 Zhiqian Wang
Zhiqian Wang Yanjun Liu
Yanjun Liu Peilin Liu
Peilin Liu Zijuan Jian1
Zijuan Jian1 Bin Tang
Bin Tang Awen Yang
Awen Yang Wenen Liu
Wenen Liu- 1Department of Clinical Laboratory, Xiangya Hospital, Central South University, Changsha, Hunan, China
- 2National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Changsha, Hunan, China
Background: The pks island and its production of the bacterial secondary metabolite genotoxin, colibactin, have attracted increasing attention. However, genomic articles focusing on pks islands in Klebsiella pneumoniae, as well as comparative genomic studies of mobile genetic elements, such as prophages, plasmids, and insertion sequences, are lacking. In this study, a large-scale analysis was conducted to understand the prevalence and evolution of pks islands, differences in mobile genetic elements between pks-negative and pks-positive K. pneumoniae, and clinical characteristics of infection caused by pks-positive K. pneumoniae.
Methods: The genomes of 2,709 K. pneumoniae were downloaded from public databases, among which, 1,422 were from NCBI and 1,287 were from the China National GeneBank DataBase (CNGBdb). Screening for virulence and resistance genes, phylogenetic tree construction, and pan-genome analysis were performed. Differences in mobile genetic elements between pks-positive and pks-negative strains were compared. The clinical characteristics of 157 pks-positive and 157 pks-negative K. pneumoniae infected patients were investigated.
Results: Of 2,709 K. pneumoniae genomes, 245 pks-positive genomes were screened. The four siderophores, type VI secretion system, and nutritional factor genes were present in at least 77.9% (191/245), 66.9% (164/245), and 63.3% (155/245) of pks-positive strains, respectively. The number and fragment length of prophage were lower in pks-positive strains than in pks-negative strains (p < 0.05). The prevalence of the IS6 family was higher in pks-negative strains than in pks-positive strains, and the prevalence of multiple plasmid replicon types differed between the pks-positive and pks-negative strains (p < 0.05). The detection rate of pks-positive K. pneumoniae in abscess samples was higher than that of pks-negative K. pneumoniae (p < 0.05).
Conclusion: The pks-positive strains had abundant virulence genes. There were differences in the distribution of mobile genetic elements between pks-positive and pks-negative isolates. Further analysis of the evolutionary pattern of pks island and epidemiological surveillance in different populations are needed.
1. Introduction
Klebsiella pneumoniae is a common opportunistic pathogen in hospitals, that can cause a variety of serious infections (Siu et al., 2012). In recent years, K. pneumoniae has become a major threat to global public health. Although many factors, including host immunity (Bengoechea and Sa Pessoa, 2019), contribute to the pathogenicity of K. pneumoniae in humans, virulence factors undoubtedly play a critical role in the pathogenesis process. K. pneumoniae includes two different phenotypes: classical K. pneumoniae (cKp) and hypervirulent K. pneumoniae (hvKp). Hypervirulent K. pneumoniae (hvKp) can cause severe infection in immunocompetent individuals with high pathogenicity and mortality (Russo and Marr, 2019). Factors contributing to the high virulence of the hvKp include capsule, siderophores, lipopolysaccharide and fimbriae (Zhu et al., 2021). Recently, pks islands, which are located on the same integrative and conjugative element as the yersiniabactin gene cluster, have been considered as a new virulence factor and have attracted more and more attention.
The pks island, also known as the clb gene island, consists of 19 genes (clbA-clbS). It was first identified in Escherichia coli IHE3034 (Nougayrède et al., 2006) and was later found in other enterobacteria, including K. pneumoniae. Colibactin is a genotoxic bacterial secondary metabolite produced by the pks island, and it can interfere with the cell cycle in eukaryotic cells and cause cell damage; consequently, the pks locus-mediated colibactin production can enhance the virulence and pathogenicity of bacteria. E. coli carrying the pks island may be involved in the development and progression of colorectal cancer and exacerbate lymphocytopenia associated with sepsis (Marcq et al., 2014; Prorok-Hamon et al., 2014; Pleguezuelos-Manzano et al., 2020). Hypervirulent K. pneumoniae carrying the pks gene island induced meningitis successfully in mice, whereas deletion of clbA gene and abolition of colibactin synthesis significantly reduced the ability of the bacterium to cause meningitis in mice (Lu et al., 2017).
Horizontal gene transfer is an essential aspect in bacterial evolution and is often associated with mobile genetic elements (MGEs) (Koonin et al., 2001; Soucy et al., 2015; Haudiquet et al., 2022). Previous studies have reported a high degree of consistency in pks islands between different bacterial species, and pks islands are characterized for being acquired by horizontal gene transfer (Putze et al., 2009). Many genomic studies focusing on E. coli have shown the horizontal propagation and evolution pattern of pks islands in E. coli and revealed the different acquisitions of pks islands (Auvray et al., 2021; Suresh et al., 2021; Wami et al., 2021). However, there are limited information focusing on pks-positive K. pneumoniae. Genomic analyses of K. pneumoniae have shown that pks islands and ybt genes can be horizontally transferred between K. pneumoniae by integrative and conjugative elements (ICEs) (Lam et al., 2018). This study aimed to comprehensively understand the prevalence and evolution of pks island in K. pneumoniae, compare the differences in mobile genetic elements between pks-positive and pks-negative K. pneumoniae, and investigate the clinical characteristics of pks-positive K. pneumoniae infection. This study will broaden our conceptual understanding of the disease-causing K. pneumoniae pks-positive strains and could facilitate the improvement of diagnosis and characterization of associated infections.
2. Materials and methods
2.1. Klebsiella pneumoniae genome sequences utilized in this study
A total of 2,709 K. pneumoniae genome sequences (1,287 complete genomes and 1,422 draft genomes) from two public databases were screened in this study. Among these sequences, 1,287 clinical strain high-quality genome sequences from a large tertiary hospital from January 2016 to July 2018 were downloaded from the China National GenBank DataBase (Chen et al., 2020).1 The remaining 1,422 K. pneumoniae genome sequences were downloaded from the NCBI database (accessed on October 13, 2022). The accession numbers of all the genes are shown in Supplementary Table S1.
2.2. Identification of pks-positive genomes
The pks sequence of K. pneumoniae strain 1,084 (CP003785.1) was used as the reference sequence. The pks-positive genomes were identified by comparing the reference sequence with the 2,709 K. pneumoniae genome using BLAST+ 2.9.0. The identity and query coverage thresholds for BLAST+ 2.9.0 were set at 90%. The identified pks-positive genomes were further confirmed using Kleborate version 2.2.0 (Lam et al., 2021).
2.3. Multi-locus sequence typing and serotype analysis
The sequence types of pks-positive K. pneumoniae were determined using MLST version 2.22.0,2 which can scan genome sequences against the traditional PubMLST typing schemes.3 Kleborate version 2.2.0 (Lam et al., 2021) was used to analyze the allelic differences of pks island and yersiniabactin determinant in each genome to determine colibactin and yersiniabactin determinant sequence types (CbST and YbST). In addition, the serotypes of pks-positive strains were identified using the kleborate version 2.2.0 (Lam et al., 2021).
2.4. Analysis of virulence genes and antibiotic resistance genes
All pks-positive and pks-negative K. pneumoniae strains were used for further analyses. Virulence and resistance genes were identified by comparison with the Virulence Factor Database (VFDB) and the Comprehensive Antibiotic Resistance Database (CARD) using abricate version 1.0.14 with default settings (Chen et al., 2005; McArthur et al., 2013). Databases used to compare virulence and resistance genes were updated to the latest version before the analysis.
2.5. Pangenome analysis of Klebsiella pneumoniae strains
All K. pneumoniae strains were annotated using Prokka version 1.14 (Seemann, 2014). The annotation files of pks-positive strains obtained in the previous step were further analyzed using Roary version 3.13.0 (Page et al., 2015) with default settings to obtain the core genes of the positive strains. Genes present in at least 99% of the strains were identified as core genes. Clusters of Orthologous Genes (COG) classification was obtained by uploading the core genes to the eggNOG (Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups) public database (Huerta-Cepas et al., 2016).
2.6. Phylogenetic analysis
The pks-positive strains were aligned to the reference genome K. pneumoniae 1,084 (CP003785.1) using Snippy version 4.6.0,5 and then the core gene SNP sites were extracted. After removing the recombinant regions using Gubbins version 2.4.1 (Croucher et al., 2015), phylogenetic trees using the maximum likelihood method were constructed using FastTree version 2.1.11 and uploaded to the iTOL website for annotation (Price et al., 2009; Letunic and Bork, 2021). A phylogenetic tree based on the pks island was constructed by aligning the genomic sequences of pks-positive strains to the pks island sequence of the reference strain. The phylogenetic tree based on core genes of all pks-positive strains and that based on the pks island were linked using the R package Phytools to construct the co-phylogenetic tree (Revell, 2012).
2.7. Identification of mobile genetic elements
The genome sequences of the strains were uploaded to the ICEFinder and ISFinder websites to predict the number and types of integrative and conjugative elements (ICEs) and insertion sequences (ISs) of K. pneumoniae strains (Siguier et al., 2006; Liu et al., 2019). The e value of the ISfinder was set to 1e-5. The gene sequences were submitted to the PHASTER website to determine the number and size of prophages (Arndt et al., 2016). The plasmid replicon type was determined using staramr version 0.8.0 (Bharat et al., 2022), which can scan the bacterial genome against the PlasmidFinder database (Carattoli et al., 2014). The percent identity threshold and percent length overlap for the PlasmidFinder results were set to 98% and 60%, respectively.
2.8. Collection of clinical information
Clinical data of 157 pks-positive and 157 pks-negative K. pneumoniae patients (random sampling) from a large tertiary hospital in China were collected by consulting the electronic medical records. Specifically, stratified random sampling was used to enroll patients in this study. Clinical data from all 157 patients with pks-positive K. pneumoniae infection in this hospital were included in the study. The number of enrolled patients with pks-negative K. pneumoniae infection was determined at a 1:1 ratio. Considering that differences in the prevalence of K. pneumoniae infection may occur in different years and in different quarters, we stratified the patients with pks-negative infection in this hospital by year and month. Then, according to the year and month distribution of pks-positive K. pneumoniae infection patients, the number of pks-negative K. pneumoniae infection enrolled patients in each year and different months of each year was determined. Within each stratum, random sampling was performed according to computer-generated random numbers. The clinical information collected included sex, age, underlying conditions, sample source, and ward distribution.
2.9. Statistical analysis
Categorical variables were analyzed using the chi-square test or Fisher’s exact test. Continuous variables were analyzed using t test or Mann–Whitney U test. Statistical analyses of all data were performed on GraphPad Prism 9 and SPSS version 26.0. p < 0.05 was considered statistically significant.
2.10. Ethics statement
This study was approved by the Central South University Ethics Committee (Changsha, Hunan Province, People’s Republic of China) with ID 202212294.
3. Results
3.1. Prevalence and distribution of pks-positive Klebsiella pneumoniae
A total of 245 pks-positive K. pneumoniae genomes were identified from 2,709 genomes; of them, 157 were from the China National GeneBank Database (CNGBdb) (Chen et al., 2020) and 88 were from the public database NCBI. The 245 pks-positive K. pneumoniae included 79.6% (195/245) from China, 10.2% (25/245) from the United States, 6.1% (15/245) from other countries, and 4.1% (10/245) with unknown background information (Figure 1). Multi-locus sequence typing showed that 58.0% (142/245) of pks-positive K. pneumoniae were ST23 and 8.2% (20/245) were ST258. ST65, ST268, and ST792 accounted for 6.9% (17/245), 6.9% (17/245), and 4.5% (11/245) of the 245 pks-positive strains, respectively. The main serotype was K1, accounting for 63.3% (155/245) of cases. K2, K20, and K107 accounted for 10.6% (26/245), 6.9% (17/245), and 7.3% (18/245) of the 245 pk-positive strains, respectively (Table 1).
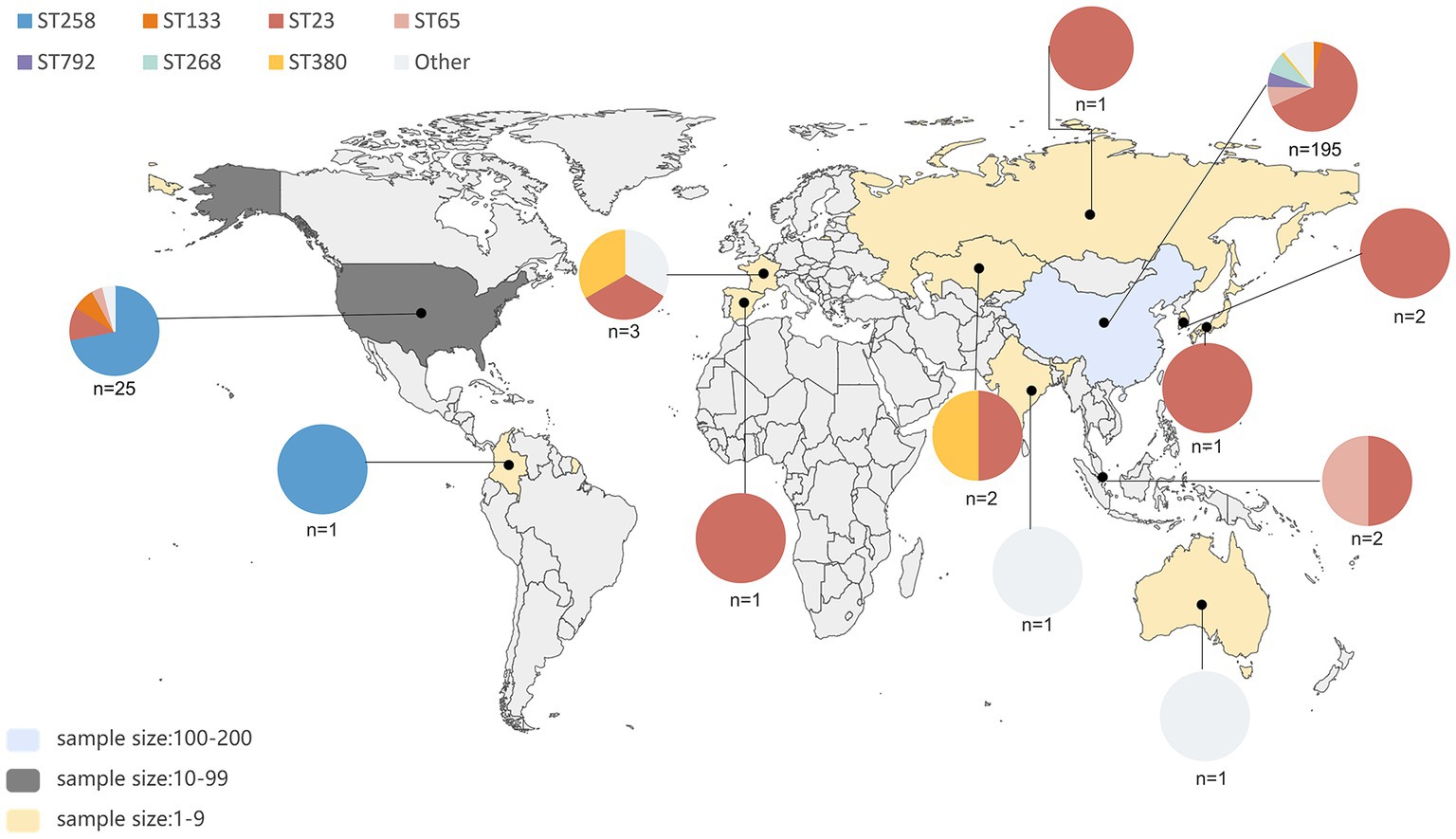
Figure 1. Geographic distribution and sequence type distribution of 235 pks-positive strains. A total of 245 pks-positive strains were screened, of which 10 had unknown background information. Sequence types and their proportions of pks-positive strains in each region are indicated by different colors in the pie chart. The number of pks-positive strains in different regions of this study is indicated by different colors, as shown in the bottom left corner.
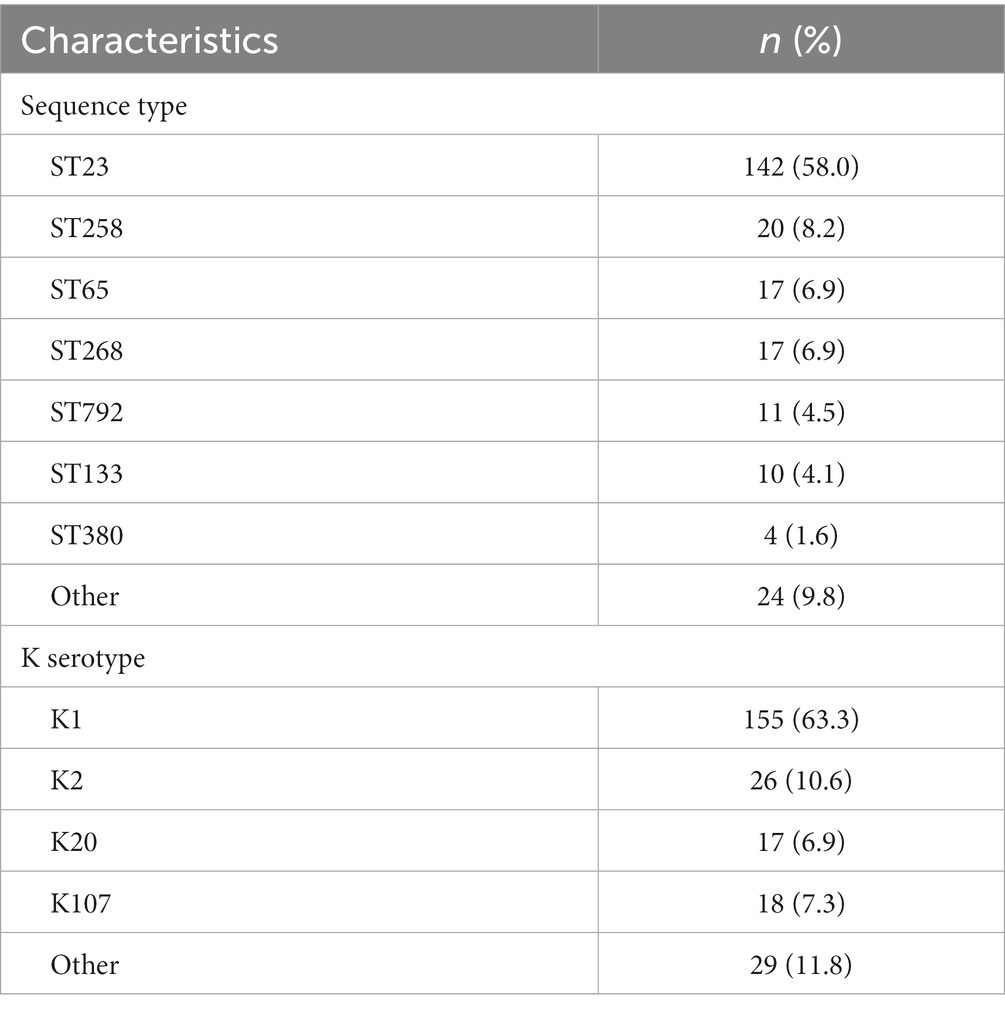
Table 1. Sequence type and serotype distribution of 245 pks-positive K. pneumoniae.
3.2. Distribution of virulence and antibiotic resistance genes in pks-positive strains
To determine the prevalence of virulence and resistance genes, we screened the genomes of 245 pks-positive K. pneumoniae (Figure 2). Virulence genes, including siderophores, type VI secretory system, type 1 and type 3 fimbriae, and nutritional factors, were investigated in this study. Yersiniabactin siderophore system genes (ybtAEPQSTUX, irp1, irp2) and enterobactin synthase genes (entABCEFS, fepABCDG) were found in at least 98.7% (242/245) of the 245 pks-positive strains. Aerobactin siderophore synthesis system genes (iucABCD) and salmochelin genes (iroBCDEN) were present in at least 77.9% (191/245) of the 245 pks-positive strains (Figure 2A). Notably, pks-positive strains showed a high prevalence of type VI secretion systems. Type VI secretion system-associated genes such as membrane complex-associated genes (tssL, tssM), substrate complex-associated genes (tssF, tssG, tssK), and tail complex-associated genes (tssA, tssB, tssC, tssD) were present in at least 66.9% (164/245) of the 245 pks-positive strains. The type 3 fimbriae gene (mrkABCDFHIJ) and type 1 fimbriae gene (fimABCEFGHIK) were present in at least 93.5% (229/245) of the 245 pks-positive strains. Nutritional factor genes (allABCDRS) were present in 63.3% (155/245) of the 245 pks-positive strains (Figure 2B).
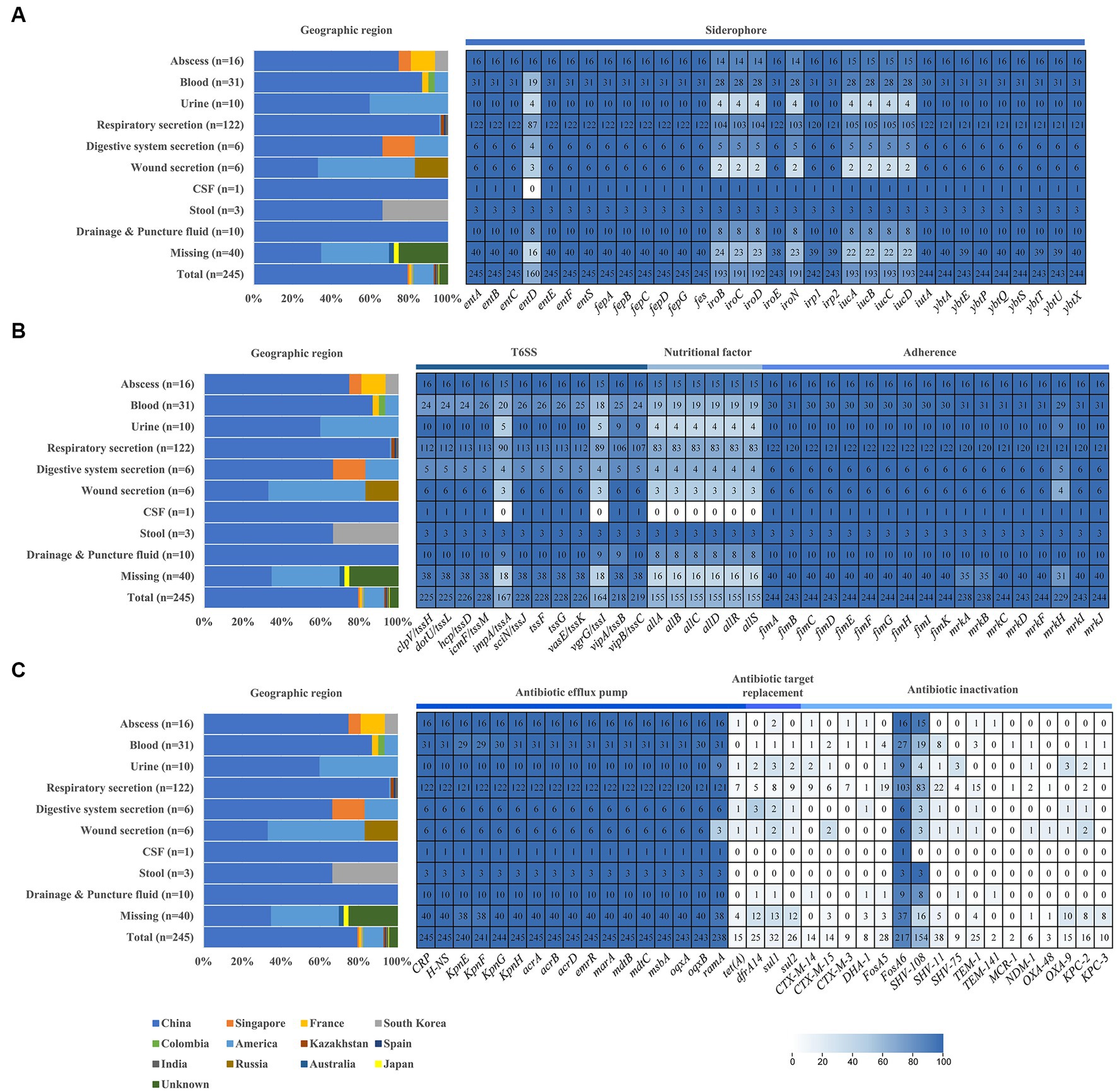
Figure 2. Distribution of geographical, virulence genes, and antibiotic resistance genes in pks-positive Klebsiella pneumoniae from different sources. (A) Siderophore associated virulence gene. (B) T6SS, nutritional factor and adherence. (C) Antibiotic resistance genes. Graphs and heatmaps show the proportion of strains, and the numbers in the heatmap boxes represent the number of strains harboring the virulence or antibiotic resistance gene.
Compared with pks-positive K. pneumoniae, the prevalence of these virulence genes in pks-negative K. pneumoniae was lower. Among 2,464 pks-negative isolates, yersiniabactin siderophore system genes (ybtAEPQSTUX, irp1, irp2), aerobactin siderophore synthesis system genes (iucABCD) and salmochelin genes (iroBCDEN) were detected in no more than 51.2% (1,261/2464), 23.0% (566/2464) and 10.5% (259/2464) of genomes, respectively. Nutritional factor genes (allABCDRS) and type VI secretory system-associated genes (impA/tssA, vgrG/tssI) were found in 2.2% (55/2464) and 30.3% (746/2464) of the 2,464 pks-negative strains, respectively (Supplementary Figure S1).
Antibiotic resistance genes associated with efflux pumps, antibiotic target replacement, and antibiotic inactivation were investigated (Figure 2C). Efflux pump genes, including CRP (global regulator), acr (aminoglycoside efflux pump), mar (multiple antibiotic resistance family), mdt (multi-drug efflux system), oqxAB (mediating quinolone resistance) and KpnEFGH, were present in at least 97.1% (238/245) of 245 pks-positive strains. Antibiotic target replacement genes (dfrA14, sul1, and sul2) were present in no more than 13.1% (32/245) of the 245 pks-positive strains. OXA-β-lactamase, NDM, KPC, and CTX-M-β-lactamase, which belong to antibiotic inactivation genes, were present in 6.5% (16/245) of the 245 pks-positive strains. More detailed results of the antibiotic-resistance genes are shown in Figure 2C.
3.3. Phylogenetic analysis and pangenome analysis
We constructed a maximum likelihood phylogenetic tree based on 245 pks-positive K. pneumoniae core genes (Figure 3). The 245 pks-positive K. pneumoniae strains have high genetic diversity and can be divided into different branches. Colibactin (clb) and yersiniabactin (ybt) determinants were strongly associated, and 99.6% (244/245) of the pks-positive strains carried both the clb and ybt genes (the first and second circles of the phylogenetic tree). In most cases, the distribution of the ybt locus and the clb locus is identical, with each ybt classification corresponding to a distinct clb classification.
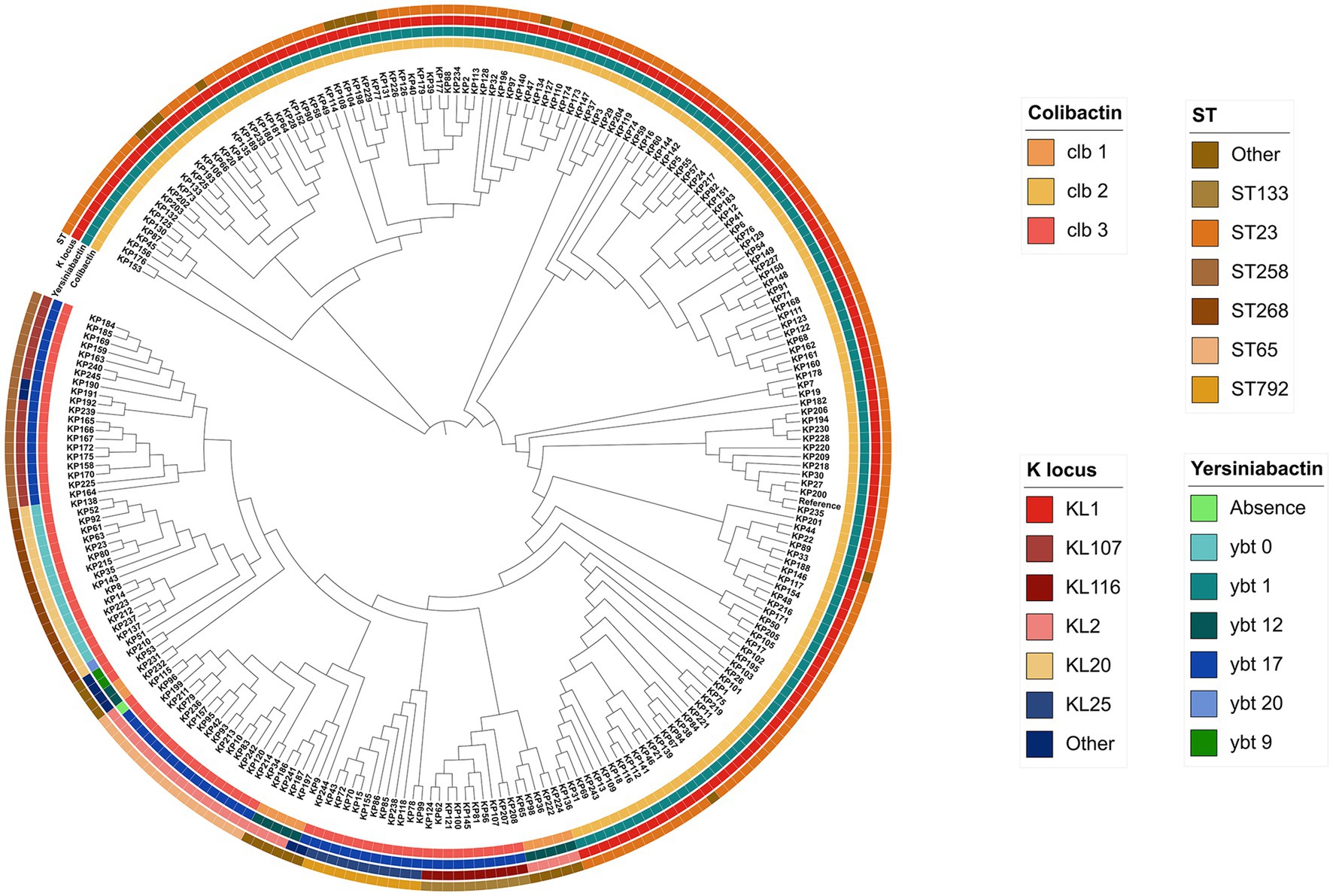
Figure 3. Phylogenetic tree based on SNP sites in core genes of 245 pks-positive strains. The outer circles from inside to outside indicate colibactin, yersiniabactin, serotype, and sequence type, respectively. Different colors in each circle represent different categories.
We also identified 3,649 core genes (genes present in at least 99% of the strains were considered core genes) in 245 pks-positive K. pneumoniae using the Roary version 3.13.0 (Page et al., 2015). A total of 99.0% (3,614/3649) of the core genes were assigned to COG categories (some genes were assigned to more than one COG category; Supplementary Figure S2). As shown in Supplementary Figure S2, the core genes belonging to the S (Function unknown) category were the most abundant (n = 728) and the K (transcription) category was the second most abundant COG assigned category (n = 370). Cell cycle control, cell division, chromosome partitioning (D), cell motility (N), and defense mechanisms (V), which are cellular processes and signals, are less represented in the core genes (n = 51, n = 38, n = 42).
3.4. Evolution of the pks locus in Klebsiella pneumoniae
To further understand the scenario of strains acquiring pks sequences, we constructed a phylogenetic tree based on the pks sequence and connected it to a phylogenetic tree based on the core genes of pks-positive K. pneumoniae (Figure 4). It can be observed that the clustering patterns of strains of clonal group (CG) 380 and CG792 lineages are consistent in the two evolutionary trees. In comparison, the clustering patterns of strains of CG65 and CG23 lineages differ in the two evolutionary trees.
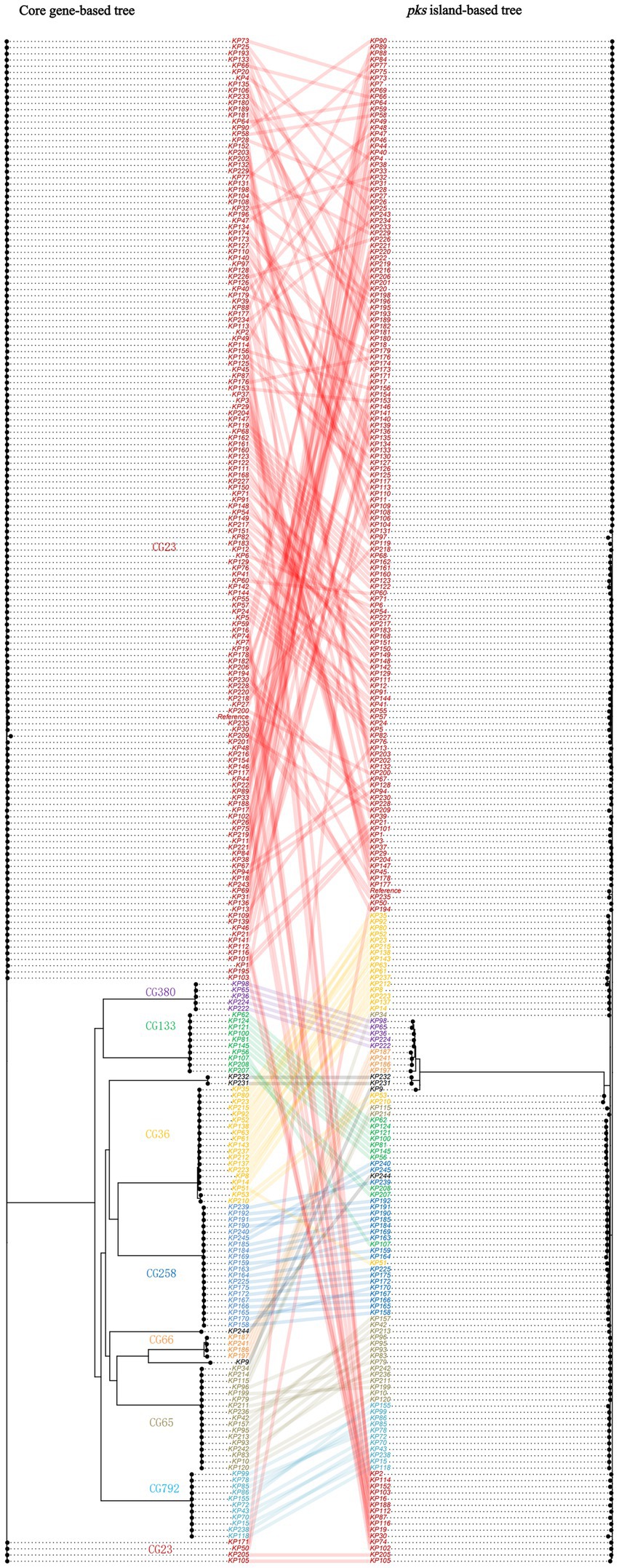
Figure 4. Co-phylogeny of pks sequences and K. pneumoniae strains. The strains belonging to the major clonal groups (CGs) are shown with colored names.
3.5. Differences between mobile genetic elements in pks-positive and pks-negative strains
To further understand the genetic background, we compared the distribution of mobile genetic elements, such as plasmids, insertion sequences (ISs), prophages, and integrative and conjugative elements (ICEs) in pks-positive and pks-negative K. pneumoniae. Considering the high accuracy of the complete genome, we focused our analysis on the mobile genetic elements of 88 pks-positive and 1,334 pks-negative K. pneumoniae complete genomes downloaded from NCBI (Figures 5–7). All 88 pks-positive strains carried at least one ICE, whereas 91.3% (1,218/1334) of pks-negative strains carried at least one ICE. However, the median number of ICEs carried by both pks-negative and pks-positive strains was 2 (Figure 5A). Prophages (intact, incomplete, and suspicious phages) were predicted in both pks-positive and pks-negative genomes. Notably, there were significant differences in the number and length of prophages between the pks-positive and pks-negative strains (Figures 5B,C). The median number of prophages in the pks-positive group was 7, while the median number of prophages in the pks-negative group was 11. Further, pks-negative K. pneumoniae appeared to carry longer prophage fragments than did pks-positive K. pneumoniae (median length 321.0 kb and 212.8 kb, respectively).
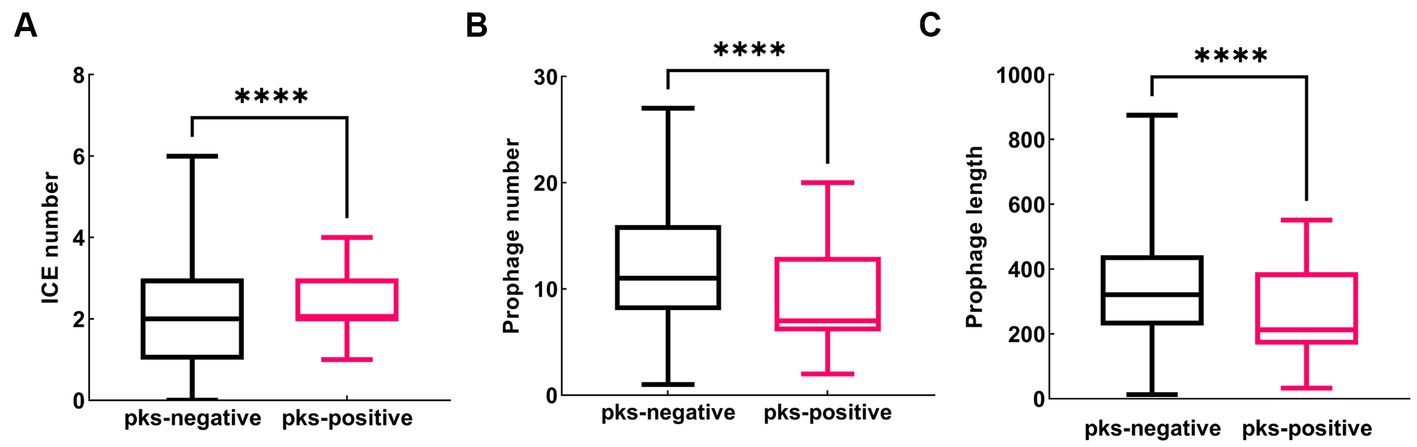
Figure 5. Distribution of predicted ICE and prophage in pks-positive and pks-negative K. pneumoniae genomes (complete genome). (A) The number of predicted ICE in the genomes of pks-positive and pks-negative strains. (B) The number of predicted prophage in the genomes of pks-positive and pks-negative strains. (C) The length of predicted prophage in the genomes of pks-positive and pks-negative strains.
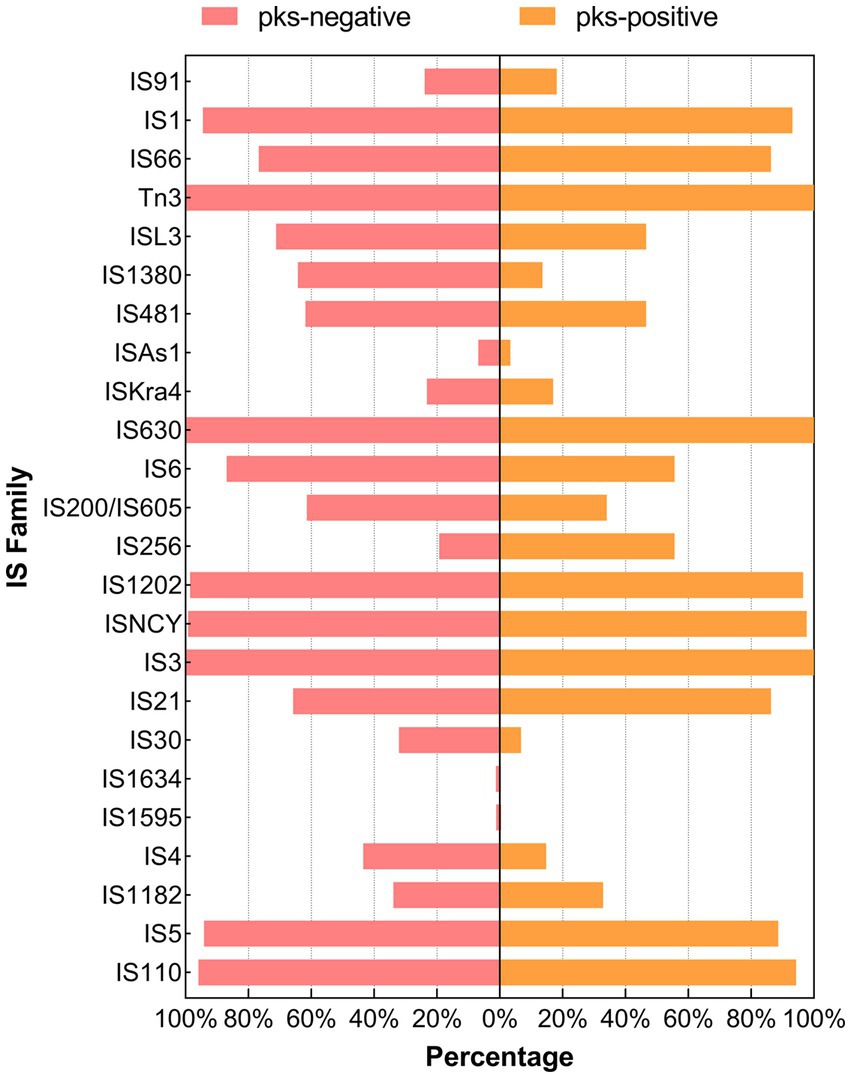
Figure 6. Distribution of the IS family between pks-positive and pks-negative K. pneumoniae (complete genome). The red color indicates that the distribution of this category in pks-positive and pks-negative strains is statistically significant.
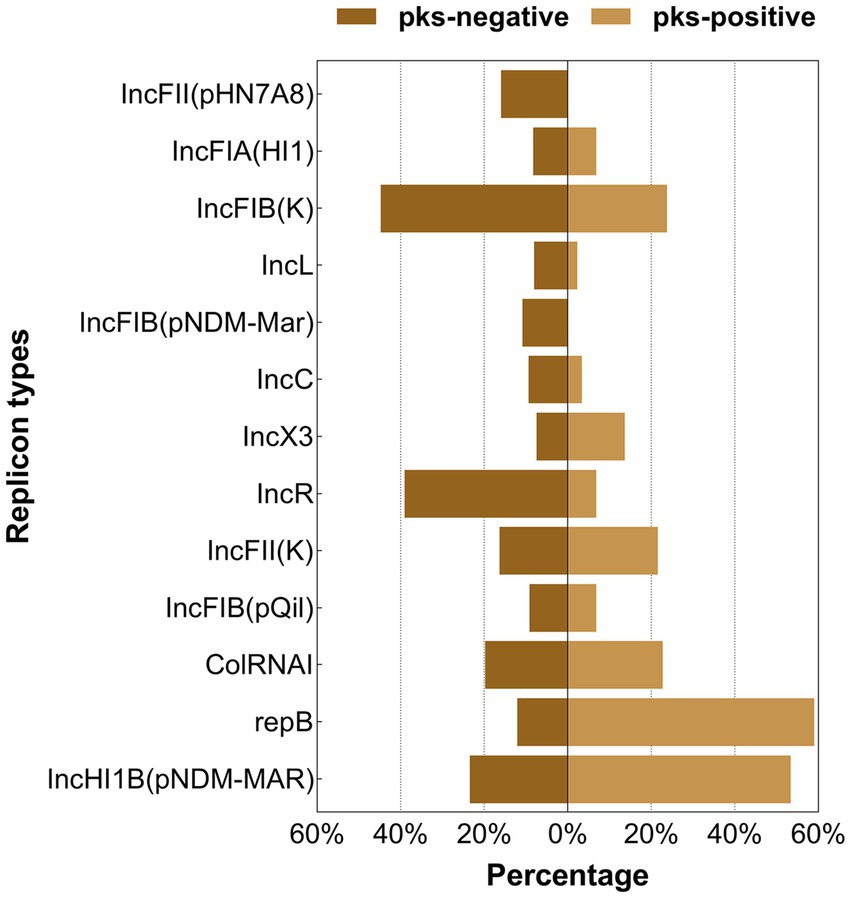
Figure 7. Distribution of the major plasmid replicon types between pks-positive and pks-negative K. pneumoniae (complete genome). The red color indicates that the distribution of this category in pks-positive and pks-negative strains is statistically significant.
We predicted 24 insertion sequence families in pks-negative strains, but only 22 in pks-positive strains (Figure 6). IS3, IS630, Tn3, IS1202, and ISNCY were present in almost all 1,442 strains, while a variety of insertion sequences, including IS6, ISL3, IS66, and IS1380, were significantly different between pks-positive and pks-negative strains (p < 0.05). In addition, we found that the pks-negative strains had more plasmid replicon types than did the pks-positive strains, which may be due to the larger number of strains contained in the pks-negative group (Supplementary Table S2). The major plasmid replicon types and their proportions in both the groups are shown in Figure 7. These plasmid replicon types included incompatibility plasmids (Inc.) and mobilizable colicin plasmid (Col) groups. RepB (59.1%, 52/88) was the most abundant plasmid replicon type in the pks-positive strains, whereas the plasmid replicon type IncFIB(K) (44.8%, 597/1334) was the most abundant in the pks-negative strains. The distribution of several types of plasmid replicons, including IncR, IncFII(pHN7A8), IncFIB(pNDM-Mar), and IncX3, differed between pks-positive and pks-negative strains (p < 0.05). The differences in mobile genetic elements between pks-positive and pks-negative strains described above were similarly observed when the study was extended from complete genomes to include both complete and incomplete genomes (Supplementary Figure S3; Supplementary Tables S3, S4; Table 2).
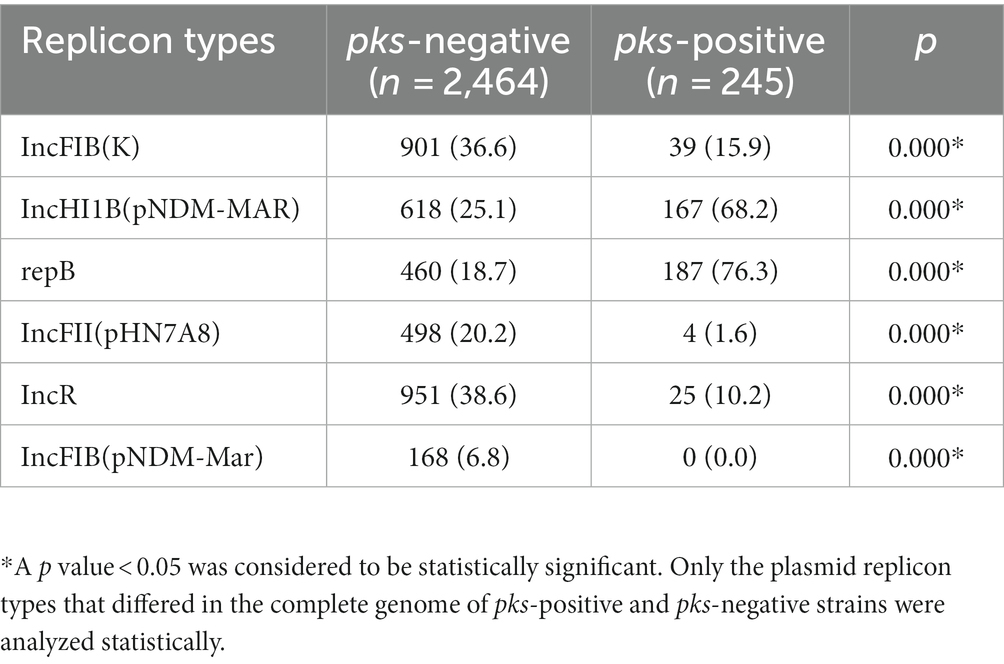
Table 2. Distribution of plasmid replicon types in pks-positive and pks-negative K. pneumoniae genomes (complete and incomplete genomes are included).
3.6. Clinical characteristics of pks-positive Klebsiella pneumoniae infection
We analyzed the clinical data of 157 pks-positive K. pneumoniae and 157 pks-negative K. pneumoniae infected patients to obtain a comprehensive picture of the risk factors for pks-positive K. pneumoniae infections (Tables 3, 4). The clinical characteristics associated with infection with pks-positive and pks-negative strains were different in several aspects. In infants younger than or equal to 1 year of age, the detection rate of pks-positive K. pneumoniae (1.9%, 3/157) was lower than that of pks-negative (12.1%, 19/157) K. pneumoniae (p < 0.05). Specimens from abscess sites were more likely to contain pks-positive strains (7.6%, 12/157) than pks-negative (1.9%, 3/157) strains (p < 0.05). Notably, the detection rate of pks-positive strains (1.9%, 3/157) was lower than that of pks-negative (11.5%, 18/157) strains in the neonatal ward (p < 0.05). This also corresponds to the lower incidence of infections caused by pks-positive K. pneumoniae in infants younger than or equal to 1 year of age, as described above. In addition, pks-positive strains (26.1%, 41/157) were more likely to be detected in patients from internal medicine than were pks-negative (14.6%, 23/157) strains (p < 0.05). Further, the statistical results suggested that patients with diabetes and cerebral infarction were more sensitive to pks-positive strains than pks-negative strains (p < 0.05).
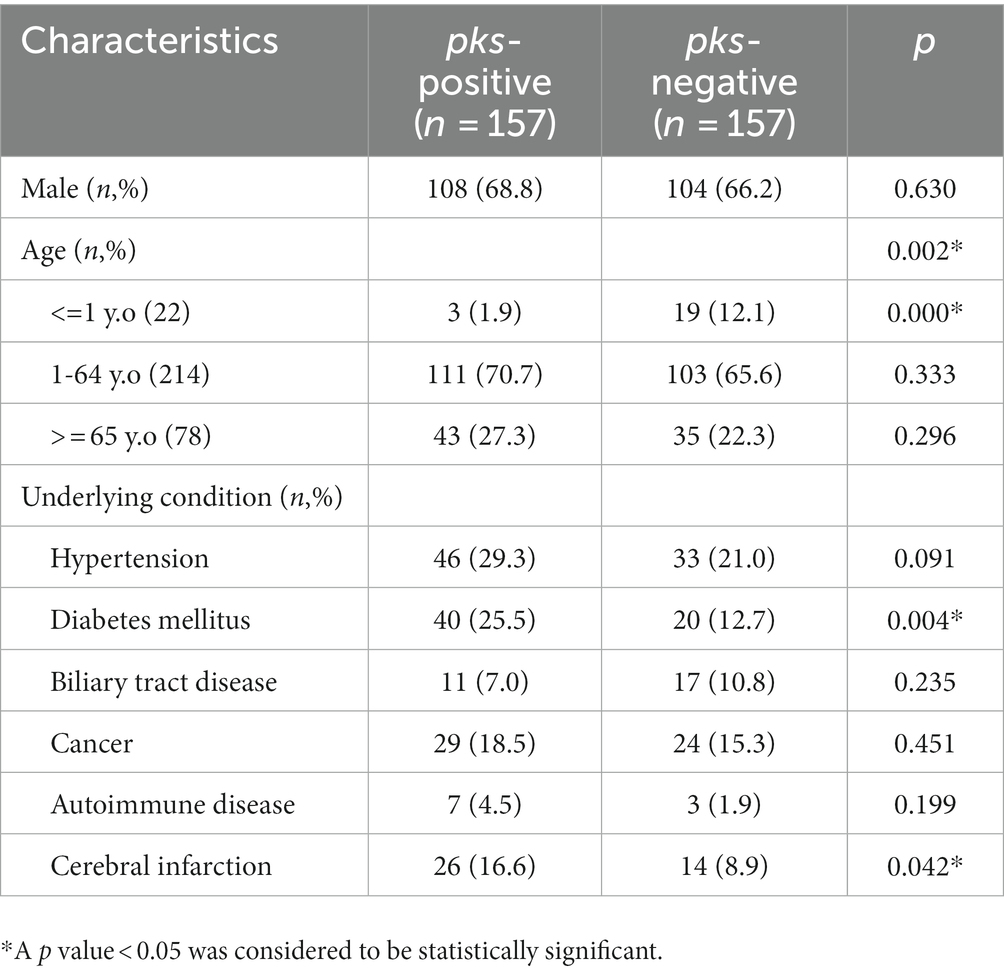
Table 3. Demographic characteristics and underlying conditions of patients with pks-positive and pks-negative K. pneumoniae infection.
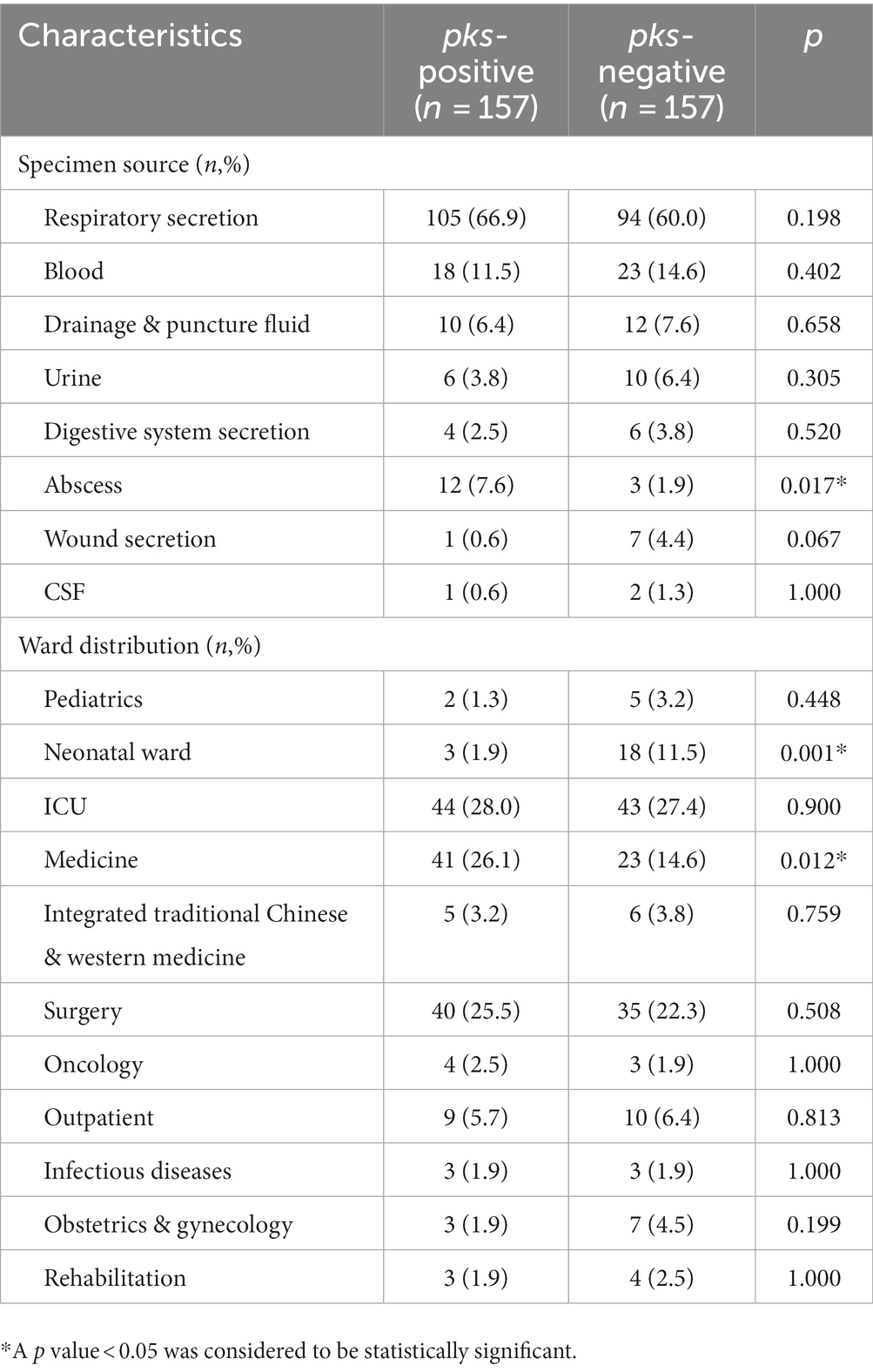
Table 4. The specimen source and ward distribution of pks-positive and pks-negative isolates.
4. Discussion
Here, a genomic analysis of K. pneumoniae genome sequences—obtained from public databases—was conducted and the clinical information of patients with pks-positive and pks-negative K. pneumoniae infections was investigated; consequently, the results provide new insights for a better understanding of the evolution and prevalence of the pks gene cluster in K. pneumoniae.
Studies have shown that K1 and ST23 are the main serotypes and sequence types of hypervirulent K. pneumoniae (hvKP) respectively (Struve et al., 2015; Shi et al., 2018). Our study found that K1 and ST23 were also the main serotypes and sequence types of pks-positive strains, which is consistent with results of previous studies (Chen et al., 2017; Lan et al., 2019; Shi et al., 2020). The sequence type of some of the pks-positive strains in our study was ST258. Notably, ST258 is one of the major sequence types of multi-drug resistant strains (Wyres et al., 2020). The presence of the pks island in the ST258 strains may reflect the unity of high virulence and resistance. In addition, 66.9% of pks-positive isolates contained type VI secretion system genes associated with hypervirulence. These suggest that the pks gene cluster and high virulence genes are closely related. A previous study showed that pks-positive K. pneumoniae have low drug resistance (Lan et al., 2019), and our study with more strains and a richer sample source demonstrated similar results. These results indicate that pks-positive strains carry abundant virulence genes and fewer antibiotic resistance genes.
To further explore the scenario under which K. pneumoniae acquired pks sequences, we constructed a phylogenetic tree. Our results showed that the clustering patterns of the CG65 and CG23 lineage strains between the two trees differed, suggesting that the pks sequences of some of these lineage strains were acquired through horizontal gene transfer. In addition, the consistency of the CG380 and CG792 lineage strains between the two trees was observed in this study. This may indicate that the most recent common ancestor of these strains acquired the pks sequences by horizontal gene transfer, and then passed the pks sequences to the offspring by vertical propagation.
Comparative genomic analysis showed that there were differences in mobile genetic elements between pks-positive and pks-negative K. pneumoniae. Our study found that the prevalence of some insertion sequences differed between the pks-positive and pks-negative strains. Among them, the IS6 family showed a higher prevalence in this study. Previous studies have shown that the IS6 family, including IS26 and IS257, plays an important role in the spread of multiple antibiotic-resistance genes (Varani et al., 2021). Therefore, we speculate that the low prevalence of IS6 among pks-positive strains may be related to their antibiotic resistance. In addition, repB is the most common replicon types in pks-positive strains. IS256 and IS21 are common insertion sequence types in pks-positive strains. However, the relationship between them and pks island needs further analysis. Notably, we found that the number and total length of prophages in pks-positive strains were significantly lower than those in pks-negative strains. Considering that phages can carry antibiotic resistance genes and virulence genes, the differences in the number and total length of prophages between pks-positive and pks-negative strains may be related to the prevalence of resistance and virulence genes among strains. In addition, Silpe et al. showed that colibactin production by pks-positive E. coli could induce prophages in bacteria (Silpe et al., 2022). These results suggest a potential complex link between pks gene islands and prophages.
Analysis of clinical characteristics showed that multiple factors were associated with pks-positive K. pneumoniae infection. Previous studies have shown a higher proportion of liver abscesses in patients infected with pks-positive strains (Shi et al., 2020). Our study found similar results, with a higher detection rate of pks-positive strains than of pks-negative strains in samples of abscess origin. It has been shown that the pks island is widely present in E. coli K1, which causes neonatal infections, and colibactin contributes to the colonization of K1 E. coli in the gastrointestinal tract (McCarthy et al., 2015). Notably, in this study, more K. pneumoniae infections in the neonatal ward were caused by pks-negative K. pneumoniae. We hypothesize that the possible reason is that the common K. pneumoniae may be sufficient to infect neonates due to their low immunity, thus causing the pks-positive K. pneumoniae to be less dominant in the infection. A study from China showed that infections caused by hypervirulent K. pneumoniae are uncommon in children (Li et al., 2021). Given the close relationship between the pks island and hypervirulent K. pneumoniae, this may also partially explain the low detection rate of pks-positive K. pneumoniae in patients younger than 1 year of age in this study. In addition, our study showed that patients infected with pks-positive K. pneumoniae had a higher proportion of diabetes and abscesses. Studies have shown that diabetes and liver abscess are strongly associated with hvKp infections (Zhang et al., 2016; Liu and Guo, 2019; Namikawa et al., 2023). This support that pks islands are closely related to hvKp and may play an important role in hvKp infections.
However, this study had certain limitations. We did not calculate the prevalence of pks islands in strains from different specimen sources because of the lack of background information from public databases and because the number of strains from different sources varied greatly. We did not collect the clinical data of all patients with pks-negative K. pneumoniae infection, but only selected some patients as study subjects by stratified random sampling. In addition, retrospective analysis of the data may also introduce bias. Finally, 79.6% of pks-positive K. pneumoniae isolates in our study were from China, which may make the conclusions of this study less generalizable.
In summary, through a large-scale analysis, we described the genomic characteristics of pks-positive K. pneumoniae and the clinical characteristics of pks-positive K. pneumoniae infection. We found for the first time that the number and fragment length of prophages were lower in pks-positive strains than in pks-negative strains, and the prevalence of the IS6 family was higher in pks-negative strains than in pks-positive strains. Plasmid replicon types differed between the pks-positive and pks-negative strains. However, further studies on the genetic background of pks islands in K. pneumoniae are needed to gain a deeper understanding of the evolution of pks islands in K. pneumoniae. In addition, more bacterial species need to be included to fully understand the transfer and spread of pks islands among different bacteria.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
WL and ZW conceived and designed the project. ZW, YL, and PL collected the genomic data and clinical data and performed the statistical analysis. ZW, BT, and AY performed the genome analysis. ZW wrote the manuscript. WL, ZW, ZJ, and QY revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (grant number 81672066) and National Key R&D Program of China (no. 2018YFC2000203).
Acknowledgments
We are grateful for resources from the High Performance Computing Center of Central South University, China.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1189120/full#supplementary-material
Footnotes
References
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Auvray, F., Perrat, A., Arimizu, Y., Chagneau, C. V., Bossuet-Greif, N., Massip, C., et al. (2021). Insights into the acquisition of the pks island and production of colibactin in the Escherichia coli population. Microb. Genom. 7:000579. doi: 10.1099/mgen.0.000579
Bengoechea, J. A., and Sa Pessoa, J. (2019). Klebsiella pneumoniae infection biology: living to counteract host defences. FEMS Microbiol. Rev. 43, 123–144. doi: 10.1093/femsre/fuy043
Bharat, A., Petkau, A., Avery, B. P., Chen, J. C., Folster, J. P., Carson, C. A., et al. (2022). Correlation between phenotypic and in silico detection of antimicrobial resistance in Salmonella enterica in Canada using Staramr. Microorganisms 10:292. doi: 10.3390/microorganisms10020292
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/aac.02412-14
Chen, F. Z., You, L. J., Yang, F., Wang, L. N., Guo, X. Q., Gao, F., et al. (2020). CNGBdb: China national GeneBank DataBase. Yi Chuan 42, 799–809. doi: 10.16288/j.yczz.20-080
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Chen, Y. T., Lai, Y. C., Tan, M. C., Hsieh, L. Y., Wang, J. T., Shiau, Y. R., et al. (2017). Prevalence and characteristics of pks genotoxin gene cluster-positive clinical Klebsiella pneumoniae isolates in Taiwan. Sci. Rep. 7:43120. doi: 10.1038/srep43120
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Haudiquet, M., de Sousa, J. M., Touchon, M., and Rocha, E. P. C. (2022). Selfish, promiscuous and sometimes useful: how mobile genetic elements drive horizontal gene transfer in microbial populations. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 377:20210234. doi: 10.1098/rstb.2021.0234
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Koonin, E. V., Makarova, K. S., and Aravind, L. (2001). Horizontal gene transfer in prokaryotes: quantification and classification. Annu. Rev. Microbiol. 55, 709–742. doi: 10.1146/annurev.micro.55.1.709
Lam, M. M. C., Wick, R. R., Watts, S. C., Cerdeira, L. T., Wyres, K. L., and Holt, K. E. (2021). A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 12:4188. doi: 10.1038/s41467-021-24448-3
Lam, M. M. C., Wick, R. R., Wyres, K. L., Gorrie, C. L., Judd, L. M., Jenney, A. W. J., et al. (2018). Genetic diversity, mobilisation and spread of the yersiniabactin-encoding mobile element ICEKp in Klebsiella pneumoniae populations. Microb. Genom. 4:e000196. doi: 10.1099/mgen.0.000196
Lan, Y., Zhou, M., Jian, Z., Yan, Q., Wang, S., and Liu, W. (2019). Prevalence of pks gene cluster and characteristics of Klebsiella pneumoniae-induced bloodstream infections. J. Clin. Lab. Anal. 33:e22838. doi: 10.1002/jcla.22838
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–w296. doi: 10.1093/nar/gkab301
Li, Y., Dong, L., Gao, W., Zhen, J., Dong, F., and Yao, K. (2021). Hypervirulent klebsiella pneumoniae infections in pediatric populations in beijing (2017-2019): clinical characteristics, molecular epidemiology and antimicrobial susceptibility. Pediatr. Infect. Dis. J. 40, 1059–1063. doi: 10.1097/inf.0000000000003253
Liu, C., and Guo, J. (2019). Hypervirulent Klebsiella pneumoniae (hypermucoviscous and aerobactin positive) infection over 6 years in the elderly in China: antimicrobial resistance patterns, molecular epidemiology and risk factor. Ann. Clin. Microbiol. Antimicrob. 18:4. doi: 10.1186/s12941-018-0302-9
Liu, M., Li, X., Xie, Y., Bi, D., Sun, J., Li, J., et al. (2019). ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 47, D660–d665. doi: 10.1093/nar/gky1123
Lu, M. C., Chen, Y. T., Chiang, M. K., Wang, Y. C., Hsiao, P. Y., Huang, Y. J., et al. (2017). Colibactin contributes to the Hypervirulence of pks(+) K1 CC23 Klebsiella pneumoniae in mouse meningitis infections. Front. Cell. Infect. Microbiol. 7:103. doi: 10.3389/fcimb.2017.00103
Marcq, I., Martin, P., Payros, D., Cuevas-Ramos, G., Boury, M., Watrin, C., et al. (2014). The genotoxin colibactin exacerbates lymphopenia and decreases survival rate in mice infected with septicemic Escherichia coli. J. Infect. Dis. 210, 285–294. doi: 10.1093/infdis/jiu071
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/aac.00419-13
McCarthy, A. J., Martin, P., Cloup, E., Stabler, R. A., Oswald, E., and Taylor, P. W. (2015). The Genotoxin Colibactin is a determinant of virulence in Escherichia coli K1 experimental neonatal systemic infection. Infect. Immun. 83, 3704–3711. doi: 10.1128/iai.00716-15
Namikawa, H., Oinuma, K. I., Yamada, K., Kaneko, Y., Kakeya, H., and Shuto, T. (2023). Predictors of hypervirulent Klebsiella pneumoniae infections: a systematic review and meta-analysis. J. Hosp. Infect. 134, 153–160. doi: 10.1016/j.jhin.2023.02.005
Nougayrède, J. P., Homburg, S., Taieb, F., Boury, M., Brzuszkiewicz, E., Gottschalk, G., et al. (2006). Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313, 848–851. doi: 10.1126/science.1127059
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pleguezuelos-Manzano, C., Puschhof, J., Rosendahl Huber, A., van Hoeck, A., Wood, H. M., Nomburg, J., et al. (2020). Mutational signature in colorectal cancer caused by genotoxic pks(+) E. coli. Nature 580, 269–273. doi: 10.1038/s41586-020-2080-8
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Prorok-Hamon, M., Friswell, M. K., Alswied, A., Roberts, C. L., Song, F., Flanagan, P. K., et al. (2014). Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 63, 761–770. doi: 10.1136/gutjnl-2013-304739
Putze, J., Hennequin, C., Nougayrède, J. P., Zhang, W., Homburg, S., Karch, H., et al. (2009). Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect. Immun. 77, 4696–4703. doi: 10.1128/iai.00522-09
Revell, L. J. (2012). Phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Russo, T. A., and Marr, C. M. (2019). Hypervirulent Klebsiella pneumoniae. Clin. Microbiol. Rev. 32, e00001–e00019. doi: 10.1128/cmr.00001-19
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shi, Q., Lan, P., Huang, D., Hua, X., Jiang, Y., Zhou, J., et al. (2018). Diversity of virulence level phenotype of hypervirulent Klebsiella pneumoniae from different sequence type lineage. BMC Microbiol. 18:94. doi: 10.1186/s12866-018-1236-2
Shi, Q., Quan, J., Lan, P., Huang, D., Zhou, J., Jiang, Y., et al. (2020). Prevalence and characteristics of pks gene cluster harbouring Klebsiella pneumoniae from bloodstream infection in China. Epidemiol. Infect. 148:e69. doi: 10.1017/s0950268820000655
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference Centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Silpe, J. E., Wong, J. W. H., Owen, S. V., Baym, M., and Balskus, E. P. (2022). The bacterial toxin colibactin triggers prophage induction. Nature 603, 315–320. doi: 10.1038/s41586-022-04444-3
Siu, L. K., Yeh, K. M., Lin, J. C., Fung, C. P., and Chang, F. Y. (2012). Klebsiella pneumoniae liver abscess: a new invasive syndrome. Lancet Infect. Dis. 12, 881–887. doi: 10.1016/s1473-3099(12)70205-0
Soucy, S. M., Huang, J., and Gogarten, J. P. (2015). Horizontal gene transfer: building the web of life. Nat. Rev. Genet. 16, 472–482. doi: 10.1038/nrg3962
Struve, C., Roe, C. C., Stegger, M., Stahlhut, S. G., Hansen, D. S., Engelthaler, D. M., et al. (2015). Mapping the evolution of Hypervirulent Klebsiella pneumoniae. mBio 6:e00630. doi: 10.1128/mBio.00630-15
Suresh, A., Shaik, S., Baddam, R., Ranjan, A., Qumar, S., Jadhav, S., et al. (2021). Evolutionary dynamics based on comparative genomics of pathogenic Escherichia coli lineages harboring polyketide synthase (pks). Island. mBio 12, e03634–e03620. doi: 10.1128/mBio.03634-20
Varani, A., He, S., Siguier, P., Ross, K., and Chandler, M. (2021). The IS6 family, a clinically important group of insertion sequences including IS26. Mob. DNA 12:11. doi: 10.1186/s13100-021-00239-x
Wami, H., Wallenstein, A., Sauer, D., Stoll, M., von Bünau, R., Oswald, E., et al. (2021). Insights into evolution and coexistence of the colibactin – and yersiniabactin secondary metabolite determinants in enterobacterial populations. Microb Genom 7:000577. doi: 10.1099/mgen.0.000577
Wyres, K. L., Lam, M. M. C., and Holt, K. E. (2020). Population genomics of Klebsiella pneumoniae. Nat. Rev. Microbiol. 18, 344–359. doi: 10.1038/s41579-019-0315-1
Zhang, Y., Zhao, C., Wang, Q., Wang, X., Chen, H., Li, H., et al. (2016). High prevalence of Hypervirulent Klebsiella pneumoniae infection in China: geographic distribution, clinical characteristics, and antimicrobial resistance. Antimicrob. Agents Chemother. 60, 6115–6120. doi: 10.1128/aac.01127-16
Keywords: Klebsiella pneumoniae, pks island, phylogeny, mobile genetic elements, clinical characteristics
Citation: Wang Z, Liu Y, Liu P, Jian Z, Yan Q, Tang B, Yang A and Liu W (2023) Genomic and clinical characterization of Klebsiella pneumoniae carrying the pks island. Front. Microbiol. 14:1189120. doi: 10.3389/fmicb.2023.1189120
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Russel Lee, National University of Singapore, SingaporeJonathan Rodriguez-Santiago, Center for Research on Infectious Diseases (CISEI), Mexico
Copyright © 2023 Wang, Liu, Liu, Jian, Yan, Tang, Yang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenen Liu, d2VuZW5saXVAMTYzLmNvbQ==