 Tiantian Wang
Tiantian Wang Xiaobin Li1,2
Xiaobin Li1,2 Jize Xu
Jize Xu- 1Agricultural College, Yanbian University, Yanji, China
- 2Agricultural College, Jilin Agricultural Science and Technology University, Jilin, China
- 3College of Landscape Architecture, Changchun University, Changchun, China
- 4College of Plant Sciences, Jilin University, Changchun, China
Green mold caused by Trichoderma spp. has become one of the most serious diseases which threatening the production of Ganoderma lingzhi. To understand the possible resistance mechanism of the G. lingzhi response to T. hengshanicum infection, we examined the G. lingzhi transcript accumulation at 0, 12, and 24 h after T. hengshanicum inoculation. The gene expression analysis was conducted on the interaction between G. lingzhi and T. hengshanicum using RNA-seq and digital gene expression (DGE) profiling methods. Transcriptome sequencing indicated that there were 162 differentially expressed genes (DEGs) at three infection time points, containing 15 up-regulated DEGs and 147 down-regulated DEGs. Resistance-related genes thaumatin-like proteins (TLPs) (PR-5s), phenylalanine ammonia-lyase, and Beta-1,3-glucan binding protein were significantly up-regulated. At the three time points of infection, the heat shock proteins (HSPs) genes of G. lingzhi were down-regulated. The down-regulation of HSPs genes led to the inhibition of HSP function, which may compromise the HSP-mediated defense signaling transduction pathway, leading to G. lingzhi susceptibility. Pathway enrichment analyses showed that the main enriched pathways by G. lingzhi after infection were sphingolipid metabolism, ether lipid metabolism, and valine, leucine and isoleucine degradation pathway. Overall, the results described here improve fundamental knowledge of molecular responses to G. lingzhi defense and contribute to the design of strategies against Trichoderma spp.
Introduction
Ganoderma lingzhi S. H. Wu, Y. Cao, and Y. C. Dai (Ganodermataceae, Agaricomycetes) is a traditional Chinese medicinal mushroom in Asia that has been used for thousands of years (Cao et al., 2012). It has good medicinal, health, and ornamental value (Wagner et al., 2003; Cao et al., 2012). The pharmacological action of G. lingzhi is based on its solid immune modulation and immune potential. The main medicinal ingredients include polysaccharides, triterpenes, peptides, proteins, adenosine, which have anti-androgen, anti-cancer, anti-diabetes, anti-hypertension, anti-melanocyte, anti-virus, and other health functions (Sanodiya et al., 2009; Qian et al., 2013; Satria et al., 2019). Currently, the main cultivation methods of G. lingzhi are substitute cultivation and basswood cultivation in China, mainly distributed in the Northeast region, the Dabie Mountains, the Southeast coast, and other places (Jin et al., 2016; Zhou et al., 2017). In recent years, the market demand for G. lingzhi has increased by 18–30% every year. In 2015, China was the world’s primary producer and exporter of G. lingzhi, with 10,000 hm2 of G. lingzhi yield and 120,000 t of spore powder, accounting for approximately 75 and 30% of the world, respectively (Jin et al., 2016; Ma, 2017; Li et al., 2021). Due to the increase of the cultivation years, some diseases of G. lingzhi have also been successively discovered. Among them, Trichoderma spp. has the characteristics of wide distribution, more kinds, rapid incidence, and strong concealment (Xu et al., 2019). Trichoderma spp. has become one of the most harmful pathogens in the cultivation of G. lingzhi, resulting in a decline in the yield and quality decline of G. lingzhi, which have caused severe economic losses to growers and restricted the development of the G. lingzhi industry (Xie and Tan, 2015; Huang et al., 2018).
Following the emergence of G. lingzhi, it is primarily controlled through the use of chemical reagents such as pesticides, which cause severe pollution to the environment and make drug resistance more likely (Fu et al., 2013; Xie et al., 2018). Based on six chemical reagents, Yan (2011) screened the bacteriostasis of different fungicides on edible fungi and Trichoderma spp. by preliminary screening, inhibition of spore germination, and mycelial germination tests. A total of 50% hymexazol has a strong bacteriostatic effect on Trichoderma spp. and a little destructive effect on various edible fungi mycelia, which can be recommended for production. Luković et al. (2021) conducted identification and fungicide screening tests on 22 isolation of Trichoderma strains. In fungicide susceptibility tests, all examined Trichoderma strains were found to be highly sensitive to prochloraz (ED 50 < 0.4 mg ⋅ mL–1) and considerably susceptible to metrafenone (ED 50 < 4 mg ⋅ mL–1). Hence, metrafenone might also be recommended to control the green mold of mushrooms. In aspects of biological control, among 50 bacterial strains isolated from mushroom compost, Bacillus subtillis B-38 inhibited Trichoderma harzianum T54 (48.08%) and Trichoderma aggressivum f. europaeum T77 (52.25%) mycelium growth in vitro. In plot trials, the incidence of the plots inoculated with the Trichoderma strains and treated with B. subtillis B-38 and B. subtilis QST 713 presented significantly lower disease incidence compared to the control, and results for disease control and yield harvested were comparable to the plots treated with prochloraz-Mn, indicating that B. subtilis B-38 and B. subtilis QST 713 could be used as suitable substitutes for chemical fungicides (Milijašević-Marčić et al., 2017; Gea et al., 2021). Although these methods can play a preventive role to a certain extent, the control effect is limited, so screening and breeding resistant varieties of G. lingzhi is the most economical and effective way to control green mold. Therefore, exploring the defense mechanism of G. lingzhi against T. hengshanicum will help to speed up the breeding process of disease-resistant G. lingzhi.
With the development of second-generation sequencing technology and molecular biology, many researchers have explored the molecular regulation mechanisms of rice bacterial blight (Sana et al., 2010), Phytophthora capsici disease (Fan et al., 2022), Nerium indicum witches’ broom disease (Wang et al., 2022), Phytophthora sojae disease (Zhu, 2018), apple alternaria blotch disease (Zhu et al., 2017), Chrysanthemum morifolium black spot (Liu L. N. et al., 2021), and wheat leaf spot (Ye et al., 2019). Bailey et al. (2013) found that the pathogen (Lecanicillium fungicola) and the host (Agaricus bisporus) changed the expression of their respective genes during the interaction through transcriptome analysis, which initially revealed the host’s defense response mechanism. Ma et al. (2021) found that the expression level of LeTLP1 was strongly induced in response to T. atroviride infection in the resistant Y3334 by transcriptome analysis and quantitative real-time polymerase chain reaction (qRT-PCR) detection. The function of LeTLP1 was verified by gene overexpression and gene silencing technology. Compared with the parent strain Y3334, LeTLP1-silenced transformants had reduced resistance relative to T. atroviride. These findings suggest that overexpression of LeTLP1 is a major mechanism for the assistance of Lentinula edodes to T. atroviride. This molecular basis provides a theoretical foundation for breeding resistant L. edodes strains. A. bisporus brown blotch disease caused by Pseudomonas tolaasii infection mainly activates the arginine and proline metabolism, cysteine and methionine metabolism, jasmonic acid (JA) biosynthesis, methane metabolism, phenylpropanoid metabolism, shikimate pathway, sulfur metabolism and signaling pathways, as well as oxidative phosphorylation pathways. Transcriptomics data combined with qPCR verification indicated that 10 differentially expressed genes (DEGs), including PIP1, MET3, AGX, PAL1, GCL, LOX 1/3, PR-like, MYB3R, UCR, and SDHB, were the most potential genes involved in the early defense. These results revealed the early defense response of A. bisporus against P. tolaasii (Yang et al., 2022). However, there has yet to be a report on how G. lingzhi responds to the pathogen Trichoderma spp. In this study, RNA-seq technology was used to analyze the transcriptome of G. lingzhi in response to T. hengshanicum infection, which has a great significance for the breeding of resistant varieties of G. lingzhi and the prevention and control of soil pollution.
Materials and methods
Trichoderma hengshanicum and Ganoderma lingzhi cultures, and inoculation method
Trichoderma hengshanicum “1009” and G. lingzhi “11GL-16” were used in all experiments and preserved in the Development and Utilization Laboratory of Fungi Resource of Jilin Agricultural Science and Technology College. G. lingzhi was grown in an edible fungus base. For G. lingzhi-back inoculation, the fruiting bodies of some growth vigor were strictly selected. T. hengshanicum isolated on potato dextrose agar (PDA) was propagated in a constant temperature incubator at 25°C for 4 days. The G. lingzhi fruiting bodies were inoculated with 5-mm-diameter mycelial blocks (from PDA culture plates). The control group inoculated PDA agar blocks without mycelium. At 0, 2, 12, and 24 h after inoculation, the G. lingzhi fruiting bodies were cut off with a sterile scalpel at a distance of 5 mm from the lesions and stored at −80°C after quick freezing with liquid nitrogen. At 0, 2, 12, and 24 h after inoculation, three fruiting bodies were taken from each replicate of each treatment group. The frozen samples were used for RNA sequencing.
RNA extraction, library construction, and sequencing
After 0, 2, 12, and 24 h of infection, more than 500 mg of fruiting bodies were collected for RNA extraction. Total RNA was extracted using a Trizol reagent kit according to the manufacturer’s protocol. RNA quality was assessed on an Agilent 2100 Bioanalyzer and checked using RNase-free agarose gel electrophoresis. Illumina MiSeq library construction was performed according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). To separate the mRNA from the total RNA, magnetic beads with poly T oligos were used. Then the enriched mRNA was fragmented into short fragments using a fragmentation buffer and reverse transcribed into cDNA with random primers. The cDNA fragments were purified with QIAquick PCR (Qiagen, Venlo, The Netherlands). Extraction Kit, end-repaired, and A base added and ligated to Illumina sequencing adapters. The ligation products were size selected by agarose gel electrophoresis, PCR amplified and sequenced using Illumina MiSeq by Personal Biotechnology Co., Ltd. (Shanghai, China).
Data filtering, de novo assembly, and gene function annotation
Reads obtained from the sequencing machines included raw reads containing adapters or low-quality bases, which would affect the following assembly and analysis. The clean reads were retrieved after trimming adapter sequences and removal of low quality (containing >50% bases with a Phred quality score < 20) using the FastQC tool. Transcriptome de novo assembly was performed with the short reads assembling program-Trinity (Grabherr et al., 2011). Firstly, a short sequence library of K-mer length was constructed using high-quality sequences. Then the short sequence was extended by the overlap of K-mer-1 length between short sequences to obtain the preliminary spliced contig sequences. Next, Chrysalis clusters related contigs that correspond to portions of alternatively spliced transcripts or otherwise unique portions of paralogous genes and then builds Bruijn graphs for each cluster of related contigs. Finally, these Bruijn graphs were processed to find the path based on the reads and paired reads in the graphs to obtain the transcripts. To comprehensively obtain gene annotation information, genes were compared with six databases, including NR (NCBI non-redundant protein sequences), Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genome (KEGG), eggNOG (evolutionary genealogy of genes: Non-supervised Orthologous Groups), Swiss-Prot, and Pfam, and the annotation situation of each database was counted.
Differentially expressed genes (DEGs) and enrichment analysis
RSEM estimated gene expression levels for each sample (Li and Dewey, 2011). The gene abundances were calculated and normalized to Reads Per kb per Million reads (RPKM). Three pairwise comparisons were made from RNA-seq data, including inoculation 0 h (CK) vs. inoculation 2 h (GT2), inoculation 0 h vs. inoculation 12 h (GT12), and inoculation 0 h vs. inoculation 24 h (GT24). DEGs was performed using DESeq2 (Love et al., 2014) software between two groups. The genes with the parameter of P-value < 0.05 and | log2FoldChange| > 1 were considered DEGs. All DEGs were mapped to GO terms in the GO database,1 gene numbers were calculated for every term, and significantly enriched GO terms in DEGs compared to the genome background were defined by hypergeometric test. The criterion for significant enrichment of GO function was P-value < 0.05. Pathway enrichment analysis identified significantly enriched metabolic pathways or signal transduction pathways in DEGs compared with the whole genome background. The corrected P-values adopted 0.05 as the threshold, and KEGG pathways meeting the above conditions were defined as significantly enriched pathways in DEGs.
Quantitative real-time polymerase chain reaction (qRT-PCR) verification
RNA-seq results were validated by selecting six DEGs to examine the consistency of their expression profiles. Total RNAs were extracted from collected G. lingzhi materials using the Trizol (Invitrogen, USA) kit according to the manufacturer’s instructions. First-strand cDNAs were synthesized by the PrimeScript™ 1st stand cDNA Synthesis Kit. The internal transcribed spacer (ITS) gene was used as an internal control. Volume for all the reactions was 20 μL; 1 μL cDNA, 10 μL 2 × SYBR real-time PCR (Applied Biosystem, Carlsbad, CA, USA), and 0.4 μL of each primer. The PCR procedure was 5 min at 95°C, followed by 40 cycles of 15 s at 95°C and 30 s at 60°C. Three biological replicates were performed per sample. The formula of 2–ΔΔCT was used to calculate gene relative expression levels.
Results
Summary of transcriptome analysis
Four sample sets, each with three biological replicates, were subjected to RNA-seq, and 12 cDNA libraries were generated: GT0-1, GT0-2, GT0-3, GT2-1, GT2-2, GT2-3, GT12-1, GT12-2, GT12-3, GT24-1, GT24-2, GT24-3. Samples were inoculated for 0 h (GT0-1, GT0-2, GT0-3) as a control. The sequencing raw data set has been deposited in National Center for Biotechnology Information Sequence Read Archive database (accession number PRJNA917261). Approximately 4.169∼5.386 million raw reads were produced for each sample, with the percentages of Q20 and Q30 being over 97.64 and 93.75%, respectively. A total of 79.7 Gb of clean data was obtained and the clean data of each sample reached more than 6.0 Gb. A transcriptome database containing 36,870,053 unigene of total length was obtained using Trinity software, with a mean length of 1,850.35 bp and GC content of 57.06%. All unigenes and transcripts obtained by transcriptome assembly were aligned with six major databases (Nr, Swiss-prot, Pfam, COG, GO, and KEGG databases).
Differentially expressed gene (DEG) analysis in Ganoderma lingzhi
Differentially expressed genes in susceptible G. lingzhi at different time points were identified using the thresholds p < 0.05 and | log2FoldChange| > 1. In response to the fungal stimulus, a total of 1,978 genes showed differential expression at three-time points after inoculation (Figure 1). A total of 754 (347 up-regulation and 407 down-regulation), 620 (259 up-regulation and 361 down-regulation), and 604 (79 up-regulation and 525 down-regulation) differential genes were observed in CK vs. GT2, CK vs. GT12, and CK vs. GT24, respectively. The Venn diagram (Figure 1) showed that both shared and unique DEGs were identified between different combinations. There were 162 shared DEGs in CK vs. GT2, CK vs. GT12, and CK vs. GT24.
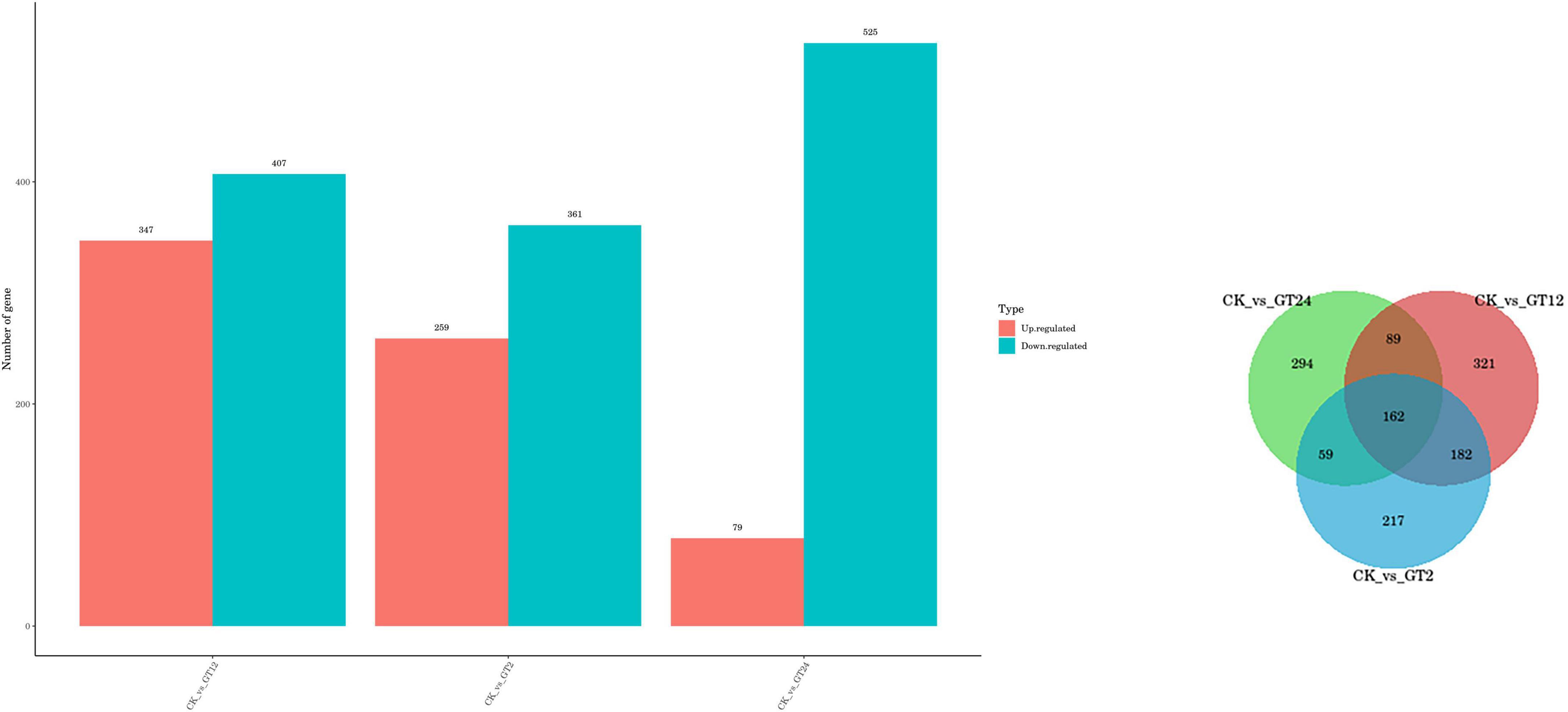
Figure 1. Differentially expressed genes (DEGs) between samples. Left: numbers of DEGs compared between two samples (CK vs. GT2, CK vs. GT12, and CK vs. GT24). DEGs are shown in red (up-regulated) and blue (down-regulated). Right: Venn diagram analysis of the DEGs in Ganoderma lingzhi after inoculation with Trichoderma hengshanicum.
Functional annotation of differentially expressed genes (DEGs)
Gene Ontology classification analysis of DEGs between CK vs. GT2, CK vs. GT12, and CK vs. GT24 was shown in Figure 2. GO had three ontologies, describing the molecular function, cellular component, and biological process of genes. At 2 h after infection, GO enrichment analysis of DEGs showed that 396, 47, and 128 GO terms were identified in biological processes, cellular components, and molecular functions, respectively. The most significant enrichment of DEGs in the biological process ontology was heterochromatin assembly by small RNA, mRNA cleavage involved in gene silencing, Wnt signaling pathway-calcium modulating pathway, and transcription (RNA-templated), whereas RNA-induced silencing complex (RISC)-loading complex and mitochondrial permeability transition pore complex occupied important positions in the cellular component ontology. The most significantly enriched molecular function ontology was catalytic activity, RNA-directed 5′-3′ RNA polymerase activity, miRNA binding, oxidoreductase activity, cofactor binding, regulatory RNA binding, nicotinamide adenine dinucleotide phosphate (NADP) binding, siRNA binding, endoribonuclease activity (cleaving siRNA-paired mRNA), endoribonuclease activity (cleaving miRNA-paired mRNA), oxidoreductase activity (acting on the CH-OH group of donors), monooxygenase activity, and iron ion binding. At 12 h after infection, GO analysis of DEGs revealed 360 entries related to biological processes, 52 cellular components, and 150 molecular functions. According to the GO annotations analysis at Level 2, the most significant enrichment of DEGs was in the oxidation-reduction process, heterochromatin assembly by small RNA, isoprenoid biosynthetic process ontology, transcription, and RNA-templated in the biological process ontology. The most significantly enriched molecular function ontology was oxidoreductase activity, catalytic activity, oxidoreductase activity (acting on CH-OH group of donors), cofactor binding, oxidoreductase activity (acting on the CH-OH group of donors, NAD or NADP as acceptor), RNA-directed 5′-3′ RNA polymerase activity, miRNA binding, coenzyme binding, and regulatory RNA binding, while myelin sheath, an intrinsic component of the membrane and integral component of membrane occupied the important positions in the cellular component ontology. At 24 h after infection, DEGs were divided into 390 functional categories according to 275 biological processes, 34 cellular components, and 81 molecular functions. The most enriched DEGs in the biological processes belonged to heterochromatin assembly by small RNA, cytoplasmic translation, response to sucrose, response to disaccharide and transcription, and RNA-templated. There were 34 terms related to cellular components, among which the most significantly enriched were cytosolic ribosome, ribosome, ribosomal subunit, cytosolic small ribosomal subunit, and small ribosomal subunit; 81 GO terms related to molecular function were identified, of which, RNA-directed 5′-3′ RNA polymerase activity and structural constituent of ribosome were the most significantly enriched metabolic pathways. In the biological process ontology, CK vs. GT2, CK vs. GT12, and CK vs. GT24 had the two most significantly enriched terms, namely, heterochromatin assembly by small RNA and transcription, RNA-templated. RNA-directed 5′-3′ RNA polymerase activity was the most significantly enriched cellular component ontology term in CK vs. GT2, CK vs. GT12, and CK vs. GT24. Five of the most significantly enriched terms in CK vs. GT2 and CK vs. GT12 were related to molecular function ontology, which were catalytic activity, oxidoreductase activity, oxidoreductase activity (acting on the CH-OH group of donors) and regulatory RNA binding, and were also enriched in CK vs. GT24.
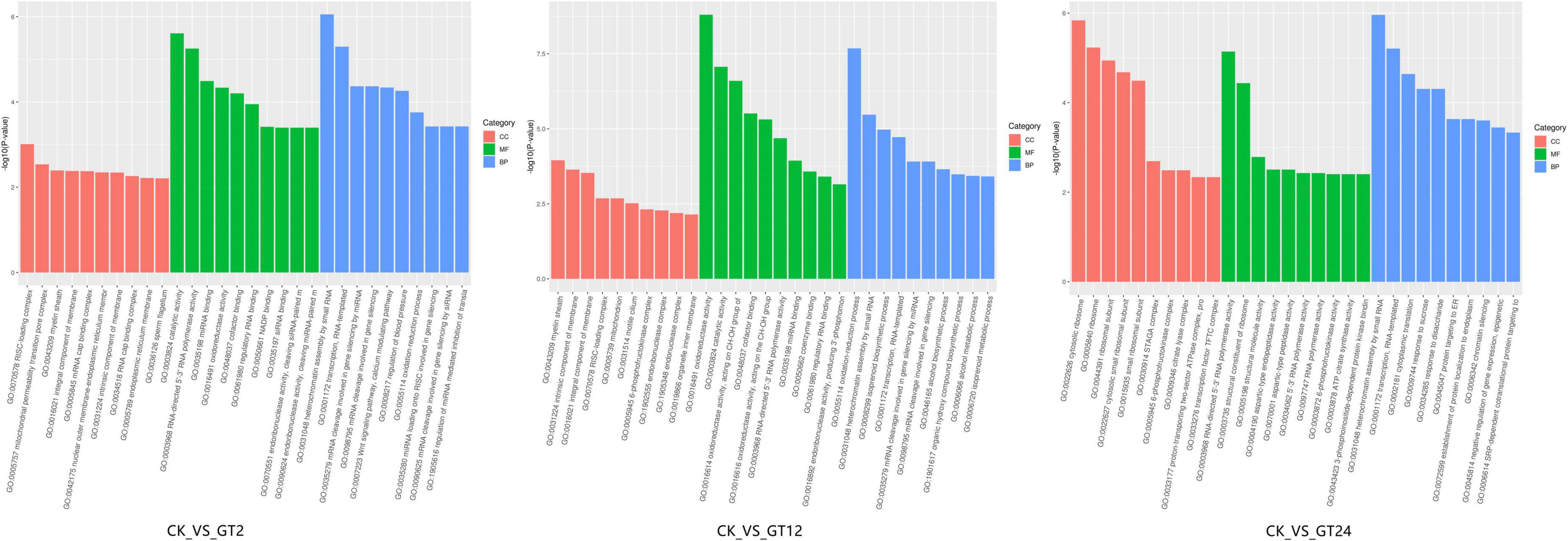
Figure 2. Gene Ontology (GO) functional enrichment analyzes of differentially expressed genes (DEGs).
To elucidate the main metabolic pathways involved in the DEGs responding to T. hengshanicum stress in G. lingzhi, the KEGG enrichment analysis was conducted in CK vs. GT2, CK vs. GT12, and CK vs. GT24. The top 20 KEGG pathways with the lowest false discovery rate (FDR) values are shown in Figure 3. The greater the richness factor, the greater the enrichment. DEGs annotated 93 metabolic pathways in CK vs. GT2, CK vs. GT12, and CK vs. GT24. Sphingolipid metabolism, ether lipid metabolism, steroid biosynthesis, and valine, leucine and isoleucine degradation were the most significantly enriched metabolic pathways in CK vs. GT2. Terpenoid backbone biosynthesis, glycine, serine and threonine metabolism, tryptophan metabolism, ascorbate, and aldarate metabolism, fatty acid degradation, arachidonic acid metabolism, glycolysis/gluconeogenesis, pyruvate metabolism, lysine degradation, steroid biosynthesis, glycerolipid metabolism, oxidative phosphorylation, phenylalanine metabolism, tyrosine metabolism, linoleic acid metabolism, histidine metabolism, valine, leucine and isoleucine degradation, folate biosynthesis, methane metabolism, and hippo signaling pathway-multiple species were the most significantly enriched metabolic pathways in CK vs. GT12. The most significantly enriched metabolic pathways in CK vs. GT24 belonged to the ribosome, sphingolipid metabolism, glycerophospholipid metabolism, ether lipid metabolism, and biosynthesis of unsaturated fatty acids. Valine, leucine and isoleucine degradation was significantly enriched in CK vs. GT2, and CK vs. GT12, and also enriched in CK vs. GT24. There were two significant metabolic pathways in CK vs. GT2 and CK vs. GT24: sphingolipid metabolism and ether lipid metabolism, which were also enriched in CK vs. GT12.
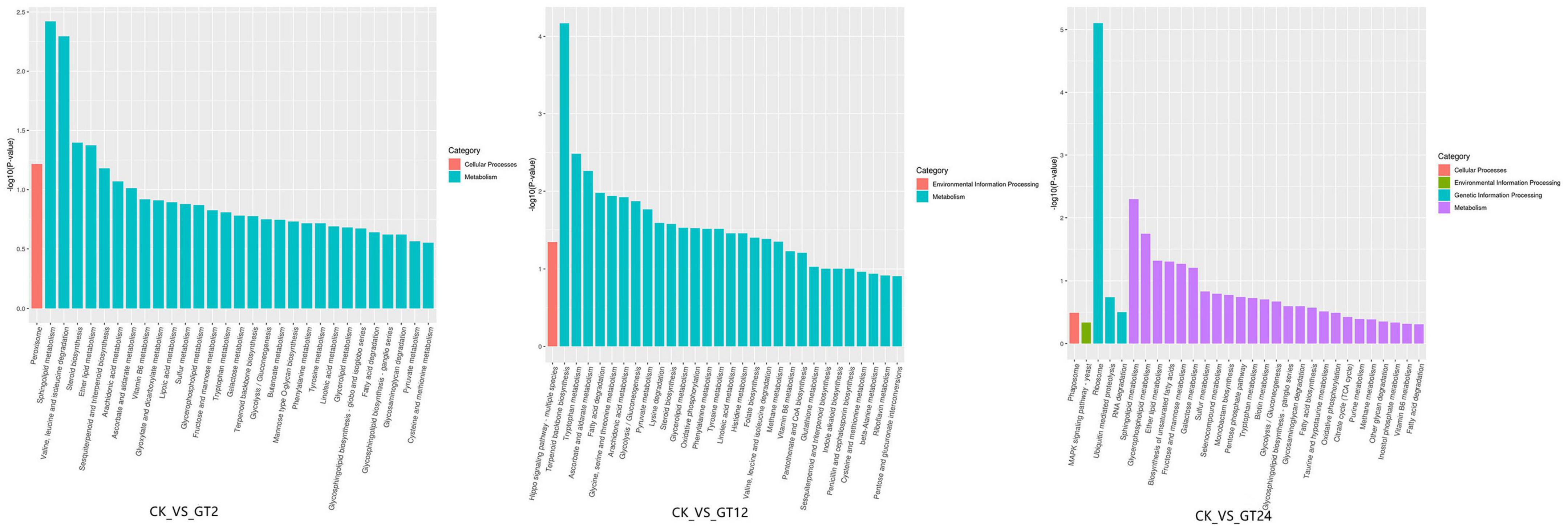
Figure 3. Kyoto Encyclopedia of Genes and Genome (KEGG) pathway enrichment analyzes of differentially expressed genes (DEGs).
qRT-PCR analysis
To confirm the reliability of the generated RNA-seq data, the expression of six DEGs was analyzed using qRT-PCR validation. Genescloud tools2 were used to visualize the results (Figure 4). Although the magnitude of differences in expression detected by qRT-PCR was not identical to those of DEGs detected by the RNA-seq results from the samples at three infection time points, the direction of the change in DEG expression was consistently using the two approaches, indicating that the results of transcriptome sequencing were highly reliable.
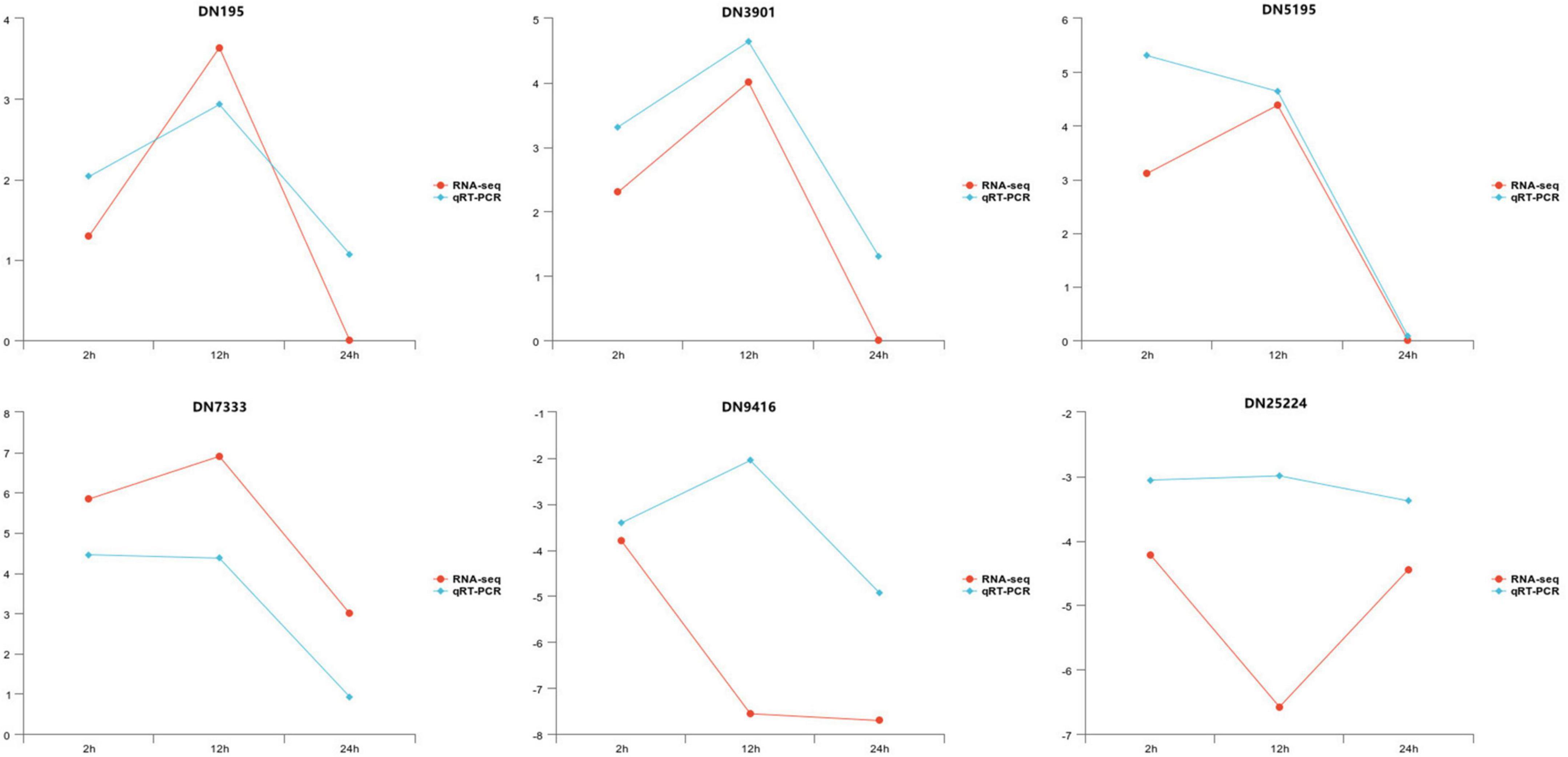
Figure 4. The relative expression level change of six selected genes from differentially expressed genes (DEGs) by quantitative real-time polymerase chain reaction (qRT-PCR).
Sphingolipid metabolism
The basic structure of biomembranes comprises various lipids, such as glycerophospholipids, sphingolipids, and sterols, and proper homeostasis of the composition of these lipids in biomembranes is extremely important for the maintenance of multiple cellular functions (Tani and Funato, 2018). Sphingolipids are essential biomembrane lipids for eukaryotic organisms. They commonly have a long-chain base (LCB) backbone. Ceramide (Cer), the hydrophobic portion of sphingolipids, comprises an LCB and a fatty acid (Dickson et al., 2006; Tani and Funato, 2018). In this study, the sphingolipid metabolism pathway of G. lingzhi was significantly enriched after T. hengshanicum infection (Figure 5). As shown in the Figure 6, two genes were up-regulated and three genes were down-regulated in the phospholipid metabolic pathway at 2 h after infection. At 12 h after infection, one gene was up-regulated and two genes were down-regulated in the phospholipid metabolic pathway. At 24 h after infection, one gene was up-regulated and four genes were down-regulated in the phospholipid metabolic pathway. This indirectly showed that T. hengshanicum infection would affect sphingolipid metabolism in G. lingzhi.
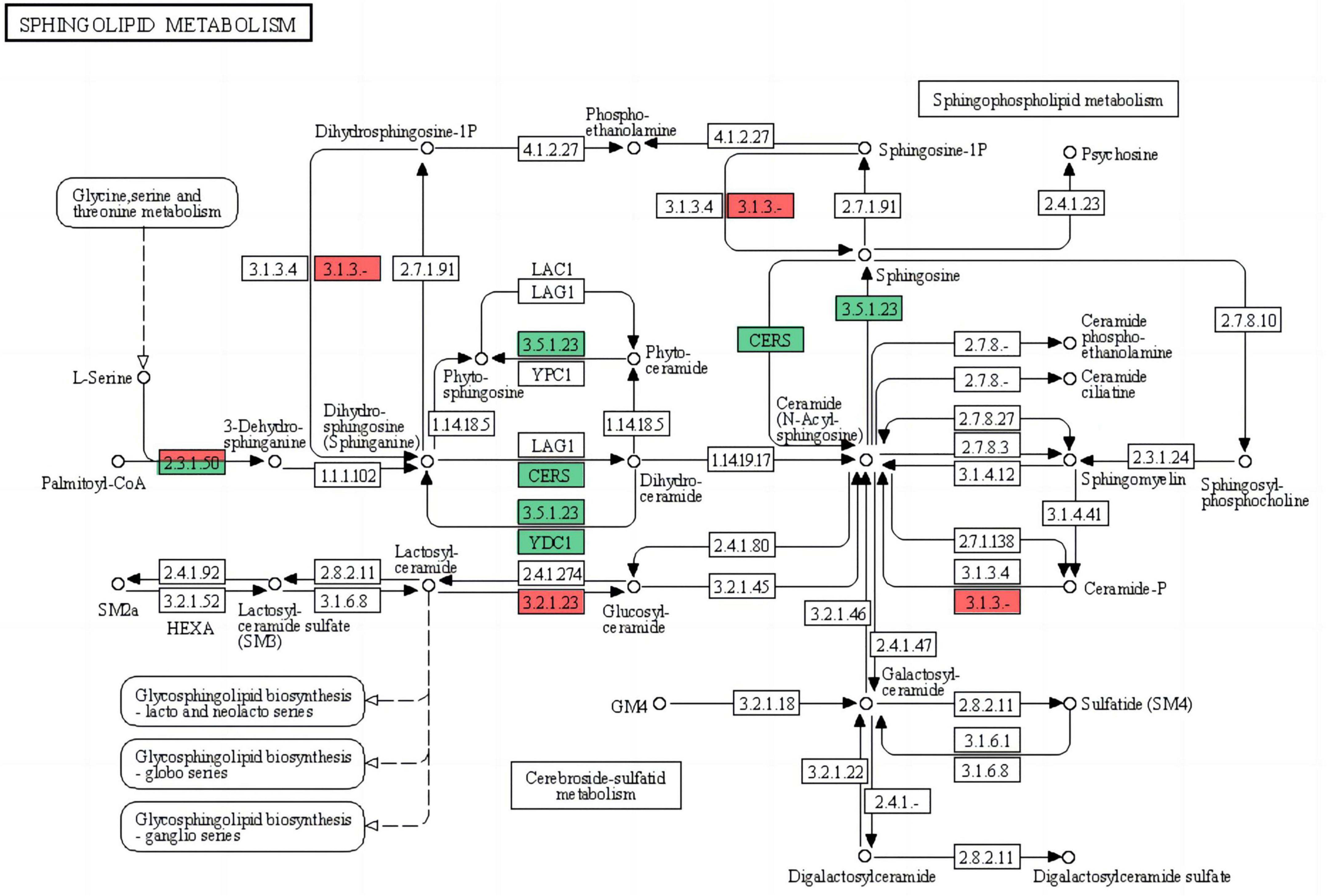
Figure 5. Differentially expressed genes (DEGs) comparison between two samples (CK vs. GT2, CK vs. GT12, and CK vs. GT24) mapped to the sphingolipid metabolism (map00600; red for up-regulated, green for down-regulated).
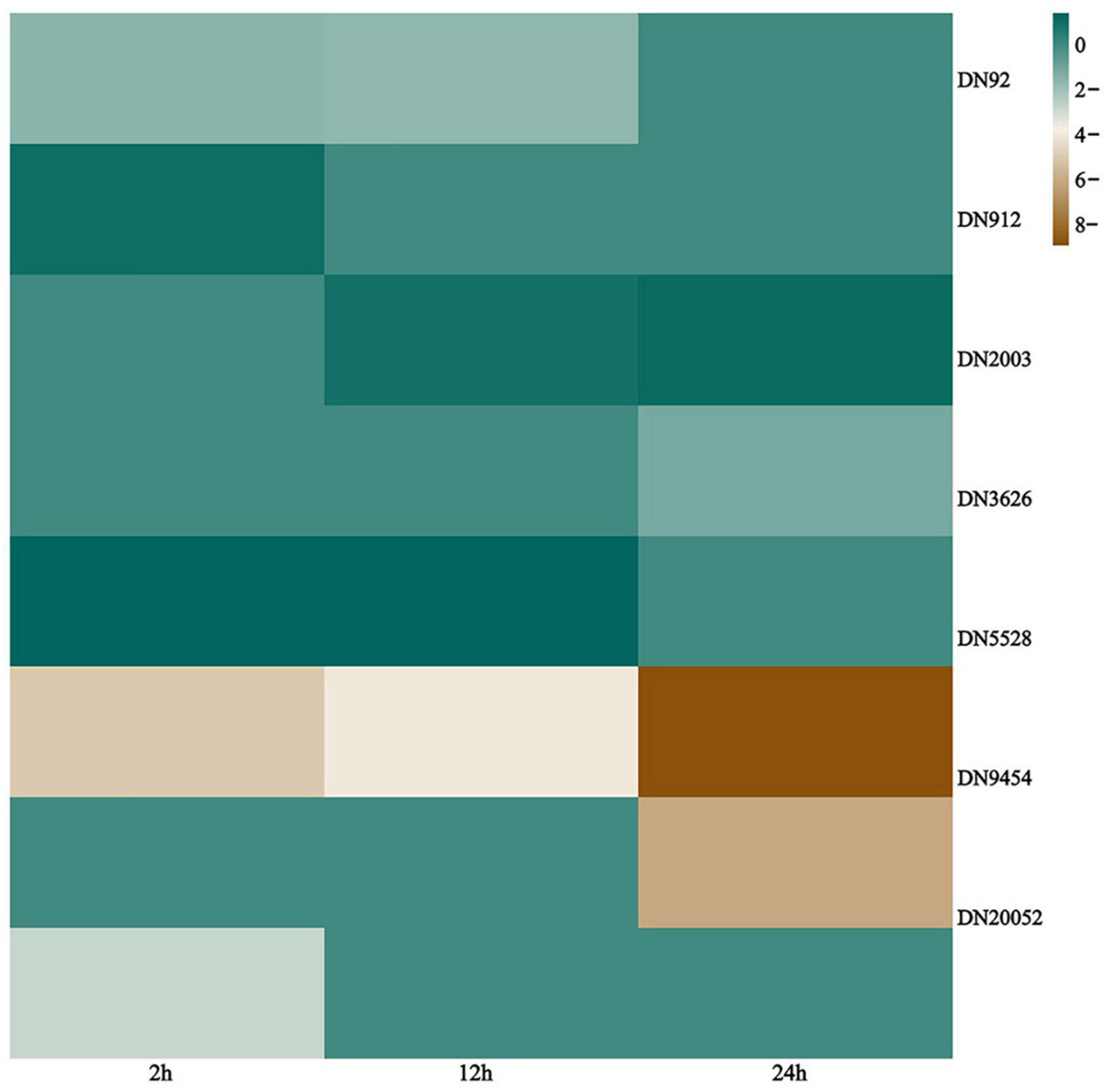
Figure 6. Heatmaps of differentially expressed genes (DEGs) involved in sphingolipid metabolism pathways. The log2Foldchange was colored using Genescloud tools (green for up-regulated, brown for down-regulated).
Pathogenesis-related proteins
Pathology-associated proteins are a class of proteins induced by plants in pathological or pathology-associated environments, initially detected from tobacco mosaic virus infection in tobacco leaves (Li, 2020). Pathogenesis-related (PR) proteins are comprised of 17 families that are normally expressed at low levels in healthy tissues but rapidly accumulate to significant levels in response to biotic or abiotic stress (Van Loon and Van Strien, 1999; Van Loon et al., 2006; Zhu et al., 2017). It was known that the timing of PR gene expression was a crucial determinant of pathogenesis. The accumulation of PR proteins is usually associated with systemic acquired resistance to a wide range of pathogens. G. lingzhi PR genes were induced in response to T. hengshanicum infection in our experiment. Our results showed that G. lingzhi up-regulated PR thaumatin-like proteins (TLPs) (PR-5s) after infection with T. hengshanicum. The PR-5s genes were up-regulated 8.6-fold at 2 h post-infection (Figure 7).
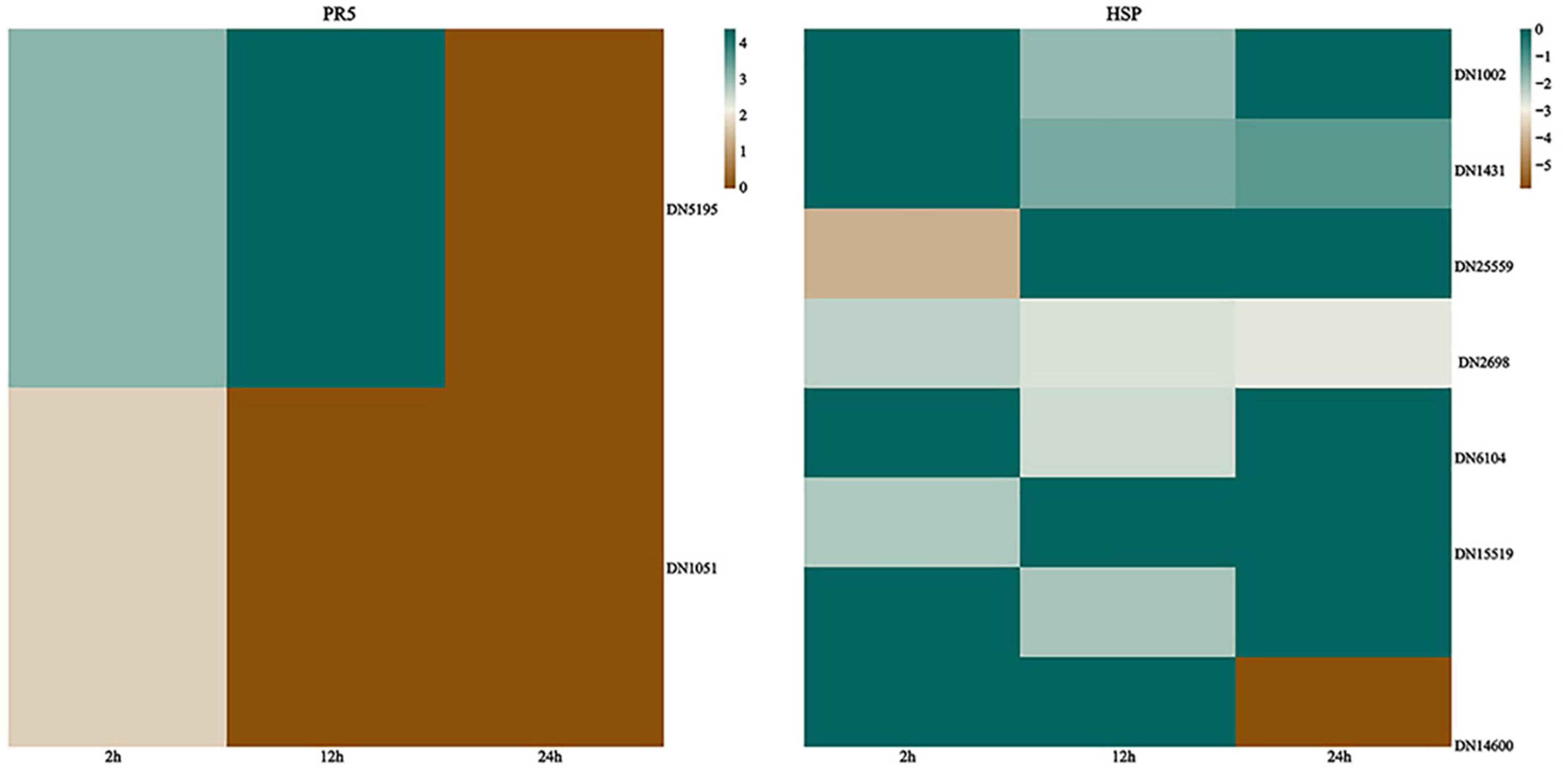
Figure 7. Left: Heatmaps of differentially expressed genes (DEGs) encoding thaumatin-like proteins (TLPs); right: Heatmap of DEGs encoding heat shock proteins (HSPs). The log2Foldchange was colored using Genescloud tools (green for up-regulated, brown for down-regulated), each horizontal row represents a DEG with its gene ID.
Heat shock proteins (HSPs) are a subset of molecular chaperones, best known because they are rapidly induced in large numbers by stress (Neumann et al., 1994; Wang et al., 2004; Scarpeci et al., 2008). These proteins are implicated in a wide variety of cellular processes as molecular chaperons, including the protection of the proteome from stress, the folding and transport of newly synthesized polypeptides, the activation of proteolysis of misfolded proteins, and the formation and dissociation of protein complexes, plays a pivotal role in the protein quality control system, ensuring the correct folding of proteins, the re-folding of misfolded proteins, and controlling the targeting of proteins for subsequent degradation. The results showed that HSPs in the G. lingzhi displayed a range of responses to T. hengshanicum. At the three time points of infection, the HSPs genes of G. lingzhi were down-regulated (Figure 7).
Discussion and conclusion
Ganoderma lingzhi has high application value in the prevention and treatment of nephritis, hypertension, and bronchitis, and has remarkable antitumor properties, and is deeply loved by people (Wu et al., 2011; Nie et al., 2013; Vitak et al., 2015; Paterson, 2016; Yan et al., 2019). With the rapid expansion of G. lingzhi cultivation, however, green mold has become one of the severe diseases threatening the production of G. lingzhi. After the fruiting bodies were infected with Trichoderma spp., lesions appeared and were covered by green mycelium. The infected fruiting bodies became deformed and withered as the disease progressed (Yan et al., 2019; Cai et al., 2020). Therefore, it is important to use disease-resistant variety to control disease that damage the quantity and quality of G. lingzhi. Currently, there are many studies on the interaction between plants and pathogens. In response to external biotic stresses, plants induce a series of immune responses, including the production of physical barriers (keratin, wax, lignin, and special stomatal structures), chemical barriers (secondary metabolites with antimicrobial properties), and molecular responses (hypersensitivity, production of reactive oxygen species, and expression of pathogen-associated genes) (Ding and Yang, 2016). Therefore, it is inevitable that macrofungi will also produce various defensive responses to resist the pathogen infection. For breeding disease-resistant G. lingzhi, it is necessary to have information on genetic variation in the hosts reaction to disease infestation. In this research, we studied the transcription of G. lingzhi at 2, 12, and 24 h after infection by T. hengshanicum, and explored the resistance genes and metabolic pathways induced by T. hengshanicum.
In this study, we selected six DEGs related to the T. hengshanicum infection of G. lingzhi for qRT-PCR verification. Our results revealed the same trend in DEG expression as established by transcriptome sequencing results, indicating high reliability of the transcriptome sequencing results in reflecting the proper expression levels of genes in G. lingzhi infected by T. hengshanicum. The six DEGs verified by qRT-PCR were the TLPs (PR-5s) gene, the phenylalanine ammonia-lyase gene, the cyanide hydratase gene, the Beta-1,3-glucan binding protein, the polyketide synthase gene, and the 6-phosphofructokinase gene. TLPs have antifungal and osmotic adjustment activities or act as an elicitor of other antifungal proteins and play an important role in the growth and development of the host and the process of stress resistance (Menu-Bouaouiche et al., 2003; Guo et al., 2016; Faillace et al., 2019; Sun et al., 2020; Liu Y. et al., 2021; Liu et al., 2022). Previous studies have shown that PR-5s is involved in the plant defense response induced by diseases and insects (Hou et al., 2018). For example, overexpression of the rice TLPs gene significantly increased the resistance of rice, and wheat to related diseases (Chen et al., 1999; Datta et al., 1999). As a result, up-regulation of PR5 expression at 2 h after infection benefits G. lingzhi in preventing a violation by T. hengshanicum. The phenylalanine ammonia-lyase gene was also up-regulated after infection. As the first rate-limiting enzyme in the phenylpropanoid metabolism pathways, phenylalanine ammonia-lyase (PALs) can catalyze the deamination of L-phenylalanine to form trans-cinnamic acid, which is a precursor of lignin, salicylic acid (SA), flavonoids, phytoalexins, as well as bioactive phenolamides via specific branch pathways, playing important roles in plant growth, development, and stress responses (Dixon et al., 2002; Zhang and Liu, 2015; You et al., 2020). For example, enhanced deposition of lignin can reinforce the plant cell wall, providing a structural barrier to pathogen spread, and the toxic phenolic precursors produced during lignin biosynthesis or polymerization can directly inhibit pathogen multiplication and movement (Tonnessen et al., 2015). The up-regulation of phenylalanine ammonia-lyase gene expression in G. lingzhi might activate the phenylpropanoid metabolic pathway in G. lingzhi and then produce some or specific related secondary metabolites to resist the infection of T. hengshanicum. Moreover, the Beta-1,3-glucan binding protein (LGBP) molecule was reported to have antibacterial, anti-biofilm, anti-inflammatory, and antioxidant properties (Iswarya et al., 2017). Many studies have shown that β-1,3-glucan binding proteins are host pattern recognition receptors (PRRs) that recognize conserved surface ligands in microorganisms designed the pathogen-associated molecule patterns (PAMPs) and have a strong affinity toward the –glucans present on the surface of bacteria and fungi, thereby activating the prophenoloxidase (proPO) activating system to elicit the invertebrate innate defense system (Zhang et al., 2016; Anjugam et al., 2017; Li S. S. et al., 2022). The results of this study revealed that T. hengshanicum infection of G. lingzhi not only up-regulated β-1,3-glucan binding proteins but also tyrosinase, and the infection site of G. lingzhi turned brown 24 h after T. hengshanicum infection, which could be due to activation of the prophenoloxidase system in G. lingzhi, resulting in the production of melanin or other secondary metabolite deposition. Intermediate sphingolipid metabolic pathways are important signal molecules closely related to cell growth, apoptosis, differentiation, senescence, stress resistance, and signaling transduction (Li J. et al., 2022). Free sphingosine binds to different substances in organisms to form Cer, sphingomyelin, and glycosphingolipids (Dickson et al., 2006; Harrison et al., 2018). In the study of Saccharomyces cerevisiae, long-chain sphingolipid bases are signaling molecules that regulate growth, responses to heat stress, cell wall synthesis and repair, endocytosis, and dynamics of the actin cytoskeleton (Dickson et al., 2006). In plants, sphingolipids were not only the main components of the plant plasmalemma, tonoplast membrane, and intima but also participated in various plant stress responses as the second messenger of plant defense mechanisms (Markham et al., 2013; Shan et al., 2019). Changes in sphingolipid content and sphingolipid/phosphorylated derivative balance in plants infected with microorganisms regulated plants to produce a resistance response (Bi et al., 2014; Yanagawa et al., 2017). Therefore, in the interaction between G. lingzhi and T. hengshanicum, G. lingzhi sphingolipids might be the signal molecules that could induce resistance G. lingzhi.
These data provided a better understanding of the mechanisms and identified potential DEGs involved in the early disease defenses of G. lingzhi against T. hengshanicum. These DEGs could be used as a screening indicator for identifying or selecting strains with high-disease-resistant properties. However, the results of this study were slightly different from those of plant-pathogen interactions. No DEGs related to plant hormones (JA, SA, and ABA, etc.) signaling transduction pathways were found in G. lingzhi infected by T. hengshanicum. In plants, SA and JA were endogenous plant hormones, that could induce the expression of pathogenicity-related proteins and systemic acquired resistance in plants, and were also recognized as a signal of plant responses to abiotic and biotic stresses. In A. bisporus, after P. tolaasii infection, JA biosynthesis and signaling transduction pathways were significantly enriched, and JA content was also detected to increase (Ma et al., 2021). Furthermore, no differential changes in genes related to cell wall synthesis were found in this study. In plants, the cuticle is the first cell wall layer encountered by a pathogen, plant pathogens must overcome the physical barrier presented by the cuticle and plant cell wall, so that the plant cell wall undergoes very large cell wall remodeling. Therefore, the changes in genes related to plant hormones and cell wall synthesis enzymes must be further studied.
In conclusion, transcriptomic analysis detected 620, 754, and 604 DEGs at 2, 12, and 24 h after infection with T. hengshanicum. Transcriptome sequencing indicated that there were 162 DEGs at three infection time points, containing 15 up-regulated DEGs and 147 down-regulated DEGs. After G. lingzhi was infected by T. hengshanicum, most of the DEGs were down-regulated at three-time points, indicating that G. lingzhi may resist the infection of T. hengshanicum mainly by down-regulating gene expression. Resistance-related genes TLPs (PR-5s) gene, phenylalanine ammonia-lyase gene, Beta-1,3-glucan binding protein were significantly up-regulated. At the three-time points of infection, the HSPs genes of G. lingzhi were down-regulated. The down-regulation of HSPs genes led to the inhibition of HSP function, which may compromise the HSP-mediated defense signaling transduction pathway, leading to G. lingzhi susceptibility. We performed GO and pathway enrichment analysis of DEGs at 2, 12, and 24 h for susceptible G. lingzhi, respectively. Four different gene sets were enriched for GO classification and KEGG enrichment, the main GO enrichment included heterochromatin assembly by small RNA and transcription, RNA-templated, RNA-directed 5′-3′ RNA polymerase activity, catalytic activity, oxidoreductase activity, oxidoreductase activity (acting on CH-OH group of donors), and regulatory RNA binding and the enriched pathways included sphingolipid metabolism, ether lipid metabolism, and valine, leucine and isoleucine degradation pathway. Although the T. hengshanicum pathogens induced resistance in G. lingzhi, such resistance could not completely prevent pathogen invasion, thereby showing disease symptoms. In conclusion, our results revealed the DEGs and metabolic pathways in the early defense response of Trichoderma spp. and provided a theoretical basis for the breeding of resistant strains of G. lingzhi.
Data availability statement
The data presented in the study are deposited in the National Center for Biotechnology Information Sequence Read Archive repository, accession number PRJNA917261.
Author contributions
TW wrote the manuscript. TW and CZ carried out experiments and collecting specimens. JX revised the manuscript and designed experiments. XL revised the manuscript and submitted the transcriptome data. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors thank the Science and Technology Department of Jilin Province (20210202080NC and 20220204066YY) and The Agricultural Key Core Technology Demonstration and Promotion (Agro-industry Technology Research System) Project of Jilin Province.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Anjugam, M., Vaseeharan, B., Iswarya, A., Amala, M., Govindarajan, M., Alharbi, N., et al. (2017). A study on β-glucan binding protein (β-GBP) and its involvement in phenoloxidase cascade in Indian white shrimp Fenneropenaeus indicus. Mol. Immunol. 92, 1–11. doi: 10.1016/j.molimm.2017.09.013
Bailey, A. M., Collopy, P. D., Thomas, D. J., Sergeant, M. R., Costa, A. M., Barker, G., et al. (2013). Transcriptomic analysis of the interactions between Agaricus bisporus and Lecanicillium fungicola. Fungal. Genet. Biol. 55, 67–76. doi: 10.1016/j.fgb.2013.04.010
Bi, F. C., Liu, Z., Wu, J. X., Liang, H., Xi, X. L., Fang, C., et al. (2014). Loss of ceramide kinase in Arabidopsis impairs defenses and promotes ceramide accumulation and mitochondrial H2O2 bursts. Plant Cell 26, 3449–3467. doi: 10.1105/tpc.114.127050
Cai, M. Z., Idrees, M., Zhou, Y., Zhang, C. L., and Xu, J. Z. (2020). First report of green mold disease caused by Trichoderma hengshanicum on Ganoderma lingzhi. Mycobiology 48, 427–430. doi: 10.1080/12298093.2020.1794230
Cao, Y., Wu, S. H., and Dai, Y. C. (2012). Species clarification of the prize medicinal Ganoderma, mushroom “Lingzhi.” Fungal Divers. 56, 49–62. doi: 10.1007/s13225-012-0178-5
Chen, W. P., Chen, P. D., Liu, D. J., Kynast, R., Friebe, B., Velazhahan, R., et al. (1999). Development of wheat scab symptoms is delayed in transgenic wheat plants that constitutively express a rice thaumatin-like protein gene. Theor. Appl. Genet. 99, 755–760. doi: 10.1007/s001220051294
Datta, K., Velazhahan, R., Oliva, O., Ona, I., Mew, T., Khush, G. S., et al. (1999). Over-expression of the cloned rice thaumatin-like protein (PR-5) gene in transgenic rice plants enhances environmental friendly resistance to Rhizoctonia solani causing sheath blight disease. Theor. Appl. Genet. 98, 1138–1145. doi: 10.1007/s001220051178
Dickson, R. C., Sumanasekera, C., and Lester, R. L. (2006). Functions and metabolism of sphingolipids in Saccharomyces cerevisiae. Prog. Lipid. Res. 45, 447–465. doi: 10.1016/j.plipres.2006.03.004
Ding, L. N., and Yang, G. X. (2016). Research advances in the mechanism and signal transduction of plant disease resistance. Biotechnol. Bull. 32, 109–117. doi: 10.13560/j.cnki.biotech.bull.1985.2016.10.013
Dixon, R. A., Achnine, L., Kota, P., Liu, C. J., Reddy, M. S., and Wang, L. J. (2002). The phenylpropanoid pathway and plantdefence-a genomics perspective. Mol. Plant Pathol. 3, 371–390. doi: 10.1046/j.1364-3703.2002.00131.x
Faillace, G. R., Turchetto-Zolet, A. C., and Guzman, F. L. (2019). Genome-wide analysis and evolution of plant thaumatin-like proteins: A focus on the origin and diversification of osmotins. Mol. Genet. Genomics 294, 1137–1157. doi: 10.1007/s00438-019-01554-y
Fan, R., Tao, X. Y., Xia, Z. Q., Sim, S., Hu, L. S., Wu, B. D., et al. (2022). Comparative transcriptome and metabolome analysis of resistant and susceptible piper species upon infection by the oomycete Phytophthora capsici. Front. Plant Sci. 23:864927. doi: 10.3389/fpls.2022.864927
Fu, S. Y., Chen, S. L., and Yan, S. Z. (2013). Colonization dynamics of endophytic bacteria in Capsicum annuum and their control effects on pepper Phytophthora blight. Chin. J. Biol. Control 29, 561–568. doi: 10.16409/j.cnki.2095-039x.2013.04.014
Gea, F. J., Navarro, M. J., Santos, M., Diánez, F., and Carrasco, J. (2021). Control of fungal diseases in mushroom crops while dealing with fungicide resistance: A review. Microorganisms 9:585. doi: 10.3390/microorganisms9030585
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Guo, J., Zhao, X., and Wang, H. L. (2016). Expression of the LePR5 gene from cherry tomato fruit induced by Cryptococcus laurentii and the analysis of LePR5 protein antifungal activity. Postharvest. Biol. Technol. 111, 337–344. doi: 10.1016/j.postharvbio.2015.09.002
Harrison, P. J., Dunn, T. M., and Campopiano, D. J. (2018). Sphingolipid biosynthesis in man and microbes. Nat. Prod. Rep. 35, 921–954. doi: 10.1039/c8np00019k
Hou, X. J., Li, X. W., Wang, Y. C., Yu, Y. C., Gao, Y., and Sun, X. L. (2018). Cloning and expression analysis of the gene encoding pathogenesis-related protein CsPR5 in the tea plant. J. Plant Prot. 45, 1078–1087. doi: 10.13802/j.cnki.zwbhxb.2018.2018937
Huang, X. W., Yan, Y. H., Zhang, T., Geng, L. J., Cheng, Z., and Xu, J. Z. (2018). Isolation, identification and rapid detection of the pathogen causing green mold on Ganoderma lingzhi. J. Plant Prot. 45, 1435–1436. doi: 10.13802/j.cnki.zwbhxb.2018.2018082
Iswarya, A., Anjugam, M., and Vaseeharan, B. (2017). Role of purified β-1, 3 glucan binding protein (β-GBP) from Paratelphusa hydrodromus and their anti-inflammatory, antioxidant and antibiofilm properties. Fish Shellfish Immunol. 68, 54–64. doi: 10.1016/j.fsi.2017.07.007
Jin, X., Liu, Z. M., Huang, Y. J., Huang, W. L., and Zheng, L. Y. (2016). Current situation and development trend of Ganoderma lucidum cultivation in China. Edib. Med. Mushrooms 24, 33–37.
Li, B., and Dewey, C. N. (2011). RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, C. D., Zhang, Y. X., Liu, Q. D., and Tang, Y. Q. (2021). Establishment of the rapid detecion system green mold on Ganoderma lingzhi caused by Trichoderma longibrachiatum. Tianjin Agric. Sci. 27, 61–65.
Li, J., Song, J. J., Zhou, Y. J., Li, G. Q., and Zhang, J. (2022). Cloning and sequence expression analysis of sphingolipid metabolic pathway gene Bcsphl1 in necrotrophic fungus Botrytis cinerea. J. Plant Prot. 49, 1046–1052. doi: 10.13802/j.cnki.zwbhxb.2022.2020317
Li, M. Y. (2020). Functional study of grape pathogenesis-related protein 4b against downy mildew. Xianyang: Northwest A&F University.
Li, S. S., Hao, Z. P., Xu, H. H., Gao, Y., Zhang, M. Y., Liang, J., et al. (2022). Silencing β-1,3-glucan binding protein enhances the susceptibility of Plutella xylostella to entomopathogenic fungus Isaria cicadae. Pest. Manage. Sci. 78, 3117–3127. doi: 10.1002/ps.6938
Liu, C., Han, L. H., Sheng, Q., and Tao, W. (2022). Study on gene evolution and codon characteristics of thaumatin-like protein family of eight plant species. Jiangsu Agric. Sci. 50, 44–51. doi: 10.15889/j.issn.1002-1302.2022.10.007
Liu, L. N., Chen, F. D., Chen, S. M., Fang, W. M., Liu, Y., and Guan, Z. Y. (2021). Dual species dynamic transcripts reveal the interaction mechanisms between Chrysanthemum morifolium and Alternaria alternata. BMC Genomics 22:523. doi: 10.1186/s12864-021-07709-9
Liu, Y., Liu, L., Asiegbu, F. O., Yang, C., Han, S., Yang, S., et al. (2021). Molecular identification and antifungal properties of four thaumatin-like proteins in spruce (Picea likiangensis). Forests 12:1268. doi: 10.3390/f12091268
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Luković, J., Milijašević-Marčić, S., Hatvani, L., Kredics, L., Szűcs, A., Vágvölgyi, C., et al. (2021). Sensitivity of Trichoderma strains from edible mushrooms to the fungicides prochloraz and metrafenone. J. Environ. Sci. Health B 56, 54–63. doi: 10.1080/03601234.2020.1838821
Ma, X. L., Fan, X. L., Wang, G. Z., Xu, R. P., Yan, L. L., Zhou, Y., et al. (2021). Enhanced expression of thaumatin-like protein gene (LeTLP1) endows resistance to Trichoderma atroviride in Lentinula edodes. Life 11:863. doi: 10.3390/life11080863
Ma, Y. (2017). Research on markering strategy of Ganoderma lucidum teain CW E-Commerce Company Ltd. Changchun: Jilin University.
Markham, J. E., Lynch, D. V., Napier, J. A., Dunn, T. M., and Cahoon, E. B. (2013). Plant sphingolipids: Function follows form. Curr. Opin. Plant Biol. 16, 350–357. doi: 10.1016/j.pbi.2013.02.009
Menu-Bouaouiche, L., Vriet, C., and Peumans, W. J. (2003). A molecular basis for the endo-β-1,3-glucanase activity of the thaumatin-like proteins from edible fruits. Biochimie 85, 123–131. doi: 10.1016/s0300-9084(03)00058-0
Milijašević-Marčić, S., Stepanović, M., Todorović, B., Duduk, B., Stepanović, J., Rekanović, E., et al. (2017). Biological control of green mould on Agaricus bisporus by a native Bacillus subtilis strain from mushroom compost. Eur. J. Plant Pathol. 148, 509–519. doi: 10.1007/s10658-016-1107-3
Neumann, D., Lichtenberger, O., Günther, D., Tschiersch, K., and Nover, L. (1994). Heat-shock proteins induce heavy-metal tolerance in higher plants. Planta 194, 360–367. doi: 10.1007/BF00197536
Nie, S. P., Zhang, H., Li, W. J., and Xie, M. Y. (2013). Current development of polysaccharides from Ganoderma: Isolation, structure and bioactivities. Bioact. Carbohydr. Diet. Fibre 1, 10–20. doi: 10.1016/j.bcdf.2013.01.001
Paterson, R. R. (2016). Ganoderma-A therapeutic fungal biofactory. Phytochemistry 67, 1985–2001. doi: 10.1016/j.phytochem.2006.07.004
Qian, J., Xu, H. B., Song, J. Y., Xu, J., Zhu, Y. J., and Chen, S. L. (2013). Genome-wide analysis of simple sequence repeats in the model medicinal mushroom Ganoderma lucidum. Gene 512, 331–336. doi: 10.1016/j.gene.2012.09.127
Sana, T. R., Fischer, S., Wohlgemuth, G., Katrekar, A., Jung, K. H., Ronald, P. C., et al. (2010). Metabolomic and transcriptomic analysis of the rice response to the bacterial blight pathogen Xanthomonas oryzae pv. oryzae. Metabolomics 6, 451–465. doi: 10.1007/s11306-010-0218-7
Sanodiya, B. S., Thakur, G. S., Baghel, R. K., Prasad, G. B., and Bisen, P. S. (2009). Ganoderma lucidum: A potent pharmacological macrofungus. Curr. Pharm. Biotechnol. 10, 717–742. doi: 10.2174/138920109789978757
Satria, D., Tamrakar, S., Suhara, H., Kaneko, S., and Shimizu, K. (2019). Mass spectrometry-based untargeted metabolomics and α-glucosidase inhibitory activity of lingzhi (Ganoderma lingzhi) during the developmental stages. Molecules 24:2044. doi: 10.3390/molecules24112044
Scarpeci, T. E., Zanor, M. I., and Valle, E. M. (2008). Investigating the role of plant heat shock proteins during oxidative stress. Plant Signal. Behav. 3, 856–857. doi: 10.4161/psb.3.10.6021
Shan, T. T., Chen, X. M., Guo, S. X., Tian, L. X., Yan, L., and Wang, X. (2019). Advances in molecular regulation of sphingolipids in plant-fungus interactions. Chin. Bull. Bot. 54, 396–404. doi: 10.11983/CBB18099
Sun, W. B., Zhou, Y., Movahedi, A., Hui, W., and Qiang, Z. G. (2020). Thaumatin-like protein (Pe-TLP) acts as a positive factor in transgenic poplars enhanced resistance to spots disease. Physiol. Mol. Plant Pathol. 112:101512. doi: 10.1016/j.pmpp.2020.101512
Tani, M., and Funato, K. (2018). Protection mechanisms against aberrant metabolism of sphingolipids in budding yeast. Curr. Genet. 64, 1021–1028. doi: 10.1007/s00294-018-0826-8
Tonnessen, B. W., Manosalva, P., Lang, J. M., Baraoidan, M., Bordeos, A., Mauleon, R., et al. (2015). Rice phenylalanine ammonia-lyase gene OsPAL4 is associated with broad spectrum disease resistance. Plant Mol. Biol. 87, 273–286. doi: 10.1007/s11103-014-0275-9
Van Loon, L. C., Rep, M., and Pieterse, C. M. (2006). Significance of inducible defense-related proteins in infected plants. Annu. Rev. Phytopathol. 44, 7.1–7.28. doi: 10.1146/annurev.phyto.44.070505.143425
Van Loon, L. C., and Van Strien, E. A. (1999). The families of pathogenesisrelated proteins, their activities, and comparative analysis of PR-1 type proteins. Physiol. Mol. Plant Pathol. 55, 85–97. doi: 10.1006/pmpp.1999.0213
Vitak, T. Y., Wasser, S. P., Nevo, E., and Sybirna, N. O. (2015). The effect of the medicinal mushrooms Agaricus brasiliensis and Ganoderma lucidum (higher Basidiomycetes) on the erythron system in normal and streptozotocin-induced diabetic rats. Int. J. Med. Mushrooms 17, 277–286. doi: 10.1615/intjmedmushrooms
Wagner, R., Mitchell, D. A., Sassaki, G. L., Amazonas, M., and Berovic, M. (2003). Current techniques for the cultivation of Ganoderma for the production Biomas, Ganoderic Acid and polysaccharides. Food Techol. Biotechnol. 41, 371–382.
Wang, S. J., Wang, S. K., Li, M., Su, Y. H., Sun, Z., and Ma, H. B. (2022). Combined transcriptome and metabolome analysis of Nerium indicum L. elaborates the key pathways that are activated in response to witches’ broom disease. BMC Plant Biol. 22:291. doi: 10.1186/s12870-022-03672-z
Wang, W. X., Vinocur, B., Shoseyov, O., and Altman, A. (2004). Role of plant heatshock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 9, 244–252. doi: 10.1016/j.tplants.2004.03.006
Wu, C. T., Lin, T. Y., Sheu, F., Hsu, H. Y., Ho, C. M., and Chen, E. I. T. (2011). Lingzhi-8 mediates p53-dependent growth arrest of lung cancer cells proliferation via the ribosomal protein S7-MDM2-p53 pathway. Carcinogenesis 32, 1890–1896. doi: 10.1093/carcin/bgr221
Xie, P. F., Guo, J. F., Zhang, C. L., and Xu, J. Z. (2018). Screening and preparation of bio-controlling bacterial agents against Ganoderma lingzhi pathogen Trichoderma pleuroticola. J. Microbiol. 38, 75–83.
Xie, Y. L., and Tan, G. J. (2015). Analysis and prevention of the reason Ganoderma lucidum infected by Trichoderma viride. Edib. Fungi China 34, 74–76. doi: 10.13629/j.cnki.53-1054.20
Xu, R., Liu, Z. H., Fu, Y. P., and Li, Y. (2019). Identification and biological characteristics of Cladobotryum mycophilum causing cobweb disease on Ganoderma lingzhi. Mycosystema 38, 669–678. doi: 10.13346/j.mycosystema.180328
Yan, Y. H. (2011). Rearch on identification of Trichoderma of mushrooms and control of Trichoderma, Mycogone cervina. Fujian. Agric. Unive.
Yan, Y. H., Zhang, C. L., Moodley, O., Zhang, L., and Xu, J. Z. (2019). Green mold caused by Trichoderma atroviride on the Lingzhi Medicinal Mushroom, Ganoderma lingzhi (Agaricomycetes). Int. J. Med. Mushrooms 21, 515–521. doi: 10.1615/IntJMedMushrooms.2019030352
Yanagawa, D., Ishikawa, T., and Imai, H. (2017). Synthesis and degradation of long-chain base phosphates affect fumonisin B1-induced cell death in Arabidopsis thaliana. J. Plant Res. 130, 571–585. doi: 10.1007/s10265-017-0923-7
Yang, X. M., Yang, K. X., Wang, X. H., Wang, Y. T., Zhao, Z. Y., and Meng, D. M. (2022). Transcriptomic analysis reveals the mechanism of bacterial disease resistance of postharvest button mushroom (Agaricus bisporus). Physiol. Mol. Plant Pathol. 122:101903. doi: 10.1016/j.pmpp.2022.101903
Ye, W., Liu, T. M., Zhang, W. M., Li, S. N., Zhu, M. Z., Li, H. H., et al. (2019). Disclosure of the molecular mechanism of wheat leaf spot disease caused by Bipolaris sorokiniana through comparative transcriptome and metabolomics analysis. Int. J. Mol. Sci. 20:6090. doi: 10.3390/ijms20236090
You, X. M., Fang, H., Wang, R. Y., Wang, G. L., and Ning, Y. S. (2020). Phenylalanine ammonia lyases mediate broad-spectrum resistance to pathogens and insect pests in plants. Sci. Bull. 65, 1425–1427. doi: 10.1016/j.tplants.2019.06.012
Zhang, X., Zhu, Y. T., Li, X. J., Wang, S. C., Li, D., Li, W. W., et al. (2016). Lipopolysaccharide and beta-1, 3-glucan binding protein (LGBP) stimulates prophenoloxidase activating system in Chinese mitten crab (Eriocheir sinensis). Dev. Comp. Immunol. 61, 70–79. doi: 10.1016/j.dci.2016.03.017
Zhang, X. B., and Liu, C. J. (2015). Multifaceted regulations of gateway enzyme phenylalanine ammonia-lyase in the biosynthesis of phenylpropanoids. Mol. Plant 8, 17–27. doi: 10.1016/j.molp.2014.11.001
Zhou, Z., Yu, M. Y., Jiang, N., Xu, X. Y., Li, F., He, L. M., et al. (2017). Current situation and future development trend of Ganoderma lucidum cultivation in China. Edib. Fungi China 36, 5–7. doi: 10.13629/j.cnki.53-1054.2017.04.002
Zhu, L. M. (2018). Transcriptomics and metabolomics analysis of soybean in response to Phytophthora sojae infection. Nanjing: Nanjing Agricultural University.
Keywords: Ganoderma lingzhi, green mold, pathway, RNA-seq, Trichoderma hengshanicum
Citation: Wang T, Li X, Zhang C and Xu J (2023) Transcriptome analysis of Ganoderma lingzhi (Agaricomycetes) response to Trichoderma hengshanicum infection. Front. Microbiol. 14:1131599. doi: 10.3389/fmicb.2023.1131599
Received: 25 December 2022; Accepted: 06 February 2023;
Published: 23 February 2023.
Edited by:
Samantha Chandranath Karunarathna, Qujing Normal University, ChinaReviewed by:
Kalani Hapuarachchi, Guizhou University, ChinaThatsanee Luangharn, Mae Fah Luang University, Thailand
Copyright © 2023 Wang, Li, Zhang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jize Xu,  eHVqejgwMkBuZW51LmVkdS5jbg==; Chunlan Zhang, NDU4NTYwOTY5QHFxLmNvbQ==
eHVqejgwMkBuZW51LmVkdS5jbg==; Chunlan Zhang, NDU4NTYwOTY5QHFxLmNvbQ==