 Qin Qi
Qin Qi Vaheesan Rajabal
Vaheesan Rajabal Timothy M. Ghaly
Timothy M. Ghaly Sasha G. Tetu
Sasha G. Tetu Michael R. Gillings
Michael R. Gillings- 1School of Natural Sciences, Macquarie University, Sydney, NSW, Australia
- 2ARC Centre of Excellence in Synthetic Biology, Macquarie University, Sydney, NSW, Australia
Bacteriophages are versatile mobile genetic elements that play key roles in driving the evolution of their bacterial hosts through horizontal gene transfer. Phages co-evolve with their bacterial hosts and have plastic genomes with extensive mosaicism. In this study, we present bioinformatic and experimental evidence that temperate and virulent (lytic) phages carry integrons, including integron-integrase genes, attC/attI recombination sites and gene cassettes. Integrons are normally found in Bacteria, where they capture, express and re-arrange mobile gene cassettes via integron-integrase activity. We demonstrate experimentally that a panel of attC sites carried in virulent phage can be recognized by the bacterial class 1 integron-integrase (IntI1) and then integrated into the paradigmatic attI1 recombination site using an attC x attI recombination assay. With an increasing number of phage genomes projected to become available, more phage-associated integrons and their components will likely be identified in the future. The discovery of integron components in bacteriophages establishes a new route for lateral transfer of these elements and their cargo genes between bacterial host cells.
1. Introduction
Bacteriophages are viruses that infect bacteria and are characterized by the sheer diversity, plasticity and mosaic nature of their genomes (Westmoreland et al., 1969; Hershey, 1971; Pedulla et al., 2003; Brüssow et al., 2004; Hatfull and Hendrix, 2011; Mavrich and Hatfull, 2017). As versatile mobile genetic elements, phages play important roles in mediating horizontal gene transfer (HGT), driving the evolution of their bacterial hosts (Zinder and Lederberg, 1952; Lennox, 1955; McDaniel et al., 2010; Villa et al., 2019) and maintaining bacterial population diversity (Buckling and Rainey, 2002; Pal et al., 2007; Rodriguez-Valera et al., 2009; Koskella and Brockhurst, 2014). Genetic materials are exchanged between phages and their bacterial hosts via three modes: generalized, specialized and lateral transduction (Zinder and Lederberg, 1952; Lennox, 1955; Parkinson, 2016; Chen et al., 2018; Chiang et al., 2019). Phage-mediated HGT can significantly alter their hosts by increasing their virulence (Wagner and Waldor, 2002), spreading genes that confer new traits, and switching between alternative bacterial lifestyles (Rice et al., 2009; Xia and Wolz, 2014). Phages co-evolve with their bacterial hosts, for example, by acquiring methyltransferases and anti-CRISPR genes that allow them to evade bacterial immune responses (Murphy et al., 2013; Landsberger et al., 2018).
Phages are broadly classified as temperate or virulent (lytic) phages (Lwoff, 1953; Adams, 1959; Howard-Varona et al., 2017; Kirsch et al., 2021). Temperate phages can switch between lytic and lysogenic cycles of replication (Barksdale and Arden, 1974; Cheong et al., 2022). During lysis, phages replicate rapidly in the bacterial cytoplasm before lysing their hosts to release progeny phages that can infect more bacteria (Howard-Varona et al., 2017). During lysogeny, latent phages exist as prophages that are integrated into bacterial genomes. Most prophage genes are thought to be silent, but some genes may be expressed and confer new phenotypes upon their hosts via lysogenic conversion (Freeman, 1951; Boyd, 1954; Holloway and Cooper, 1962; Howard-Varona et al., 2017). In contrast, virulent phages, also known more specifically as obligate lytic phages, can only undergo lytic cycles of infection (Brüssow, 2005).
Phages can contribute to the spread of antimicrobial resistance (AMR) genes (Colavecchio et al., 2017; Debroas and Siguret, 2019; Cook et al., 2021), but the extent to which this occurs in natural settings is subject to ongoing debate (Enault et al., 2017; Borodovich et al., 2022). A recent study demonstrated for the first time that AMR genes of class 1 integrons can be found in naturally occurring phage-plasmids that exhibit hybrid properties of both temperate phages and plasmids (Moura de Sousa et al., 2021; Pfeifer et al., 2022). Integrons are genetic elements that can capture, express and re-arrange mobile gene cassettes in diverse bacterial phyla (Stokes and Hall, 1989; Mazel, 2006; Gillings, 2014; Escudero et al., 2015; Ghaly et al., 2020, 2021a). Each complete integron has an integron-integrase gene (intI), which encodes a site-specific tyrosine recombinase that catalyses the capture and excision of exogenous gene cassettes into the integron recombination site, attI (Hall and Stokes, 1993; Recchia et al., 1994; Escudero et al., 2015). Gene cassettes are circular, non-replicative genetic elements that typically carry an open reading frame (ORF) and a cassette recombination site (attC) that can be recombined with attI or other attC sites in integron gene cassette arrays. Class 1 integrons, which are associated with a highly conserved integron-integrase gene (intI1) and a well-defined attI1 recombination site, are disseminated by mobile genetic elements such as plasmids and transposons (Stokes et al., 1997; Partridge et al., 2000; Gillings et al., 2008). Generally, the spread of class 1 integrons correlates with antimicrobial use and anthropogenic pollution (Gillings et al., 2015; Gillings, 2017). The recent discovery of class 1 integrons in phage-plasmids suggests a previously unrecognized route through which phages can mediate the spread of gene cassette-associated AMR genes (Pfeifer et al., 2022).
In this study, we detect complete integrons or integron components in phage genomes, including examples of CALINs (clusters of attCs lacking an associated integron-integrase) in virulent phages that infect environmental bacterial species. Using attC x attI recombination assays in Escherichia coli, we showed that phage-borne attC sites can be recognized by IntI1 and integrated into the paradigmatic attI1 site of class 1 integrons. These results suggest that phage-borne gene cassettes have the potential to be recruited into bacterial integrons and vice versa, which represents an underexplored route of HGT between phages and bacteria.
2. Materials and methods
2.1. Bioinformatic detection of integrons and attC sites in phage genomes
All bacteriophage genomes with the descriptions “complete genomes” or “genomic sequences” were downloaded from the NCBI Genome database (last accessed on May 1, 2022). “Unverified,” “partial” or “incomplete” phage genomes were excluded. IntegronFinder 2.0 was used to detect complete integrons, CALINs (clusters of attCs lacking an associated integron-integrase) and In0 elements (integron-integrase that does not carry any gene cassettes) in the 10,705 downloaded phage genomes, using default parameters (Neron et al., 2022). In addition, every phage genome was screened for integron attC sites using a previously described HattCI + INFERNAL pipeline (Nawrocki and Eddy, 2013; Pereira et al., 2016; Ghaly et al., 2022a). For the HattCI + INFERNAL analysis, the minimum bit-score threshold was set to 20, and a minimum of two predicted attC sites were required in each candidate CALIN according to previously published methods (Ghaly et al., 2022a). All predicted attC sites that were located within 3 kB of another attC site were used in subsequent analysis. Annotations of attC sites were based on the results from both sets of bioinformatic predictions (i.e., IntegronFinder and HattCI + INFERNAL). ORF annotations were based on IntegronFinder results and the original GenBank annotations for the respective phages from NCBI. An in-house script attC-taxa.sh1 was used to classify the phage-associated attC sites according to sequence and structural homologies of chromosomal attC sequences in 11 bacterial taxa (Ghaly et al., 2022a).
2.2. Construction of attC donor strains for attC x attI1 suicide conjugation assays
To confirm that a panel of virulent phage-borne attC sites can be recognized by class 1 integron integrase (IntI1) and recombined into the attI1 site, the bottom strands of six attC sites (two each from T. jenkinsii phage TJE1, Pseudoalteromonas marina phage PH101 and Polaribacter phage P12002L) were cloned into the mobilisable suicide vector pJP5603 (Riedel et al., 2013). The attCaadA7 is frequently used as a benchmark positive control sequence in attC x attI1 suicide conjugation assays (Bouvier et al., 2005; Biskri et al., 2005; Vit et al., 2021). Overlapping long primers (synthesized by Sigma-Aldrich, United States) were annealed and ligated to the XbaI/BamHI restriction sites of pJP5603 (Supplementary Table S3). The ligation products were electroporated into the DAP auxotrophic E. coli WM3064 λpir strain. The attC donor strains were then isolated on LB agar selective media containing kanamycin (Km) and diaminopimelic acid (DAP). To generate attC donor plasmids that deliver the top strands of the respective attC sites, the same pairs of overlapping long primers were annealed and ligated to the XbaI/BamHI restriction sites of the pJP5603rev vector with its oriT in an inverted orientation relative to that of the pJP5603 parental vector (Ghaly et al., 2022b).
2.3. Quantification of attC x attI1 recombination frequencies
To quantify the recombination frequencies between candidate phage-borne attC sites and attI, we performed an attC x attI recombination assay similar to those used in previous works (Bouvier et al., 2005, 2009; Vit et al., 2021). An L-arabinose-inducible, intI1-expressing E. coli recipient strain from a previous study (Ghaly et al., 2022b; UB5201 + pBAD24::intI1 + pACYC184::attI1; carbenicillin (Cb) and chloramphenicol (Cm) resistant) was individually filter-mated in DAP-supplemented LB broth with one of the seven attC donor strains (Km resistant; attC sequences shown in Supplementary Table S3). IntI1 expression was induced using L-arabinose at 2 mg/ml or suppressed with D-glucose at 10 mg/ml in the negative control set. After 6 h of incubation at 37°C, the resuspended conjugation mix was plated on DAP-free LB agar containing Km. This allowed negative selection of the donor strain (which is not viable in the absence of DAP) and positive selection of the recombinant recipient clones that acquired Km-resistance following co-integration with the attC donor plasmids. To quantify the number of potential recipient strain cells, the resuspended conjugation mix was also plated onto DAP-free LB agar containing Cb.
Recombination frequencies were calculated by dividing the colony forming units (CFU) of Km-resistant recombinants by that of the total number of Cb-resistant recipients after 2 days of incubation. The assays were performed in three biological replicates, and recombination frequencies were calculated as the mean of the three independent experiments. One-way ANOVA was used to compare the means of the recombination frequencies for the bottom strands. To verify the sequence of the attI1/attC recombination junctions, colony PCR was performed on eight randomly chosen colonies per conjugation per biological replicate using GoTaq polymerase (Promega Inc., United States) and the primer pairs pACYC_F (5’-GAACCTTCGAAAAACCGCCC-3′) / M13R (5’-GCGGATAACAATTTCACACAGG-3′) and pACYC_R (5’-ACAGTACTGCGATGAGTGGC-3′) / M13F (5’-TGTAAAACGACGGCCAGT-3′). The thermocycler settings for the colony PCR were as follows: 95°C for 5 min (cell lysis and initial denaturation), 35 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, and 72°C for 5 min. The PCR products were Sanger sequenced for four recombinant colonies per conjugation set (Macrogen Inc., South Korea).
2.4. Identification of phages with plasmid replicons
PlasmidFinder (v2.1.6) was used to screen for plasmid replicons in phage genomes that contain integrons or integron components (Carattoli et al., 2014). The BLASTn method with default parameters was used to search the phage genomes against the nucleotide sequences of 116 replicons in the current version of the PlasmidFinder database (last accessed on December 12, 2022).
3. Results and discussion
We screened 10,705 bacteriophage genomes from the NCBI Genome database for integrons and attC recombination sites using previously described IntegronFinder and HattCI + INFERNAL bioinformatic pipelines (Nawrocki and Eddy, 2013; Pereira et al., 2016; Ghaly et al., 2021b; Neron et al., 2022). We detected 3 complete integrons and an integron-integrase without gene cassettes (ln0 element) residing in temperate phage genomes (Table 1), and 17 CALINs obtained from virulent, temperate, and uncharacterized phages (Table 2 and Supplementary Table S1). The genetic contexts of the predicted integrons and CALINs shown in Figures 1, 2 in the phage genomes are summarized in Supplementary Table S2. The annotations for all the predicted integrons and integron components in these phage genomes are available in Supplementary File S1.
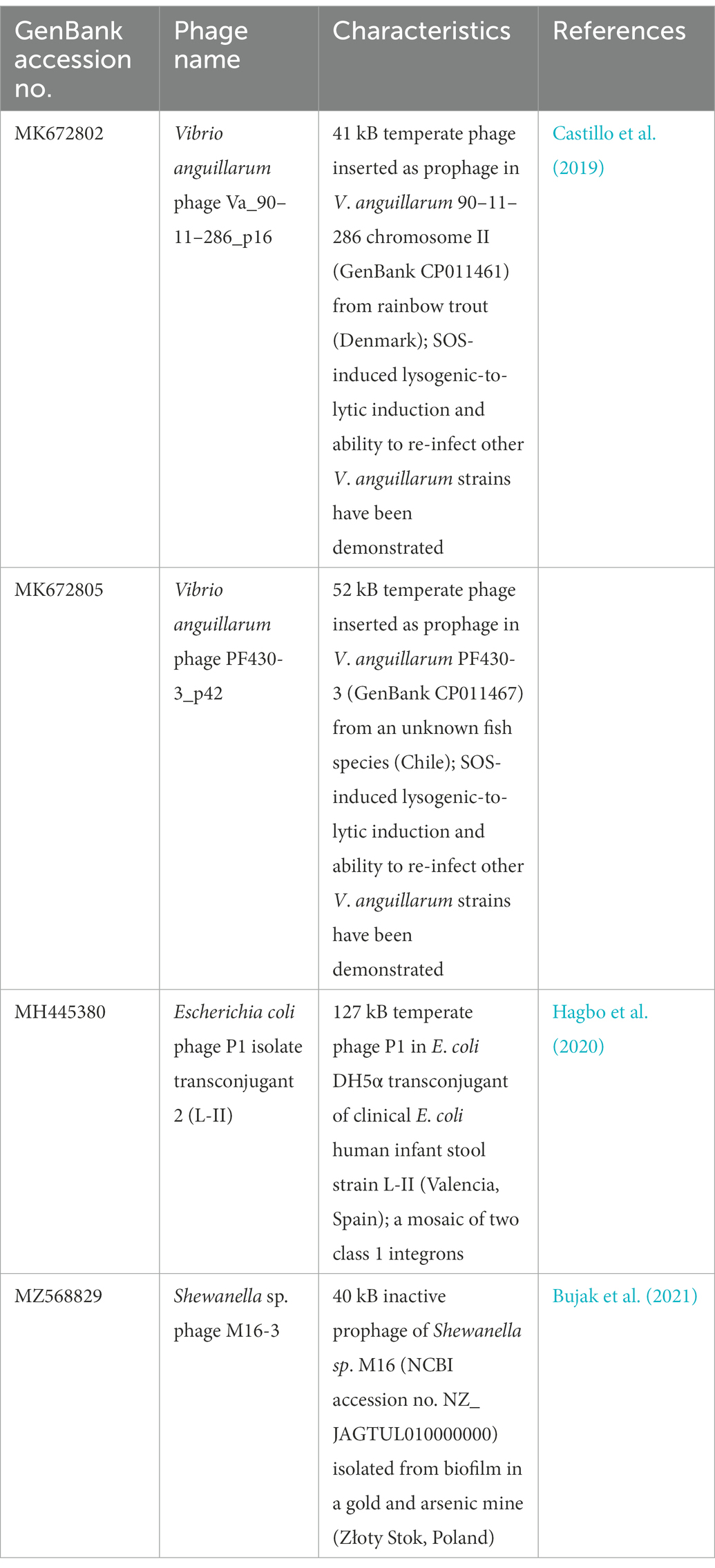
Table 1. Temperate phage genomes with complete integrons and In0 element (integron-integrase that does not carry gene cassettes) as predicted by IntegronFinder (Neron et al., 2022).
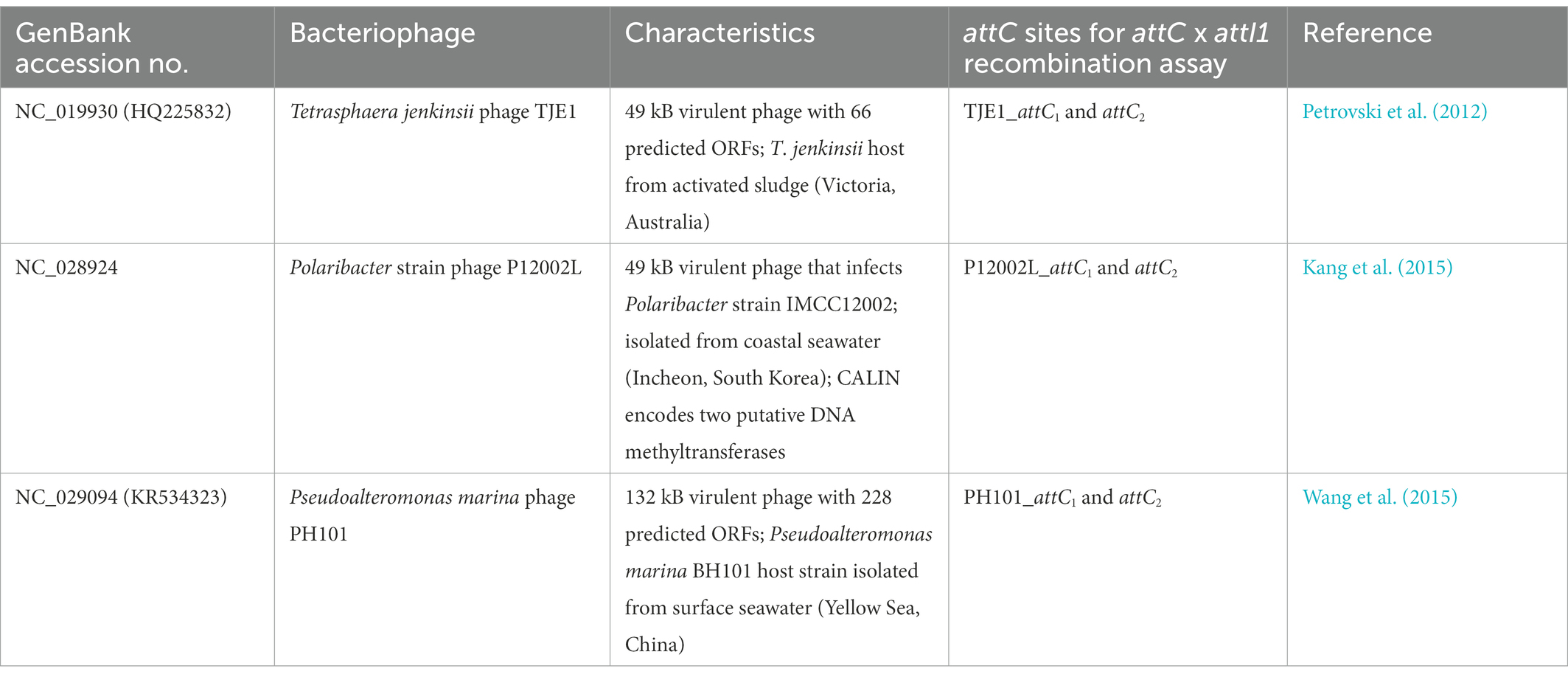
Table 2. Experimentally isolated and characterized virulent phages that contain CALINs with predicted gene cassettes and attC sites.
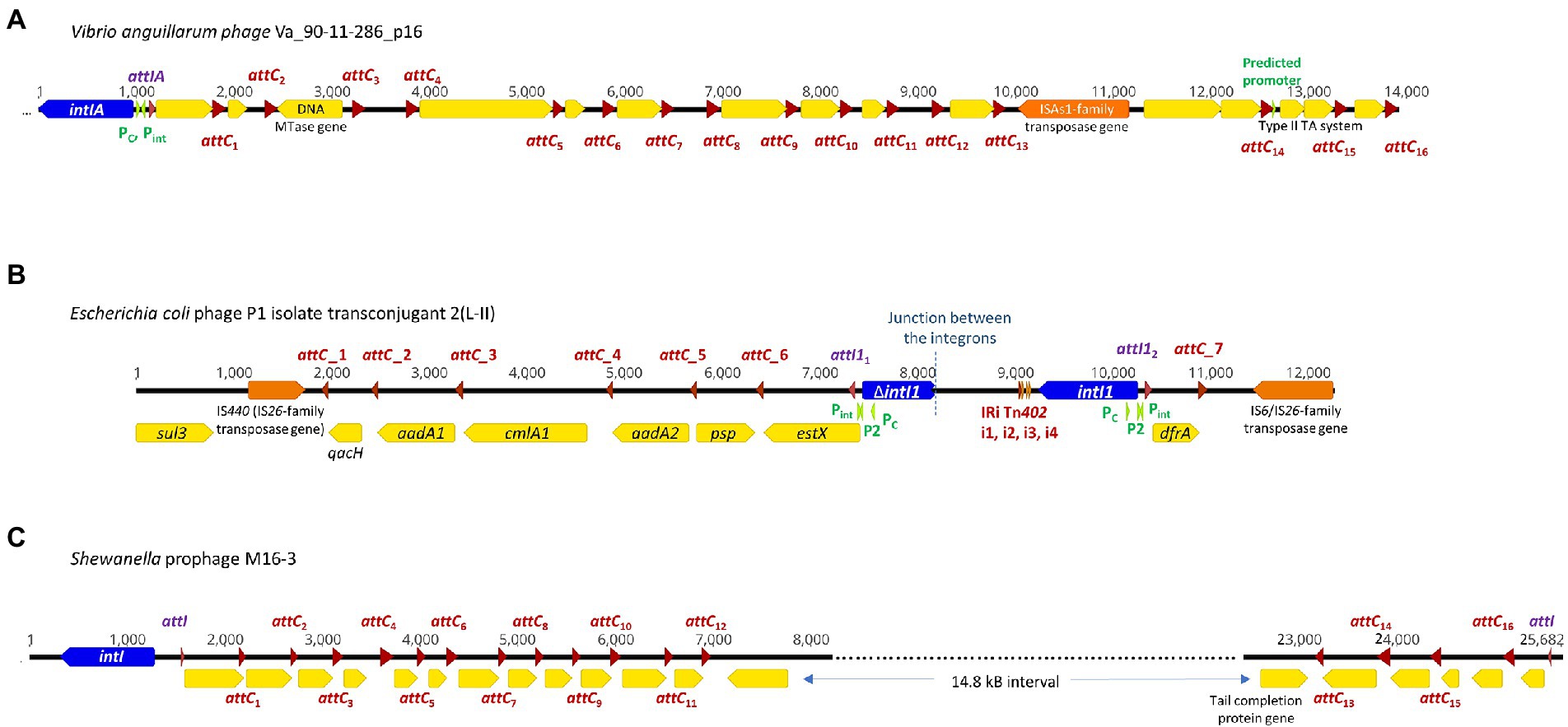
Figure 1. Complete integrons and integron-integrase that does not carry gene cassettes (In0 element) were predicted in four temperate phage genomes. The integron-integrase genes are in blue, the attC sites are represented by red arrows, and attI1 sites by purple arrows. The open reading frames (ORFs) are shown in yellow, and the promoters in green. ORFs encoding predicted DNA methyltransferases (MTase) and toxin-antitoxin modules are specified. Nucleotide numbers are shown above each schematic diagram. (A) The functional V. anguillarum phage Va_90–11–286_p16 carries a complete Vibrio-type integron; its gene cassette array is a subset of the chromosomal integron in the host genome. The double-ORF encoding a type II toxin-antitoxin system has its own predicted promoter within that gene cassette. (B) E. coli phage P1 isolate transconjugant 2 (L-II) contains fragments of two class 1 integrons, with their boundary represented by the dotted line. attC_1, 2, 4 and 5 can be classified as the Xanthomonadales type (Ghaly et al., 2021b). (C) Shewanella sp. prophage M16-3 contains a complete integron and a CALIN, each with its own attI site and integron gene cassette arrays.
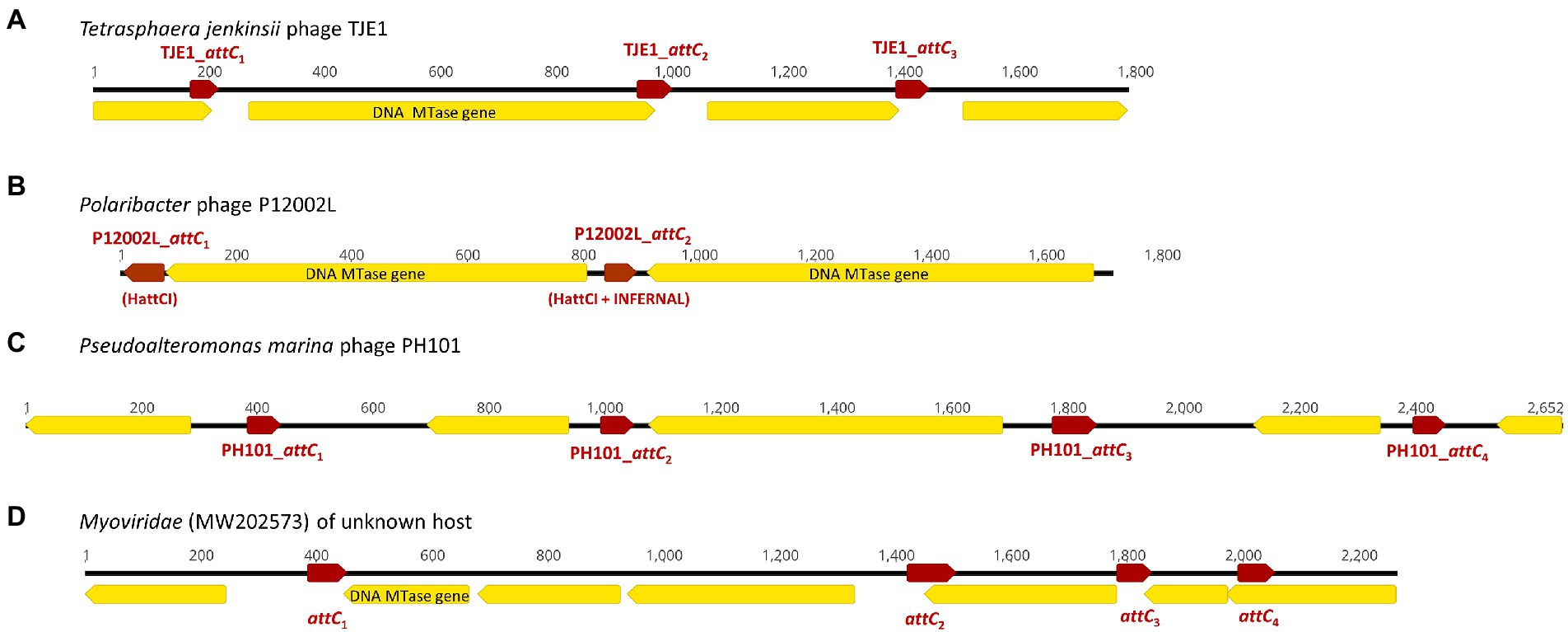
Figure 2. Examples of CALINs found in both virulent and temperate phages with structures that resemble integron gene cassette arrays in bacterial integrons. The open reading frames (ORFs) are shown in yellow, while the attC sites are represented by red arrows. ORFs encoding predicted DNA methyltransferases (MTase) are annotated. (A) CALIN in the virulent Tetrasphaera jenkinsii phage TJE1 contains 4 ORFs and 3 attC sites with the typical structure of an integron gene cassette array. (B) Two predicted DNA MTase genes were found in the CALIN of the virulent Polaribacter sp. phage P12002L. The attC sites were predicted with HattCI (P12002L_attC1) and HattCI + INFERNAL (P12002L_attC2). The latter showed attC x attI1 recombination activity. (C) CALIN in the virulent Pseudoalteromonas marina phage PH101. (D) CALIN in a Myroviridae phage (GenBank MW202573) of unknown bacterial host with a putative DNA MTase gene.
3.1. Discovery of fragments of sedentary chromosomal integrons in fully functional phages of Vibrio anguillarum
Integrons can be mobile through their association with mobile genetic elements, such as transposons and plasmids, or sedentary and located in the chromosomes (Loot et al., 2017). Sedentary chromosomal integrons tend to have attC sites that are highly conserved in nucleotide sequences and secondary structures (Rowe-Magnus et al., 2003; Ghaly et al., 2021b). The gene cassettes in sedentary chromosomal integrons also tend to carry genes of hitherto unknown functions (Vaisvila et al., 2001; Gillings et al., 2005; Boucher et al., 2007). Vibrio anguillarum phages Va_90–11–286_p16 and Va_PF430-3_p42 exemplify the ability of functional temperate phages to capture a segment of integron or integron component from sedentary chromosomal integrons. Their host V. anguillarum is a Gram-negative pathogen that causes vibriosis in many aquaculture species of fish, crustaceans, and molluscs (Frans et al., 2011). Mitomycin C induced prophage induction has been demonstrated for both phages, which allowed both phages to be purified and subsequently whole-genome sequenced (Castillo et al., 2019). They were further experimentally verified to be functional and can re-infect a variety of other V. anguillarum strains (Castillo et al., 2019).
Phage Va_90–11–286_p16 (Table 1) is a prophage in chromosome II of the highly virulent V. anguillarum strain 90–11–286 (Castillo et al., 2017). Intriguingly, its chromosomal excision locations are flanked by two predicted attC sites, each of which contains the same 68 bp perfect direct repeat (Supplementary Figure S1A). The location of prophage excision at two attC sites could potentially be explained by an increase in integron-integrase activities triggered by the SOS response, which also contributes to prophage induction (Cambray et al., 2011). The direct repeat (DR) at the 5′-end of the prophage insertion site coincides with the 3′-end boundary of a chromosomal CALIN downstream of a truncated integron-integrase gene (ΔintIA) in the host chromosome. The DR at the 3′-end of the prophage is found within a sedentary chromosomal integron (SCI) in the host chromosome. The complete integron captured by the prophage is a subset of the much longer sedentary chromosomal integron (SCI) as predicted by IntegronFinder (Neron et al., 2022). Its integron-integrase (IntIA) shares 75% pairwise amino acid identity with that of V. cholera O1 biovar El Tor strain N16961, while its cassette array comprises mostly single-ORF gene cassettes encoding proteins of unknown functions (Figure 1A). As expected, all the attC sites are of the Vibrionales type (Ghaly et al., 2021a). A predicted type II RelE/ParE-family endoribonuclease toxin gene and its cognate Phd/YefM-family antitoxin gene are found in a double-ORF operon, which has its own putative promoter. In mobile genetic elements, toxin-antitoxin (TA) systems are primarily considered to be addiction modules that prevent the loss of mobile elements from their host cells (Hayes, 2003). The roles of chromosomal TA systems are more diverse, and less well understood (Hayes, 2003; Saavedra De Bast et al., 2008; Van Melderen, 2010). In the chromosomal integron of V. cholerae, TA systems contribute to the stabilization of gene cassettes (Iqbal et al., 2015). The attC sites found in phage Va_90–11–286_p16 shared a high degree of sequence similarity but were not identical (Supplementary Figure S2).
Phage Va_PF430-3_p42 is a prophage in chromosome II of V. anguillarum strain PF430-3 and is associated with an integron-integrase without any gene cassettes (In0 element; Supplementary Figure S1B). The IntIA of phage Va_PF430-3_p42 is identical to that of Va_90–11–286_p16 in amino acid sequence. Prophage excision likely occurred at the 5′-end of the integron-integrase (intIA) gene in the chromosomal integron of the Vibrio host (Supplementary Figure S1B), resulting in the capture of an “orphan” intIA gene by the phage.
3.2. Mosaic of class 1 integrons in an Escherichia coli P1 phage
Unlike most temperate phages, P1 phages exist as circular, autonomous plasmids during lysogeny instead of integrating as prophages into their host chromosomes (Łobocka et al., 2004). A mosaic of two distinct class 1 integrons was found in E. coli phage P1 isolate transconjugant 2 (L-II; Figure 1B). The clinical E. coli strain from which this P1 phage originated also harboured IncI1, IncFIB, and ColE plasmids (Hagbo et al., 2020). Phage P1 isolate transconjugant 2 contains a plasmid replicon that shares 98.6% nucleotide sequence identity with that found in a p0111-type plasmid (NCBI accession no. AP010962; Carattoli et al., 2014). In Figure 1B, the left-hand portion of the map corresponds to the remnant of a sul3/IS440-associated class 1 integron. Although its IntI1 gene is truncated, all the other features remain intact. The class 1 integron gene cassette array estX-psp-aadA2-cmlA1-aadA1-qacH has previously been found in other Enterobacteriaceae plasmids of clinical origin (Chah et al., 2010; Chang et al., 2011). The right-hand side of the map in Figure 1B is a complete class 1 integron with an intact intI1 and a short integron gene cassette array. The non-coding region downstream of intI1 contains the inverted repeat (IRi) and four associated short repeats (i1-i4) that are characteristic of the Tn402 transposon, and the 3′-end of gene cassette array has been invaded by an IS6/IS26-family transposase gene (Kamali-Moghaddam and Sundström, 2001).
Although this P1 phage had transferred from its original E. coli clinical host strain to a DH5α transconjugant strain, it is not known if it displays all the properties of a functional phage (Hagbo et al., 2020). For a temperate phage to be considered fully functional, it needs to be able to switch from lysogeny to lysis, infect another bacterial cell, repeat the process when infecting a third host cell (Boeckman et al., 2022; Pfeifer et al., 2022). P1 phages are regarded as phage-plasmids that display characteristics of both phages and plasmids. The prevalence of phage-plasmids such as P1, N15 and SSU5 has been estimated to be as high as ~7% amongst all sequenced plasmids (Moura de Sousa et al., 2021). A recent study discovered phage-plasmids carrying class 1 integrons and provided experimental evidence that these phages have the ability to infect/re-infect other bacterial strains and confer AMR phenotypes (Pfeifer et al., 2022). With a growing recognition that many plasmids are in fact phage-plasmids, it is anticipated that more functional and integron-associated temperate phages will be uncovered in the future.
3.3. Complete integron and clusters of attCs lacking an integron-integrase in an inactive prophage of a Shewanella strain
We detected a complete integron and a CALIN in a prophage M16-3 (Figure 1C) in the chromosome of the Shewanella species strain M13 (NCBI accession no. NZ_JAGTUL010000000), which was isolated from a gold and arsenic mine. The DNA sequence of M16-3 has been verified by Sanger sequencing (Bujak et al., 2021). Due to a premature stop codon in its phage integrase gene, M16-3 is thought to be a cryptic prophage (Bujak et al., 2021). Integron-associated prophages and prophage-like regions have also been observed in the chromosomes of K. pneumoniae, E. coli, and P. aeruginosa strains (Kondo et al., 2021). In all these cases, it is not clear though if the association between the prophages, complete integrons and/or CALINs occurred before or after the prophages became cryptic.
The intact integron-integrase (IntI) found in phage M16-3 shares 95.3% pairwise amino acid identity with that of Shewanella oneidensis MR-1 (GenBank NC_004347; Drouin et al., 2002). A Shewanella-type attI site (5’-AAACGCGCATGCGCACTAATAAAATGTT-3′), which differs from that of S. oneidensis MR-1 by a single nucleotide (Drouin et al., 2002), can be found at the start of both the gene cassette array of the complete integron and of the CALIN. Putative phage-specific genes, including those encoding phage tail, capsid scaffolding, and terminase proteins, can be found in the regions flanking the CALIN.
3.4. The discovery of clusters of attCs lacking an integron-integrase in virulent phages
We discovered 17 CALINs in temperate, virulent, and uncharacterized phages (Table 2 and Supplementary Table S1). Each CALIN in our dataset contains a minimum of two predicted attC sites. The gene cassettes in virulent phages Tetrasphaera jenkinsii TJE1 and Pseudoalteromonas marina PH101 are oriented in the same direction, which is the structure most typical of bacterial gene cassette arrays (Figure 2). We also observed attC sites with divergent directions in the Polaribacter strain phage P12002L and multiple ORFs between adjacent attC sites in the Myroviridae phage of unknown host. These observations could potentially be attributed to molecular mechanisms that generate mosaicism in phage genomes.
We focused on the attC sites in three virulent phages that have been isolated from their environmental bacterial hosts for further experimental validation (Table 2; Figures 2A–C). The three bacterial hosts occur in different environments. T. jenkinsii is a Gram-positive, filamentous bacterial species that causes bulking in activated sludge of wastewater treatment plants (Petrovski et al., 2012). The Polaribacter species and P. marina hosts are found in marine ecosystems such as in surface seawater (Kang et al., 2015; Wang et al., 2015). We demonstrated that five out of the six attC sites from these virulent phages can be successfully recognized by the class 1 integron-integrase (IntI1) and incorporated into the canonical attI1 recombination site. Two adjacent attC sites were selected from each phage for their ability to integrate into attI1 (Table 2). Using a previously described conjugation-based attC x attI1 recombination assay (Bouvier et al., 2005; Nivina et al., 2020; Ghaly et al., 2022b), we showed that gene cassette insertion can take place following the transfer of single-stranded suicide vector DNA with phage-borne attC sites (bottom strands) into an intI1-expressing E. coli recipient strain that also harbours an attI1 site (Figure 3A). With the exception of P12002L_attC1, the attI1/attC junctions in the five co-integrant plasmids formed through fusion between the attI1-bearing recipient plasmid and the phage-borne attC donor plasmids were verified by Sanger sequencing, which confirmed that the correct R-R’ box (5′-G|TTRRRY-3′) sequences were obtained (Figure 4). For the attC bottom strands, we found no significant difference in the mean recombination frequencies of the successfully integrated phage-borne attC sites and the bacterial attCaadA7 positive control (Figure 3B, one-way ANOVA: p = 0.189, F5, 12 = 1.79). The mean recombination frequencies of the successfully integrated attC top strands were lower than those of the respective bottom strands, which is consistent with previous observations that attC bottom strands are more recombinogenic than their top strands during attC x attI1 integration (Bouvier et al., 2005; Nivina et al., 2016).
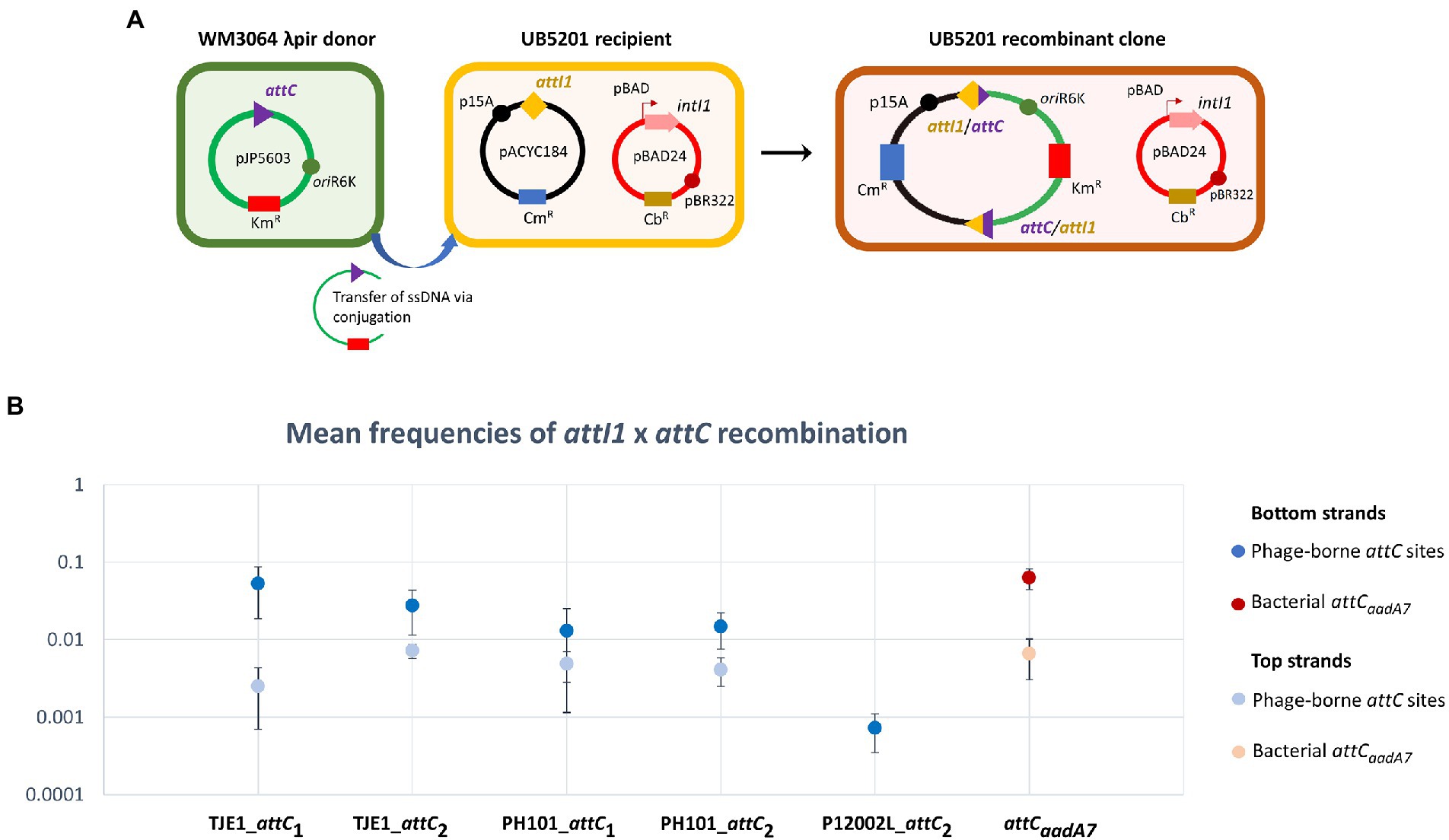
Figure 3. A panel of attC sites from virulent phages can be successfully recognized by the class 1 integron-integrase (IntI1) and incorporated into the canonical attI1 recombination site. (A) Schematic of the principles of the attC x attI recombination assay. During conjugation, the attC-containing pJP5603 plasmid is delivered into the UB5201 strain, which carries an L-arabinose inducible intI1 gene and an attI1 site on plasmids pBAD24 and pACYC184, respectively. The donor suicide vector cannot replicate within the recipient cells. It can only persist and confer kanamycin resistance following successful attC x attI recombination to form a co-integrate plasmid in the recombinant clone. (B) Mean recombination frequencies (± standard error of the mean) between attI1 and five virulent phage-borne attC sites. The bacterial attCaadA7 site was used as the positive control. Mean recombination frequencies were calculated from three independent sets of biological replicates. No statistically significant difference in mean recombination frequencies was detected across the six successfully integrated bottom strands of attC sites (one-way ANOVA: p = 0.189, F5, 12 = 1.79). The mean recombination frequencies of the five successfully integrated top strands of attC sites were lower than those of the bottom strands. No recombinant clones were obtained for the top strand of P12002L_attC2.
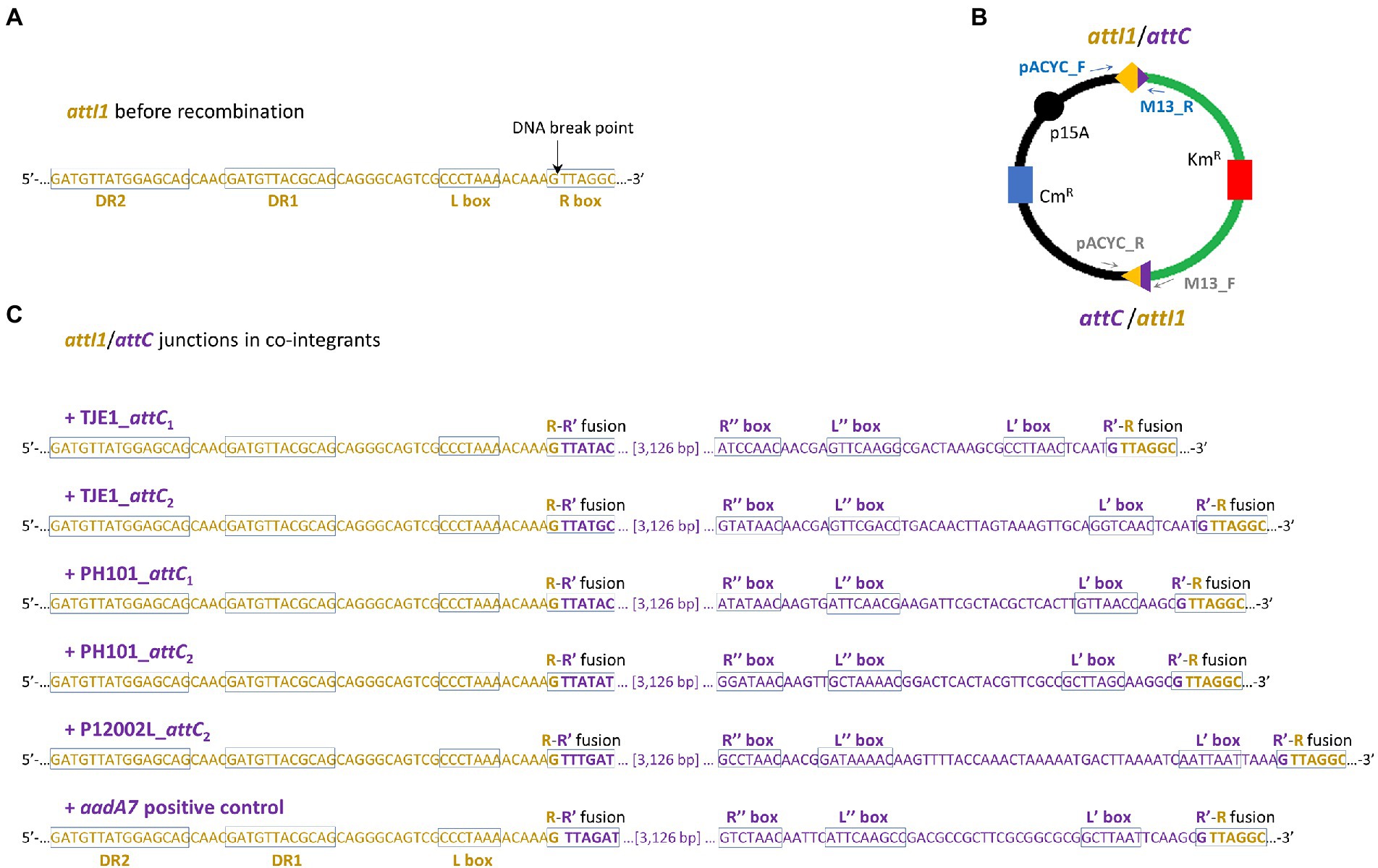
Figure 4. Two attI1/attC junctions per co-integrant plasmid were formed between the attI1-bearing recipient plasmid and the attC-donor plasmids. (A) DNA sequence of the attI1 site in the UB5201 recipient strain with L and R boxes (core IntI1 binding sites) and direct repeats DR1 and DR2. DNA cleavage occurs at the insertion points indicated by the black arrows during attC x attI recombination. (B) Schematic of the two recombination junctions in co-integrant plasmids following attC x attI recombination assay. The two pairs of PCR primers used to verify the two recombination junctions are shown as blue and grey arrows (schematic not drawn to scale). (C) DNA sequences at attC/attI junctions in co-integrant plasmids following attI1 recombination with five virulent phage-borne attC sites and the bacterial attCaadA7 positive control. The nucleotide bases belonging to attI1 and attC are shown in dark orange and purple, respectively. The recombined R-R’ boxes at the two attC/attI junctions are shown in the rectangles. The inverted repeats of the respective attC sites (L’, L,” R’, and R”) are all annotated according to HattCI predictions (Pereira et al., 2016).
We analyzed the genetic contexts of the predicted integrons/CALINs in the genomes of the phages in Figures 1, 2 and summarized their boundaries in the phage genomes in Supplementary Table S2. Notably, the CALIN in T. jenkinsii TJE1 was found downstream of a predicted transcriptional terminator (5’-TGTTAGACCGATCGGTCCAAC-3′), which contains two inverted 5’-GTTRRRY-3′ sequences (underlined). attC sites are also thought to contain weak transcriptional terminators (Engelstadter et al., 2016). Inverted or multiple 5’-GTTRRRY-3′ motifs can also be seen at the boundaries of the complete integron/CALIN in Shewanella sp. phage M16-3. These observations raise the possibility that the similarities between these repeated motifs and the R’/R” boxes that are frequently found in attC sites could potentially have contributed to their incorporation into the phage genomes via mechanisms such as off-target homologous recombination and illegitimate recombination events (Morris et al., 2008; Bertrand et al., 2019; Pfeifer et al., 2021).
In total, phage-associated gene cassettes carried five predicted DNA methyltransferases (MTases) with significant PFAM matches corresponding to three conserved domains (N6_N4_Mtase, Methyltransf_11 and Methyltransf_16; Supplementary Table S4). In Bacteria, DNA MTases are often accompanied by their cognate restriction endonucleases to form restriction-modification (R-M) systems; restriction endonucleases cleave foreign DNA from invading phages and other mobile genetic elements, while their cognate MTases methylate specific nucleotide bases in bacterial DNA to protect them from DNA cleavage. Some bacterial R-M systems are known to be encoded within integron gene cassettes (Buongermino Pereira et al., 2020; Ghaly et al., 2021a). In temperate and virulent phages, “orphan” DNA MTases that protect phages from bacterial restriction endonucleases have been described (Labrie et al., 2010; Murphy et al., 2013). This observation suggests that phages with integron recombination sites have access to gene cassettes that could confer fitness advantages by overcoming bacterial phage defence mechanisms. Their function is to methylate phage DNA to negate the activity of host encoded restriction endonucleases which recognise the same sequences (Murphy et al., 2014).
In conclusion, we identified integrons and their components in 21 different phages recovered from diverse bacterial host species from different ecosystems. Although the currently observed frequency of association between integrons/integron components and phage genomes is not high, our findings suggest that carriage of integrons in phage genomes could occur across diverse niches and bacterial hosts. While there has been renewed interest in phage biology in recent years, there are still relatively few whole-genome sequenced phages that are available to search for evidence of integrons and CALINs. Additionally, scanning for integrons in prophages from bacterial genome sequences is computationally challenging, since prophage contigs in whole-genome sequencing data are often split by insertion sequences, repetitive sequences and sequences that are homologous to other bacterial genes (Vale et al., 2017; Arndt et al., 2019). With growing interest in phage biology leading to increased rates of genome sequencing, together with improvements in prophage prediction algorithms and long-read DNA sequencing technologies, more integron-associated phage genomes could be identified in the future (Cook et al., 2021).
Here we established that a panel of phage-borne attC sites are genuine integron recombination sites that can be recognized and recruited by the bacterial class 1 integron. It has been well demonstrated that integrons and gene cassettes transfer among Bacteria via mobile genetic elements such as plasmids and transposons. The finding of operational attC sites in virulent phage genomes establishes the potential for phages to also mediate dissemination of integron gene cassettes between bacterial lineages. This mode of HGT has not been fully appreciated and demonstrates that synergies between different types of mobile element could be an important driver of gene transfer.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://doi.org/10.5061/dryad.z08kprrh5.
Author contributions
QQ, ST, and MG conceptualization of study. QQ and TG bioinformatic analysis. QQ and VR experimental investigation and validation. MG and ST project management and supervision. QQ writing the original draft of the manuscript. QQ, VR, TG, ST, and MG reviewing and editing the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Australian Research Council Discovery Project DP200101874 to MG and ST.
Acknowledgments
We thank Steve Petrovski (La Trobe University, Australia) for the E. coli strains and plasmids used in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1091391/full#supplementary-material
Footnotes
References
Adams, M. H. (1959). Bacteriophages. New York, Interscience Publishers, doi: 10.5962/bhl.title.6966.
Arndt, D., Marcu, A., Liang, Y., and Wishart, D. S. (2019). PHAST, PHASTER and PHASTEST: tools for finding prophage in bacterial genomes. Brief. Bioinform. 20, 1560–1567. doi: 10.1093/bib/bbx121
Barksdale, L., and Arden, S. B. (1974). Persisting bacteriophage infections, lysogeny, and phage conversions. Annu. Rev. Microbiol. 28, 265–300. doi: 10.1146/annurev.mi.28.100174.001405
Bertrand, C., Thibessard, A., Bruand, C., Lecointe, F., and Leblond, P. (2019). Bacterial NHEJ: a never ending story. Mol. Microbiol. 111, 1139–1151. doi: 10.1111/mmi.14218
Biskri, L., Bouvier, M., Guérout, A.-M., Boisnard, S., and Mazel, D. (2005). Comparative study of class 1 integron and Vibrio cholerae superintegron integrase activities. J. Bacteriol. 187, 1740–1750. doi: 10.1128/JB.187.5.1740-1750.2005
Boeckman, J., Korn, A., Yao, G., Ravindran, A., Gonzalez, C., and Gill, J. (2022). Sheep in wolves' clothing: temperate T7-like bacteriophages and the origins of the Autographiviridae. Virology 568, 86–100. doi: 10.1016/j.virol.2022.01.013
Borodovich, T., Shkoporov, A. N., Ross, R. P., and Hill, C. (2022). Phage-mediated horizontal gene transfer and its implications for the human gut microbiome. Gastroenterol. Rep. 10:goac012. doi: 10.1093/gastro/goac012
Boucher, Y., Labbate, M., Koenig, J. E., and Stokes, H. W. (2007). Integrons: mobilizable platforms that promote genetic diversity in bacteria. Trends Microbiol. 15, 301–309. doi: 10.1016/j.tim.2007.05.004
Bouvier, M., Demarre, G., and Mazel, D. (2005). Integron cassette insertion: a recombination process involving a folded single strand substrate. EMBO J. 24, 4356–4367. doi: 10.1038/sj.emboj.7600898
Bouvier, M., Ducos-Galand, M., Loot, C., Bikard, D., and Mazel, D. (2009). Structural features of single-stranded integron cassette attC sites and their role in strand selection. PLoS Genet. 5:e1000632. doi: 10.1371/journal.pgen.1000632
Brüssow, H. (2005). Phage therapy: the Escherichia coli experience. Microbiology 151, 2133–2140. doi: 10.1099/mic.0.27849-0
Brüssow, H., Canchaya, C., and Hardt, W. D. (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68, 560–602. doi: 10.1128/MMBR.68.3.560-602.2004
Buckling, A., and Rainey, P. B. (2002). The role of parasites in sympatric and allopatric host diversification. Nature 420, 496–499. doi: 10.1038/nature01164
Bujak, K., Decewicz, P., Rosinska, J. M., and Radlinska, M. (2021). Genome study of a novel virulent phage vB_SspS_KASIA and mu-like prophages of Shewanella sp. M16 provides insights into the genetic diversity of the Shewanella virome. Int. J. Mol. Sci. 22:1070. doi: 10.3390/ijms222011070
Buongermino Pereira, M., Österlund, T., Eriksson, K. M., Backhaus, T., Axelson-Fisk, M., and Kristiansson, E. (2020). A comprehensive survey of integron-associated genes present in metagenomes. BMC Genomics 21:495. doi: 10.1186/s12864-020-06830-5
Cambray, G., Sanchez-Alberola, N., Campoy, S., Guerin, É., Da Re, S., González-Zorn, B., et al. (2011). Prevalence of SOS-mediated control of integron integrase expression as an adaptive trait of chromosomal and mobile integrons. Mob. DNA 2:6. doi: 10.1186/1759-8753-2-6
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Castillo, D., Alvise, P. D., Xu, R., Zhang, F., Middelboe, M., and Gram, L. (2017). Comparative genome analyses of Vibrio anguillarum strains reveal a link with pathogenicity traits. mSystems 2:17. doi: 10.1128/mSystems.00001-17
Castillo, D., Andersen, N., Kalatzis, P. G., and Middelboe, M. (2019). Large phenotypic and genetic diversity of prophages induced from the fish pathogen Vibrio anguillarum. Viruses 11:983. doi: 10.3390/v11110983
Chah, K. F., Agbo, I. C., Eze, D. C., Somalo, S., Estepa, V., and Torres, C. (2010). Antimicrobial resistance, integrons and plasmid replicon typing in multiresistant clinical Escherichia coli strains from Enugu state, Nigeria. J. Basic Microbiol. 50, S18–S24. doi: 10.1002/jobm.200900325
Chang, C. Y., Lu, P. L., Lin, C. C., Lee, T. M., Tsai, M. Y., and Chang, L. L. (2011). Integron types, gene cassettes, antimicrobial resistance genes and plasmids of Shigella sonnei isolates from outbreaks and sporadic cases in Taiwan. J. Med. Microbiol. 60, 197–204. doi: 10.1099/jmm.0.022517-0
Chen, J., Quiles-Puchalt, N., Chiang, Y. N., Bacigalupe, R., Fillol-Salom, A., Chee, M. S. J., et al. (2018). Genome hypermobility by lateral transduction. Science 362, 207–212. doi: 10.1126/science.aat5867
Chiang, Y. N., Penadés, J. R., and Chen, J. (2019). Genetic transduction by phages and chromosomal islands: the new and non-canonical. PLoS Pathog. 15:e1007878. doi: 10.1371/journal.ppat.1007878
Cheong, K. H., Wen, T., Benler, S., Koh, J. M., and Koonin, E. V. (2022). Alternating lysis and lysogeny is a winning strategy in bacteriophages due to Parrondo's paradox. Proc. Natl. Acad. Sci. U. S. A. 119:e2115145119. doi: 10.1073/pnas.2115145119
Colavecchio, A., Cadieux, B., Lo, A., and Goodridge, L. D. (2017). Bacteriophages contribute to the spread of antibiotic resistance genes among foodborne pathogens of the Enterobacteriaceae family - a review. Front. Microbiol. 8:1108. doi: 10.3389/fmicb.2017.01108
Cook, R., Brown, N., Redgwell, T., Rihtman, B., Barnes, M., Clokie, M., et al. (2021). Infrastructure for a PHAge reference database: identification of large-scale biases in the current collection of cultured phage genomes. PHAGE 2, 214–223. doi: 10.1089/phage.2021.0007
Debroas, D., and Siguret, C. (2019). Viruses as key reservoirs of antibiotic resistance genes in the environment. ISME J. 13, 2856–2867. doi: 10.1038/s41396-019-0478-9
Drouin, F., Mélançon, J., and Roy, P. H. (2002). The IntI-like tyrosine recombinase of Shewanella oneidensis is active as an integron integrase. J. Bacteriol. 184, 1811–1815. doi: 10.1128/JB.184.6.1811-1815.2002
Enault, F., Briet, A., Bouteille, L., Roux, S., Sullivan, M. B., and Petit, M. A. (2017). Phages rarely encode antibiotic resistance genes: a cautionary tale for virome analyses. ISME J. 11, 237–247. doi: 10.1038/ismej.2016.90
Engelstadter, J., Harms, K., and Johnsen, P. J. (2016). The evolutionary dynamics of integrons in changing environments. ISME J. 10, 1296–1307. doi: 10.1038/ismej.2015.222
Escudero, J. A., Loot, C., Nivina, A., and Mazel, D. (2015). The integron: adaptation on demand. Microbiol. Spectr. 3:MDNA3-0019-2014.
Frans, I., Michiels, C. W., Bossier, P., Willems, K. A., Lievens, B., and Rediers, H. (2011). Vibrio anguillarum as a fish pathogen: virulence factors, diagnosis and prevention. J. Fish Dis. 34, 643–661. doi: 10.1111/j.1365-2761.2011.01279.x
Freeman, V. J. (1951). Studies on the virulence of bacteriophage-infected strains of Corynebacterium diphtheriae. J. Bacteriol. 61, 675–688. doi: 10.1128/jb.61.6.675-688.1951
Ghaly, T. M., Geoghegan, J. L., Tetu, S. G., and Gillings, M. R. (2020). The peril and promise of integrons: beyond antibiotic resistance. Trends Microbiol. 28, 455–464. doi: 10.1016/j.tim.2019.12.002
Ghaly, T. M., Gillings, M. R., Penesyan, A., Qi, Q., Rajabal, V., and Tetu, S. G. (2021a). The natural history of integrons. Microorganisms 9:2212. doi: 10.3390/microorganisms9112212
Ghaly, T. M., Penesyan, A., Pritchard, A., Qi, Q., Rajabal, V., Tetu, S. G., et al. (2022a). Methods for the targeted sequencing and analysis of integrons and their gene cassettes from complex microbial communities. Microb. Genom. 8:788. doi: 10.1099/mgen.0.000788
Ghaly, T. M., Tetu, S. G., and Gillings, M. R. (2021b). Predicting the taxonomic and environmental sources of integron gene cassettes using structural and sequence homology of attC sites. Commun. Biol. 4:946. doi: 10.1038/s42003-021-02489-0
Ghaly, T. M., Tetu, S. G., Penesyan, A., Qi, Q., Rajabal, V., and Gillings, M. R. (2022b). Discovery of integrons in Archaea: platforms for cross-domain gene transfer. Sci. Adv. 8:eabq6376. doi: 10.1126/sciadv.abq6376
Gillings, M., Boucher, Y., Labbate, M., Holmes, A., Krishnan, S., Holley, M., et al. (2008). The evolution of class 1 integrons and the rise of antibiotic resistance. J. Bacteriol. 190, 5095–5100. doi: 10.1128/JB.00152-08
Gillings, M. R. (2014). Integrons: past, present, and future. Microbiol. Mol. Biol. Rev. 78, 257–277. doi: 10.1128/MMBR.00056-13
Gillings, M. R. (2017). Class 1 integrons as invasive species. Curr. Opin. Microbiol. 38, 10–15. doi: 10.1016/j.mib.2017.03.002
Gillings, M. R., Gaze, W. H., Pruden, A., Smalla, K., Tiedje, J. M., and Zhu, Y. G. (2015). Using the class 1 integron-integrase gene as a proxy for anthropogenic pollution. ISME J. 9, 1269–1279. doi: 10.1038/ismej.2014.226
Gillings, M. R., Holley, M. P., Stokes, H. W., and Holmes, A. J. (2005). Integrons in Xanthomonas: a source of species genome diversity. Proc. Natl. Acad. Sci. U. S. A. 102, 4419–4424. doi: 10.1073/pnas.0406620102
Hagbo, M., Ravi, A., Angell, I. L., Sunde, M., Ludvigsen, J., Diep, D. B., et al. (2020). Experimental support for multidrug resistance transfer potential in the preterm infant gut microbiota. Pediatr. Res. 88, 57–65. doi: 10.1038/s41390-019-0491-8
Hall, R. M., and Stokes, H. W. (1993). Integrons: novel DNA elements which capture genes by site-specific recombination. Genetica 90, 115–132. doi: 10.1007/BF01435034
Hatfull, G. F., and Hendrix, R. W. (2011). Bacteriophages and their genomes. Curr. Opin. Virol. 1, 298–303. doi: 10.1016/j.coviro.2011.06.009
Hayes, F. (2003). Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301, 1496–1499. doi: 10.1126/science.1088157
Hershey, A. D. (1971). The bacteriophage lambda. Cold Spring Harbor, N.Y., Cold Spring Harbor Laboratory.
Holloway, B. W., and Cooper, G. N. (1962). Lysogenic conversion in Pseudomonas aeruginosa. J. Bacteriol. 84, 1321–1324. doi: 10.1128/jb.84.6.1321-1324.1962
Howard-Varona, C., Hargreaves, K. R., Abedon, S. T., and Sullivan, M. B. (2017). Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 11, 1511–1520. doi: 10.1038/ismej.2017.16
Iqbal, N., Guérout, A. M., Krin, E., Le Roux, F., and Mazel, D. (2015). Comprehensive functional analysis of the 18 Vibrio cholerae N16961 toxin-antitoxin systems substantiates their role in stabilizing the superintegron. J. Bacteriol. 197, 2150–2159. doi: 10.1128/JB.00108-15
Kamali-Moghaddam, M., and Sundström, L. (2001). Arrayed transposase-binding sequences on the ends of transposon Tn5090/Tn402. Nucleic Acids Res. 29, 1005–1011. doi: 10.1093/nar/29.4.1005
Kang, I., Jang, H., and Cho, J. C. (2015). Complete genome sequences of bacteriophages P12002L and P12002S, two lytic phages that infect a marine Polaribacter strain. Stand. Genomic Sci. 10:82. doi: 10.1186/s40793-015-0076-z
Kirsch, J. M., Brzozowski, R. S., Faith, D., Round, J. L., Secor, P. R., and Duerkop, B. A. (2021). Bacteriophage-bacteria interactions in the gut: from invertebrates to mammals. Annu. Rev. Virol. 8, 95–113. doi: 10.1146/annurev-virology-091919-101238
Kondo, K., Kawano, M., and Sugai, M. (2021). Distribution of antimicrobial resistance and virulence genes within the prophage-associated regions in nosocomial pathogens. mSphere 6:e0045221. doi: 10.1128/mSphere.00452-21
Koskella, B., and Brockhurst, M. A. (2014). Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 38, 916–931. doi: 10.1111/1574-6976.12072
Labrie, S. J., Samson, J. E., and Moineau, S. (2010). Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–327. doi: 10.1038/nrmicro2315
Landsberger, M., Gandon, S., Meaden, S., Rollie, C., Chevallereau, A., Chabas, H., et al. (2018). Anti-CRISPR phages cooperate to overcome CRISPR-Cas immunity. Cells 174, 908–916.e12. doi: 10.1016/j.cell.2018.05.058
Lennox, E. S. (1955). Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1, 190–206. doi: 10.1016/0042-6822(55)90016-7
Loot, C., Nivina, A., Cury, J., Escudero, J. A., Ducos-Galand, M., Bikard, D., et al. (2017). Differences in integron cassette excision dynamics shape a trade-off between evolvability and genetic capacitance. MBio 8:16. doi: 10.1128/mBio.02296-16
Łobocka, M. B., Rose, D. J., Plunkett, G., Rusin, M., Samojedny, A., Lehnherr, H., et al. (2004). Genome of bacteriophage P1. J. Bacteriol. 186, 7032–7068. doi: 10.1128/JB.186.21.7032-7068.2004
Mavrich, T. N., and Hatfull, G. F. (2017). Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2:17112. doi: 10.1038/nmicrobiol.2017.112
Mazel, D. (2006). Integrons: agents of bacterial evolution. Nat. Rev. Microbiol. 4, 608–620. doi: 10.1038/nrmicro1462
McDaniel, L. D., Young, E., Delaney, J., Ruhnau, F., Ritchie, K. B., and Paul, J. H. (2010). High frequency of horizontal gene transfer in the oceans. Science 330:50. doi: 10.1126/science.1192243
Morris, P., Marinelli, L. J., Jacobs-Sera, D., Hendrix, R. W., and Hatfull, G. F. (2008). Genomic characterization of mycobacteriophage Giles: evidence for phage acquisition of host DNA by illegitimate recombination. J. Bacteriol. 190, 2172–2182. doi: 10.1128/JB.01657-07
Moura de Sousa, J. A., Pfeifer, E., Touchon, M., and Rocha, E. P. C. (2021). Causes and consequences of bacteriophage diversification via genetic exchanges across lifestyles and bacterial taxa. Mol. Biol. Evol. 38, 2497–2512. doi: 10.1093/molbev/msab044
Murphy, J., Klumpp, J., Mahony, J., O'Connell-Motherway, M., Nauta, A., and van Sinderen, D. (2014). Methyltransferases acquired by lactococcal 936-type phage provide protection against restriction endonuclease activity. BMC Genomics 15:831. doi: 10.1186/1471-2164-15-831
Murphy, J., Mahony, J., Ainsworth, S., Nauta, A., and van Sinderen, D. (2013). Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl. Environ. Microbiol. 79, 7547–7555. doi: 10.1128/AEM.02229-13
Nawrocki, E. P., and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Neron, B., Littner, E., Haudiquet, M., Perrin, A., Cury, J., and Rocha, E. P. C. (2022). IntegronFinder 2.0: identification and analysis of integrons across bacteria, with a focus on antibiotic resistance in Klebsiella. Microorganisms 10:700. doi: 10.3390/microorganisms10040700
Nivina, A., Escudero, J. A., Vit, C., Mazel, D., and Loot, C. (2016). Efficiency of integron cassette insertion in correct orientation is ensured by the interplay of the three unpaired features of attC recombination sites. Nucleic Acids Res. 44, 7792–7803. doi: 10.1093/nar/gkw646
Nivina, A., Grieb, M. S., Loot, C., Bikard, D., Cury, J., Shehata, L., et al. (2020). Structure-specific DNA recombination sites: design, validation, and machine learning-based refinement. Sci. Adv. 6:eaay2922. doi: 10.1126/sciadv.aay2922
Parkinson, J. S. (2016). Classic spotlight: the discovery of bacterial transduction. J. Bacteriol. 198, 2899–2900. doi: 10.1128/JB.00635-16
Pal, C., Maciá, M. D., Oliver, A., Schachar, I., and Buckling, A. (2007). Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 450, 1079–1081. doi: 10.1038/nature06350
Partridge, S. R., Recchia, G. D., Scaramuzzi, C., Collis, C. M., Stokes, H. W., and Hall, R. M. (2000). Definition of the attI1 site of class 1 integrons. Microbiology 146, 2855–2864. doi: 10.1099/00221287-146-11-2855
Pedulla, M. L., Ford, M. E., Houtz, J. M., Karthikeyan, T., Wadsworth, C., Lewis, J. A., et al. (2003). Origins of highly mosaic mycobacteriophage genomes. Cell 113, 171–182. doi: 10.1016/S0092-8674(03)00233-2
Pereira, M. B., Wallroth, M., Kristiansson, E., and Axelson-Fisk, M. (2016). HattCI: fast and accurate attC site identification using hidden Markov models. J. Comput. Biol. 23, 891–902. doi: 10.1089/cmb.2016.0024
Petrovski, S., Tillett, D., and Seviour, R. J. (2012). Isolation and complete genome sequence of a bacteriophage lysing Tetrasphaera jenkinsii, a filamentous bacteria responsible for bulking in activated sludge. Virus Genes 45, 380–388. doi: 10.1089/cmb.2016.0024
Pfeifer, E., Bonnin, R. A., and Rocha, E. P. C. (2022). Phage-plasmids spread antibiotic resistance genes through infection and lysogenic conversion. MBio 13:e0185122. doi: 10.1128/mbio.01851-22
Pfeifer, E., Moura de Sousa, J. A., Touchon, M., and Rocha, E. P. C. (2021). Bacteria have numerous distinctive groups of phage-plasmids with conserved phage and variable plasmid gene repertoires. Nucleic Acids Res. 49, 2655–2673. doi: 10.1093/nar/gkab064
Recchia, G. D., Stokes, H. W., and Hall, R. M. (1994). Characterization of specific and secondary recombination sites recognized by the integron DNA integrase. Nucleic Acids Res. 22, 2071–2078. doi: 10.1093/nar/22.11.2071
Rice, S. A., Tan, C. H., Mikkelsen, P. J., Kung, V., Woo, J., Tay, M., et al. (2009). The biofilm life cycle and virulence of Pseudomonas aeruginosa are dependent on a filamentous prophage. ISME J. 3, 271–282. doi: 10.1038/ismej.2008.109
Riedel, T., Rohlfs, M., Buchholz, I., Wagner-Dobler, I., and Reck, M. (2013). Complete sequence of the suicide vector pJP5603. Plasmid 69, 104–107. doi: 10.1016/j.plasmid.2012.07.005
Rodriguez-Valera, F., Martin-Cuadrado, A. B., Rodriguez-Brito, B., Pasić, L., Thingstad, T. F., Rohwer, F., et al. (2009). Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 7, 828–836. doi: 10.1038/nrmicro2235
Rowe-Magnus, D. A., Guerout, A.-M., Biskri, L., Bouige, P., and Mazel, D. (2003). Comparative analysis of superintegrons: engineering extensive genetic diversity in the Vibrionaceae. Genome Res. 13, 428–442. doi: 10.1101/gr.617103
Saavedra De Bast, M., Mine, N., and Van Melderen, L. (2008). Chromosomal toxin-antitoxin systems may act as anti-addiction modules. J. Bacteriol. 190, 4603–4609. doi: 10.1128/JB.00357-08
Stokes, H. W., and Hall, R. M. (1989). A novel family of potentially mobile DNA elements encoding site-specific gene-integration functions: integrons. Mol. Microbiol. 3, 1669–1683. doi: 10.1111/j.1365-2958.1989.tb00153.x
Stokes, H. W., O'Gorman, D. B., Recchia, G. D., Parsekhian, M., and Hall, R. M. (1997). Structure and function of 59-base element recombination sites associated with mobile gene cassettes. Mol. Microbiol. 26, 731–745. doi: 10.1046/j.1365-2958.1997.6091980.x
Vale, F. F., Nunes, A., Oleastro, M., Gomes, J. P., Sampaio, D. A., Rocha, R., et al. (2017). Genomic structure and insertion sites of Helicobacter pylori prophages from various geographical origins. Sci. Rep. 7:42471. doi: 10.1038/srep42471
Vaisvila, R., Morgan, R. D., Posfai, J., and Raleigh, E. A. (2001). Discovery and distribution of super-integrons among Pseudomonads. Mol. Microbiol. 42, 587–601. doi: 10.1046/j.1365-2958.2001.02604.x
Van Melderen, L. (2010). Toxin-antitoxin systems: why so many, what for? Curr. Opin. Microbiol. 13, 781–785. doi: 10.1016/j.mib.2010.10.006
Villa, T. G., Feijoo-Siota, L., Rama, J. R., Sánchez-Pérez, A., and Viñas, M. (2019). in Horizontal gene transfer between bacteriophages and bacteria: Antibiotic resistances and toxin production. Horizontal gene transfer: Breaking borders between living kingdoms. eds. T. G. Villa and M. Viñas (Cham: Springer International Publishing), 97–142.
Vit, C., Richard, E., Fournes, F., Whiteway, C., Eyer, X., Lapaillerie, D., et al. (2021). Cassette recruitment in the chromosomal integron of Vibrio cholerae. Nucleic Acids Res. 49, 5654–5670. doi: 10.1093/nar/gkab412
Wagner, P. L., and Waldor, M. K. (2002). Bacteriophage control of bacterial virulence. Infect. Immun. 70, 3985–3993. doi: 10.1128/IAI.70.8.3985-3993
Wang, D. B., Sun, M. Q., Shao, H. B., Li, Y., Meng, X., Liu, Z. Y., et al. (2015). Characterization and genome sequencing of a novel bacteriophage PH101 infecting Pseudoalteromonas marina BH101 from the Yellow Sea of China. Curr. Microbiol. 71, 594–600. doi: 10.1007/s00284-015-0896-5
Westmoreland, B. C., Szybalski, W., and Ris, H. (1969). Mapping of deletions and substitutions in heteroduplex DNA molecules of bacteriophage lambda by electron microscopy. Science 163, 1343–1348. doi: 10.1126/science.163.3873.1343
Xia, G., and Wolz, C. (2014). Phages of Staphylococcus aureus and their impact on host evolution. Infect. Genet. Evol. 21, 593–601. doi: 10.1016/j.meegid.2013.04.022
Keywords: integrons, bacteriophages, horizontal gene transfer, mobile genetic elements, gene cassette-associated recombination sites, bacteria–phage coevolution
Citation: Qi Q, Rajabal V, Ghaly TM, Tetu SG and Gillings MR (2023) Identification of integrons and gene cassette-associated recombination sites in bacteriophage genomes. Front. Microbiol. 14:1091391. doi: 10.3389/fmicb.2023.1091391
Edited by:
Alicja Wegrzyn, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, PolandReviewed by:
Dariusz Bartosik, University of Warsaw, PolandLima-Mendez Gipsi, Institut de Louvain des Sciences et Technologies Biomoléculaires, Université Catholique de Louvain, Belgium
Copyright © 2023 Qi, Rajabal, Ghaly, Tetu and Gillings. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qin Qi, ✉ cWluLnFpQG1xLmVkdS5lZHU=