 Luo-Yan Zhang
Luo-Yan Zhang Hong Yu1
Hong Yu1 Jin Xu
Jin Xu- 1Yunnan Academy of Biodiversity, Southwest Forestry University, Kunming, China
- 2Key Laboratory for Forest Resources Conservation and Utilization in the Southwest Mountains of China, Ministry of Education, Southwest Forestry University, Kunming, China
- 3School of Ecology and Environment, Yunnan University, Kunming, China
Mating may promote microbial diversity through sexual transmission, while mating-induced immune responses may decrease it. Therefore, the study of mating-induced microbiomes changes under different mating systems is informative to unravel its biological relevance and evolutionary significance. Here, we studied the microbiomes in a community context within the abdomen of Spodoptera frugiperda females using 16S rDNA sequences by setting virgin females, and females mated once, twice, or thrice with the same or different males. Alpha and beta diversities revealed that mating significantly affected the composition of microbiomes in S. frugiperda females, wherein virgin females have the highest diversity, followed by one-time mated females and females mated with multiple males, while females mated repeatedly with the same male showed the lowest diversity. The low diversity in females mated repeatedly with the same male may be due to lower sexual transmission as only mated with one mate and higher immune response from repeated matings. Functional prediction by FAPROTAX and literature searching found 17 possible pathogens and 12 beneficial microbiomes. Multiple mating turned over the abundance of pathogens and beneficial microbes, for example, Enterococcus and Lactobacillus spp. (beneficial) showed higher abundance in virgin females while Morganella and Serratia spp. (pathogens) showed higher abundance in females mated with multiple males. These results suggest that mating causes a decline in the diversity of symbiotic microbiomes and promiscuity incurs a higher pathogen abundance in S. frugiperda females, which may be the result of sexual transmission of bacterial strains and immune responses targeting members of the microbiomes. To our knowledge, we demonstrate microbiomes changes in female insects under virgin and different mating regimes for the first time.
Introduction
Animals live and evolve in a world dominated by microbes, from which they host a diversity of microbiomes on and in their bodies. It is now widely accepted that microbiomes can be an integral part of the host phenotype, which may influence the health, physiology, development, behavior, and reproduction of the host, and potentially influence the host genome and be a part of the host genome (Schoenmakers et al., 2019; Bellinvia et al., 2020a,b; Rowe et al., 2021). A wide range of studies so far have been performed to reveal the role of microbiomes on host skin, oral, and gut from perspectives of ecology, adaption, and evolution (Dillon and Dillon, 2004; Voirol et al., 2018; Rowe et al., 2021). However, reproductive microbiomes or reproduction-related microbiomes are relatively neglected and less known (Bellinvia et al., 2020a; Rowe et al., 2021). Reproductive microbiomes, including bacteria, fungi, viruses, protozoans, and unicellular algae, living in or on any structure, organ, fluid, or tissue of a host, typically make contact with the gametes or reproductive tract or organs of another individual through mating and spawning (Rowe et al., 2021).
Studies in mammals, particularly human, have demonstrated that a large number of sexually transmitted microbes have negative effects on the health and survival of hosts (Schoenmakers et al., 2019; Rowe et al., 2021). Fitness tests indicated that these microbes often can have significant effects (but not negative always) on the reproductive performance and success of males and females (Schoenmakers et al., 2019; Rowe et al., 2021). Evidence, in male mammals, increasingly recognized that some of these microbes may decrease sperm quality and fertility (Fraczek et al., 2013; Schoenmakers et al., 2019; Rowe et al., 2021) and, in female mammals, demonstrated that some microbes are associated with female health and reproduction, including sexually transmitted infections, preterm birth, infertility, and low fecundity (Lockhart et al., 1996; Ravel et al., 2011; Chen et al., 2017; Schoenmakers et al., 2019; Marconi et al., 2020). Studies in human also found that the typically dominated vaginal microbe, Lactobacillus spp., can be “healthy” vaginal microbiomes, which are oxygen-tolerant anaerobes that exhibit antimicrobial activity against a range of vaginal pathogens, probably by physically barring against pathogen adhesion, production of lactic acid, and stimulating host defense responses (Ravel et al., 2011; Chen et al., 2017; Tachedjian et al., 2017; Younes et al., 2018). Studies in this field are relatively limited in insects. A study in the bedbug showed that exposuring males to polymicrobial mixture (such as Acinetobacter, Alcaligenes, Bacillus, and Staphylococcus) significantly increased sperm mortality (up to 40%) (Otti et al., 2013). In some insects including some lepidopterans, Wolbachia infection may lead to increased survival and reproduction (Chen et al., 2012; Moriyama et al., 2015; Voirol et al., 2018), while some Enterococcus species, such as E. faecalis, may have a negative impact on the fecundity of fruit flies (Akami et al., 2019; Noman et al., 2021). Moreover, studies also found that reproductive-related microbiomes play a diversity of roles in the reproduction process of animals, including insects, which is related to sexual selection, sperm competition, genetic compatibility, sexual conflict, and speciation (Schoenmakers et al., 2019; Rowe et al., 2021). However, most studies examined bacteria effects with only one or a few types of bacteria (Rowe et al., 2021). One possible reason is that these microbes are large in quantity and species, and most of them are hard to cultivate or uncultivable (Bellinvia et al., 2020a). While the study of individual microbes is fundamental and informative, it is important to understand the microbiomes in a community context (Schoenmakers et al., 2019; Rowe et al., 2021).
Recent progress on high-throughput sequencing and bioinformatics has revolutionized our understanding of symbiotic microbiomes. In the field of reproductive microbiomes, a few studies have demonstrated the bacterial communities of the reproductive organs in insects using 16S rDNA sequencing and subsequent bioinformatic analysis (Wang et al., 2014; Otti et al., 2017; Bellinvia et al., 2020a,b). In the citrus fruit fly, Bactrocera minax, sequencing revealed that female ovary has the highest diversity in microbiomes, followed by male testis, and the bacterial diversity of reproductive organs is higher than that of the gut (Wang et al., 2014). Recent studies in the common bedbug, Cimex lectularius, showed that genital microbiomes varied between populations and the sexes and mating changes the genital microbiomes, suggesting that bacteria are sexually transmitted and these bacteria might play an important role in shaping the evolution of reproductive traits (Bellinvia et al., 2020a,b). Future studies based on these new findings and technologies are likely to provide deeper insights in this field by investigating the transmission dynamics between males and females.
Mating can cause major changes in female physiology and behavior. Studies have shown that mating has a positive effect on female reproductive activities, which may negatively affect female maintenance (such as immunity responses) and survival due to trade-offs on resource utilization by females, or male suppression of female immunity to promote sperm storage and egg fertilization in females (reviewed in Schwenke et al., 2016; Wigby et al., 2019). However, studies have also shown that mating may upregulate the female's immune response due to the transfer of foreign materials and mating caused infections (Delbare et al., 2017; Okada et al., 2017; Oku et al., 2019; Ahmed-Braimah et al., 2021). These different discoveries may stem from different mating systems. Females of polygamous species should have higher postmating immunity if sexually transmitted infection is the major factor driving female postmating costs (Oku et al., 2019). Therefore, it is imperative and informative to study reproductive microbiomes as a whole in a community context in different mating systems (such as polygamy or polyandry) and under different mating conditions (such as repeated mating or multiple mating). Male mating with more than one female is known as polygamy, while female mating with more than one male is known as polyandry. Multiple mating (mating with more than one mate) and repeated mating (mating repeatedly with the same mate) are common in both sexes in insects, birds, and mammals (Andersson, 1994).
As mentioned above, studies have studied the microbiomes in the reproductive organs and guts in mated and virgin insects (Wang et al., 2014; Bellinvia et al., 2020a,b). However, in addition to reproductive organs and guts, symbiotic microbiomes also exist in other tissues or organs, either intracellularly or extracellularly, such as in the fat body and hemolymph (Szklarzewicz and Michalik, 2017). All these symbiotic microbiomes are under the surveillance of insect immune system and thus are likely to be affected by the mating-induced defense responses (Whitlow, 2004; Okada et al., 2017; Oku et al., 2019). Therefore, in this study, we studied the symbiotic microbiomes in the abdomen of Spodoptera frugiperda females by evaluating microbiomes as a whole in a community context. To test the above signified hypotheses, we used virgin females as controls, treat other females to mate once, twice, or thrice with the same (repeated mating) or different males (multiple mating), and then measured the exact changes on microbiomes in female abdomen using 16S rDNA sequencing. The evolutionary significance of reproductive microbiomes and interactions between host physiology and microbial communities were further explored based on these data.
The fall armyworm, Spodoptera frugiperda (Lepidoptera: Noctuidae), is native to tropical and subtropical regions from Argentina to the United States in the Western Hemisphere (Kumar et al., 2009). S. frugiperda is a long-distance migratory pest, but there was no report on its distribution outside the Americas before 2015. This pest was first discovered in Africa in 2016 (Abrahams et al., 2017) and then invaded India and some other areas in Asia in 2018 (FAO, 2019; CABI, 2020). Soon thereafter, this pest was discovered in Yunnan Province of China at the end of 2018 and then quickly spread northward to the vast areas of China (Guo et al., 2019; Xu et al., 2019; Zhang et al., 2019). This pest mainly damages corn and wheat in China, with the annual potential loss being $17,286–52,143 m for corn (Qin et al., 2020) and $15,571–90,143 m for wheat (Xu et al., 2020), respectively. Symbiotic microbiomes play imperative roles in the survival and reproduction of the host, in addition to the interactions of insect pests, host plants, and natural enemies (Ferrari and Vavre, 2011; Perilla-Henao and Casteel, 2016; Beck and Vannette, 2017). Therefore, modifying symbiotic microbiomes is a potential management strategy for the control of agricultural insect pests (Perilla-Henao and Casteel, 2016; Beck and Vannette, 2017).
Materials and Methods
Insect Rearing and Sampling
Spodoptera frugiperda larvae were collected in July 2019 on corn plants in the field near Zhanyi town in Yunnan Province, China. The insects were reared on the artificial diet (Li et al., 2006) under 28 ± 1°C and 60–80% relative humidity with 14:10-h light:dark photoperiod, in the IPM Laboratory of Southwest Forestry University (Kunming, China). This insect has 11 generations in the laboratory before this study that was performed in January 2021. All insects used for sequencing are reared using the same method under the same condition as above, which minimized the random effects from the insect populations and environment.
To ensure virginity, male and female pupae were sexed based on morphological characteristics (Dong et al., 2019) and then were caged separately. Adult eclosion was recorded daily, and the eclosion day was recorded as the 1st day since eclosion (Figure 1). Females were allowed to mate once, twice, and thrice with the same or different males within the 4th to 6th day since eclosion. A female mated more than once with the same mate was recognized as repeated mating, while a female mated more than once with different mates was recognized as multiple mating. Virgin females were used as controls. Three replicates were used for each treatment, and six females were used for each replicate. Before abdomen sampling, the whole body of females was rinsed twice with sterile water and was surface-sterilized in 75% ethanol for 90 s and then rinsed twice again using sterile water. The abdomens were then cut from the sterilized females using sterile scissors and were stored at – 80°C until use.
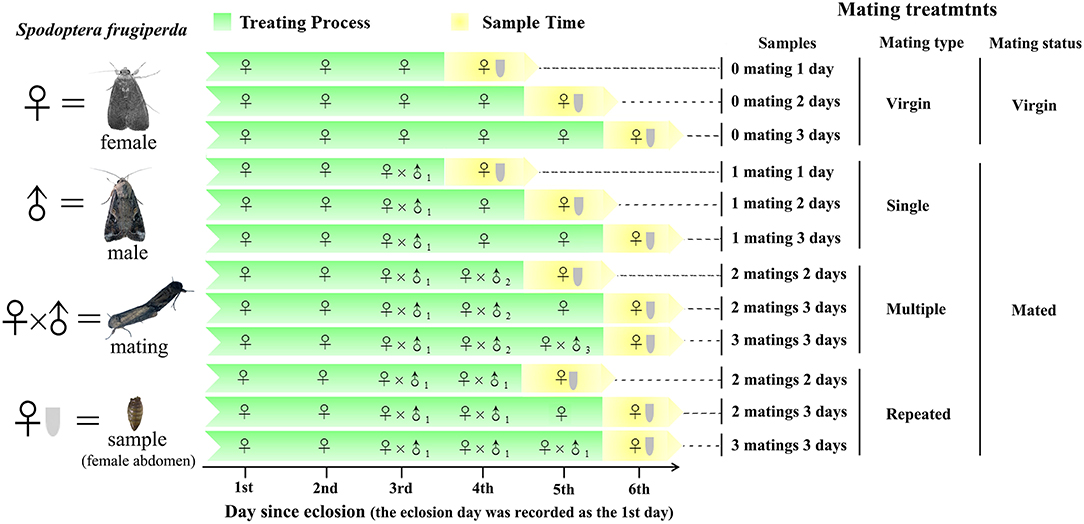
Figure 1. Experimental design and sampling methods. Single refer to one-time mated females, in which 1 mating 1 day (n = 3), 1 mating 2 days (n = 3), and 1 mating 3 days (n = 3) refer to one-time mated females sampled at 1, 2, and 3 d postmating (since the first mating), respectively. Multiple refer to females mated with multiple males (one male one mating), in which 2 matings 2 days (n = 3), 2 matings 3 days (n = 3), and 3 matings 3 days (n = 3) refer to twice and thrice mated females sampled at 2 and 3 d postmating (since the first mating), respectively. Repeated refer to females mated repeatedly with the same male, in which 2 matings 2 days (n = 3), 2 matings 3 days (n = 3), and 3 matings 3 days (n = 3) refer to twice and thrice mated females sampled at 2 and 3 d postmating (since the first mating), respectively. Virgin refer to unmated females, in which 0 mating 1 day (n = 3), 0 mating 2 days, and 0 mating 3 days (n = 3) refer to virgin females sampled at 1, 2, and 3 d postmating (relative to the above mating treatments), respectively.
DNA Extraction and 16S rDNA Sequencing
Total genome DNA was extracted from samples using CTAB method (Doyle and Doyle, 1987). The 16S rDNA regions were amplified via PCR with the V3–V4 primers (515F: 5′-GTGYCAGCMGCCGCGGTAA-3′; 806R:5′-GGACTACNNGGGTATCTAAT-3′) and with Phusion High-Fidelity PCR Master Mix (New England Biolabs, USA). The PCR products were analyzed using 2% agarose gel electrophoresis and were purified using a GeneJET Gel Extraction Kit (Thermo Scientific, USA). Sequencing libraries were generated using a TruSeq DNA PCR-Free Library Preparation Kit (Illumina, USA), according to the manufacturer's recommendations. Library quality was assessed on a Qubit Fluorometer (Thermo Scientific, USA) and an Agilent Bioanalyzer 2100 system (Agilent Technologies, USA). Finally, the library was sequenced on an Illumina NovaSeq PE250 platform (Novogene Bioinformatics Technology Co., Ltd., Beijing, China). Double-distilled water (ddH2O) was used as a negative control for DNA extraction and PCR amplification. Electrophoresis did not show any band or smudge in the negative control. No negative controls were used for library construction and sequencing, and no positive controls were used for DNA extraction or sequencing. The obtained raw data were deposited into the NCBI Sequence Read Archive (SRA) database (Accession No.: PRJNA742857).
Data Analysis
Clean reads were obtained by removing chimera sequences and low-quality reads from the raw reads. Uparse software (Uparse v7.0.1001, http://drive5.com/uparse/) (Edgar, 2013) was used for subsequent sequence analysis. The sequences with ≥ 97% similarity were assigned to the same OTUs. The representative sequence for each OTU was screened for further annotation using Silva 138 (https://www.arb-silva.de/) (Wang et al., 2007) based on the Mothur algorithm.
The QIIME (Version 1.7.0) (Caporaso et al., 2010) was used for the analysis of alpha diversity to reveal the complexity of species within samples. The differences of Shannon diversity indices between different mating treatments (Figure 1) were analyzed using an ANOVA followed by LSD tests with Bonferroni correction for multiple comparisons.
Beta diversity was applied to assess the differences of microbial community between treatments. The significance of differences between treatments was tested using non-parametric multivariate analysis of variance (NPMANOVA) based on Bray–Curtis metrics and then visualized accordingly using the principal coordinates analysis (PCoA) based on Bray–Curtis metrics. NPMANOVA was performed using R (Version 4.0.3) (Team, 2020) with vegan (Dixon, 2003) and phyloseq (Mcmurdie and Holmes, 2013) packages.
The linear discriminant analysis (LDA) of effect size (http://huttenhower.sph.harvard.edu/galaxy/) was used to determine OTUs that discriminate among the populations with an LDA score is more than 3.0. A cladogram was also constructed to show the LDA results.
FAPROTAX (Louca et al., 2016) was used to predict the potential functional annotation of taxa in different size fractions, which predicts functions of uncultured prokaryotes from the known functions of cultured bacterial genera.
Results
Sequencing and Quality Control
By 16S rDNA gene sequencing using the Illumina NovaSeq platform, ~70,000 clean reads were obtained from each of the 36 sequenced libraries, with the average length being 424–429 bp (Supplementary Table 1). The percentages of Q20 and Q30 of all samples' clean reads ranged from 96.60 to 97.53% and from 90.62 to 92.56%, respectively. These sequences were clustered into 2005 OTUs (Supplementary Table 2). The rarefaction analysis showed a saturating number of OTUs (Supplementary Figure 1), which indicates an adequate sequencing output for all samples.
Diversity Indices of Bacterial OTUs
Analysis of variance based on Shannon diversity metrics indicated that the OTU abundance was significantly different between virgin and mated females (ANOVA: F1,34 = 11.8, P = 0.002; Figure 2A), among females of different mating types (ANOVA: F4,32 = 8.808, P < 0.0001; Figure 2B) and among different samples (ANOVA: F11,24 = 5.061, P < 0.0001; Figure 2C). Within different mating types (Figure 2B), post-hoc pairwise comparison by LSD test (Supplementary Table 3) showed that virgin females (Virgin) have the highest abundance (P < 0.05), followed by one-time mated females (Single) and females mated with multiple males (Multiple) (P < 0.05), while females mated repeatedly with the same male (Repeated) showed the lowest abundance (P < 0.05). Within different samples (Figure 2C), post-hoc LSD test (Supplementary Table 4) also showed a similar decline trend on abundance, that is, samples of Virgin showed the highest abundance, followed by samples of Single, and then samples of Multiple, while samples from Repeated showed the lowest diversity (P < 0.05).
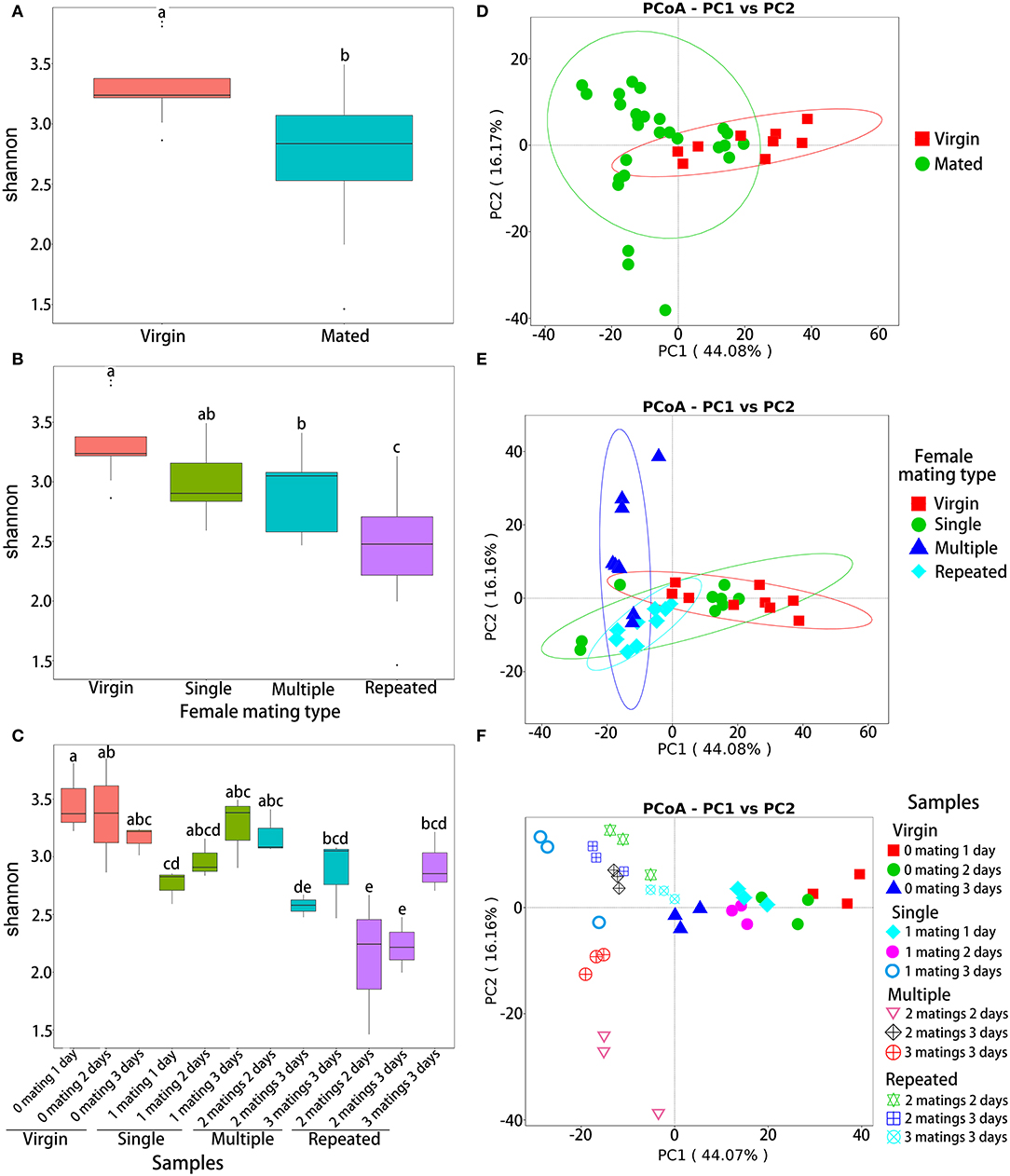
Figure 2. Diversity indices of bacterial OTUs in S. frugiperda females with different mating treatments. Shannon diversity indices of virgin and mated females (A), females with different mating types (B), and all samples (C). PCoA ordination based on Bray–Curtis distances of virgin and mated females (D), females with different mating types (E), and all samples (F). In each of the subgraphs of (a) to (c), bars with different letters are significantly different (P < 0.05).
Beta-diversity analysis based on Bray–Curtis distance (illustrated by PCoA) also showed significant variances in the composition of OTUs between virgin and mated females (NPMANOVA: F1,34 = 3.00, R2= 0.081, P = 0.019; Figure 2D), among females of different mating types (NPMANOVA: F3,32 = 6.506, R2= 0.379, P < 0.001; Figure 2E) and among different samples (NPMANOVA: F11,24 = 14.036, R2= 0.865, P < 0.001; Figure 2F).
Taxonomy Assignment
The obtained 2005 OTUs (Supplementary Table 2, Supplementary Figure 2) were classified into 33 phyla (Supplementary Table 5, Figure 3), 81 classes (Supplementary Table 6), 203 orders (Supplementary Table 7), 286 families (Supplementary Table 8), 447 genera (Supplementary Table 9, Figure 4A), and 194 species (Supplementary Table 10, Figure 4B). Within the obtained 2005 OTUs, 605 of them were shared (common OTUs) by females of different mating types (Supplementary Figure 2). The abundance pattern at the phylum level showed that Proteobacteria are the predominant bacterial phylum in S. frugiperda, with Multiple showing the highest percentage (90.94%), followed by Repeated with 88.99%, then Single with 86.38%, and Virgin showing the lowest percentage (82.14%) (Figure 3). The second and third dominant Phyla are Firmicutes and Bacteroidota, and both of them showed the highest abundance in virgin females (Figure 3).
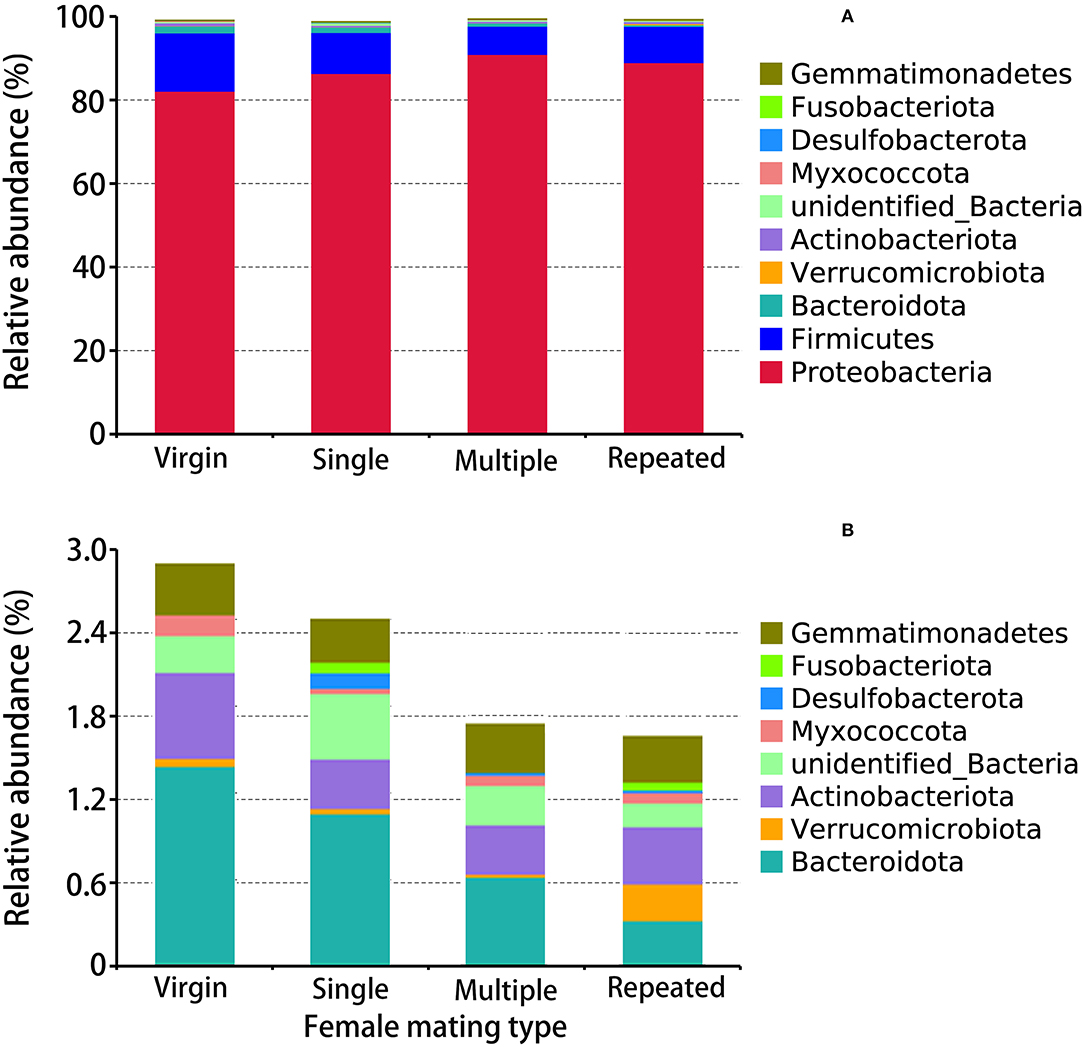
Figure 3. Taxonomy assignment of bacterial OTUs at the phylum level in S. frugiperda females of different mating types. (A) The abundance pattern of the top 10 phyla. (B) A clearer display of the relative abundance of the 3rd to the 10th phylum.
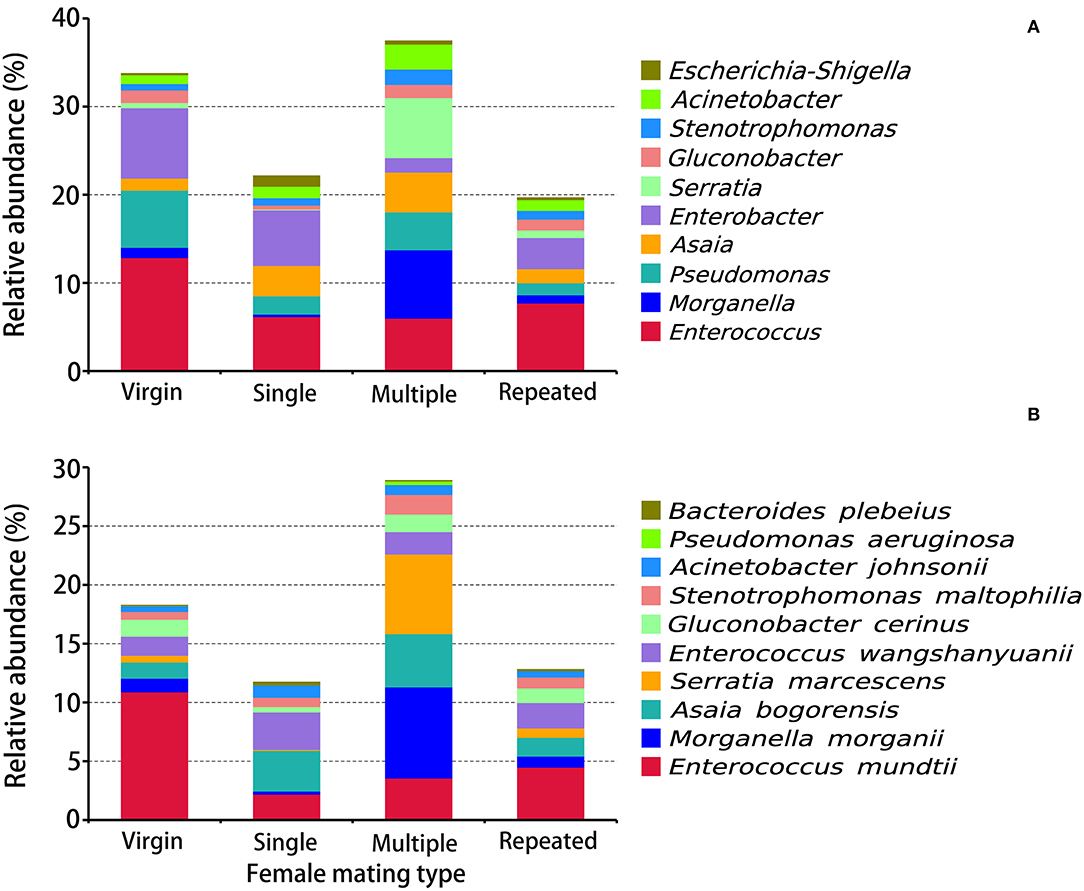
Figure 4. Taxonomy assignment of bacterial OTUs at the genus and species levels in S. frugiperda females of different mating types. (A) The abundance pattern at the genus level (top 10). (B) The abundance pattern at the species level (top 10).
OTUs on the genus level (Figure 4A) indicated that Enterococcus (12.88%), Enterobacter (7.99%), and Pseudomonas (6.48%) are the first three dominant genera in Virgin, while in Single are Enterobacter (6.30%), Enterococcus (6.20%), and Asaia (3.47%), in Multiple are Morganella (7.76%), Serratia (6.82%), and Enterococcus (6.01%), and in Repeated are Enterococcus (7.72%), Enterobacter (3.54%), and Asaia (1.59%), indicating remarkable difference on bacterial composition in females from different mating types. The prominent difference on the genus level within females from different mating types is that Morganella (7.76%) and Serratia (6.82%) are the most dominant genera in Multiple, while in the other mating types their abundance is much lower (0.09–1.17%) (Figure 4A).
OTU clustering into the species level (Figure 4B) disclosed that Enterococcus mundtii showed a higher abundance in Virgin (10.90%) than in the other mating types (2.19–4.50%), whereby Morganella morganii and Serratia marcescens showed higher abundance in Multiple (7.76 and 6.67%) than in the other mating types (0.26–1.16 and 0.09–0.80%).
Functional Prediction Using FAPROTAX
FAPROTAX was used to evaluate the potential functional differences between different mating types. The predominant function is chemoheterotrophy (responsible for an average 11.33–17.92% of the total annotated functions) in all mating types, followed by fermentation (6.08–12.17%), animal parasites or symbionts (4.66% to 7.76%), and aerobic chemoheterotrophy (3.66–5.94%), and Virgin showed relative higher abundant in all of these functions (Figure 5A, Supplementary Figure 3). The most variable function predicted by FAPROTAX is human pathogens (Figure 5A, Supplementary Figure 3), where Multiple showed the highest abundance on this function (6.94%), followed by Repeated (2.39%), and then Virgin (1.82%), and Single showed the lowest abundance on this function (1.53%).
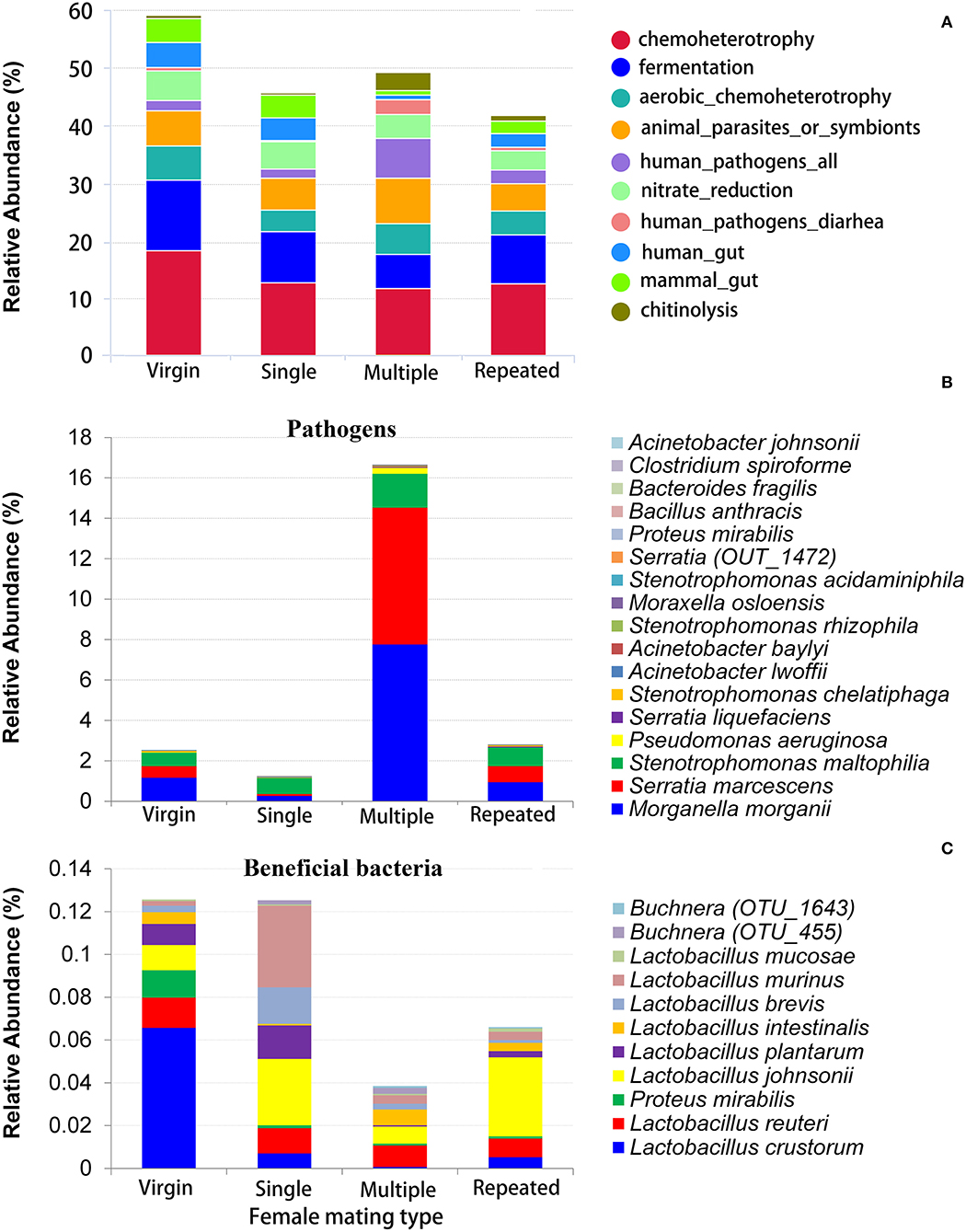
Figure 5. Functional prediction of bacteria in S. frugiperda females of different mating types. (A) The relative abundance of dominate bacterial functional groups (top 10) distribution predicted by FAPROTAX; (B) the relative abundance of possible pathogens; (C) the relative abundance of possible beneficial bacteria (bacteria that are beneficial for their hosts).
Pathogen and Beneficial Bacteria Profiles
Based on the above taxonomy assignment and functional prediction, the possible pathogens and beneficial bacteria were further evaluated based on evidence from published studies. A total of 17 animal pathogens were found, with three of them being insect pathogens and others being pathogens of human and other animal taxa (Table 1). Twelve beneficial bacterial species were found, including eight species from Lactobacillus, two species from Buchnera, one species from Proteus, and one species from Enterococcus (Table 1).
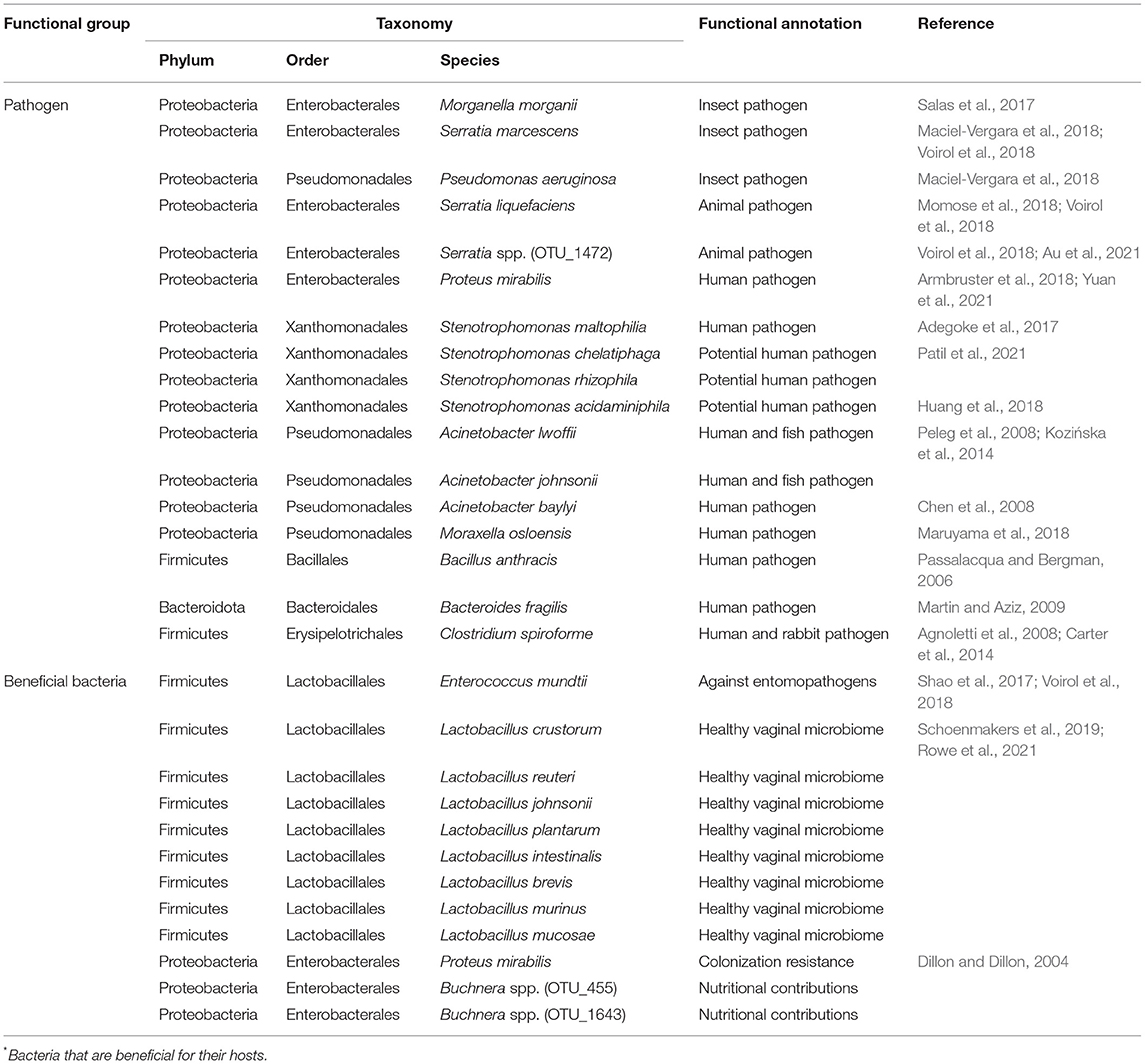
Table 1. Possible pathogens and beneficial bacteria* profiles in S. frugiperda.
Statistics indicated that the Multiple has the most abundance on pathogens (16.65%), which is more than five times than that of the other mating types (1.26–2.81%) (Figure 5B). On the contrary, Multiple showed the lowest abundance on beneficial bacteria (see Figure 4B for Enterococcus mundtii and Figure 5C for other beneficial bacteria).
Linear discriminant analysis (LDA) also showed remarkable differences on the number and taxon of microbial biomarkers between different mating types. More microbiomes were detected as important biomarkers in Virgin and Multiple than in Single and Repeated, with one beneficial bacteria (Enterococcus mundtii) showing in the Virgin while two pathogens (Morganella morganii and Serratia marcescens) in the Multiple (Figure 6). The OTUs represented in each population also were illustrated by phylogenetic levels from phylum to genus in the cladogram (Supplementary Figure 4).
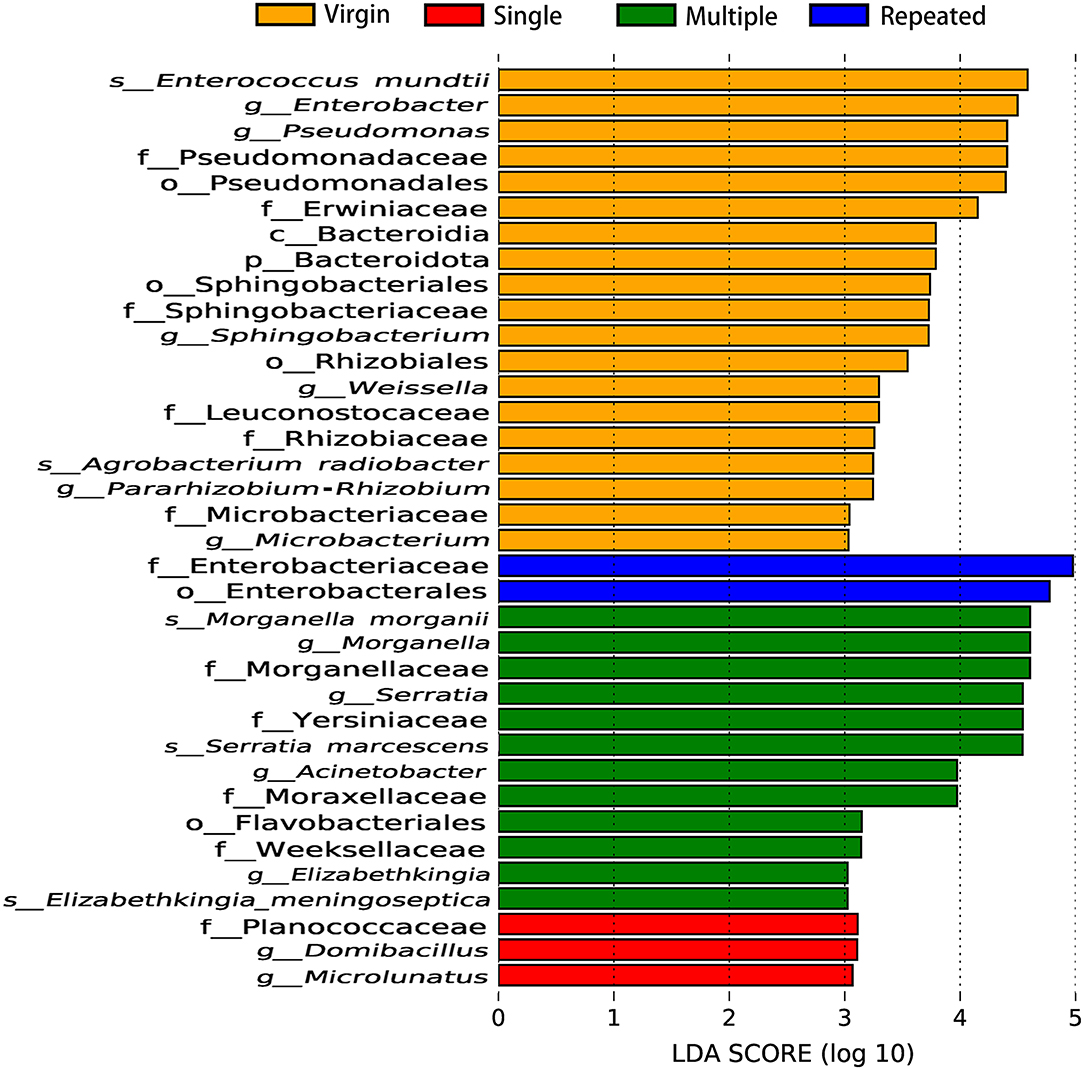
Figure 6. Important biomarkers of bacterial communities in S. frugiperda females of different mating types revealed by linear discriminant analysis (LDA).
Discussion
The microbiomes associated with animal reproduction are opportunistic and/or sexually transmittable, which may affect host immune defense and future colonization of other microorganisms. Only a few studies have studied the microbiomes in the reproductive organs and guts in insects (Wang et al., 2014; Otti et al., 2017; Bellinvia et al., 2020a,b) and tested the effect of mating on the composition of microbiomes (Bellinvia et al., 2020a,b). In this study, we considered the symbiotic microbiomes in the abdomen as a whole in a community context and tested the diversity and abundance of microbiomes under different mating conditions (Figure 1).
Beta-diversity analysis based on Bray–Curtis distance showed significant variances in the composition of OTUs in females with different mating statuses (Figures 2D–F), suggesting that mating significantly affected the composition of microbiomes in the abdomen of S. frugiperda females. The composition of genital microbiomes of females should be more affected by the sexual transmission of bacterial strains, via male genitalia and the ejaculate, and by bacteria transferred from the cuticle to the reproductive duct during and after mating (Lange et al., 2013; Reinhardt et al., 2015). Moreover, these invading bacteria may enter the hemolymph and other organs via reproductive duct and copulatory wounds (Lange et al., 2013; Reinhardt et al., 2015; Bellinvia et al., 2020a). Previous studies in a number of insect species have found pronounced differences in the microbiomes of reproductive organs between females and males (Wang et al., 2014; Otti et al., 2017; Bellinvia et al., 2020a,b). Bellinvia et al. (2020a,b) further demonstrated that bacteria are sexually transmitted in the common bedbug, C. lectularius. However, results in C. lectularius show that mating did not cause significant effect on alpha diversity of bacteria in the reproductive organs (Bellinvia et al., 2020b). In this study, alpha diversity indicated that the OTU diversity was significantly different in S. frugiperda females with different mating statuses, and mated females showed significant lower diversity than virgin females (Figure 2A) and in more details, virgin females have the highest diversity, followed by one-time mated females and females mated with multiple males, while females mated repeatedly showed the lowest diversity (Figures 2B,C). On the one hand, mating is likely to promote the diversity of microbes due to sexual transmission (Bellinvia et al., 2020a,b). On the other hand, mating may upregulate female immune responses (Oku et al., 2019), particularly in females with polygamous mating systems (Okada et al., 2017; Oku et al., 2019), which may affect the composition and diversity of microbes inside their bodies. In our study, females mated repeatedly with the same mates showed the lowest diversity (Figures 2B,C), which may be due to lower sexual transmission as only mated with one male but higher immune response from repeated matings.
In this study, the obtained 2005 OTUs were classified into 33 phyla, with Proteobacteria, Firmicutes, and Bacteroidota being the first three dominant phyla (Figure 3). Matings further increased the abundance of Proteobacteria (up to 90%) but decreased the abundance of Firmicutes and Bacteroidota (Figure 3). In the citrus fruit fly, Bactrocera minax, 16S rDNA sequencing revealed that Proteobacteria were dominant in gut and reproductive organs (Wang et al., 2014). Other studies have also found an abundance of Proteobacteria in various insect species, such as the sawfly Cephalcia chuxiongica (Yu et al., 2021), the desert locust Schistocerca gregaria (Dillon et al., 2010), the Lutzomyia sand fly (Sant'anna et al., 2012), and two ground beetles (Lundgren et al., 2007). Proteobacteria may play important roles in insects, such as help insects to fix nitrogen and preventing the establishment or proliferation of pathogenic bacteria (Dillon and Dillon, 2004; Dixon and Kahn, 2004; Behar et al., 2005). OTUs clustering into the genus (Figure 4A) also revealed significant differences in females from different mating types. For example, Morganella and Serratia are the most dominant genus in multiple mated females, while in other mating types their abundances are much lower. Further, Enterococcus mundtii showed a higher abundance in virgin females than in females from the other mating types, while Morganella morganii and Serratia marcescens showed higher abundances in multiple mated females than in females from the other mating types (Figure 4B). Species from Enterococcus may play a protective role against insect pathogens in the lepidopteran intestine (Voirol et al., 2018). For example, E. faecalis found in the gypsy moth can acidify its local environment to facilitate it colonize alkaline niches, which may help to host against pathogenic toxins that are activated in alkaline conditions, such as those from Bacillus thuringiensis (Broderick et al., 2004). E. mundtii is a highly abundant bacterium in the gut of S. littoralis, which is known to produce antimicrobial compounds that act against gram-positive pathogens (such as Listeria), but not against resident gut bacteria (Shao et al., 2017; Voirol et al., 2018). M. morganii is a lethal pathogen of Mexican fruit fly larvae (Salas et al., 2017).
Some common gut bacterial inhabitants can be beneficial or detrimental depending on the community composition of the gut. Serratia spp. is one of such bacteria, which is a genus known to be pathogenic in many animals including insects (Chadwick et al., 1990; Ishii et al., 2014; Momose et al., 2018; Voirol et al., 2018). For example, the SM1 strain of S. marcescens was highly toxic to the black-winged termite, Odontotermes formosanus, probably through the production of insecticidal protease and metalloproteinases (Fu et al., 2021). Serratia spp. are commonly reported in lepidopterans, while their roles are unclear in terms of detrimental or beneficial effects (Broderick et al., 2004). Future studies should evaluate whether such bacteria exert no negative effects or provide benefits on insect health depending on the composition of the entire insect microbial community and whether they switch from a symbiont to a systemic pathogen when the environmental condition is altered, such as postmating.
Functional prediction by FAPROTAX also disclosed a most variable on the function of human pathogens all in S. frugiperda females from different mating types, where females mated with multiple males showed higher abundance on this function than females from other mating types (Figure 5A, Supplementary Figure 3).
Due to the turnover on beneficial and detrimental microbiomes between virgin and multiple mated females found by taxonomy assignment and functional prediction by FAPROTAX, the possible pathogens and beneficial bacteria were further determined based on evidence from previous studies. The FAPROTAX and evidence-based double checking ensure the credibility of the functional evaluation of bacteria. Three insect pathogenic species and 14 pathogenic species from human and other animal taxa were verified by previous published studies (Table 1). Twelve beneficial bacterial species from four genera, Lactobacillus, Buchnera, Proteus, and Enterococcus, were also verified by published studies (Table 1). Buchnera has been implicated in various nutritional and non-nutritional functions in insects, such as provisioning of essential amino acids and nitrogen recycling (Douglas, 1998; Dillon and Dillon, 2004). Studies in human found that the Lactobacillus spp. are beneficial vaginal microbiomes, which may function in barring against pathogen adhesion, production of lactic acid against the colonization of pathogens, and stimulating host defense responses (Ravel et al., 2011; Chen et al., 2017; Tachedjian et al., 2017; Younes et al., 2018). The function of Lactobacillus spp. in insects is still unclear and thus warrants further studies.
Statistics showed that females mated with multiple males have the highest abundance on pathogens (more than five times than that of females from the other mating types) (Figure 5B) while they have the lowest abundance on beneficial bacteria (Figures 4B, 5C) in comparison with that of females from the other mating types. Moreover, LDA demonstrates that the Enterococcus mundtii (a beneficial bacterium) is one of the important biomarkers in virgin females, whereas Morganella morganii and Serratia marcescens (pathogens) are two of the important biomarkers in multiple mated females (Figure 6), further supporting the hypothesis that promiscuity turned over the abundance of beneficial and detrimental microbiomes in S. frugiperda females.
However, whether and how these possible pathogens and beneficial bacteria affect the health and reproduction of S. frugiperda still needs to be validated by further studies. Moreover, future studies by comparing microbiomes of reproductive organs and non-reproductive organs of both sexes under different mating conditions are expected to achieve deeper insight in this field.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
L-YZ and HYu collected the insects. L-YZ, D-YF, and JX designed the study. L-YZ, HYu, and D-YF dissected the insects and extracted DNA. L-YZ, SY, HYe, and JX analyzed the data. L-YZ, JX, and HYe wrote the paper. All authors read and approved the final manuscript.
Funding
This study was jointly supported by projects from Science and Technology Planning Project in Key Areas of Yunnan Province (202001BB050002), Basic Research Projects of Yunnan Province (202201AS070025), Joint Special Project of Yunnan Province for Agricultural Basic Research (2018FG001-002), and National Natural Science Foundation Program of P.R. China (31760635, 31560606).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.878856/full#supplementary-material
Supplementary Figure 1. Rarefaction curves represent observed OTUs in different samples.
Supplementary Figure 2. The Venn (A) and petals (B) diagrams of OTUs from different mating types.
Supplementary Figure 3. The heat map of bacterial functions (top 35) in the FAPROTAX database for S. frugiperda females. The samples are grouped according to the similarity of each other. Different colors indicate the relative abundance of groups in the individual samples, wherein red represents the function with higher abundance, and blue represents the function with lower abundance in the corresponding sample.
Supplementary Figure 4. Cladogram of the LDA results, where rings from the inner to outer indicate phylum, class, order, family, and genus, respectively.
References
Abrahams, P., Bateman, M., Beale, T., Clottey, V., Cock, M., Colmenarez, Y., et al. (2017). Fall armyworm: impacts and implications for Africa. Outlooks Pest. Manag. 28, 196–201. doi: 10.1564/v28_oct_02
Adegoke, A. A., Stenstrom, T. A., and Okoh, A. I. (2017). Stenotrophomonas maltophilia as an emerging ubiquitous pathogen: looking beyond contemporary antibiotic therapy. Front. Microbiol. 8, 2276. doi: 10.3389/fmicb.2017.02276
Agnoletti, F., Ferro, T., Guolo, A., Cocchi, M., Drigo, I., Bacchin, C., et al. (2008). “Clostridium Spiroforme Drug Susceptibility”, in: The 9th World Rabbit Congress. Verona: World Rabbit Science.
Ahmed-Braimah, Y. H., Wolfner, M. F., and Clark, A. G. (2021). Differences in postmating transcriptional responses between conspecific and heterospecific matings in Drosophila. Mol. Biol. Evol. 38, 986–999. doi: 10.1093/molbev/msaa264
Akami, M., Ren, X. M., Qi, X. W., Mansour, A., Gao, B. L., Cao, S., et al. (2019). Symbiotic bacteria motivate the foraging decision and promote fecundity and survival of Bactrocera dorsalis (Diptera: Tephritidae). BMC Microbiol. 19, 229. doi: 10.1186/s12866-019-1607-3
Armbruster, C. E., Mobley, H. L. T., and Pearson, M. M. (2018). Pathogenesis of Proteus mirabilis infection. EcoSal Plus. 8:ESP-0009-2017. doi: 10.1128/ecosalplus.ESP-0009-2017
Au, C. W. H., Yap, D. Y. H., Chan, J. F. W., Yip, T. P. S., and Chan, T. M. (2021). Exit site infection and peritonitis due to Serratia species in patients receiving peritoneal dialysis: epidemiology and clinical outcomes. Nephrology 26, 255–261. doi: 10.1111/nep.13813
Beck, J. J., and Vannette, R. L. (2017). Harnessing insect-microbe chemical communications to control insect pests of agricultural systems. J. Agric. Food Chem. 65, 23–28. doi: 10.1021/acs.jafc.6b04298
Behar, A., Yuval, B., and Jurkevitch, E. (2005). Enterobacteria-mediated nitrogen fixation in natural populations of the fruit fly Ceratitis capitata. Mol. Ecol. 14, 2637–2643. doi: 10.1111/j.1365-294X.2005.02615.x
Bellinvia, S., Johnston, P. R., Mbedi, S., and Otti, O. (2020a). Mating changes the genital microbiome in both sexes of the common bedbug Cimex lectularius across populations. Proc. Biol. Sci. 287, 20200302. doi: 10.1098/rspb.2020.0302
Bellinvia, S., Johnston, P. R., Reinhardt, K., and Otti, O. (2020b). Bacterial communities of the reproductive organs of virgin and mated common bedbugs, Cimex lectularius. Ecol. Entomol. 45, 142–154. doi: 10.1111/een.12784
Broderick, N. A., Raffa, K. F., Goodman, R. M., and Handelsman, J. (2004). Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl. Environ. Microbiol. 70, 293–300. doi: 10.1128/AEM.70.1.293-300.2004
CABI (2020). Invasive Species Compendium: Fall Armyworm Portal. Kenya: CABI Africa. Available online at: https://www.cabi.org/isc/fallarmyworm (accessed November 26, 2020).
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7, 335–336. doi: 10.1038/nmeth.f.303
Carter, G. P., Cheung, J. K., Larcombe, S., and Lyras, D. (2014). Regulation of toxin production in the pathogenic clostridia. Mol. Microbiol. 91, 221–231. doi: 10.1111/mmi.12469
Chadwick, J. S., Caldwell, S. S., and Chadwick, P. (1990). Adherence patterns and virulence for Galleria mellonella larvae of isolates of Serratia marcescens. J. Invertebr. Pathol. 55, 133–134. doi: 10.1016/0022-2011(90)90044-7
Chen, C., Song, X., Wei, W., Zhong, H., Dai, J., Lan, Z., et al. (2017). The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 8, 875. doi: 10.1038/s41467-017-00901-0
Chen, S.-J., Lu, F., Cheng, J.-A., Jiang, M.-X., and Way, M. O. (2012). Identification and biological role of the endosymbionts Wolbachia in rice water weevil (Coleoptera: Curculionidae). Environ. Entomol. 41, 469–477. doi: 10.1603/EN11195
Chen, T.-L., Siu, L.-K., Lee, Y.-T., Chen, C.-P., Huang, L.-Y., Wu, R. C.-C., et al. (2008). Acinetobacter baylyi as a pathogen for opportunistic infection. J. Clin. Microbiol. 46, 2938–2944. doi: 10.1128/JCM.00232-08
Delbare, S. Y. N., Chow, C. Y., Wolfner, M. F., and Clark, A. G. (2017). Roles of female and male genotype in post-mating responses in Drosophila melanogaster. J. Hered. 108, 740–753. doi: 10.1093/jhered/esx081
Dillon, R. J., and Dillon, V. M. (2004). The gut bacteria of insects: nonpathogenic interactions. Annu. Rev. Entomol. 71–92. doi: 10.1146/annurev.ento.49.061802.123416
Dillon, R. J., Webster, G., Weightman, A. J., and Charnley, A. K. (2010). Diversity of gut microbiota increases with aging and starvation in the desert locust. Antonie van Leeuwenhoek 97, 69–77. doi: 10.1007/s10482-009-9389-5
Dixon, P. (2003). VEGAN, a package of R functions for community ecology. J. Veg. Sci. 14, 927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x
Dixon, R., and Kahn, D. (2004). Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2, 621–631. doi: 10.1038/nrmicro954
Dong, Q.-J., Zhou, J.-C., Zhu, K.-H., Z.-,T, Z., and Dong, H. (2019). A simple method for identifiying sexuality of Spodoptera frugiperd (J. E. Smith) pupae and adults. Plant Prot. 45, 96–98. doi: 10.16688/j.zwbh.2019367
Douglas, A. E. (1998). Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. Annu. Rev. Entomol. 43, 17–37. doi: 10.1146/annurev.ento.43.1.17
Doyle, J., and Doyle, J. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissues. Phytochemical Bulletin 19, 11–15.
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
FAO (2019). FAO Statement on Fall Armyworm in Sri Lanka. Available online at: http://www.fao.org/srilanka/news/detail-events/en/c/1177796/ (accessed November 26, 2020).
Ferrari, J., and Vavre, F. (2011). Bacterial symbionts in insects or the story of communities affecting communities. Philos. Trans. R Soc. Lond. B Biol. Sci. 366, 1389–1400. doi: 10.1098/rstb.2010.0226
Fraczek, M., Szumala-Kakol, A., Dworacki, G., Sanocka, D., and Kurpisz, M. (2013). In vitro reconstruction of inflammatory reaction in human semen: effect on sperm DNA fragmentation. J. Reprod. Immunol. 100, 76–85. doi: 10.1016/j.jri.2013.09.005
Fu, R., Luo, J., Feng, K., Lu, X., and Tang, F. (2021). Termite-killing components in Serratia marcescens (SM1). J. For. Res. 32, 1739–1744. doi: 10.1007/s11676-020-01172-0
Guo, Z. B., Jiang, R. X., Tang, Y. L., Gu, R. C., Li, Q. Y., Xing, T., et al. (2019). Identification of new islolates of gut bacteria of Spodoptera frugiperda feeding on sorghum in Chongqing area. J. Southwest University 41, 9–16. doi: 10.13718/j.cnki.xdzk.2019.09.002
Huang, Y.-T., Chen, J.-M., Ho, B.-C., Wu, Z.-Y., Kuo, R. C., and Liu, P.-Y. (2018). Genome sequencing and comparative analysis of Stenotrophomonas acidaminiphila reveal evolutionary insights into sulfamethoxazole resistance. Front. Microbiol. 9, e1013. doi: 10.3389/fmicb.2018.01013
Ishii, K., Adachi, T., Hamamoto, H., and Sekimizu, K. (2014). Serratia marcescens suppresses host cellular immunity via the production of an adhesion-inhibitory factor against immunosurveillance cells. J. Biol. Chem. 289, 5876–5888. doi: 10.1074/jbc.M113.544536
Kozińska, A., Pazdzior, E., Pekala, A., and Niemczuk, W. (2014). Acinetobacter johnsonii and Acinetobacter lwoffii- the emerging fish pathogens. Bull. Vet. Inst. Pulawy 58, 193–199. doi: 10.2478/bvip-2014-0029
Kumar, M., Gupta, G. P., and Rajam, M. V. (2009). Silencing of acetylcholinesterase gene of Helicoverpa armigera by siRNA affects larval growth and its life cycle. J. Insect Physiol. 55, 273–278. doi: 10.1016/j.jinsphys.2008.12.005
Lange, R., Reinhardt, K., Michiels, N. K., and Anthes, N. (2013). Functions, diversity, and evolution of traumatic mating. Biol. Rev. 88, 585–601. doi: 10.1111/brv.12018
Li, W., Zou, W. J., and Wang, L. H. (2006). The bionomics and control of Prodenia litura in Kunming. Southwest China J Agric Sci. 19, 85–89. doi: 10.16213/j.cnki.scjas.2006.01.021
Lockhart, A. B., Thrall, P. H., and Antonovics, J. (1996). Sexually transmitted diseases in animals: ecological and evolutionary implications. Biol. Rev. 71, 415–471. doi: 10.1111/j.1469-185X.1996.tb01281.x
Louca, S., Parfrey, L. W., and Doebeli, M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. doi: 10.1126/science.aaf4507
Lundgren, J. G., Lehman, R. M., and Chee-Sanford, J. (2007). Bacterial communities within digestive tracts of ground beetles (Coleoptera: Carabidae). Ann. Entomol. Soc. America 100, 275–282. doi: 10.1603/0013-8746(2007)100275:BCWDTO2.0.CO;2
Maciel-Vergara, G., Jensen, A. B., and Eilenberg, J. (2018). Cannibalism as a possible entry route for opportunistic pathogenic bacteria to insect hosts, exemplified by Pseudomonas aeruginosa, a pathogen of the giant mealworm Zophobas morio. Insects 9, 88. doi: 10.3390/insects9030088
Marconi, C., El-Zein, M., Ravel, J., Ma, B., Lima, M. D., Carvalho, N. S., et al. (2020). Characterization of the vaginal microbiome in women of reproductive age from 5 regions in Brazil. Sex. Transm. Dis. 47, 562–569. doi: 10.1097/OLQ.0000000000001204
Martin, T., and Aziz, H. (2009). Bacteroides fragilis: a case study of bacteremia and septic arthritis. Clin. Lab. Sci. 22, 131–135. doi: 10.29074/ascls.22.3.131
Maruyama, Y., Shigemura, T., Aoyama, K., Nagano, N., and Nakazawa, Y. (2018). Bacteremia due to Moraxella osloensis: a case report and literature review. Braz. J. Infect. Dis. 22, 60–62. doi: 10.1016/j.bjid.2017.10.008
Mcmurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217. doi: 10.1371/journal.pone.0061217
Momose, T., Masutani, S., Oshima, A., Kawasaki, H., Tanaka, R., Iwamoto, Y., et al. (2018). First pediatric case of infective endocarditis caused by Serratia Liquefaciens. Int. Heart J. 59, 1485–1487. doi: 10.1536/ihj.17-595
Moriyama, M., Nikoh, N., Hosokawa, T., and Fukatsu, T. (2015). Riboflavin provisioning underlies Wolbachia's fitness contribution to its insect host. Mbio 6, e01732–e01715. doi: 10.1128/mBio.01732-15
Noman, M. S., Shi, G., Liu, L.-J., and Li, Z.-H. (2021). Diversity of bacteria in different life stages and their impact on the development and reproduction of Zeugodacus tau (Diptera: Tephritidae). Insect Sci. 28, 363–376. doi: 10.1111/1744-7917.12768
Okada, K., Suzaki, Y., Sasaki, R., and Katsuki, M. (2017). Fitness costs of polyandry to female cigarette beetle Lasioderma serricorne. Behav. Ecol. Sociobiol. 71, 86. doi: 10.1007/s00265-017-2316-2
Oku, K., Price, T. A. R., and Wedell, N. (2019). Does mating negatively affect female immune defences in insects? Anim. Bio. 69, 117–136. doi: 10.1163/15707563-20191082
Otti, O., Deines, P., Hammerschmidt, K., and Reinhardt, K. (2017). Regular wounding in a natural system: bacteria associated with reproductive organs of bedbugs and their quorum sensing abilities. Front. Immunol. 8, 1855–1855. doi: 10.3389/fimmu.2017.01855
Otti, O., Mctighe, A. P., and Reinhardt, K. (2013). In vitro antimicrobial sperm protection by an ejaculate-like substance. Funct. Ecol. 27, 219–226. doi: 10.1111/1365-2435.12025
Passalacqua, K. D., and Bergman, N. H. (2006). Bocillus anthracis: interactions with the host and establishment of inhalational anthrax. Future Microbiol. 1, 397–415. doi: 10.2217/17460913.1.4.397
Patil, P. P., Kumar, S., Kaur, A., Midha, S., Bansal, K., and Patil, P. B. (2021). Global transcriptome analysis of Stenotrophomonas maltophilia in response to growth at human body temperature. Microb. Genom. 7:000600. doi: 10.1099/mgen.0.000600
Peleg, A. Y., Seifert, H., and Paterson, D. L. (2008). Acinetobacter baumannii: Emergence of a successful pathogen. Clinical Microbiology Reviews 21, 538–582. doi: 10.1128/CMR.00058-07
Perilla-Henao, L. M., and Casteel, C. L. (2016). Vector-borne bacterial plant pathogens: interactions with Hemipteran insects and plants. Front. Plant Sci. 7, 1163. doi: 10.3389/fpls.2016.01163
Qin, Y., Yang, D., Kang, D., Zhao, Z., Zhao, Z., Yang, P., et al. (2020). Potential economic loss assessment of maize industry caused by fall armyword (Spodoptera frugiperda) in china. Plant Pro. 46, 69–73. doi: 10.16688/j.zwbh.2019528
Ravel, J., Gajer, P., Abdo, Z., Schneider, G. M., Koenig, S. S. K., Mcculle, S. L., et al. (2011). Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. U.S.A. 108, 4680–4687. doi: 10.1073/pnas.1002611107
Reinhardt, K., Anthes, N., and Lange, R. (2015). Copulatory wounding and traumatic insemination. Cold Spring Harb. Perspect. Biol. 7, a017582. doi: 10.1101/cshperspect.a017582
Rowe, M., Veerus, L., Trosvik, P., Buckling, A., and Pizzari, T. (2021). The reproductive microbiome: an emerging driver of sexual selection, sexual conflict, mating systems, and reproductive isolation. Trends Ecol. Evol. 36, 98–98. doi: 10.1016/j.tree.2020.10.015
Salas, B., Conway, H. E., Schuenzel, E. L., Hopperstad, K., Vitek, C., and Vacek, D. C. (2017). Morganella morganii (Enterobacteriales: Enterobacteriaceae) is a lethal pathogen of Mexican fruit fly (Diptera: Tephritidae) larvae. Fla. Entomol. 100, 743–751. doi: 10.1653/024.100.0422
Sant'anna, M. R. V., Darby, A. C., Brazil, R. P., Montoya-Lerma, J., Dillon, V. M., Bates, P. A., et al. (2012). Investigation of the bacterial communities associated with females of Lutzomyia sand fly species from south America. PLoS ONE 7, e42531. doi: 10.1371/journal.pone.0042531
Schoenmakers, S., Steegers-Theunissen, R., and Faas, M. (2019). The matter of the reproductive microbiome. Obstet. Med. 12, 107–115. doi: 10.1177/1753495X18775899
Schwenke, R. A., Lazzaro, B. P., and Wolfner, M. F. (2016). Reproduction-immunity trade-offs in insects. Annu. Rev. Entomol. 61, 239–256. doi: 10.1146/annurev-ento-010715-023924
Shao, Y., Chen, B., Sun, C., Ishida, K., Hertweck, C., and Boland, W. (2017). Symbiont-derived antimicrobials contribute to the control of the lepidopteran gut microbiota. Cell Chem. Biol. 24, 66–75. doi: 10.1016/j.chembiol.2016.11.015
Szklarzewicz, T., and Michalik, A. (2017). Transovarial transmission of symbionts in insects. Oocytes 63, 43–67. doi: 10.1007/978-3-319-60855-6_3
Tachedjian, G., Aldunate, M., Bradshaw, C. S., and Cone, R. A. (2017). The role of lactic acid production by probiotic Lactobacillus species in vaginal health. Res. Microbiol. 168, 782–792. doi: 10.1016/j.resmic.2017.04.001
Team, R. C. (2020). “R: A Language and Environment for Statistical Computing”. Vienna, Austria: R Foundation for Statistical Computing.
Voirol, L. R. P., Frago, E., Kaltenpoth, M., Hilker, M., and Fatouros, N. E. (2018). Bacterial symbionts in Lepidoptera: their diversity, transmission, and impact on the host. Front. Microbiol. 9, 556. doi: 10.3389/fmicb.2018.00556
Wang, A., Yao, Z., Zheng, W., and Zhang, H. (2014). Bacterial communities in the gut and reproductive organs of Bactrocera minax (Diptera: Tephritidae) based on 454 pyrosequencing. PLoS ONE 9, e106988. doi: 10.1371/journal.pone.0106988
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Whitlow, C. B. (2004). Bacterial sexually transmitted diseases. Clin. Colon. Rectal. Surg. 17, 209–214. doi: 10.1055/s-2004-836940
Wigby, S., Suarez, S. S., Lazzaro, B. P., Pizzari, T., and Wolfner, M. F. (2019). Sperm success and immunity. Curr. Top. Dev. Biol. 135, 287–313. doi: 10.1016/bs.ctdb.2019.04.002
Xu, L. N., Hu, B. J., Su, X. Y., Qi, R. D., Su, W. H., Qiu, K., et al. (2019). Genetic analysis of the fall armyworm Spodoptera frugiperda invaded in Anhui province. Plant Prot. 45, 47–53. doi: 10.16688/j.zwbh.2019348
Xu, Y., Li, Z., Chen, J., Li, Z., and Qin, Y. (2020). Assessment of potential economic loss of wheat industry caused by the fall armyworm Spodoptera frugiperda in China. J. Plant. Prot. 47, 740–746. doi: 10.13802/j.cnki.zwbhxb.2020.2020810
Younes, J. A., Lievens, E., Hummelen, R., Van Der Westen, R., Reid, G., and Petrova, M. I. (2018). Women and their microbes: the unexpected friendship. Trends Microbiol. 26, 16–32. doi: 10.1016/j.tim.2017.07.008
Yu, H., Du, C.-M., Shi, M.-R., Feng, L., Fu, D.-Y., Xu, J., et al. (2021). The diversity and function of intestinal microorganisms in four geographic Cephalcia chuxiongica (a pine defoliator) populations. J. Appl. Entomol. 145, 394–405. doi: 10.1111/jen.12858
Yuan, F., Huang, Z., Yang, T., Wang, G., Li, P., Yang, B., et al. (2021). Pathogenesis of Proteus mirabilis in catheter-associated urinary tract infections. Urol. Int. 105, 354–361. doi: 10.1159/000514097
Keywords: Spodoptera frugiperda, reproductive microbiomes, mating, sexually transmitted microbes, pathogen, beneficial bacteria
Citation: Zhang L-Y, Yu H, Fu D-Y, Xu J, Yang S and Ye H (2022) Mating Leads to a Decline in the Diversity of Symbiotic Microbiomes and Promiscuity Increased Pathogen Abundance in a Moth. Front. Microbiol. 13:878856. doi: 10.3389/fmicb.2022.878856
Received: 18 February 2022; Accepted: 14 April 2022;
Published: 12 May 2022.
Edited by:
Chih-Horng Kuo, Academia Sinica, TaiwanReviewed by:
Anke Kloock, New York University, United StatesShahid Karim, University of Southern Mississippi, United States
Oliver Otti, Technical University of Dresden, Germany
Copyright © 2022 Zhang, Yu, Fu, Xu, Yang and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Xu, eHVqaW4yNzk4QDEyNi5jb20=; Hui Ye, aHVpeWUxOTZAMTYzLmNvbQ==