comprised primarily of Firmicutes related to the Lactobacillales and Clostridiales (Gong et al., 2002; Lan et al., 2002; Lu et al., 2003a; Zhu and Joerger, 2003). The ileal and cecal communities become segregated quickly, the composition of each becomes unique, and community diversity peaks in broiler chickens near the time when they are processed at 49 days of age (Lu et al., 2003a). Chicks exposed to a mature intestinal microbiota at hatch rapidly develop high community diversity (Lee et al., 2006; Pedroso et al., 2016) and are resistant to Salmonella colonization (Nurmi and Rantala, 1973; Nurmi et al., 1992; Nakamura et al., 2002). In fact, the microbiota from chickens, as young as 21 days of age, seeded in newly hatched chicks dramatically reduces Salmonella abundance in 1-week old layer chickens (Varmuzova et al., 2016). These findings are the basis of the concept and practice of competitive exclusion.
Humans, regardless of age, can present gastroenteritis upon consumption of Salmonella-contaminated water, milk, or food. While susceptible mouse strains are commonly used as an animal model for understanding Salmonella pathogenesis, these infected animals present a lymphoid-associated enteric fever instead of gastroenteritis (Santos et al., 2001). However, mice administered streptomycin, prior to challenge, develop inflammation of the colon with Salmonella infection (Hapfelmeier et al., 2004). Streptomycin treatment decreases abundance of Firmicutes and increases Salmonella abundance in challenged mice (Sekirov et al., 2008) indicating that the composition of the intestinal microbiota therefore has a profound effect on pathogen behavior. A decline in intestinal species diversity favors enteropathogen colonization (Antharam et al., 2013; Lone et al., 2013; Stanley et al., 2014; Zhang et al., 2015) and disease (Antharam et al., 2013; McMurtry et al., 2015; Rodriguez et al., 2015; Singh et al., 2015). Why are chicks protected from enteropathogen colonization when seeded at hatch with intestinal microbiota from adults versus juveniles? What changes in the intestinal microbiota as animals age causes this colonization resistance? Microbiome diversity may ensure the presence of sufficient competitors or antagonists to block pathogen colonization and persistence.
In order to study these hypotheses, a molecular ecology approach was used to reveal the intestinal community structure relative to Salmonella abundance in chickens. Community diversity and abundance of some species correlated with Salmonella abundance in the chicken cecum. Several Firmicutes, particular clostridial species, were positively associated with Salmonella abundance, but others correlated with low Salmonella abundance. It appears that species diversity may be key to understanding pathogen exclusion in the intestine.
Materials and Methods
Salmonella Colonization
A total of 100, 1-day-old, specific pathogen-free, white leghorn chickens (Charles River Laboratories; Wilmington, MA, United States) were placed in one of five HEPA-filtered, isolator units (20 birds per unit). Each unit has wire mesh floors to reduce re-exposure due to coprophagy, and received feed and water ad libitum up to 42 days of age. Chick box liners and the inside of isolator units were swabbed for Salmonella with milk-soaked, 3MTM Sponge Stick (3M; St. Paul, MN, United States) as previously described (Liljebjelke et al., 2005). Birds and their environment were culture-negative for Salmonella on the day of placement of chickens in isolator unit. Chickens were reared on a commercial, non-medicated, pelleted, starter feed throughout the course of the study. At 2 days of age, chicks were orally inoculated with 1.1 × 106 CFU/0.1 ml of Salmonella Typhimurium SL1344. Salmonella inoculum was prepared by streaking SL1344 onto Tryptic Soy Agar (Thermo Fisher Scientific; Pittsburg, PA, United States) which was subsequently used to inoculate 5 ml Luria-Bertani (LB) broth (Provence and Curtiss, 1994) in sterile, capped, 13 × 100 mm glass tubes (Thermo Fisher Scientific). The broth culture was incubated overnight, static at 37°C. The overnight culture was diluted 1/10 in sterile saline. The Salmonella challenge inoculum was serially diluted 10-fold in buffered saline gelatin (Provence and Curtiss, 1994) and plated onto LB agar to determine bacterial cell density. The number of birds per unit was maintained at a stocking density reflective of commercial standards; culling birds periodically as they grew to maintain this stocking density. At 21, 28, 35, and 42 days of age, one bird from each of the five isolator units was collected (n = 5), euthanized and the ceca were aseptically removed. The number given to each sample (1–5) corresponds to the isolator unit from which the bird was collected. Cecal contents were collected and homogenized in pH 7.0 phosphate-buffer saline (1:10 w/v). Between days 35 and 42, one bird died unexpectantly (isolator 5) and therefore the last time point only had four subjects, instead of five, left for analysis.
DNA Extraction
Bacterial cells, present in the cecal contents of 19 samples, were lysed using beads, solution 1 and IRS of Mo Bio Soil DNA extraction kit (Mo Bio Laboratories Inc., Carlsbad, CA, United States) by vortexing at maximum speed for 40 min (Lu et al., 2003b). Lysates were treated with sodium dodecyl sulfate (0.5%) and proteinase K (0.1 μg/ml) and incubated at 37°C for 30 min. Samples were extracted twice with an equal volume of phenol-chloroform-isoamyl alcohol (25:24:1) and once with chloroform-isoamyl alcohol (24:1). DNAse-free, RNAse (20 μl) was added to each sample and incubated at 37°C for 15 min. DNA was concentrated with a 0.6 volume of isopropanol, and the DNA pellet was resuspended in sterile water. The quality and quantity of DNA was assessed by agarose gel electrophoresis.
qPCR
Quantitative PCR was used to determine the amount of S. Typhimurium in cecal contents. A 5-μl aliquot of the lysate was diluted 1:4 in TE buffer [10 mM Tris, 0.1 mM EDTA (pH 8.0)] and used as a template in the qPCR assay. qPCR was performed using SYBR green master mix (Bio-Rad, Hercules, CA, United States) and the MJ Research Chomo4 real-time 4-color 96-well PCR system (Bio-Rad). qPCR data was analyzed by the relative standard curve method (Fey et al., 2004). Serial diluted DNA from S. Typhimurium SL1344 was used as a standard. The virulence gene invA (Daum et al., 2002) was used as the target amplicon to estimate Salmonella abundance and the gene ttr (Malorny et al., 2004) was used as an internal reference for data normalization.
PCR Amplification of Cecal 16S rRNA Libraries
The bacterial primers 27F YM + 3 and 515R-NK (5-CCG CNG CKG CTG GCA C-3), targeting the regions V3 and V6, were used. The primer 27f-YM3 is four parts 27f-YM (5-AGA GTT TGA TYM TGG CTCA G), plus one part each of primers specific for the amplification of Bifidobacteriaceae (27f-Bif, 5-AGGG TTC GAT TCT GGC TCA G), Borrelia (27f-Bor, 5-AGA GTT TGA TCC TGG CTT AG), and Chlamydiales (27f-Chl, 5-AGA ATT TGA TCT TGG TTC AG) sequences (Acosta-Martinez et al., 2008; Garcia et al., 2011). The primers were synthesized with a sequencing adaptor and a specific 8-nucleotide barcode (Hamady et al., 2008) and were a gift from Dr. William Whitman (University of Georgia). A 10 μl PCR mixture was prepared with 20 mM of each primer, 100 ng of DNA template and 9 μl of Platinum Taq DNA Polymerase (Invitrogen, Carlsbad, CA, United States). PCR amplification of the bacterial 16S rRNA genes was conducted after an initial denaturation at 95°C for 3 min followed by 20 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 68°C for 60 s. The final extension was carried out at 68°C for 4 min. PCR amplifications were done using Idaho Rapid Cycler thermocycler (Idaho Technology). DNA extracted from S. Typhimurium SL1344 was used as a control. PCR products were visualized by electrophoresis on 1% agarose gels, stained with SYBR Green Dye (Invitrogen) and ∼550 bp amplicons were excised from the gel. Amplicons obtained from 3 replicates of the same samples were pooled together. Products were purified from the agarose gel initially using the Qiagen QIAquick Gel Extraction Kit (Qiagen, Valencia, CA, United States), followed by the Agencourt AMpure magnetic beads (Beckman Coulter, Brea, CA, United States). Purified amplicons were resuspended in water, and the quality of the fragment was assessed by agarose gel electrophoresis and the concentration was measured with a Beckman DU640 spectrophotometer (Beckman Instruments, Fullerton, CA, United States). Barcoded 16S amplicon samples were submitted to the University of Georgia Genomics Facility for pyrosequencing using a 454 GS-FLX Titanium sequencing in accordance with established methods.
Processing, Assembly, and Analysis of Cecal 16S rRNA Libraries
All sequence processing was performed using MOTHUR software version 1.37.1 (Schloss et al., 2009). Sequences containing more than eight homopolymers nucleotides, and mismatched or ambiguous bases were removed. High-quality sequences were aligned against the SILVA database. UCHIME software was used to identify and remove chimeric sequences (Edgar et al., 2011). Operational taxonomic units (OTUs) were assigned at a 97% identity using the furthest-neighbor algorithm, and taxonomic assignments were made using the Ribosomal Database Project taxonomy – RDP (Cole et al., 2009). The error rate was assessed using the control sample, and the group was eliminated from our dataset for subsequent analysis (Schloss et al., 2009). Representative sequences of each OTU were classified using BLASTN (Altschul et al., 1990). All 16S rRNA sequence data is publicly available through National Center for Biotechnology Information (NCBI) the GenBank database under accession numbers KX913959 to KX914443. Rarefaction curves were produced as described by Hughes et al. (2001). The diversity indexes Chao, Shannon, Inverse Simpson and Smith Wilson Evenness indexes were calculated using MOTHUR. Chao index estimates the number of species (OTUs) comprising the microbial community, Shannon index determines how uniformly 16S rRNA sequences are spread into the different OTUs (Hill et al., 2003), and the Simpson index is an indication of the richness in a community with a uniform evenness that would have the same level of diversity. The inverted Simpson index was used to ensure that an increase in the reciprocal index reflects an increase in diversity (Magurran, 1988). A Venn diagram representing shared and unique OTUs was drawn for cecal communities with the two highest (1.4 × 109, 1.7 × 109 CFU/g) and lowest (PCR-negative) Salmonella abundance; representing permissive and exclusive communities, respectively (Shade and Handelsman, 2012).
Statistical Analysis
Cecal communities were compared using Metastats (White et al., 2009) which is based on a non-parametric t-test, used to identify OTUs associated with carriage status (p < 0.05). Significant OTUs, with abundance higher than 0.001% in the community (Stanley et al., 2014) were identified in all experimental samples. The correlation coefficient and r-squared calculated and trend lines were drawn using Excel (Langer and Microsoft Corporation, 2007).
Results
Salmonella Abundance in the Ceca of Experimentally Infected, Commercial Broiler Chickens
Salmonella abundance in the ceca was determined using qPCR for birds 21–42 days of age. The average Salmonella abundances were 6.8 × 108 (range of 1.0 × 107–1.6 × 109), 1.2 × 107 (range of 2.8 × 105–4.0 × 107), 1.7 × 107 (range of 0–7.3 × 107), and 5.1 × 104 (range of 0–2.0 × 105) CFU/g of ceca for broiler chickens at 21, 28, 35, and 42 days of age, respectively (Figure 1 and Table 1).
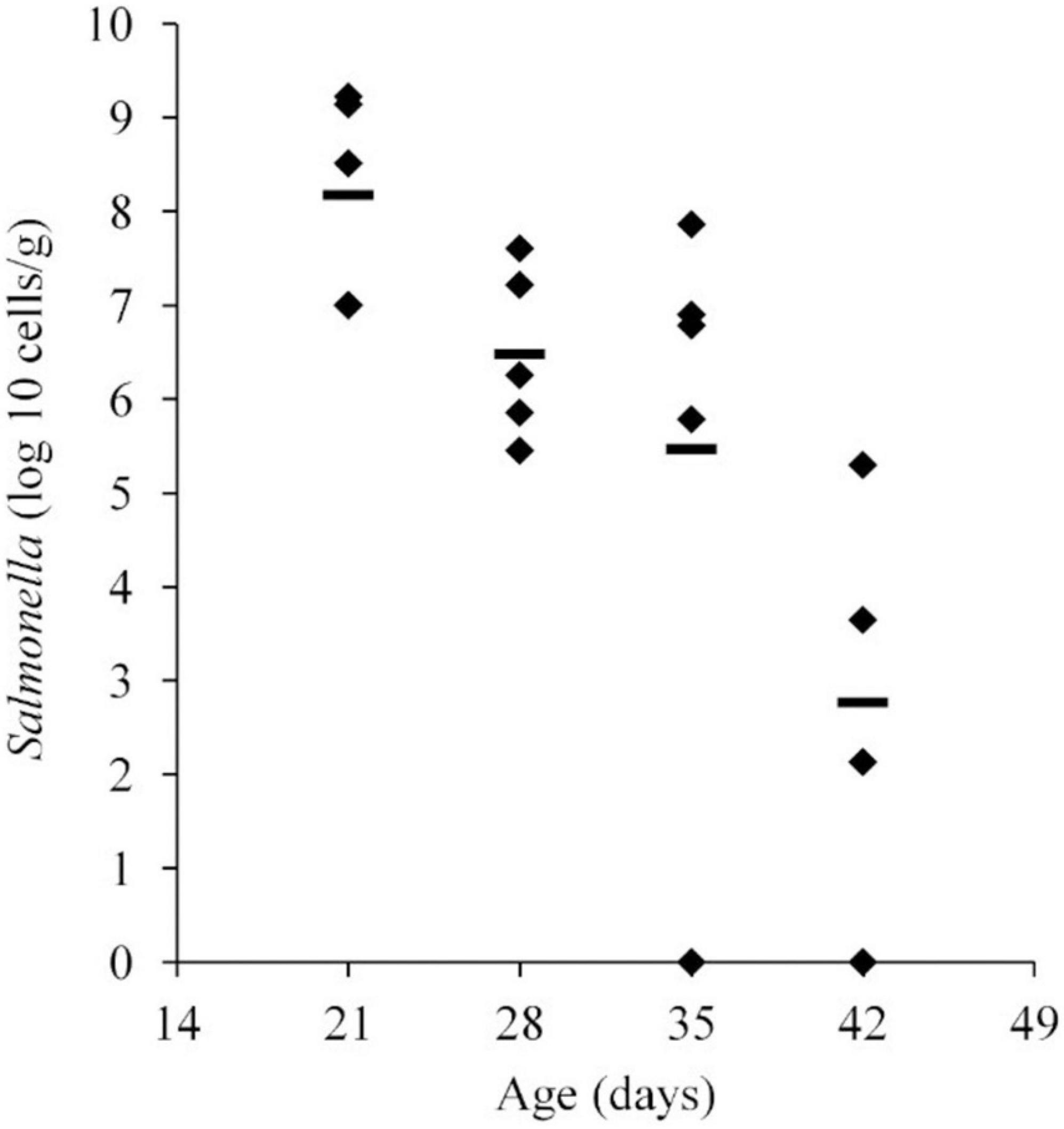
Figure 1. Salmonella abundance in cecal samples collected from chickens at 21, 28, 35, and 42 days old. Chickens were inoculated with 1.1 × 106 CFU of Salmonella Typhimurium at 2 days of age. The mean abundance is indicated by the black line.
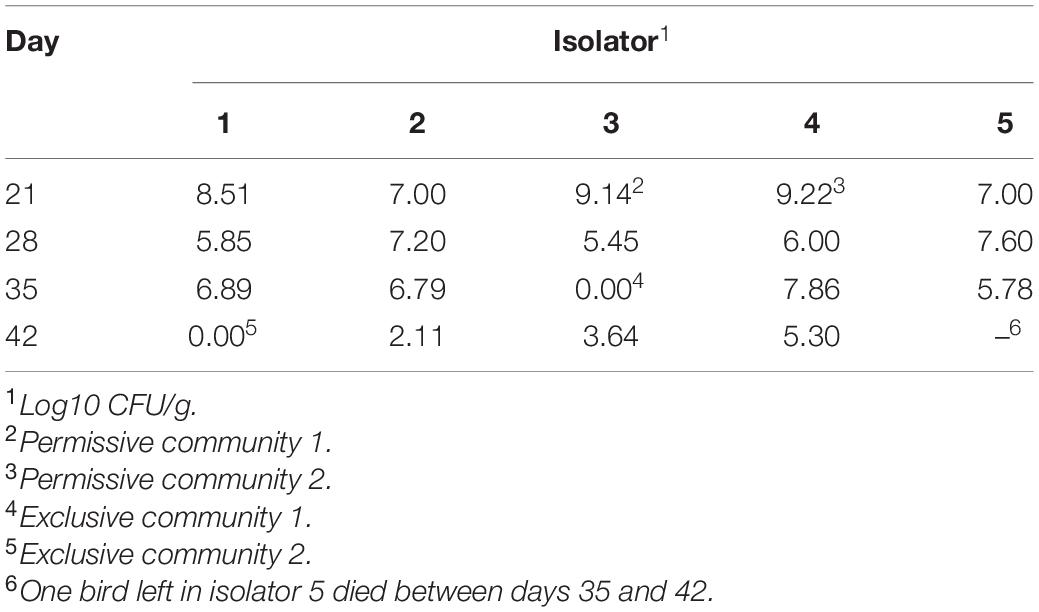
Table 1. Salmonella abundance in chicken ceca at 21, 28, 35, and 42 days old.
Salmonella Abundance Decreases in the Chicken Ceca With an Increase in Community Diversity
A detailed data analysis was focused first on cecal communities with the two highest and two lowest Salmonella abundances; representing permissive and exclusive communities, respectively. Differences observed between exclusive and permissive communities were later applied across all samples. Cecal communities where no Salmonella was detected by PCR were designated as Exclusive communities 1 and 2; for days 35 (35-3) and 42 (42-1), respectively (See Table 1). Similarly, cecal samples with the two highest Salmonella abundance on day 21 (Permissive community 1: 1.4 × 109; Permissive community 2: 1.7 × 109 CFU/g) were designated as Salmonella permissive communities (See Table 1). Permissive community 1 and Exclusive community 2 resulted from birds removed from the same isolator. The region V3-V6 of the bacterial 16S rRNA was sequenced from the 19 cecal samples containing variable abundances of Salmonella. A total of 212,990 high quality filtered sequences were randomly selected; 11,210 16S rRNA sequences per cecal community. The sequences clustered into 485 OTUs. The average Good’s coverage, a method for estimating the percentage of the total species represented, was 99.7% ± 0.1% (mean ± SD); suggesting a sufficient sampling of the cecal bacterial communities. Salmonella exclusive cecal communities showed rarefaction curves with higher diversity compared to the permissive cecal communities (Figure 2). There were additional differences between exclusive and permissive communities as measured using several diversity parameters (Table 2). The number of OTUs, representing different bacterial species, was significantly (χ2 < 0.001) lower in permissive communities (70 ± 8) compared to exclusive communities (117 ± 24). Similarly, Chao index was significantly (χ2 < 0.001) lower in permissive communities (92.65 ± 0.21) in comparison to exclusive communities (172.05 ± 48.01). Species diversity was higher in the Salmonella exclusive cecal communities by the Shannon index (2.661 ± 0.20 versus 2.331 ± 0.06) and inverse Simpson index (7.277 ± 2.146 versus 7.103 ± 0.213). However, evenness was slightly higher in permissive communities (0.536 ± 0.006 versus 0.522 ± 0.006) by the Smith Wilson index.
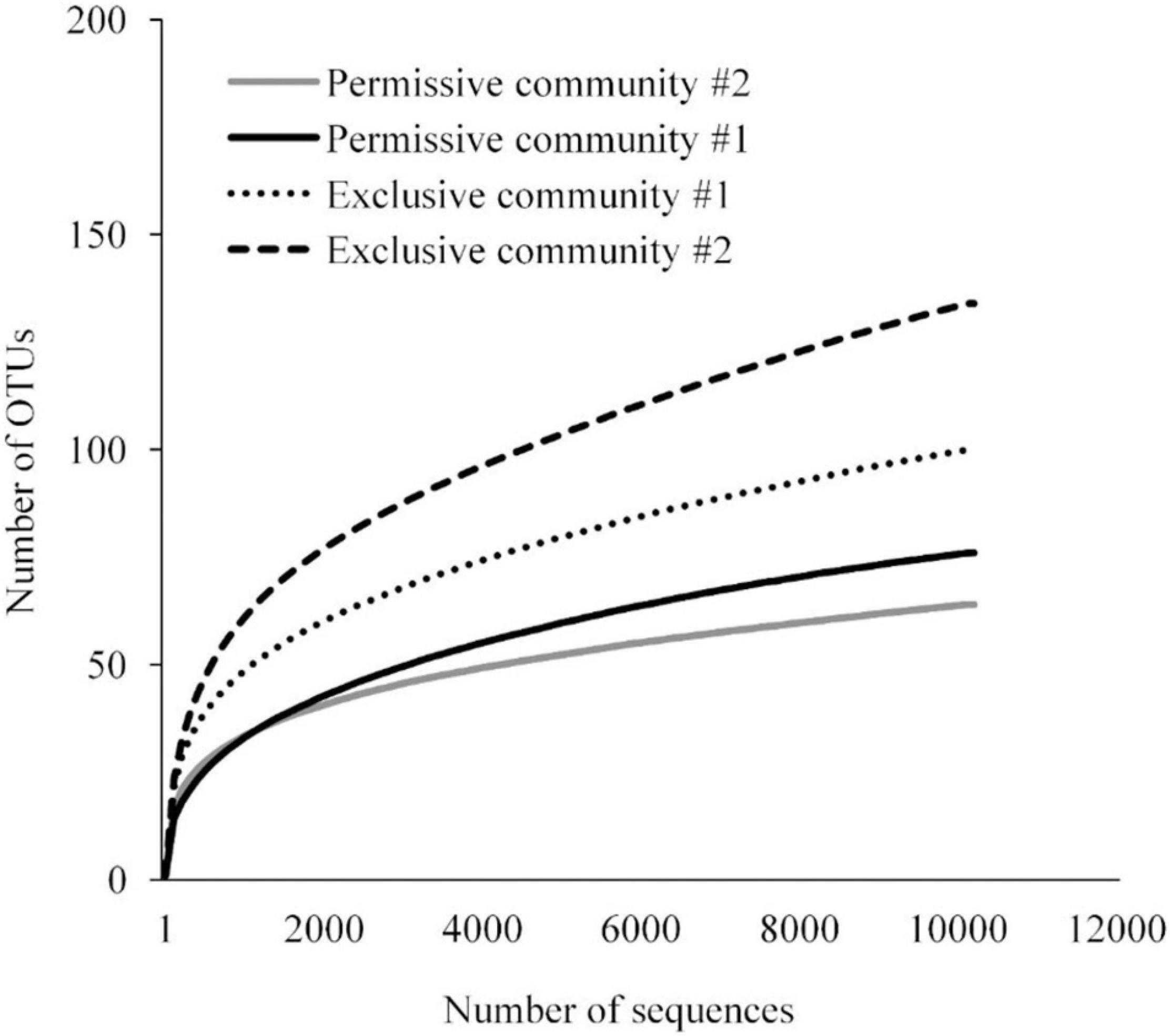
Figure 2. Rarefaction curve for bacterial 16S rRNA OTUs at 97% of similarity for Salmonella exclusive and permissive cecal communities.
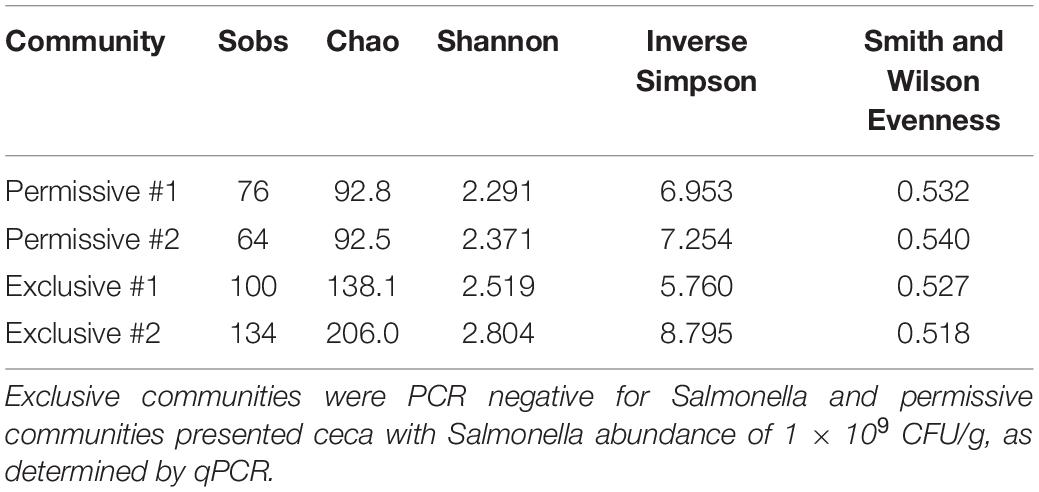
Table 2. Diversity indices for Salmonella exclusive or permissive cecal communities.
When these diversity indices were applied across all samples, a statistically significant negative correlation was observed between Salmonella abundance in the chicken ceca (expressed in logs) and the number of OTUs (P < 0.01), Chao index (P < 0.01), and Shannon index (P < 0.01) (n = 19) (Figure 3). While there appeared to be a negative correlation between Salmonella abundance and diversity, as measured by the inverse Simpson index, this correlation was not significant by the Pearson correlation coefficient (p = 0.18). Evenness presented a statistically significant positive correlation with the Salmonella cecal abundance (P < 0.02).
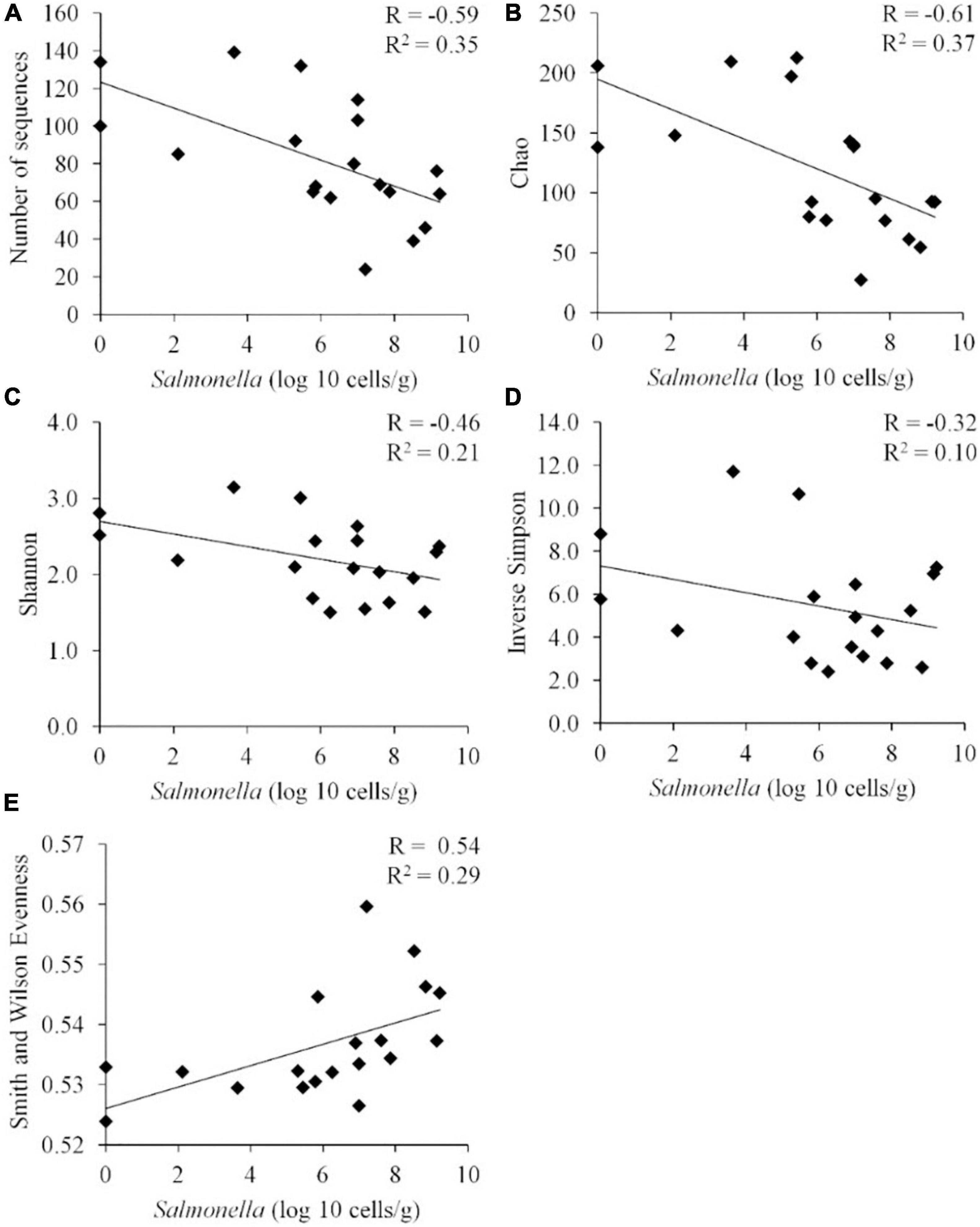
Figure 3. Correlation between the Salmonella abundance and community diversity for all cecal communities (n = 19). Community diversity was measured by number of unique OTUs (A) (P < 0.01), Chao index (B) (P < 0.01), Shannon index (C) (P < 0.01), inverted Simpson index (D) (p = 0.18), and Smith Wilson evenness index (E) (P < 0.02). R, Pearson correlation coefficient; and R2, coefficient of determination.
Species Composition of Salmonella Exclusive and Permissive Cecal Communities and the Identification of Bacterial Species That May Influence Salmonella Abundance
Averaging across samples, the most abundant phyla observed in the cecal microbiota were Firmicutes (96.9%) and Proteobacteria (0.6%); and members of Clostridiales accounted for 90% of the total community 16S rRNA sequences (n = 193,003). The most abundant bacterial groups present in the ceca were unclassified Lachnospiraceae (37.2%), Roseburia (20.5%), Clostridium XI (13.5%), Clostridium XIVa (5.0%), Blautia (4.0%), unclassified Firmicutes (3.1%), unclassified Ruminococcaceae (3.0%), unclassified Bacteria (2.4%), unclassified Clostridiales (2.1%), Faecalibacterium (1.4%), Oscillibacter (0.5%), Enterobacteriaceae (0.5%), unclassified Erysipelotrichaceae (0.5%), unclassified Bacillales (0.4%), Flavonifractor (0.2%), and Enterococcus (0.2%).
The Salmonella exclusive community had a larger proportion of Faecalibacterium (P < 0.05), Turicibacter (P < 0.02), and unclassified Firmicutes (P < 0.005); and less Blautia (P < 0.01), and unclassified Lachnospiraceae (P < 0.05) compared to the permissive community. Twenty-six OTUs unique to exclusive communities were identified (Figure 4). These OTUs represent 6.99% of the total sequences for these cecal communities (n = 1,569). A smaller number of OTUs were unique to the permissive communities; 7 OTUs that represented 0.25% of the total 16S rRNA sequences (n = 58) (Table 2). A more diverse set of OTUs were observed in exclusive communities. The OTUs observed in permissive communities were affiliated with the phyla Firmicutes and Proteobacteria, while OTUs observed in exclusive communities belonged to the Firmicutes, Proteobacteria and Actinobacteria phyla. OTUs observed in permissive communities were related to the Enterobacteriaceae, Enterococcaceae, Lachnospiraceae, and Clostridiaceae families, while OTUs presented in exclusive communities were related to Hyphomicrobiaceae, Eggerthellaceae, Clostridiaceae, Eubacteriaceae, Oscillospiraceae, Ruminococcaceae, and Lachnospiraceae families (Table 3).
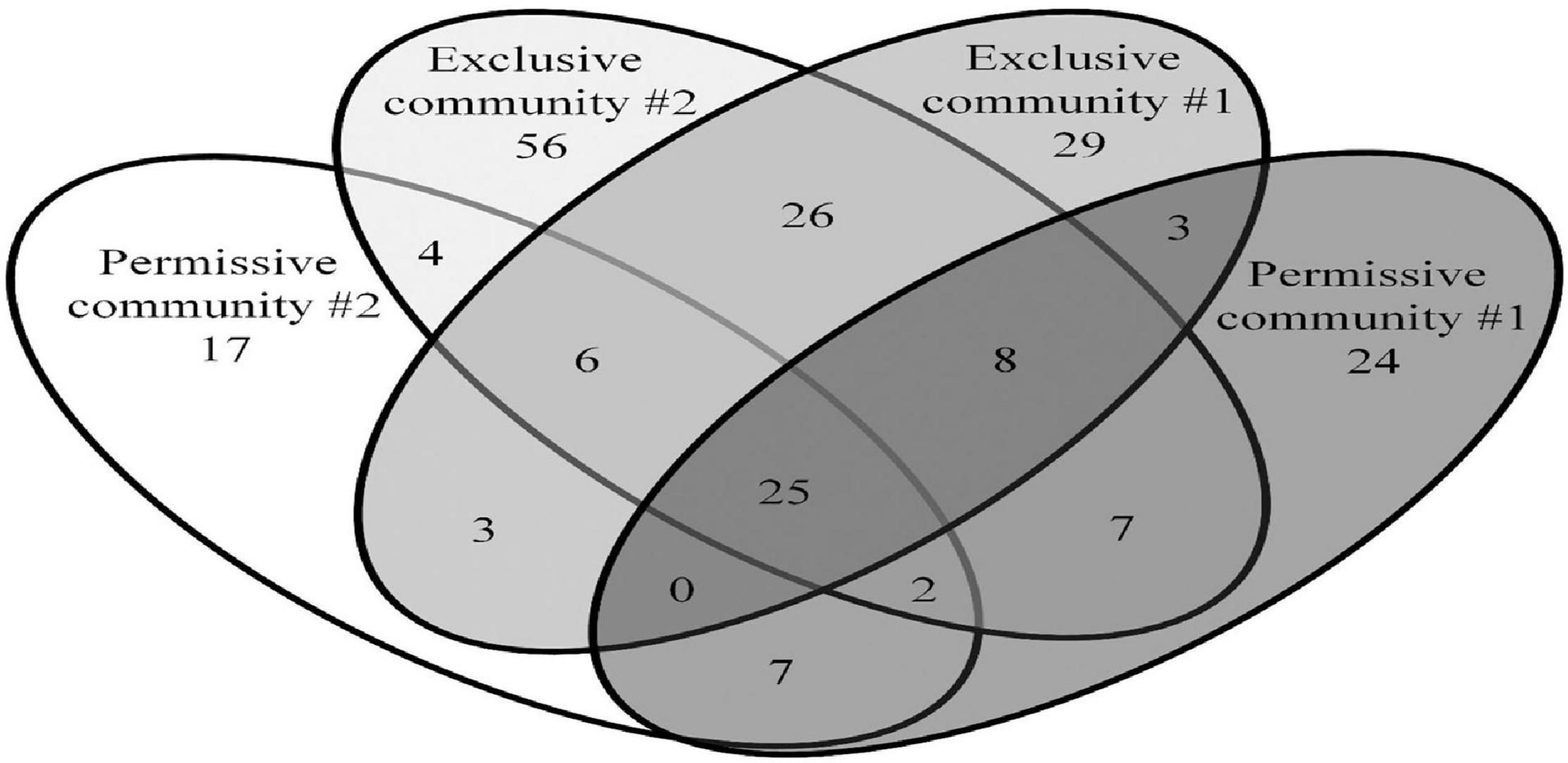
Figure 4. Shared and unique OTUs observed in Salmonella exclusive and permissive cecal communities. Numbers below groups indicate the number of OTUs.

Table 3. OTUs unique to Salmonella exclusive or permissive cecal communities.
Metastats (Schloss et al., 2009) was used to quantify differences between groups and identify OTUs that most strongly influence the differences observed between the Salmonella permissive and exclusive communities (p < 0.05). Only OTUs with total abundance higher than 0.001% were considered in this analysis. Seven OTUs were identified (Figure 5). There was a greater frequency of OTUs related to Eubacterium biforme (OTU 79, 93% of similarity to RDP database), Roseburia hominis (OTU 6, presenting 97% of similarity), Robinsoniella peoriensis (OTU 4, 98% of similarity), and Roseburia hominis (OTU 3, 97% of similarity) in the Salmonella permissive communities. Of these 4 OTUs, there was a positive correlation between Roseburia hominis OTU 3 (97%, P < 0.01), and OTU 6 (97%, P < 0.05) with Salmonella abundance across all samples (n = 19) (Figure 6). These 4 OTUs accounted for 24.7% of the total 16S rRNA sequences for cecal samples with 109 Salmonella cells/g and just 7.0% of the total sequences for Salmonella-negative cecal samples. Ethanoligenens harbinense (OTU 31, 95%), Oscillibacter ruminantium (OTU 28, 94%) and Faecalibacterium prausnitzii (OTU 10, 98%) were more abundant in the Salmonella exclusive cecal communities. There was a negative correlation between the presence of Faecalibacterium prausnitzii (OTU 10, 97% of similarity to RDP, P < 0.05), Oscillibacter ruminantium (OTU 28, 95%, P < 0.001) and Ethanoligenens harbinense (OTU 31, 95%, P < 0.01) with Salmonella abundance across all samples (n = 19) (Figure 7); and accounted for 6.1% of total 16S rRNA sequences for Salmonella, PCR-negative cecal samples. These OTUs represented just 0.04% of total sequences in cecal samples with 109 Salmonella cells/g.
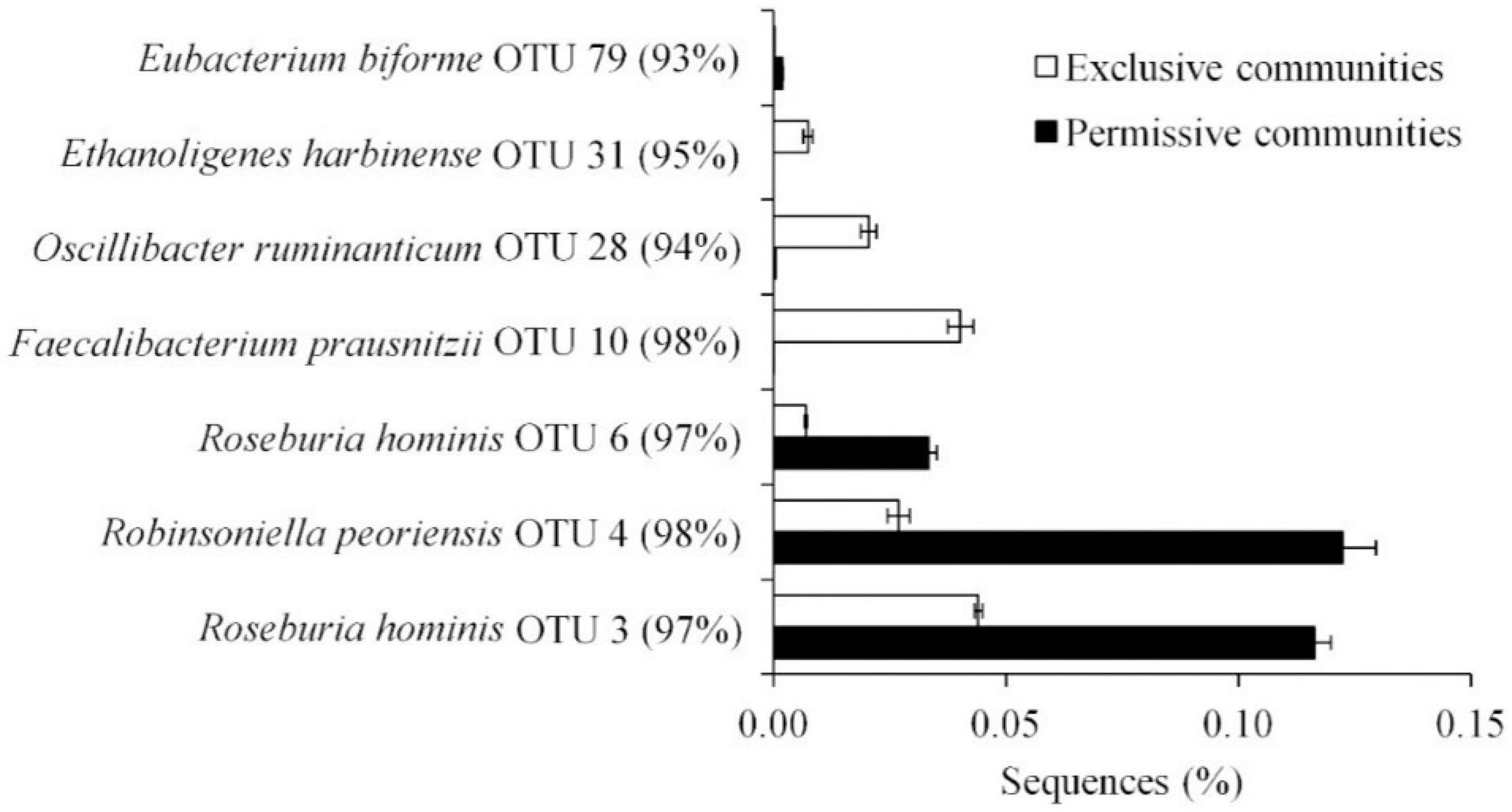
Figure 5. OTUs representing more than 0.01% of the total sequences and significantly (P < 0.05) associated with Salmonella exclusive and permissive cecal communities identified by Metastats.
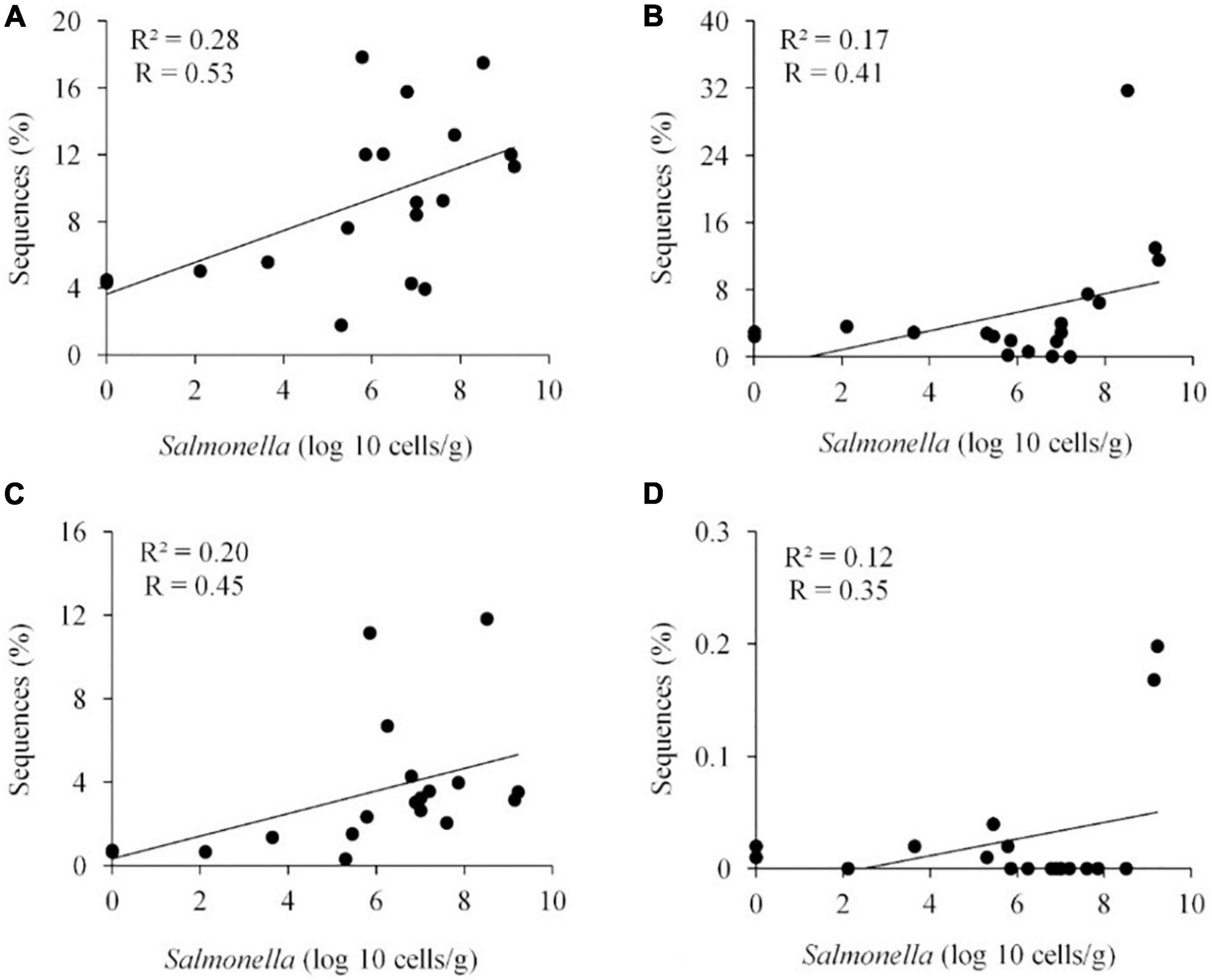
Figure 6. Positive correlation between the presence of specific OTUs and Salmonella abundance in the ceca (n = 19). Roseburia hominis OTU 3, 97% of similarity to RDP, P < 0.01 (A); Robinsoniella peoriensis OTU 4, 98%, NS (B); Roseburia hominis OTU 6, 97%, P < 0.05 (C); and Eubacterium biforme OTU 79, 93%, NS (D). R, Pearson correlation coefficient; and R2, coefficient of determination.
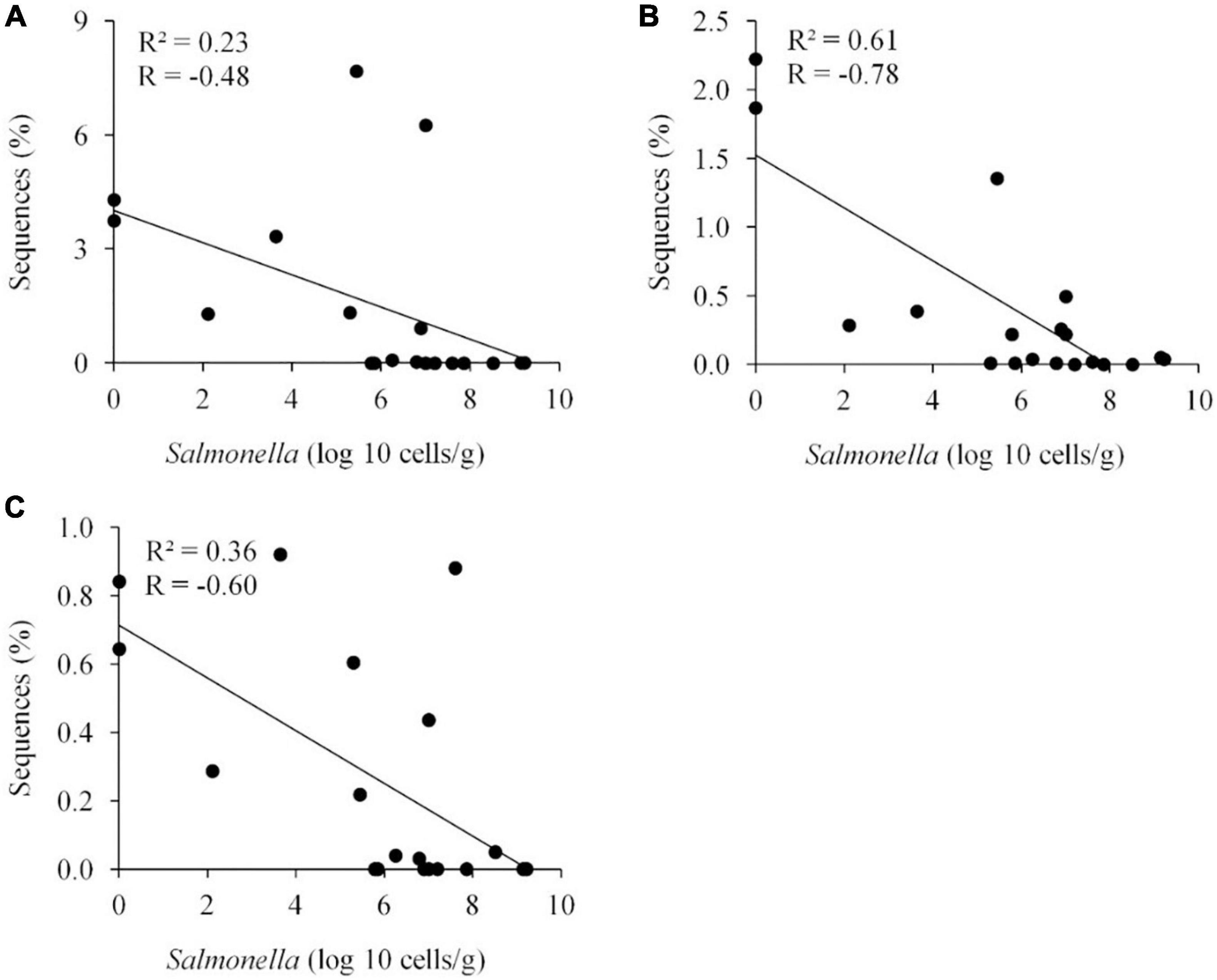
Figure 7. Negative correlation between the presence of specific OTUs and Salmonella abundance in the ceca (n = 19). Faecalibacterium prausnitzii OTU 10, 98%, P < 0.05 (A); Oscillibacter ruminantium OTU 28, 95%, P < 0.001 (B); and Ethanoligenens harbinense OTU 31, 95%, P < 0.01 (C). R, Pearson correlation coefficient; and R2, coefficient of determination.
Discussion
Unlike neonatal mammals, chicks seldom present symptoms of illness unless they are administered a large Salmonella challenge dose, at day of hatch. However, chicks orally administered Salmonella at 2 days of age exhibit little if any disease symptoms, and Salmonella abundance rapidly increases during the 1st week to 106–107 CFU/g and remains at these levels for 2–3 weeks. However, by the 4th week of age, there is a substantial decline in Salmonella abundance (Cheng et al., 2015). The composition of the intestinal community is also in flux up to the 3rd week of age when a distinct, stable community structure develops in the chicken ileum and ceca at approximately the same time that Salmonella abundance decreases (Lu et al., 2003a). Consequently, intestinal microbiota from chickens 21 days of age or older prevents Salmonella colonization in 8-day old layer chickens (Varmuzova et al., 2016). In this study, a wide difference in Salmonella abundance was observed in chickens at 35 and 42 days of age where 20% of birds were negative. This allowed the opportunity to compare intestinal community composition in birds with a range of Salmonella abundance.
Intestinal community diversity appears to adversely affect Salmonella abundance in chickens. Animals with highly diverse intestinal communities have been shown to be resistant to pathogen colonization and disease (Kamada et al., 2013). Bacterial diversity especially appears to be a significant factor affecting enteropathogen prevalence and abundance for shiga-toxin producing Escherichia coli (STEC) in cattle (Xu et al., 2014; Chopyk et al., 2016) and soil (van Elsas et al., 2012), C. difficile in humans (Antharam et al., 2013; Zhang et al., 2015), C. perfringens in chickens (Stanley et al., 2014), and Campylobacter jejuni in mice (Lone et al., 2013). In addition, community diversity seems to be an important predictor of intestinal health (Antharam et al., 2013; Wills et al., 2014; McMurtry et al., 2015; Rodriguez et al., 2015; Singh et al., 2015). A negative correlation between Salmonella abundance and evenness was also observed in the distribution of bacterial species. Evenness represents the degree to which species are distributed within a population. A lower evenness score for the Salmonella permissive community infers that a few bacterial species dominate the community. Similar findings have been reported for Salmonella in pigs and diarrheal illnesses in horses where lower evenness scores were associated with pathogen prevalence, abundance and symptoms of disease (Bearson et al., 2013; Rodriguez et al., 2015).
Intestinal community diversity appears to be a function of age, increasing as birds mature (Lu et al., 2003a; Crhanova et al., 2011; Videnska et al., 2013; Azcarate-Peril et al., 2018). The greatest diversity appears to be in the cecum, an intestinal compartment where Salmonella persists in the chicken (Azcarate-Peril et al., 2018). It was important to have all the birds exposed to the same treatment in order to control the possibility that Salmonella infection itself may alter the cecal community composition and species abundance (Videnska et al., 2013; Azcarate-Peril et al., 2018; Mon et al., 2020). With the exception of He et al. (2003), most studies did not observe a negative correlation between cecal community diversity and Salmonella abundance or prevalence (Crhanova et al., 2011; Videnska et al., 2013; Azcarate-Peril et al., 2018; Mon et al., 2020). In fact, one study observed an increase in species richness with Salmonella infection (Mon et al., 2020). It has been shown that changes in the intestinal community composition may result in a proteobacterial bloom favoring conditions of Salmonella proliferation (Singh et al., 2015; Zhang et al., 2015). Salmonella can also induce inflammation which reduces its obligate anaerobic competitors while providing it with additional nutrients for growth (Winter et al., 2010; Thiennimitr et al., 2011; Rivera-Chavez et al., 2016). While Salmonella may change the gut microbiome in chickens, it appears to involve a mechanism distinct from mammals (Rimet et al., 2019). However, these studies focused on chickens less than 21 days of age, early in chicken intestinal community development (Lu et al., 2003a). Varmuzova et al. (2016) demonstrated that only the intestinal microbiota from birds 21 days of age or older could reduce Salmonella colonization.
Specific genera and species were identified that correlated with Salmonella abundance in the chicken cecum. The majority of these OTUs were related to Clostridiales, the most abundant group of the chicken ceca (Lu et al., 2003a), with six OTUs identified as Ethanoligenens harbinense, Oscillibacter ruminantium, Faecalibacterium prausnitzii, Roseburia hominis, Eubacterium biforme, and Robinsoniella peoriensis. These genera or species have also been associated with enteropathogen colonization or intestinal health in other studies (Antharam et al., 2013; Miquel et al., 2013; Rossi et al., 2014; Stanley et al., 2014; Xu et al., 2014; Thibodeau et al., 2015; Knoll et al., 2016). Others have also noted absence or reduced abundance of F. prausnitzii and member species of the clostridial families (Collins et al., 1994) Lachnospiraceae (XIV) and Ruminococcaceae (III/IV) in birds colonized with Salmonella (Pourabedin et al., 2017; Liu et al., 2018; Khan and Chousalkar, 2020; Ding et al., 2021). Several of these species, associated with exclusive communities but absent from the Salmonella permissive community, may play an anti-inflammatory role in maintaining intestinal homeostasis and health (Wu and Wu, 2012; Miquel et al., 2013). Inflammation results in the production of tetrathionate and other metabolites which can be used to enhance Salmonella growth and thereby improve its persistence, and spread (Winter et al., 2010; Thiennimitr et al., 2011). However, these clostridial species also are likely to produce short chain fatty-acids (SCFA) such as butyrate, which reduces inflammation in the chicken intestine (Wu et al., 2016). Butyrate also represses expression of the Salmonella cell-invasion locus in SPI1 (Gantois et al., 2006) reducing its ability to elicit intestinal inflammation (Rivera-Chavez et al., 2016). Clostridiales abundance in the avian intestine may explain why birds are more resistant than mammals to Salmonella gastroenteritis.
Interestingly, several intestinal species may also have a positive impact on enteropathogen colonization (Xu et al., 2014; Thibodeau et al., 2015). Conceptually more emphasis is placed on pathogen exclusive species and mechanisms that explain their inhibitory effects, ignoring possible synergism between the pathogen and pathogen-permissive species as an alternate explanation behind competitive exclusion. The chicken intestinal microbiota can also produce fermentation end-products and other metabolites that can be metabolically exploited by Salmonella (Cheng et al., 2015). The metabolism of permissive species may result in cooperation with the pathogen, while the exclusive species may compete with the enteropathogen for these metabolites or out-compete the permissive species within the ecosystem. In either scenario, Salmonella would be unable to thrive in the chicken intestine.
While other studies have reported negative or positive correlations between intestinal community composition, abundance of specific genera/species and Salmonella abundance or prevalence (Videnska et al., 2013; Azcarate-Peril et al., 2018), this has been a tenuous association as reflected in comparisons among studies (Azcarate-Peril et al., 2018; Ma et al., 2020; Mon et al., 2020) and trials (Videnska et al., 2013). The exclusive genera/species, identified in this study, have been associated with Salmonella exclusion in some studies (Pourabedin et al., 2017; Liu et al., 2018; Khan and Chousalkar, 2020; Ding et al., 2021), but absent in others (Videnska et al., 2013; Azcarate-Peril et al., 2018; Ma et al., 2020; Mon et al., 2020). The genera/species, identified in this study, that negatively correlate with Salmonella are also absent in a competitive exclusion product known to effectively reduce Salmonella colonization in poultry (Pedroso et al., 2016). But the dominant member genera and species, present in this competitive exclusion product, are transient in chickens fed the product (Pedroso et al., 2016) indicating that the mechanism of competitive exclusion is complex. The contradictions may reflect the inherent nature of community diversity, which ensures there are always protagonists present to prevent enteropathogen colonization or illness in the animal population.
Conclusion
Increased bacterial diversity and the composition of the cecal microbiota adversely affected Salmonella colonization in chickens. The isolation of the bacterial species associated with pathogen abundance are necessary to better understand the microbe-microbe interactions that exclude or permit pathogen persistence and a better understanding of the mechanism of competitive exclusion. Moreover, this work provides the fundamental first step toward the development of next generation, direct fed microbials that target and exclude enteropathogens from poultry.
Data Availability Statement
All 16S rDNA sequence data is publicly available through National Center for Biotechnology Information (NCBI) the GenBank database under accession numbers KX913959 to KX914443.
Ethics Statement
The animal study was reviewed and approved by the University of Georgia Animal Care and Use and Procedures Committee.
Author Contributions
ML, AP, and JM: conceptualization and writing—review and editing. AP: methodology, formal analysis, and data curation. AP and JM: writing—original draft preparation. ML: supervision and project administration. ML and JM: funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the United States Department of Agriculture (2009-03561; VA-160130).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acosta-Martinez, V. S. K., Dowd, S. E., Sun, Y., and Allen, V. (2008). Tag encoded pyrosequencing analysis of bacterial diversity in a single soil type as affacted by management and land use. Soil Biol. Biochem. 40, 2762-2770.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Antharam, V. C., Li, E. C., Ishmael, A., Sharma, A., Mai, V., Rand, K. H., et al. (2013). Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol. 51, 2884–2892. doi: 10.1128/jcm.00845-13
Azcarate-Peril, M. A., Butz, N., Cadenas, M. B., Koci, M., Ballou, A., Mendoza, M., et al. (2018). An attenuated Salmonella enterica serovar typhimurium strain and galacto-oligosaccharides accelerate clearance of Salmonella infections in poultry through modifications to the gut microbiome. Appl. Environ. Microbiol. 84:e02526-17.
Baumler, A. J., Tsolis, R. M., Ficht, T. A., and Adams, L. G. (1998). Evolution of host adaptation in Salmonella enterica. Infect. Immun. 66, 4579–4587.
Bearson, S. M., Allen, H. K., Bearson, B. L., Looft, T., Brunelle, B. W., Kich, J. D., et al. (2013). Profiling the gastrointestinal microbiota in response to Salmonella: low versus high Salmonella shedding in the natural porcine host. Infect. Genet. Evol. 16, 330–340. doi: 10.1016/j.meegid.2013.03.022
Cheng, Y., Pedroso, A. A., Porwollik, S., Mcclelland, M., Lee, M. D., Kwan, T., et al. (2015). rpoS-Regulated core genes involved in the competitive fitness of Salmonella enterica serovar Kentucky in the intestines of chickens. Appl. Environ. Microbiol. 81, 502–514. doi: 10.1128/aem.03219-14
Chopyk, J., Moore, R. M., Dispirito, Z., Stromberg, Z. R., Lewis, G. L., Renter, D. G., et al. (2016). Presence of pathogenic Escherichia coli is correlated with bacterial community diversity and composition on pre-harvest cattle hides. Microbiome 4;9.
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., et al. (2009). The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145.
Collins, M. D., Lawson, P. A., Willems, A., Cordoba, J. J., Fernandez-Garayzabal, J., Garcia, P., et al. (1994). The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int. J. Syst. Bacteriol. 44, 812–826. doi: 10.1099/00207713-44-4-812
Crhanova, M., Hradecka, H., Faldynova, M., Matulova, M., Havlickova, H., Sisak, F., et al. (2011). Immune response of chicken gut to natural colonization by gut microflora and to Salmonella enterica serovar enteritidis infection. Infect. Immun. 79, 2755–2763. doi: 10.1128/iai.01375-10
Daum, L. T., Barnes, W. J., Mcavin, J. C., Neidert, M. S., Cooper, L. A., Huff, W. B., et al. (2002). Real-time PCR detection of salmonella in suspect foods from a gastroenteritis outbreak in kerr county, Texas. J. Clin. Microbiol. 40, 3050–3052. doi: 10.1128/jcm.40.8.3050-3052.2002
Ding, J., Zhou, H., Luo, L., Xiao, L., Yang, K., Yang, L., et al. (2021). Heritable gut microbiome associated with Salmonella enterica serovar pullorum infection in chickens. mSystems 6:e01192-20..
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fey, A., Eichler, S., Flavier, S., Christen, R., Hofle, M. G., and Guzman, C. A. (2004). Establishment of a real-time PCR-based approach for accurate quantification of bacterial RNA targets in water, using Salmonella as a model organism. Appl. Environ. Microbiol. 70, 3618–3623. doi: 10.1128/aem.70.6.3618-3623.2004
Galan, J. E., and Curtiss, R. III (1989). Cloning and molecular characterization of genes whose products allow Salmonella typhimurium to penetrate tissue culture cells. Proc. Natl. Acad. Sci. U.S.A. 86, 6383–6387. doi: 10.1073/pnas.86.16.6383
Gantois, I., Ducatelle, R., Pasmans, F., Haesebrouck, F., Hautefort, I., Thompson, A., et al. (2006). Butyrate specifically down-regulates salmonella pathogenicity island 1 gene expression. Appl. Environ. Microbiol. 72, 946–949. doi: 10.1128/aem.72.1.946-949.2006
Garcia, S. L., Jangid, K., Whitman, W. B., and Das, K. C. (2011). Transition of microbial communities during the adaption to anaerobic digestion of carrot waste. Bioresour. Technol. 102, 7249–7256. doi: 10.1016/j.biortech.2011.04.098
Gong, J., Forster, R. J., Yu, H., Chambers, J. R., Sabour, P. M., Wheatcroft, R., et al. (2002). Diversity and phylogenetic analysis of bacteria in the mucosa of chicken ceca and comparison with bacteria in the cecal lumen. FEMS Microbiol. Lett. 208, 1–7. doi: 10.1111/j.1574-6968.2002.tb11051.x
Hamady, M., Walker, J. J., Harris, J. K., Gold, N. J., and Knight, R. (2008). Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods 5, 235–237. doi: 10.1038/nmeth.1184
Hapfelmeier, S., Ehrbar, K., Stecher, B., Barthel, M., Kremer, M., and Hardt, W. D. (2004). Role of the Salmonella pathogenicity island 1 effector proteins SipA, SopB, SopE, and SopE2 in Salmonella enterica subspecies 1 serovar typhimurium colitis in streptomycin-pretreated mice. Infect. Immun. 72, 795–809. doi: 10.1128/iai.72.2.795-809.2004
He, Y., Yang, Y., Dong, Y., Ito, K., and Zhang, B. (2020). Highly nutritious diet resists Salmonella Typhimurium infections by improving intestinal microbiota and morphology in broiler chickens. Poult. Sci. 99, 7055–7065.
Hill, T. C., Walsh, K. A., Harris, J. A., and Moffett, B. F. (2003). Using ecological diversity measures with bacterial communities. FEMS Microbiol. Ecol. 43, 1–11. doi: 10.1111/j.1574-6941.2003.tb01040.x
Hughes, J. B., Hellmann, J. J., Ricketts, T. H., and Bohannan, B. J. (2001). Counting the uncountable: statistical approaches to estimating microbial diversity. Appl. Environ. Microbiol. 67, 4399–4406. doi: 10.1128/aem.67.10.4399-4406.2001
Jung, H. C., Eckmann, L., Yang, S. K., Panja, A., Fierer, J., Morzycka-Wroblewska, E., et al. (1995). A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J. Clin. Invest. 95, 55–65. doi: 10.1172/jci117676
Kamada, N., Chen, G. Y., Inohara, N., and Nunez, G. (2013). Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 14, 685–690. doi: 10.1038/ni.2608
Khan, S., and Chousalkar, K. K. (2020). Salmonella typhimurium infection disrupts but continuous feeding of Bacillus based probiotic restores gut microbiota in infected hens. J. Anim. Sci. Biotechnol. 11:29.
Knoll, R. L., Forslund, K., Kultima, J. R., Meyer, C. U., Kullmer, U., Sunagawa, S., et al. (2016). Gut microbiota differs between children with inflammatory bowel disease and healthy siblings in taxonomic and functional composition–a metagenomic analysis. Am. J. physiol. Gastrointest. Liver Physiol. 312, G327-G339.
Lan, P. T., Hayashi, H., Sakamoto, M., and Benno, Y. (2002). Phylogenetic analysis of cecal microbiota in chicken by the use of 16S rDNA clone libraries. Microbiol. Immunol. 46, 371–382. doi: 10.1111/j.1348-0421.2002.tb02709.x
Langer, M., and Microsoft Corporation (2007). Microsoft Office Excel 2007 for Windows. Berkeley, CA: Peachpit Press.
Lee, M. D., Lu, J., Harmon, B., Hofacre, C. L., and Maurer, J. J. (2006). “Molecular basis for AGP effects in poultry,” in Antimicrobial Growth Promoters: Where Do We Go From Here?, eds D. Barug, J. De Jong, A. K. Kies, and M. W. A. Verstegen (Wageningen: Wageningen Academic Pub), 149–163.
Lichtensteiger, C. A., and Vimr, E. R. (2003). Systemic and enteric colonization of pigs by a hilA signature-tagged mutant of Salmonella choleraesuis. Microb. Pathog. 34, 149–154. doi: 10.1016/s0882-4010(02)00196-1
Liljebjelke, K. A., Hofacre, C. L., Liu, T., White, D. G., Ayers, S., Young, S., et al. (2005). Vertical and horizontal transmission of salmonella within integrated broiler production system. Foodborne Pathog. Dis. 2, 90–102. doi: 10.1089/fpd.2005.2.90
Liu, L., Lin, L., Zheng, L., Tang, H., Fan, X., Xue, N., et al. (2018). Cecal microbiome profile altered by Salmonella enterica, serovar Enteritidis inoculation in chicken. Gut Pathog. 10:34.
Lone, A. G., Selinger, L. B., Uwiera, R. R., Xu, Y., and Inglis, G. D. (2013). Campylobacter jejuni colonization is associated with a dysbiosis in the cecal microbiota of mice in the absence of prominent inflammation. PLoS One 8:e75325. doi: 10.1371/journal.pone.0075325
Lu, J., Idris, U., Harmon, B., Hofacre, C., Maurer, J. J., and Lee, M. D. (2003a). Diversity and succession of the intestinal bacterial community of the maturing broiler chicken. Appl. Environ. Microbiol. 69, 6816–6824. doi: 10.1128/aem.69.11.6816-6824.2003
Lu, J., Sanchez, S., Hofacre, C., Maurer, J. J., Harmon, B. G., and Lee, M. D. (2003b). Evaluation of broiler litter with reference to the microbial composition as assessed by using 16S rRNA and functional gene markers. Appl. Environ. Microbiol. 69, 901–908. doi: 10.1128/aem.69.2.901-908.2003
Ma, B., Mei, X., Lei, C., Li, C., Gao, Y., Kong, L., et al. (2020). enrofloxacin shifts intestinal microbiota and metabolic profiling and hinders recovery from Salmonella enterica subsp. enterica serovar typhimurium infection in neonatal chickens. mSphere 5, e00725-20.
Magurran, A. E. (1988). Ecological Diversity and Its Measurement. Princeton, NJ: Princeton University Press.
Malorny, B., Paccassoni, E., Fach, P., Bunge, C., Martin, A., and Helmuth, R. (2004). Diagnostic real-time PCR for detection of Salmonella in food. Appl. Environ. Microbiol. 70, 7046–7052. doi: 10.1128/aem.70.12.7046-7052.2004
McMurtry, V. E., Gupta, R. W., Tran, L., Blanchard, E. E. T., Penn, D., Taylor, C. M., et al. (2015). Bacterial diversity and Clostridia abundance decrease with increasing severity of necrotizing enterocolitis. Microbiome 3:11.
Miquel, S., Martin, R., Rossi, O., Bermudez-Humaran, L. G., Chatel, J. M., Sokol, H., et al. (2013). Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 16, 255–261. doi: 10.1016/j.mib.2013.06.003
Mon, K. K. Z., Zhu, Y., Chanthavixay, G., Kern, C., and Zhou, H. (2020). Integrative analysis of gut microbiome and metabolites revealed novel mechanisms of intestinal Salmonella carriage in chicken. Sci. Rep. 10:4809.
Nakamura, A., Ota, Y., Mizukami, A., Ito, T., Ngwai, Y. B., and Adachi, Y. (2002). Evaluation of aviguard, a commercial competitive exclusion product for efficacy and after-effect on the antibody response of chicks to Salmonella. Poul. Sci. 81, 1653–1660. doi: 10.1093/ps/81.11.1653
Nurmi, E., Nuotio, L., and Schneitz, C. (1992). The competitive exclusion concept: development and future. Int. J. Food microbiol. 15, 237–240. doi: 10.1016/0168-1605(92)90054-7
Nurmi, E., and Rantala, M. (1973). New aspects of Salmonella infection in broiler production. Nature 241, 210–211. doi: 10.1038/241210a0
Pedroso, A. A., Batal, A. B., and Lee, M. D. (2016). Effect of in ovo administration of an adult-derived microbiota on establishment of the intestinal microbiome in chickens. Am. J. Vet. Res. 77, 514–526. doi: 10.2460/ajvr.77.5.514
Pourabedin, M., Chen, Q., Yang, M., and Zhao, X. (2017). Mannan- and xylooligosaccharides modulate caecal microbiota and expression of inflammatory-related cytokines and reduce caecal Salmonella Enteritidis colonisation in young chickens. FEMS Microbiol. Ecol. 93:fiw226. doi: 10.1093/femsec/fiw226
Provence, D. L., and Curtiss, R. III (1994). “Gene Transfer in Gram-Negative Bacteria,” in Methods for General and Molecular Bacteriology, eds P. Gerhardt, R. G. E. Murray, W. A. Wood, and N. R. Krieg (Washington, DC: ASM Press), 317–364.
Rimet, C. S., Maurer, J. J., Berghaus, R. D., Jordan, B. J., Da Silva, L. H. A., Stabler, L. J., et al. (2019). The contribution of eimeria coinfection and intestinal inflammation to cecal colonization and systemic spread of Salmonella typhimurium deficient in tetrathionate reductase or type III secretion systems Salmonella pathogenicity island 1 or 2. Avian Dis. 63, 559–567. doi: 10.1637/aviandiseases-d-19-00082
Rivera-Chavez, F., Zhang, L. F., Faber, F., Lopez, C. A., Byndloss, M. X., Olsan, E. E., et al. (2016). Depletion of butyrate-producing clostridia from the gut microbiota drives an aerobic luminal expansion of Salmonella. Cell Host Microbe 19, 443–454. doi: 10.1016/j.chom.2016.03.004
Rodriguez, C., Taminiau, B., Brevers, B., Avesani, V., Van Broeck, J., Leroux, A., et al. (2015). Faecal microbiota characterisation of horses using 16 rdna barcoded pyrosequencing, and carriage rate of clostridium difficile at hospital admission. BMC Microbiol. 15:181. doi: 10.1186/s12866-015-0514-5
Rossi, G., Pengo, G., Caldin, M., Palumbo Piccionello, A., Steiner, J. M., Cohen, N. D., et al. (2014). Comparison of microbiological, histological, and immunomodulatory parameters in response to treatment with either combination therapy with prednisone and metronidazole or probiotic VSL#3 strains in dogs with idiopathic inflammatory bowel disease. PLoS One 9:e94699. doi: 10.1371/journal.pone.0094699
Sanchez, S., Hofacre, C. L., Lee, M. D., Maurer, J. J., and Doyle, M. P. (2002). Animal sources of salmonellosis in humans. J. Am. Vet. Med. Assoc. 221, 492–497. doi: 10.2460/javma.2002.221.492
Santos, R. L., Zhang, S., Tsolis, R. M., Kingsley, R. A., Adams, L. G., and Baumler, A. J. (2001). Animal models of Salmonella infections: enteritis versus typhoid fever. Microbes Infection 3, 1335–1344. doi: 10.1016/s1286-4579(01)01495-2
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-09
Sekirov, I., Tam, N. M., Jogova, M., Robertson, M. L., Li, Y., Lupp, C., et al. (2008). Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect. Immun. 76, 4726–4736. doi: 10.1128/iai.00319-08
Shade, A., and Handelsman, J. (2012). Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12. doi: 10.1111/j.1462-2920.2011.02585.x
Singh, P., Teal, T. K., Marsh, T. L., Tiedje, J. M., Mosci, R., Jernigan, K., et al. (2015). Intestinal microbial communities associated with acute enteric infections and disease recovery. Microbiome 3:45.
Stanley, D., Wu, S. B., Rodgers, N., Swick, R. A., and Moore, R. J. (2014). Differential responses of cecal microbiota to fishmeal, eimeria and clostridium perfringens in a necrotic enteritis challenge model in chickens. PLoS One 9:e104739. doi: 10.1371/journal.pone.0104739
Thibodeau, A., Fravalo, P., Yergeau, E., Arsenault, J., Lahaye, L., and Letellier, A. (2015). Chicken caecal microbiome modifications induced by Campylobacter jejuni colonization and by a non-antibiotic feed additive. PLoS One 10:e0131978. doi: 10.1371/journal.pone.0131978
Thiennimitr, P., Winter, S. E., Winter, M. G., Xavier, M. N., Tolstikov, V., Huseby, D. L., et al. (2011). Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. U.S.A. 108, 17480–17485. doi: 10.1073/pnas.1107857108
van Elsas, J. D., Chiurazzi, M., Mallon, C. A., Elhottova, D., Kristufek, V., and Salles, J. F. (2012). Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc. Natl. Acad. Sci. U.S.A. 109, 1159–1164. doi: 10.1073/pnas.1109326109
Varmuzova, K., Kubasova, T., Davidova-Gerzova, L., Sisak, F., Havlickova, H., Sebkova, A., et al. (2016). Composition of gut microbiota influences resistance of newly hatched chickens to Salmonella Enteritidis infection. Front. Microbiol. 7:957. doi: 10.3389/fmicb.2016.00957
Videnska, P., Sisak, F., Havlickova, H., Faldynova, M., and Rychlik, I. (2013). Influence of Salmonella enterica serovar Enteritidis infection on the composition of chicken cecal microbiota. BMC Vet. Res. 9:140. doi: 10.1186/1746-6148-9-140
Watson, P. R., Galyov, E. E., Paulin, S. M., Jones, P. W., and Wallis, T. S. (1998). Mutation of invH, but not stn, reduces Salmonella-induced enteritis in cattle. Infect. Immun. 66, 1432–1438. doi: 10.1128/iai.66.4.1432-1438.1998
White, J. R., Nagarajan, N., and Pop, M. (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5:e1000352. doi: 10.1371/journal.pcbi.1000352
Wills, E. S., Jonkers, D. M., Savelkoul, P. H., Masclee, A. A., Pierik, M. J., and Penders, J. (2014). Fecal microbial composition of ulcerative colitis and Crohn’s disease patients in remission and subsequent exacerbation. PLoS One 9:e90981. doi: 10.1371/journal.pone.0090981
Winter, S. E., Thiennimitr, P., Winter, M. G., Butler, B. P., Huseby, D. L., Crawford, R. W., et al. (2010). Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429.
Wu, H. J., and Wu, E. (2012). The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 3, 4–14. doi: 10.4161/gmic.19320
Wu, Y., Zhou, Y., Lu, C., Ahmad, H., Zhang, H., He, J., et al. (2016). Influence of butyrate loaded clinoptilolite dietary supplementation on growth performance, development of intestine and antioxidant capacity in broiler chickens. PLoS One 11:e0154410. doi: 10.1371/journal.pone.0154410
Xu, Y., Dugat-Bony, E., Zaheer, R., Selinger, L., Barbieri, R., Munns, K., et al. (2014). Escherichia coli O157:H7 super-shedder and non-shedder feedlot steers harbour distinct fecal bacterial communities. PLoS One 9:e98115. doi: 10.1371/journal.pone.0098115
Zhang, L., Dong, D., Jiang, C., Li, Z., Wang, X., and Peng, Y. (2015). Insight into alteration of gut microbiota in Clostridium difficile infection and asymptomatic C. difficile colonization. Anaerobe 34, 1–7. doi: 10.1016/j.anaerobe.2015.03.008
Keywords: Salmonella, chicken – broiler, intestine, diversity, Clostridia
Citation: Pedroso AA, Lee MD and Maurer JJ (2021) Strength Lies in Diversity: How Community Diversity Limits Salmonella Abundance in the Chicken Intestine. Front. Microbiol. 12:694215. doi: 10.3389/fmicb.2021.694215
Received: 12 April 2021; Accepted: 24 May 2021;
Published: 15 June 2021.
Edited by:
Yosra A. Helmy, The Ohio State University, United StatesReviewed by:
Mary Anne Amalaradjou, University of Connecticut, United StatesJuan D. Latorre, University of Arkansas, United States
Copyright © 2021 Pedroso, Lee and Maurer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John J. Maurer, amptYXVyZXJAdnQuZWR1
†Present address: Adriana A. Pedroso, Ventura Coastal, LLC, Tipton, CA, United States