 Jaime Rosas-Díaz1
Jaime Rosas-Díaz1 Alejandra Escobar-Zepeda2†
Alejandra Escobar-Zepeda2† Libertad Adaya1
Libertad Adaya1 Jorge Rojas-Vargas1
Jorge Rojas-Vargas1 Diego Humberto Cuervo-Amaya1
Diego Humberto Cuervo-Amaya1 Ayixon Sánchez-Reyes3
Ayixon Sánchez-Reyes3 Liliana Pardo-López1*
Liliana Pardo-López1*- 1Departamento de Microbiología Molecular, Instituto de Biotecnología, Universidad Nacional Autoónoma de México, Cuernavaca, Mexico
- 2Unidad Universitaria de Secuenciación Masiva y Bioinformática, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Cuernavaca, Mexico
- 3Cátedras Conacyt – Instituto de Biotecnología, Universidad Nacional Autónoma de México, Cuernavaca, Mexico
Paenarthrobacter sp. GOM3, which is a strain that represents a new species-specific context within the genus Paenarthrobacter, is clearly a branched member independent of any group described thus far. This strain was recovered from marine sediments in the Gulf of Mexico, and despite being isolated from a consortium capable of growing with phenanthrene as a sole carbon source, this strain could not grow successfully in the presence of this substrate alone. We hypothesized that the GOM3 strain could participate in the assimilation of intermediate metabolites for the degradation of aromatic compounds. To date, there are no experimental reports of Paenarthrobacter species that degrade polycyclic aromatic hydrocarbons (PAHs) or their intermediate metabolites. In this work, we report genomic and experimental evidence of metabolic benzoate, gentisate, and protocatechuate degradation by Paenarthrobacter sp. GOM3. Gentisate was the preferred substrate with the highest volumetric consumption rate, and genomic analysis revealed that this strain possesses multiple gene copies for the specific transport of gentisate. Furthermore, upon analyzing the GOM3 genome, we found five different dioxygenases involved in the activation of aromatic compounds, suggesting its potential for complete remediation of PAH-contaminated sites in combination with strains capable of assimilating the upper PAH degradation pathway. Additionally, this strain was characterized experimentally for its pathogenic potential and in silico for its antimicrobial resistance. An overview of the potential ecological role of this strain in the context of other members of this taxonomic clade is also reported.
Introduction
Paenarthrobacter adv. Paene, of which the translation from Latin is “almost” or “nearly” (Glare, 1968) (almost Arthrobacter), is a genus of gram-positive bacteria proposed recently from the reclassification of six species, namely, Paenarthrobacter aurescens, Paenarthrobacter histidinolovorans, Paenarthrobacter ilicis, Paenarthrobacter nicotinovorans, Paenarthrobacter nitroguajacolicus, and Paenarthrobacter ureafaciens, which belong to the Arthrobacter genus and, until now, have remained the only species with validated names (Busse, 2016). Most species of this genus have been isolated from soil, and they have a common peptidoglycan type A3α (Lys–Ala–Thr–Ala A11.17); a quinone system that contains menaquinone MK-9(H2); a polar lipid profile with the presence of diphosphatidylglycerol, phosphatidylglycerol, phosphatidylinositol, dimannosylglyceride, and monogalactosyldiacylglycerol; and a G+C content in genomic DNA from 61.3 to 63.6 (mol%) (Busse, 2016; Busse and Wieser, 2018).
Members of this genus have been reported to have interesting metabolic capabilities to degrade xenobiotics used in industrial applications and bioremediation. Examples include the degradation of the aromatic amine atrazine and similar compounds used as herbicides by P. aurescens TC1 (Strong et al., 2002), the utilization of nitroaromatic compounds by P. nitroguajacolicus (Kotoućková et al., 2004), nicotine metabolism by P. nicotinovorans (Baitsch et al., 2001), and nylon derivative degradation by P. ureafaciens KI72 (Takehara et al., 2017). Nevertheless, to date, there is no experimental evidence that strains of this genus are capable of degrading aromatic compounds, such as polycyclic aromatic hydrocarbons (PAHs) or their intermediate metabolites.
Aromatic compounds can be defined as organic molecules formed by one or more aromatic rings. The shape of the ring provides structural and chemical stability due to a symmetric system of π electrons, which makes them recalcitrant compounds in the environment (Vogt et al., 2011). These aromatic compounds are found in nature as lignin, amino acids and tannins, although other sources also include anthropogenic activities, such as agricultural (herbicides and insecticides), industrial (solvents, detergents and petroleum), and domestic sources (Cinar, 2004). Many of these compounds that are released to the environment are principal environmental pollutants due to their toxicity to living beings (Cao et al., 2009). In the environment, these compounds can be subjected to diverse physical, chemical, and biological phenomena that lead to their transformation, elimination or transport to other environmental compartments through evaporation, dilution, precipitation, lixiviation, sequestration, abiotic reactions (hydrolysis, photooxidation, or chemical oxidation), bioaccumulation, or microbial biodegradation (Hassanshahian et al., 2015).
The aerobic bacterial biodegradation of aromatic compounds has been divided into upper pathways, which begin from the transformation of the original compound into a few central intermediates (catechol, gentisate, and protocatechuate), and lower pathways, in which the aromatic ring of the intermediates is cleaved, producing into intermediary metabolites (acetyl-CoA, succinyl-CoA, and pyruvate) (Fuchs et al., 2011). However, it is well-documented that in some cases, during the degradation of compounds, such as PAHs, where there are multiple upper pathways involved, it is possible to detect intermediate metabolites as a result of incomplete biodegradation. This suggests that biodegradation occurs mainly in several upper pathways and only in some lower pathways (Luan et al., 2006) and that the production of intermediate metabolites is a common phenomenon observed in the partial degradation of PAHs (Cinar, 2004; Dastgheib et al., 2012).
The superior routes of degradation consist of one oxidation catalyzed by nonheme Rieske iron oxygenases, flavoproteins and soluble di-Fe monooxygenases, which are responsible for the activation and subsequent degradation of the aromatic ring (Gibson and Parales, 2000) and are classified as monooxygenases (hydroxylases) or dioxygenases (Parales and Resnick, 2004; Huijbers et al., 2014). Monooxygenases catalyze the cleavage of the oxygen-oxygen bond of O2, inserting an oxygen atom within the aromatic ring and forming phenols and later catechols (Ladino-Orjuela et al., 2016). Dioxygenases, on the other hand, carry out the dihydroxylation of the aromatic ring, resulting in the formation of a cis-dihydrodiol, which is rearomatized toward an intermediate diol by the action of a dehydrogenase (Mallick et al., 2011). The transformation of these structurally diverse aromatic compounds through peripheral metabolic pathways yields key intermediates, such as catechol (1,2-dihydroxybenzene), protocatechuate (3,4-dihydroxybenzoate), and gentisate (2,5-dihydroxybenzoate), which are subsequently channeled through a few central (lower) pathways into cellular metabolism (Cao et al., 2009). Once in the lower pathways, these catabolites are metabolized by intra- or extradiol dioxygenase enzymes, each of which can cleave the aromatic ring via the intradiol (ortho) and extradiol (meta) routes, respectively (Fritsche and Hofrichter, 2008). The intradiol reaction that takes place between the two hydroxyl groups is carried out by a dioxygenase using Fe(II) as a cofactor (Guzik et al., 2013). The extradiol reaction that occurs at the carbon-carbon bond adjacent to one of the hydroxyl groups is carried out by dioxygenases using Fe(III) as a cofactor (Suenaga et al., 2014).
Despite the many reports about aromatic compound degradation by bacteria, from PAHs to central intermediates, there is no study that evaluates the bacterial degradation capability of mixtures of central intermediates (benzoate, gentisate, and protocatechuate), which is how aromatic compounds are found in nature (Landry and Tremblay, 2012).
In the present study, we report the isolation of a bacterium from the southwestern Gulf of Mexico named GOM3, which, according to genomics and experimental results, is capable of degrading central intermediates related to the lower degradation pathway of aromatic compounds. The ecological role of this strain and a first approach to its pathogenicity have also been reported. To date, taxonomic analyses suggest that this strain represents a novel genomospecies of the genus Paenarthrobacter, which would make it the first species of this genus isolated from marine sediments.
Materials and Methods
Sample Collection and Strain Isolation
To obtain bacterial isolates, 1 g of marine sediment from station S03 collected in June 2015 (18°44′24.0′′N 94°30′00.0′′W, 275 m depth) was added to 50 mL of minimum medium composed of the following compounds (g⋅L–1): Na2SO4, 0.183; CaCl2, 0.073; NH4Cl, 0.267; MgCl2⋅6H2O, 0.16; NaMoO4⋅2H2O, 0.0002; FeSO4, 0.005; NaCl, 11.68; K2HPO4, 0.8; KH2PO4, 0.2; and phenanthrene, 0.01% w/v, as the sole carbon source. The cultures were incubated at 200 rpm and 30°C for 3 months, and every 30 days, the cultures were transferred to fresh minimum medium.
For the isolation of strains from consortia enriched with phenanthrene as the sole carbon source, serial dilutions were prepared up to 10–6, and 100 μL of the strain was plated in solid EDM containing the following compounds (g⋅L–1): NaCl, 23.6; KCl, 0.64; MgCl2⋅6H2O, 4.53; MgSO4⋅7H2O, 5.94; CaCl2⋅2H2O, 0.98; agar, 15; tryptone, 5; and yeast extract, 2.5. The Petri dishes were incubated for 7 days at 30°C. The strains were cultured in minimal medium (described previously) with 0.01% phenanthrene (w/v), and only strain GOM3 showed slight growth.
Nucleic Acid Extraction and Genome Sequencing
An isolated colony from strain GOM3 was grown overnight at 30°C and 180 rpm in 25 mL of LB medium in a 125 mL flask, and total DNA was extracted using a Quick-DNATM Miniprep Kit from Zymo Research (Irvine, CA, United States) following the kit instructions. Sequencing was performed on an Illumina NextSeq 500 platform following a paired-end protocol of 75 cycles (San Diego, CA, United States) at the Unidad Universitaria de Secuenciación Masiva y Bioinformática, UNAM, México. Polymerase chain reaction (PCR) was performed using Taq polymerase with the BAC primers F27 and R1492 for 16S rDNA (Heuer et al., 1997 referenced by Monciardini et al., 2002). The PCR product was purified with DNA Clean & Concentrator-25 from Zymo Research and subsequently sequenced by Sanger in the DNA Synthesis and Sequencing Unit of the Institute of Biotechnology, UNAM, México.
Genome Assembly and Refinement
After adapter sequence trimming and control quality filtering, the paired-end reads obtained from Illumina sequencing were used for de novo genome assembly with Velvet v1.2.10 (Zerbino, 2010), with a k-mer size of 63. To improve the quality of the assembly, we used REAPR v1.0.18 (Hunt et al., 2013) for correcting misassembled scaffolds, BESST v2.2.5 (Sahlin et al., 2014) for scaffolding, GapFiller v1-10 (Nadalin et al., 2012) for filling gaps between scaffolds and iCORN2 v0.95 (Otto et al., 2010) for correcting errors in scaffolds. Contamination and the completeness of the final version of the assembly were evaluated using the CheckM v1.0.12 toolkit (Parks et al., 2015).
Taxonomic Identification and Phylogenetic Analysis
For the first taxonomic identification, the 16S gene sequence obtained by Sanger was assembled by CAP in BioEdit (Hall, 1999) and annotated on Blast (McGinnis and Madden, 2004) using the 16S ribosomal RNA sequences for the Bacteria and Archaea database (consulted on June 03, 2021). To obtain phylogenetic neighbors with standing in nomenclature, the genome of Paenarthrobacter sp. strain GOM3 was compared against a custom database containing all type strain genomes available in the NCBI assembly portal (https://www.ncbi.nlm.nih.gov/assembly, consulted on April 01, 2021) (Sánchez-Reyes and Fernández-López, 2021) via the MASH algorithm (Ondov et al., 2016). The twenty types strains with the smallest MASH distances were chosen for further analysis via average nucleotide identity (ANI) determination with fastANI (Jain et al., 2018), digital DNA-DNA hybridization (dDDH) (Meier-Kolthoff et al., 2013) and a phylogenomic reconstruction with a set of 92 bacterial core genes included in UBCG software (Na et al., 2018). For the phylogenomic reconstruction, we also added 27 RefSeq genomes for the Paenarthrobacter lineage on NCBI (consulted on April 29, 2021). The final version of the genome assembly was used for taxonomic labeling against the Genome Taxonomy Database using GTDBtk v1.3.0 (Chaumeil et al., 2019).
Inference of Putative Protein Functions and Pangenomics Analysis
Functional annotation was performed on CDSs predicted from contig sequences using GeneMark V4.32 (Besemer et al., 2001). Genes translated to proteins were submitted to the RAST server for functional inference (Aziz et al., 2008). This annotation was complemented by the following prediction of specific genomic features: (1) antimicrobial resistance gene prediction using the Resistance Genes Identifier (RGI) pipeline v5.1.1 with the CARD database v3.1.0. (Alcock et al., 2019), (2) analysis of secondary metabolite biosynthetic gene clusters using antiSMASH v5.1.2 (Blin et al., 2019) with the option of “relaxed” strictness, and 3) PATRIC v3.6.9 annotation for virulence factor identification (Wattam et al., 2014).
Twelve genomes annotated as Paenarthrobacter species were representatives in the GTDB database (NCBI accession IDs: P. sp000526335 GCF_000526335, P. sp002979775 GCF_002979775, P. sp001512285 GCF_001512285, P. sp006964045 GCF_006964045, P. sp000281065 GCF_000281065, P. aurescens_A GCF_000014925, P. aurescens GCF_006538985, P. nicotinovorans_A GCF_000514015, P. sp002224285 GCF_002224285, P. sp900106835 GCF_900106835, P. sp001423565 GCF_001423565, and P. ureafaciens GCF_004028095). An assembly of Paenarthrobacter sp. HW13 (IMG/ER study ID: Gs0118559), which is a strain reported to harbor aromatic degradation genes (Moraes et al., 2018), was also used, and the closest strains according to GBDP distances (NCBI accession IDs: P. aurescens NBRC12136 GCA_006538985, P. ureafaciens DSM20126 GCA_004028095) were used to conduct pangenomics analysis using the Panaroo tool v1.2.4 (Tonkin-Hill et al., 2020) with the following parameters: –clean-mode strict; –alignment core; –core_threshold 1; –refind_prop_match 0.75; and –merge_paralogs. The aim of this analysis was to determine the uniqueness of this new species in terms of gene diversity and its functions in the context of close reference genomes. Sequences of proteins detected as unique to the strain of interest were mapped within the Kyoto Encyclopedia of Genes and Genomes (KEGG) database through their Automatic Annotation Server (KAAS v2.1) using the single-directional best hit (SBH) method (Moriya et al., 2007). Additionally, an enrichment of metabolic pathways in biochemical cycle analysis was performed using all the amino acid sequences in Multigenomic Entropy Based Score (MEBS) software (De Anda et al., 2017), with a restrictive false discovery rate (FDR) of 0.0001.
Experimental Evidence of Aromatic Compound Degradation
An overnight culture of GOM3 was grown in LB medium at 30°C and 180 rpm. A beginning aliquot was calculated to result in an OD600 nm near 0.1 in 5 mL of fresh minimum medium with benzoate, gentisate or protocatechuate individually at a concentration of 0.1%. When the mixtures of substrates were used, we adjusted each compound to 0.05% in pairwise mixtures and to 0.033% when the mixture contained all three compounds (concentrations are expressed as % w/v). Due to the acidic nature of these compounds, they were previously neutralized with 5 M NaOH to pH 7, and all media were passed through 0.45 μm filters. The experiments were carried out in biological triplicates. The aromatic compound concentration was measured with two different strategies. In the presence of one or two aromatic compounds, we used UV microplates in a Synergy 2 Multi-Mode de BioTek microplate lector. Benzoate, gentisate, and protocatechuate were measured at 230 nm (Cabel et al., 2000), 330 and 260 nm (Robbins, 2003), respectively. Due to absorbance interference from mixed substrates, when the compounds were in a mixture, we used an Agilent 1220 high-performance liquid chromatography model equipped with a Phenomenex Luna 5 μm C18 column (100 Å 150 mm × 3 mm). We used a gradient method for separating each compound, and the absorbance was read at the same wavelength described previously. To calculate the concentration, we employed a calibration curve.
The specific growth rate (μ) and duplication time were estimated when strain GOM3 was in an exponential growth phase. Both kinetic parameters were estimated using equations described by Widdel (2007). For the specific growth rate, we used the following equation:
where μ is the specific growth rate, OD is the optical density, and t is the time.
To calculate the duplication time, we used the following equation:
where μ is the specific growth rate and td is the duplication time.
Finally, to measure the volumetric consumption rate, we plotted time against the OD and computed the line slope when the compound concentration decreased.
Pathogenicity Assessment
The strain was grown in minimum medium with 3 g⋅L–1 peptone and 3 g⋅L–1 yeast extract for 20 h at 30°C. The cells were harvested by centrifugation, washed three times with 10 mM sterile MgSO4 and resuspended in sterile phosphate buffered saline (PBS). Galleria mellonella was used as a pathogenicity model for pathogenicity assays (Cools et al., 2019). To determine the health index (HI) (Loh et al., 2013), a group of 10 larvae was injected in triplicate with 100 CFU/10 μL following the protocol reported by Cools et al. (2019) and incubated at 30°C in the dark. The larvae were monitored for 5 days. One hundred CFU/10 μL Pseudomonas aeruginosa ATCC 27853 strain and 100 CFU/10 μL Escherichia coli DH5α were used as positive and negative controls, respectively. To calculate the median lethal dose (LD50), triplicate larval groups (n = 15) were injected with 102, 104, 106, 108, and 1010 CFU/10 μL strain. The larvae were incubated at 30°C and scored as dead or alive 48 h post-infection. Larvae were considered dead if they displayed no movement even with stimulation. The LD50 was calculated using a probit regression model with the “ecotox” library in R (Hlina et al., 2019).
Results
Genome Sequencing and Bioinformatic Analysis
The refined version of the genomic assembly had a 63.2% GC content and a length of 4.4 Mbp encompassed in 50 fragments. Nonetheless, the N50 value was 2.3 Mbp, and the L50 was seven contigs. This assembly was nearly complete (99.71%) and contained a low proportion of contaminating sequences, representing 0.39% of the assembly according to the CheckM tool. In total, 4,140 genes were predicted. A complete operon of rRNA containing 16S, 5S, and 23S was found, and a total collection of 52 genes for tRNAs was identified. This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JADIXW000000000. The version described in this paper is version JADIXW010000000.
Taxonomic Identification and Intergenomic Distance Inference Through ANI
The 16S rDNA sequence of the GOM3 strain had the highest identity (95.07%) with P. nitroguajacolicus strain G2-1 16S (accession number NR_027199.1), with a query coverage of 98%. Whole-genome similarity metrics allowed us to assess the taxonomic circumscription for strain-allowed Paenarthrobacter, Pseudoarthrobacter, or Arthobacter genomes, suggesting a continuum of diversity among this strain and its closest relatives (Table 1). Additionally, the estimation of dDDH notably indicated the absence of a species-specific relationship within the Paenarthrobacter/Arthrobacter/Pseudoarthrobacter clades. Therefore, we concluded that Paenarthrobacter sp. GOM3 is a novel species within the Paenarthrobacter genus (Micrococcaceae family).
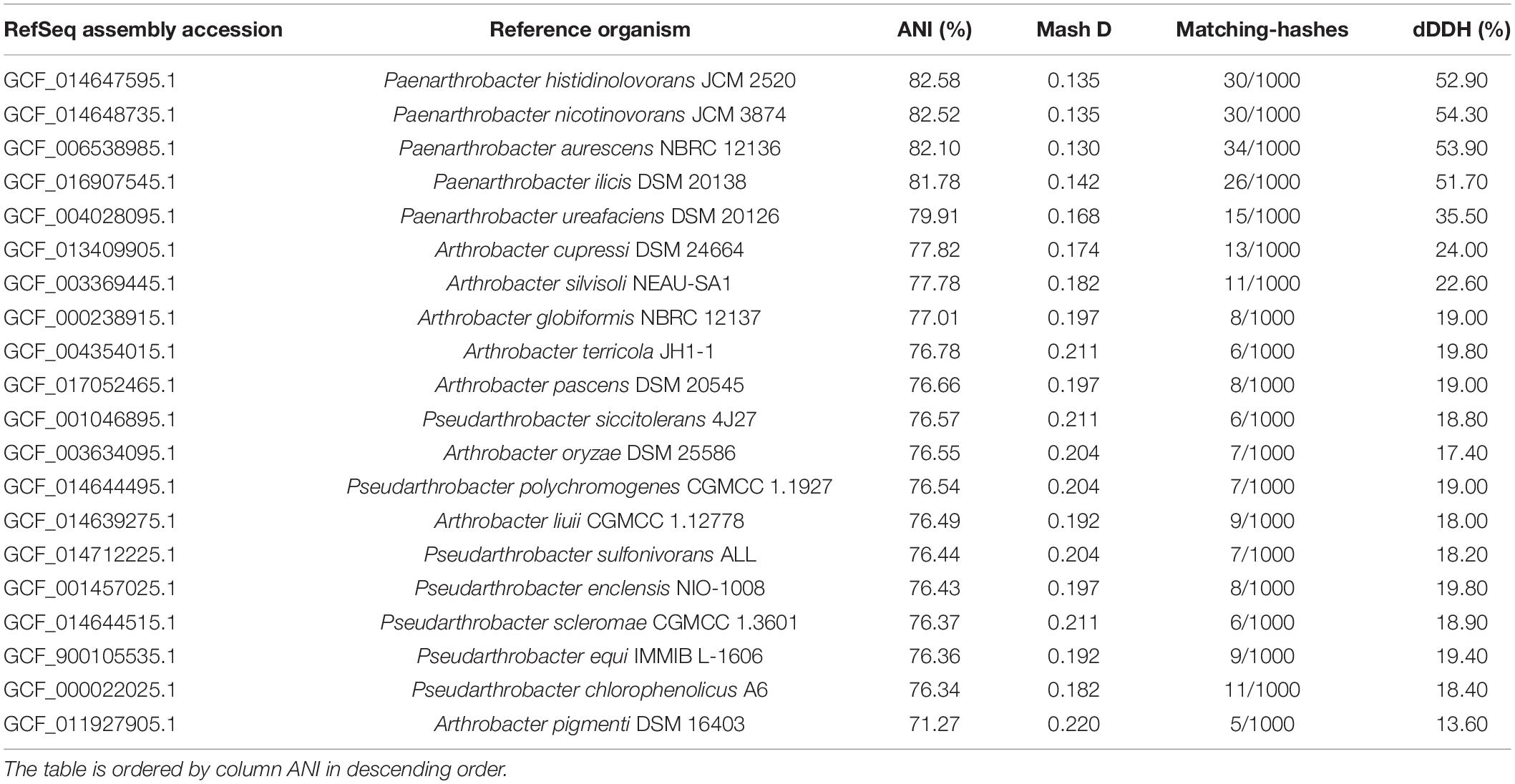
Table 1. Pairwise comparisons of Paenarthrobacter sp. GOM3 against closets type strain genomes according to several overall genome relatedness indexes (Mash D, ANI, and digital DNA-DNA hybridization (dDDH).
A phylogenomic approach using 92 single-copy genes extracted from query and neighboring genomes with UBCG software also confirmed that Paenarthrobacter sp. GOM3 is a novel species identified by clustering as an independent branch within the Paenarthrobacter-type species clade (Figure 1) close to Paenarthrobacter ilicis DSM 20138. The coherent separation of the Paenarthrobacter/Arthrobacter/Pseudoarthrobacter groups into three independent clades is highly supported by the gene support indices (GSIs).
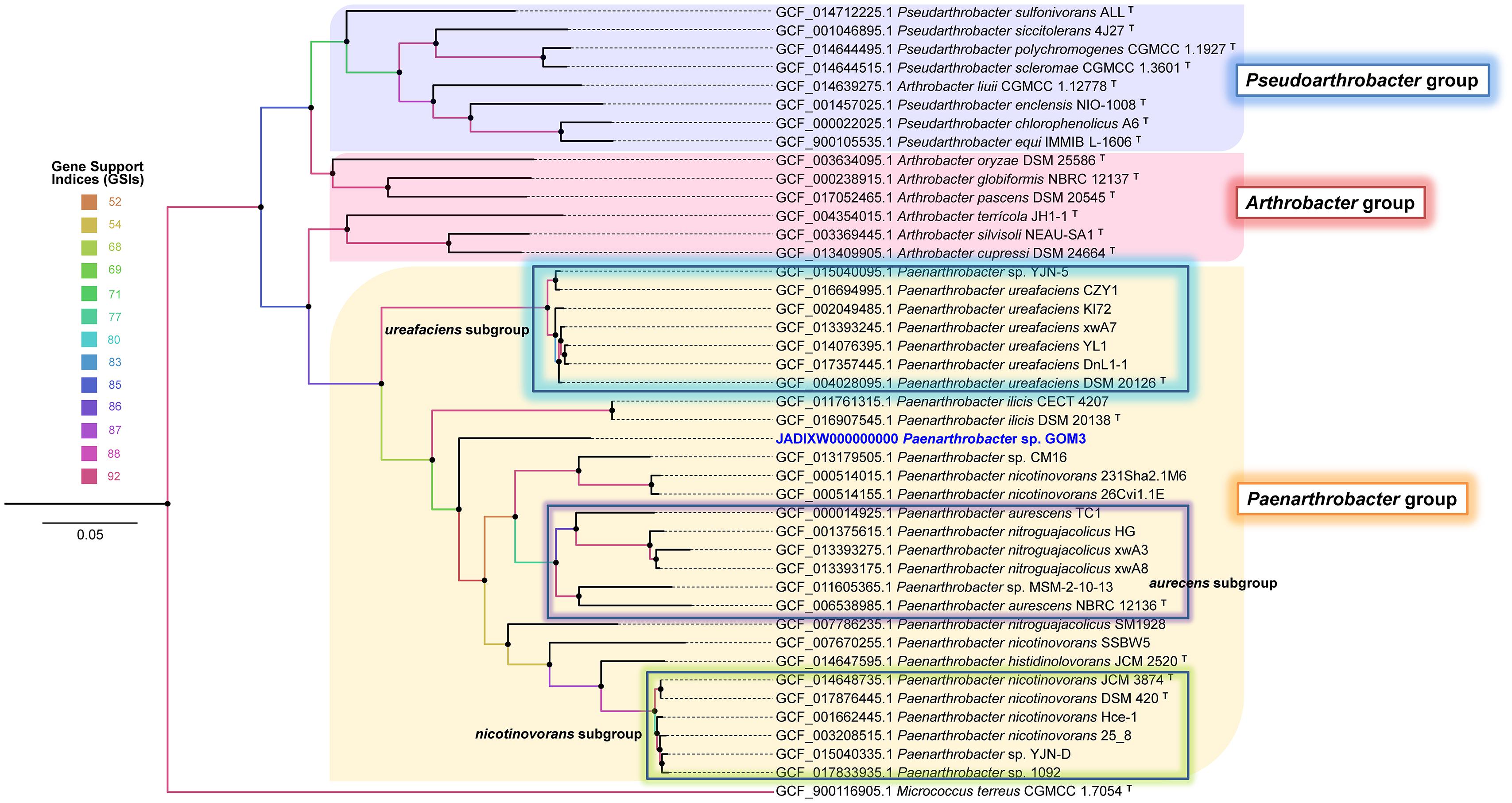
Figure 1. Phylogenomic unrooted tree inferred with 92 housekeeping bacterial core genes in the UBCG pipeline. Gene support indices (GSIs) are represented in color scale per node. Bar 0.05 substitution per site. The Micrococcus terreus CGMCC 1.7054T genome was used as an outgroup.
Ecological Role of Paenarthrobacter sp. GOM3
According to the Genome Taxonomy DataBase (GTDB), our assembly is classified at the genus level as Paenarthrobacter. The closest relative to our strain is Paenarthrobacter sp. 001423565, which was isolated from an Arabidopsis leaf (NCBI:txid1736274). Twelve representative genomes belonging to Paenarthrobacter species were used for pangenomic analysis. The assemblies of P. aurescens NCBR 12136, P. ureafaciens DSM 20126 and Paenarthrobacter sp. strain HW13 were also included.
We observed a core of genes comprising 2,046 sequences out of the 13,402 sequences forming the pangenome. Paenarthrobacter sp. GOM3 has 565 unique genes, most of which have unknown functions (71.3%) according to PROKKA annotation. Nonetheless, relevant potential functions were found in these unique genes when mapping vs. the KEGG database, such as catechol and benzoate/toluate dioxygenases for aromatic compound degradation, and a collection of permeases for a variety of sugars and other substrates, such as shikimate and glycerol, and ions, such as nickel and cyanate (Supplementary Table 1).
To better understand the ecological role of this strain, the mechanisms of biochemical pathways involving S, C, O, Fe, and N were evaluated based on amino acid sequences for each strain in the pangenomic analysis. This analysis revealed that the highest score for our strain corresponded to the nitrogen cycle (Pfam entropy score 12.302), indicating that these bacteria could use nitrogen compounds, such as ammonia or nitrates, as energy sources. The second highest score corresponded to oxygen (score 4.493), agreeing with the aerobic nature of the strain, and the third corresponded to carbon (score 1.241), suggesting that this strain can degrade methane methyl compounds, such as methylamine, a common compound in the marine ecosystem. A negative score for sulfur (score -0.644) indicated that this Paenarthrobacter did not have all the pathways of sulfur cycle.
Comparing our strain with the 16 strains from the pangenomic group, a heatmap was generated to identify the completeness of specific pathways in the reference genomes, revealing potential metabolic feature completeness (Figure 2). Ten metabolic pathways with 100% completeness were present in almost all the samples, including some reactions of the nitrogen cycle, such as ammonia assimilation (I), nitrate reduction (V and VI), superpathway ammonia assimilation and nitrate reductase (nirBD) enzyme. Other pathways with 100% completeness included sulfoacetaldehyde degradation and the widespread rhodanase enzyme involved in detoxification of cyanide. There was a reduced number of genes involved in methane compounds or nitrogen fixation. Pfam domains involved in tetrathionate S4O6–2 (ttrABC) and S0 (sreABC) reduction were only present in two strains, Paenarthrobacter sp. GOM3 and Paenarthrobacter sp. HW13, with 100% completeness, indicating that they likely use tetrathionate and elemental sulfur as a source of energy and electron acceptors. We could not identify the key enzyme marker for methane metabolism in any of the strains, e.g., the methyl-coenzyme M reductase (MCR) complex.
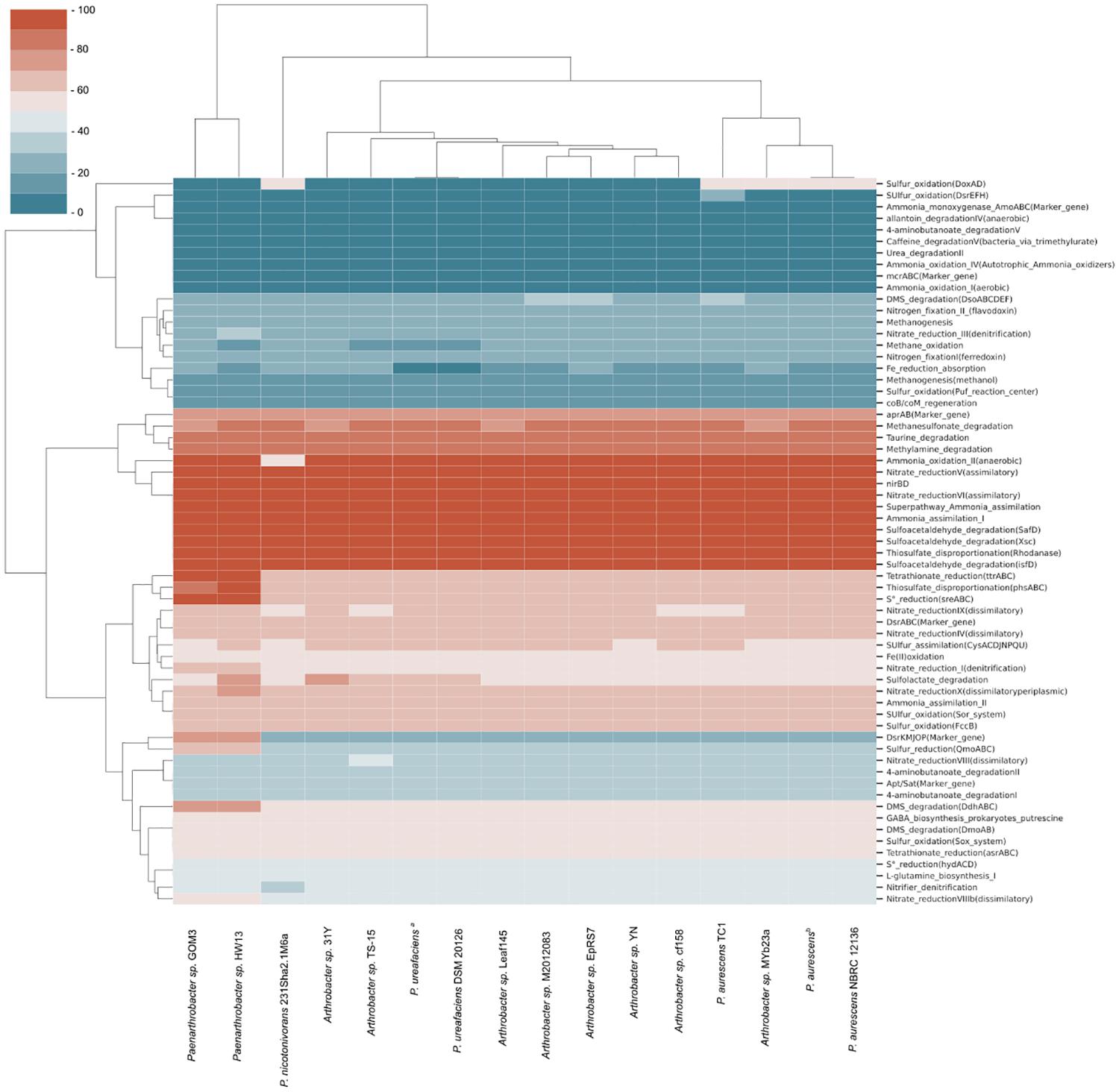
Figure 2. Metabolic completeness of C, O, N, S, and Fe cycles of strains related to pangenomic analysis. The most complete pathways in biogeochemical cycles are marked in red, and the least complete pathways shift to blue. P. ureafaciensa corresponds to RefSeq assembly accession GCF_006538985, and P. aurescensb corresponds to RefSeq assembly accession GCF_004028095.
Functional Annotation Analysis and Experimental Support
Genome annotation of Paenarthrobacter sp. GOM3 showed genes related to the degradation of aromatic compounds, such as benzoate, gentisate, and protocatechuate (Supplementary Tables 2–4, respectively).
In the case of benzoate, this strain has catabolic benABCD genes that convert benzoate into catechol (Carrington et al., 1994) and catBCA genes that transform catechol into 3-oxoadipate-enol-lactone (Houghton et al., 1995). In addition, GOM3 possesses a benK gene from the major facilitator superfamily (MFS) reported as a transporter of benzoate into the cell (Collier et al., 1997). Additionally, in this genomic context, a transcriptional regulator belonging to the LysR family was found (Maddocks and Oyston, 2008; Figure 3A). To verify the capacity of GOM3 to use the aromatic metabolic pathways found in its genome, this strain was grown in the presence of benzoate as the only carbon source, demonstrating that these genes are functional. Strain GOM3 had a specific growth rate (μ) of 0.087 ± 0.002 h–1 and a volumetric consumption rate of 0.027 ± 0.002 g/L h–1 (Supplementary Table 5), although it had a lag phase of 8 h in which it did not grow and the benzoate concentration did not decrease (Figure 3D, see benzoate growth kinetics and substrate degradation).
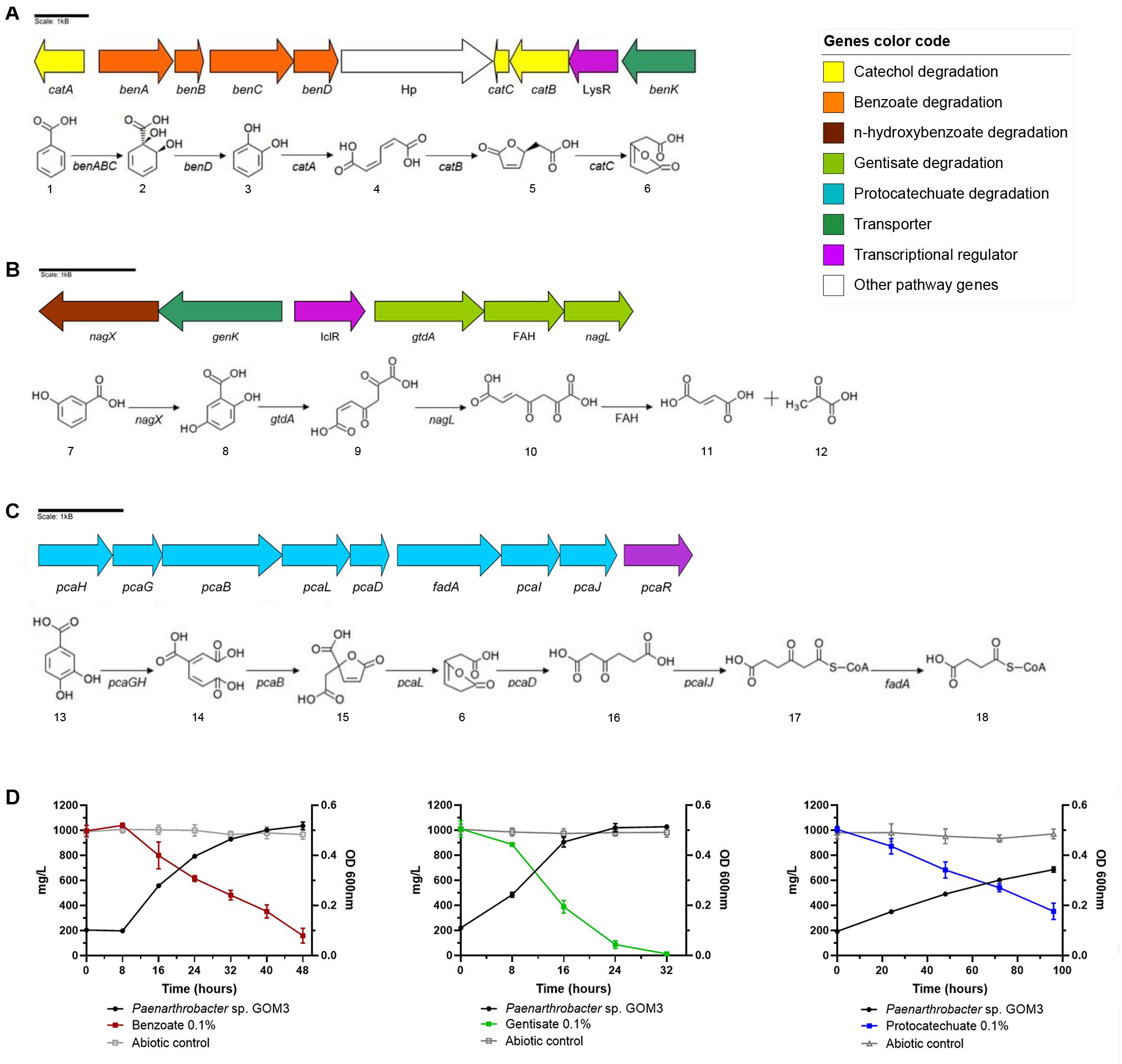
Figure 3. Genomic and experimental evidence of aromatic compound degradation by Paenarthrobacter sp. GOM3. Degradation pathways and genomic context are shown for three aromatic compounds: (A) benzoate, (B) gentisate, and (C) protocatechuate. (D) Growth kinetics and decrease in its corresponding compound concentration. Whiskers represent the standard deviation of three biological replicates. Metabolic compounds: (1) benzoate; (2) cis-1,2-dihydroxycyclohexa-3,5-diene-1-carboxylate; (3) catechol; (4) cis,cis-muconate; (5) (+)-muconolactone; (6) 3-oxoadipate enol-lactone; (7) 3-hydroxybenzoate; (8) gentisate; (9) 3-maleylpyruvate; (10) 3-fumarylpyruvate; (11) fumarate; (12) pyruvate; (13) protocatechuate; (14) beta-carboxy-cis,cis-muconate; (15) gamma-carboxymuconolactone; (16) 3-oxoadipate; (17) 3-oxoadipyl-CoA; and (18) succinyl-CoA. Concentrations of each aromatic compound in abiotic controls in gray.
Two catabolic genes related to the gentisate degradation pathway were found in the genome—gentisate 1,2 dioxygenase gtdA and maleylpyruvate isomerase nagL (Zhou et al., 2001)—and in the vicinity of these genes, we identified a transporter genK (Xu et al., 2012), plus three more copies in the genome and a transcriptional regulator that belongs to the isocitrate lyase regulator type (IclR type) (Molina-Henares et al., 2006). Moreover, we found the nagX gene, which encodes a putative n-hydroxybenzoate hydroxylase that converts 3-hydroxybenzoate into gentisate (Park et al., 2007), and a gene that encodes a protein of the fumarylacetoacetate hydrolase family (Figure 3B). The μ of this strain was similar to that observed for the benzoate substrate (0.089 ± 0.003 h–1); however, it did not show a lag phase in the presence of gentisate (see Figure 3D, gentisate growth kinetics and substrate degradation). The volumetric consumption rate of gentisate was 0.039 ± 0.002 g/L h–1 (Supplementary Table 5).
Genes related to protocatechuate catabolism were also identified in the GOM3 genome responsible for converting this aromatic compound into the intermediary metabolite succinyl-CoA (Kowalchuk et al., 1994; Figure 3C). In the same genomic context, we found the transcriptional regulator pcaR (Romero-Steiner et al., 1994). In comparison with the other two substrates, the growth phase was more prolonged (see Figure 3D, protocatechuate growth kinetics and substrate degradation), and the μ and volumetric rate consumption values were lower, 0.02 ± 0.002 h–1 and 0.007 ± 0.001 g/L h–1, respectively (Supplementary Table 5).
In addition, we tested the degradation capacity of GOM3 in mixtures of aromatic compounds. We observed that benzoate and gentisate (B/G) mix substrates were degraded simultaneously (Figure 4A), but the volumetric consumption rate of gentisate (0.028 ± 0.002 g/L h–1) was greater than that of benzoate (0.013 ± 0.001 g/L h–1), with a Wilcoxon test P-value of 0.05. The degradation of both substrates was complete after 40 h (Figure 4A). The specific growth rate in this mix was 0.085 ± 0.003 h–1.
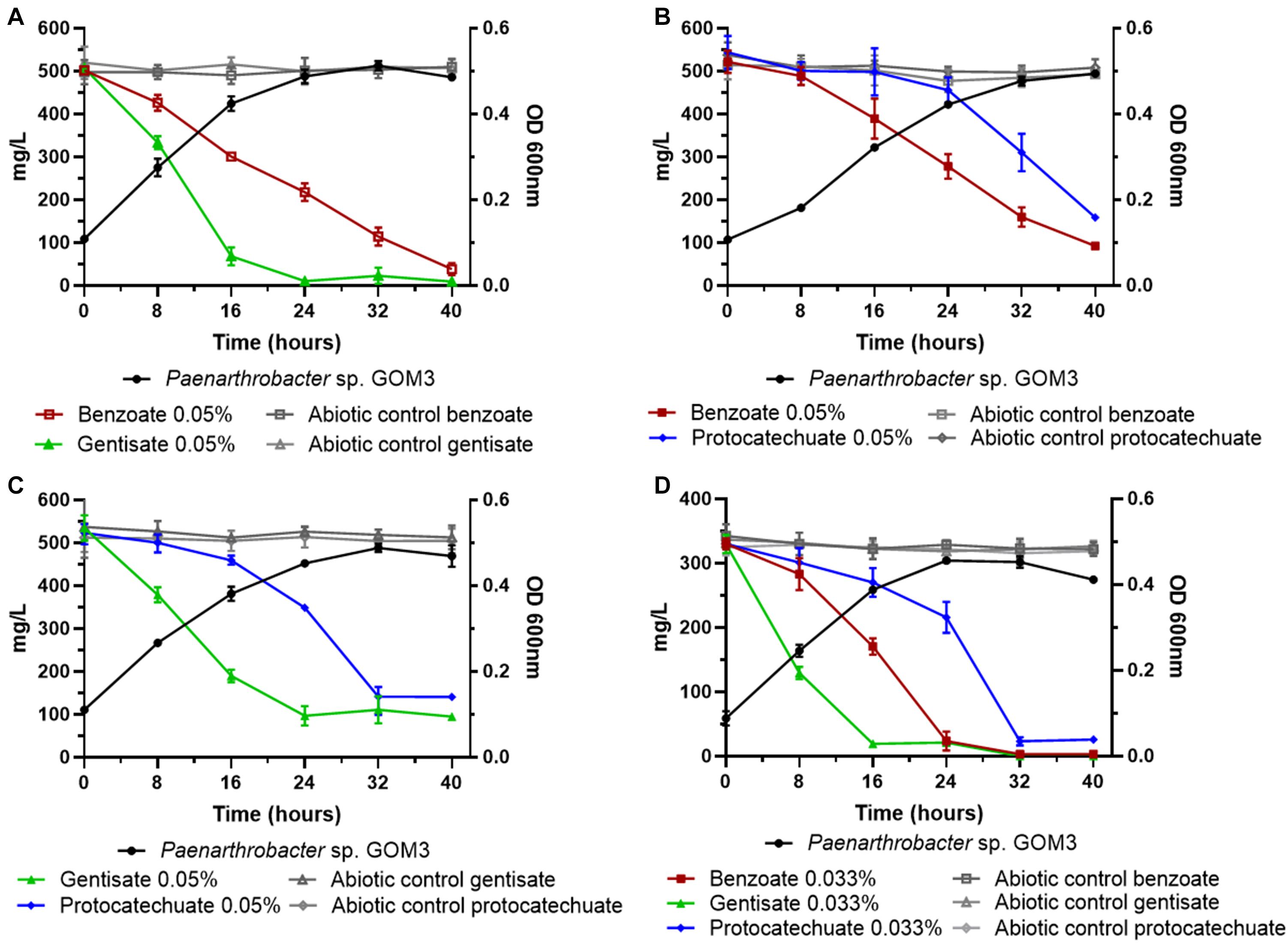
Figure 4. Substrate decrease and growth kinetics of Paenarthrobacter sp. GOM3 in the presence of different mixtures of aromatic compounds (A) benzoate and gentisate, (B) benzoate and protocatechuate, (C) gentisate and protocatechuate and (D) the three compounds. Whiskers represent the standard deviation of three biological replicates. Aromatic compound in abiotic controls in gray.
In the mixture of benzoate and protocatechuate (B/P), we observed that the benzoate concentration decreased after 16 h, while the protocatechuate concentration started decreasing after 24 h (Figure 4B); however, both substrates had similar volumetric consumption rates (Wilcoxon test P = 0.13) (Supplementary Table 6), and the μ for this mix was 0.069 ± 0.002 h–1.
When gentisate and protocatechuate were mixed (G/P), the volumetric consumption rate of gentisate was higher than that of protocatechuate (Wilcoxon test P = 0.05) (Figure 4C and Supplementary Table 6), and the μ value of the mix was 0.077 ± 0.003 h–1.
Finally, when all three aromatic compounds were present, we observed that all substrate concentrations decreased simultaneously (Figure 4D), but at different rates. The compound with the highest volumetric consumption rate was gentisate (0.019 ± 0.001 g/L h–1), followed by benzoate (0.013 ± 0.001 g/L h–1) and protocatechuate (0.009 g/L h–1) (global Wilcoxon test P = 0.027).
Pathogenicity Assessment
Some strains of the Arthrobacter genus are reported as opportunistic human pathogens (Huang et al., 2005). These strains are associated with clinical features, such as bacteremia, periodontitis, and endocarditis (Bernasconi et al., 2004). Because our strain is related to this genus (Figure 1), a first glimpse of its pathogenicity is shown below. An HI assay with 100 CFU/10 μL inoculum injected into G. mellonella showed that Paenarthrobacter sp. GOM3 was able to cause infection at a lower level than the pathogen P. aeruginosa ATCC 27853, in contrast with the negative controls (Supplementary Figure 1A). Using the doses described in the section “Materials and Methods,” a dose-response curve at 48 h was obtained (Supplementary Figure 1A). With 106 CFU, nearly 40% of the larval population was dead, 90% died with 108 CFU exposure, and 100% died with 1010 CFU exposure. The LD50 calculated for our strain was 2.29 × 106 CFU at 48 h, with a confidence level of 0.95. In contrast with the P. aeruginosa LD50 of 5–17 CFU between 24 and 48 h (Miyata et al., 2003; Sonnleitner et al., 2003; Andrejko et al., 2014), the E. coli LD50 between 1 and 5 × 104 CFU at 120 h (Alghoribi et al., 2014), and the Klebsiella pneumoniae LD50 of 1 × 104 CFU at 72 h (Insua et al., 2013), the LD50 value of our strain is higher, indicating that the GOM3 strain is less harmful than recognized pathogens.
This sign of stress is related to possible pathogenic mechanisms of our strain and the presence of some virulence factors in its genome, according to PATRIC annotation, such as the ilvD (78% amino acid identity) and icl (79% identity) genes. These two genes do not have an obvious association with virulence mechanisms; however, experimental evidence supports that they are linked with virulence attenuation in Mycobacterium tuberculosis when the gene is inactivated, which makes them a good target for drug and vaccine development (Muñoz-Elías and McKinney, 2005; Singh et al., 2011). Additionally, it is suggested that our strain may have other virulence factors, with identities of approximately 60%, such as cspA (68%) for survival, groL1 (60%) for adherence, TetR (59%) for regulation, and ideR (57%) for iron uptake.
In addition, we performed an in silico screening for genes related to antimicrobial resistance to assess the potential of GOM3 to succeed in the presence of this kind of drug. Two antimicrobial resistance genes were found in the Paenarthrobacter sp. GOM3 genome using the RGI pipeline. One gene was annotated as an ATP-binding cassette ribosomal protection protein capable of providing protection to a variety of drug classes, such as macrolide, lincosamide, streptogramin, tetracycline, oxazolidinone, phenicol, and pleuromutilin antibiotics. The other gene was annotated as a resistance-nodulation-cell division (RND) antibiotic efflux pump, with potential function against macrolide, monobactam, tetracycline, and aminocoumarin antibiotics. Further inspection of PROKKA functional annotation yielded a list of the following genes associated with antimicrobial resistance: 11 copies of fosfomycin resistance protein AbaF (abaF); 3 copies of linearmycin resistance ATP-binding protein LnrL (lnrL); 2 copies of putative multidrug resistance protein MdtD (mdtD), multidrug resistance protein Stp (stp), and multidrug resistance protein 3 (bmr3); and one copy of tetracycline resistance protein class C (tetA), antiseptic resistance protein (qacA), multidrug resistance protein NorM (norM), multidrug resistance protein MdtL (mdtL), putative multidrug resistance protein EmrY (emrY), daunorubicin/doxorubicin resistance ATP-binding protein DrrA (drrA), and bicyclomycin resistance protein (bcr).
Discussion
The GOM3 strain represents a new species-specific context within the Paenarthrobacter genus (for which there are only six species described) and is grouped into an independent branch, with early divergence within it. In addition to phylogenetics, several lines of genomic evidence support this observation, highlighting the low nucleotide-level genomic similarities (ANI < 95% dDDH < 70%), which constitute standards in current microbial classification schemes, especially in the definition of genomospecies (Figure 1 and Table 1). The phylogenetically closest strains are P. ilicis DSM 20138, Paenarthrobacter sp. CM16, and P. nicotinovorans strains 231Sha2.1M6 and 26Cvi1.1E, and the latter two strains were probably misclassified at the species level since they clustered outside the nicotinovorans subgroup (see the highlighted green square in Figure 1). Although the taxonomic description of the GOM3 strain is beyond the scope of this paper, our phylogenomic reconstruction supports that the Paenarthrobacter clade is monophyletic and that it has undergone significant radiation with coherently defined genomic groups (P. urefaciens, P. aurecens, P. nicotinovorans subgroups, and the species P. ilicis and P. histidinolovorans were less populated at the genomic level but well-separated from the other groups). The GOM3 strain is clearly an independently branched member of any group described thus far. Although the CM16 strain was isolated from a halophytic endophytome, specifically from roots of the coastal plant Cakile maritima, the GOM3 strain of this study represents the first marine Paenarthrobacter isolated at seafloor depth.
The GOM3 strain was recuperated from marine sediment obtained to a depth of 275 m in the Gulf of Mexico cultured with phenanthrene. Afterward, the strain was successfully isolated, but when its growth and degradation capacity in the presence of phenanthrene were tested, minimal growth was observed, and there were no significant changes in the consumption of hydrocarbons (Supplementary Figure 2). In a similar study performed by Wang et al. (2018), different strains belonging to species of the genera Halomonas, Chromohalobacter, Thalassospira, and Alcanivorax were isolated from a consortium enriched with phenanthrene, none of which could grow individually in the presence of this aromatic compound. These authors suggest that these strains probably play an important role in the lower pathways of PAH degradation since they found genes related to the degradation of central intermediaries in their genomes.
There are no experimental reports of Paenarthrobacter species that degrade benzoate, gentisate or protocatechuate. However, Meng et al. (2017) found genes related to the degradation of benzoate in the genome of Paenarthrobacter nicotinovorans Hce-1 isolated from a polluted hyaluronic acid solution. In another study, Moraes et al. (2018) identified eight gene clusters related to aromatic degradation (including gentisate, protocatechuate, and catechol) in a novel Paenarthrobacter sp. HW13 strain isolated from a lignin-degrading consortium.
The benzoate degradation pathway has been commonly found in hydrocarbon-degrading marine bacteria in the Gulf of Mexico (Muriel-Millán et al., 2019; Rodríguez-Salazar et al., 2021). Raggi et al. (2020) reported the presence of recognized hydrocarbon-degrading bacteria and their metabolic potential through aerobic and anaerobic pathways in sediment samples collected from depths of 1,320, 2,966, and 3,010 m. The high diversity of Rieske oxygenases observed in the northern samples indicates potential for the degradation of diverse aromatic compounds, such as benzoate, benzene, toluene, phthalate, naphthalene, or biphenyl under aerobic conditions. Notably, Actinobacteria, in general, is not a dominant group in Gulf of Mexico sediments, and the Paenarthrobacter (or Arthrobacter) genus, in particular, is detected in very low abundance by 16S rRNA metaprofiling and is not part of the Gulf of Mexico taxonomic baseline (Godoy-Lozano et al., 2018).
Analyzing the genome of GOM3, we found five different dioxygenases, namely, 1,2-dioxygenase, catechol 1-dioxygenase, catechol 2,3-dioxygenase, protocatechuate 3,4-dioxygenase and gentisate 1,2-dioxygenase, involved in the activation of aromatic compounds. These enzymes are responsible for the GOM3 strain being able to grow in the presence of benzoate, gentisate and protocatechuate, with gentisate being the preferred substrate and exhibiting the highest volumetric consumption rate (Figures 3D, 4 and Supplementary Table 6). Genomic analysis revealed that this strain possesses four copies of the genK gene encoding the specific transporter gentisate. A remarkable observation within the genus is that all type strains use p-hydroxybenzoate as a carbon source but do not appear to assimilate the parent compound benzoic acid, suggesting that the activating enzyme benzoate 4-monooxygenase (EC: 1.14.14.92) is missing from these strains. However, the GOM3 strain possesses two genes encoding enzymes related to benzoate degradation via hydroxylation: a 4-hydroxybenzoate 3-monooxygenase [NAD(P)H] and two copies of 3-hydroxybenzoate 4-monooxygenase.
According to our experimental results, GOM3 has a substrate preference for gentisate, as this substrate has the highest consumption rate in every mix and when tested independently. However, the lack of a diauxic growth pattern in cultures with a mix of two or three carbon sources (Figure 4) suggests that the GOM3 strain could consume the three molecules at the same time, as seen in their simultaneous concentration decrease. The growth rate differences for each carbon source (gentisate, benzoate, and protocatechuate) could be attributed to several reasons. The possible greater gentisate influx could be due to the four copies of the genK transporter gene in contrast to one copy of the benK benzoate transporter and no copies of a specific (pcaK) protocatechuate transporter. A possible explanation for this redundancy in the gentisate pathway could be that this pathway produces fumarate and pyruvate. The former can be used to produce malate, and the latter can be used to produce oxaloacetate, feeding, and restarting the TCA cycle, as seen in P. nicotinovorans pAO1 (Mihăşan et al., 2021). The IclR regulator in the gentisate operon could regulate the glyoxylate shunt (Molina-Henares et al., 2006) to produce succinate before inhibition due to an increased malate concentration. On the other hand, the degradation of both benzoate and protocatechuate produces acetyl-CoA and succinyl-CoA. In this case, there is no direct access to the glyoxylate shunt, and the production of the other TCA cycle components could be slower, explaining the lag phase seen in benzoate and protocatechuate culture kinetics. The lack of a transporter for protocatechuate would suggest that its pathway catabolizes intracellular protocatechuate produced as an intermediary of other pathways.
Notably, we observed that protocatechuate is degraded by GOM3 more quickly in the presence of the other aromatic compounds. Some studies have found that in mixtures of aromatic compounds, synergic effects can be originated, such as the Sagittula stellata E-37 strain, which increased their growth rate in a mixture of benzoate/p-hydroxybenzoate (Gulvik and Buchan, 2013). In the case of gentisate degradation, we did not find the enzyme responsible for the subsequent reaction to transform 3-fumarylpyruvate (or acylpyruvate hydrolase and 3-fumarylpyruvate hydrolase); however, Pircher et al. (2011) found in vitro that a fumarylacetate hydrolase (FAH) also has acylpyruvate hydrolase activity, which could indicate that the enzyme belonging to the FAH family found in the gentisate pathway of Paenarthrobacter sp. GOM3 (Figure 3B) could convert fumarylpyruvate into fumarate and pyruvate. Further studies to understand the regulation of the related pathways in the GOM3 strain are required.
As part of the characterization of GOM3 as a new species, we compared the pathways involved in biogeochemical cycles of different Arthrobacter and Paenarthrobacter strains (Figure 2), resulting in very similar profiles across these strains. This result agrees with the sources of these bacteria, the majority of which were obtained from soil samples or sediment of natural springs. The analysis revealed an enrichment of key enzymes for nitrate reduction, ammonia assimilation and degradation of certain sulfur compounds. The nitrate reduction potential is consistent with metabolism under anaerobic or microaerophilic conditions (Jetten, 2008; Raggi et al., 2020), where nitrate is the second preferred electron acceptor after oxygen (Hensen et al., 2006). In particular, for the GOM3 strain, assimilatory nitrate reduction to ammonia or ammonification would consist of the internalization of nitrate/nitrite with MFS membrane transporters (Nrt), conversion of nitrate to nitrite by nitrate reductase (nasA) and conversion from nitrite to ammonia by the nitrite reductase (NADH) large subunit (nirBD). This ammonium is further converted to glutamine by glutamine synthetase (glnA) and is later incorporated in glutamate metabolism. In our analysis, there was no evidence of enzymes involved in nitrification reactions that convert ammonium back to nitrite/nitrate, as neither PFam was relevant for nitrogen fixation (Figure 2), indicating that the main source of nitrogen for the analyzed strains was environmental nitrate. Interestingly, the GOM3 and HW13 strains were the only strains with complete PFam domains involved in elemental sulfur reduction and tetrathionate reduction. Although the GOM3 and HW13 strains were isolated from different sources, namely, marine sediment and sugarcane plantation soil, respectively, both probably use H2 or organic substrates as electron donors to yield S0 and S4O6–2 as energy sources (Caspi et al., 2012). The GOM3 and HW13 strains were the only strains with 75% PFam completeness for dissimilatory sulfate reduction and oxidation, which is a predominant terminal pathway of organic matter mineralization in the anoxic seabed (Jørgensen et al., 2019).
In addition to these biochemical capabilities, we observed a unique battery of permeases in GOM3 that internalize a variety of substrates (Supplementary Table 1). The most prevalent permease was a peptide/nickel transport system with 14 related genes (KEGG orthology K02031-K02035) belonging to pathway ko02024 of quorum sensing (QS). Gram-positive bacteria use autoinduced peptides instead of N-acyl homoserine lactones (AHLs) as signaling molecules. Peptide autoinducers require specialized transport mechanisms to be excreted and imported back into the cell by a two-component system mechanism (Miller and Bassler, 2001). In species of the genus Arthrobacter, quorum-quenching activity in cross phylum interactions through the production of the depsipeptide arthroamide has been reported (Igarashi et al., 2015). In nonribosomal peptide synthetase systems, depsipeptide bonds are formed by nucleophilic attack on thioesterase-bound substrates (McClure et al., 2016). The prediction of biosynthetic gene clusters for secondary metabolites in GOM3 revealed the presence of regions for a nonribosomal peptide synthetase system, suggesting that this strain could have quorum-quenching activity. However, further analysis and experimental support are needed to corroborate this statement.
The experimental and genomic characterization of novel bacteria gives us the opportunity to explore the potential of hydrocarbon degradation that remains unexplored and that could be applied in bioremediation. Rational bacterial consortia design requires an understanding of their metabolic pathways and mechanisms that facilitate hydrocarbon degradation (i.e., chemotaxis, biofilm formation, biosurfactant production, and efflux pumps), as well as intercellular relationships (Xu et al., 2018). Individual draft genomes of hydrocarbon-degrading bacteria are key to the rational design of synthetic consortia with positive metabolic cooperativity. To achieve this goal, it is important to select not only the most efficient bacteria but also bacteria with different mechanisms for hydrocarbon degradation that can complement each other in a synthetic consortium.
In this way, we consider that Paenarthrobacter sp. GOM3 is a strain that can participate in a consortium with complex hydrocarbon-degrading strains with substrates, such as polyaromatics, resins, or asphaltenes (Pseudomonas, Cycloclasticus, and Bacillus) and with aliphatic degrading bacteria (Alcanivorax, Rhodococcus, and Dietzia) (Xu et al., 2018). There are no previous reports wherein this strain has been used in the bioremediation of contaminated sites; thus, exploring its biodegradation capacity is important.
As we are considering GOM3 as a promising strain for use in bioremediation, we explored its pathogenic potential using a lepidopteran model, G. mellonella, which is a good model for testing human pathogens (Hernandez et al., 2019). Our study shows that GOM3 reduced the larval health status in contrast with negative controls but not to the same level as the lower HI score for P. aeruginosa ATCC 27853. These differences could be related to the presence of virulence factors and pathogenic mechanisms, as was observed in experiments of extraintestinal pathogenic E. coli using larvae, wherein a relatively high virulence gene number killed larvae faster than a low virulence gene number (Tsai et al., 2016). Our calculated LD50 also supports this HI result. A higher number of CFUs of GOM3 are needed to kill half the population of larvae than of other pathogenic bacteria. To date, there are no LD50 reports of any opportunistic Arthrobacter pathogens. However, according to PATRIC annotation, the opportunistic pathogen Arthrobacter strain A. woluwensis DSM 10495 (Genome ID 156980.3) (84.21% ANI compared with GOM3) has the same ilc and ilvD virulence factors as our strain. If there is any interest in better understanding the virulence mechanisms of this strain, it will be necessary to perform future pathogenicity studies, such as establishing the activation of stress responses and repair mechanisms in larvae or applying murine model testing.
Conclusion
Paenarthrobacter sp. GOM3 is a new species in the genus and is the first marine strain isolated on the seafloor, where its abundance is very low. There are few studies of the degradation of PAHs since the lower metabolic pathways are almost always ignored; however, the sequencing of its genome allowed the identification of enzymes that participate in the degradation of aromatic compounds. This is the first report experimentally demonstrating that a strain of this genus can grow in the presence of aromatic compounds, such as benzoate, protocatechuate, and gentisate, with the latter being the preferred carbon source. The GOM3 strain also has metabolic potential for nitrate reduction to ammonia, elemental sulfur reduction and tetrathionate reduction, as well as genes for QS. Additionally, we experimentally showed that the GOM3 strain can cause infection in the G. mellonella model, but it is less harmful than recognized pathogens and bears a wide range of antimicrobial resistances according to genomic analysis.
Due to the versatility in the metabolism of the GOM3 strain in the degradation of intermediate metabolites and the fact that it is not pathogenic, this strain could well be used in bioremediation as part of a degrading consortium in sites contaminated with aromatic hydrocarbons.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/ Supplementary Material.
Author Contributions
JR-D, AE-Z, AS-R, and LP-L contributed to the design and implementation of the research. JR-D, AE-Z, AS-R, and JR-V performed the computational and bioinformatic analyses. JR-D, LA, JR-V, and DC-A performed the experiments and analyzed the data. LP-L coordinated the group, managed the resources, and guaranteed their availability to perform the experiments and analysis. All authors contributed to the writing of the manuscript and approved the final version.
Funding
This research was funded by the National Council of Science and Technology of Mexico – Mexican Ministry of Energy- Hydrocarbon Trust, project 201441, and PAPIIT-DGAPA IN207019. This is a contribution of the Gulf of Mexico Research Consortium (Consorcio de Investigacioìn del Golfo de Meìxico (CIGoM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge the PEMEX specific request to the Hydrocarbon Fund to address the environmental effects of oil spills in the Gulf of Mexico. We also acknowledge Marcela Ayala and Rosa Román Miranda for suggestions and experimental support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.713702/full#supplementary-material
Supplementary Figure 1 | Paenarthrobacter sp. GOM3 in G. mellonella infection model. (A) Health index (HI) scores of wax larvae over 5 days post infection with 100 UFC/10 μL. P. aeruginosa ATCC 27853 exhibited an HI score of zero after 24 h (yellow line). The marine strain had a lower HI than the negative controls. (B) Dose-response curve with a range of Paenarthrobacter sp. GOM3 doses after 48 h of infection (n = 15). The gray area shows the confidence limits using the probit analysis with the “ecotox” library in R.
Supplementary Figure 2 | Percentage of PAH degraded after 49 days by Paenarthrobacter sp. GOM3 growing in minimum medium with phenanthrene 0.02%. Assay made by duplicate.
Supplementary Table 1 | Relevant functional annotations of unique genes detected in Paenarthrobacter sp. GOM3 by pangenomic analysis.
Supplementary Table 2 | Genomic context of genes involved in the degradation of benzoate inside Paenarthrobacter sp. GOM3 genome and the percentage of identity of these genes with other strains.
Supplementary Table 3 | Genomic context of genes involved in the degradation of gentisate inside Paenarthrobacter sp. GOM3 genome and the percentage of identity of these genes with other strains.
Supplementary Table 4 | Genomic context of genes involved in the degradation of protocatechuates inside Paenarthrobacter sp. GOM3 genome and the percentage of identity of these genes with other strains.
Supplementary Table 5 | Mean and standard deviation of kinetic parameters of Paenarthrobacter sp. GOM3 growing in different aromatic compounds individually.
Supplementary Table 6 | Mean and standard deviation of kinetic parameters of Paenarthrobacter sp. growing in different mixtures of aromatic compounds.
Supplementary Table 7 | Mean and standard deviation of final concentrations of each aromatic compound in abiotic controls.
References
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2019). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525.
Alghoribi, M. F., Gibreel, T. M., Dodgson, A. R., Beatson, S. A., and Upton, M. (2014). Galleria Mellonella infection model demonstrates high lethality of ST69 and ST127 uropathogenic E. Coli. PLoS One 9:e101547. doi: 10.1371/journal.pone.0101547
Andrejko, M., Zdybicka-Barabas, A., and Cytryñska, M. (2014). Diverse effects of Galleria Mellonella infection with entomopathogenic and clinical strains of Pseudomonas Aeruginosa. J. Invertebr. Pathol. 115, 14–25. doi: 10.1016/j.jip.2013.10.006
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Baitsch, D., Sandu, C., Brandsch, R., and Igloi, G. L. (2001). Gene cluster on pAO1 of arthrobacter nicotinovorans involved in degradation of the plant alkaloid nicotine: cloning, purification, and characterization of 2,6-dihydroxypyridine 3-hydroxylase. J. Bacteriol. 183, 5262–5267. doi: 10.1128/jb.183.18.5262-5267.2001
Bernasconi, E., Valsangiacomo, C., Peduzzi, R., Carota, A., Moccetti, T., and Funke, G. (2004). Arthrobacter woluwensis subacute infective endocarditis: case report and review of the literature. Clin. Infect. Dis. 38, e27–e31.
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Busse, H. J. (2016). Review of the taxonomy of the genus arthrobacter, emendation of the genus arthrobacter sensu lato, proposal to reclassify selected species of the genus arthrobacter in the novel genera Glutamicibacter gen. nov., Paeniglutamicibacter gen. nov., Pseudoglutamicibacter gen. nov., Paenarthrobacter gen. nov. and Pseudarthrobacter gen. nov., and emended description of arthrobacter roseus. Int. J. Syst. Evol. Microbiol. 66, 9–37. doi: 10.1099/ijsem.0.000702
Busse, H. J., and Wieser, M. (2018). “Paenarthrobacter,” in Bergey’s Manual of Systematics of Archaea and Bacteria, ed. W. B. Whitman (Hoboken, NJ: John Wiley & Sons, Inc.) doi: 10.1002/9781118960608.gbm01436
Cabel, A. C. Jr., Aragon, G. M., Trazona, R. G., and Magdadaro, M. C. (2000). “Application of sodium benzoate in surface water flow measurements in mindanao geothermal production field, philippines,” in Proceedings of the World Geothermal Congress, (Kyushu), 1019–1024.
Cao, B., Nagarajan, K., and Loh, K. C. (2009). Biodegradation of aromatic compounds: current status and opportunities for biomolecular approaches. Appl. Microbiol. Biotechnol. 85, 207–228. doi: 10.1007/s00253-009-2192-4
Carrington, B., Lowe, A., Shaw, L. E., and Williams, P. A. (1994). The lower pathway operon for benzoate catabolism in biphenyl-utilizing Pseudomonas Sp. strain ic and the nucleotide sequence of the bphe gene for catechol 2,3-dioxygenase. Microbiology 140(Pt 3), (March): 499–508.,Google Scholar
Caspi, R., Billington, R., Ferrer, L., Foerster, H., Fulcher, C. A., Keseler, I. M., et al. (2012). The MetaCyc database of metabolic pathways and enzymes and the biocyc collection of pathway/genome databases. Nucleic Acids Res. 40, D742–D753.
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P., and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927.
Cinar, O. (2004). Biodegradation of central intermediate compounds produced from biodegradation of aromatic compounds. Bioprocess Biosyst. Eng. 26, 341–345. doi: 10.1007/s00449-004-0364-2
Collier, L. S., Nichols, N. N., and Neidle, E. L. (1997). benK Encodes a hydrophobic permease-like protein involved in benzoate degradation by Acinetobacter Sp. Strain ADP1. J. Bacteriol. 179, 5943–5946. doi: 10.1128/jb.179.18.5943-5946.1997
Cools, F., Torfs, E., Aizawa, J., Vanhoutte, B., Maes, L., and Caljon, G. (2019). Optimization and characterization of a Galleria mellonella larval infection model for virulence studies and the evaluation of therapeutics against Streptococcus Pneumoniae. Front. Microbiol. 10:311.
Dastgheib, S. M., Amoozegar, M. A., Khajeh, K., Shavandi, M., and Ventosa, A. (2012). Biodegradation of polycyclic aromatic hydrocarbons by a halophilic microbial consortium. Appl. Microbiol. Biotechnol. 95, 789–798. doi: 10.1007/s00253-011-3706-4
De Anda, V., Zapata-Peñasco, I., Poot-Hernandez, A. C., Eguiarte, L. E., Contreras-Moreira, B., and Souza, V. (2017). MEBS, a software platform to evaluate large (meta)genomic collections according to their metabolic machinery: unraveling the sulfur cycle. GigaScience 6, 1–17. doi: 10.1096/fasebj.2020.34.s1.02797
Fritsche, W., and Hofrichter, M. (2008). “Aerobic degradation by microorganisms,” in Biotechnology, eds H.-J. Rehm and G. Reed (Weinheim: Wiley-VCH Verlag GmbH), 144–167. doi: 10.1002/9783527620999.ch6m
Fuchs, G., Boll, M., and Heider, J. (2011). Microbial degradation of aromatic compounds - from one strategy to four. Nat. Rev. Microbiol. 9, 803–816. doi: 10.1038/nrmicro2652
Gibson, D. T., and Parales, R. E. (2000). Aromatic Hydrocarbon Dioxygenases in Environmental Biotechnology. Curr. Opin. Biotechnol. 11, 236–243. doi: 10.1016/s0958-1669(00)00090-2
Glare, P. G. W. (1968). Oxford Latin Dictionary. Available online at: https://ixtheo.de/Record/1123707774 (accessed July 1, 2021).
Godoy-Lozano, E. E., Escobar-Zepeda, A., Raggi, L., Merino, E., Gutierrez-Rios, R. M., Juarez, K., et al. (2018). Bacterial diversity and the geochemical landscape in the Southwestern Gulf of Mexico. Front. Microbiol. 9:2528.
Gulvik, C. A., and Buchan, A. (2013). Simultaneous catabolism of plant-derived aromatic compounds results in enhanced growth for members of the roseobacter lineage. Appl. Environ. Microbiol. 79, 3716–3723. doi: 10.1128/aem.00405-13
Guzik, U., Hupert-Kocurek, K., and Wojcieszyñska, D. (2013). “Intradiol dioxygenases — the key enzymes in xenobiotics degradation,” in Biodegradation of Hazardous and Special Products, eds R. Chamy and F. Rosenkranz (Rijeka: IntechOpen).
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucl. Acids. Symp. Ser. 41, 95–98.
Hassanshahian, M., Abarian, M., and Cappello, S. (2015). “Biodegradation of aromatic compounds”, in Biodegradation and Bioremediation of Polluted Systems - New Advances and Technologies, eds R. Chamy and F. Rosenkranz (IntechOpen). doi: 10.5772/59459
Hensen, C., Zabel, M., and Schulz, H. N. (2006). “Benthic cycling of oxygen, nitrogen and phosphorus,” in Marine Geochemistry, eds H. D. Schulz and M. Zabel (Berlin: Springer), 207–240. doi: 10.1007/3-540-32144-6_6
Hernandez, R. J., Hesse, E., Dowling, A. J., Coyle, N. M., Feil, E. J., Gaze, W. H., et al. (2019). Using the wax moth larva Galleria Mellonella infection model to detect emerging bacterial pathogens. PeerJ 6:e6150. doi: 10.7717/peerj.6150
Heuer, H., Krsek, M., Baker, P., Smalla, K., and Wellington, E. M. (1997). Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl. Environ. Microbiol. 63, 3233–3241. doi: 10.1128/aem.63.8.3233-3241.1997
Hlina, B. L., Birceanu, O., Robinson, C. S., Dhiyebi, H., and Wilkie, M. P. (2019). Ecotox. Ottawa, CA: ECOTOX.
Houghton, J. E., Brown, T. M., Appel, A. J., Hughes, E. J., and Ornston, L. N. (1995). Discontinuities in the evolution of Pseudomonas putida cat genes. J. Bacteriol. 177, 401–412. doi: 10.1128/jb.177.2.401-412.1995
Huang, Y., Zhao, N., He, L., Wang, L., Liu, Z., You, M., et al. (2005). Arthrobacter scleromae Sp. Nov. Isolated from human clinical specimens. J. Clin. Microbiol. 43, 1451–1455. doi: 10.1128/jcm.43.3.1451-1455.2005
Huijbers, M. M., Montersino, S., Westphal, A. H., Tischler, D., and van Berkel, W. J. (2014). Flavin dependent monooxygenases. Arch. Biochem. Biophys. 544, 2–17.
Hunt, M., Kikuchi, T., Sanders, M., Newbold, C., Berriman, M., and Otto, T. D. (2013). REAPR: a universal tool for genome assembly evaluation. Genome Biol. 14, R47.
Igarashi, Y., Yamamoto, K., Fukuda, T., Shojima, A., Nakayama, J., Carro, L., et al. (2015). Arthroamide, a cyclic depsipeptide with quorum sensing inhibitory activity from Arthrobacter Sp. J. Nat. Prod. 78, 2827–2831. doi: 10.1021/acs.jnatprod.5b00540
Insua, J. L., Llobet, E., Moranta, D., Pérez-Gutiérrez, C., Tomás, A., Garmendia, J., et al. (2013). Modeling Klebsiella Pneumoniae pathogenesis by infection of the wax moth Galleria mellonella. Infect. Immun. 81, 3552–3565. doi: 10.1128/iai.00391-13
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90k prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114.
Jetten, M. S. M. (2008). The microbial nitrogen cycle. Environ. Microbiol. 10, 2903–2909. doi: 10.1111/j.1462-2920.2008.01786.x
Jørgensen, B. B., Findlay, A. J., and Pellerin, A. (2019). The biogeochemical sulfur cycle of marine sediments. Front. Microbiol. 10:849.
Kotoućková, L., Schumann, P., Durnová, E., Spröer, C., Sedláćek, I., Neća, J., et al. (2004). Arthrobacter Nitroguajacolicus Sp. Nov., a novel 4-nitroguaiacol-degrading actinobacterium. Int. J. Syst. Evol. Microbiol. 54(Pt 3), 773–777. doi: 10.1099/ijs.0.02923-0
Kowalchuk, G. A., Hartnett, G. B., Benson, A., Houghton, J. E., Ngai, K. L., and Ornston, L. N. (1994). Contrasting patterns of evolutionary divergence within the acinetobacter calcoaceticus pca operon. Gene 146, 23–30. doi: 10.1016/0378-1119(94)90829-x
Ladino-Orjuela, G., Gomes, E., da Silva, R., Salt, C., and Parsons, J. R. (2016). Metabolic pathways for degradation of aromatic hydrocarbons by bacteria. Rev. Environ. Contam. Toxicol. 237, 105–121. doi: 10.1007/978-3-319-23573-8_5
Landry, C., and Tremblay, L. (2012). Compositional differences between size classes of dissolved organic matter from freshwater and seawater revealed by an HPLC-FTIR system. Environ. Sci. Technol. 46, 1700–1707. doi: 10.1021/es203711v
Loh, J. M., Adenwalla, N., Wiles, S., and Proft, T. (2013). Galleria Mellonella larvae as an infection model for group A Streptococcus. Virulence 4, 419–428.
Luan, T. G., Yu, K. S., Zhong, Y., Zhou, H. W., Lan, C. Y., and Tam, N. F. (2006). Study of metabolites from the degradation of polycyclic aromatic hydrocarbons (pahs) by bacterial consortium enriched from mangrove sediments. Chemosphere 65, 2289–2296. doi: 10.1016/j.chemosphere.2006.05.013
Maddocks, S. E., and Oyston, P. C. F. (2008). Structure and function of the LysR-Type Transcriptional Regulator (LTTR) family proteins. Microbiology 154(Pt 12), 3609–3623. doi: 10.1099/mic.0.2008/022772-0
Mallick, S., Chakraborty, J., and Dutta, T. K. (2011). Role of oxygenases in guiding diverse metabolic pathways in the bacterial degradation of low-molecular-weight polycyclic aromatic hydrocarbons: a review. Crit. Rev. Microbiol. 37, 64–90. doi: 10.3109/1040841x.2010.512268
McClure, R. A., Goering, A. W., Ju, K. S., Baccile, J. A., Schroeder, F. C., and Metcalf, W. W. (2016). Elucidating the rimosamide-detoxin natural product families and their biosynthesis using metabolite/gene cluster correlations. ACS Chem. Biol. 11, 3452–3460. doi: 10.1021/acschembio.6b00779
McGinnis, S., and Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25.
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Meng, J., Sun, X., Li, S., and Liang, H. (2017). Draft Genome Sequence of Paenarthrobacter Nicotinovorans Hce-1. Genome Announc. 5, e00727–17.
Mihăşan, M., Boiangiu, R. Ş, Guzun, D., Babii, C., Aslebagh, R., and Channaveerappa, D. (2021). Time-dependent analysis of Paenarthrobacter nicotinovorans pao1 nicotine-related proteome. ACS Omega 6, 14242–14251. doi: 10.1021/acsomega.1c01020
Miller, M. B., and Bassler, B. L. (2001). Quorum sensing in bacteria. Ann. Rev. Microbiol. 55, 165–199.
Miyata, S., Casey, M., Frank, D. W., Ausubel, F. M., and Drenkard, E. (2003). Use of the Galleria mellonella caterpillar as a model host to study the role of the type iii secretion system in Pseudomonas Aeruginosa pathogenesis. Infect. Immun. 71, 2404–2413. doi: 10.1128/iai.71.5.2404-2413.2003
Molina-Henares, A. J., Krell, T., Eugenia Guazzaroni, M., Segura, A., and Ramos, J. L. (2006). Members of the IclR family of bacterial transcriptional regulators function as activators And/or repressors. FEMS Microbiol. Rev. 30, 157–186. doi: 10.1111/j.1574-6976.2005.00008.x
Monciardini, P., Sosio, M., Cavaletti, L., Chiocchini, C., and Donadio, S. (2002). New PCR primers for the selective amplification of 16S rDNA from different groups of actinomycetes. FEMS Microbiol. Ecol. 42, 419–429. doi: 10.1016/s0168-6496(02)00353-7
Moraes, E. C., Alvarez, T. M., Persinoti, G. F., Tomazetto, G., Brenelli, L. B., Paixão, D. A. A., et al. (2018). Lignolytic-consortium omics analyses reveal novel genomes and pathways involved in lignin modification and valorization. Biotechnol. Biofuels 11, 1–16.
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185.
Muñoz-Elías, E. J., and McKinney, J. D. (2005). Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med. 11, 638–644. doi: 10.1038/nm1252
Muriel-Millán, L. F., Rodríguez-Mejía, J. L., Godoy-Lozano, E. E., Rivera-Gómez, N., Gutierrez-Rios, R. M., Morales-Guzmán, D., et al. (2019). Functional and genomic characterization of a Pseudomonas Aeruginosa strain isolated from the southwestern gulf of mexico reveals an enhanced adaptation for long-chain alkane degradation. Front. Mar. Sci. 6:572.
Na, S. I., Kim, Y. O., Yoon, S. H., Ha, S. M., Baek, I., and Chun, J. (2018). UBCG: up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56, 280–285. doi: 10.1007/s12275-018-8014-6
Nadalin, F., Vezzi, F., and Policriti, A. (2012). GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinformatics 13 Suppl 14:S8.
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using minhash. Genome Biol. 17:132.
Otto, T. D., Sanders, M., Berriman, M., and Newbold, C. (2010). Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics 26, 1704–1707. doi: 10.1093/bioinformatics/btq269
Parales, R. E., and Resnick, S. M. (2004). “Aromatic hydrocarbon dioxygenases,” in Biodegradation and Bioremediation, eds A. Singh and O. P. Ward (Berlin: Springer), 175–195. doi: 10.1007/978-3-662-06066-7_8
Park, M., Jeon, Y., Jang, H. H., Ro, H. S., Park, W., Madsen, E. L., et al. (2007). Molecular and biochemical characterization of 3-hydroxybenzoate 6-hydroxylase from Polaromonas naphthalenivorans CJ2. Appl. Environ. Microbiol. 73, 5146–5152. doi: 10.1128/aem.00782-07
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pircher, H., Straganz, G. D., Ehehalt, D., Morrow, G., Tanguay, R. M., and Jansen-Dürr, P. (2011). Identification of human fumarylacetoacetate hydrolase domain-containing protein 1 (FAHD1) as a novel mitochondrial acylpyruvase. J. Biol. Chem. 286, 36500–36508. doi: 10.1074/jbc.m111.264770
Raggi, L., García-Guevara, F., Godoy-Lozano, E. E., Martínez-Santana, A., Escobar-Zepeda, A., Gutierrez-Rios, R. M., et al. (2020). Metagenomic profiling and microbial metabolic potential of perdido fold belt (NW) and campeche knolls (SE) in the Gulf of Mexico. Front. Microbiol. 11:1825.
Robbins, R. J. (2003). Phenolic acids in foods: an overview of analytical methodology. J. Agric. Food Chem. 51, 2866–2887. doi: 10.1021/jf026182t
Rodríguez-Salazar, J., Loza, A., Ornelas-Ocampo, K., Gutierrez-Rios, R. M., and Pardo-López, L. (2021). Bacteria from the Southern Gulf of Mexico: baseline, diversity, hydrocarbon-degrading potential and future applications. Front. Mar. Sci. 8:232.
Romero-Steiner, S., Parales, R. E., Harwood, C. S., and Houghton, J. E. (1994). Characterization of the pcaR regulatory gene from Pseudomonas putida, which is required for the complete degradation of p-hydroxybenzoate. J. Bacteriol. 176, 5771–5779. doi: 10.1128/jb.176.18.5771-5779.1994
Sahlin, K., Vezzi, F., Nystedt, B., Lundeberg, J., and Arvestad, L. (2014). BESST–efficient scaffolding of large fragmented assemblies. BMC Bioinformatics 15:281.
Sánchez-Reyes, A., and Fernández-López, M. G. (2021). Mash sketched reference dataset for genome-based taxonomy and comparative genomics. Preprints doi: 10.20944/preprints202106.0368.v1
Singh, V., Chandra, D., Srivastava, B. S., and Srivastava, R. (2011). Downregulation of Rv0189c, encoding a dihydroxyacid dehydratase, affects growth of mycobacterium tuberculosis in vitro and in mice. Microbiology 157(Pt 1), 38–46. doi: 10.1099/mic.0.042358-0
Sonnleitner, E., Hagens, S., Rosenau, F., Wilhelm, S., Habel, A., Jäger, K. E., et al. (2003). Reduced virulence of a Hfq mutant of Pseudomonas Aeruginosa O1. Microb. Pathog. 35, 217–228. doi: 10.1016/s0882-4010(03)00149-9
Strong, L. C., Rosendahl, C., Johnson, G., Sadowsky, M. J., and Wackett, L. P. (2002). Arthrobacter aurescens TC1 metabolizes diverse S-triazine ring compounds. Appl. Environ. Microbiol. 68, 5973–5980. doi: 10.1128/aem.68.12.5973-5980.2002
Suenaga, H., Mizuta, S., Miyazaki, K., and Yaoi, K. (2014). Diversity of extradiol dioxygenases in aromatic-degrading microbial community explored using both culture-dependent and culture-independent approaches. FEMS Microbiol. Ecol. 90, 367–379.
Takehara, I., Kato, D. I., Takeo, M., and Negoro, S. (2017). Draft genome sequence of the nylon oligomer-degrading bacterium arthrobacter sp. strain KI72. Genome Announc. 5, e00217–17.
Tonkin-Hill, G., MacAlasdair, N., Ruis, C., Weimann, A., Horesh, G., Lees, J. A., et al. (2020). Producing polished prokaryotic pangenomes with the panaroo pipeline. Genome Biol. 21:180.
Tsai, C. J., Loh, J. M., and Proft, T. (2016). Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing. Virulence 7, 214–229. doi: 10.1080/21505594.2015.1135289
Vogt, C., Kleinsteuber, S., and Richnow, H. H. (2011). Anaerobic benzene degradation by bacteria. Microb. Biotechnol. 4, 710–724. doi: 10.1111/j.1751-7915.2011.00260.x
Wang, C., Huang, Y., Zhang, Z., and Wang, H. (2018). Salinity effect on the metabolic pathway and microbial function in phenanthrene degradation by a halophilic consortium. AMB Express 8:67. doi: 10.1186/s13568-018-0594-3
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591.
Widdel, F. (2007). Theory and measurement of bacterial growth. Di Dalam Grundpraktikum Mikrobiologie 4, 1–11.
Xu, X., Liu, W., Tian, S., Wang, W., Qi, Q., Jiang, P., et al. (2018). Petroleum hydrocarbon-degrading bacteria for the remediation of oil pollution under aerobic conditions: a perspective analysis. Front. Microbiol. 9: 2885.
Xu, Y., Wang, S. H., Chao, H. J., Liu, S. J., and Zhou, N. Y. (2012). Biochemical and molecular characterization of the gentisate transporter GenK in Corynebacterium glutamicum. PLoS One 7:e38701. doi: 10.1371/journal.pone.0038701
Zerbino, D. R. (2010). Using the velvet de novo assembler for short-read sequencing technologies. Curr. Protoc. Bioinformatics 11:Unit11.5.
Keywords: marine bacteria, aromatic compounds, hydrocarbon-degrading bacteria, Gulf of Mexico, bioprospection
Citation: Rosas-Díaz J, Escobar-Zepeda A, Adaya L, Rojas-Vargas J, Cuervo-Amaya DH, Sánchez-Reyes A and Pardo-López L (2021) Paenarthrobacter sp. GOM3 Is a Novel Marine Species With Monoaromatic Degradation Relevance. Front. Microbiol. 12:713702. doi: 10.3389/fmicb.2021.713702
Received: 23 May 2021; Accepted: 12 July 2021;
Published: 03 August 2021.
Edited by:
Pratyoosh Shukla, Banaras Hindu University, IndiaReviewed by:
Monali C. Rahalkar, Agharkar Research Institute, IndiaXiaojing Li, Agro-Environmental Protection Institute (CAAS), China
Onruthai Pinyakong, Chulalongkorn University, Thailand
Rup Lal, University of Delhi, India
Copyright © 2021 Rosas-Díaz, Escobar-Zepeda, Adaya, Rojas-Vargas, Cuervo-Amaya, Sánchez-Reyes and Pardo-López. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liliana Pardo-López, bGlsaWFuYS5wYXJkb0BpYnQudW5hbS5teA==
†Present address: Alejandra Escobar-Zepeda, Host-Microbiota Interactions Laboratory, Wellcome Sanger Institute, Hinxton, United Kingdom; European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Hinxton, United Kingdom