 Bailin An
Bailin An Hongbin Zhang3†
Hongbin Zhang3† Fengcai Zhu
Fengcai Zhu Lunbiao Cui
Lunbiao Cui- 1National Health Commission (NHC) Key Laboratory of Enteric Pathogenic Microbiology, Jiangsu Provincial Center for Disease Control and Prevention, Nanjing, China
- 2College of Pharmacy, Nankai University, Tianjin, China
- 3Jiangyin City Center for Disease Control and Prevention, Wuxi, China
- 4School of Public Health, Nanjing Medical University, Nanjing, China
Salmonella spp. is one of the most common foodborne disease-causing pathogens that can cause severe diseases in very low infectious doses. Rapid and sensitive detecting Salmonella spp. is advantageous to the control of its spread. In this study, a conserved short fragment of the Salmonella invA gene was selected and used to design primers and specific crRNA (CRISPR RNA) for establishing a one-tube and two-step reaction system for Salmonella spp. detection, by combining recombinase polymerase amplification (RPA) with CRISPR-Cas13a (Clustered Regularly Interspaced Short Palindromic Repeats associated protein 13a) cleavage. The established one-tube RPA-Cas13a method can complete the detection within 20 min and the two-step RPA-Cas13a method detection time within 45 min. The designed primers were highly specific to Salmonella spp. and had no cross-reaction with the other nine diarrheal bacteria. The one-tube RPA-Cas13a could detect the Salmonella genome with the limit of 102 copies, which was the same as real-time polymerase chain reaction (PCR), but less sensitive than two-step RPA-Cas13a (100 copies). The detection results of one-tube or two-step RPA-Cas13a and real-time PCR were highly consistent in clinical samples. One-tube RPA-Cas13a developed in this study provides a simple, rapid, and specific detection method for Salmonella spp. While two-step assay was more sensitive and suitable for samples at low abundance.
Introduction
Salmonella, a gram-negative bacillus, is the most common diarrheal pathogenic bacteria and infects millions of people across the world every year, with the most common clinical manifestations being acute gastroenteritis, encephalitis, pericarditis, sepsis, and even death (Hugas and Beloeil, 2014; Ferrari et al., 2019). According to statistics by the World Health Organization, the number of outbreaks of different food-borne intestinal diseases since 2010 has reached 582 million, with nearly 350,000 deaths, including 52,000 Salmonella cases (World Health Organization [WHO], 2016)]. In the United States, the Centers for Disease Control and Prevention (CDC) estimated food was one of the main sources of Salmonella infection; Salmonella caused approximately 1.35 million infections, 26,500 hospitalizations, and 420 deaths every year (CDC, 2020). In the European Union, European Centre for Disease Prevention and Control reported 91,857 Salmonella infection cases in humans, and Salmonella caused 30.7% of all food-borne outbreaks during 2018 (European Food Safety Authority, & European Centre for Disease Prevention and Control, 2020). In China, the previous study has estimated the incidence of non-typhoid salmonellosis at 626.6 cases per 100,000 people (Yue et al., 2020). Timely screening of Salmonella is the key to prevent and control diarrheal disease outbreaks. To date, the detection methods for Salmonella, including traditional biochemical culture, immune testing, and molecular biological approaches, are represented by polymerase chain reaction (PCR)/real-time PCR. These methods are time-consuming, or poor in specificity or of low sensitivity, and sometimes require expensive instruments for laboratory setup. Especially, many diarrheal pathogenic bacteria, including Salmonella, can cause severe infectious diseases at low infectious doses. More sensitive and specific Salmonella detection methods are urgently needed.
Over the past year, researchers have applied the latest recombinase polymerase amplification (RPA)/recombinase aided amplification (RAA) techniques to achieve amplification for Salmonella. However, the detection of amplification products by agarose gel electrophoresis showed poor sensitivity, and fluorescent probes for visual detection were expensive. A typical detection of pathogenic Salmonella was reported by Zhang et al. (2017) using the real-time fluorescence RAA detection system, but the detection limit was 103 copies/μL, equivalent to ∼5 fg plasmid DNA. An optical biosensor reported by Zheng et al. (2020) could detect 1.8 × 101 copies/μL but was not useful in POCT (point-of-care testing).
The CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR associated protein) system is a sophisticated adaptive immune system, and the different CRISPR-Cas nucleases display specific cleavage activity when target-specific crRNA recognizes the DNA target (Bhaya et al., 2011; Jackson et al., 2014; Kirchner and Schneider, 2015). In recent years, researchers have found that the cleavage activity of CRISPR-Cas nucleases could be used not only as a programmable tool for gene editing but also for in vitro nucleic acid detection (Arslan et al., 2014; Zhou et al., 2020). In this study, we established a one-tube and a two-step reaction system for Salmonella spp. detection by combining RPA with CRISPR-Cas13a cleavage, which provided a sensitive and convenient molecular detection method for the early diagnosis of diarrheal diseases of Salmonella infection.
Materials and Methods
Cas13a Protein Expression and Purification
Cas13a was purified as referred to in previous studies (East-Seletsky et al., 2016, 2017; Su et al., 2020). Briefly, the Cas13a gene in plasmid pC019-LwCas13a from Leptotrichia wadei (Addgene, United States) was subcloned into the psmarti vector (xhoI restriction site), and the correct plasmid was transformed into BL21(DE3) (General Biosystem, China). The expression of the target protein was induced at different temperatures (15 and 37°C) and different concentrations of IPTG (0.2 and 1.0 mM) (Amresco, United States). The Cas13a protein was purified by Ni-NTA (Smart-Lifesciences, China) using the 6 × His Tag antibody and horseradish peroxidase conjugate (Invitrogen, United States). After the addition of SUMO Protease (General Biosystem, China) to remove the fusion SUMO label, and the purified target protein was dialyzed into protein buffer [50 mM Tris, pH 7.5, 600 mM NaCl, 5% (vol/vol) glycerol, and 2 mM DTT].
Strains, Clinical Samples, and Nucleic Acid Extraction
Salmonella and other diarrheal pathogenic bacteria (Staphylococcus aureus, Listeria monocytogenes, Enterococcus faecalis, Shigella flexneri, Vibrio parahemolyticus, Escherichia coli O157, Yersinia enterocolitica, Campylobacter jejuni, Vibrio vulnificus) were provided by Jiangsu Provincial Center for Disease Control and Prevention. A total of 84 clinical stool samples that were selected for clinical validation of the method were collected from different hospitals in Jiangsu Province in 2019. The nucleic acid was extracted by EX-DNA Bacterial Genomic Extraction Kit (Tianlong, China) on a fully automatic nucleic acid extractor (Tianlong, China).
Preparation of Primers, Probes, and crRNA
Salmonella-specific invA gene was retrieved from GenBank, and the conservative sequence region was selected to design RPA amplification primers using Primer 5 software, and Primer-BLAST software was used to verify the specificity of the primer sequence. The designed primers, probes, and oligonucleotides were synthesized by Sangon Biotech (Shanghai, China). The oligonucleotides containing T7 promoter, repeat, and spacer sequences were annealed with a T7 primer. Then, the crRNA was synthesized by incubating at 42°C for 2 h with T7 RNA polymerase (TaKaRa, China). The synthesized crRNA was digested with DNase I (TaKaRa, China) at 37°C for 1 h followed by purification with RNA rapid concentration and purification kit (Sangon Biotech, China) according to the manufacturer’s protocol. The concentration of crRNA was quantified using Qubit 2.0 (Invitrogen, United States). All nucleic acid sequences used in this study are shown in Table 1.

Table 1. RPA primers, crRNA, and RNA reporter probe sequences.
Collateral Cutting Capacity of CRISPR-Cas13a
The amplicon of the RPA reaction was used as the template for transcription in vitro with T7 RNA polymerase, and the transcription product was purified to obtain the Salmonella target RNA. Then, the target RNAs were diluted gradiently (100–105 pM). The synthesized oligonucleotide Salmonella-RPA-R was used as a non-target control (500 ng/μL). Verification of CRISPR-Cas13a cutting activity was completed as follows: 5 × reaction buffer 5 μL (final concentration 20 mM HEPES, 60 mM NaCl, 6 mM MgCl2, pH 6.8), 25 mM NTP mix 1 μL (ATP, GTP, CTP, UTP, Bio Lab, China), Recombinant RNase Inhibitor (TaKaRa, China) 0.5 μL, 1.2 μM crRNA 0.6 μL, 10 μM RNA-probe 0.5 μL, 1 μM Cas13a 1 μL, or 103 pM target RNA or non-target control 2 μL, adding RNase-free ddH2O to 25 μL. The reaction was conducted at 39°C for 30 min, and RNase A was used as the positive control.
Two-Step RPA-Cas13a
In the two-step RPA-Cas13a assay, basic RPA reactions were first conducted according to the instructions of the TwistAmp Basic Kit (Twist Dx, Cambridge, United Kingdom). Each RPA reaction was carried out in a 50 μL reaction volume containing Primer Free Rehydration buffer 29.5 μL, 0.24–0.96 μM forward and reverse primers each 0.5 μL, target DNA template 2 μL, and RNase-free ddH2O 15 μL. The reaction mixes were vortexed and spun briefly, and then 280 mM magnesium acetate (MgOAc) 2.5 μL was added and mixed well to start a reaction in fluorescence detector F1620 (Qitian, China) at 37, 39, and 41°C for 20 min. Then, a 25 μL CRISPR-Cas13a reaction system was performed with 5 × reaction buffer 5 μL, 25 mM NTP mix 1 μL, Recombinant RNase Inhibitor 0.5 μL, 1.2 μM Salmonella-crRNA 0.6 μL, 10 μM RNA-probe 0.5 μL, T7 RNA Polymerase 0.3 μL, 1 μM Cas13a 1 μL, and added RNase-free ddH2O to 23 μL, and RPA products 2 μL. The fluorescent signals were collected at 39°C for 30 min in fluorescence detector F1620.
One-Tube RPA-Cas13a
The one-tube RPA-Cas13a assay combined RPA with CRISPR-Cas13a in a one-tube reaction system. Briefly, the 50 μL one-tube reaction system consisted of Primer Free Rehydration buffer 29.5 μL, 25 mM NTP mix 4 μL, Recombinant RNase Inhibitor 4 μL, T7 RNA Polymerase 1 μL, 0.24–0.96 μM primers each 0.5 μL, 1.2 μM crRNA 1 μL, 10 μM RNA-probe 1 μL, 1 μM Cas13a 2 μL, target DNA template 2 μL, and RNase-free ddH2O 1 μL. The reaction mixes were vortexed and spun briefly, and then 280 mM MgOAc 2.5 μL was added to the tube cap and was centrifuged into the reaction solution. Fluorescent signals were collected at 37, 39, and 41°C for 2 h in fluorescence detector F1620.
Sensitivity and Specificity of One-Tube and Two-Step RPA-Cas13a
To evaluate the minimum detection limit, serial dsDNA standards were prepared as follows: using the extracted nucleic acid of Salmonella as a template, a 50 μL LA Taq PCR (TaKaRa, China) containing LA Taq 0.5 μL, 2.5 μM dNTP 4 μL, 10 × buffer 5 μL, 10 μM forward/reverse primer each 1 μL, and RNase-free ddH2O 37.5 μL was performed. The PCR amplification product was purified by High Pure PCR Product Purification Kit (Roche, CH). The dsDNA quantification was performed in a Qubit digital fluorimeter using the Qubit dsDNA BR Assay (Invitrogen, Waltham, United States). The dsDNA copy number was then determined using the following formula: {[6.02 × 1014 × dsDNA concentration (ng/μL) × 10–9]}/[DNA in length × 660]. The gradiently diluted dsDNA standards (107∼10–1 copies/μL) were detected by the one-tube and two-step RPA-Cas13a. We also chose real-time PCR (Hein et al., 2006) (TaKaRa, China) as the reference test. The specificity of the one-tube and two-step RPA-Cas13a was tested by using nucleic acid extracted from diarrheal pathogenic bacteria strains: Salmonella, S. aureus, L. monocytogenes, E. faecalis, S. flexneri, V. parahemolyticus, E. coli O157, Y. enterocolitica, C. jejuni, and V. vulnificus.
Real-Time Polymerase Chain Reaction
Real-time PCR was performed on the LightCycler 2.0 instrument in which premix Ex TaqTM (Takara, China) was used (Hein et al., 2006). The reaction was performed as follows: 95°C for 30 s, followed by 40 cycles of 95°C for 5 s and then 60°C for 20 s.
Consistency Comparison Using Real-Time Polymerase Chain Reaction
Genomic DNAs from 84 clinical stool samples were selected for a clinical comparison experiment. All samples were tested with the two-step, one-tube RPA-Cas13a, and real-time PCR assay (Hein et al., 2006). The consistency of the three methods above was compared using SPSS 24.0 software. κ-tests were used for consistency analysis.
Results
Collateral Cutting Capacity of CRISPR-Cas13a
A significant fluorescent signal in the reactions of the positive control within was verified with RNase A. Compared with the detection of Cas13a for target RNA and non-target RNA control, the results showed that Cas13a could achieve specific detection for target RNA of Salmonella in the presence of its corresponding crRNA. Furthermore, there was no detection signal generated for Cas12a compared to Cas13a. The detection signal of CRISPR-Cas13a for 103 pM was almost identical to that of the positive control (Figure 1). The results of different dilutions for RNA standards showed that the minimum test limit was 10 pM (Figure 2).
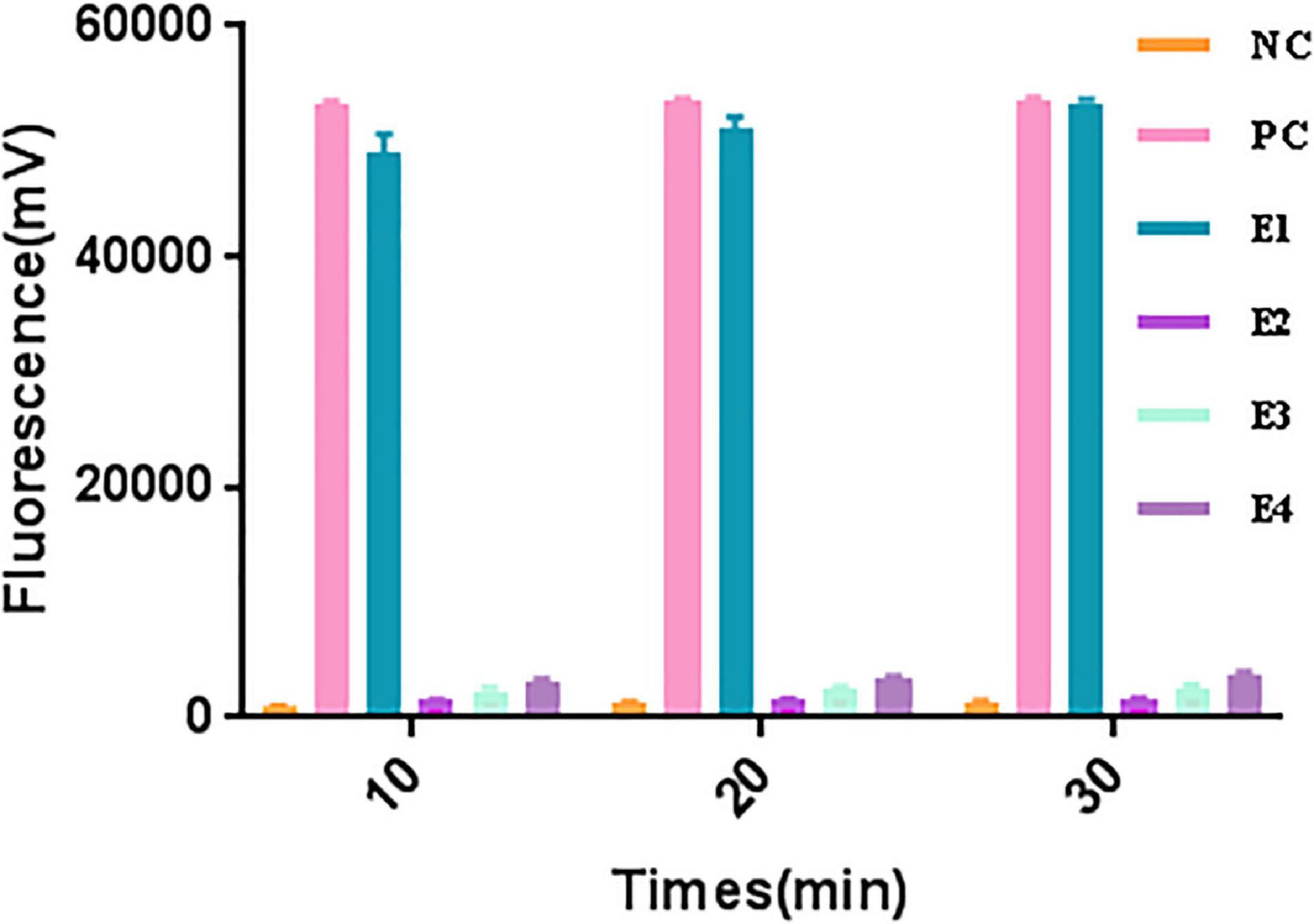
Figure 1. The collateral cutting activity of CRISPR-Cas13a. NC, negative control (RNase-free ddH2O); PC, positive control (RNase A); E1, Cas13a with 103 pM target RNA; E2, Cas13a with 103 pM non-target control; E3, Cas12a with 103 pM target RNA; E4, Cas13a with non-crRNA.
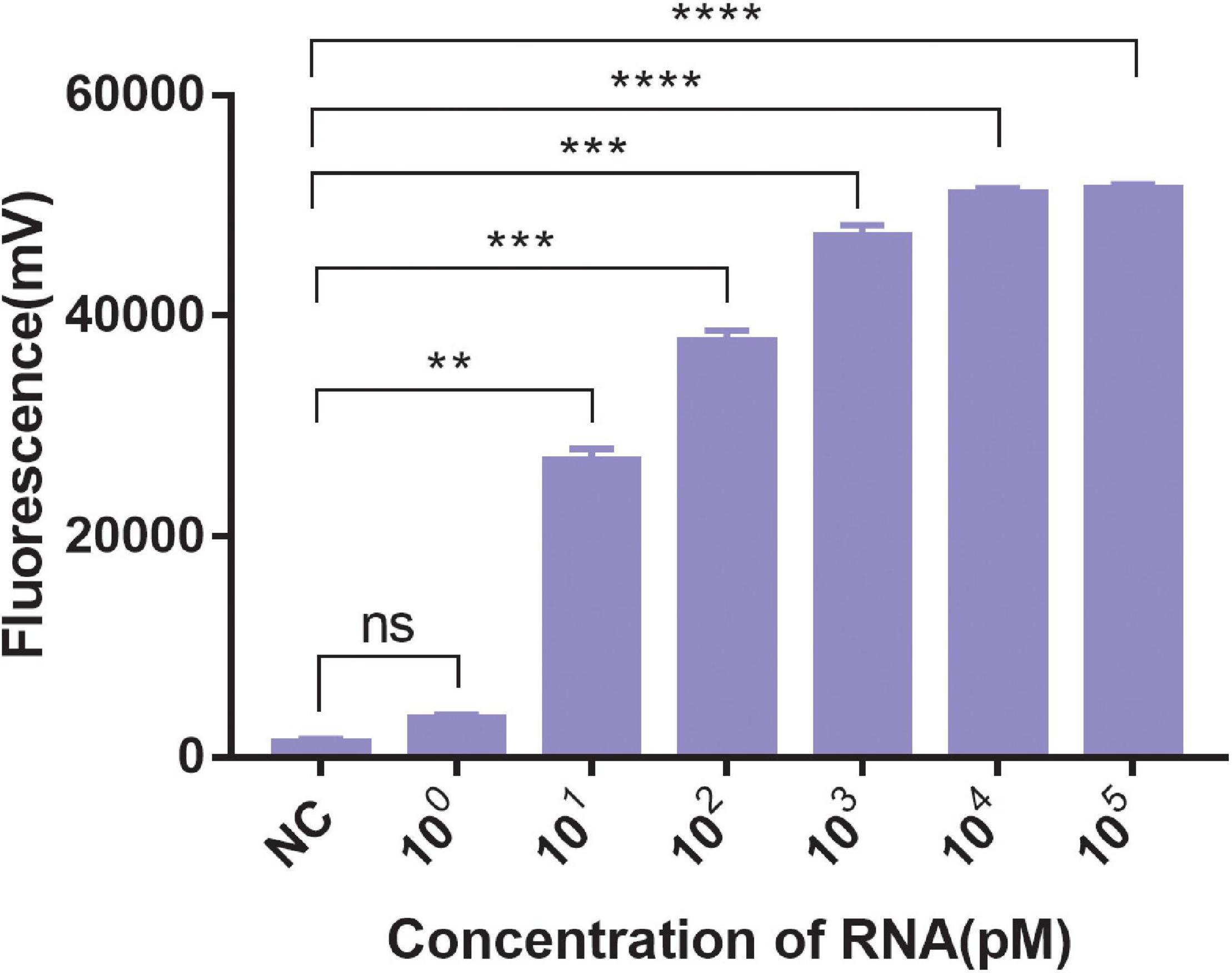
Figure 2. The sensitivity of CRISPR-Cas13a. 100∼105 pM were the concentrations of RNA standards. NC, negative control (RNase-free ddH2O). Unpaired two-tailed t-test was used to analyze the difference from NC. **p < 0.01; ***p < 0.001; ****p < 0.0001. ns, not significant.
Optimization of Two-Step RPA-Cas13a Detection System
We first tested the best RPA amplification temperature using 0.48 μM primer concentration. The results showed the highest fluorescent signals were obtained at 39°C in all three Cas13a detecting time points (Figure 3A). Therefore, 39°C was selected as the best RPA amplification temperature. Then, the best primer concentrations were explored at 39°C. The optimal primer concentration of 0.48 μM was found (Figure 3B). Therefore, we chose 0.48 μM and 39°C as the best RPA primer concentration and amplification temperature.
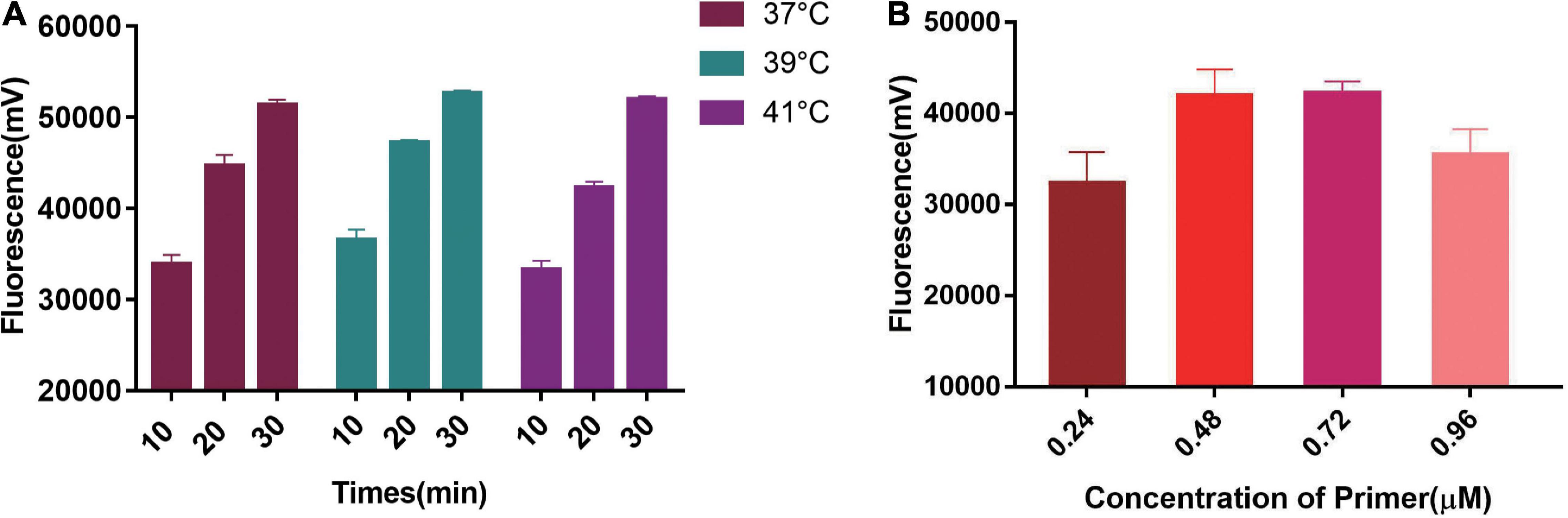
Figure 3. Single factor screening results of two-step RPA-Cas13a detection. (A) The effect of temperatures, 37, 39, and 41°C represented the temperature of the RPA reaction. (B) The effect of RPA primer concentration.
Optimization of One-Tube RPA-Cas13a Detection System
We first tested the different temperature (37, 39, 41°C) in a one-tube RPA-Cas13a detection system and found that 39°C had the highest fluorescent signals. The detection signal grew stronger from 10 to 90 min and reached its peak at 90 min, whereas the negative control had no visible detection signal (Figure 4A). We then explored the optimal primer concentration under 39°C and found the highest fluorescent signals were obtained at 0.48 μM primer concentrations. Therefore, the optimized conditions of one-tube RPA-Cas13a were 39°C with 0.48 μM primer concentration (Figure 4B).
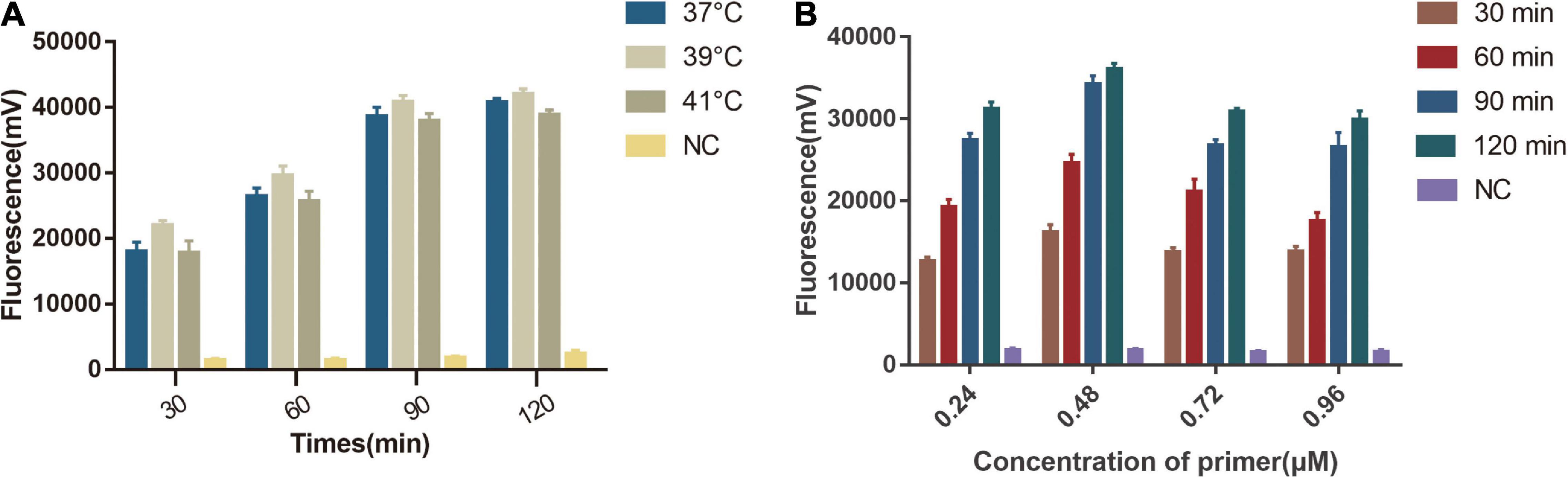
Figure 4. Single factor screening results of one-tube RPA-Cas13a detection. (A) The effect of temperature on one-tube RPA-Cas13a. 37, 39, and 41°C represented the temperature of the one-tube RPA-Cas13a reaction. NC, negative control (RNase-free ddH2O). (B) The effect of primer concentration on one-tube RPA-Cas13a. 30, 60, 90, and 120 min represented the time of one-tube RPA-Cas13a reaction. NC, negative control (RNase-free ddH2O).
Sensitivity and Specificity of One-Tube and Two-Step RPA-Cas13a
As shown in Figure 5A, the limit of detection of one-tube RPA-Cas13a for Salmonella was 102 copies/μL, which showed the same sensitivity to real-time PCR (102 copies/μL, Figure 6) and slightly lower than that of two-step RPA-Cas13a (100 copies/μL, Figure 5B). The whole detection time of one-tube RPA-Cas13a for 102 copies/μL was only 20 min (Figure 5A), which was shorter than those for two-step RPA-Cas13a (RPA: 20 min, transfer time: 5 min, and CRISPR-Cas13a: 20 min, Figure 5B) and real-time PCR (60 min or much longer, Figure 6). For specificity assessment, whether one-tube RPA-Cas13a at 60 min (Figure 7A) or two-step RPA-Cas13a at 30 min (Figure 7B), only the Salmonella showed detection signals among the 10 common diarrheal bacteria, and no cross-reactions were found.
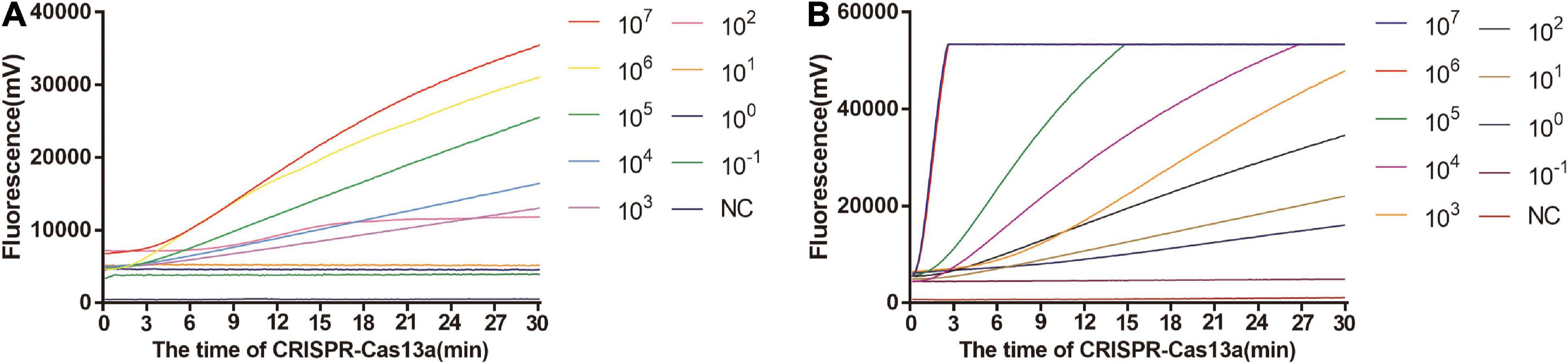
Figure 5. The sensitivity of one-tube and two-step RPA-Cas13a. (A) The sensitivity of one-tube RPA-Cas13a. (B) The sensitivity of two-step RPA-Cas13a. 107∼10– 1 (copies/μL) represented the concentration of Salmonella dsDNA standards. NC, negative control (RNase-free ddH2O).
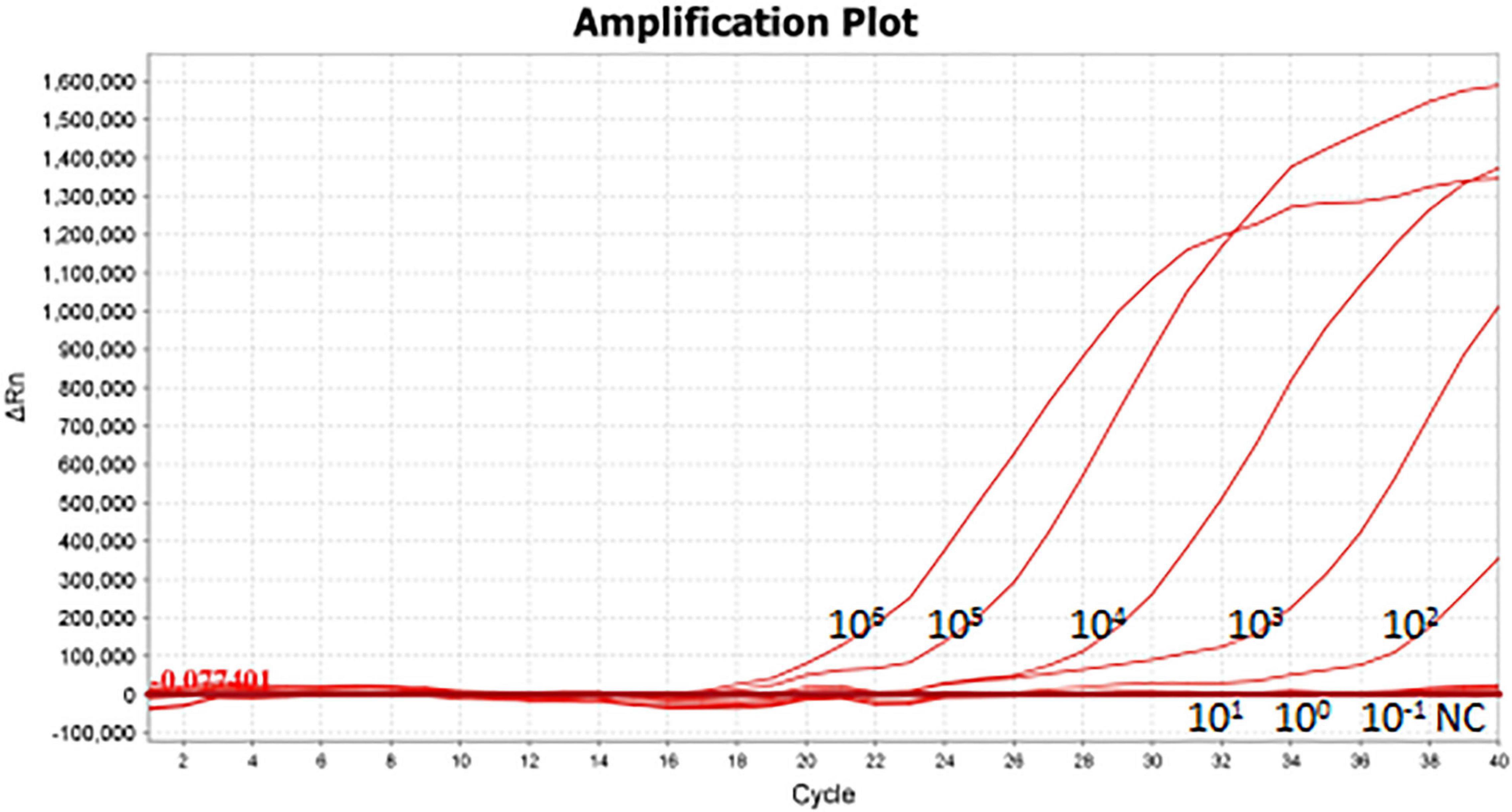
Figure 6. The sensitivity of real-time PCR 106∼10– 1 (copies/μL) represented the concentration of Salmonella dsDNA standards. NC, negative control (RNase-free ddH2O).
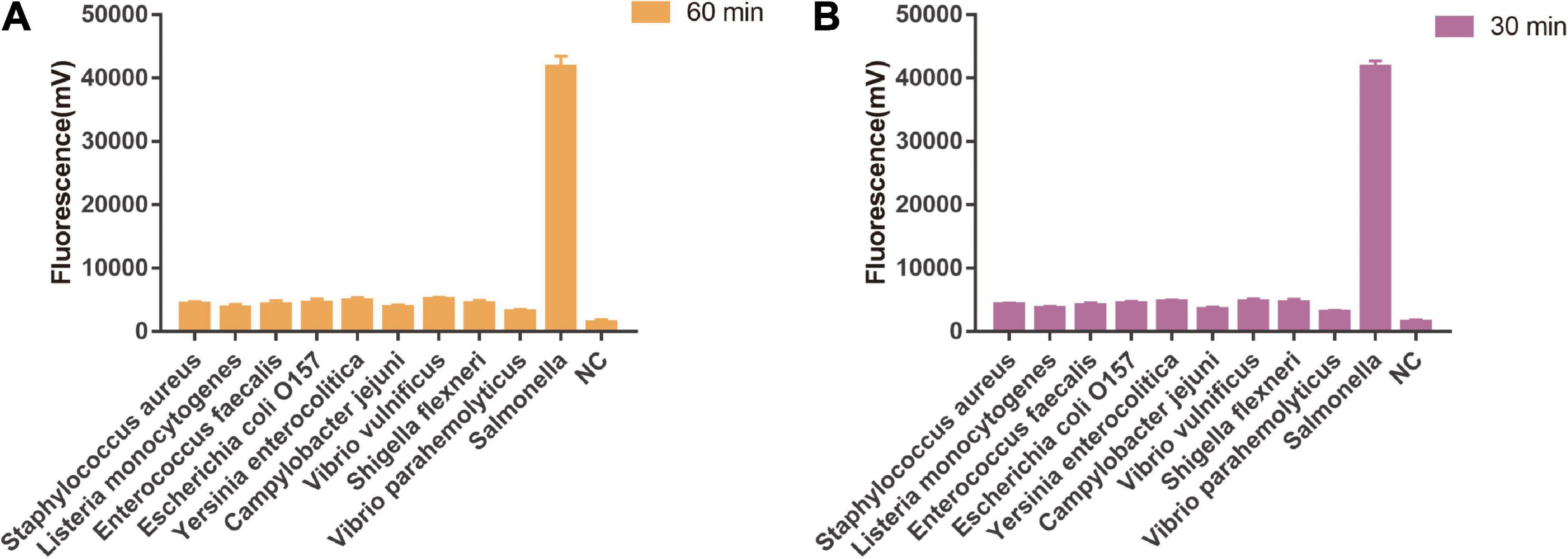
Figure 7. The specificity of one-tube and two-step RPA-Cas13a. (A) The specificity of one-tube RPA-Cas13a. 60 min was the time of one-tube RPA-Cas13a. NC, negative control (RNase-free ddH2O). (B) The specificity of two-step RPA-Cas13a. 30 min was the time of CRISPR-Cas13a reaction. NC, negative control (RNase-free ddH2O).
Consistency Comparison Using Real-Time Polymerase Chain Reaction
For evaluating the applicability of developed one-tube and two-step RPA-Cas13a for Salmonella in clinical stool samples, a total of 84 clinical stool samples from different hospitals were selected and tested by real-time PCR, two-step RPA-Cas13a, and one-tube RPA-Cas13a. Sixteen samples were detected positive for Salmonella by real-time PCR. Besides those 16 samples, three additional samples were judged as positive by two-step RPA-Cas13a in which one was also detected positive by one-tube RPA-Cas13a. However, the signal intensity was significantly lower than that of the other 16 positive samples but significantly higher than that of the negative control sample. At the same time, those three samples were also identified by Sanger sequencing technology and determined to be positive for Salmonella. The two-step RPA-Cas13a detection results are in good consistency with real-time PCR (κ = 0.892), the one-tube RPA-Cas13a detection results are also in good consistency with real-time PCR (κ = 0.962), and the statistical results are shown in Table 2.
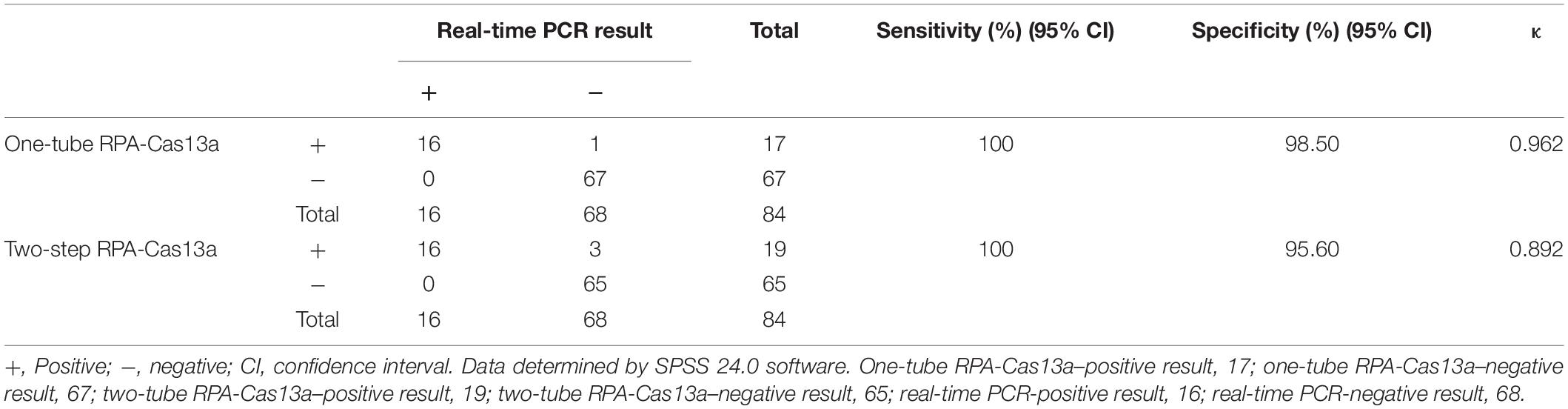
Table 2. Clinical samples consistency comparison using three detection methods.
Discussion
Currently, the isothermal amplification of nucleic acid technology has been used for the rapid pathogen detection of infectious diseases because of its high specificity, efficiency, and simplicity and plays an increasingly important role in promoting field detection and control of pathogen diseases. But the isothermal amplification of nucleic acid technology has its disadvantages, such as the LAMP (loop-mediated isothermal amplification) having an intricate primer design and non-specific reactions. Compared with other isothermal amplification technologies, RPA/RAA can be performed near ambient temperature (37–42°C) and has higher amplification efficiencies in recent years. The RPA process relies on three core enzymes, the recombinant enzymes uvsX and uvsY, SSB gp32 (Single-stranded DNA-binding protein), and Bsu DNA polymerase helping target regions of the template that can exponentially amplify within 20 min (Ahmed et al., 2015; Ma et al., 2019). Most importantly, the RPA/RAA has a fairly simple primer design. Commonly, the detection platforms most used for post-RPA detection are fluorescence RPA/RAA, gel electrophoresis, or LFD (lateral flow dipstick), but these methods require an expensive design for the probe, low sensitivity, or lack of specificity. It is necessary to establish a more sensitive and specific detection method that can be combined with RPA.
In recent years, studies have found that the collateral cutting activity of CRISPR-Cas13a is activated by a specific crRNA that targets a specific single-stranded RNA sequence, and collateral cutting activity can then cleave nearby non-specific RNAs (Knott et al., 2017; Zuo et al., 2017). Based on this principle, non-specific RNA reporting probes can add to the reaction system to achieve specific detection of target molecules (Abudayyeh et al., 2016; Gootenberg et al., 2017). The feature of collateral cutting activity has been used in the field of nucleic acid detection (Shen et al., 2020), among which the most representative is the SHERLOCK (Specific High-sensitivity Enzymatic Reporter UnLOCKing) by combining RPA with CRISPR-Cas13a (Myhrvold et al., 2018). This method provides a new direction for developing nucleic acid detection technology for pathogenic microbes, with the advantages of simplicity, rapidity, sensitivity, and specificity. The current detection based on SHERLOCK is mainly a two-step method. The one-tube method has been reported, but its sensitivity is insufficient.
The T7 promoter sequence is introduced into the RPA amplification primer, and the RPA amplification products are transcribed into a large amount of RNA by T7 RNA polymerase, which improves the sensitivity of detecting Salmonella targets. The results showed that two-step RPA-Cas13a is more sensitive than real-time PCR. Its detection limit for Salmonella can reach 100 copies/μL, One-tube RPA-Cas13a could achieve the same detection sensitivity as real-time PCR with 102 copies/μL. In addition, the crRNA design of CRISPR-Cas13a is relatively simple, requiring only 24 bases. The target-specific crRNA would further increase the detection specificity. The specificity results showed that there is no cross-reaction between the RPA-Cas13a of Salmonella and the other nine diarrheal pathogenic bacteria. Compared with the fluorescence probes used in RPA/RAA assay, the CRISPR non-specific detection probe is pretty easy to design in the present study and cost saving. In addition, both RPA and CRISPR detection can be conducted on a simple constant temperature equipment, which greatly reduces the requirements for equipment and personnel. The one-tube RPA-Cas13a detection process can detect Salmonella within 20 min, while two-step RPA-Cas13a costs 45 min, which was shorter compared with LAMP and real-time PCR method. The influence of clinical samples on RPA-Cas13a detection performance was verified: the Salmonella detection rate of the two-step or one-tube RPA-Cas13a detection method was highly consistent as that of real-time PCR and could even detect clinical stool samples at low infectious doses, whereas three samples with low doses cannot be detected by real-time PCR. Of the three low infectious dose samples, one can detect by one-step assay, and three can detect by two-step assay. Although the two-step RPA-Cas13a assay is more sensitive, it is also more easily contaminated. Some intervention measures, such as using oily non-interfering substances to seal the liquid surface, the use of specific reagent tubes, and so on, can be used to reduce or avoid the contamination, which would help this assay to be used in routine clinical trials. We recommend one-tube RPA-Cas13a assay as the rapid POCT method for Salmonella in rural hospitals with resource-poor settings, whereas two-step RPA-Cas13a for low abundance detection.
Conclusion
In this study, a novel molecular diagnostic method based on CRISPR-Cas13a combined with RPA was successfully demonstrated for rapid, sensitive, and specific detection of Salmonella and provides a new approach for the rapid screening of clinical samples in hospitals.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by the Jiangsu Provincial Center for Disease Control and Prevention. The ethics committee waived the requirement of written informed consent for participation.
Author Contributions
BA: methodology establishment, data sorting, and analysis, visualization, writing—original draft. HZ: methodology establishment, data sorting, writing—review and editing, and software. XS: methodology establishment, data sorting, and analysis. YuG and TW: methodology establishment. YiG: funding acquisition and writing—review and editing. FZ: project administration and supervision. LC: conceptualization, funding acquisition, project administration, validation, and writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
The study was supported in part by the Key Research and Development Project of Jiangsu Province (BE2019761), the Natural Science Foundation of Jiangsu Province (BK20211373), the Jiangsu Provincial Key Medical Discipline of Epidemiology (ZDXKA2016008), and “333”Projects of Jiangsu Province (BRA2017552).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abudayyeh, O. O., Gootenberg, J. S., Konermann, S., Joung, J., Slaymaker, I. M., Cox, D., et al. (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353:aaf5573. doi: 10.1126/science.aaf5573
Ahmed, A., Pranav, P., Oumar, F., Sasikanya, T., Doris, H., Ponpan, M., et al. (2015). Recombinase polymerase amplification assay for rapid diagnostics of dengue infection. PLoS One 10:e0129682. doi: 10.1371/journal.pone.0129682
Arslan, Z., Hermanns, V., Wurm, R., Wagner, R., and Pul, U. (2014). Detection and characterization of spacer integration intermediates in type I-E CRISPR–Cas system. Nucleic Acids Res. 42, 7884–7893. doi: 10.1093/nar/gku510
Bhaya, D., Davison, M., and Barrangou, R. (2011). CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 45, 273–297. doi: 10.1146/annurev-genet-110410-132430
CDC (2020). Centers for Disease Control and Prevention Salmonella. Available online at: https://www.cdc.gov/Salmonella/index.html [Accessed Nov 3, 2020].
East-Seletsky, A., O’Connell, M. R., Burstein, D., Knott, G. J., and Doudna, J. A. (2017). RNA targeting by functionally orthogonal type VI-A CRISPR-Cas enzymes. Mol. Cell 66, 373–383.e3.
East-Seletsky, A., O’Connell, M. R., Knight, S. C., Burstein, D., Cate, J., Tjian, R., et al. (2016). Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273. doi: 10.1038/nature19802
European Food Safety Authority, & European Centre for Disease Prevention and Control (2020). The European Union Summary Report on Antimicrobial Resistance in zoonotic and indicator bacteria from humans, animals and food in 2017/2018. EFSA J. 18:e06007. doi: 10.2903/j.efsa.2020.6007
Ferrari, R. G., Rosario, D., Neto, A. C., Borges, S., and Junior, C. (2019). Worldwide epidemiology of salmonella serovars in animal-based foods: a meta-analysis. Appl. Environ. Microbiol. 85:e591. doi: 10.1128/AEM.00591-19
Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. doi: 10.1126/science.aam9321
Hein, I., Flekna, G., Krassnig, M., and Wagner, M. (2006). Real-time PCR for the detection of Salmonella spp. in food: an alternative approach to a conventional PCR system suggested by the FOOD-PCR project. J. Microbiol. Methods 66, 538–547. doi: 10.1016/j.mimet.2006.02.008
Hugas, M., and Beloeil, P. (2014). Controlling Salmonella along the food chain in the European Union – progress over the last ten years. Euro Surveill. 19:20804. doi: 10.2807/1560-7917.es2014.19.19.20804
Jackson, R. N., Wiedenheft, B., Oost, J., and Westra, E. R. (2014). Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 12, 479–492. doi: 10.1038/nrmicro3279
Kirchner, M., and Schneider, S. (2015). CRISPR-Cas: from the bacterial adaptive immune system to a versatile tool for genome engineering. Angew. Chem. Int. Edn. 54, 13508–13514. doi: 10.1002/anie.201504741
Knott, G. J., East-Seletsky, A., Cofsky, J. C., Holton, J. M., Charles, E., O’Connell, M. R., et al. (2017). Guide-bound structures of an RNA-targeting A-cleaving CRISPR-Cas13a enzyme. Nat. Struct. Mol. Biol. 24, 825–833. doi: 10.1038/nsmb.3466
Ma, L., Zeng, F., Huang, B., Zhu, Y., and Guo, P. (2019). Point-of-care diagnostic assay for rapid detection of porcine deltacoronavirus using the recombinase polymerase amplification method. Transbound. Emerg. Dis. 66, 1324–1331. doi: 10.1111/tbed.13155
Myhrvold, C., Freije, C. A., Gootenberg, J. S., Abudayyeh, O. O., Metsky, H. C., Durbin, A. F., et al. (2018). Field-deployable viral diagnostics using CRISPR-Cas13. Science 360, 444–448. doi: 10.1126/science.aas8836
Shen, J., Zhou, X., Shan, Y., Yue, H., Huang, R., Hu, J., et al. (2020). Sensitive detection of a bacterial pathogen using allosteric probe-initiated catalysis and CRISPR-Cas13a amplification reaction. Nat. Commun. 11:267. doi: 10.1038/s41467-019-14135-9
Su, X., Ge, Y., Zhang, Q., Lin, Z., Ehsan, M., Xiao, X., et al. (2020). Rapid detection of Staphylococcus aureus by CRISPR-Cas13a combined with recombinase aided amplification(RAA). J. Pathog. Biol. 159, 11–16. doi: 10.13350/j.cjpb.200302
World Health Organization [WHO] (2016). WHO Estimates of the Global Burden of Foodborne Diseases: Foodborne Disease Burden Epidemiology Reference Group 2007-2015. Available online at: https://apps.who.int/bitstream/10665/199350/1/9789241565165_eng.pdf [Accessed March 1, 2016].
Yue, M., Song, H., and Bai, L. (2020). Call for special issue papers: food safety in china: current practices and future needs. Foodborne Pathog. Dis. 17:529. doi: 10.1089/fpd.2020.29013.cfp5
Zhang, X., Guo, L., Ma, R., Cong, L., Wu, Z., Wei, Y., et al. (2017). Rapid detection of Salmonella with recombinase aided amplification. J. Microbiol. Methods 139, 202–204. doi: 10.1016/j.mimet.2017.06.011
Zheng, L., Cai, G., Qi, W., Wang, S., and Lin, J. (2020). Optical biosensor for rapid detection of salmonella typhimurium based on porous gold@platinum nanocatalysts and a 3D fluidic chip. ACS Sens. 5, 65–72. doi: 10.1021/acssensors.9b01472
Zhou, J., Yin, L., Dong, Y., Peng, L., Liu, G., Man, S., et al. (2020). CRISPR-Cas13a based bacterial detection platform: sensing pathogen Staphylococcus aureus in food samples. Anal. Chim. Acta 1127, 225–233. doi: 10.1016/j.aca.2020.06.041
Keywords: Salmonella, foodborne disease, RPA, CRISPR-Cas13a, molecular detection
Citation: An B, Zhang H, Su X, Guo Y, Wu T, Ge Y, Zhu F and Cui L (2021) Rapid and Sensitive Detection of Salmonella spp. Using CRISPR-Cas13a Combined With Recombinase Polymerase Amplification. Front. Microbiol. 12:732426. doi: 10.3389/fmicb.2021.732426
Received: 29 June 2021; Accepted: 14 September 2021;
Published: 18 October 2021.
Edited by:
Xuejun Ma, Chinese Center for Disease Control and Prevention, ChinaReviewed by:
Jesús Navas, University of Cantabria, SpainZhiyang Li, Nanjing Drum Tower Hospital, China
Copyright © 2021 An, Zhang, Su, Guo, Wu, Ge, Zhu and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lunbiao Cui, bGJjdWlAanNjZGMuY24=
†These authors have contributed equally to this work