 Arif Hussain1†
Arif Hussain1† Razib Mazumder1†
Razib Mazumder1† Abdullah Ahmed1Umme Saima1
Abdullah Ahmed1Umme Saima1 Jody E. Phelan2
Jody E. Phelan2 Susana Campino2Dilruba Ahmed3
Susana Campino2Dilruba Ahmed3 Md Asadulghani4
Md Asadulghani4 Taane G. Clark2
Taane G. Clark2 Dinesh Mondal1*
Dinesh Mondal1*- 1Laboratory Sciences and Services Division, International Centre for Diarrhoeal Disease Research, Bangladesh (icddr,b), Dhaka, Bangladesh
- 2Department of Infection Biology, London School of Hygiene and Tropical Medicine, London, United Kingdom
- 3Clinical Microbiology and Immunology Laboratory, Laboratory Sciences and Services Division, International Centre for Diarrhoeal Disease Research, Bangladesh, Dhaka, Bangladesh
- 4Biosafety and BSL3 Laboratory, Biosafety Office, International Centre for Diarrhoeal Disease Research, Bangladesh, Dhaka, Bangladesh
Klebsiella pneumoniae is recognized as an urgent public health threat because of the emergence of difficult-to-treat (DTR) strains and hypervirulent clones, resulting in infections with high morbidity and mortality rates. Despite its prominence, little is known about the genomic epidemiology of K. pneumoniae in resource-limited settings like Bangladesh. We sequenced genomes of 32 K. pneumoniae strains isolated from patient samples at the International Center for Diarrhoeal Disease Research, Bangladesh (icddr,b). Genome sequences were examined for their diversity, population structure, resistome, virulome, MLST, O and K antigens and plasmids. Our results revealed the presence of two K. pneumoniae phylogroups, namely KpI (K. pneumoniae) (97%) and KpII (K. quasipneumoniae) (3%). The genomic characterization revealed that 25% (8/32) of isolates were associated with high-risk multidrug-resistant clones, including ST11, ST14, ST15, ST307, ST231 and ST147. The virulome analysis confirmed the presence of six (19%) hypervirulent K. pneumoniae (hvKp) and 26 (81%) classical K. pneumoniae (cKp) strains. The most common ESBL gene identified was blaCTX-M-15 (50%). Around 9% (3/32) isolates exhibited a difficult-to-treat phenotype, harboring carbapenem resistance genes (2 strains harbored blaNDM-5 plus blaOXA-232, one isolate blaOXA-181). The most prevalent O antigen was O1 (56%). The capsular polysaccharides K2, K20, K16 and K62 were enriched in the K. pneumoniae population. This study suggests the circulation of the major international high-risk multidrug-resistant and hypervirulent (hvKp) K. pneumoniae clones in Dhaka, Bangladesh. These findings warrant immediate appropriate interventions, which would otherwise lead to a high burden of untreatable life-threatening infections locally.
Highlights
-Klebsiella pneumoniae is a major contributor of antimicrobial resistance crisis worldwide.
-It is a major public health threat presenting a therapeutic challenge.
-Despite its prominence.
-There is an acute scarcity of data on the major lineages within K. pneumoniae isolates from Bangladesh which has high infectious disease burden.
-We report the presence of hypervirulent Klebsiella pneumoniae (hvKp) with high virulence score, in addition to classical K. pneumoniae (cKp) isolates.
-We have also reported the predominance of international high-risk multidrug-resistant clones that were associated with high AMR rates.
-In addition to these strains, we identified strains with difficult-to-treat (DTR) resistance phenotypes that were completely resistant to all first line antimicrobial agents.
-Results of this study has set the ground for further whole genome-based surveillance studies to closely monitor the evolutionary trends in K. pneumoniae in Bangladesh that would help in initiating stringent actions for the development of control strategies.
Introduction
Klebsiella pneumoniae is a notorious pathogen that has been implicated in a high number of high-risk infections and higher rates of resistance to multiple antimicrobial agents, including the last resort drugs (Paczosa and Mecsas, 2016). K. pneumoniae is a significant human pathogen acknowledged by the World Health Organization (WHO) and the US Centers for Disease Control and Prevention (CDC) as one of the priority pathogens exhibiting high propensities to acquire and spread resistance to multiple antimicrobial classes (Holt et al., 2015; Wyres et al., 2020). It is responsible for a wide range of hospital-acquired and community-acquired infections, including pneumonia, meningitis, wound infections, bacteremia, gastrointestinal and urinary tract infections (Musicha et al., 2019). The global spread of successful clonal lineages of K. pneumoniae is associated with the spread of extended-spectrum β-lactamase (ESBLs) and carbapenemases (Magiorakos et al., 2012). It is a major public health threat, presenting an extreme therapeutic challenge (Magiorakos et al., 2012). Classical K. pneumoniae (cKP) strains are primarily restricted in causing infections among immunocompromised patients (Paczosa and Mecsas, 2016). However, the emergence and rapid dissemination of hypervirulent (hvKP) strains have broadened the scope of the susceptible population, including healthy and immunocompetent individuals (Paczosa and Mecsas, 2016). Compared to the classical (cKP) strains, the hvKP strains are mostly associated with infections that originate from the community (Yu et al., 2006). The virulence in the hvKP is contributed by the capsule (K1, K2, K20 capsular types), lipopolysaccharide (LPS) (rmpA and rmpA2 regulatory genes), and siderophores (aerobactin) (Russo and Marr, 2019). The hvKP strains have further evolved by acquiring carbapenem resistance; these are referred to as carbapenem-resistant hypervirulent K. pneumoniae -hvKP (CR-hvKp) (Okanda et al., 2021; Sundaresan et al., 2022; Hussain et al., 2023). They are responsible for a serious public health crisis as they do not just represent hypervirulence and multi-drug resistance but exhibit high rates of transmission (Sundaresan et al., 2022). Genomic analysis of such strains remains an active area of research. K. pneumoniae are usually classified by their sequence types (STs); strains of certain STs have expanded clonally and distributed worldwide. They are called high-risk clones (Wyres and Holt, 2016; Wyres et al., 2019). Bacterial whole-genome sequencing studies have identified distinct phylogroups associated with K. pneumoniae; they include KpI, KpII and KpIII. These have been later classified as individual species- KpI as K. pneumoniae, KpII as K. quasipneumoniae and KpIII as K. variicola (Holt et al., 2015). The conventional microbiological methods are unable to distinguish between these Klebsiella species (Saxenborn et al., 2021).
Recent WGS studies on global collection of strains from hospital outbreaks and community-acquired K. pneumoniae infections provided a genomic framework for clonal diversity, antimicrobial resistance and virulence factors associated with this pathogen (Bowers et al., 2015; Holt et al., 2015; Fostervold et al., 2022). These studies have identified a wide spectrum of diversity, reinforced the diversification of K. pneumoniae into three phylogroups; KpI, KpII and KpIII and identified the role of hypervirulent and MDR clonal groups such as CG258, CG307, CG14 and CG15 in the nationwide and global dissemination of ESBL and carbapenem resistance (Bowers et al., 2015; Chung The et al., 2015; Holt et al., 2015; Fostervold et al., 2022). A nationwide AMR surveillance study in Bangladesh reported K. pneumoniae to be the third most abundant organism recovered from the clinical specimens, preceded by E. coli and pseudomonas species (Habib et al., 2021). Other in-country reports suggest the proportion of MDR K. pneumoniae has increased to over 80% (Ahmed et al., 2019; Aminul et al., 2021; Habib et al., 2021); this has driven the use of carbapenem as a drug of choice for treating MDR K. pneumoniae infections (Habib et al., 2021). Consequently, high carbapenem resistance rates were also reported from Bangladesh (Habib et al., 2021). In one study, up to 90% of isolates were found resistant to imipenem (Habib et al., 2021). Another study on a collection of isolates from a tertiary hospital in Dhaka, Bangladesh, reported the emergence of colistin resistance in a successful K pneumoniae sequence type 15 (Farzana et al., 2020). A study conducted by icddr,b between 2014 and 2017 showed that the increase in K. pneumoniae resistance rates has directly impacted the mortality in young children suffering concomitantly from bacteremia and pneumonia (Chisti et al., 2021).
Despite the increasing number of studies on K. pneumoniae nationally and globally, there is limited know-how on the genomic characteristics of such isolates from Bangladesh, as few studies have included the isolates from this region. Most in-country studies have analyzed hospital outbreak isolates focusing on MDR phenotypes and characterized them using conventional methods. Thus, there is a need for high-resolution studies on K. pneumonia isolates, including both resistant and susceptible ones. This would improve our understanding of population dynamics of community origin K. pneumonia beyond a handful of well-known clones.
This study used clinical K. pneumoniae isolates cultured from urine and pus specimens from a referral diagnostic center (icddr,b) in Dhaka, Bangladesh. We used a genomics-based approach to analyze the genetic diversity/relatedness, antimicrobial resistance and the virulome of the 32 clinical K. pneumoniae isolates to elucidate the genome dynamics of K. pneumoniae isolates, identifying the dominant/significant clones and evaluating their association with clinically relevant AMR and virulence determinants.
Materials and methods
Clinical isolates
We employed 32 K. pneumoniae isolates in this study. These were collected from a referral clinical microbiology laboratory at the International Center for Diarrheal Disease Research, Bangladesh (icddr,b), Dhaka, Bangladesh. The 32 K. pneumoniae isolates comprise 13 isolates recovered in August 2019, 12 isolates from September 2019 and 6 isolates from October 2019. The isolates from each month represent around 5% of K. pneumoniae isolates cultured from patient samples. The remaining one isolate archived at the Genome Center originating from the same setting was also used in this study. The isolates were randomly collected with minimum criteria that included: (i) Originating from different patients; (ii) Excluding samples from hospitalized patients; (iii) Phenotypically confirmed as K. pneumoniae by the Vitek-2 bacterial identification system. We employed disk diffusion tests to evaluate the antimicrobial susceptibility of the study isolates as per the CLSI guideline. The following 20 antibiotics were tested against the isolates: ampicillin, nitrofurantoin, ceftazidime, ceftriaxone, cefuroxime, cefixime, amikacin, ciprofloxacin, cotrimoxazole, nalidixic, cefepime, piperacillin/tazobactam, amoxycillin/clavulanic acid, gentamicin, tigecycline, cefoperazone/sulbactam, ertapenem, meropenem, imipenem and colistin. MDR was defined as resistance to at least one antimicrobial agent representing three antimicrobial classes. Difficult-to-treat (DTR) phenotype denotes resistance to all β-lactam generations, including carbapenems and fluoroquinolone. Extensively drug resistance (XDR) was defined as resistance to all antibiotic categories except to one or two antibiotic categories.
Whole genome sequencing
Bacterial genomic DNA extraction was done from single-colony cultures using the QIAamp DNA MINI kit per the manufacturer’s instructions. DNA QC and quantification were determined using the NanoDrop spectrophotometer (Thermo Fisher Scientific, United States) and Qubit 2.0 Fluorometer (Life Technologies), respectively (Mazumder et al., 2020b). The library preparation was performed utilizing the Nextera XT DNA library preparation kit (Illumina). The normalized and pooled library was subjected to 150-base paired-end reads sequencing using the Mid-output v2.5 sequencing kit. Sequencing was performed at the icddr,b Genome Center using the Nextseq500 platform (Mazumder et al., 2021).
Bioinformatic analysis
The high-quality reads were used for de novo assembly using SPAdes 3.11.1 (Bankevich et al., 2012). We performed the quality checks of the genome assemblies using QUAST 5.0.2 (Gurevich et al., 2013). The resulting genome assemblies were subjected to the Prokka 1.14.6 (Seemann, 2014) software for annotation.
The resistome of K. pneumoniae isolates was obtained by analyzing the presence of antimicrobial resistance genes and plasmid replicons using ResFinder 4.1 (Zankari et al., 2017) and plasmidFinder (Carattoli et al., 2014) hosted by the Center for Genomic Epidemiology (CGE). We used an identity cut-off of 98% and a coverage threshold of 80%.
Virulence genes were identified using a custom database of 28 K. pneumoniae virulence genes derived from the Virulence Factor Database (VFDB) (Chen et al., 2005) and known virulence genes from NCBI. Gene presence/absence was evaluated by BLASTn analysis with a cut-off coverage of 80% and a cut-off identity of 96%. Genes were classified functionally as; type I fimbriae (fimD, fimK, fimH, and fimC), type III fimbriae (mrkD, mrkJ, mrkF, mrkC, mrkA, and mrkI), siderophores/yersiniabactin (iutA, entB, ybtS, iucA, ybtA, irp1, irp2, and fyuA), regulator of mucoid phenotype [p-rmpA (accession No. KJ469368.1), p-rmpA2 (accession No. S64176.1)], salmochelin (iroB, iroC, iroD, and iroN), allantoin utilization (alls), type IV pili (pilW), type IV secretion system (clpV/tssH), metabolite transporter [peg-344 (accession No. MZ245622.1)] and colibactin [pks gene cluster (accession No. AM229678.1)]. Isolates were defined as hypervirulent (hvKp) K. pneumoniae if positive for ≥1 biomarkers including; peg-344, iroB, iucA, rmpA, and rmpA2 as suggested by the report from hypervirulent K. pneumoniae investigator group (Russo et al., 2018).
Additionally, isolates were typed using the Kaptive database (Wick et al., 2018) to identify the polysaccharide capsule (k antigen) and lipopolysaccharide (O antigen). The ST of each strain was assessed by submitting the genome assemblies to MLST 2.0 at Center for Genomic Epidemiology (CGE).
Core and pan-genome analysis
We performed the core and pan-genome analysis of 32 K. pneumoniae genomes. The GFF files obtained from Prokka (Seemann, 2014) were used as input for Roary (Page et al., 2015). The default settings were employed: core-gene threshold, 99%; the maximum number of clusters, 50,000 and inflation value for the MCL algorithm was: 1.5. A multi-fasta alignment was created using PRANK (option-e in roary). RaxML (Stamatakis, 2014) was used for constructing a phylogenetic tree from the aligned fasta file generated by Roary. The GAMA distribution of rates were used to evaluate the final tree, the robustness of the clades was verified using 100 bootstrap repetitions. The core-genome phylogeny and pangenome fingerprints of the isolates, along with their metadata, were visualized by Phandango (Hadfield et al., 2018). Snippy (Bush et al., 2020) was used to find SNPs among the genomes. Klebsiella pneumoniae strain KP64 (NZ_AP018750.1) was used as a reference genome. Gubbins (Croucher et al., 2015) was used to filter true point mutations from those arising from recombination.
Statistical analysis
Statistical comparisons between proportions were computed using Fisher’s exact two-tailed test. The non-parametric Mann–Whitney U test was used to calculate the statistical analysis for the aggregate virulence score. SPSS version 10.0 was used for all statistical calculations. Statistical significance was defined as p values ≤0.05.
Results
Population structure of Klebsiella pneumoniae
The genome features, associated metadata, and accession details are provided in Table 1. Pan-genome analysis of the 32 K. pneumoniae sequences predicted an average of 5,043 genes per genome, resulting in a pan-genome consisting of 12,778 genes. We identified 3,226 core and 9,552 accessory genes in our genome collection. A roary matrix was constructed from 32 K. pneumoniae genomes to demonstrate the genetic relatedness at the core genome level (Figure 1). A considerable correlation was observed between the phylogenetic clustering and some of the sequence types such as ST985, ST65, ST14, ST420, ST35 and ST15 and their K-types (K2, K20, K39 and K62) indicating presence of monophyletic clades corresponding to the above described STs. The strain GCKP36 exhibited the highest variation in core and accessory gene content compared to the rest of the genomes. WGS species identification confirmed that the strain GCKP36 (3%) was K. quasipneumoniae while the rest were K. pneumoniae (97%). The SNP analysis of 32 strains revealed 187,851 SNPs. The majority of Bangladeshi strains belonged to KpI phylogroup (K. pneumoniae) [97% (31/32)], whereas only one isolate was KpII (K. quasipneumoniae) [3% (1/32)]. None of the strains in our collection belonged to KpIII phylogroup (Klebsiella variicola). The isolate of KpII (GCKP36) exhibited the highest number of SNPs (221,163) compared to the rest of the strains belonging to KpI lineage. It showed the longest branch length (indicating its divergence from KpI strains); however, it did not form a separate branch with the rest of the genomes as the number of genomes belonging to this lineage was only one. Like the core-genome-based phylogeny, the core genome-SNP-based phylogenetic tree clustered the strains having identical STs associated with identical metadata, particularly STs such as ST420 and ST985 strongly correlated with the serogroups and MDR phenotype and strains belonging to ST420 and ST65 showed correlation with hypervirulence genotype. Strains belonging to ST35 did not show any substantial correlation (Figure 2). Surprisingly, the strains belonging to ST14 formed singletons. They were inconsistent with the serogroups and MDR phenotype data (Figure 2).
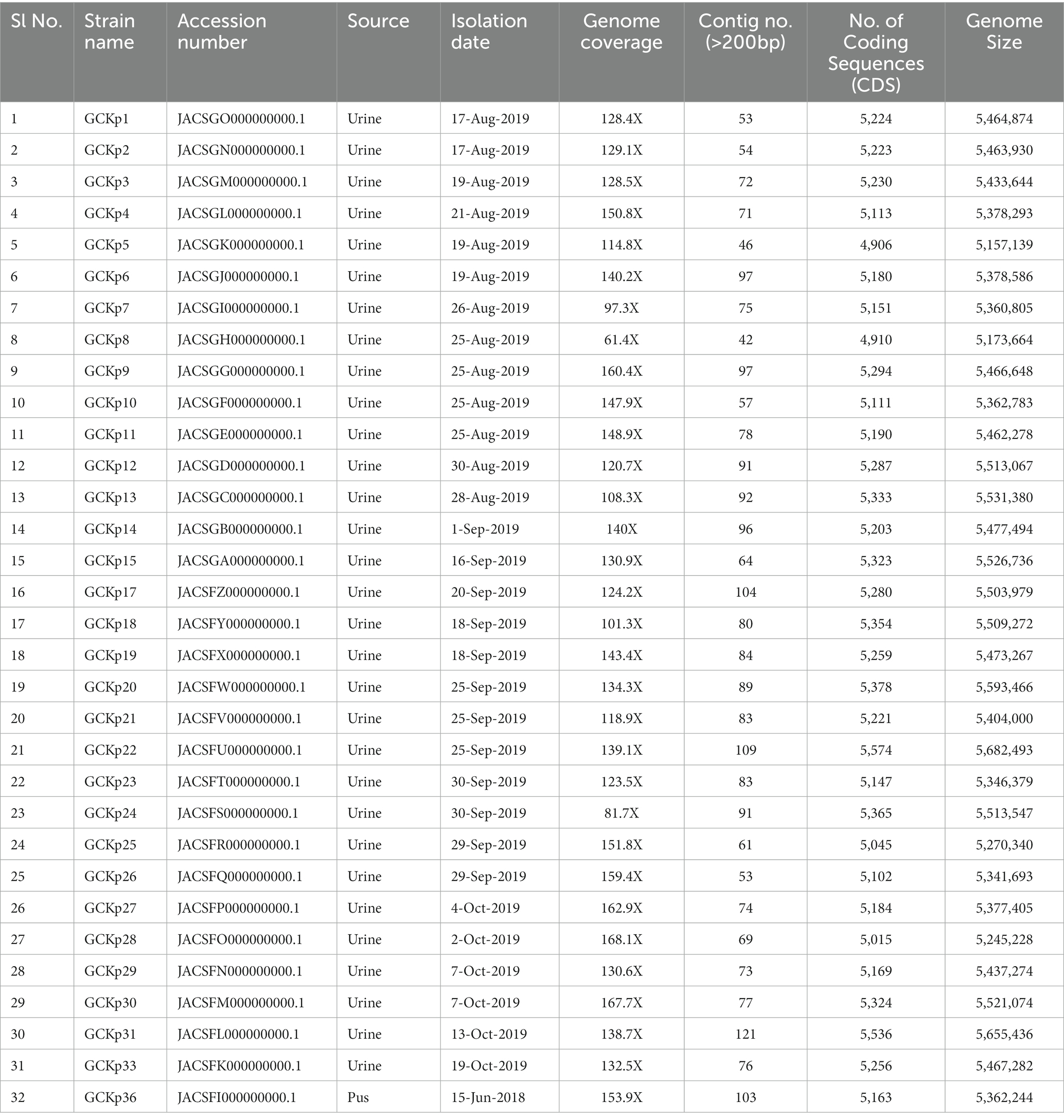
Table 1. Genome features and metadata of 32 K. pneumoniae isolates from Bangladesh.
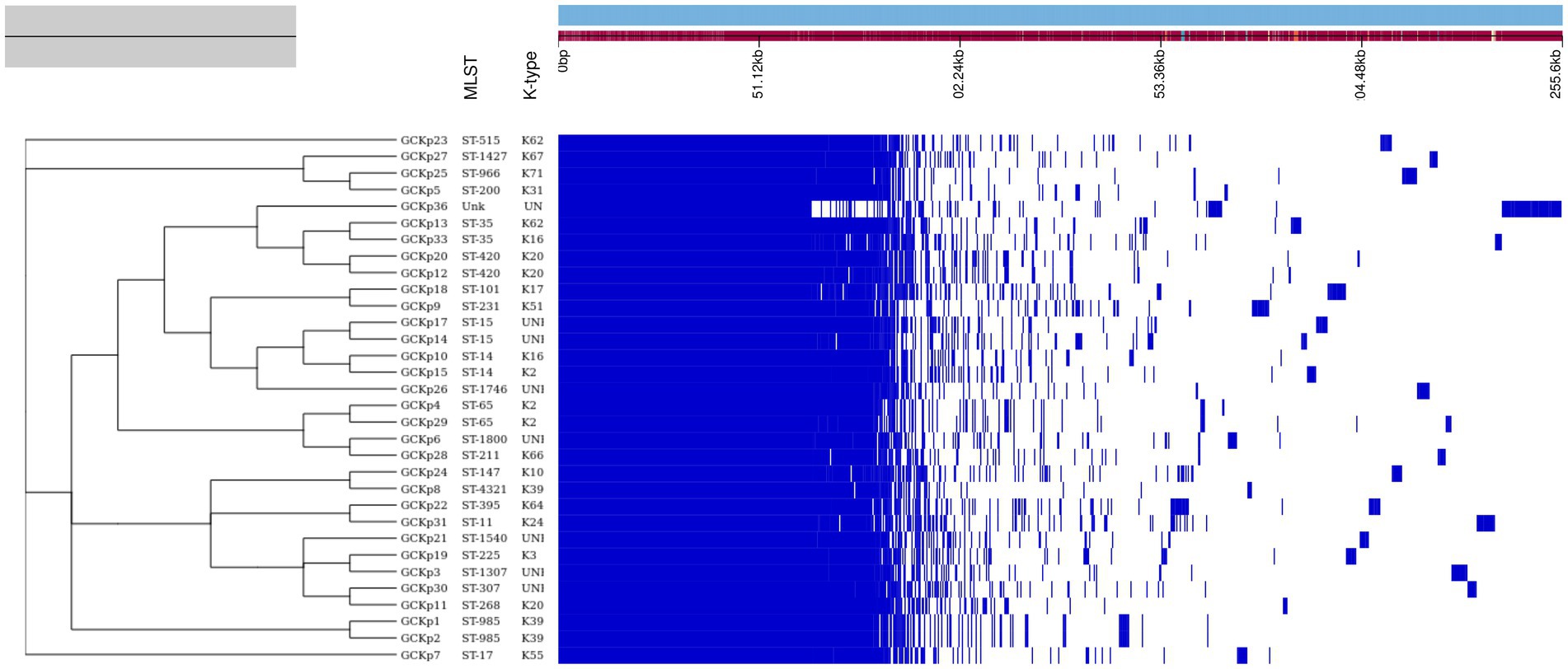
Figure 1. The core genome-based phylogenetic tree of 32 K. pneumoniae isolates compared to the metadata (STs and capsular polysaccharide) and a matrix with the presence and absence of core and accessory genes.
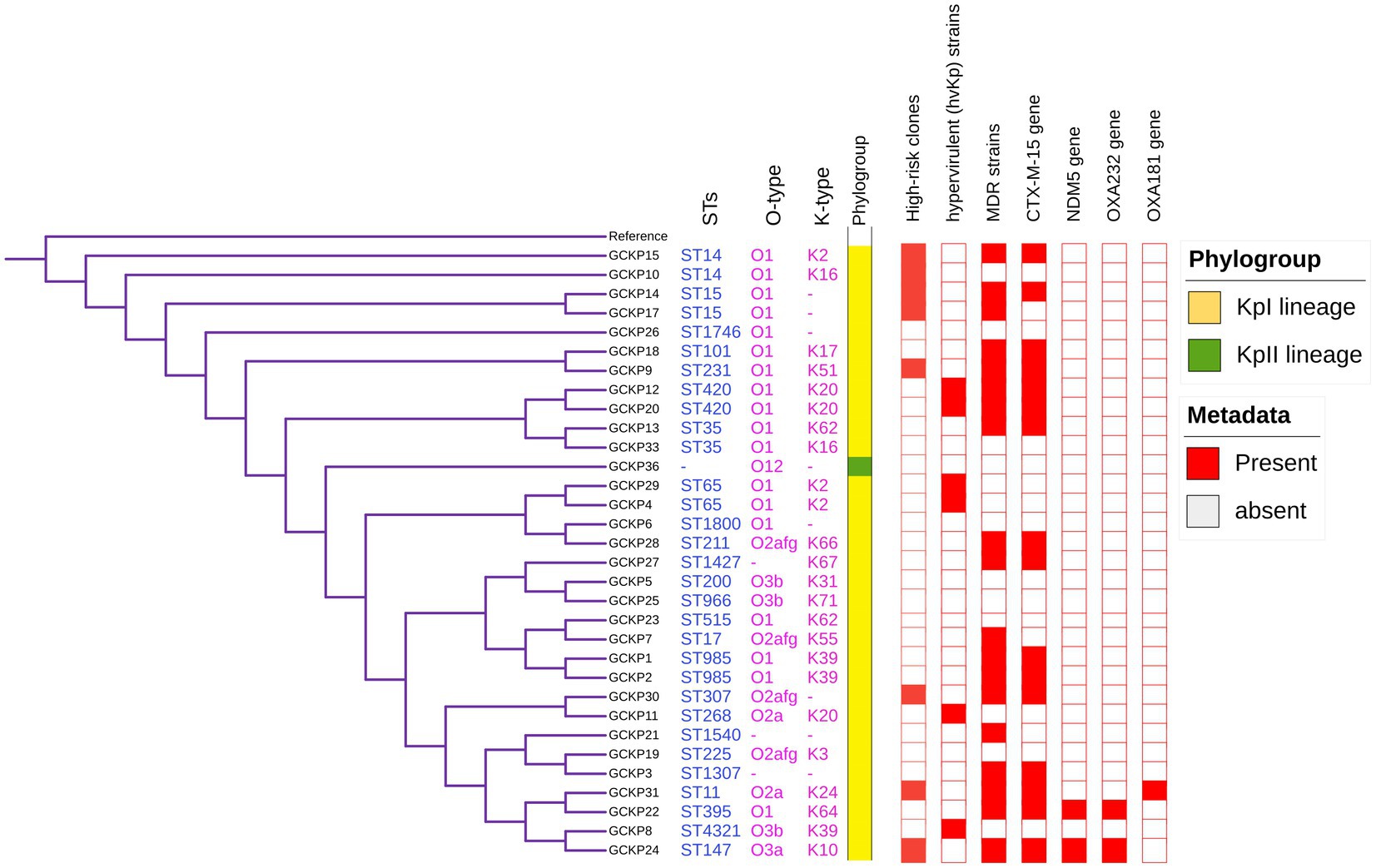
Figure 2. Core-genome SNP-based phylogenetic tree of 32 K. pneumoniae isolates analyzed from Dhaka, Bangladesh. The tree is annotated with the STs, O antigen, capsular polysaccharide, phylogroups, high-risk groups, hvKp strains, MDR status, presence of blaCTX-M-15, blaNDM5, blaOXA232, and blaOXA181 genes.
Isolates typing
The degree of K. pneumoniae sequence diversity was further analyzed by typing STs and surface-exposed polysaccharides (O and K antigens). We identified 26 diverse STs among 32 K. pneumoniae isolates in our collection that included six STs associated with high-risk clones, namely ST11 (3%), ST14 (6%), ST15 (6%), ST307 (3%), ST231 (3%) and ST147 (3%) (Figure 2). Overall, six STs represented more than one isolate which includes, ST985 (2/32), ST65 (2/32), ST14(2/32), ST420(2/32), ST35(2/32) and ST15 (2/32). The remaining 20 STs were represented by a single isolate, as shown in Figure 1. Overall, a total of six O antigens were detected; the most prevalent O type was O1 (56.2%), followed by O2afg (13%) and O2a (6.2%). Sixteen different K-serotypes were identified in all strains. K antigens that were represented in more than one isolate comprised K2 (9%), K20 (9%), K16 (6%) and K62 (6%) (Figure 2). Among these, the two capsular types, K2 and K20 were mainly associated with hypervirulence genotype. The remaining 12 capsular types were singletons (Figure 2).
Antimicrobial resistance and resistome of Klebsiella pneumoniae strains
None of the 32 K. pneumoniae strains was completely susceptible to the 20 antibiotics tested. The K. pneumoniae isolates showed the highest resistance to ampicillin (100%), followed by nitrofurantoin (94%), ceftazidime (60%) and cefixime (59%). Moderately resistant to ceftriaxone (56%), cefuroxime (56%), ciprofloxacin (53%), cotrimoxazole (50%) and nalidixic acid (50%). A varying portion of isolates were resistant to cefepime (44%), piperacillin/tazobactam (37%), amoxycillin/clavulanic acid (31%), gentamicin (28%), tigecycline (25%), cefoperazone/sulbactam (25%) and amikacin (13%). The strains were least resistant to carbapenems [ertapenem, meropenem and imipenem (9% each)] and colistin (6%). Over half (56%) of the K. pneumoniae strains were multi-drug resistant (MDR).
Our WGS analysis identified a total of 65 unique acquired AMR genes conferring resistance to different classes of antimicrobial agents with an average of 11 genes per genome (Table 2).
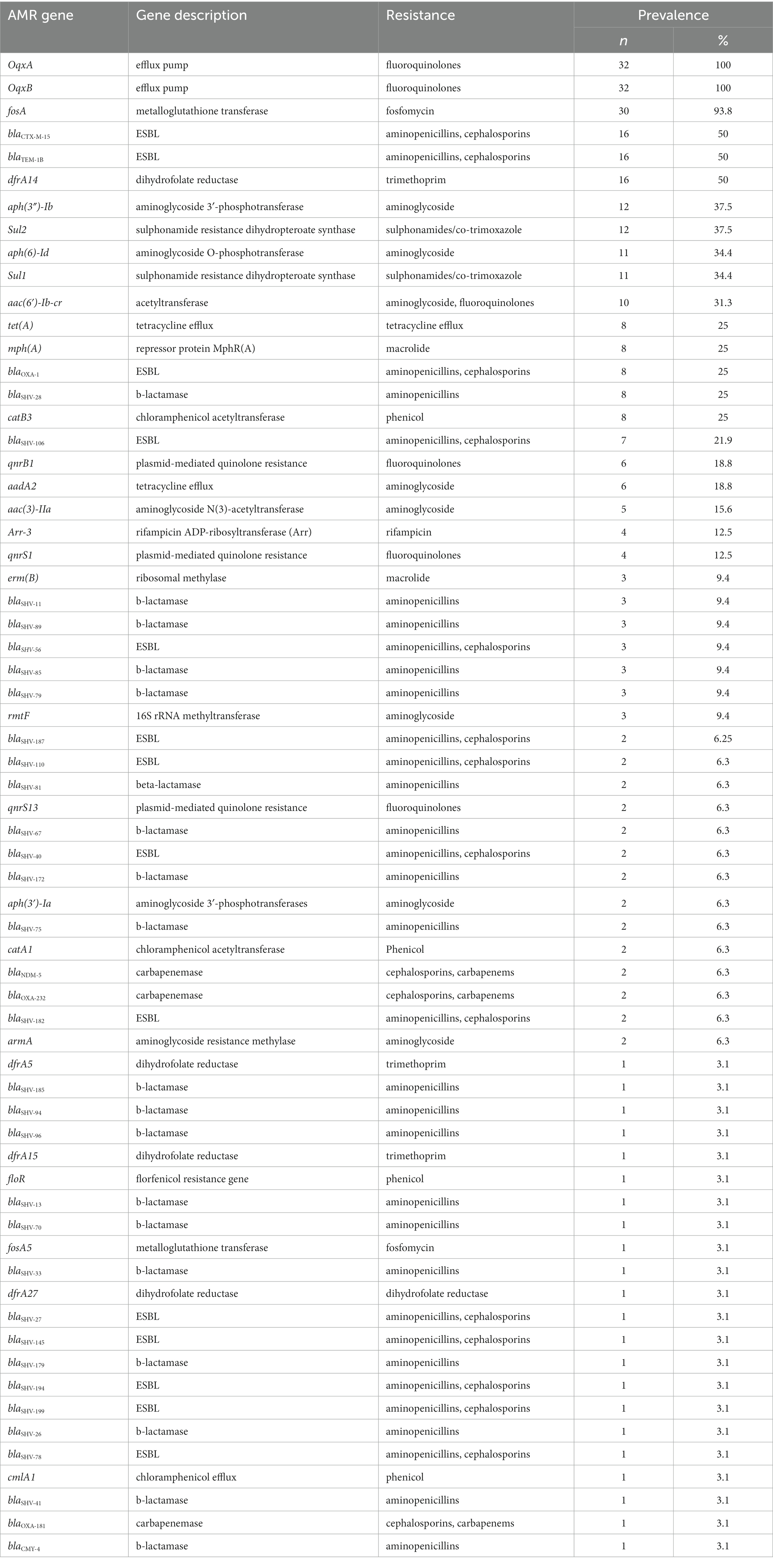
Table 2. Distribution of AMR genes among 32 clinical K. pneumoniae isolates from Dhaka, Bangladesh.
β-Lactam resistance
We identified 35 genes conferring resistance to β-lactam antibiotics. Among all the identified β-lactam genes, blaCTX-M-15 (16/32) and blaTEM-1B (16/32) were predominant. We also detected the presence of several allelic variants (29) of blaSHV gene (Table 2). We observed a 100% concordance between the presence of ESBL genes (blaCTX-M-15 and blaTEM-1B) and cephalosporin resistance, particularly a significant association with ceftriaxone and ceftazidime antibiotics (p < 0.001). Sequence analysis identified that the blaCTX-M-15 genes were mostly present on plasmid DNA [14/16 (88%)] rather than on chromosomes [2/16 (13%)] (Table 3). It was majorly associated with three plasmid replicons, IncFII, followed by IncFIB and Col440I (Figure 3). The genetic environment of blaCTX-M-15 in most strains comprises the insertion element ISEcp1. Other insertion sequences identified were ISKra4 and IS6100 adjacent to the blaCTX-M-15 gene. All blaCTX-M-15 positive strains demonstrated the MDR phenotype and were affiliated with diverse sequence types (STs). We detected three genes encoding carbapenem resistance, including blaNDM-5 (6%), blaOXA-232 (6%) and blaOXA-181 (3%). Two strains concomitantly harbored both blaNDM-5 and blaOXA-232 genes. One strain harbored the blaOXA-181 gene. All three strains carrying carbapenem resistance genes showed a difficult-to-treat resistance (DTR) phenotype, as these strains were resistant to all β-lactams, together with carbapenems and fluoroquinolone agents tested. In fact, they were resistant to all antibiotics tested (19/20) except for colistin. These three strains also coharbored blaCTX-M-15 and blaTEM-1B genes.
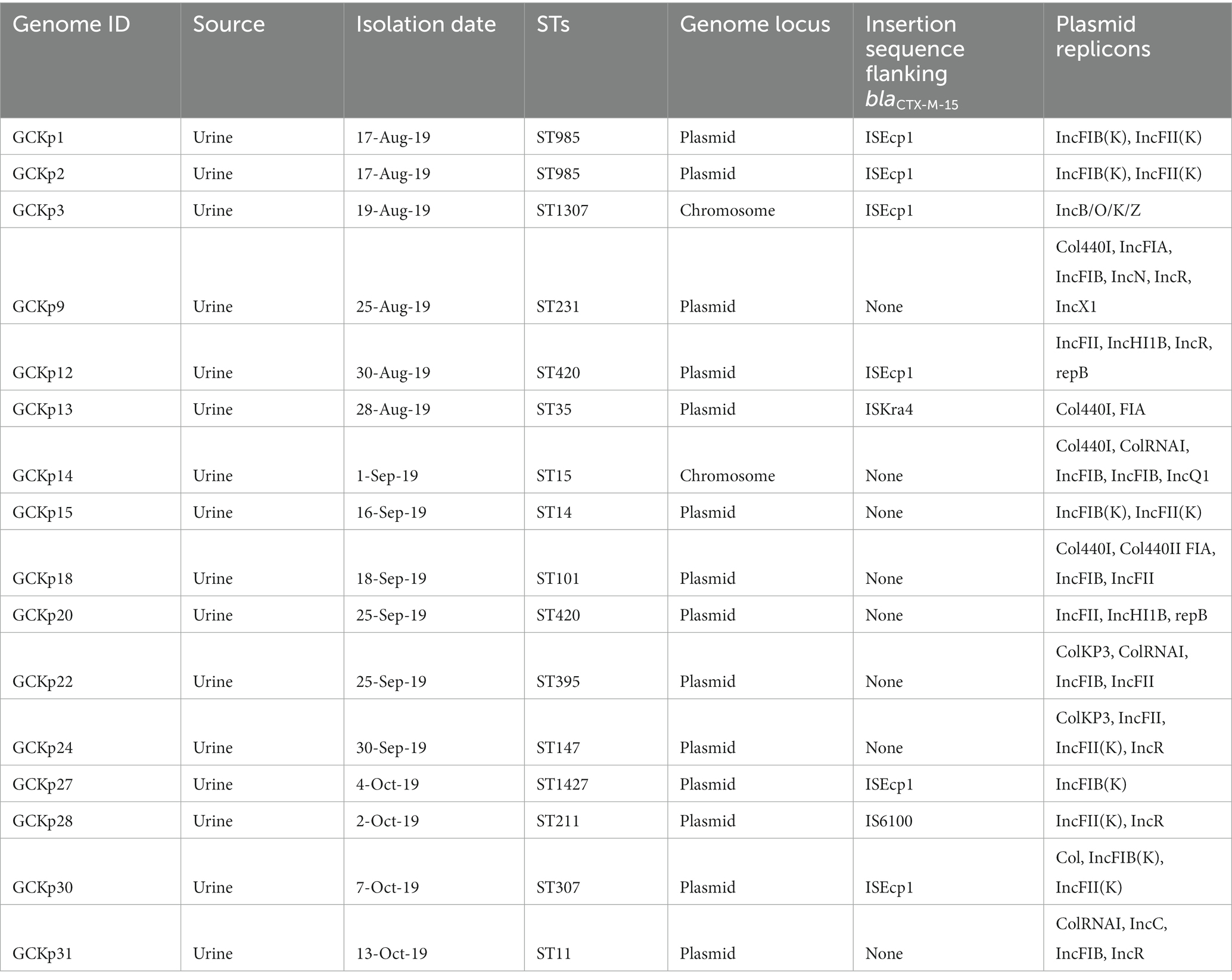
Table 3. Characteristics of CTX-M-15 associated clinical K. pneumoniae strains from Dhaka, Bangladesh.
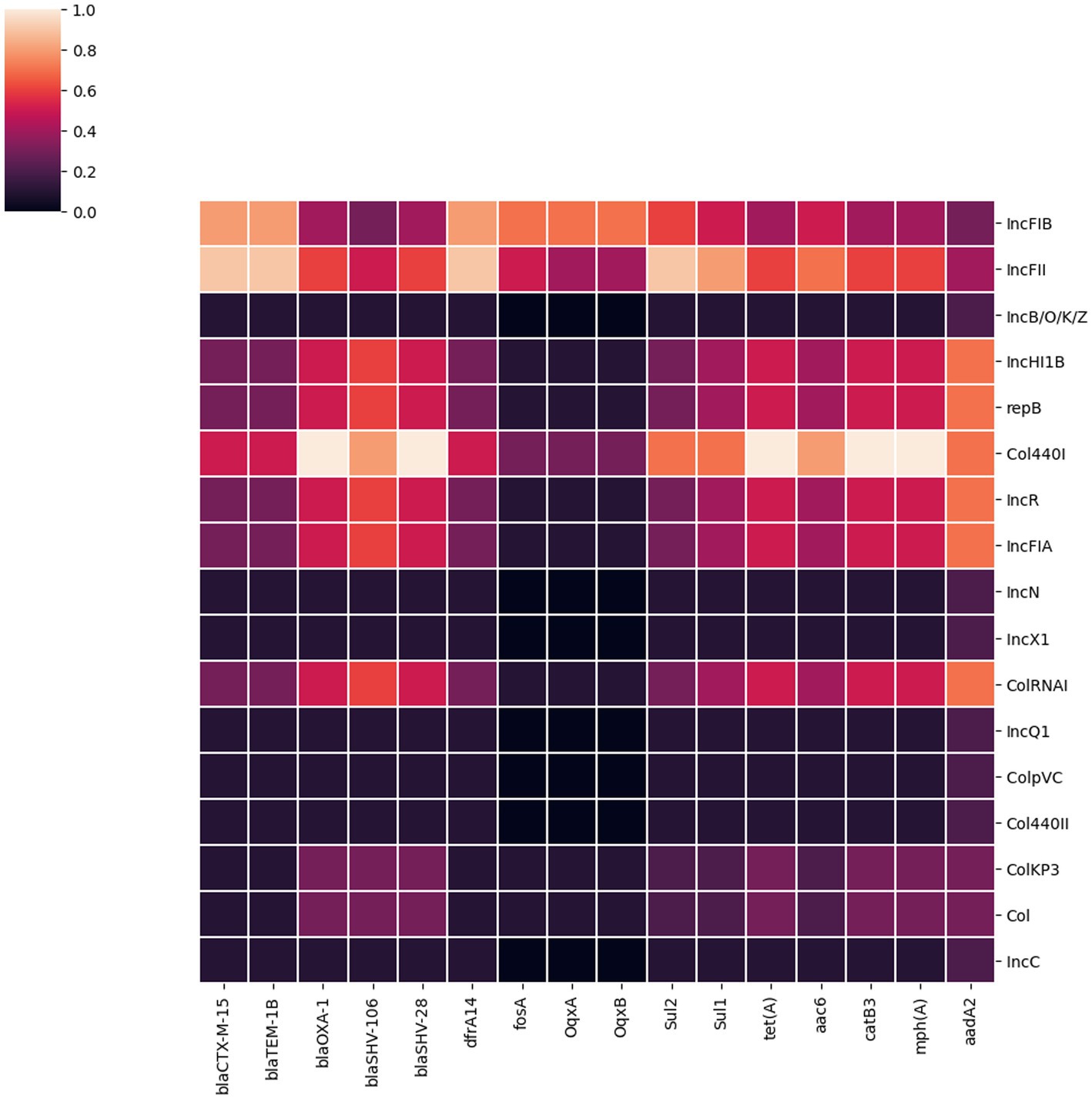
Figure 3. A heatmap illustrating the association between plasmid replicon types and the major AMR genes present in 32 K. pneumoniae isolates. It portrays that most AMR genes in K. pneumoniae isolates from Dhaka were associated with IncFIB, IncFII and Col440I plasmids.
Aminoglycoside resistance
We identified eight genes associated with aminoglycoside resistance. These include aph(3″)-Ib (37.5%), aph(6)-Id (35%), aac(6′)-Ib-cr (31.3%), aadA2 (18.8%), aac(3)-IIa (15.6%), rmtF (9.4%), aph(3′)-Ia (6.3%) and armA (6.3%). Out of these eight genes, the gene aac(6′)-Ib-cr was significantly associated with the gentamicin resistance phenotype (p < 0.001). There was no association between these genes and amikacin resistance phenotype.
Fluoroquinolone resistance
We screened for mutations in genes responsible for quinolone resistance. We detected substitutions at codon positions S80I (six isolates), S83Y (two isolates) in the amino acid sequence of parC and S83I (two isolates), S83F (three isolates) and D87A (five isolates) in the amino acid sequence of gyrA. Seven isolates had mutations in both the quinolone resistance genes gyrA and ParC. The ciprofloxacin and nalidixic acid resistance was associated with all the above-identified mutations in gyrA and parC. In addition to this we identified acquired AMR genes encoding for fluoroquinolone resistance such as oqxA/oqxB [32/32 (100%) isolates], qnrB1 [6/32 (3.5%) isolates] and qnrS [6/32 (3.5%) isolates].
Plasmid incompatibility groups
We identified 17 different plasmid incompatibility groups among 32 K. pneumoniae isolates (including FIB, FII, B/O/K/Z, Col44I, HI1B, repB, R, FIA, N, X1, ColRNA, Q1, ColPVC, Col440II, ColKP3, ColC) (Figure 3). FIB (66%), FII (44%) and Col440I (25%) were the predominant plasmid replicons across all 32 isolates. Consequently, the majority of AMR genes were associated with these three plasmid replicons (FIB, FII and Col440I) as shown in Figure 3.
Virulome of Klebsiella pneumoniae strains
Figure 4 shows the distribution of virulence factors associated with adherence, iron uptake, nutritional factor, secretion system, metabolism regulator, and mucoid phenotype among the 32 K. pneumoniae isolates. All (28) the virulence genes were detected in at least one K. pneumoniae isolate. Seven genes (fimD, fimC, fimH, fimK, iutA, entB, and iroN) exhibited prevalence rates of >90%. Six genes belonging to the type 3 fimbriae (mrkA, mrkC, mrkD, mrkF, mrkI, and mrkJ) and a gene belonging to type IV secretion system (clp/tssH) exhibited >80% prevalence and the rest of the genes exhibited <50% prevalence rates. For the four salmochelin genes, the order was: iroN (97%), iroB (22%), iroD (22%) and iroC (16%). Further, according to the aforementioned criteria, 6 (19%) strains in our collection were qualified as hypervirulent K. pneumoniae (hvKp) strains. Consequently, the remaining 26 strains were denoted as classical K. pneumoniae strains (cKp). Five out of the six hvKp strains were associated with all the five biomarkers of hypervirulence [peg-344 (metabolite transporter), iroB; iucA (siderophores) p-rmpA; p-rmpA2 (regulator of mucoid phenotype)]. One hvKp strain (GCKP8) harbored only one (iroB) hvKp-associated biomarker. One hvkp strain (GCKP11) was also positive for colibactin gene (pks gene cluster) rest of the strains (31 strains) were all negative for colibactin gene. Interesting, among the six hvKp strains, we identified two MDR (blaCTX-M-15 -positive) hypervirulent K. pneumoniae (MDR-hvKp) strains, while four (4/6) of the hvKp strains had non-MDR phenotypes. The two MDR-hvKp strains belonged to ST 420, serotype O1 antigen, and K20 capsular type. In contrast to the cKP strains, the hvKp strains had higher virulence prevalence, as the aggregate virulence score [median (range)] for hvKp strains [25 (18–27)] was higher than the cKp strains [15 (9–20)]. Moreover, the mean rank of virulence genes was significantly higher in hvKp strains than in cKp strains (p < 0.05).
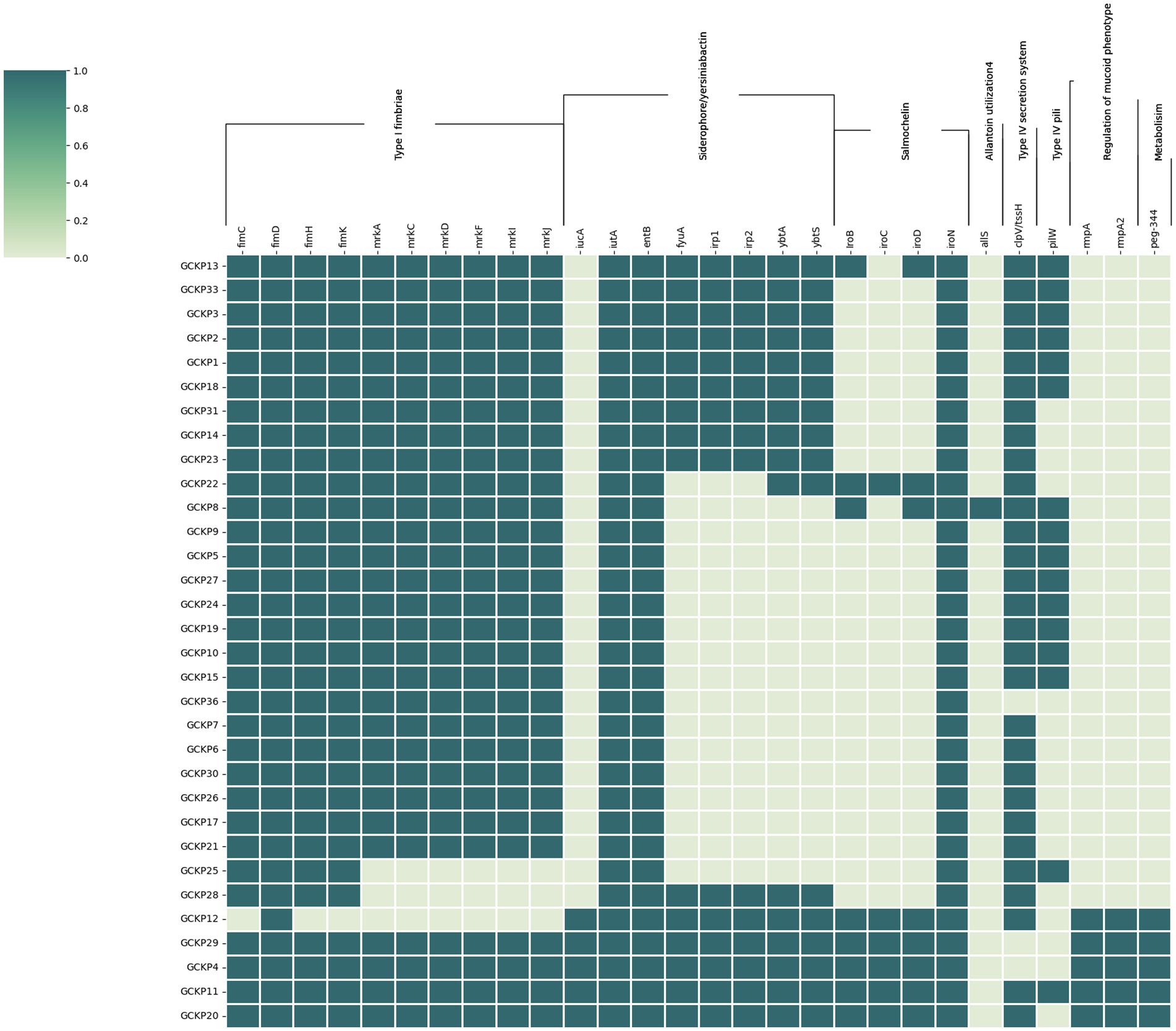
Figure 4. The virulome of 32 K. pneumoniae isolates from Dhaka: the virulence genes (n = 28) in K. pneumoniae were screened using VFDB and BLASTn analysis of known targets. The dark colored blocks indicate the presence of virulence genes.
Discussion
Klebsiella pneumoniae is a significant pathogen worldwide because of its link with a growing number of infections and high rates of AMR. It is a predominant community and hospital-acquired pathogen often associated with high-risk patients. However, data on the genomics epidemiology of such strains from Bangladesh is very sparse. To expand our understanding of the genomics of K. pneumoniae in Dhaka, Bangladesh, we performed an exhaustive molecular characterization of 32 K. pneumoniae clinical isolates obtained from a referral diagnostic center at icddr,b, Dhaka, Bangladesh, between August to October 2019. We used a genomics-based approach to analyze the genetic diversity/relatedness, antimicrobial resistance and the virulome of the study isolates.
We have shown that the population of K. pneumoniae isolates from Bangladesh reflects the global population structure of K. pneumoniae. However, the isolates from this study exhibited only 2 of 3 K. pneumoniae lineages comprising KpI and KpII. The KpIII lineage was not detected in our collection; this could be because the study isolates were sampled from a single site and for a short time frame (3 months). However, an earlier study has reported the outbreak of multi-drug resistant infections of KpIII lineage (K. variicola) in Bangladesh (Farzana et al., 2019). The pan-genome and core genome analysis revealed reduced nucleotide diversity in the study isolates.
In this study, 26 STs were identified in the genomes analyzed. In particular, we have identified six international high-risk multidrug-resistant clones consisting of ST14, ST15, ST11, ST307, ST231, and ST147. Globally, several studies have reported that these STs were associated with high rates of AMR, including MDR and extensively drug-resistant (XDR) strains of K. pneumoniae (Navon-Venezia et al., 2017; Mancini et al., 2018). The predominance of well-known international high-risk clones, including the clonal groups CG258 (ST11) and CG15 (ST14, ST15), is not unique to this study. However, in a previous study, some of these clones were described to be associated with hospital-acquired infections among carbapenem-resistant strains (Okanda et al., 2021). Since no a priori selection of resistant phenotypes was performed in this study, this indicates a strong prevalence of high-risk clones among K. pneumoniae associated with community-acquired infections. All strains [25% (8/32)] affiliated with these high-risk clones were found to be MDR and harbored ESBL genes such as blaCTX-M-15 and/or blaTEM-1B, except for one strain of ST14. The plasmid replicons FIA and FII were enriched in these strains.
In addition to the high-risk multidrug-resistant clones described above, we identified six (19%) hypervirulent K. pneumoniae (hvKp) strains in our collection. hvKp strains have gained increased attention globally, as these often infect healthy individuals from the community causing invasive and metastatic infections (Decré et al., 2011; Pomakova et al., 2011; Siu et al., 2011). The hvKp strains in this study exhibited high virulence scores compared to the classical K. pneumoniae (cKp) strains. The six hvKp strains represented all 3 months of the sampling period. These six hvKp strains comprised of 2 ST 420: K20, 2 ST65: K2, 1 ST268: K20 and 1 ST4321: K39. Previous studies have reported the presence of hvKp strains among 3 of 4 STs (ST420, ST65 and ST268) identified in this study (Shen et al., 2020; Eger et al., 2021). However, the strain with ST4321 was assigned as hvKp in this study, harboring only one hypervirulence biomarker. The presence of hypervirulence biomarker in sequence type ST4321 described here indicates that hvKp strains are evolving across various clonal types. The majority of the hvKp strains were affiliated with O1 antigen and K20 capsular polysaccharides. IncHI1B and repB plasmid replicons were predominant in hvKp strains. The two hvKp strains associated with ST420 were MDR and positive for ESBL genes (blaCTX-M-15 and blaTEM-1B). This is, to the best of our knowledge, the first report of hvKp strains in Bangladesh.
Another significant group of strains observed in this study comprised three K. pneumoniae strains (9%) that were positive for carbapenem resistance genes. Two of the three carbapenem-resistant strains were concomitantly positive for blaNDM-5 and blaOXA-232 and another strain was positive for blaOXA-181 (a variant of blaOXA48). All these three strains were extensively drug-resistant (XDR), exhibiting a difficult-to-treat phenotype. The three strains belonged to ST395:K64, ST147:K10 and ST11:K24. Earlier studies described these three STs harboring carbapenem resistance genes as epidemic clones responsible for significant morbidity and mortality (Logan and Weinstein, 2017). The three strains exhibited an average of 18 (out of 28 genes tested) virulence genes representing type I fimbriae, siderophores/yersiniabactin, salmochelin and type IV secretion system, indicating grave consequences of infection with such strains.
In general, the ESBL genotypes were also prevalent in strains other than the high-risk clones and carbapenem-resistant strains, indicating high propensities of K. pneumoniae to acquire and spread ESBL genes. We found a link between the presence of key AMR genes and a small number of plasmid replicons, such as IncFIB, IncFII and Col440I. This suggests that a few plasmids can play an important role in spreading AMR genes. Especially, the genetic mechanisms for acquiring and spreading ESBL and carbapenem resistance are widespread in the study setting (Mazumder et al., 2020a, 2021). Any selective pressure exerted by the indiscriminate use of a reserve group of antibiotics would lead to the prevalence of XDR strains in the population. Overall, the phylogenetic analysis and STs identified revealed a heterogenous distribution of the K. pneumoniae strains. The lone strain belonging to the KpII lineage (GCKP36) (K. quasipneumoniae) was susceptible to most antibiotics and consequently had fewer resistance genes. The strain was untypeable for MLST and K types and had a moderate number of virulence genes. This strain was misidentified as K. pneumoniae using the Vitek-2 system. However, a recent report has shown that strains of K. quasipneumoniae were no less virulent than K. pneumoniae strains (Long et al., 2017).
The first limitation of this investigation is that the Dhaka isolates originated from a single location. The second limitation is that a small number of isolates were sampled for a short duration (3 months only). The third limitation is that the hypervirulence is determined only on the basis of molecular markers previously reported. These limitations prevent the generalizability of the study findings to the country level. However, the parallels between our findings and those of other research in the region (Sherchan et al., 2020; Sundaresan et al., 2022) including the similarities in population structure, emergence of resistant and hypervirulent lineages indicate that this study is most probably representative of the genomic epidemiology of K. pneumoniae in Bangladesh. The main advantage of this study is that we have not selected the isolates intending to enrich for resistance or virulence phenotypes or genotypes. This approach allowed us to describe the population structure and various features associated with K. pneumonia in the community-acquired infections in this setting.
In conclusion, our analysis of 32 genomes of K. pneumoniae revealed the abundance of ESBL K. pneumoniae. We have shown that the K. pneumoniae population in Dhaka, Bangladesh is similar to other settings globally. We strongly suggest the detection of several major high-risk multidrug-resistant clonal lineages such as ST14, ST15, ST11, ST307, ST231 and ST147 in our collection. We confirm a moderately high prevalence (19%) of hypervirulent K. pneumoniae (hvKp), which is of particular concern that needs to be addressed immediately. The presence of difficult-to-treat carbapenem-producing K. pneumoniae strains needs more attention to prevent its dissemination. The consistency in population structure (prevalence of high-risk and hypervirulent lineages) with global isolates puts Bangladesh in the global context in terms of exchange of significant epidemiological K. pneumoniae clones, and these may cause untreatable aggressive infections in a country where the health care systems have very limited antibiotic options available for treatment.
The hypervirulent K. pneumoniae (hvKp) report and the over-representation of the international high-risk multidrug-resistant clones warrant WGS-based surveillance to closely monitor the evolutionary trends and convergence of virulence and resistance among K. pneumoniae strains in Bangladesh.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, JACSGO000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGN000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGM000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGL000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGK000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGJ000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGI000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGH000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGG000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGF000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGE000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGD000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGC000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGB000000000.1, https://www.ncbi.nlm.nih.gov/, JACSGA000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFZ000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFY000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFX000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFW000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFV000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFU000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFT000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFS000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFR000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFQ000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFP000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFO000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFN000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFM000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFL000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFK000000000.1, https://www.ncbi.nlm.nih.gov/, JACSFI000000000.1.
Author contributions
AH and RM designed the study, drafted the manuscript, and performed genome sequencing. AH, AA, and RM carried out the bioinformatics analyzes and interpretation of results and prepared tables and figures. US provided technical assistance in microbiology and sequencing work. JP, SC, MA, DA, TC, and DM contributed to the discussions and reviewed the manuscript. DM supervised the study. All authors have read and approved the final manuscript.
Acknowledgments
icddr,b is grateful to the governments of Bangladesh, Canada, Sweden, and the United Kingdom for providing core/unrestricted support for its operation and research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed, I., Rabbi, M. B., and Sultana, S. (2019). Antibiotic resistance in Bangladesh: a systematic review. Int. J. Infect. Dis. 80, 54–61. doi: 10.1016/j.ijid.2018.12.017
Aminul, P., Anwar, S., Molla, M. M. A., and Miah, M. R. A. (2021). Evaluation of antibiotic resistance patterns in clinical isolates of Klebsiella pneumoniae in Bangladesh. Biosaf. Health 3, 301–306. doi: 10.1016/j.bsheal.2021.11.001
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bowers, J. R., Kitchel, B., Driebe, E. M., MacCannell, D. R., Roe, C., Lemmer, D., et al. (2015). Genomic analysis of the emergence and rapid global dissemination of the clonal group 258 Klebsiella pneumoniae pandemic. PLoS One 10:e0133727. doi: 10.1371/journal.pone.0133727
Bush, S. J., Foster, D., Eyre, D. W., Clark, E. L., de Maio, N., Shaw, L. P., et al. (2020). Genomic diversity affects the accuracy of bacterial single-nucleotide polymorphism–calling pipelines. Gigascience 9, 1–21. doi: 10.1093/gigascience/giaa007
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Chisti, M. J., Harris, J. B., Carroll, R. W., Shahunja, K. M., Shahid, A. S. M. S. B., Moschovis, P. P., et al. (2021). Antibiotic-resistant bacteremia in young children hospitalized with pneumonia in Bangladesh is associated with a high mortality rate. Open Forum Infect. Dis. 8:ofab260. doi: 10.1093/ofid/ofab260
Chung The, H., Karkey, A., Pham Thanh, D., Boinett, C. J., Cain, A. K., Ellington, M., et al. (2015). A high-resolution genomic analysis of multidrug-resistant hospital outbreaks of Klebsiella pneumoniae. EMBO Mol. Med. 7, 227–239. doi: 10.15252/emmm.201404767
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Decré, D., Verdet, C., Emirian, A., Le Gourrierec, T., Petit, J. C., Offenstadt, G., et al. (2011). Emerging severe and fatal infections due to Klebsiella pneumoniae in two university hospitals in France. J. Clin. Microbiol. 49, 3012–3014. doi: 10.1128/JCM.00676-11
Eger, E., Heiden, S. E., Becker, K., Rau, A., Geisenhainer, K., Idelevich, E. A., et al. (2021). Hypervirulent Klebsiella pneumoniae sequence type 420 with a chromosomally inserted virulence plasmid. Int. J. Mol. Sci. Artic. 22:9196. doi: 10.3390/ijms22179196
Farzana, R., Jones, L. S., Barratt, A., Rahman, M. A., Sands, K., Portal, E., et al. (2020). Emergence of Mobile Colistin resistance (mcr-8) in a highly successful Klebsiella pneumoniae sequence type 15 clone from clinical infections in Bangladesh. mSphere 5:e00023. doi: 10.1128/mSphere.00023-20
Farzana, R., Jones, L. S., Rahman, M. A., Andrey, D. O., Sands, K., Portal, E., et al. (2019). Outbreak of Hypervirulent multidrug-resistant Klebsiella variicola causing high mortality in neonates in Bangladesh. Clin. Infect. Dis. 68, 1225–1227. doi: 10.1093/cid/ciy778
Fostervold, A., Hetland, M. A. K., Bakksjø, R., Bernhoff, E., Holt, K. E., Samuelsen, Ø., et al. (2022). A nationwide genomic study of clinical Klebsiella pneumoniae in Norway 2001–15: introduction and spread of ESBLs facilitated by clonal groups CG15 and CG307. J. Antimicrob. Chemother. 77, 665–674. doi: 10.1093/jac/dkab463
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Habib, Z. H., Binte Golam Rasul, S., Alam, M. A., Bably, N. N., Khan, I. A., Rizvi, S. M. S., et al. (2021). The findings of antimicrobial resistance surveillance in Bangladesh (2016-2020). medRxiv 2021:21251710. doi: 10.1101/2021.06.12.21251710
Hadfield, J., Croucher, N. J., Goater, R. J., Abudahab, K., Aanensen, D. M., and Harris, S. R. (2018). Phandango: an interactive viewer for bacterial population genomics. Bioinformatics 34, 292–293. doi: 10.1093/bioinformatics/btx610
Holt, K. E., Wertheim, H., Zadoks, R. N., Baker, S., Whitehouse, C. A., Dance, D., et al. (2015). Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. U. S. A. 112, E3574–E3581. doi: 10.1073/pnas.1501049112
Hussain, A., Mazumder, R., Asadulghani, M., Clark, T. G., and Mondal, D. (2023). Combination of virulence and antibiotic resistance: a successful bacterial strategy to survive under hostile environments. Bact. Surviv. Hostile Environ. 153, 101–117. doi: 10.1016/B978-0-323-91806-0.00004-7
Logan, L. K., and Weinstein, R. A. (2017). The epidemiology of Carbapenem-resistant Enterobacteriaceae: The impact and evolution of a global menace. J. Infect. Dis. 215, S28–S36. doi: 10.1093/infdis/jiw282
Long, S. W., Linson, S. E., Ojeda Saavedra, M., Cantu, C., Davis, J. J., Brettin, T., et al. (2017). Whole-genome sequencing of human clinical Klebsiella pneumoniae isolates reveals misidentification and misunderstandings of Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae. mSphere 2:e00290. doi: 10.1128/mSphereDirect.00290-17
Magiorakos, A. P., Srinivasan, A., Carey, R. B., Carmeli, Y., Falagas, M. E., Giske, C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Mancini, S., Poirel, L., Tritten, M. L., Lienhard, R., Bassi, C., and Nordmann, P. (2018). Emergence of an MDR Klebsiella pneumoniae ST231 producing OXA-232 and RmtF in Switzerland. J. Antimicrob. Chemother. 73, 821–823. doi: 10.1093/jac/dkx428
Mazumder, R., Abdullah, A., Ahmed, D., and Hussain, A. (2020a). High prevalence of blactx-m-15 gene among extended-spectrum β-lactamase-producing Escherichia coli isolates causing extraintestinal infections in Bangladesh. Antibiotics 9:796. doi: 10.3390/antibiotics9110796
Mazumder, R., Abdullah, A., Hussain, A., Ahmed, D., and Mondal, D. (2020b). Draft genome sequence of Chromobacterium violaceum RDN09, isolated from a patient with a wound infection in Bangladesh. Microbiol. Resour. Announc. 9:e00957. doi: 10.1128/mra.00957-20
Mazumder, R., Hussain, A., Abdullah, A., Islam, M. N., Sadique, M. T., Muniruzzaman, S. M., et al. (2021). International high-risk clones among extended-spectrum β-lactamase–producing Escherichia coli in Dhaka. Bangladesh. Front. Microbiol. 12:2843. doi: 10.3389/fmicb.2021.736464
Musicha, P., Msefula, C. L., Mather, A. E., Chaguza, C., Cain, A. K., Peno, C., et al. (2019). Genomic analysis of Klebsiella pneumoniae isolates from Malawi reveals acquisition of multiple ESBL determinants across diverse lineages. J. Antimicrob. Chemother. 74, 1223–1232. doi: 10.1093/jac/dkz032
Navon-Venezia, S., Kondratyeva, K., and Carattoli, A. (2017). Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol. Rev. 41, 252–275. doi: 10.1093/femsre/fux013
Okanda, T., Haque, A., Koshikawa, T., Islam, A., Huda, Q., Takemura, H., et al. (2021). Characteristics of Carbapenemase-producing Klebsiella pneumoniae isolated in the intensive care unit of the largest tertiary Hospital in Bangladesh. Front. Microbiol. 11:3357. doi: 10.3389/fmicb.2020.612020
Paczosa, M. K., and Mecsas, J. (2016). Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 80, 629–661. doi: 10.1128/MMBR.00078-15
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pomakova, D. K., Hsiao, C. B., Beanan, J. M., Olson, R., MacDonald, U., Keynan, Y., et al. (2011). Clinical and phenotypic differences between classic and hypervirulent Klebsiella pneumonia: an emerging and under-recognized pathogenic variant. Eur. J. Clin. Microbiol. Infect. Dis. 31, 981–989. doi: 10.1007/S10096-011-1396-6
Russo, T. A., and Marr, C. M. (2019). Hypervirulent Klebsiella pneumoniae. Available at: https://journals.asm.org/journal/cmr (Acrcessed November 1, 2021).
Russo, T. A., Olson, R., Fang, C. T., Stoesser, N., Miller, M., MacDonald, U., et al. (2018). Identification of biomarkers for differentiation of hypervirulent klebsiella pneumoniae from classical K. pneumoniae. J. Clin. Microbiol. 56:e00776. doi: 10.1128/JCM.00776-18
Saxenborn, P., Baxter, J., Tilevik, A., Fagerlind, M., Dyrkell, F., Pernestig, A. K., et al. (2021). Genotypic characterization of clinical Klebsiella spp. isolates collected from patients with suspected community-onset Sepsis, Sweden. Front. Microbiol. 12:1091. doi: 10.3389/fmicb.2021.640408
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shen, P., Berglund, B., Chen, Y., Zhou, Y., Xiao, T., Xiao, Y., et al. (2020). Hypervirulence markers among non-ST11 strains of Carbapenem–and multidrug-resistant Klebsiella pneumoniae isolated from patients with bloodstream infections. Front. Microbiol. 11:1199. doi: 10.3389/fmicb.2020.01199
Sherchan, J. B., Tada, T., Shrestha, S., Uchida, H., Hishinuma, T., Morioka, S., et al. (2020). Emergence of clinical isolates of highly carbapenem-resistant Klebsiella pneumoniae co-harboring blaNDM-5 and blaOXA-181 or −232 in Nepal. Int. J. Infect. Dis. 92, 247–252. doi: 10.1016/j.ijid.2020.01.040
Siu, L. K., Fung, C. P., Chang, F. Y., Lee, N., Yeh, K. M., Koh, T. H., et al. (2011). Molecular typing and virulence analysis of serotype K1 Klebsiella pneumoniae strains isolated from liver abscess patients and stool samples from noninfectious subjects in Hong Kong, Singapore, and Taiwan. J. Clin. Microbiol. 49, 3761–3765. doi: 10.1128/JCM.00977-11
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies bioinformatics | Oxford academic. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sundaresan, A. K., Vincent, K., Mohan, G. B. M., and Ramakrishnan, J. (2022). Association of sequence types, antimicrobial resistance and virulence genes in Indian isolates of Klebsiella pneumoniae: a comparative genomics study. J. Glob. Antimicrob. Resist. 30, 431–441. doi: 10.1016/j.jgar.2022.05.006
Wick, R. R., Heinz, E., Holt, K. E., and Wyres, K. L. (2018). Kaptive web: user-friendly capsule and lipopolysaccharide serotype prediction for Klebsiella genomes. J. Clin. Microbiol. 56. doi: 10.1128/JCM.00197-18
Wyres, K. L., Hawkey, J., Hetland, M. A. K., Fostervold, A., Wick, R. R., Judd, L. M., et al. (2019). Emergence and rapid global dissemination of CTX-M-15-associated Klebsiella pneumoniae strain ST307. J. Antimicrob. Chemother. 74, 577–581. doi: 10.1093/jac/dky492
Wyres, K. L., and Holt, K. E. (2016). Klebsiella pneumoniae population genomics and antimicrobial-resistant clones. Trends Microbiol. 24, 944–956. doi: 10.1016/j.tim.2016.09.007
Wyres, K. L., Nguyen, T. N. T., Lam, M. M. C., Judd, L. M., Van Vinh Chau, N., Dance, D. A. B., et al. (2020). Genomic surveillance for hypervirulence and multi-drug resistance in invasive Klebsiella pneumoniae from south and Southeast Asia. Genome Med. 12, 1–16. doi: 10.1186/s13073-019-0706-y
Yu, W. L., Ko, W. C., Cheng, K. C., Lee, H. C., Ke, D. S., Lee, C. C., et al. (2006). Association between rmpA and magA genes and clinical syndromes caused by Klebsiella pneumoniae in Taiwan. Clin. Infect. Dis. 42, 1351–1358. doi: 10.1086/503420
Zankari, E., Allesøe, R., Joensen, K. G., Cavaco, L. M., Lund, O., and Aarestrup, F. M. (2017). PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 72, 2764–2768. doi: 10.1093/jac/dkx217
Keywords: Hypervirulent Klebsiella pneumoniae, high risk Klebsiella pneumoniae clone, Klebsiella pneumoniae from low and middle income country, omics, molecular epidemiology
Citation: Hussain A, Mazumder R, Ahmed A, Saima U, Phelan JE, Campino S, Ahmed D, Asadulghani M, Clark TG and Mondal D (2023) Genome dynamics of high-risk resistant and hypervirulent Klebsiella pneumoniae clones in Dhaka, Bangladesh. Front. Microbiol. 14:1184196. doi: 10.3389/fmicb.2023.1184196
Edited by:
Miklos Fuzi, Independent Researcher, Budapest, HungaryReviewed by:
M. John Albert, Kuwait University, KuwaitYing-Tsong Chen, National Chung Hsing University, Taiwan
Copyright © 2023 Hussain, Mazumder, Ahmed, Saima, Phelan, Campino, Ahmed, Asadulghani, Clark and Mondal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dinesh Mondal, ZGluNjNkQGljZGRyYi5vcmc=
†These authors have contributed equally to this work and share first authorship