 T. G. Sumithra1
T. G. Sumithra1 Krupesha S. R. Sharma1*
Krupesha S. R. Sharma1* Suja Gangadharan1
Suja Gangadharan1 Gayathri Suresh1
Gayathri Suresh1 Vishnu Prasad1P. V. Amala1P. Sayooj1Ambarish P. Gop2
Vishnu Prasad1P. V. Amala1P. Sayooj1Ambarish P. Gop2 M. K. Anil2Prasanna Kumar Patil3
M. K. Anil2Prasanna Kumar Patil3 Gopalakrishnan Achamveetil1
Gopalakrishnan Achamveetil1- 1Marine Biotechnology Division, Indian Council of Agricultural Research (ICAR)-Central Marine Fisheries Research Institute, Kochi, India
- 2Vizhinjam Regional Centre of ICAR-Central Marine Fisheries Research Institute, Thiruvananthapuram, India
- 3Aquatic Animal Health and Environment Division, ICAR-Central Institute of Brackishwater Aquaculture, Chennai, India
Information on unintended effects of therapeutic exposure of antibiotics on the fish gut microbiome is a vital prerequisite for ensuring fish and environmental health during sustainable aquaculture production strategies. The present study forms the first report on the impact of florfenicol (FFC), a recommended antibiotic for aquaculture, on the gut microbiome of snubnose pompano (Trachinotus blochii), a high-value marine aquaculture candidate. Both culture-dependent and independent techniques were applied to identify the possible dysbiosis and restoration dynamics, pointing out the probable risks to the host and environment health. The results revealed the critical transient dysbiotic events in the taxonomic and functional metagenomic profiles and significant reductions in the bacterial load and diversity measures. More importantly, there was a complete restoration of gut microbiome density, diversity, functional metagenomic profiles, and taxonomic composition (up to class level) within 10–15 days of antibiotic withdrawal, establishing the required period for applying proper management measures to ensure animal and environment health, following FFC treatment. The observed transient increase in the relative abundance of opportunistic pathogens suggested the need to apply proper stress management measures and probiotics during the period. Simultaneously, the results demonstrated the inhibitory potential of FFC against marine pathogens (vibrios) and ampicillin-resistant microbes. The study pointed out the possible microbial signatures of stress in fish and possible probiotic microbes (Serratia sp., Methanobrevibacter sp., Acinetobacter sp., and Bacillus sp.) that can be explored to design fish health improvisation strategies. Strikingly, the therapeutic exposure of FFC neither caused any irreversible increase in antibiotic resistance nor promoted the FFC resistant microbes in the gut. The significant transient increase in the numbers of kanamycin-resistant bacteria and abundance of two multidrug resistance encoding genes (K03327 and K03585) in the treated fish gut during the initial 10 days post-withdrawal suggested the need for implementing proper aquaculture effluent processing measures during the period, thus, helps to reduce the spillover of antibiotic-resistant microbes from the gut of the treated fish to the environment. In brief, the paper generates interesting and first-hand insights on the implications of FFC treatment in the gut microbiome of a marine aquaculture candidate targeting its safe and efficient application in unavoidable circumstances. Implementation of mitigation strategies against the identified risks during the initial 15 days of withdrawal period is warranted to ensure cleaner and sustainable aquaculture production from aquatic animal and ecosystem health perspectives.
Introduction
Aquaculture is the fastest-growing food-producing sector globally and makes a significant contribution to poverty alleviation, food security, and income generation across the globe (Walker and Winton, 2010). Since most of the development in the aquaculture sector has occurred during the last 50 years, the sustainability of aquaculture practices, both in terms of economics and environmental health, has evolved into a growing concern (Boyd et al., 2020). The increasing reliance on farmed fish for human nutrition, as well as the persistent and growing challenge of infectious diseases often leads to a heavy reliance on the use of antibiotics for prophylactic or therapeutic measures in the aquaculture industry (Schmidt et al., 2017). Excessive or indiscriminate antibiotics use forms a significant constraint in achieving sustainable aquaculture production. Antibiotic exposure can cause several dysbiotic events in the gut microbiota of the host, with a considerable influence on host immunity, development, nutrition, and health (Pennycook and Scanlan, 2021). Studies in terrestrial animals, including humans, have demonstrated the wide-ranging implications of antibiotic therapy on the ecology and evolution of the gut microbiota, with pronounced effects on host’s health and welfare (Pennycook and Scanlan, 2021). The current research interest on gut dysbiotic events focuses mainly on mammalian models, while studies on non-mammalian models such as the fish are scarce. Furthermore, the role of the fish microbiome in host’s health is less established to date (Schmidt et al., 2017). The effects of antibiotic administration on the fish gut microbiome and the possible outcomes are still under-study (Kokou et al., 2020). The corresponding information on major antimicrobials in tropical marine fish species is completely lacking. The information on the impacts of antibiotics on the gastrointestinal microbiota of aquaculture candidate fish species can be directly applied to exploring sustainable aquaculture production strategies (Navarrete et al., 2008).
The application of antibiotics may cause the emergence of antibiotic resistance in gut microbes of the host, raising concerns about consumer and environmental safety through the contamination by aquaculture wastes carrying antibiotic-resistant bacteria (Cabello et al., 2013). Studies have demonstrated that the dysbioses in the gut microbiome in response to antibiotic treatment led to the emergence of antibiotic resistance in the gut (Shoemaker et al., 2001; Francino, 2016). Due to the vast density of bacterial cells and species richness, the gut microbiota is likely to be prone to horizontal gene transfer leading to the spread of antimicrobial-resistant genes between bacterial taxa in addition to their spread to incoming pathogenic microorganisms (Whittle et al., 2002; Sommer et al., 2009). In brief, the research on antibiotic effects on the gut microbiome is critical from agricultural and ecological perspectives and needs to be soon addressed to achieve sustainable aquaculture practices (Kokou et al., 2020).
The snubnose pompano (Trachinotus blochii) is one of the promising candidates among the different high-value marine tropical finfishes with fast growth rates and high market demand, therefore, are recommended for sustainable marine aquaculture practices (FAO et al., 2021). Incidences of infectious diseases, especially vibriosis, hamper many successful farming practices of pompano (Liu et al., 2004; Yu et al., 2018), forcing the farmers to rely on the use of different antimicrobials to treat or prevent the diseases. There are only four recommended antimicrobial compounds for aquaculture purposes, viz. sulfamerazine, oxytetracycline, sulfadimethoxine-ormetoprim, and florfenicol (FFC) (U.S. Food and Drug Administration [USFDA], 2008). Of these, FFC is the preferred antimicrobial since it has never been used in human medicine (Committee for veterinary medicinal products, 2001). The clinical efficacy of FFC through oral administration has been ratified in different farmed marine fish species against fish pathogens (Feng et al., 2018; San Martín et al., 2019). Similarly, while limited information is available on the effects of FFC treatment on the fish gut microbiome (Abdelhamed et al., 2019), no corresponding information is available on the marine fish gut microbiome. Given the above facts, the present study was envisaged to ascertain the impact of the therapeutic exposure of FFC on the gut microbiome of T. blochii through a combination of culture-dependent and culture-independent approaches with the following objectives: (1) to evaluate the influence of FFC exposure in T. blochii on the gut microbiota through both culture-dependent and independent techniques; (2) to understand the restoration dynamics of the gut microbiome following FFC treatment by evaluating the changes in cultivable bacterial density, diversity measures of 16S rRNA amplicon-based metagenomics, and taxonomic microbial composition; (3) to outline the emergence of antimicrobial resistance in the gut microbes during FFC therapeutic interventions; and (4) to identify the prospective gut microbial biomarkers of FFC treatment in T. blochii.
Materials and Methods
Experimental Fish
Healthy snubnose pompano juveniles, with an average weight of 12 ± 0.62 g, were used in the present study. Fish (12 ± 0.62 g) were brought from the marine aquaculture facility of the Vizhinjam Regional Centre of ICAR-Central Marine Fisheries Research Institute (ICAR-CMFRI) and acclimatized for seven days in oval-shaped fibre reinforced plastic (FRP) tanks containing 700 L of de-chlorinated and continuously aerated water (temperature: 29.8 ± 0.54°C; pH: 7.5 ± 0.7; salinity: 18 ± 1.4). Fish were fed with floating pellet feed (Nutrila from Growel) at 5% biomass during acclimatization. The animals were allowed to acclimatize for seven days. Active feeding was observed after 2 days of stocking. Water quality parameters (ammonia, nitrite, and nitrate) were maintained to the optimal levels through 20% daily water exchange and siphoning out of the waste materials throughout the experimental period.
All the experiments involving live fish were done adhering to animal research reporting of in vivo experiments (ARRIVE) guidelines (du Sert et al., 2020), the guidelines of EU Directive 2010/63/EU for animal experiments (2019), and the U.K. Animals Scientific Procedures Act (1986). “The animal study was reviewed and approved by the ICAR-CMFRI, Kochi, India (CIBA/AINP-FH/2020-21).
Preparation of Medicated Feed
The FFC medicated feed was prepared by surface coating the drug (Amit et al., 2017) onto a commercial.8 mm pellet feed (Nutrila from Growel). The proximate composition of the feed on a dry matter basis was 52% protein, 12% fat, and 1.5% fiber. The required dose of FFC (10 mg/kg biomass/day) was achieved by mixing 53 mg of FFC powder (Tokyo Chemical Industry, Japan), with 500 μL of fish oil, and then, coating uniformly onto 100 g of commercial feed. The mixture was kept for 5 min until the FFC oil mixture was evenly distributed on the pellets. The prepared feed was then dried at 300°C for 1 h. The control/non-medicated feed was processed in the same manner with surface coating only with fish oil.
Experimental Design
For the experiments, the animals were randomly divided into two groups, viz. treatment and control groups. Each group was maintained in triplicate FRP tanks (25 fish per tank) containing 700 L of filtered water (salinity: 18%; temperature: 29°C) with continuous aeration. In the treatment group, fish were fed with FFC medicated feed at 10 mg/kg biomass for 10 days. After 10 days of medicated feeding, the fish in the treatment group were fed with non-medicated feed. Fish in the control group were fed with non-medicated feed throughout the experimental period. All the fish were fed at 2% of body mass per day, and it was confirmed that the fish had consumed all the feed. Fish were monitored daily for 30 days after the commencement of experiments. When fishes were removed from a tank for sampling, the quantity of feed administered to the tank was adjusted proportionally to maintain the therapeutic dose at 10 mg/kg of body weight (Soto et al., 2010).
Sampling
Four fish/time points/tanks were randomly sampled at 5-day intervals from the commencement of the experiment. The external surface of the fish was cleaned using 70% ethanol to avoid surface microbial contamination. After opening the ventral surface, the entire gut was aseptically removed using clamps to prevent the release of intestinal contents. Gut samples of two fish from each tank were pooled and used for culture-dependent microbiological analysis. Gut samples from the remaining two fish in each tank were pooled and used for metagenomics analysis.
Culture-Dependent Microbiological Analysis
The gut, along with the intestinal contents of each pool, was resuspended as 1 g/mL in sterile phosphate-buffered saline (PBS) and homogenized. Serial 10-fold dilutions of each homogenate were prepared and spread on Tryptic Soy Agar (TSA) and thiosulphate citrate bile salt sucrose agar (TCBS) plates supplemented with 1% sodium chloride (Himedia, India) in duplicates and incubated at 30°C for 48 h under aerobic conditions. The total viable count was expressed as the number of colony-forming units (CFU) per gram (Hovda et al., 2007). The viable counts of presumptive vibrios (mesophilic Vibrionaceae and other closely related vibrios) were enumerated after 48 h of incubation on TCBS agar (Bolinches et al., 1988). Further, each homogenate was added to six other selective culture plates containing either one of the six antibiotics, namely ampicillin, oxytetracycline, kanamycin, FFC, enrofloxacin, and meropenem (Guardabassi et al., 2002). The final concentration was 50 μg/mL for each antibiotic and 30 μg/mL for FFC (Kenzaka et al., 2006).
Genomic DNA Isolation
For isolating the total microbiome DNA, the Qiamp stool kit (Qiagen) was utilized with certain modifications for reducing host DNA contamination (Wanka et al., 2018; Bruggeling et al., 2021). Initially, the gut, along with the intestinal contents of each pool, was homogenized in PBS (pH 7.4). The homogenate was then centrifuged at 1,000 rpm for 10 min. The supernatant was taken as a source of loosely associated bacteria with the target tissues. The bacteria strongly associated with the tissues were separated using a detergent solution (0.9% saline with 1% (w/v) Triton X 100) and collected with a pipette (Bruggeling et al., 2021). These solutions (representing loosely and strongly associated bacteria of the target tissues) were mixed and centrifuged at 12,000 rpm for 10 min. The pellet representing loosely and strongly associated bacteria (Wanka et al., 2018; Bruggeling et al., 2021) was further processed for DNA isolation using DNeasy Blood and Tissue Kit (Qiagen) following the manufacturer’s protocol. The concentrations of DNA were measured using the Qubit Fluorimeter (V.3.0) and preserved at –20°C until needed.
Amplification and Next-Generation Sequencing
The hypervariable V3–V4 region of the prokaryotic 16SrRNA gene from the total bacterial DNA (10 ng) was amplified using the primers, viz. Pro341F (5′-CCTACGGGNBGCASCAG-3′) and Pro805R (5′-GACTACNVGGGTATCTAATCC-3′) (Takahashi et al., 2014). The amplified product was gel-purified to remove non-specific amplification, if there is any. Metagenomic library preparation was done using the NEBNext Ultra DNA library preparation kit (New England Biolabs) using equimolar quantities of PCR amplicon (5 ng). The library quantity and quality were estimated in Agilent 2200 TapeStation. The sequencing was then performed on an Illumina HiSeq 2500 platform (2 × 300 paired-end sequencings) (AgriGenome Labs Private Limited, Kochi, India). The high-quality samples were refined and used for metagenomics analysis.
Metagenomics Analysis
The raw reads generated were demultiplexed and evaluated for quality using the FastQC tool (version 0.11.8) with default parameters. The base quality (Phred Score; Q), adapter dimers, GC content, base composition, and ambiguous bases (apart from A, T, G, and C) were thoroughly scrutinized. The forward and reverse primer sequences were maintained to get all the possible 16S rRNA gene sequence information. Further downstream analysis was done using the Quantitative Insights into Microbial Ecology pipeline (QIIME2™ version 2021.4.0) (Bolyen et al., 2019). Demultiplexed pair-end reads were merged, filtered, and denoised using the Divisive Amplicon Denoising Algorithm 2 (DADA2) (Callahan et al., 2016). An alpha rarefaction curve was then generated to ensure that the relation between the read depth and new taxon detection approached an asymptote in all the samples. The Naive Bayesian classifier against the SILVA database version 138 was then applied to assign the taxonomic information on the obtained amplicon sequence variants (ASVs), in which operational taxonomic units (OTUs) were clustered by 99% homology. The ANCOM plugin was used to calculate the relative abundance of each taxonomic level within the samples. The diversity measures were estimated in QIIME2 using the core metrics pipeline. The PICRUSt2 tool was explored to predict metagenome functions, viz. KEGG orthologs and pathways (Douglas et al., 2020).
Statistical Analysis
The normality and homogeneity of variance of different data sets were initially checked using the Shapiro–Wilk test and the Levene test, respectively. One-way ANOVA followed by Tukey’s HSD test was used to compare the viable counts of bacteria in the various culture media between different days of antibiotic exposure with P values of <0.05 and <0.01 set to represent significant and highly significant differences, respectively. The OTU abundance of the taxa accounting for >0.01% was used to create the relative abundance plot. The α-diversity measures in terms of OTU richness, evenness, abundance-based coverage estimator (ACE), chao1, Shannon index, and Simpson index were then calculated using the Past software (version 3.5.2) (Hammer et al., 2001). Differences in α-diversity indices in microbial diversity on different days of antibiotic exposures were determined with an ANOVA/Krusswallis test based on the normality of the data. The similarity index of different days of antibiotic exposures was calculated using the Bray–Curtis distance method and compared through PERMANOVA analysis in PAST 3.5.2 software. The average Bray–Curtis similarity index of each group was analyzed through the hierarchical clustering via paired group UPGMA algorithm in the PAST software (Hammer et al., 2001) to graphically represent the species complexity between days. The relative abundance data of the gut microbes at each taxonomic level on different days was compared with the control fish using the independent T-test/the Mann–Whitney U test based on the normality of the data. These analyses were performed using SPSS (version 16), where P-value of <0.05 was set to represent the significant difference. Contributions of each taxonomy assigned bacterial OTU to the difference between gut bacterial communities of the treated fish group and the control group at different days of treatment were analyzed with the Similarity Percentage (SIMPER) analysis using the PAST software. The OTU abundance data accounting for >0.1% contribution to the dissimilarity in SIMPER analysis was used to generate the heatmap. The results of the PICRUSt2 analysis were also applied in the SIMPER analysis to determine the KEGG genes and pathways, which contributed most to the discrimination of samples in each day compared to the control group. The abundance data of KEGG genes and pathways contributing to >0.1% dissimilarity in SIMPER analysis on each day was used to generate the heat map and compared with the control fish using the independent T-test/the Mann–Whitney U test based on the normality of the data.
Results
Survival Rates
The fish in both the control and treatment groups showed 100% survival throughout the experimental period. Further, no clinical abnormalities were observed throughout the study period.
Enumeration of Total Cultivable Bacteria and Presumptive Vibrios in the Gut
The results of the enumeration of gut microbes during different days of FFC exposure are presented as log colony forming units (log10 CFU ± SE) per gram of gut tissue (Figure 1). The gut of the control fish showed 6.1 ± 0.05 and 5.38 ± 0.07 cultivable bacteria in ZMA and TCBS, respectively. The Tukey post hoc test revealed that there was no significant difference in the CFU value between different days of the experiment within the control fish. However, there was a significant difference (P < 0.05) in the CFU values of the FFC treated fish between different days. The least bacterial count in both the media was observed in FFC treated fish on zero-day post-withdrawal (11th day after the initiation of the treatment) (P < 0.001). Further, the total viable bacterial counts on the fifth-day post-initiation and fifth and 10th-day post-withdrawal of FFC treatment were significantly lower than the control fish (P < 0.05) (Figure 1A). The presumptive vibrio counts on the fifth-day post-initiation and fifth-day post-withdrawal of FFC treatment were significantly lower than the control fish (P < 0.05) (Figure 1B). In other words, the total viable count and presumptive vibrio counts were similar to the control group (P > 0.05) by 10 and 5 days, respectively, post-withdrawal of FFC treatment.
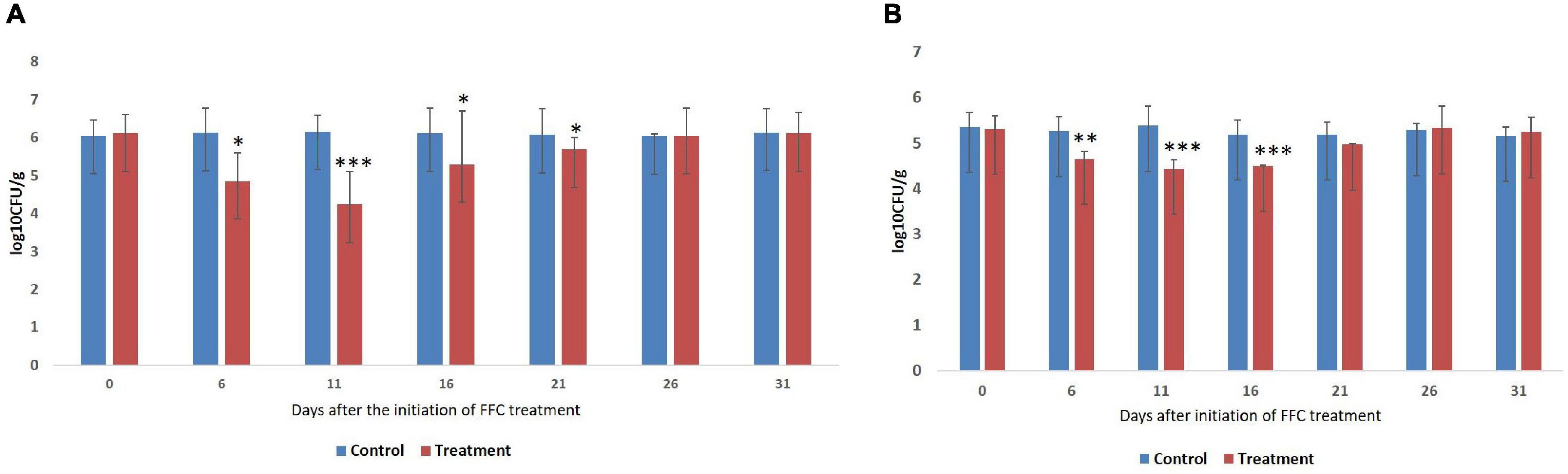
Figure 1. Enumeration of gut bacteria during different days of therapeutic exposure. (A) Enumeration of total viable counts of gut bacteria. (B) Enumeration of presumptive vibrio counts. Average log10 CFU per gram of gut tissue ± SE is shown in Y-axis. P-values less than 0.05, 0.01, and 0.001 are summarized with one, two, and three asterisks respectively, to represent the significant difference levels compared to the control animals. CFU, colony-forming units; FFC, florfenicol.
Enumeration of Antimicrobial-Resistant Gut Bacteria
The gut of the control fish revealed the presence of bacteria having resistance to ampicillin and kanamycin (Figure 2), with significantly higher numbers of ampicillin-resistant bacteria. During the FFC therapeutic course (fifth-day post-initiation), ampicillin-resistant microbes could not be detected in the treated group. Further, the numbers of ampicillin-resistant gut bacteria were significantly lower (P < 0.05) than the control fish after 20 days post-withdrawal of the treatment. There was a significant (P < 0.05) but transient increase in the numbers of kanamycin-resistant bacteria in the FFC treated fish than in the control group at five days post-initiation, and 5- and 10-days post-withdrawal of FFC treatment. More importantly, there was no growth in the FFC supplemented media in both control and the FFC treated fish even at zero-day post-withdrawal of treatment.
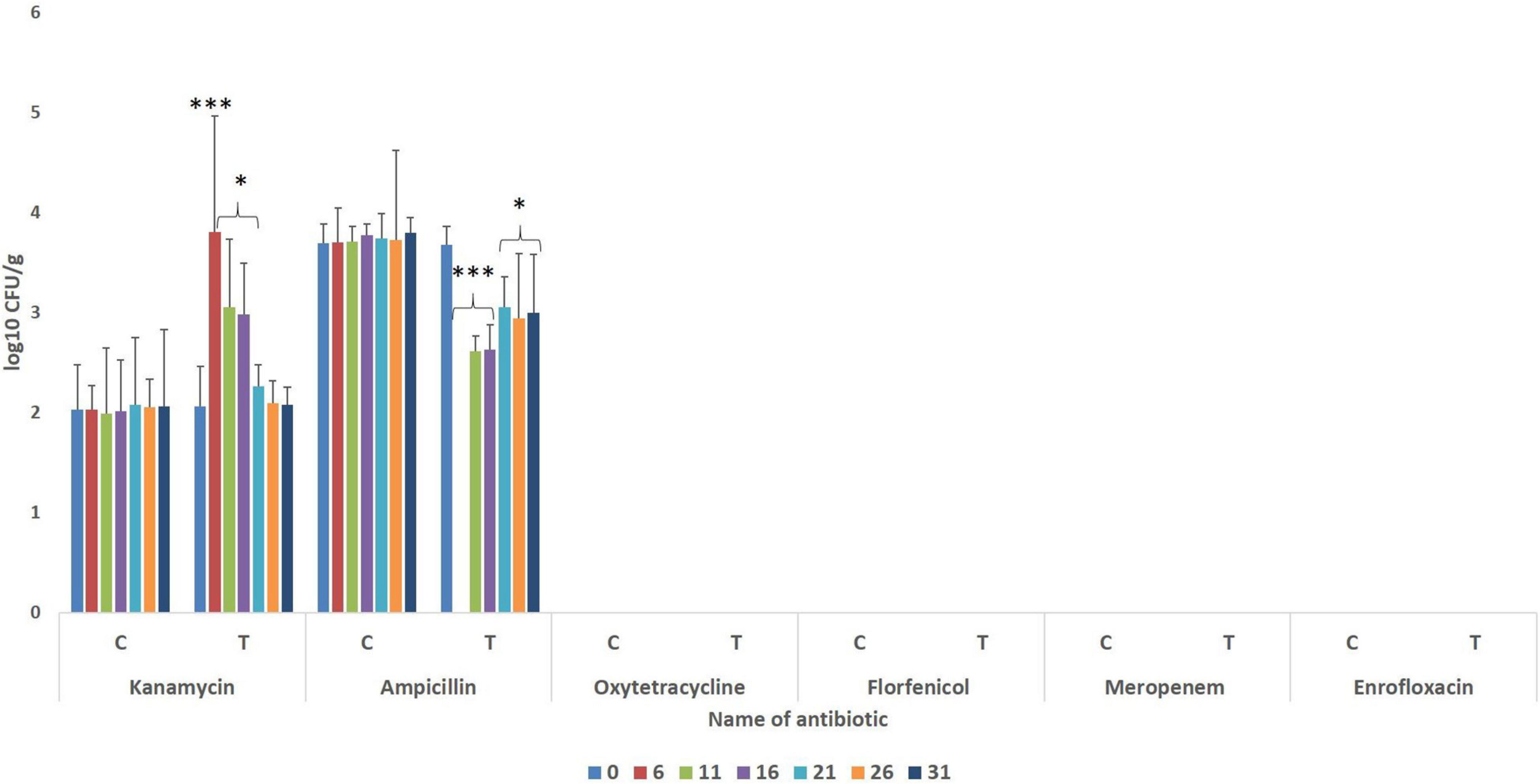
Figure 2. Enumeration of antibiotic-resistant bacteria during different days of FFC exposure. Average log10 CFU per gram of gut tissue ± SE is shown in Y-axis. P-values less than 0.05, 0.01, and 0.001 are summarized with one, two, and three asterisks, respectively, to represent the significant difference levels compared to the control animals. Different days after initiation of FFC treatment are shown in different color bars. CFU, colony-forming units; FFC, florfenicol; C, control fish; T, FFC treated fish.
Metagenomic Library Preparation and Sequencing
The parameters recorded during metagenomic library preparation and sequencing are shown in Supplementary Table 1. All the samples were qualified for library preparation and sequencing. A total of 9, 31, 971 reads of 16S rRNA sequence were achieved following the quality filtering through DADA2. The numbers of merged reads varied from 1,209 to 10,105. The α-rarefaction curve displayed that a sequencing depth of ∼1,095 was sufficient to capture the maximum diversity, so that the features at this depth were used for the analysis. The metagenomic data sets of the present study are deposited as Sequence Read Archive (SRA) data (Accession numbers: SRR16990455–SRR16990471) under the Bio project Accession No. PRJNA780352 in the National Center for Biotechnology Information database.
Microbial Diversity in the Gut of Snubnose Pompano Juveniles
Taxonomic assignment of OTUs showed two identified distinct domains [Bacteria (91.66%), Archaea, (0.48%), and unassigned (7.94%)], 7 identified distinct phyla, 9 identified distinct classes, 12 identified distinct orders, 14 identified distinct families, and 20 identified distinct genera with ≥0.01% relative abundance. Proteobacteria (72.73%) occupied the maximum relative abundance among the identified phyla, followed by Firmicutes (13.64%), Euryarchaeota (9.83%), Deinococcus-Thermus (1.83%), Acidobacteria (1.54%), Thaumarchaeota (0.29%), and Dependentiae (0.14%) (Figure 3A). There were 11 distinct genera (≥0.01% relative abundance) within Proteobacteria in the order of Serratia sp. > Unassigned Enterobacteriaceae > Ralstonia sp. > Enterobacter sp. > Unassigned Burkholderiaceae > Acinetobacter sp. > Pseudomonas sp. > Unassigned γ-Proteobacteria > Curvibacter sp. > Pandoraea sp. > Erwinia sp. Even though the phylum Firmicutes was the second most dominant phylum after Proteobacteria, only two dominant genera could be identified, which were Bacillus sp > Brevibacillus sp. There were three identified genera in the phylum Euryarchaeota in the order of Methanobrevibacter sp. > Uncultured marine group II Euryarchaeote > Halomarine sp. Only one dominant genus, viz. Meiothermus sp., Pyrinomanas sp., and Candidatus Nitrosopelagicus sp., could be identified from the phylum Deinococcus-Thermus, Acidobacteria, and Thaumarchaeota, respectively.
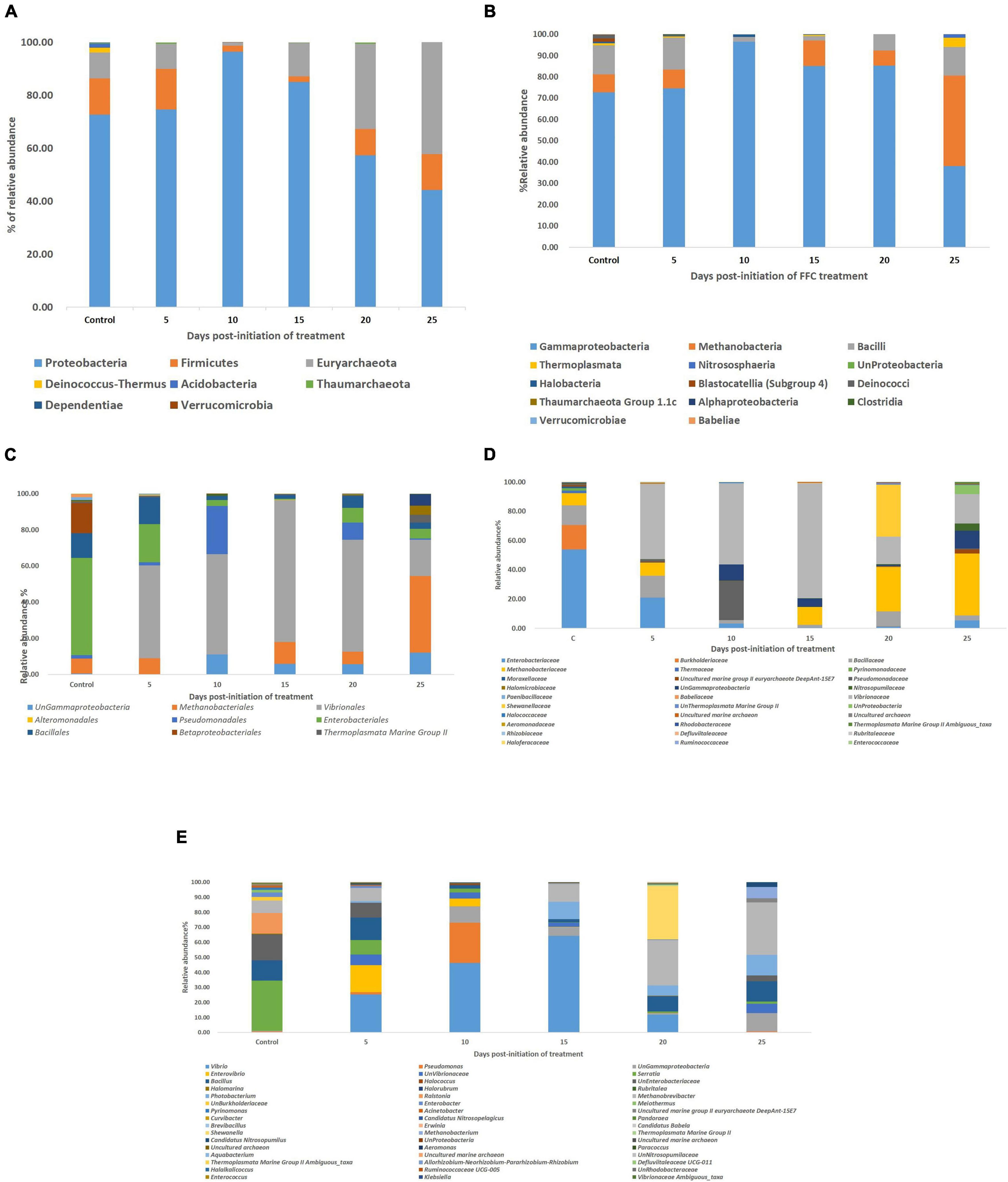
Figure 3. Impact of FFC therapeutic exposure on the gut microbiome taxonomy. (A) At phylum level; (B) at class level; (C) at order level; (D) at family level; (E) at genus level. FFC, florfenicol.
Dynamics of Microbial Diversity Measures Following Therapeutic Exposure of Florfenicol
Diversity analysis (α-diversity) of gut microbial communities revealed significant changes in all the diversity measures, except evenness and Simpson index, between different days (P < 0.05). All the gut microbial diversity measures were statistically similar in the FFC treated fish on the 5th day from the initiation of treatment and on the 15th day post-withdrawal to that of the control fish. The diversity measures of the control group, and FFC treated fish at 5 days from the initiation of treatment and on the 15th day post-withdrawal were found to be significantly higher than other groups (Figure 4). In other words, the fish belonging to zero, 5-, and 10-days post-withdrawal of FFC treatment had significantly lower diversity measures of the gut microbiome than the control group. Furthermore, within the treatment group, there was no significant difference (P > 0.05) in the gut microbial diversity measures on zero, 5-, and 10-days post-withdrawal of FFC treatment. The pattern in the dynamics of different diversity measures following the FFC treatment is shown in Figure 4. All the diversity measures were significantly decreased following FFC therapy and became similar to the control group at 15 days post-withdrawal. In other words, the differences in the gut microbial diversity measures became statistically indistinguishable in comparison to the control group on the 15th day post-withdrawal. PERMANOVA analysis based on the Bray–Curtis similarity index also demonstrated that the FFC treatment had a significant impact on the gut microbial communities of T. blochii (P = 0.05, F value = 1.312, Permutation number = 9,999, Total sum of squares = 6.04). The hierarchical clustering based on the average Bray-Curtis similarity index of each day showed that gut microbial communities of the fish on the 15th day post-withdrawal were clustered along with the control (Cluster II), while the others formed a distinct independent cluster (Cluster I) away from the first group (Figure 5).
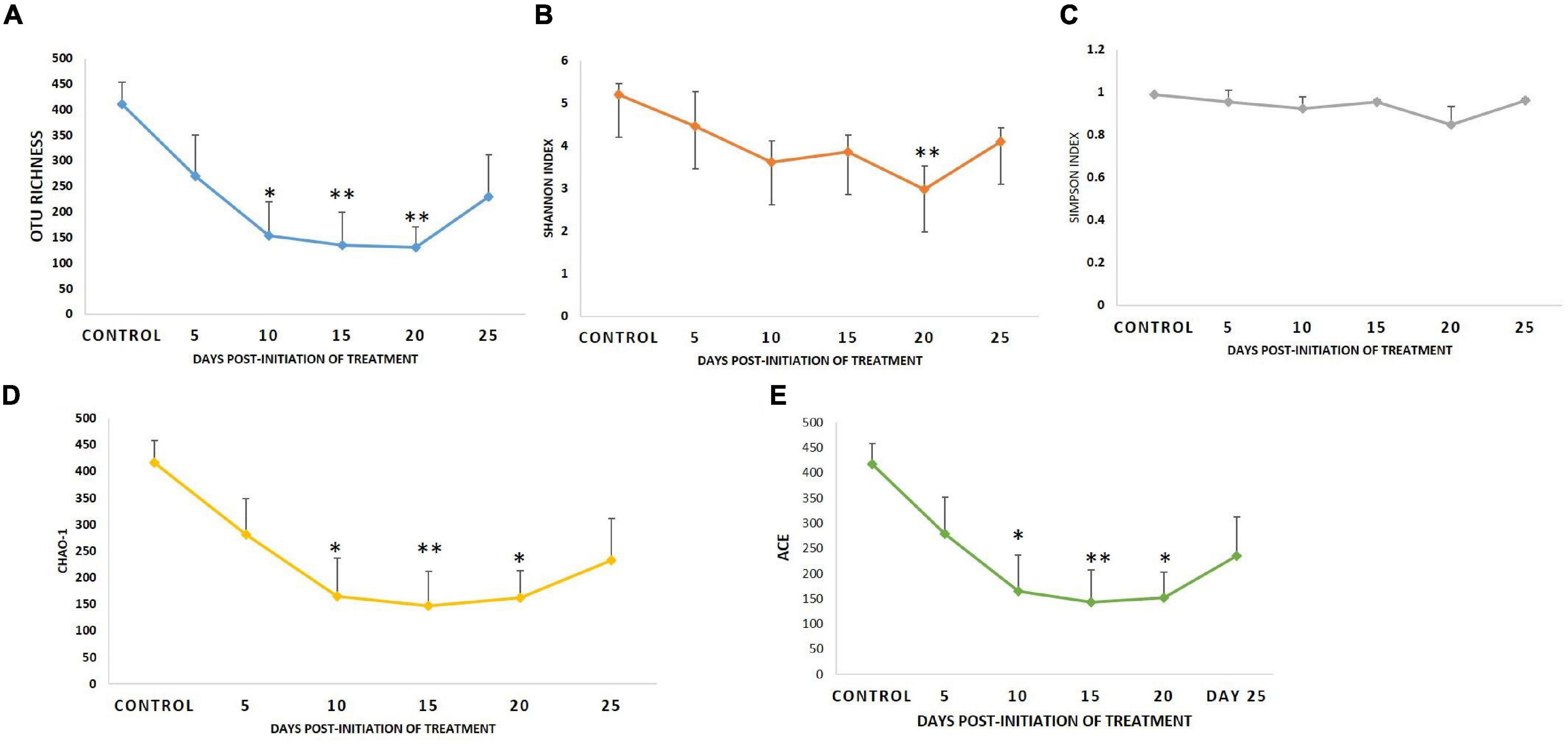
Figure 4. Dynamics of microbial diversity measures following therapeutic exposure of FFC. (A) Dynamics of OTU richness. (B) Dynamics of Shannon index. (C) Dynamics of Simpson index. (D) Dynamics of Chao-1. (E) Dynamics of abundance-based coverage estimator. P-values less than 0.05, 0.01, and 0.001 are summarized with one, two, and three asterisks, respectively, to represent the significant difference levels from the control animals.
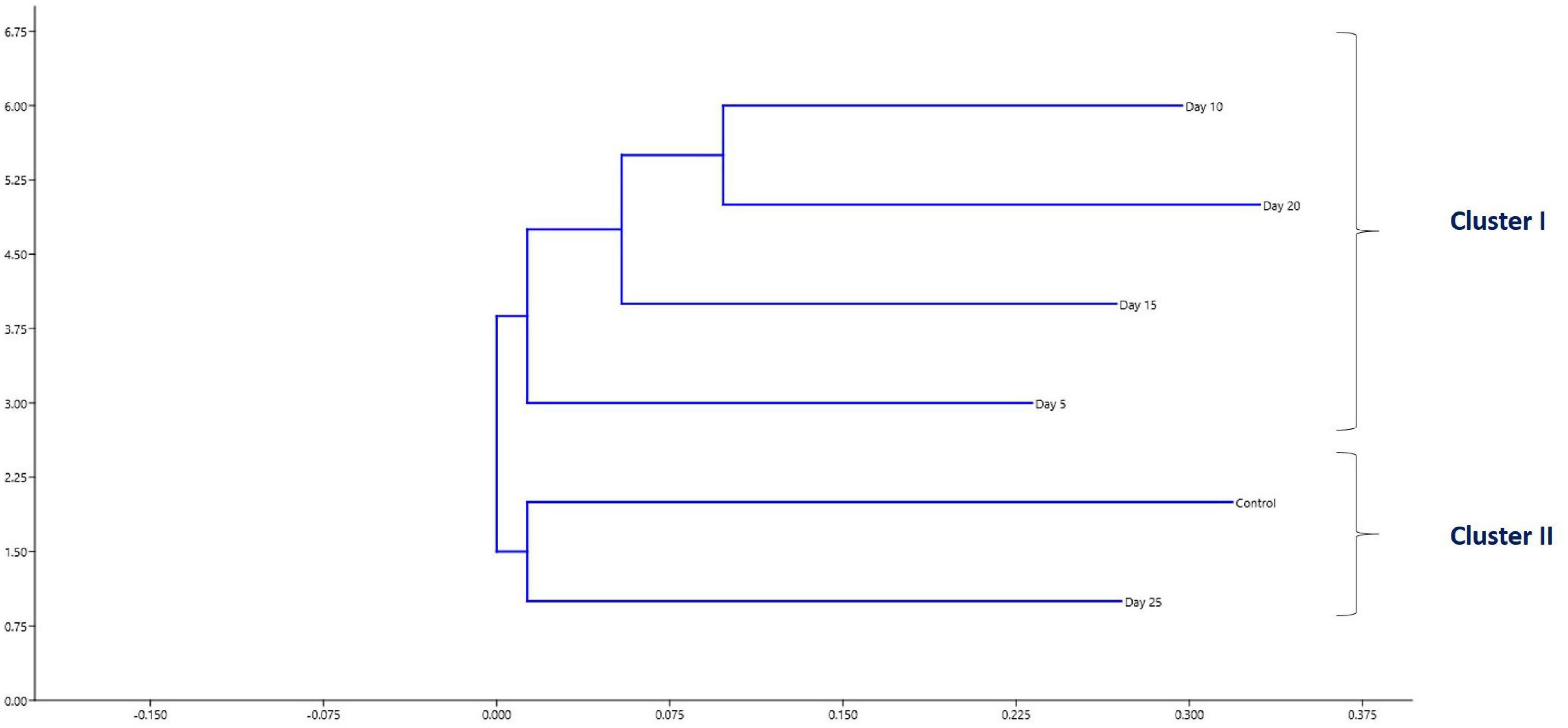
Figure 5. Hierarchical clustering based on the average Bray-Curtis similarity index.
Impact of FFC Therapeutic Exposure on the Gut Microbiome Taxonomy
There were significant (P < 0.05) changes in the relative abundance of certain gut microbial taxon following FFC exposures compared to the control group (Figure 3). Further, there were considerable variations between the individual fish belonging to the same exposure period. Nevertheless, there was no significant difference between the treatment and control group in the relative abundance of microbes at the domain level on any day of FFC exposure. At the phylum level, the shift in microbiome communities was largely driven by OTUs assigned to the phylum Proteobacteria (Figure 3). The relative abundance of Proteobacteria was significantly increased at zero-day post-withdrawal, leading to the reduced representation of the other two phyla, namely Euryacrcheota and Firmicutes (Figure 6A). The changes in the phyla level became similar to the control group (p < 0.05) from 10 days post-withdrawal. At the class level, there was a significant (P < 0.05) transient increase and decrease in the relative abundance of γ-Proteobacteria and Methanobacteria, respectively, at zero-day post-withdrawal. The relative abundance of the class, Bacilli was also reduced at zero-day post-withdrawal (Figure 6B). The changes in different classes became similar to the control group at 15 days post-withdrawal. Among the major changes in the order level, the decrease in the relative abundance of Enterobacteriales and increase in Vibrionales remained even on the 15th day post-withdrawal (Figure 6C). At the family level, the relative abundance of Enterobacteriaceae and Moraxellaceae remained significantly at a lower level than the control fish at 15 days post-withdrawal (Figure 6D). At the genus level, there were several transient changes, however, the relative abundance of Serratia sp. and Acinetobacter sp. remained significantly at a lower level even at 15 days post-withdrawal (Figure 6E). It was striking to note that the relative abundance of Vibrio sp. showed an increasing trend from five days post-initiation of FFC treatment, reached the maximum level at five days post-withdrawal, and then showed a decreasing trend (Figure 6E). The details of the major changes in the gut microbial taxonomy are briefly represented in Table 1.
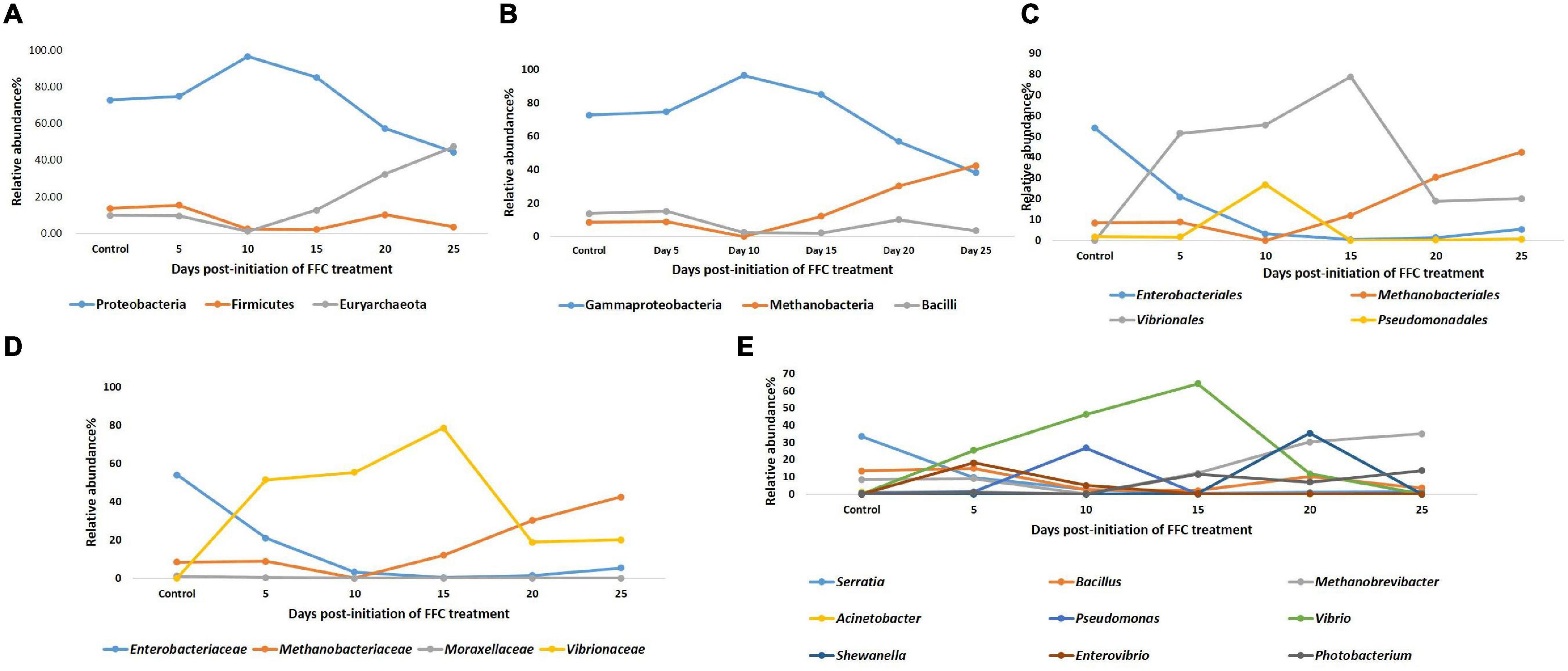
Figure 6. Dynamics of microbial taxon whose relative abundance was significantly altered by FFC treatment. (A) At phylum level; (B) at class level; (C) at order level; (D) at family level; (E) at genus level.
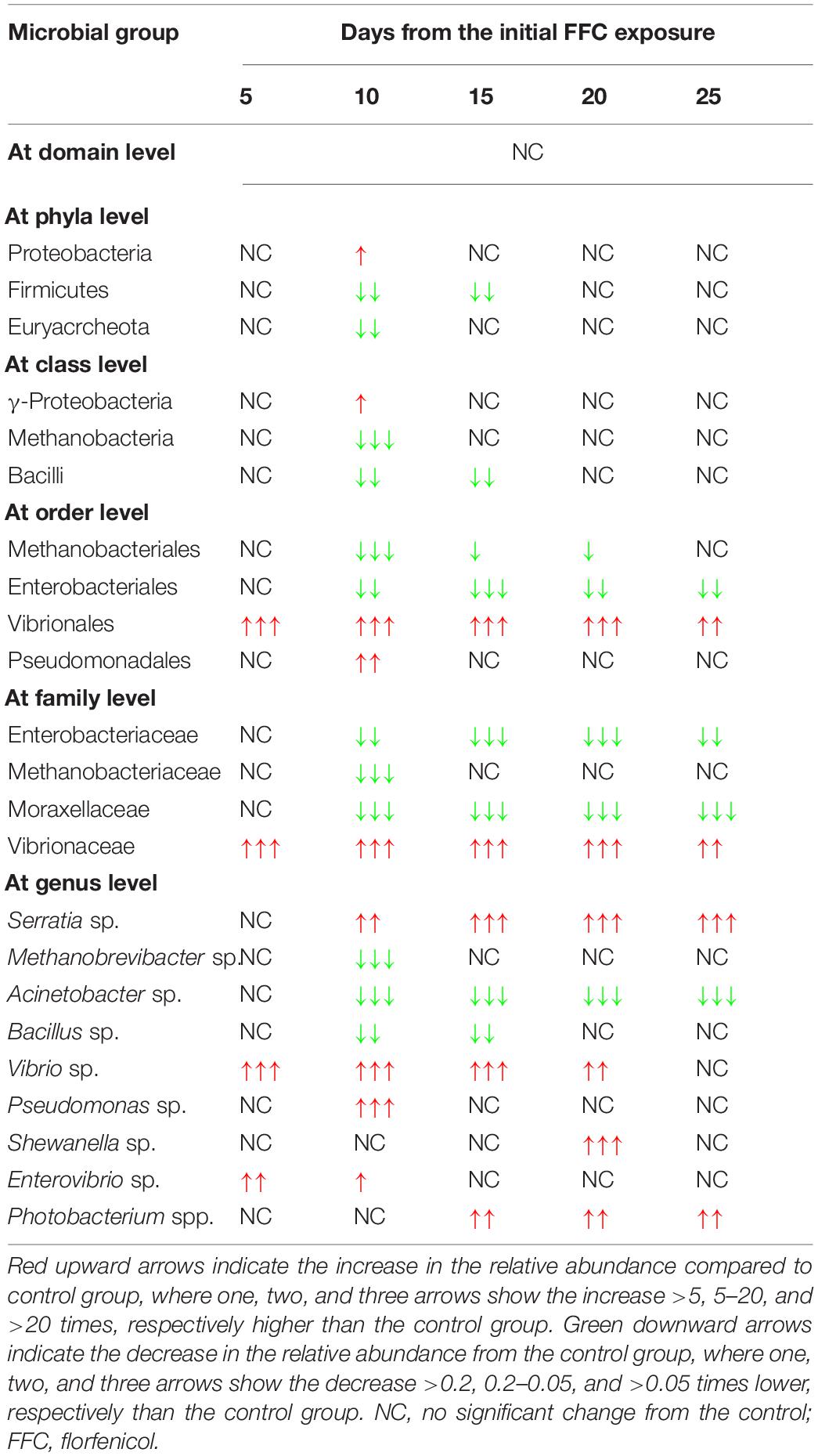
Table 1. Major changes from the control group in the gut microbial taxonomy following therapeutic exposure to florfenicol (FFC).
In the SIMPER analysis, there were 15, 14, 13, 13, and 16 differentiating identified genus-level taxa in the FFC-treated fish group compared to the control fish (>0.1% contribution to the dissimilarity) at 5, 10-, 15-, 20-, and 25-days post-initiation of the treatment (Figure 7). On the 5th day, the maximum dissimilarity in the treated fish group in comparison to the control group was caused by an increased abundance of Enterovibrio sp. and Vibrio sp. and decreased abundance of Serratia sp. At zero-day post-withdrawal, the maximum dissimilarity was caused by an increased abundance of Vibrio sp. and Pseudomonas sp. and decreased abundance of Serratia sp. At five-day post-withdrawal, the maximum dissimilarity of the FFC-treated fish compared to the control was caused by an increased abundance of Vibrio spp. and decreased abundance of Serratia spp. The maximum dissimilarity compared to the control fish at 10-day post-withdrawal was contributed by the increased abundance of Shewanella sp. and decreased abundance of Serratia sp. On the 15th day post-withdrawal, the increased abundance of Photobacterium sp. and decreased abundance of Serratia sp. contributed to the maximum dissimilarity in the FFC treated fish group compared to the control fish.
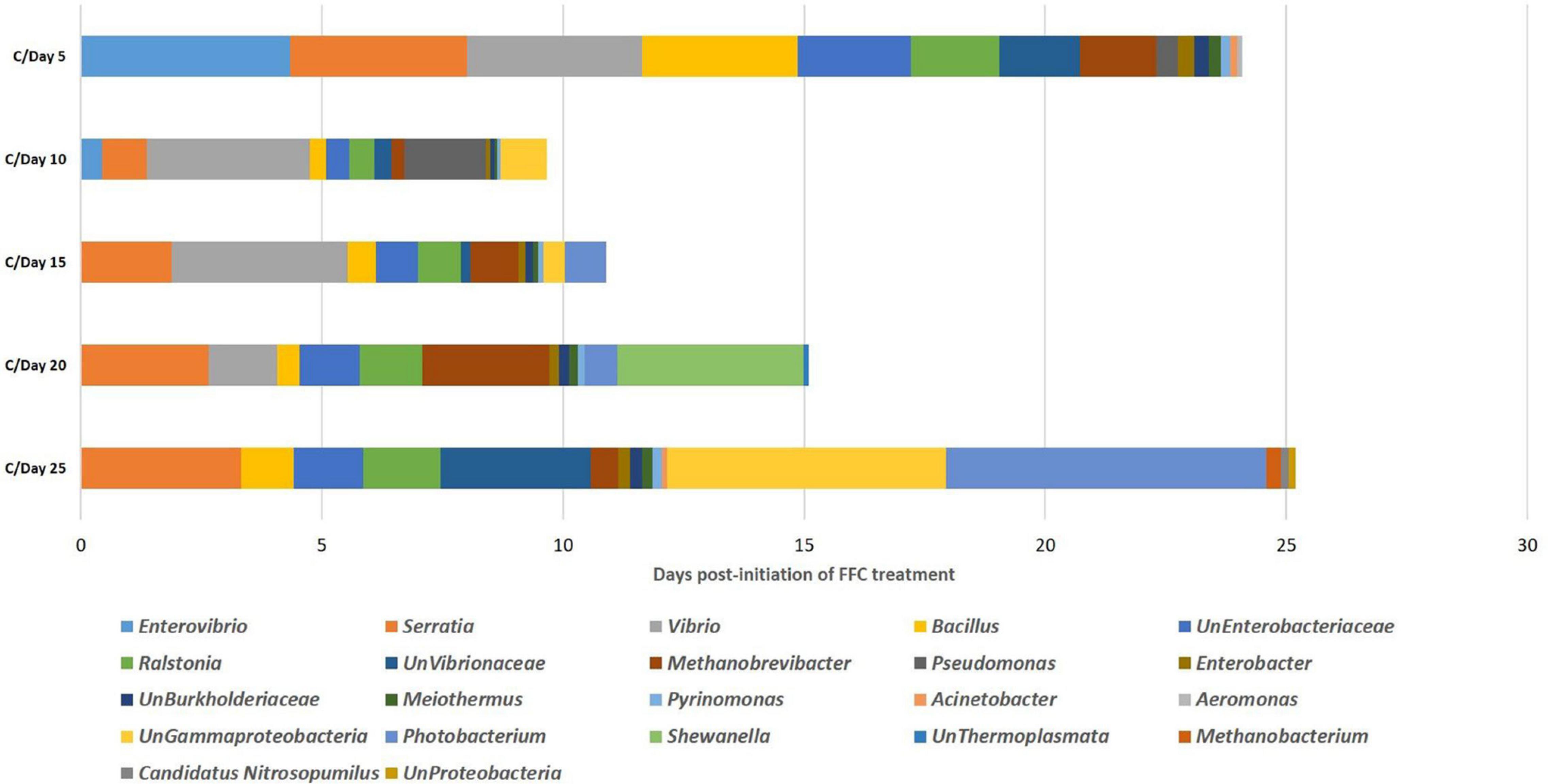
Figure 7. Similarity percentages analysis showing the microbial taxon contributing to the discrimination between different days of FFC exposure.
Impact of Florfenicol Therapeutic Exposure on the Functional Metagenomics of Gut Microbiome
There were 62, 51, 56, 114, and two differentiating KEGG genes at 5, 10, 15, 20, and 25 days, respectively, of therapeutic exposure with >0.1% contribution. Of which, the abundances of 45 and 44 KEGG genes were significantly different in the FFC treated fish group compared to the control group, at zero and five days of the FFC withdrawal (Table 2). However, none of the genes were significantly different in the treated group in comparison to the control group at five days post-initiation of treatment, and 10 and 15 days of FFC withdrawal. In other words, the abundance of KEGG genes returned to the control group level by 10 days of FFC withdrawal. At zero-day post-withdrawal, the maximum significant dissimilarity compared to the control group was caused by the increased abundance of K03406 (methyl-accepting chemotaxis protein), K02030 (polar amino acid transport system substrate-binding protein), K00059 (3-oxoacyl-[acyl-carrier protein] reductase), K02014 (Ferrichrome outer membrane transporter), and K02015 (iron ABC transporter permease). Further, the significant increases in the abundances of two multidrug-resistant proteins, viz. K03327 and K03585 at zero-day post-withdrawal were noteworthy. On the fifth day post-withdrawal, the maximum significant dissimilarity in comparison to the control group was caused by the increased abundance of K03406 (methyl-accepting chemotaxis protein), K02004 (Efflux ABC transporter permease protein), K02003 (ABC transporter system ATP binding protein), and K02015 (iron ABC transporter permease). It was interesting to note that there were no significant changes in the abundance of the genes involved in multidrug/antibiotic resistance from the fifth-day post-withdrawal onward.
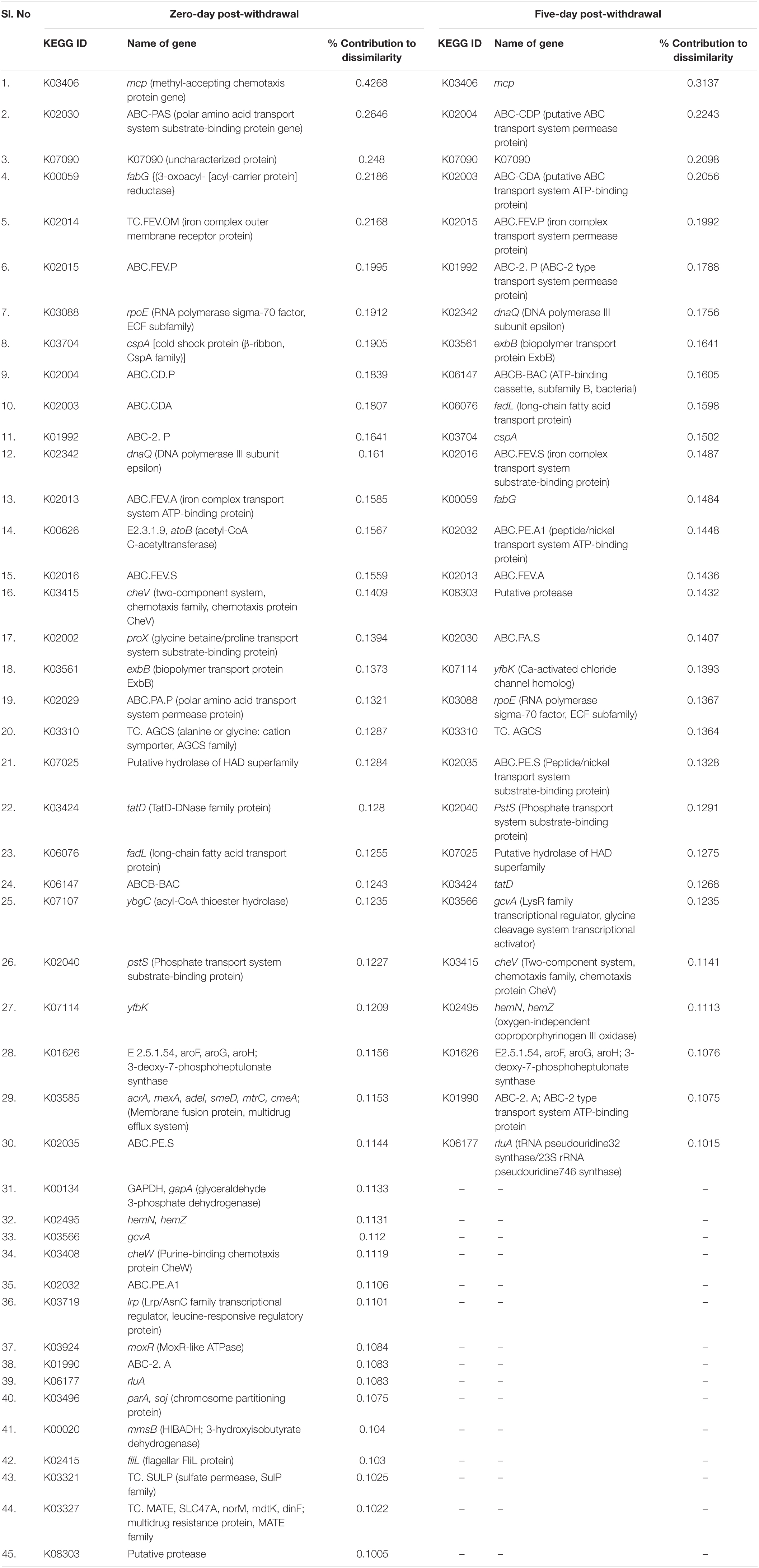
Table 2. Details of KEGG genes whose abundances were significantly different compared to the control fish.
There were 235, 205, 207, 206, and 174 differentiating KEGG pathways at 5, 10, 15, 20, and 25 days of therapeutic exposure with >0.1% contribution, respectively. Of which, the abundances of 97 and 173 KEGG pathways were significantly different in comparison to the control group, at zero and five days of the FFC withdrawal (Table 2). However, the abundances of KEGG pathways at five days post-initiation of treatment, and 10 and 15 days of FFC post-withdrawal were similar to that of the control group (P > 0.05). In other words, the abundance of KEGG pathways also became similar to the control group by 10 days post-withdrawal of FFC treatment. At zero-day post-withdrawal, the maximum significant dissimilarity compared to the control was caused by the increased abundance of PWY3781 (aerobic respiration/cytochrome c), PWY7663 (gondoate biosynthesis: anaerobic), FAS YN-ELONG-PWY (fatty acid biosynthesis), FAO PWY (fatty acid oxidation), and PWY 7664 (oleate biosynthesis IV anaerobic). On the fifth day post-withdrawal, the maximum significant dissimilarity compared to the control was caused by the increased abundance of PWY7663 (gondoate biosynthesis: anaerobic), FAO PWY (fatty acid oxidation), and PWY 5989 (stearate biosynthesis II).
Discussion
Antibiotics play a significant role in treating and controlling bacterial diseases, which are a major impediment to the economic sustainability of aquaculture (Pridgeon, 2012). The antibiotic treatment options are critically crucial for farmed tropical marine fishes like snubnose pompano (T. blochii), where alternative prophylactic strategies such as vaccines are completely absent. Contrariwise, cutting-edge research in terrestrial animals has amply demonstrated the adverse effect of antibiotic treatment in disrupting the healthy gut microbiome of the host (Holman et al., 2019). As the gut microbiome serves several vital biological and physiological functions for the host, understanding the intended and unintended consequences of antibiotic treatment on the gut microbiome is vital to support the overall health and welfare of the farmed animals (Payne et al., 2021). The antibiotic treatment can also cause the emergence of antimicrobial resistance (AMR) in gut microbes, raising additional concerns about consumer and environmental safety (Cabello et al., 2013). The effect of antibiotic treatment on the gut microbiome of marine fish has not been studied, so far, despite its increasing relevance from both agricultural and ecological perspectives. In this context, the present study was envisaged to ascertain the impact of recommended therapeutic dose of FFC, one of the FDA-recommended antimicrobial compounds for aquaculture use, on the gut microbiome of T. blochii. The study forms the first report on the unintentional consequences of an approved antibiotic treatment on the gut microbiome of a marine fish species.
Preliminary evaluation using culture-dependent methods showed that the FFC treatment significantly (P < 0.05) reduced the gut bacterial density even from 5 days post-initiation of the treatment. This was an expected result, as feeding antibiotics were shown to inhibit the normal intestinal microbiota of fish (Gaskins et al., 2002). There was a two log-reduction in the total viable bacterial count compared to the control fish gut at zero-day post-withdrawal. A similar observation was reported in zebrafish, where colistin and vancomycin treatment reduced the total viable count by 3–5 logs and 2 logs, respectively (Brugman et al., 2009). More importantly, in the present study, the total viable bacteria and presumptive vibrios became similar to the control group by the 10th and 5th-day post-FFC withdrawal, respectively. These results suggested that the therapeutic exposure to FFC could induce significant short-term changes to the cultivable gut microbiome, followed by its restoration at the 10th-day post-withdrawal. The findings were supported by Kim et al. (2019), who observed the restoration of the cultivable bacterial load in olive flounder (Paralichthys olivaceus) on the 10th day after treatment with the therapeutic dose of oxytetracycline and amoxicillin.
The evaluation using 16SrRNA amplicon-based metagenomics approach was followed for a detailed understanding of the treatment consequences on the gut microbiome. The results of α-diversity metrics showed that the FFC-treated fish had significantly lower diversity and richness measures of gut microbiome than the control group at 0-, 5-, and 10-days post-withdrawal of FFC-medicated feed. Earlier studies on antibiotic-induced perturbations in the commensal microbes of the freshwater fish gut and aquatic environments have also revealed the reduction in the gut microbial diversity measures following either FFC or oxytetracycline treatment (He et al., 2011; Navarrete et al., 2008; Kim et al., 2019; Wang et al., 2019; Zeng et al., 2019). Using hierarchical clustering, the visual representation of β−diversity measures reflected a clear separation between FFC-fed and control samples. More strikingly, both alpha and beta diversity measures showed a similarity between the gut microbiome of the FFC-fed and control fish at 15 days post-withdrawal, illustrating the restoration of the gut microbiome at 15 days of post-withdrawal of medicated feed in the test groups. While the difference in the gut bacterial density between FFC-treated and control fish was evident from the fifth-day post-initiation of treatment in culture-dependent methods, and no dissimilarity was observed in culture-independent methodologies based on α- and β-diversity measures. The ineptitude of DNA-based culture-independent methodologies to differentiate between the dead and viable bacteria in the gut can be attributed to the above observation. In support of our findings, Reikvam et al. (2011) reported that DNA-based methodologies could not detect the reduction in microbial load as early as those observed by culture-dependent techniques.
To shed more light on the FFC treatment-induced gut microbial dysbiosis, the changes in the relative abundance of different OTUs were analyzed. The results revealed certain significant changes in the relative abundance of different gut microbial taxons. At the phylum level, there was a significant increase in the relative abundance of Proteobacteria in the FFC-treated group, along with a significant reduction in the relative abundance of Euryacrcheota and Firmicutes. Increased abundance of Proteobacteria and decreased abundance of Firmicutes in the fish gut and aquaculture environment of FFC-treated freshwater fish have also been reported in earlier studies (Abdelhamed et al., 2019; Zeng et al., 2019). The studies on transgenic fast-growing common carp (Cyprinus carpio L.) showed that the increased relative abundance of Firmicutes could confer a fast growth to the fish (Li et al., 2013). Even though the effects of dietary FFC on the weight gain of fish were not recorded in the present study, Gaikowski et al. (2013) demonstrated a significant reduction in the bodyweight of FFC-treated tilapia (Oreochromis sp.). Taken together, the decreased relative abundance of Firmicutes observed in the present study might be a novel explanation for the reductions in body weight of FFC-treated tilapia observed by Gaikowski et al. (2013) and warrant future investigation to confirm the hypothesis. Another interesting observation in the microbial shift during downstream analysis was the significant reduction in the relative abundance of Enterobacteriaceae. As many members of Enterobacteriaceae have been reported to benefit from the host metabolic activity and nutrient utilization (Wu et al., 2012), the observed decrease can be another possible novel explanation for the reductions in bodyweight of the FFC-treated fish reported in previous studies.
At the class level, there was a significant transient increase in the relative abundance of γ-Proteobacteria and decreased abundance of Bacilli at zero-day post-withdrawal but became similar to the control group from fifth-day post-withdrawal in the case of γ-Proteobacteria and 10th-day post-withdrawal in the case of Bacilli. The γ-Proteobacteria comprise most opportunistic fish pathogens and tend to increase after exposure to different stressors in fish (Austin and Austin, 2007; Boutin et al., 2013; Webster et al., 2021). The results reinforced the earlier hypothesis of Webster et al. (2021) that an increased abundance of γ-Proteobacteria in the fish gut can be a common signature of certain stress exposure. On further downstream analysis, the reduced abundance of Firmicutes was linked to a reduction in the abundance of Bacillus sp. Whereas, the increased abundance of γ-Proteobacteria was related to an increase in the abundance of Vibrio sp., Enterovibrio sp., Photobacterium sp., Pseudomonas sp., and Shewanella sp. As these genera represent significant opportunistic marine fish pathogens (Mohamad et al., 2019), the transient rise observed in their relative abundance for a period of five-day post-withdrawal of the FFC suggested the possible increase in the susceptibility of the treated fish to different opportunistic diseases caused by them. The results warrant applying health management measures, like probiotics during the FFC withdrawal period, to reverse the observed negative effect as done by Schmidt et al. (2017), following streptomycin treatment in a freshwater fish species, Poecilia sphenops. In this context, it is noteworthy that the microbes belonging to Vibrionaceae can grow in TCBS agar (Bolinches et al., 1988), and the observed reduction of the bacterial count on the TCBS agar through culture-dependent mehods in the present study (from fifth-day post-initiation to the fifth-day post-withdrawal of FFC treatment), showed that the increase observed in Vibrionaceae was only a relative increase. In short, the results showed that FFC treatment caused a reduction in all the gut microbial counts but there was a clear shift in the gut microbiome toward well-known putative pathogens. In support of our findings, the shifts in the gut microbiome toward well-known putative pathogens following different antibiotic treatments were reported in the earlier studies on freshwater fish (Zhou et al., 2018; Sáenz et al., 2019; Payne et al., 2021). Simultaneously, the observed reduction in the bacterial count on TCBS agar showed that FFC has good antagonistic activity against the major marine fish pathogens (belonging to Vibrionacetheae family).
Further, different OTUs affiliated with Serratia sp., Methanobrevibacter sp., Acinetobacter sp., and Bacillus sp. showed reduction for a brief period in the FFC-treated fish gut. The possible role of Methanobrevibacter sp. in fiber digestibility in swine (Niu et al., 2015) and the potential probiotic activity of Acinetobacter sp. and Bacillus sp. in fish (Pandey et al., 2011; Tarnecki et al., 2019; Kuebutornye et al., 2020) have been reported. The observed reversal of their abundance within 20 days post-FFC treatment suggested the transient nature of dysbiosis following the antibiotic treatment. The gut microbiome research in humans has demonstrated that the use of certain probiotic strains could reduce antibiotic-associated diseases (Szajewska and Kołodziej, 2015). Accordingly, the reduced specific microbial signatures in the present study must be explored in the future for developing microbial management measures/probiotic dietary supplements to improve fish health during the antibiotic withdrawal period.
An important observation from the present study was the complete restoration of the gut microbiome in terms of cultivable bacterial load, diversity measures of metagenomics, functional metagenomic profiles, and taxonomic composition up to class level within 10–15 days of FFC withdrawal. However, at the lower taxonomic level (order, family, and genus), microbial composition remained changed at 15 days post-withdrawal. More specifically, the changes observed in the relative abundance of Vibrionaceae (Vibrio sp. and Photobacterium sp.), Enterobacteriaceae (Serratia sp.), and Moraxellaceae (Acinetobacter sp.) remained at 15 days of post-withdrawal. Even though restoration of cultivable gut microbial count by 10 days post-withdrawal of antibiotic treatment was reported by Kim et al. (2019), no previous study has evaluated the restoration dynamics of the gut microbial composition following antibiotic treatment in fish. In similar studies on human and mice gut microbiome, the bacterial load and diversity measures reversed over time after antibiotic therapy (Dethlefsen et al., 2008; Looft and Allen, 2012; Michelle et al., 2019), which supports findings. The implication of the observed short and long-term changes in the gut microbiome profiles on the long-term host’s health and metabolism are also warranted in the future and may enable the development of new therapeutic/prophylactic strategies. Further, the impact of FFC on the gills and skin microbiome, intestinal integrity, and health and immunity indices of fish, as well as the forces that shaped the restoration of gut microbiome composition during the post-withdrawal period, are interesting topics for future investigation.
To have more information on gut microbial dysbiosis in terms of AMR, the enumeration of gut bacteria in different antibiotic-embedded plates was done. Interestingly, the FFC treatment could significantly reduce the numbers of ampicillin-resistant microbes in the gut of treated fish, which persisted even after 20 days post-withdrawal of the treatment. The results suggested that FFC treatment can be used against infections caused by ampicillin-resistant microbes. In accordance with our results, Maaland et al. (2015) pointed out that FFC and chloramphenicol can be used to treat infections caused by extended-spectrum β-lactamase producing bacteria. More strikingly, it was noted that the therapeutic exposure to FFC did not induce the emergence of FFC resistant bacteria in the gut of the treated fish. The observation further confirms the concept that the excessive/indiscriminate use of antibiotics, not the therapeutic dose, increases the likelihood of resistance acquisition by the gut microbiota (Francino, 2016). However, there was a significant transient increase in the numbers of kanamycin-resistant bacteria in the FFC treated fish up to 10 days post-withdrawal of the treatment, suggesting that an indirect transient selection of kanamycin-resistant bacteria occurred as a secondary effect of FFC treatment. Even though kanamycin treatment was shown to promote short-term resistance against streptomycin, tetracycline, and ampicillin (Chen et al., 2009), the interaction between FFC and kanamycin has not been studied to date. To find a probable reason for the observation, the results of PICRUSt (Langille et al., 2013) were thoroughly scrutinized. The results revealed a set of gut microbial KEGG genes and pathways related to different transporter systems, cell metabolism, biosynthesis, cell motility, SOS response, and extracellular structure, which were significantly altered by the FFC treatment. Among these, the significant but transient increase in the abundances of two multidrug-resistant proteins, viz. K03327 (a multidrug resistance protein belonging to the MATE family) and K03585 (membrane fusion protein belonging to multidrug efflux system) at zero-day post-withdrawal were noteworthy. The results suggest that FFC treatment could transiently select multidrug resistance due to the increase in multiple efflux pump systems. In consonance with our findings, Looft et al. (2012) found that several resistance genes unrelated to the exposed antibiotic were enriched in the swine gut microbiome following antibiotic exposure. More importantly, the changes in the abundance of all the KEGG genes and pathways of gut microbes were only transient and became similar to the control group by the 10th-day post-withdrawal. Altogether, the results suggested that the therapeutic dose of FFC did not cause an irreversible increase in the antibiotic resistance profiles of the gut microbiota. Furthermore, it did not promote the FFC resistant microbes in the gut of the treated fish. However, the transient increase in the numbers of kanamycin-resistant microbes and the abundance of multidrug resistance encoding genes observed in the present study warrant certain bona fide strategies for processing aquaculture effluents during the first 10 days post-withdrawal period of antibiotic therapy for avoiding the probable emergence and dispersal of antimicrobial-resistant bacteria in the environments. Further, the results warrant future investigations on the changes in the frequency of different AMR genes and applications of shotgun metagenomic methods to profile whole microbial genomes to have a thorough understanding of the impacts of FFC use in aquaculture on the dispersal of AMR phenomenon in the environment.
Conclusion
The present study forms the first report on the modulation and restoration dynamics of gut microbiota following the oral therapeutic dosing of FFC in a high-value marine aquaculture candidate fish species. The results showed a complete restoration of the gut microbiome within 10-15 days of FFC withdrawal except for a few changes in the lower microbial composition. The results highlighted the need for implementing better stress-management measures during the initial days of the withdrawal period. The study also pointed out the possible microbial signatures of stress in the fish and possible probiotic microbes that can be explored to design fish health improvisation strategies during the withdrawal period. The results also suggested that the therapeutic exposure to FFC did not cause an irreversible increase in the antibiotic resistance profiles of the gut microbiota. Further, it did not promote the FFC resistant microbes in the gut of the treated fish. In brief, the paper generates interesting insights on the implications of FFC treatment in a marine fish species, targeting its applications to formulate the identified risk minimization strategies during sustainable aquaculture practices. Further, the results emphasize the need to implement better infectious disease management measures in aquaculture facilities and recommend restriction of the antimicrobial treatment for the inevitable situations with the specified therapeutic dose and duration only.
Data Availability Statement
The data presented in the study are deposited in the NCBI repository, as Sequence Read Archive (SRA) data (accession numbers: SRR16990455–SRR16990471) under the Bio project accession no. PRJNA780352.
Ethics Statement
The animal study was reviewed and approved by the Institute Animal Ethics Committee of ICAR-CMFRI, Kochi, India (Grant No: CIBA/AINP-FH/2020-21).
Author Contributions
KS conceptualized the presented idea, supervised the project, and acquired financial support for the project leading to this publication. TS and KS supervised the findings, analyzed the results, and wrote the manuscript. GS performed the bioinformatics analysis of the data. SG and VP conducted the experiments, sampling, and sample processing. PA, PS, and MA provided technical support to carry out the experiments. PP and AG provided critical feedback while drafting the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The funding for this research was provided by the Indian Council of Agricultural Research (ICAR) under the All India Network Project on Fish Health (Grant No. CIBA/AINP-FH/2015–16).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to express gratitude to the Director, and the Head, Marine Biotechnology Division, ICAR-CMFRI for the facilities provided. GS acknowledges the Department of Biotechnology funded project “E.G. Silas Centre of Excellence and Innovation (EGS - CoEI) in Marine Fish Microbiome and Nutrigenomics” (BT/AAQ/3/SP28267/2018) for the fellowship. SG and VP acknowledge ICAR funded All India Network Project on Fish Health for the fellowship. PA acknowledges CSIR-UGC for the research fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2022.881275/full#supplementary-material
References
Abdelhamed, H., Ozdemir, O., Waldbieser, G., Perkins, A. D., Lawrence, M. L., and Karsi, A. (2019). Effects of florfenicol feeding on diversity and composition of the intestinal microbiota of channel catfish (Ictalurus punctatus). Aquac. Res. 50, 3663–3672. doi: 10.1111/are.14325
Amit, R., Subodh, S. G., and Aklakur, M. (2017). Prospects of medicated feed in aquaculture. Nutr. Food Sci. Int. J. 3:555617. doi: 10.19080/NFSIJ.2017.03.555617
Austin, B., and Austin, D. A. (2007). Bacterial Fish Pathogens. Cham: Springer International Publishing. doi: 10.1007/978-3-319-32674-0
Bolinches, J., Romalde, J. L., and Toranzo, A. E. (1988). Evaluation of selective media for isolation and enumeration of vibrios from estuarine waters. J. Microbiol. Methods 8, 151–160. doi: 10.1016/0167-7012(88)90016-4
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Boutin, S., Bernatchez, L., Audet, C., and Derôme, N. (2013). Network analysis highlights complex interactions between pathogen, host and commensal microbiota. PLoS One 8:e84772. doi: 10.1371/journal.pone.0084772
Boyd, C. E., D’Abramo, L. R., Glencross, B. D., Huyben, D. C., Juarez, L. M., Lockwood, G. S., et al. (2020). Achieving sustainable aquaculture: historical and current perspectives and future needs and challenges. J. World Aquac. Soc. 51, 578–633. doi: 10.1111/jwas.12714
Bruggeling, C. E., Garza, D. R., Achouiti, S., Mes, W., Dutilh, B. E., and Boleij, A. (2021). Optimized bacterial DNA isolation method for microbiome analysis of human tissues. Microbiologyopen 10:e1191. doi: 10.1002/mbo3.1191
Brugman, S., Liu, K., Lindenbergh–Kortleve, D., Samsom, J. N., Furuta, G. T., Renshaw, S. A., et al. (2009). Oxazolone-induced enterocolitis in zebrafish depends on the composition of the intestinal microbiota. Gastroenterology 137, 1757–1767.e1. doi: 10.1053/j.gastro.2009.07.069
Cabello, F. C., Godfrey, H. P., Tomova, A., Ivanova, L., Dölz, H., Millanao, A., et al. (2013). Antimicrobial use in aquaculture re-examined: its relevance to antimicrobial resistance and to animal and human health. Environ. Microbiol. 15, 1917–1942. doi: 10.1111/1462-2920.12134
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chen, L. X., He, S., Li, C., and Ryu, J. (2009). Sublethal kanamycin induced cross resistance to functionally and structurally unrelated antibiotics. J. Exp. Microbiol. Immunol. 13, 53–57.
Committee for veterinary medicinal products (2001). Florfenicol-Summary Report. The European Agency for the Evaluation of Medicinal Products. Available Online at: https://www.ema.europa.eu/en/documents/mrl-report/florfenicol-summary-report-1-committee-veterinary-medicinal-products_en.pdf [accessed January 30, 2022].
Dethlefsen, L., Huse, S., Sogin, M. L., and Relman, D. A. (2008). The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6:e280. doi: 10.1371/journal.pbio.0060280
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
du Sert, N. P., Hurst, V., Ahluwalia, A., Alam, S., Avey, M. T., Baker, M., et al. (2020). The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. Exp. Physiol. 105, 1459–1466. doi: 10.1113/EP088870
EU Directive 2010/63/EU for animal experiments (2019). Legislation for the Protection of Animals Used for Scientific Purposes. Available Online at: https://ec.europa.eu/environment/chemicals/lab_animals/legislation_en.htm [accessed January 17, 2021].
Feng, J.-B., Ruan, H.-T., Chen, H.-G., Luo, J.-Z., and Dong, J.-D. (2018). Pharmacokinetics of florfenicol in the orange-spotted grouper, Epinephelus coioides, following oral administration in warm seawater. J. World Aquac. Soc. 49, 1058–1067. doi: 10.1111/jwas.12509
Francino, M. P. (2016). Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances. Front. Microbiol. 6:1543. doi: 10.3389/fmicb.2015.01543
Gaikowski, M. P., Wolf, J. C., Schleis, S. M., Tuomari, D., and Endris, R. G. (2013). Safety of florfenicol administered in feed to tilapia (Oreochromis sp.). Toxicol. Pathol. 41, 639–652. doi: 10.1177/0192623312463986
Gaskins, H. R., Collier, C. T., and Anderson, D. B. (2002). Antibiotics as growth promotants: mode of action. Anim. Biotechnol. 13, 29–42. doi: 10.1081/ABIO-120005768
Guardabassi, L., Lo Fo Wong, D. M., and Dalsgaard, A. (2002). The effects of tertiary wastewater treatment on the prevalence of antimicrobial resistant bacteria. Water Res. 36, 1955–1964. doi: 10.1016/S0043-1354(01)00429-8
Hammer, Ø., Harper, D. A. T., and Ryan, P. D. (2001). PAST: paleontological statistics software package for education and data analysis. Palaeontol. Electron. 4, 1–9.
He, S., Zhou, Z., Meng, K., Zhao, H., Yao, B., Ringø, E., et al. (2011). Effects of dietary antibiotic growth promoter and Saccharomyces cerevisiae fermentation product on production, intestinal bacterial community, and nonspecific immunity of hybrid tilapia (Oreochromis niloticus female × Oreochromis aureus male)1. J. Anim. Sci. 89, 84–92. doi: 10.2527/jas.2010-3032
Holman, D. B., Yang, W., and Alexander, T. W. (2019). Antibiotic treatment in feedlot cattle: a longitudinal study of the effect of oxytetracycline and tulathromycin on the fecal and nasopharyngeal microbiota. Microbiome 7:86. doi: 10.1186/s40168-019-0696-4
Hovda, M. B., Lunestad, B. T., Fontanillas, R., and Rosnes, J. T. (2007). Molecular characterization of the intestinal microbiota of farmed Atlantic salmon (Salmo salar L.). Aquaculture 272, 581–588. doi: 10.1016/j.aquaculture.2007.08.045
Kenzaka, T., Yamaguchi, N., Utrarachkij, F., Suthienkul, O., and Nasu, M. (2006). Rapid identification and enumeration of antibiotic resistant bacteria in urban canals by microcolony-fluorescence in situ hybridization. J. Health Sci. 52, 703–710. doi: 10.1248/jhs.52.703
Kim, A., Kim, N., Roh, H. J., Chun, W.-K., Ho, D. T., Lee, Y., et al. (2019). Administration of antibiotics can cause dysbiosis in fish gut. Aquaculture 512:734330. doi: 10.1016/j.aquaculture.2019.734330
Kokou, F., Sasson, G., Mizrahi, I., and Cnaani, A. (2020). Antibiotic effect and microbiome persistence vary along the European seabass gut. Sci. Rep. 10:10003. doi: 10.1038/s41598-020-66622-5
Kuebutornye, F. K. A., Abarike, E. D., Lu, Y., Hlordzi, V., Sakyi, M. E., Afriyie, G., et al. (2020). Mechanisms and the role of probiotic Bacillus in mitigating fish pathogens in aquaculture. Fish Physiol. Biochem. 46, 819–841. doi: 10.1007/s10695-019-00754-y
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Li, X., Yan, Q., Xie, S., Hu, W., Yu, Y., and Hu, Z. (2013). Gut microbiota contributes to the growth of fast-growing transgenic common carp (Cyprinus carpio L.). PLoS One 8:e64577. doi: 10.1371/journal.pone.0064577
Liu, X., Chen, Y., Wang, X., and Ji, R. (2004). Foodborne disease outbreaks in China from 1992 to 2001 national foodborne disease surveillance system. Wei Sheng Yan Jiu 33, 725–727.
Looft, T., and Allen, H. K. (2012). Collateral effects of antibiotics on mammalian gut microbiomes. Gut Microbes 3, 463–467. doi: 10.4161/gmic.21288
Looft, T., Johnson, T. A., Allen, H. K., Bayles, D. O., Alt, D. P., Stedtfeld, R. D., et al. (2012). In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. U.S.A. 109, 1691–1696. doi: 10.1073/pnas.1120238109
Maaland, M. G., Mo, S. S., Schwarz, S., and Guardabassi, L. (2015). In vitro assessment of chloramphenicol and florfenicol as second-line antimicrobial agents in dogs. J. Vet. Pharmacol. Ther. 38, 443–450. doi: 10.1111/jvp.12204
Michelle Ng, K., Aranda-Díaz, A., Tropini, C., Frankel, M. R., Van Treuren, W., O’Loughlin, C. T., et al. (2019). Recovery of the gut microbiota after antibiotics depends on host diet, community context, and environmental reservoirs. Cell Host Microbe 26, 650–665.e4. doi: 10.1016/j.chom.2019.10.011
Mohamad, N., Amal, M. N. A., Yasin, I. S. M., Zamri Saad, M., Nasruddin, N. S., Al-saari, N., et al. (2019). Vibriosis in cultured marine fishes: a review. Aquaculture 512:734289. doi: 10.1016/j.aquaculture.2019.734289
Navarrete, P., Mardones, P., Opazo, R., Espejo, R., and Romero, J. (2008). Oxytetracycline treatment reduces bacterial diversity of intestinal microbiota of Atlantic salmon. J. Aquat. Anim. Health 20, 177–183. doi: 10.1577/H07-043.1
Niu, Q., Li, P., Hao, S., Zhang, Y., Kim, S. W., Li, H., et al. (2015). Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci. Rep. 5:9938. doi: 10.1038/srep09938
Pandey, A., Naik, M., and Dubey, S. K. (2011). Biological characterization of marine fish pathogen, Acinetobacter sp. strain An 2 producing antibacterial metabolites. J. Scienfic Ind. Res. 70, 135–141.
Payne, C. J., Turnbull, J. F., MacKenzie, S., and Crumlish, M. (2021). Investigating the effect of an oxytetracycline treatment on the gut microbiome and antimicrobial resistance gene dynamics in Nile Tilapia (Oreochromis niloticus). Antibiotics 10:1213. doi: 10.3390/antibiotics10101213
Pennycook, J. H., and Scanlan, P. D. (2021). Ecological and evolutionary responses to antibiotic treatment in the human gut microbiota. FEMS Microbiol. Rev. 45:fuab018. doi: 10.1093/femsre/fuab018
Pridgeon, J. W. (2012). Major bacterial diseases in aquaculture and their vaccine development. CAB Rev. Perspect. Agric. Vet. Sci. Nutr. Nat. Resour. 7, 1–16. doi: 10.1079/PAVSNNR20127048
Reikvam, D. H., Erofeev, A., Sandvik, A., Grcic, V., Jahnsen, F. L., Gaustad, P., et al. (2011). Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS One 6:e17996. doi: 10.1371/journal.pone.0017996
San Martín, B., Fresno, M., Cornejo, J., Godoy, M., Ibarra, R., Vidal, R., et al. (2019). Optimization of florfenicol dose against Piscirickettsia salmonis in Salmo salar through PK/PD studies. PLoS One 14:e0215174. doi: 10.1371/journal.pone.0215174
Sáenz, J. S., Marques, T. V., Barone, R. S. C., Cyrino, J. E. P., Kublik, S., Nesme, J., et al. (2019). Oral administration of antibiotics increased the potential mobility of bacterial resistance genes in the gut of the fish Piaractus mesopotamicus. Microbiome 7:24. doi: 10.1186/s40168-019-0632-7
Schmidt, V., Gomez-Chiarri, M., Roy, C., Smith, K., and Amaral-Zettler, L. (2017). Subtle microbiome manipulation using probiotics reduces antibiotic-associated mortality in fish. mSystems 2:e00133-17. doi: 10.1128/mSystems.00133-17
Shoemaker, N. B., Vlamakis, H., Hayes, K., and Salyers, A. A. (2001). Evidence for extensive resistance gene transfer among Bacteroides spp. and among Bacteroides and other genera in the human colon. Appl. Environ. Microbiol. 67, 561–568. doi: 10.1128/AEM.67.2.561-568.2001
Sommer, M. O. A., Dantas, G., and Church, G. M. (2009). Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 325, 1128–1131. doi: 10.1126/science.1176950
Soto, E., Endris, R. G., and Hawke, J. P. (2010). In Vitro and in vivo efficacy of florfenicol for treatment of Francisella asiatica infection in tilapia. Antimicrob. Agents Chemother. 54, 4664–4670. doi: 10.1128/AAC.00206-10
Szajewska, H., and Kołodziej, M. (2015). Systematic review with meta-analysis: Lactobacillus rhamnosus GG in the prevention of antibiotic-associated diarrhoea in children and adults. Aliment. Pharmacol. Ther. 42, 1149–1157. doi: 10.1111/apt.13404
Takahashi, S., Tomita, J., Nishioka, K., Hisada, T., and Nishijima, M. (2014). Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next-generation sequencing. PLoS One 9:e105592. doi: 10.1371/journal.pone.0105592
Tarnecki, A. M., Wafapoor, M., Phillips, R. N., and Rhody, N. R. (2019). Benefits of a Bacillus probiotic to larval fish survival and transport stress resistance. Sci. Rep. 9:4892. doi: 10.1038/s41598-019-39316-w
U.K. Animals Scientific Procedures Act (1986). U.K. Animals Scientific Procedures Act. Available Online at: https://www.legislation.gov.uk/ukpga/1986/14/contents [accessed January 17, 2020].
U.S. Food and Drug Administration [USFDA] (2008). Approved Aquaculture Drugs. Available Online at: https://www.fda.gov/animal-veterinary/aquaculture/approved-aquaculture-drugs [accessed January 30, 2022].
Walker, P. J., and Winton, J. R. (2010). Emerging viral diseases of fish and shrimp. Vet. Res. 41:51. doi: 10.1051/vetres/2010022
Wang, E., Yuan, Z., Wang, K., Gao, D., Liu, Z., and Liles, M. R. (2019). Consumption of florfenicol-medicated feed alters the composition of the channel catfish intestinal microbiota including enriching the relative abundance of opportunistic pathogens. Aquaculture 501, 111–118.
Wanka, K. M., Damerau, T., Costas, B., Krueger, A., Schulz, C., and Wuertz, S. (2018). Isolation and characterization of native probiotics for fish farming. BMC Microbiol. 18:119. doi: 10.1186/s12866-018-1260-2
Webster, T. M. U., Consuegra, S., and Garcia de Leaniz, C. (2021). Early life stress causes persistent impacts on the microbiome of Atlantic salmon. Comp. Biochem. Physiol. Part D Genomics Proteomics 40:100888. doi: 10.1016/j.cbd.2021.100888
Whittle, G., Shoemaker, N. B., and Salyers, A. A. (2002). The role of Bacteroides conjugative transposons in the dissemination of antibiotic resistance genes. Cell. Mol. Life Sci. 59, 2044–2054. doi: 10.1007/s000180200004
Wu, S., Wang, G., Angert, E. R., Wang, W., Li, W., and Zou, H. (2012). Composition, diversity, and origin of the bacterial community in grass carp intestine. PLoS One 7:e30440. doi: 10.1371/journal.pone.0030440
Yu, Q., Liu, M., Li, F., Yang, W., Lu, Z., Wu, S., et al. (2018). Identification and characterization of marine pathogenic vibrios in cultured golden pompano (Trachinotus ovatus) in Guangxi, China. Ann. Mar. Sci. 2, 016–019. doi: 10.17352/ams.000010
Zeng, S., Hou, D., Liu, J., Ji, P., Weng, S., He, J., et al. (2019). Antibiotic supplement in feed can perturb the intestinal microbial composition and function in Pacific white shrimp. Appl. Microbiol. Biotechnol. 103, 3111–3122. doi: 10.1007/s00253-019-09671-9
Keywords: fish health, environment, antimicrobial resistance, kanamycin, antibiotics, postwithdrawal
Citation: Sumithra TG, Sharma KSR, Gangadharan S, Suresh G, Prasad V, Amala PV, Sayooj P, Gop AP, Anil MK, Patil PK and Achamveetil G (2022) Dysbiosis and Restoration Dynamics of the Gut Microbiome Following Therapeutic Exposure to Florfenicol in Snubnose Pompano (Trachinotus blochii) to Aid in Sustainable Aquaculture Production Strategies. Front. Microbiol. 13:881275. doi: 10.3389/fmicb.2022.881275
Received: 22 February 2022; Accepted: 30 March 2022;
Published: 30 May 2022.
Edited by:
Stanley Chun Kwan Lau, Hong Kong University of Science and Technology, Hong Kong SAR, ChinaReviewed by:
Jodi Woan-Fei Law, Monash University Malaysia, MalaysiaAtte Von Wright, University of Eastern Finland, Finland
Copyright © 2022 Sumithra, Sharma, Gangadharan, Suresh, Prasad, Amala, Sayooj, Gop, Anil, Patil and Achamveetil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Krupesha S. R. Sharma, a3J1cGVzaHNoYXJtYUBnbWFpbC5jb20=