 Bin Yang
Bin Yang Wenzheng Bao
Wenzheng Bao Baitong Chen
Baitong Chen- 1School of Information Science and Engineering, Zaozhuang University, Zaozhuang, China
- 2School of Information and Electrical Engineering, Xuzhou University of Technology, Xuzhou, China
- 3Xuzhou No.1 People’s Hospital, Xuzhou, China
In order to screen the disease-related compounds of a traditional Chinese medicine prescription in network pharmacology research accurately, a new virtual screening method based on flexible neural tree (FNT) model, hybrid evolutionary method and negative sample selection algorithm is proposed. A novel hybrid evolutionary algorithm based on the Grammar-guided genetic programming and salp swarm algorithm is proposed to infer the optimal FNT. According to hypertension, diabetes, and Corona Virus Disease 2019, disease-related compounds are collected from the up-to-date literatures. The unrelated compounds are chosen by negative sample selection algorithm. ECFP6, MACCS, Macrocycle, and RDKit are utilized to numerically characterize the chemical structure of each compound collected, respectively. The experiment results show that our proposed method performs better than classical classifiers [Support Vector Machine (SVM), random forest (RF), AdaBoost, decision tree (DT), Gradient Boosting Decision Tree (GBDT), KNN, logic regression (LR), and Naive Bayes (NB)], up-to-date classifier (gcForest), and deep learning method (forgeNet) in terms of AUC, ROC, TPR, FPR, Precision, Specificity, and F1. MACCS method is suitable for the maximum number of classifiers. All methods perform poorly with ECFP6 molecular descriptor.
Introduction
Computer-aided drug design (CADD) has gradually become an indispensable emerging technology in the research and development of a new drug (Leelananda and Steffen, 2016; Tong et al., 2019; Maia et al., 2020). CADD technology reduces the capital, time, and labor cost of drug development and greatly improves the efficiency of the research and development of new drug (Gomeni et al., 2001). Virtual screening is one of the important comprehensive technical means in CADD, which is a process of discovering new ligands on the basis of biological structure based on the computer methods (Guasch et al., 2016; Olubiyi et al., 2020; Rajguru et al., 2020). It is a new technology and method for innovative drug research. By using the high-speed computing of computer, a small number of potential active compounds are screened from a large number of candidate compounds, so as to greatly reduce the blindness of subsequent experimental verification. In the future, virtual screening technology will become an important means to explore the relevant biochemical space because of its many advantages, such as high efficiency, high speed, low cost, and so on (Zaslavskiy et al., 2019; Guo et al., 2021; Maddah et al., 2021; Selvaraj et al., 2021; Yang et al., 2021).
In the past decade, virtual screening has been applied to the medical and the pharmaceutical researches widely (Meng et al., 2011; Bajusz et al., 2017). The most commonly used virtual screening method is molecular docking, and the software involved contains AutoDock, SLIDE, DOCK, Flex X, etc. (Morris et al., 1996; Kellenberger et al., 2004; Taufer et al., 2005). Fischer et al. (2021) utilized virtual screening method to screen 25, 56, 750 compounds in order to make the analysis about the binding of small molecules to translationally controlled tumor protein. Baxter et al. (2000) utilized molecular docking to screen ligand-receptor complexes in virtual database and tabu search method was utilized to assist this work. Talluri (2021) utilized Vina and SMINA to make molecular docking to predict potential drugs for the treatment of Corona Virus Disease 2019 (COVID-19). Zhou et al. (2016) screened the compounds of Chicory, which were bundled with concentrated nucleoside transporter 2 (CNT2) in order to validate that CNT2 as the potential target of chicory could reduce the absorption of purine nucleosides in the intestine. Meenakumari et al. (2019) made docking analysis between 17 coumarin derivatives and carbonic anhydrase IX (CAIX) to screen the ligands. Thiyagarajan et al. (2016) made molecular docking between the 3D structures of focal adhesion kinase and S6 kinase and 60 natural compounds to obtain the new specific inhibitors, and the findings could provide help for the treatment of tumorigenesis and metastasis.
In order to improve the time and accuracy of virtual screening, some machine learning methods have been utilized to assist or replace molecular docking (Berishvili et al., 2018; Zaki et al., 2021). Wang et al. (2016) proposed a new virtual screening based on ensemble learning and SVM to tackle with protein-ligand in action fingerprint. Zhang Y. et al. (2019) investigated the performances of 8 classifiers containing decision tree (DT), KNN, SVM, random forest (RF), extremely randomizer tree, AdaBoost, gradient boosting tree, and XGBoost with ACC inhibitor data for the researches of drug design and discovery. Zhang et al. (2017) proposed a new scoring function based on machine learning to screen the compounds targeting the viral neuraminidase protein so as to make anti-influenza therapy. Chen et al. (2011) proposed a ligand screening algorithm based SVM to discovery lead compounds. Bustamam et al. (2021) proposed a dipeptidyl peptidase-4 (DPP-4) inhibitors identification method based on Rotation Forest and Deep Neural Network with the fingerprint datasets for the treatment of type 2 diabetes mellitus. Zheng et al. (2020) utilized Naïve Bayesian and recursive partitioning to select the important active chemical components from many compounds in Xiaoshuan Tongluo formula with ECFP_6 and MACCS feature sets for treating stroke.
Virtual screening of disease-related compounds can narrow the scope of analysis in network pharmacology research. In this paper a new virtual screening method based on flexible neural tree (FNT) model is proposed to screen the disease-related active compounds. A novel hybrid evolutionary algorithm based on Grammar-guided genetic programming and salp swarm algorithm is proposed to infer the structure and parameters in each FNT model. The 3 diseases (hypertension, diabetes, and COVID-19) related compounds are searched from the up-to-date literatures. The unrelated compounds are selected by negative sample selection algorithm from DUD-E website. About 4 kinds of molecular descriptors (ECFP6, MACCS, Macrocycle, and RDKit) are utilized to numerically characterize the chemical structures of related and unrelated compounds of diseases, respectively. We make the investigation about the performances of these 4 molecular descriptors.
Materials and Methods
Flexible Neural Tree Model
In order to solve the automatic design problem of artificial neural network, FNT was proposed, which is a hierarchical, multilayer, and irregular artificial neural network (Chen et al., 2012). FNT can transform a single and fixed neural network model into a special tree model that can change flexibly between various levels. It could overcome the difficulty of structural optimization of common neural network, have strong adaptive ability for various classification and prediction problems, and obtain high classification and prediction accuracy. In this paper, FNT is proposed to predict active disease-related compounds. An example of structure of FNT model is showed in Figure 1. AFNT includes input layer, several hidden layers and output layer. The nodes in the input layer are created randomly from terminal set T = {x1, x2,…,xn}. The nodes in the hidden layers are selected randomly from terminal set and operator set F = { + 2, + 3,…, + n}. The output layer contains one node.
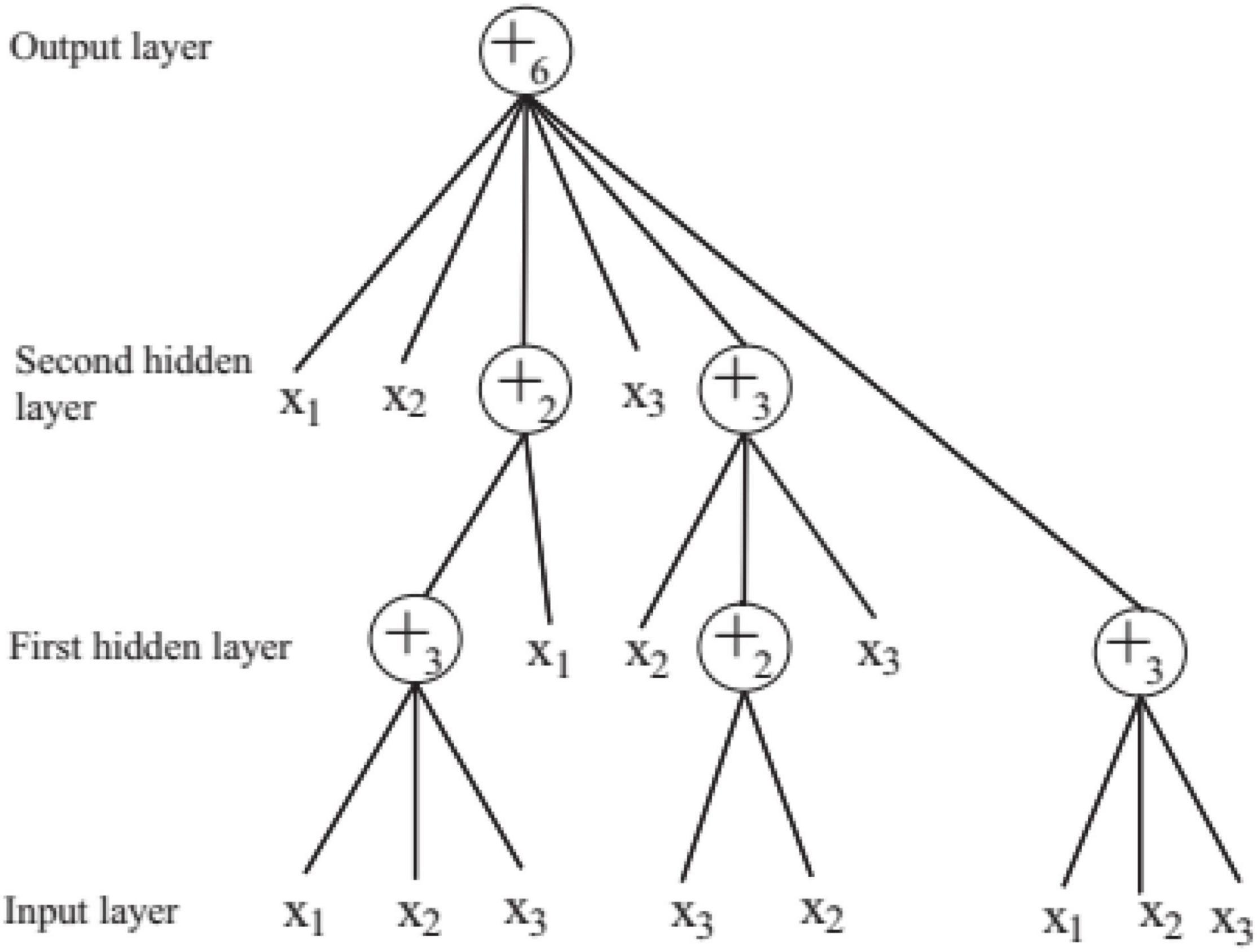
Figure 1. An example of flexible neural tree.
In FNT, each layer is randomly generated according to the operation set and terminal set. The maximum depth of tree is set in advance. If an operator instruction + n is selected, n branches are created randomly from set T and F, which are terminal variables and operators. And n weights are generated randomly. If a terminal variable is selected, the corresponding branch is terminated. When FNT is created randomly, the depth of FNT could not exceed the maximum depth. + n is depicted in Figure 2 and is calculated as follows.
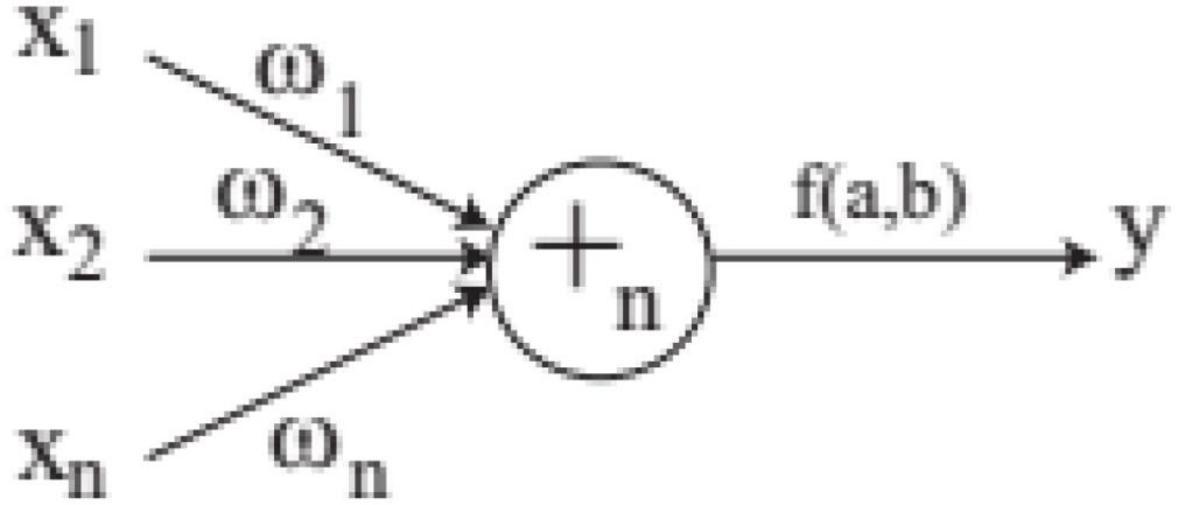
Figure 2. A flexible neuron operator.
The final output of + n is calculated by activation function, which is given as follows.
Where an and bn are parameters of activation function.
Model Optimization Algorithm
Grammar-Guided Genetic Programming
Grammar-guided genetic programming (GGGP) was proposed in order to overcome the shortcomings of genetic programming (Wu and Chen, 2007). In this paper, GGGP is utilized to search the optimal structure of FNT model. In GGGP, context-free grammar (CFG) model is utilized to guide the evolutionary process of GP in order to search the optimal solution faster.
The CFG model contains a quadruple, which is represented as G = {N, T, P,∑}, where N is non-terminal symbol set, T is terminal symbol set, P is production rule set and ∑ is beginning symbol set. The 4 sets satisfy the conditions: N⋂T = ϕ and ∑ ∈ N. An element in production rule set is represented as x→y, where x ∈ N, and y ∈ N⋃T. Assuming that terminal set and operator set are set as T = {x1, x2,…,xn}, and F = { + 2, + 3}, 4 sets of CFG model are defined:N = {s, exp, var, op2, op3}, T = { + 2, + 3, x1, x2, …, xn}, ∑ = {s}, and P is represented with Eq. (3) or Eq. (4).
Generate the initial population randomly. When generating each individual tree, the non-terminal node S is started with. Then the subtree of each non-terminal node is derived in top-down and left-right order according to the rules of the syntax model. When all non-terminal nodes in the tree have sub-trees, stop the derivation process of the tree, and then judge the depth of the tree. If the depth is greater than the predefined maximum depth, the tree is considered invalid, and a tree is regenerated after deletion. If the depth is less than the maximum depth, the tree is considered and can be saved to the population. Then 3 genetic operators (replication, crossover, and mutation) are utilized to generate a new population in the iteration process.
Salp Swarm Algorithm
The Salp swarm algorithm (SSA) is a new swarm optimization algorithm proposed by Mirjalili et al. (2017). The main idea of SSA comes from simulating the group behavior of salp chain (Babaei et al., 2020; Ren et al., 2021). In this algorithm, salp chain is divided into 2 groups: leader and follower. The leader is at the head of the salp chain, and the followers are at the back of the chain. In each iteration, the leader directs the followers to move in a chain toward the food. In the process of moving, the leader makes global search, while the follower makes full local search, which greatly avoid falling into local optimization. The leader’s leadership role for the followers behind will be weaker and weaker. The followers behind will not blindly move toward the leader, which could maintain the diversity of the population. Therefore, this movement mode makes the salp chain have a strong ability of global search and local development. Because of its simple implementation, fast convergence speed, and easy computer implementation, SSA is utilized to optimize the parameters of FNT model. The SSA is given as follows in detailed.
(1) Initialize the population. Suppose that population size is m, the dimension is n, the upper bound of the search space is , the lower bound is . The positions of salp population are created randomly by the following equation.
(2) Give the fitness values of population according to the fitness function defined in advanced. In the iteration process, the position of the food is not clear, so the fitness values of all individual salps are calculated and sorted. And the position of salp with the optimal fitness value is set as the current food position, which is set as F = {F1, F2,…, Fn}.
(3) Positions of leader and followers are updated. The leader is responsible for searching food to lead the moving direction of the whole group. The position of the leader is updated as follows (Chen and Mu, 2021).
Where and Fi are the i-th positions of leader (the first salp) and food. c2 and c3 are random number. c1 is the convergence factor in SSA, which could play the role of balancing global search and local development. c1 is calculated as follows.
Where t is the current generation and T is the maximum generation.
The positions of the followers are updated according to Newton’s laws of motion, which is defined as follows.
Where a is acceleration. The difference between two adjacent iterations is 1 and v0 = 0, so Eq. (8) could be defined as follows.
(4) Update the fitness values of new population and the position of food. If the end condition is satisfied, algorithm is stopped; otherwise go to step (3).
Screen Disease-Related Compounds by Our Proposed Method
Virtual screening is needed in the research of network pharmacology to select the disease-related compounds. In this paper, a novel virtual screening method based on FNT, hybrid evolutionary method and negative sample selection algorithm is proposed, which is depicted in Figure 3.
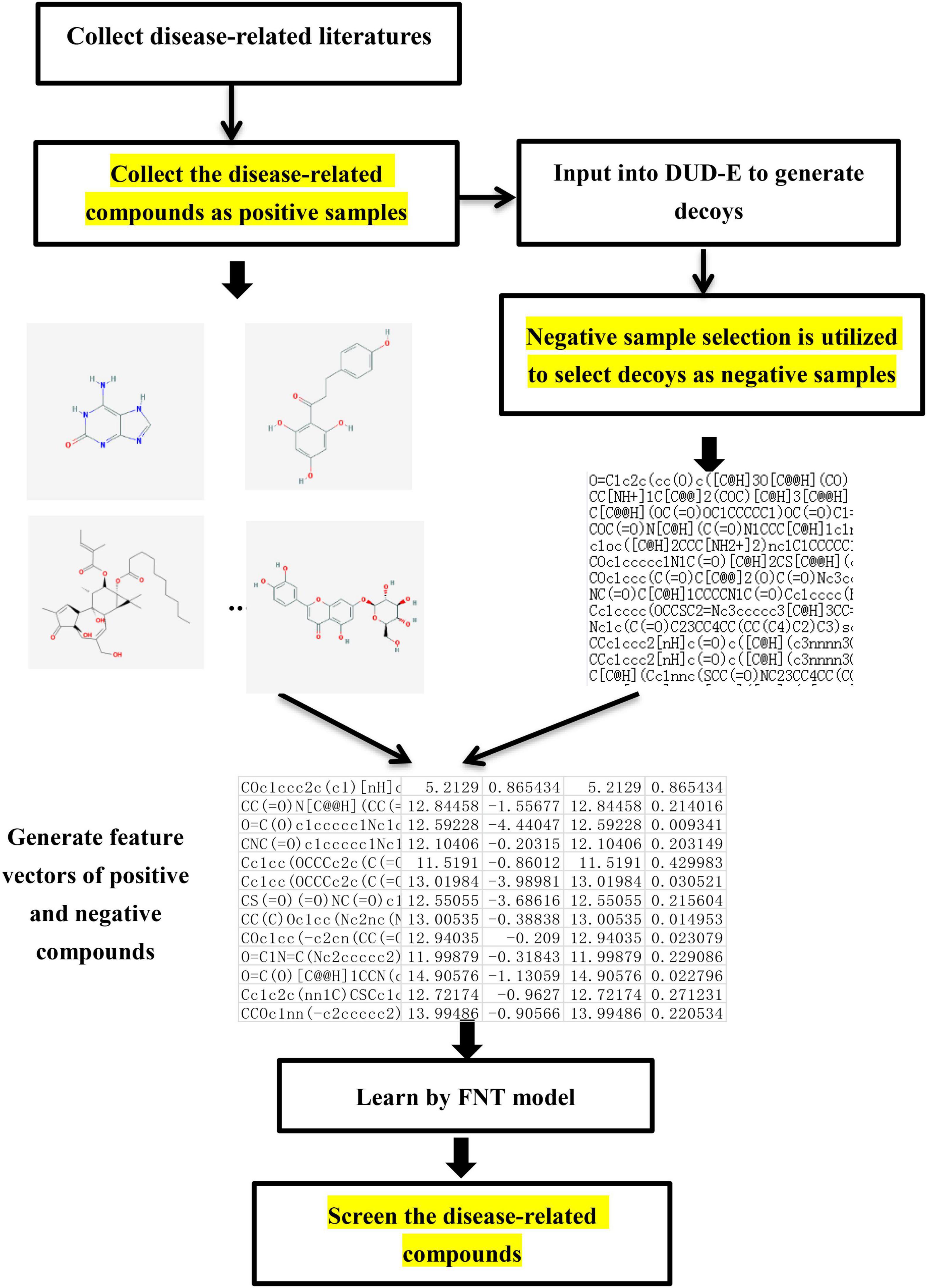
Figure 3. The flowchart of screening disease-related compounds algorithm.
(1) Disease-related compound dataset collection. Search the up-to-date literatures for treating diseases according to the name of disease. By consulting these literatures with data mining method, the active compounds for the treatment of the disease are collected as the positive compound samples. In order to generate the unrelated compounds, the positive compounds are input into DUD-E database to generate the corresponding decoys, which are set as negative samples (Mysinger et al., 2012). There are too many decoys generated compared to the number of positive samples. In order to balance the proportion of positive samples and negative samples, negative sample selection based on Tanimoto index (Algorithm 1) is presented to choose a certain number of decoys that are quite different from the positive sample set. Tanimoto index could measure the distance between the 2 compounds, which can measure the similarity between 2 sets (Klekota et al., 2005), which can solve the relationship between 0 and 1 well. The greater Tanimoto index is, the higher the similarity of 2 sets is. The Tanimoto index of 2 sets A and B is calculated as followed.
Algorithm 1. Negative sample selection algorithm.

(2) Screening process. The related and unrelated molecules collected are all chemical structures. To facilitate the compounds collected inputting into flexible neural tree model, 4 kinds of molecular descriptors (ECFP6, MACCS, Macrocycle, and RDKit) are utilized to numerically characterize the chemical structure of each compound (Todeschini and Consonni, 2009). ECFP6 contains 2,048 features, which denotes all possible molecular routes retrieved from the atom according to radius 3 and each bit denotes whether the special stator structure exists. MACCS contains 166 molecular characteristic sites, such as ISOTOPE, ATOMIC NO, 4M RING, and GROUP VIII. Macrocycle contains 1,613 features, which refer the information about the ring-size, sugars, and ester functional groups. RDK it contains 208 features, such as number of valence electros, number of radical electrons, charge information, and number of Aliphatic Carbocycles. Cross-validation method is utilized to divide the training and testing datasets to test the performance of our proposed method. With the feature vector of each compound in the training dataset as the input, flexible neural tree model is utilized to train with the feature datasets. A hybrid evolutionary method based on grammar-guided genetic programming and salp swarm algorithm is proposed to search the optimal structure and parameters of FNT model. For the unknown compounds of testing dataset, the feature vectors are used as the input of the optimal FNT model to obtain the output results. If the result is higher than 0.5, the compound is identified to be disease-related; otherwise, it is unrelated.
Experiment Results and Discussion
In order to test the effectiveness of our method, the important compounds were collected, which were involved in the treatment of hypertension, diabetes, and COVID-19. The related compounds of these 3 diseases are regarded as positive samples and the numbers of samples are 67, 124, and 88, respectively. Negative sample selection method is utilized to select the inactive compounds about hypertension, diabetes and COVID-19, and the numbers of negative samples are 134, 248, and 176, respectively. The 4 kinds of molecular descriptors (ECFP6, MACCS, Macrocycle, and RDKit) are utilized to numerically characterize related and unrelated compounds of diseases, respectively.
The 10-cross validation method is utilized to test the performance of our method. SVM (Hearst et al., 1998), RF (Breiman, 2001), AdaBoost (Collins et al., 2002), decision tree (DT) (Safavian and Landgrebe, 1991), GBDT (Zhang B. et al., 2019), KNN, logical regression (LR) (Collins et al., 2002), gc Forest (Zhou and Feng, 2017), forgeNet (Kong and Yu, 2020), and Naive Bayes (NB) (Kim et al., 2006)are also utilized to identify disease-related compounds of three diseases. In our method, operator set is set as F = { + 2, + 3, + 4, + 5}, population size is set as 30 and the maximum depth of tree is set as 5. In SVM, linear kernel function is selected. In RF, the number of trees is set as 100. In GBDT, the number of regression trees is set as 200. In DT, CART algorithm is utilized. The parameters of other algorithms are set by default. The AUC performances of 11 methods with the datasets about hypertension, diabetes, and COVID-19 are shown in Figures 4–6, respectively. From Figure 4, it could be seen that with ECFP6, Macrocycle, and RDKit methods, our method has the highest AUC performances among 11 methods. With MACCS method, the AUC values obtained by our method and RF are very close to 1.0, which are 0.999889 and 0.997772, respectively. For Figure 5, in terms of AUC, it could be clearly seen that our method performs best with ECFP6, MACCS, and RDKit methods. With Macrocycle feature method, our method, gcForest, and SVM could obtain the better AUC values than other 8 methods, which are 1, 0.99803, and 0.998435, respectively. By the comparison of these 3 methods, our method performs best, which show that our method is a good classifier for disease-compound identification problem. For Figure 6, with ECFP6molecular descriptor, our method and SVM could obtain the higher AUC values than other 9 methods, which are 0.996901 and 0.99703. With other molecular descriptors, our method could obtain the better performances, which are equal to or very close to 1.0.
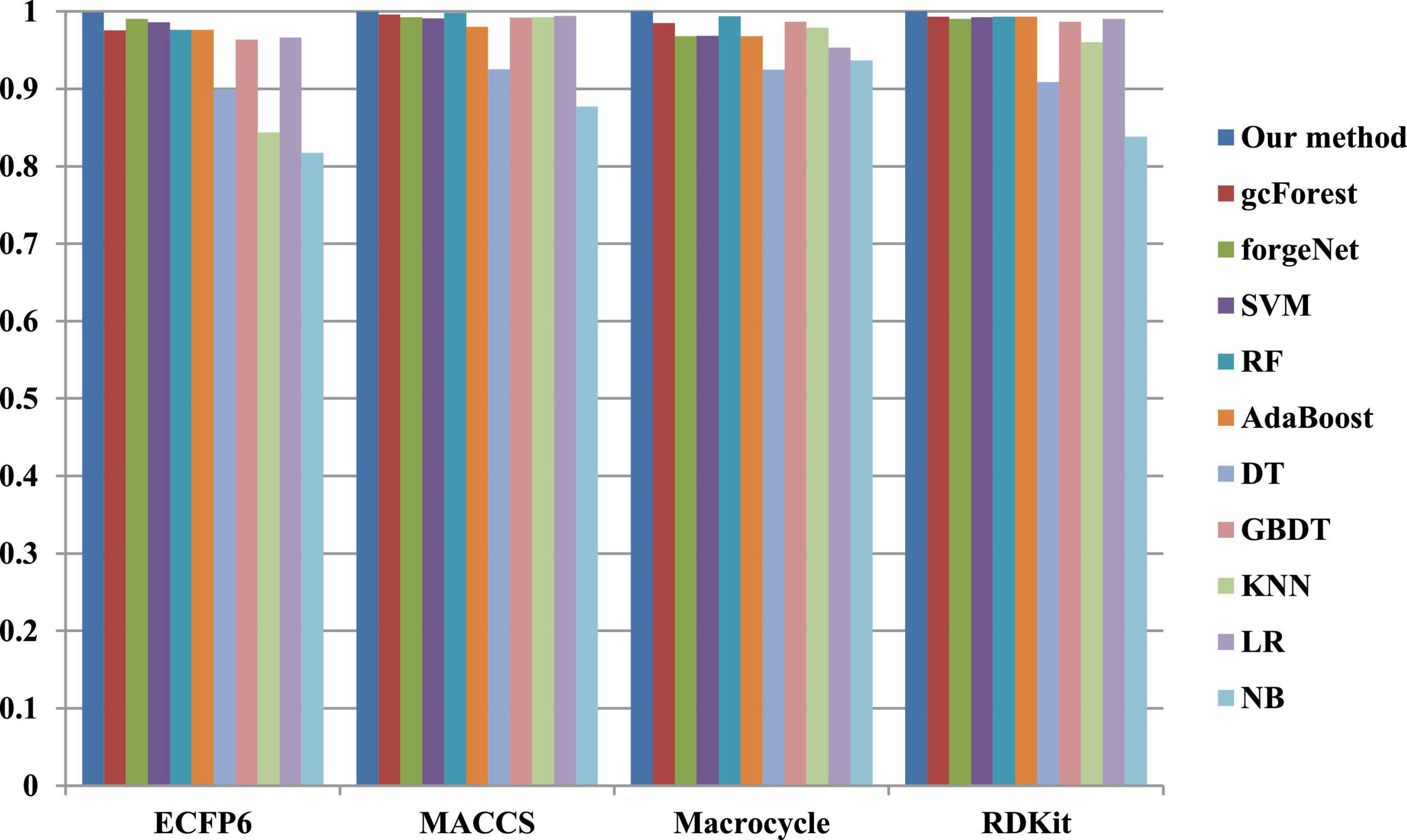
Figure 4. AUC performances of 11 methods with hypertension dataset.
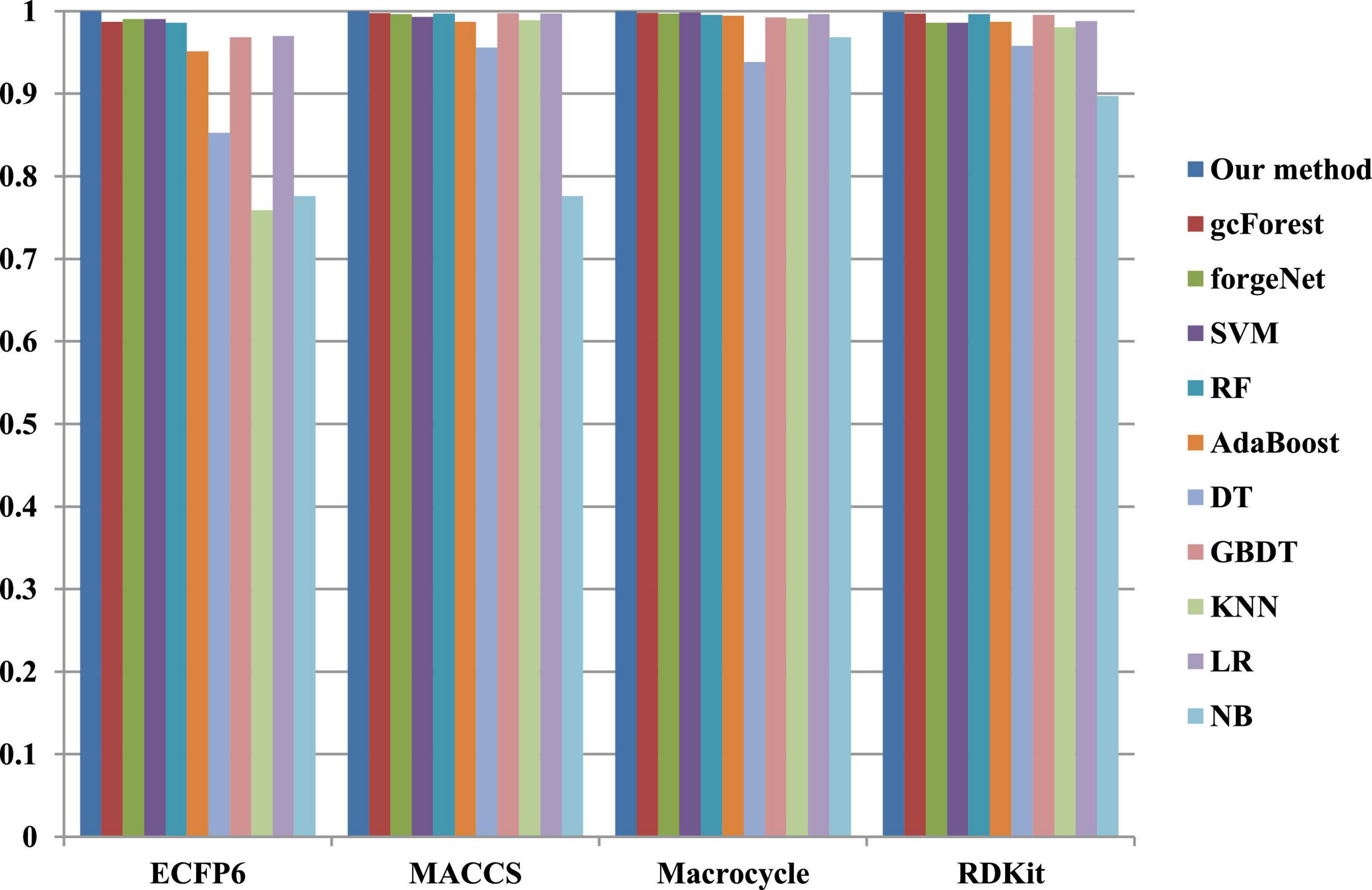
Figure 5. AUC performances of 11 methods with diabetes dataset.
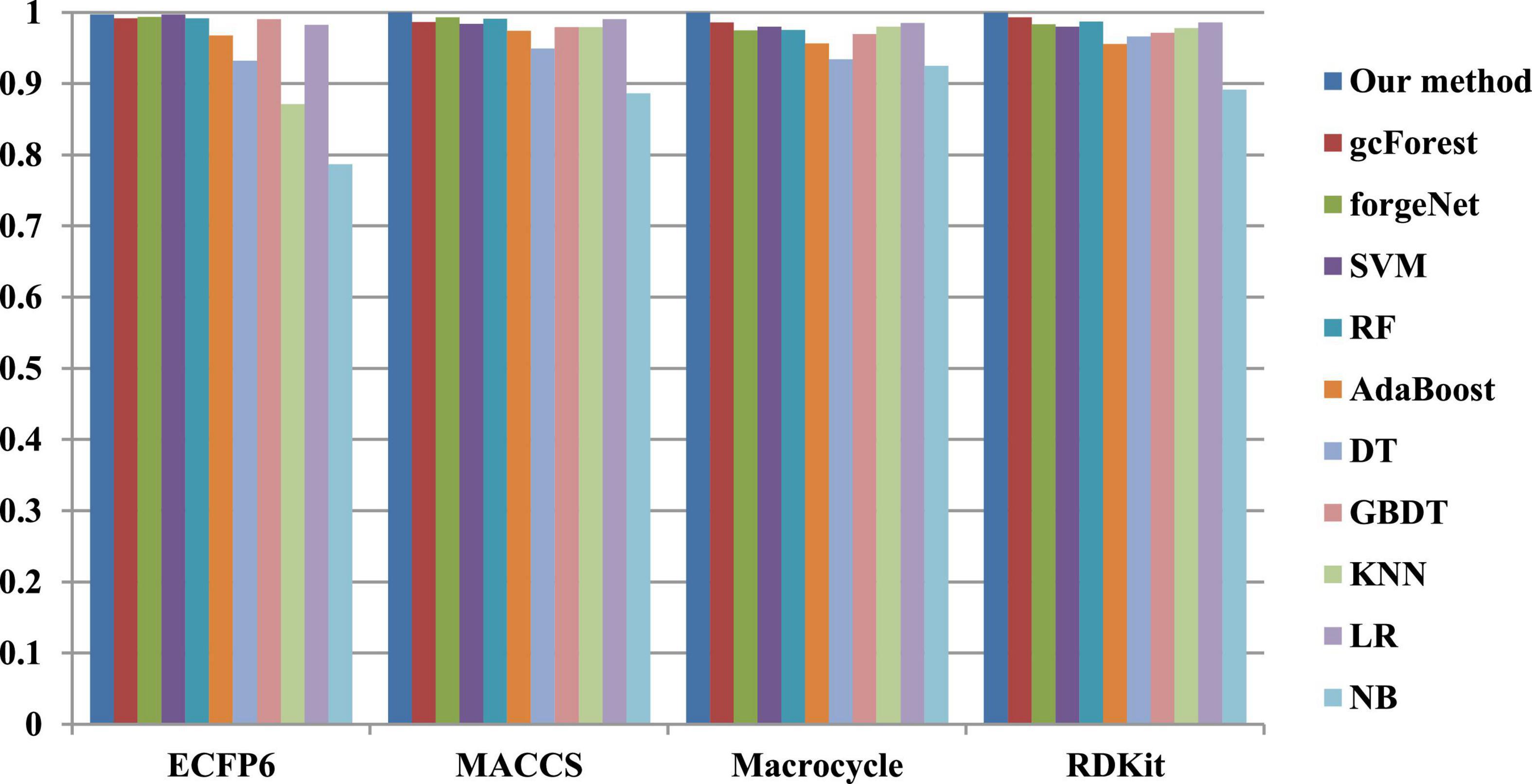
Figure 6. AUC performances of 11 methods with COVID-19 dataset.
TPR, FPR, Precision, Specificity, and F1 are also utilized to test the performances of 11 methods for compound identification about 3 diseases. TPR denotes the ratio of true disease-related compounds identified against all true disease-related ones. FPR denotes the ratio of disease-related compounds identified erroneously against all true disease-unrelated ones. Precision denotes the ratio of true disease-related compounds identified against all disease-related ones identified. Specificity is the ratio of true disease-unrelated compounds identified against all true disease-unrelated ones. F1 could evaluate a classifier comprehensively with Precision and Recall. TPR, FPR, Precision, Specificity, and F1performances of11 methods with the datasets about hypertension, diabetes and COVID-19 are listed in Tables 1–3, respectively. In Table 1, with ECFP6 method, our method has the highest TPR performance among 11 classifiers, which shows that our method could identify more true disease-related compounds. In terms of FPR, Precision and Specificity, forgeNet and RF perform best, which reveal that all the true disease-unrelated compounds are identified. But our method could obtain the highest F1 performance. Overall our method could obtain the more accurate identification results. With MACCS, Macrocycle, and RDKit, our method could obtain the best performances of TPR, FPR, Precision, Specificity, and F1.
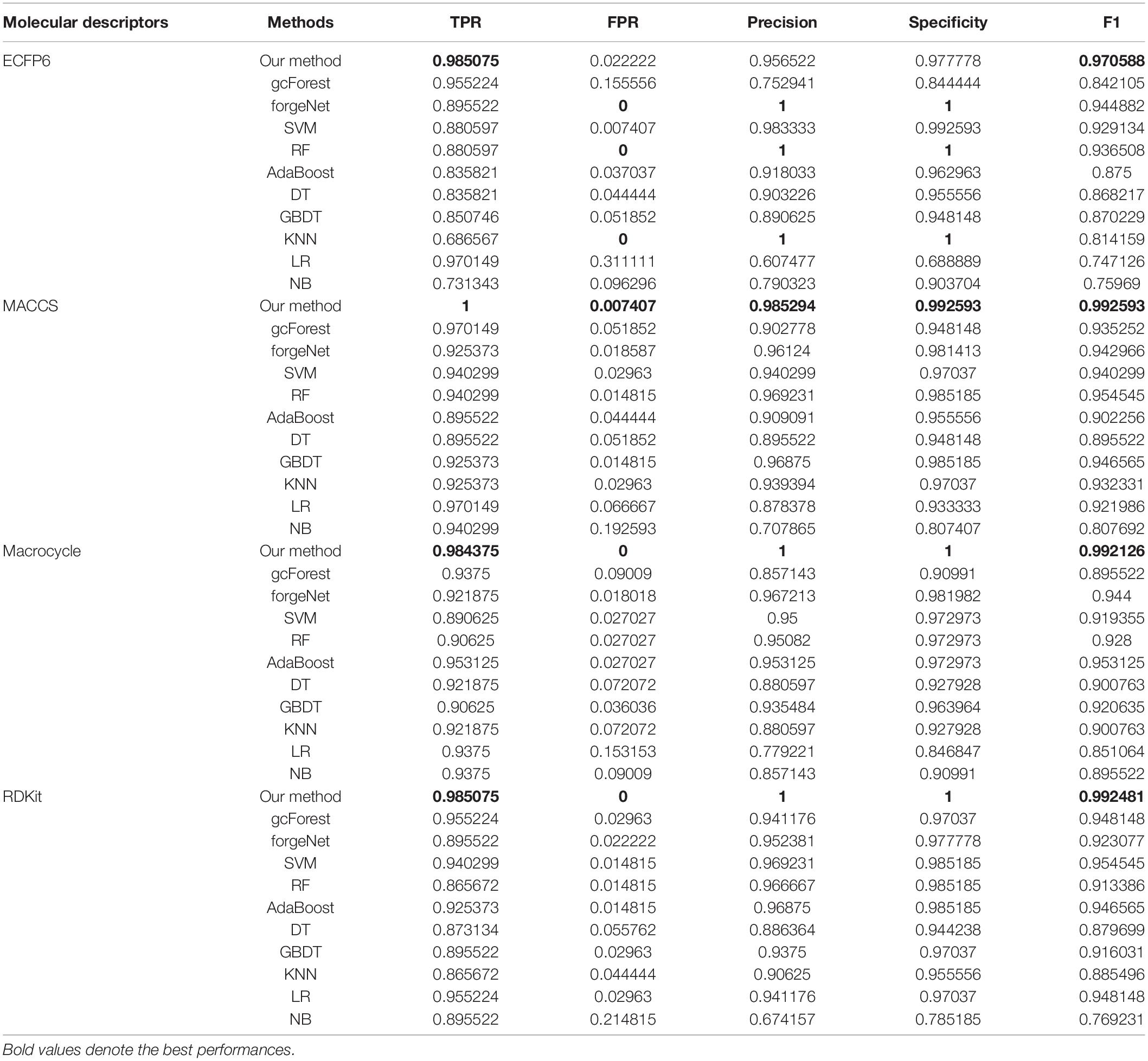
Table 1. Prediction performances of 11 methods with hypertension dataset.
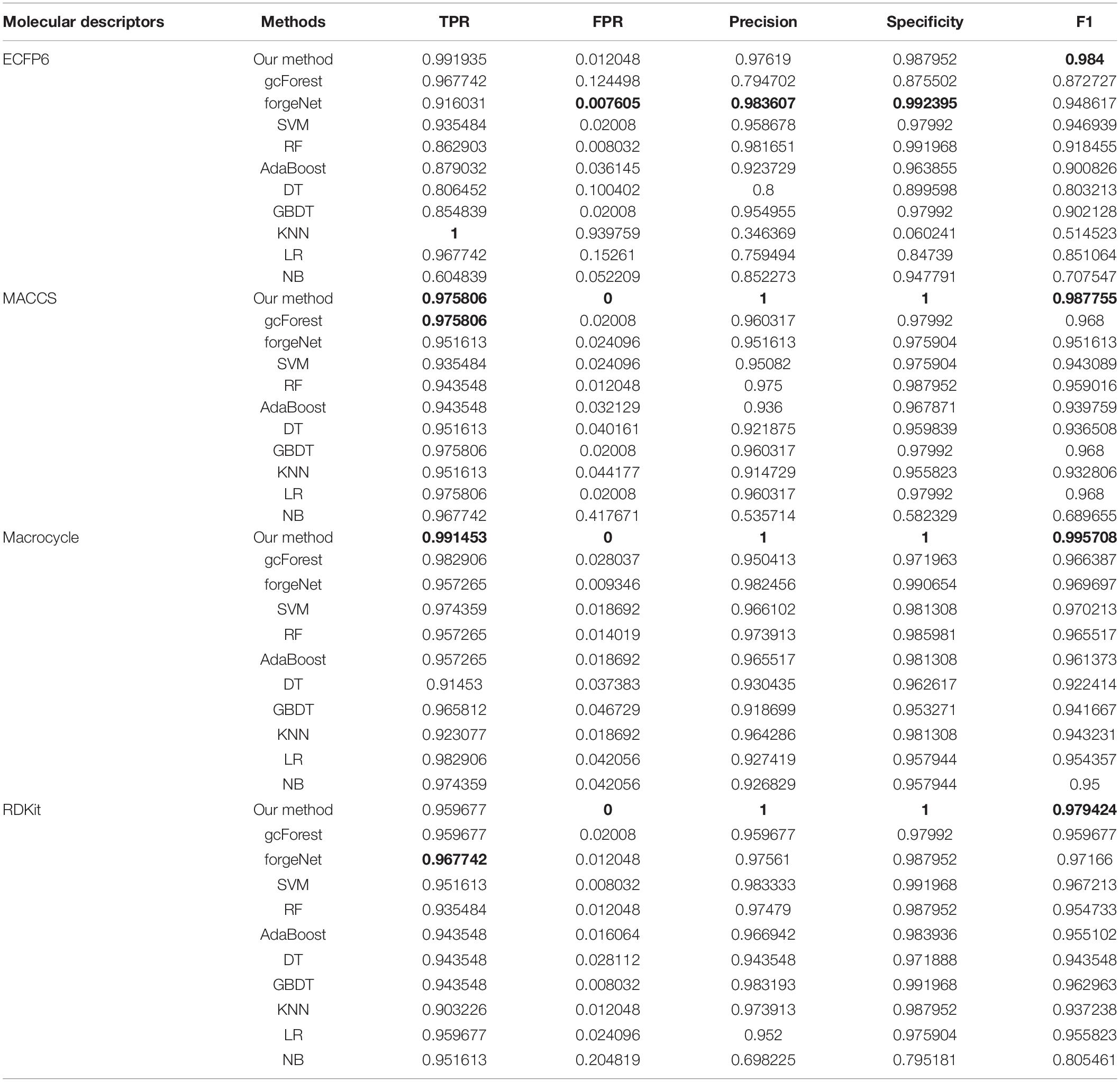
Table 2. Prediction performances of 11 methods with diabetes dataset.
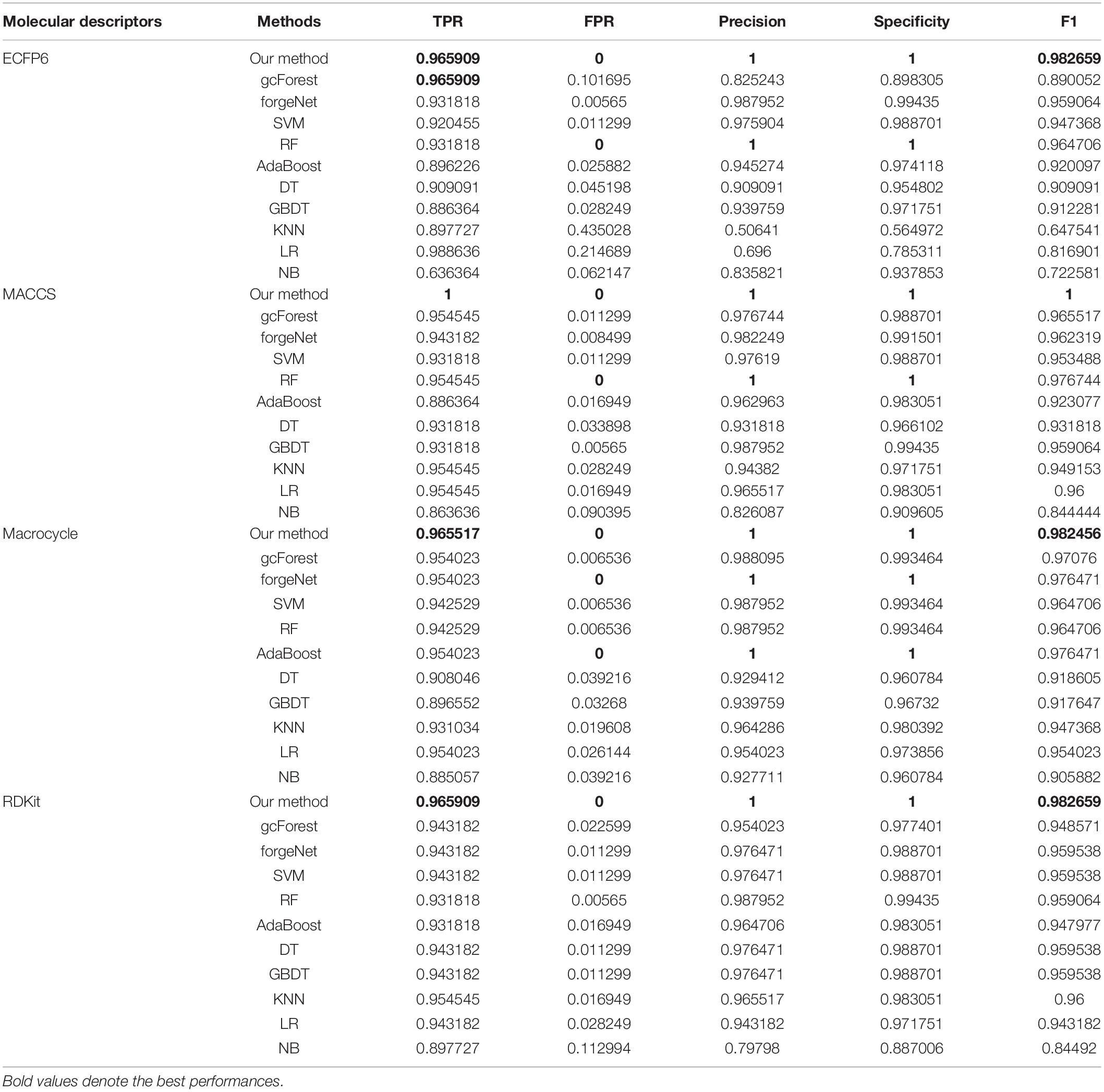
Table 3. Prediction performances of 11 methods with COVID-19 dataset.
In Table 2, with ECFP6 method, KNN has the highest TPR performance among 11 classifiers, which is 1.0. The result shows that KNN could identify all true disease-related compounds. In terms of FPR, Precision, and Specificity, forgeNet perform better than other 10 methods. But our method could also obtain the highest F1 performance. Overall our method could obtain the more accurate identification results. With MACCS and Macrocycle, our method could obtain the best performances of TPR, FPR, Precision, Specificity, and F1. With RDKit, our method performs best in terms of FPR, Precision, Specificity, and F1, while forgeNet could obtain the best TPR performance. For Table 3, our method performs best with 4 kinds of molecular descriptors in terms of 5 criterions. All results show that our method could predict disease-related compounds more accurately than gcForest, forgeNet, SVM, RF, AdaBoost, DT, GBDT, KNN, LR, and NB.
According to the performances of 11 methods with the datasets from 3 diseases and 4 molecular descriptors, 11 methods are ranked. For each molecular descriptor, the averaged ranking results of each method are listed in Table 4. From Table 4, we can see that our method, gcforest, forgenet, RF, GDBT, and LR perform best with MACCS feature set, while SVM and DT perform best with RDKit feature set. AdaBoost, KNN and NB perform better with Mordred feature set than the other 3 feature sets. All methods perform poorly with ECFP6 molecular descriptor. The results also show that the different molecular descriptors of compounds are suitable for the different classifiers and the ranking results can provide the guidance for each classifier to choose the appropriate molecular descriptor to solve the problem in the future. On the whole, MACCS method is suitable for the maximum number of classifiers. In future research, MACCS method can be preferred for a new classifier.
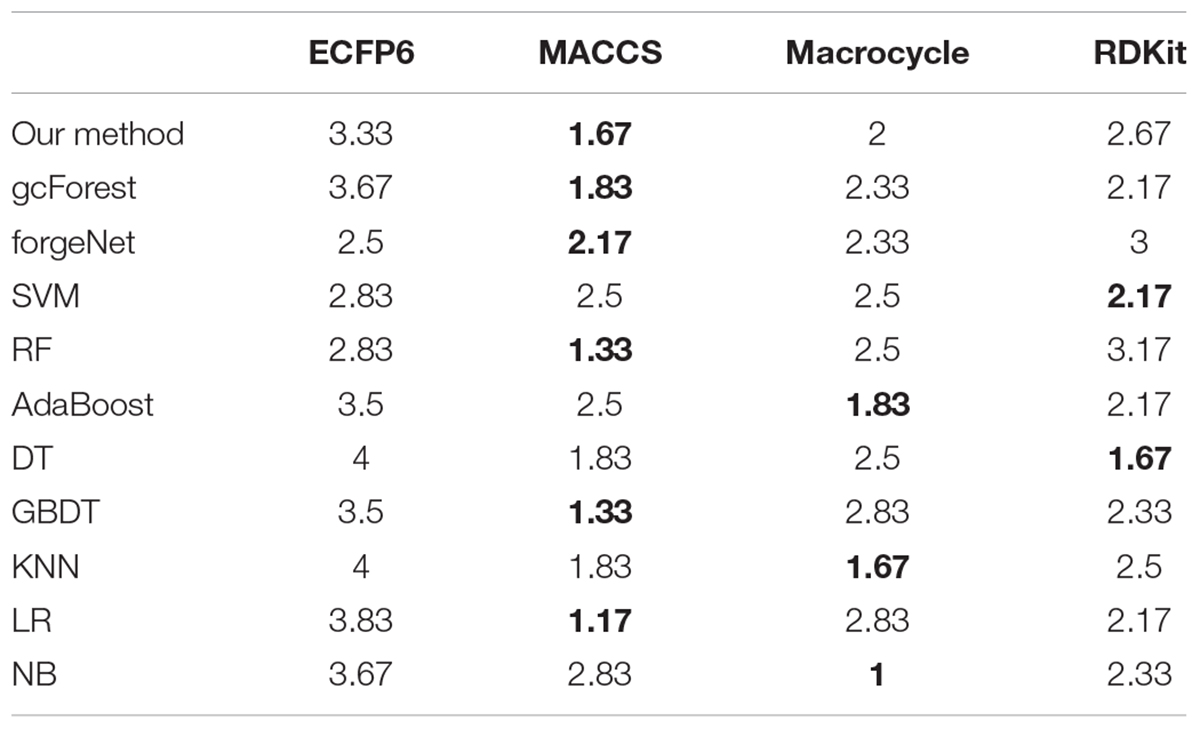
Table 4. Averaged ranking scores of 11 methods with 3 datasets.
We investigate the performances of our method with different ratios of positive and negative samples. The 8 kinds of ratios (1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:8, and 1:10) are selected and COVID-19 dataset is utilized. The identification results are depicted in Figure 7. From Figure 7, it could be seen that when the ratios are 1:1, 1:2, 1:3, and 1:4, our method could have the better ROC and AUC performances. The excessive imbalance of data may affect the classification performance of the algorithm.
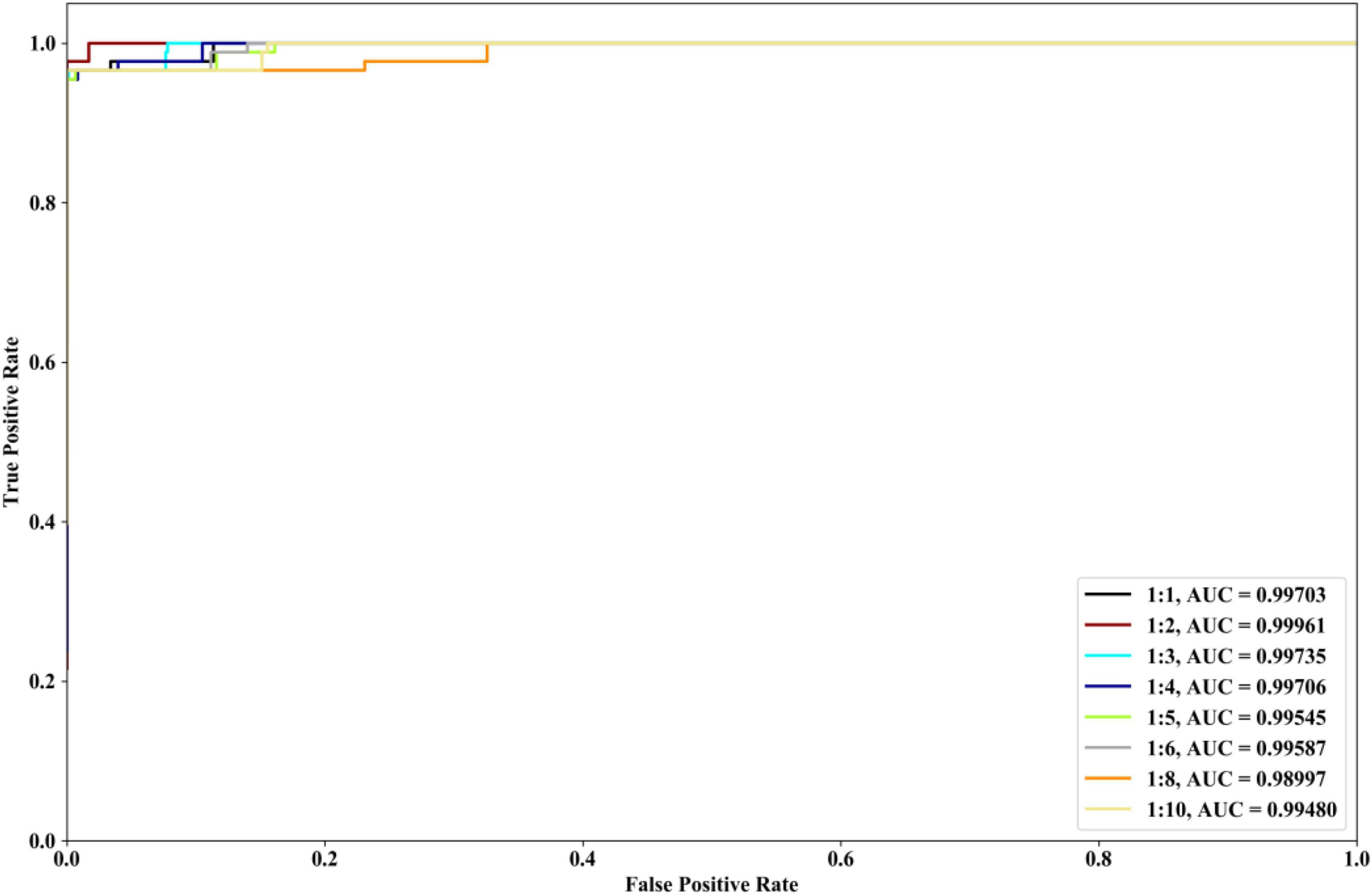
Figure 7. Performances of our method with COVID-19 dataset and the different ratios of positive and negative samples.
Conclusion
In order to sort the candidate compounds in a traditional Chinese medicine prescription and narrow the scope of analysis in network pharmacology research accurately, this paper proposes a new virtual screening method based on flexible neural tree (FNT) model, hybrid evolutionary method, and negative sample selection algorithm to screen the disease-related active compounds. 3 diseases (hypertension, diabetes, and Corona Virus Disease 2019) related compounds are collected from the up-to-date literatures. The unrelated compounds are selected by negative sample selection algorithm from DUD-E website. 4 kinds of molecular descriptors (ECFP6, MACCS, Macrocycle, and RDKit) are utilized to characterize the features of related and unrelated compounds of diseases, respectively. The experiment results show that our proposed method performs better than classical classifiers (SVM, RF, AdaBoost, DT, GBDT, KNN, LR, and NB), up-to-date classifier (gcForest) and deep learning method (forgeNet) in terms of AUC, ROC, TPR, FPR, Precision, Specificity, and F1.
We also investigate the performances of 11 methods with 4 kinds of molecular descriptors. The results show that our method, gcforest, forgenet, RF, GDBT, and LR perform best with MACCS feature set, while SVM and DT perform best with RDKit feature set, AdaBoost, KNN and NB perform best with Mordred feature set. With ECFP6 molecular descriptor all methods perform poorly.
In the paper, our proposed method has been successfully applied to hypertension, diabetes, and Corona Virus Disease. In the future, our method will be utilized to identify other chronic disorders related compounds, such as cancers, coronary heart disease, and rheumatoid disease.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
WB conceived the method and wrote the main manuscript text. BY designed the method and conducted the experiments. All authors reviewed the manuscript.
Funding
This work was supported by the talent project of “Qingtan scholar” of Zaozhuang University, the Natural Science Foundation of China (Nos. 61702445 and 61902337), Jiangsu Provincial Natural Science Foundation (No. SBK2019040953), Natural Science Fund for Colleges and Universities in Jiangsu Province (No. 19KJB520016), Young talents of Science and Technology in Jiangsu, Youth Innovation Team of Scientific Research Foundation of the Higher Education Institutions of Shandong Province, China (No. 2019KJM006), the Key Research Program of the Science Foundation of Shandong Province (No. ZR2020KE001), the fundamental Research Funds for the Central Universities (2020QN89), Xuzhou Science and Technology Plan Project (KC19142 and KC21047), the Ph.D. research startup foundation of Zaozhuang University (No. 2014BS13), and Zaozhuang University Foundation (No. 2015YY02).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Babaei, F., Lashkari, Z. B., Safari, A., Farrokhifar, M., Salehi, J., et al. (2020). Salp swarm algorithm-based fractional-order PID controller for LFC systems in the presence of delayed EV aggregators. IET Electr. Syst. Transport. 10, 259–267. doi: 10.1049/iet-est.2019.0076
Bajusz, D., Ferenczy, G. G., and Keser, G. M. (2017). Structure-Based Virtual Screening Approaches in Kinase-Directed Drug Discovery. Curr. Topics Med. Chem. 17, 2235–2259. doi: 10.2174/1568026617666170224121313
Baxter, C. A., Murray, C. W., Waszkowycz, B., Li, J., Sykes, R. A., Bone, R. G., et al. (2000). New approach to molecular docking and its application to virtual screening of chemical databases. J. Chem. Inform. Comput. Sci. 40, 254–262. doi: 10.1021/ci990440d
Berishvili, V. P., Voronkov, A. E., Radchenko, E. V., Palyulin, V. A., et al. (2018). Machine Learning Classification Models to Improve the Docking-based Screening: a Case of PI3K-Tankyrase Inhibitors. QSAR Combinator. Sci. 37:e1800030. doi: 10.1002/minf.201800030
Bustamam, A., Hamzah, H., Husna, N. A., Syarofina, S., Dwimantara, N., Yanuar, A., et al. (2021). Artificial intelligence paradigm for ligand-based virtual screening on the drug discovery of type 2 diabetes mellitus. J. Big Data 8:74. doi: 10.1186/s40537-021-00465-3
Chen, Y. F., Hsu, K. C., Lin, P. T., Hsu, D. F., Kristal, B. S., Yang, J. M., et al. (2011). LigSeeSVM: ligand-based virtual screening using support vector machines and data fusion. Int. J. Comput. Biol. Drug Design 4, 274–289. doi: 10.1504/IJCBDD.2011.041415
Chen, Y. H., Yang, B., and Meng, Q. (2012). Small-time scale network traffic prediction based on flexible neural tree. Appl. Soft Comput. 12, 274–279. doi: 10.1016/j.asoc.2011.08.045
Collins, M., Schapire, R. E., and Singer, Y. (2002). Logistic Regression, AdaBoost and Bregman Distances. Mach. Learn. 48, 253–285. doi: 10.1023/A:1013912006537
Fischer, N., Seo, E. J., Abdelfatah, S., Fleischer, E., Klinger, A., Efferth, T., et al. (2021). A novel ligand of the translationally controlled tumor protein (TCTP) identified by virtual drug screening for cancer differentiation therapy. Invest. N. Drugs 39, 914–927. doi: 10.1007/s10637-020-01042-w
Gomeni, R., BaniM, D., Angeli, C., Corsi, M., and Bye, A. (2001). Computer-assisted drug development (CADD): an emerging technology for designing first-time-in-man and proof-of-concept studies from preclinical experiments. Eur. J. Pharmaceut. Sci. 13, 261–270. doi: 10.1016/S0928-0987(01)00111-7
Guasch, L., Zakharov, A. V., Tarasova, O. A., Poroikov, V. V., Liao, C., Nicklaus, M. C., et al. (2016). Novel HIV-1 Integrase Inhibitor Development by Virtual Screening Based on QSAR Models. Curr. Topics Med. Chem. 16, 441–448. doi: 10.2174/1568026615666150813150433
Guo, S., Xie, H., Lei, Y., Liu, B., Zhang, L., Xu, Y., et al. (2021). Discovery of Novel Inhibitors Against Main Protease (Mpro) of SARS-CoV-2 via Virtual Screening and Biochemical Evaluation. Bioorgan. Chem. 110:104767. doi: 10.1016/j.bioorg.2021.104767
Hearst, M. A., Dumais, S. T., Osman, E., Platt, J., and Scholkopf, B. (1998). Support Vector Machines. IEEE Intell. Syst. 13, 18–28. doi: 10.1109/5254.708428
Kellenberger, E., Rodrigo, J., Muller, P., and Rognan, D. (2004). Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 57, 225–242. doi: 10.1002/prot.20149
Kim, S. B., Han, K. S., Rim, H. C., and Myaeung, S. H. (2006). Some Effective Techniques for Naive Bayes Text Classification. IEEE Transac. Knowledge Data Eng. 18, 1457–1466. doi: 10.1109/TKDE.2006.180
Klekota, J., Brauner, E., and Schreiber, S. L. (2005). Identifying Biologically Active Compound Classes Using Phenotypic Screening Data and Sampling Statistics. J. Chem. Inform. Modeling 45, 1824–1836. doi: 10.1021/ci050087d
Kong, Y., and Yu, T. (2020). forgeNet: a graph deep neural network model using tree-based ensemble classifiers for feature graph construction. Bioinformatics 36, 3507–3515. doi: 10.1093/bioinformatics/btaa164
Leelananda, S. P., and Steffen, L. (2016). Computational methods in drug discovery. Beilstein J. Organ. Chem. 12, 2694–2718. doi: 10.3762/bjoc.12.267
Maddah, M., Bahramsoltani, R., Yekta, N. H., Rahimi, R., Aliabadi, R., Pourfath, M., et al. (2021). Proposing high-affinity inhibitors from Glycyrrhiza glabra L. against SARS-CoV-2 infection: virtual screening and computational analysis. N. J. Chem. 45, 15977–15995. doi: 10.1039/D1NJ02031E
Maia, E. H. B., Assis, L. C., de Oliveira, T. A., da Silva, A. M., and Taranto, A. G. (2020). Structure-Based Virtual Screening: from Classical to Artificial Intelligence. Front. Chem. 8:343. doi: 10.3389/fchem.2020.00343
Meenakumari, K., Bupesh, G., Vasanth, S., Vasu, C. A., Pandian, K., Prabhu, K., et al. (2019). Molecular docking based virtual screening of carbonic anhydrase IX with coumarin (a cinnamon compound) derived ligands. Bioinformation 15, 744–749. doi: 10.6026/97320630015744
Meng, X. Y., Zhang, H. X., Mezei, M., and Cui, M. (2011). Molecular Docking: a Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided Drug Design 7, 146–157. doi: 10.2174/157340911795677602
Mirjalili, S., Gandomi, A. H., Mirjalili, S. Z., Saremi, S., Faris, H., Mirajalili, M. S., et al. (2017). Salp swarm algorithm: a bio-inspired optimizer for engineering design problems. Adv. Eng. Soft. 114, 163–191. doi: 10.1016/j.advengsoft.2017.07.002
Morris, G. M., Goodsell, D. S., Huey, R., and Olson, A. J. (1996). Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J. Mol. Recogn. 10, 293–304. doi: 10.1007/BF00124499
Mysinger, M. M., Carchia, M., Irwin, J. J., and Shoichet, B. K. (2012). Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 55:6582. doi: 10.1021/jm300687e
Olubiyi, O. O., Olagunju, M., Keutmann, M., Loschwitz, J., and Strodel, B. (2020). High Throughput Virtual Screening to Discover Inhibitors of the Main Protease of the Coronavirus SARS-CoV-2. Molecules 25:3193. doi: 10.3390/molecules25143193
Rajguru, T., Bora, D., and Modi, M. K. (2020). Combined CADD and Virtual Screening to Identify Novel Nonpeptidic Falcipain-2 Inhibitors. Curr. Comput. Drug Design 17, 579–588. doi: 10.2174/1573409916666200701213526
Ren, H., Li, J., Chen, H., Li, C. Y., et al. (2021). Adaptive levy-assisted salp swarm algorithm: analysis and optimization case studies. Mathemat. Comput. Simul. 181, 380–409. doi: 10.1016/j.matcom.2020.09.027
Safavian, S. R., and Landgrebe, D. (1991). A survey of decision tree classifier methodology. IEEE Transac. Syst. Man, Cybernet. 21, 660–674. doi: 10.1109/21.97458
Selvaraj, C., Panwar, U., Dinesh, D. C., Boura, E., Singh, P., Dubey, V. K., et al. (2021). Microsecond MD Simulation and Multiple-Conformation Virtual Screening to Identify Potential Anti-COVID-19 Inhibitors Against SARS-CoV-2 Main Protease. Front. Chem. 8:595273. doi: 10.3389/fchem.2020.595273
Talluri, S. (2021). Molecular Docking and Virtual Screening based prediction of drugs for COVID-19. Comb Chem. High Throughput Screen 24, 716–728. doi: 10.2174/1386207323666200814132149
Taufer, M., Crowley, M., Price, D. J., Chien, A. A., and Brooks, C. L. III, et al. (2005). Study of a highly accurate and fast protein-ligand docking method based on molecular dynamics. Concurr. Comput. 14, 1627–1641. doi: 10.1002/cpe.949
Thiyagarajan, V., Lin, S. H., Chang, Y. C., Weng, C. F., et al. (2016). Identification of novel FAK and S6K1 dual inhibitors from natural compounds via ADMET screening and molecular docking. Biomed. Pharmacother. 80, 52–62. doi: 10.1016/j.biopha.2016.02.020
Todeschini, R., and Consonni, V. (2009). Molecular Descriptors for Chemoinformatics. Weinheim: Wiley-VCH. doi: 10.1002/9783527628766
Tong, J., Qin, S., and Jiang, G. (2019). 3D-QSAR Study of Melittin and Amoebapore Analogues by CoMFA and CoMSIA Methods. Chin. J. Struct. Chem. 2, 201–210.
Wang, M. Y., Peng, L., and Qiao, P. L. (2016). The Virtual Screening of the Drug Protein with a Few Crystal Structures Based on the Adaboost-SVM. Comput. Math Methods Med. 2016:4809831. doi: 10.1155/2016/4809831
Wu, P., and Chen, Y. (2007). “Grammar Guided Genetic Programming for Flexible Neural Trees Optimization,” in Advances in Knowledge Discovery and Data Mining. PAKDD 2007. Lecture Notes in Computer Science(), Vol. 4426, eds Z. H. Zhou, H. Li, and Q. Yang (Berlin, Heidelber: Springer).
Yang, Z., Zhou, Y., and Zhong, L. (2021). Discovery of BAZ1A bromodomain inhibitors with the aid of virtual screening and activity evaluation. Bioorganic Med. Chem. Lett. 33:127745. doi: 10.1016/j.bmcl.2020.127745
Zaki, M. E. A., Alhussain, S. A., Masand, V. H., Akasapu, S., Bajaj, S. O., Ghosh, A., et al. (2021). Identification of Anti-SARS-CoV-2 Compounds from Food Using QSAR-Based Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Analysis. Pharmaceuticals 14:357. doi: 10.3390/ph14040357
Zaslavskiy, M., Jégou, S., and Tramel, E. W. (2019). ToxicBlend: virtual screening of toxic compounds with ensemble predictors. Computat. Toxicol. 10, 81–88. doi: 10.1016/j.comtox.2019.01.001
Zhang, B., Ren, J., Cheng, Y., Wang, B., Wei, Z., et al. (2019). Health Data Driven on Continuous Blood Pressure Prediction based on Gradient Boosting Decision Tree Algorithm. IEEE ACCESS 7, 32423–32433. doi: 10.1109/ACCESS.2019.2902217
Zhang, L., Ai, H. X., Li, S. M., Qi, M. Y., Zhao, J., Zhao, Q., et al. (2017). Virtual screening approach to identifying influenza virus neuraminidase inhibitors using molecular docking combined with machine-learning-based scoring function. Oncotarget 8, 83142–83154. doi: 10.18632/oncotarget.20915
Zhang, Y., Wang, Y., Zhou, W., Fan, Y., Zhao, J., Zhu, L., et al. (2019). A combined drug discovery strategy based on machine learning and molecular docking. Chem. Biol. Drug Design 93, 685–699. doi: 10.1111/cbdd.13494
Zheng, Y., Kong, L., Jia, H., Zhang, B., Wang, Z., Xu, L., et al. (2020). Network pharmacology study on anti-stroke of Xiaoshuan Tongluo formula based on systematic compound-target interaction prediction models. Acta Pharmaceut. Sin. 55, 256–264.
Zhou, Y., Zhang, B., Lin, Z. J., Zhang, X. M., Li, F., Wang, H. G., et al. (2016). Virtual screening for components in Chicory combined with CNT2 target based on molecular docking. Zhongguo Zhong Yao Za Zhi 41, 3962–3967.
Keywords: virtual screening, network pharmacology, flexible neural tree, grammar-guided genetic programming, salp swarm algorithm
Citation: Yang B, Bao W and Chen B (2022) Disease-Ligand Identification Based on Flexible Neural Tree. Front. Microbiol. 13:912145. doi: 10.3389/fmicb.2022.912145
Received: 15 March 2022; Accepted: 06 May 2022;
Published: 06 June 2022.
Edited by:
Liang Wang, Xuzhou Medical University, ChinaReviewed by:
Chun-Chun Wang, China University of Mining and Technology, ChinaChandrabose Selvaraj, Alagappa University, India
Copyright © 2022 Yang, Bao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenzheng Bao, YmFvd3o1NTU1NUAxMjYuY29t