Abstract
This study aimed to improve knowledge of raptor microbiomes by providing the first description of tracheal and cloacal bacterial diversity of Eastern Imperial Eagles (Aquila heliaca). To date, only few studies are available and they are carried out mainly on captive birds. The Eastern Imperial Eagle is species of significant conservation concern and, therefore, characterization microbiota contributes valuable information to the field of avian microbiology and aids in conservation efforts for this threatened species, moreover, identification of avian and human pathogens within microbial communities and evaluation of potential threats to birds, humans, and other species are crucial for sustainably balancing the wellbeing of ecosystems, 3,500 OTUs were identified from each sample supported by ∼2.8 Million sequence reads. The tracheal and cloacal microbiomes were dominated by Gammaproteobacteria (67.5%), Bacilli (43.8%), and Negativicutes (22.0%). We detected dissimilarities between cloacal (unique 440 OTUs) and tracheal (337 unique OTUs) samples, and significant evidence of moderate positive monotonic relationship between cloacal and tracheal bacterial communities. No significant differences between individuals from different nests. Aquila heliaca can serve as an indicator of presence of bacterial species in its respective habitats. Efforts aiming at protection of red-listed birds may not presently prioritize microbiome considerations but integrating microbiome research into conservation strategies could yield significant benefits.
Introduction
The Eastern Imperial Eagle (Aquila heliaca) is a threatened species in Georgia. Up to 20 pairs breed in the east of the country (Galvez et al., 2005; Javakhishvili et al., 2020; Budagashvili, 2024). The species is listed as vulnerable (VU) (IUCN, 2016) and is declining over the entire breeding range (BirdLife, 2023). The main causes of mortality are poaching, collisions with power lines, habitat degradation, and the reduction of the habitual prey (Rudnick et al., 2008; Bragin et al., 2021; Budagashvili, 2024). Despite concerted legislative efforts, the regressive trend persists.
While biological and ecological studies have shed light on various aspects of A. heliaca’s life history (Hák et al., 2007; Katzner et al., 2014; Bekmansurov et al., 2015; Bragin et al., 2021), the microbial landscape within this species remains unexplored. The prevalence and incidence of infectious diseases in these birds remain poorly understood. Characterization and monitoring of microbiomes is emerging as a tool in conservation, particularly for the management of endangered species (Bahrndorff et al., 2016; West et al., 2019; Dallas and Warne, 2023). This approach provides insights into the relationships between the microbial communities inhabiting various species and their environment, offering valuable data that can help guide conservation strategies (Song et al., 2020; Dallas and Warne, 2023). As a result, the role of microbiomes in conservation biodiversity and supporting species at risk of extinction is being increasingly recognized. Undertaking relevant surveillance for diseases that may pose a threat to populations of birds of prey is a key aspect of preparing national or regional raptor conservation and management strategies (Kovács and Williams, 2012). Monitoring diseases and assessing their role in the effective conservation of Aquila heliaca is widely acknowledged by scientists working on the conservation of the species (Heredia, 1996; Horváth et al., 2006; Budagashvili, 2024). Although studies on microbiome of wildlife are increasing, most of these studies have focused on mammals (Colston and Jackson, 2016; Moeller and Sanders, 2020; Jeong et al., 2021), whereas bird species remain an exception (Araghi et al., 2015; Grond et al., 2018; Capunitan et al., 2020Zhao et al., 2022).
The interactions between different taxa within a microbiome are complex and multidirectional, and actively influence the overall health and wellbeing of the host species. Microbial composition is host specific and undergoes dynamic change over host’s lifetime, shaped by various factors: environmental conditions, dietary patterns and physiological state (Sekirov et al., 2010). The microbial community inhabiting in an organism plays a fundamental role in key ecological processes that are essential for resilience and survival. Studies indicate that it is associated with disease resistance, also influences host lifespan, reproductive success, and adaptive capacities to changes in the environment (Casadevall and Pirofski, 2000; Muegge et al., 2011; Stenkat et al., 2014). Mycotic, bacterial and viral species are beneficial, while some contribute to essential physiological processes and host adaptations, others cause infectious diseases and are identified as a threat of biodiversity (Daszak et al., 2000; Brilhante et al., 2012; West et al., 2019; Cunningham et al., 2017). The interconnection between microbial ecology and host health, necessitates a holistic approach to the conservation of threatened species (Bordenstein and Theis, 2015; Bahrndorff et al., 2016). Disease outbreaks can lead to disruption of ecological interactions, affect reproductive capabilities, alter predator-prey dynamics by affecting the health and behavior of species, decline population sizes, and even drive species to extinction (Smith et al., 2006; Hohenlohe et al., 2021). For endangered or vulnerable species, these microbial interactions are especially significant, as they can directly affect their ability to cope with environmental stressors and diseases. This underscores the importance of studying microbial communities within threatened species, as a lack of knowledge can have far-reaching consequences, thus, a deeper understanding of microbiome dynamics offers a promising avenue for enhancing conservation efforts by supporting the health, resilience, and long-term survival of the species.
Besides well-assessed various molecular methods (target sequencing, whole genome sequencing), to date we have a poor understanding of the evolutionary and ecological processes relevant to microbiome composition and function, especially in wild organisms and non-model taxa. Compounding these challenges is the difficulty in collecting biological samples, as rare niches in remote geographical locations present obstacles to efficient sampling. Microbiome studies of threatened species are of increasing concern for development management strategies and survival success of the species (Bahrndorff et al., 2016; West et al., 2019; Dallas and Warne, 2023). Incorporating such studies into conservation strategies can enhance our understanding of the complex interplay between organisms and their microbial communities, ultimately contributing to the successful management and survival.
Birds of prey cover hundreds of kilometers every year during migration and tend to spread emerging infectious diseases (Movalli et al., 2018). Raptors, due to their role as both predators and scavengers, can become reservoirs for a range of potentially zoonotic agents (Vidal et al., 2017; Rossi et al., 2021; Vogt, 2022). Obligatory migrant Eastern Imperial Eagle might be carriers of microbial pathogens and be potential spreaders to other species, even in humans e.g., droppings containing strains of Clostridium and Campylobacter spp. (Brilhante et al., 2012; Jurado-Tarifa et al., 2016). Studies of bacterial infections and carriage rates in raptors are limited and are focused on disease outbreaks in domestic birds and pet birds due to the economic value of the industry. The lack of interest and low commercial value have resulted in a limited understanding of the role of Aquila species as reservoirs and zoonotic disease vectors. Understanding the potential for the spread of avian bacterial pathogens from migrant raptors could provide a valuable model for estimating the risks associated with disease transmission among wild birds and other taxa. Focus on spreading bacteria in poultry has restrictions, as findings from domestic birds may not capture the full spectrum of transmission routes and other types of avian pathogens might not be screened.
The 16S rRNA sequencing method is a marker gene approach and comprehensively identifies all culturable and non-culturable as well as known and unknown microorganisms present in the upper respiratory tract and in the digestive system and hence, provides a better taxonomic resolution and genomic information on the microbial communities of host organisms (Dick et al., 2009; Johnson et al., 2019).
In Georgia, between 2016 and 2021, 18 chicks of Eastern Imperial Eagle were captured and tagged with GPS telemetry devices, 14 of these chicks died within the first year after hatching (cause of death undetermined) (Budagashvili, 2024). The primary objective of the study was to investigate the microbiome in nestling chicks in order to screen for pathogenic bacteria, which could potentially explain the high mortality rate among juvenile eagles. Given the inherent difficulty of catching and sampling wild (free-living) raptors, to minimize potential disturbances and the stress associated with handling, we analyzed tracheal and cloacal swabs from chicks. Although fecal samples are good sources for intestinal microbiome studies (Gao et al., 2021), the arid habitat of A. heliaca resulted in limited fecal material in nests, making chick sampling difficult and stressful.
Understanding the microbiome of juvenile Eastern Imperial Eagles is crucial, as it can offer insights into the health of these birds, potential pathogenic bacteria they may harbor, and how their microbiomes might influence their overall development and survival. In the first weeks of life, chicks of the Eastern Imperial Eagle primarily rely on regurgitated food provided by their parents. This feeding behavior not only ensures that the chicks’ nutritional needs are met, but also plays a crucial role in the establishment of their initial microbiome through the inoculation of microbiota from parents to offspring (Wang et al., 2018; Dietz et al., 2019; Bodawatta et al., 2022a).
By analyzing the bacterial composition of nestling chicks, we can gain valuable insights—though not an exact match—into the microbial communities, especially pathogenic organisms, present in their parents. This approach is particularly useful because studying adult, free-living raptors in their natural habitat presents significant challenges. As a result, examining the microbiota of the chicks provides an indirect, yet informative, glimpse into the microbial composition of the parent birds. By characterizing the microbiomes of the tracheal and cloacal environments in A. heliaca, we can identify avian and human prospective pathogens, assess the occurrence of microbial diseases and evaluate potential threats to the species, to humans and other birds. Considering how ecological factors, nesting sites, microclimate and age, might influence microbial communities, we can estimate indicators of the overall health of the eagles and provide recommendations for targeted conservation strategies for the population in Georgia.
Materials and methods
Sample collection
Six A. heliaca nests along the Iori River in a semi-arid habitat were sampled, representing 30% of the known Georgian population. Distances between nests ranged from 3.7 to 96.2 km. Seven chicks, approximately 8 weeks old and showing no visible health complications, were sampled from these nests. Sterile swabs were used to collect tracheal and cloacal samples, which were placed separately in sterile phosphate-buffered saline (PBS, pH 7.0). All samples were frozen before laboratory processing. No birds were killed or injured during sampling.
Nucleic acid extraction and 16S rRNA amplification
Nucleic acids were extracted from tracheal and cloacal swabs using a MagMAX total nucleic acid isolation kit (Thermo Fisher Scientific) according to the manufacturer’s instructions, with a final 60 μL of elution volume. We determined the sex of each chick by amplifying the CHD1 gene fragment according to Fridolfsson and Ellegren (1999). Samples obtained from the trachea and cloaca were amplified and sequenced separately. The complete 1,500 bp 16S rRNA gene was amplified with primer pairs: CCTAYGGGNNGCNGCAG and GACTACNVGGGTMTCTAATCC (Patel, 2001). PCR was carried out with final concentrations of 1 μM of each primer, 0.3 mM dNTPs, 1.5 mM Mg2 +, 1 unit of Q5 High-Fidelity DNA Polymerase (Promega Inc.) and 1X buffer. The cycling conditions were as follows: initial denaturation at 95°C for 3 min followed by 30 cycles at 95°C for 40 s, 55°C 40 s, 72°C for 2 min. The amplicons were verified via 1% agarose gel electrophoresis, with SybrSafe staining. Each PCR reaction was repeated three times. The amplicons were mixed to ensure amplification of a substantial proportion of all possible microbial taxa.
16S rRNA amplicons were fragmented via enzymatic digestion using an NEBNext Ultra II FS kit according to the manufacturer’s instructions. The fragmented DNA was sized 350-650 bp segments using AMPure XP beads. DNA libraries were repaired, and Illumina-specific adapter sequences were ligated to each fragment. The samples were dual indexed and a second size selection step was performed. Purified DNA libraries were quantified using Tape Station DS1000 high sensitivity reagents (Agilent Technologies) and the samples were diluted to a concentration of 2 nM concentration. For the internal control, 1% PHiX DNA was used. The 12 pM denatured libraries were sequenced with a MiSeq Reagent Kit v3 (600-cycle) kit according to the manufacturer’s protocol.
Data analysis
The Illumina base calling pipeline v1.40 was used for fluorescence image analysis and FasTQ formatted sequence data were obtained. Read-based taxonomic classification was performed with the EDGE web based graphic interface (Li et al., 2017) and Geneious prime.1 The sequences were quality controlled using FastQC v.0.12.1 (Andrews, 2010). Low quality reads, adapters, indexes and primers were removed with Trimmomatic v. 0.39 (Bolger et al., 2014) and BBduk tools (Bushnell et al., 2017). Taxonomic assignment of operational taxonomic units (OTUs) was carried out with EDGE bioinformatics web based analysis (Li et al., 2017). We filtered all OTUs with a minimum of 10 sequence reads. We prepared a separate data set and included all reads related to the potential notable animal or human diseases OTUs. We removed mitochondrial 16S rRNA sequences using the prune_taxa function. Alpha diversity was calculated using the Chao 1 index and Shannon measure with absolute abundance of reads in phyloseq v. 1.46.0 (McMurdie and Holmes, 2013). Differences between the microbiota of cloacal and tracheal samples of each individual were estimated using the Anosim function and Mann-Whitney test, and correlations between nesting sites and microbiome composition were measured with Mantel statistics VEGAN v. 2.4-6 (Oksanen et al., 2013). Venn diagrams were drawn using the web tool.2
We measured Spearman’s rank-order correlation for detection strength and direction of association between ranked variables: abundance of OTUs in cloacal and tracheal samples of all individuals with cocor v. 1.1-3 (Diedenhofen and Musch, 2015). The presence of potential zoonotic pathogens was estimated according to the CDC pathogen list (Benskin et al., 2009; Vogt, 2023).
Results
Among the seven birds, four were females and three were males. Approximately 3,500 OTUs were identified from each sample supported by ∼2.8 Million (M) sequence reads. Filtering OTUs with a minimum of 50 reads per taxon resulted in a subset of 1,075 OTUs for bacterial and archaeal domains for further analysis procedures. This selection ensured us to have a robust dataset, focusing on OTUs with sufficient read coverage for reliable analysis. Overall, 1,075 species belonged to 193 families, 51 classes, and 24 phyla. Most taxa were classified as Pseudomonadota (42.6%), Bacillota (22%), and Actinemicetota (14.49%) phyla. No significant differences in bacterial composition were observed between sexes.
The relative abundance of taxonomic classes in the bacterial community is shown in Figure 1. The most abundant bacterial classes in the tracheal and cloacal samples of A. heliaca were Gammaproteobacteria (67.5%), Bacilli (43.8%), and Negativicutes (22.0%). The following classes were detected in higher proportions in the cloaca: Negativicutes (16.69%), Clostridia (16.1%), and Actinomycetes (6.21%), while Fusobacteria (15.4%), Bacteroidia (9.6%), and Betaproteobacteria (9.6%) were detected in the tracheal samples.
FIGURE 1
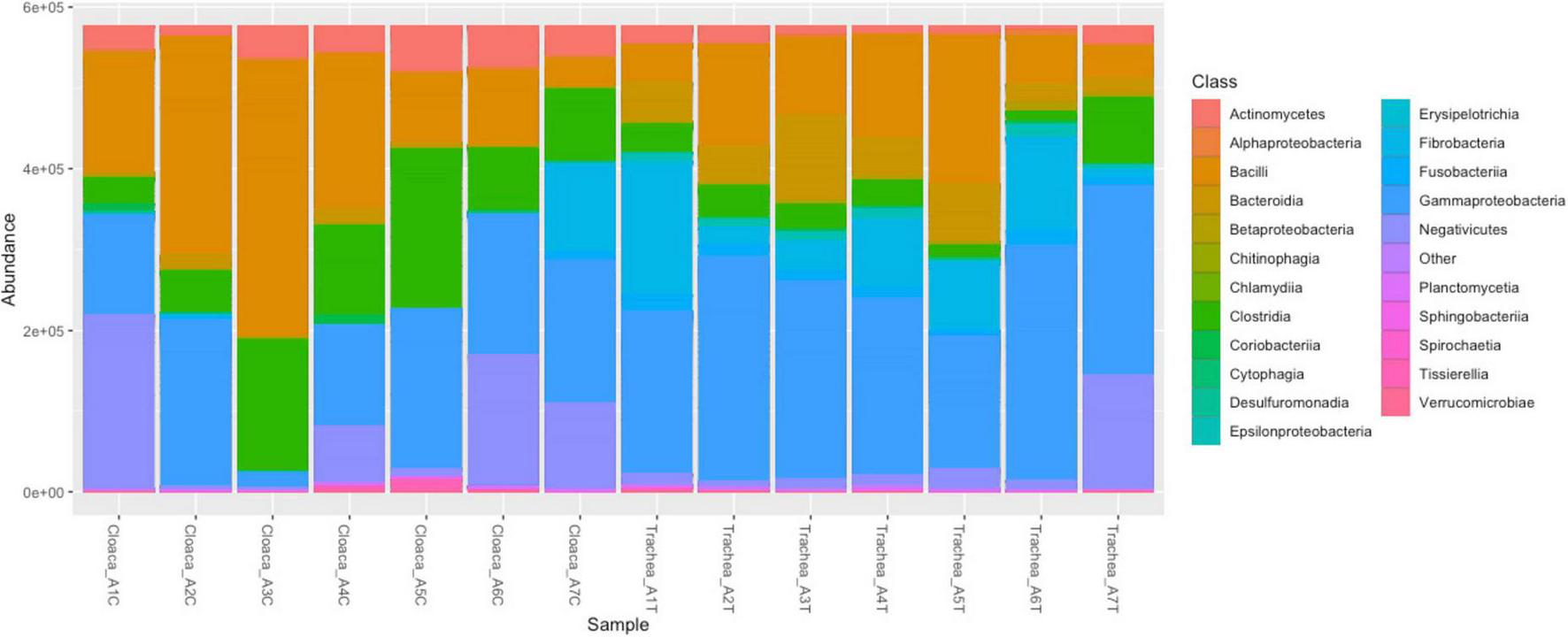
Composition of bacterial classes in cloacal and tracheal samples of Aquila heliacal. Abundances are measured using bray-curtis distances. Each color corresponds to a bacterial class, and the vertical bars represent the abundance of species at the class level. The X-axis denotes the samples.
The richness of the bacterial community is reflected by Chao 1 index, which ranges between 500 and 1,075 in different samples. The Shannon index, showing the bacterial community diversity, ranged from 3.0 to 4.0 (Figure 2). Comparison of tracheal and cloacal bacterial communities revealed a significant difference according to the Kruskal-Wallis test (49.56, p = 0); Mann-Whitney U-Test (p = 0.001). The tested individuals shed distinctive microbial communities in their digestive and respiratory tracts, and dissimilarity was measured with Anosim R 0.45; P < 0.005 (Figure 3a). The length of the bows represents the level of heterogeneity, and the width represents the number of screening species. Among the identified taxa 440 OTUs were unique for the trachea and 337 OTUs were unique for the cloaca, while 21.5% species were shared by cloacal and tracheal communities, Venn diagram (Figure 3b). Analysis of cumulative taxon abundances in the trachea and cloaca indicated significant evidence of moderate positive monotonic relationship between cloacal and tracheal bacterial communities (Spearman’s rank correlation coefficient rho = 0.67, p < 2.2e −16).
FIGURE 2
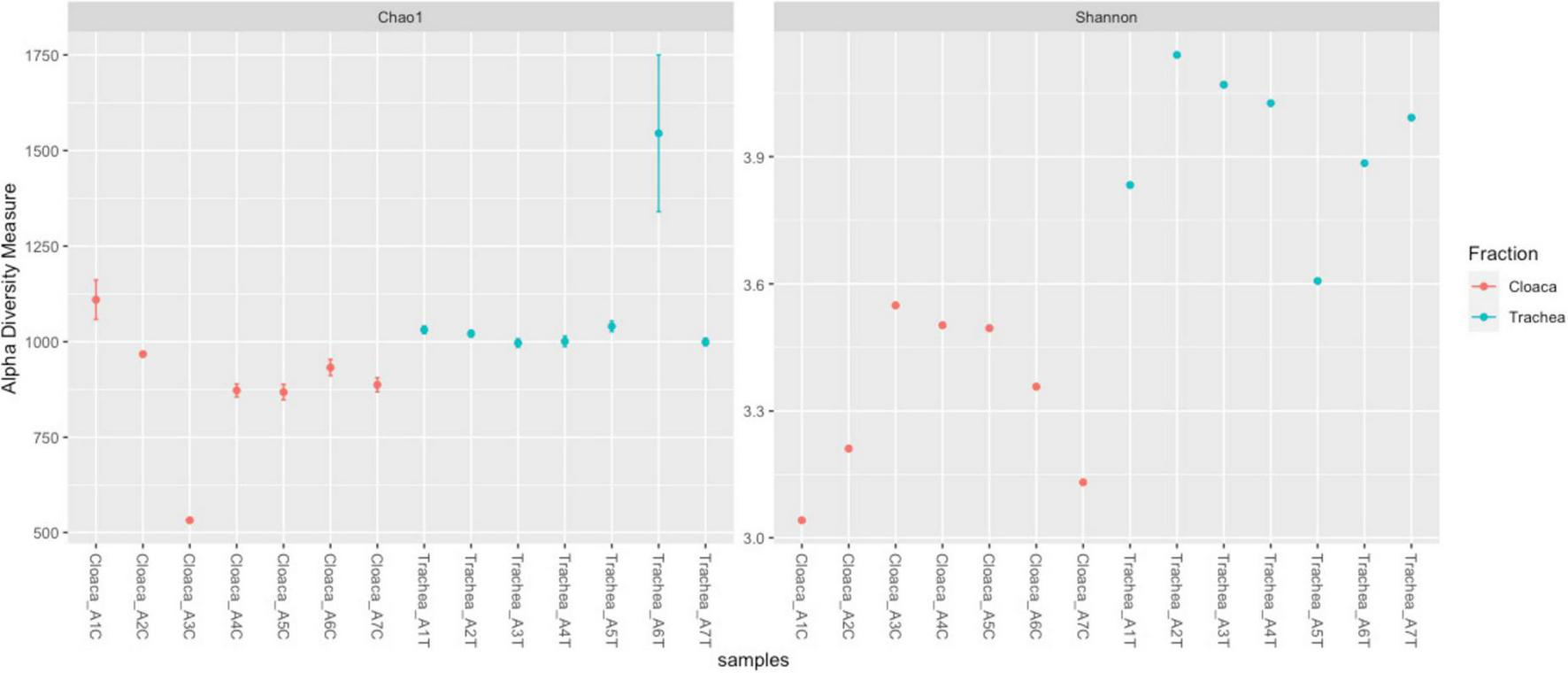
Alpha diversity plot visualizing differences in microbiota structure across samples. The Y-axis represents alpha diversity measures, while the X-axis distinguishes between samples (red for cloacal, blue for tracheal). Diversity is assessed using Chao1 and Shannon indexes.
FIGURE 3
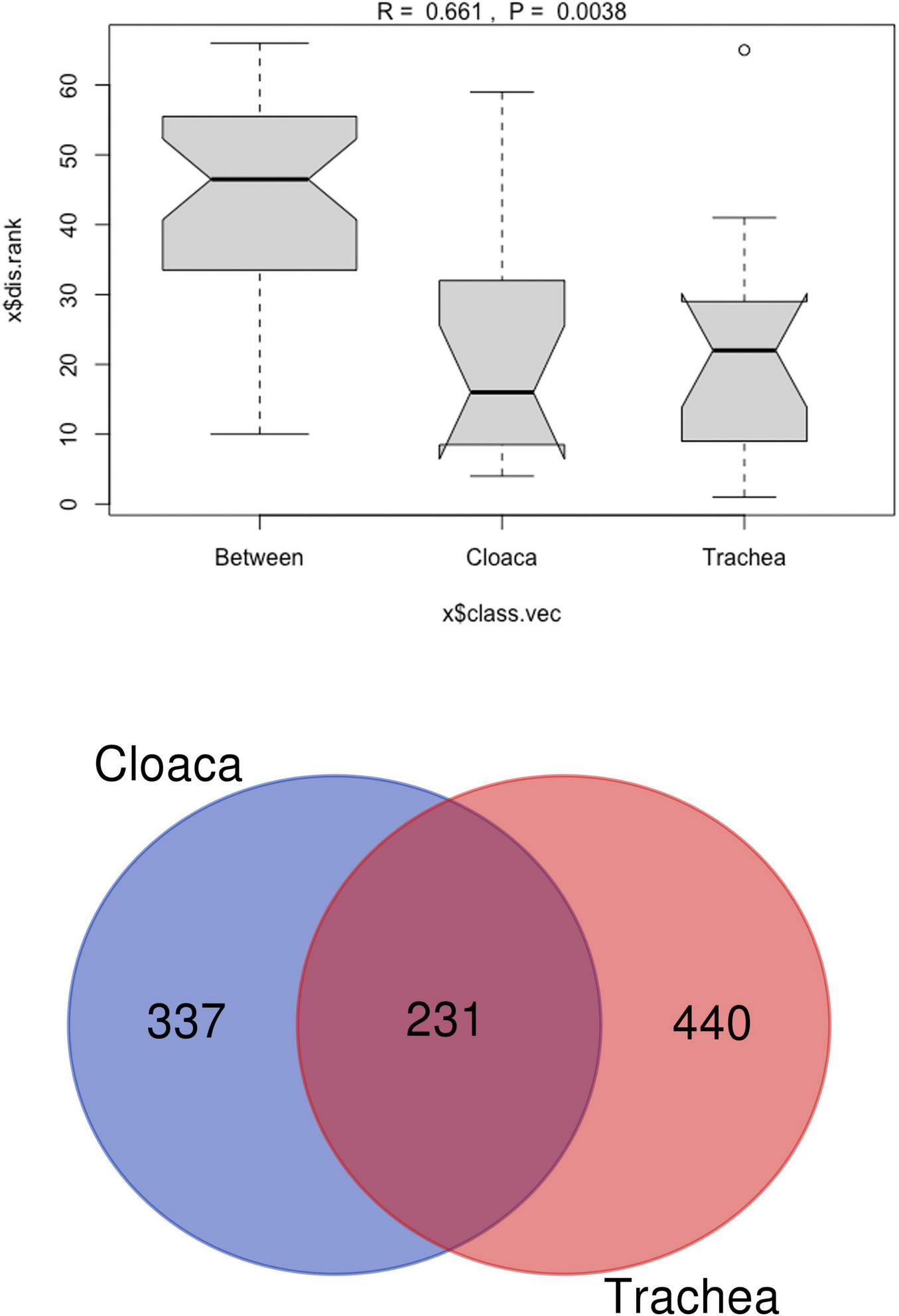
(a) Analysis of similarity (ANOSIM) plot testing dissimilarity between cloacal and tracheal bacterial communities in A. heliaca samples. The Y-axis represents the distance rank between samples, and the X-axis represents the dissimilarity results between both groups. (b) Venn diagram for cloacal and tracheal bacterial species in Eastern Imperial Eagle samples. The Venn diagram shows the total and overlapping (purple) species in the cloaca (blue) and trachea (pink).
No statistically significant differences in microflora composition were detected between individuals from different nests when tracheal and cloacal samples were compared separately (Anosim tracheal samples R = 0.39, p = 0.2; Anosim cloacal samples R = 0.14, p = 0.5). However, the bacterial composition in tracheal samples showed a significant correlation with geographic distance (Mantel statistic r = 0.75, p = 0.03), whereas no significant correlation was found in cloacal samples (Mantel statistic r = −0.04, p = 0.4).
Among the identified bacterial species, 382 have the potential to cause notable human and wildlife diseases. The most abundant bacterial species was Alteromonas australica, represented by 1 million reads. The tracheal and cloacal microbiomes were dominated by Staphylococcus aureus, Megamonas funiformis, and Ornithobacterium rhinotracheale. The following pathogenic species were detected in the cloaca only: Staphylococcus simulans, Acinetobacter radioresistens, Fastidiosipila sanguinis, Trueperella bialowiezensis, and Mycoplasma iowae. Pathogens such as Moraxella cuniculi, Moraxella ovis, Pasteurella aerogenes, Bacteroides vulgatus, Campylobacter gracilis, Mannheimia varigena, Massilia oculi, Mycoplasma phocidae, Fusobacterium ulcerans, Weeksella virosa, Mycoplasma maculosum, Morganella, Morganii, Helicobacter pylori, Neisseria canis, Xanthomonas vasicola, and Yersinia enterocolitica originate from the trachea and was not detected in cloacal samples.
In our dataset, we identified four out of five species from the HACEK group, (Haemophilus parainfluenzae, Aggregatibacter aphrophilus, Cardiobacterium hominis, and Eikenella corrodens) of gram-negative bacteria associated with infective endocarditis in humans (Bläckberg et al., 2021; Khaledi et al., 2022). We identified widely known zoonotic bacterial species, such as Salmonella spp., Klebsielle spp., Campilobacter spp., and Chlamydia psittaci. Host-specific mammalian pathogens include Bacillus anthracis, Mycobacterium leprae, Brucella abortus, B. canis, B. melitensis, Streptococcus pyogenes, Vibrio cholerae, V. mimicus, and Halophilic Vibrio cincinnatiensis.
Discussion
We present the first insights into the microbiome inventory of Eastern Imperial Eagles. The species is VU worldwide (IUCN, 2016). We characterized the tracheal and cloacal microbiomes of 30% of the Georgian population using the complete 16S rRNA gene. Taxonomic assignment of bacterial taxa using the 16S rRNA sequencing method is a cornerstone of taxonomic identification, as highlighted by recent studies (Jeong et al., 2021; Abellan-Schneyder et al., 2021; Hung et al., 2022). Although the number of wildlife microbiome studies is growing, the majority concentrate on mammals (Colston and Jackson, 2016; Moeller and Sanders, 2020), commercially significant birds, and captive-bred avian species (Zhao et al., 2022). Studies investigating the microbiomes of free-living raptors face limitations such as small population sizes, uncontrolled environmental factors (including diet, microhabitats, age, and season), and the difficulties associated with capturing and sampling (Oliveira et al., 2020).
In our study, we identified approximately 3,500 OTUs per sample from ∼2.8 million sequence reads. After filtering for OTUs with a minimum of 50 reads per taxon, we retained 1,075 OTUs, representing bacterial and archaeal domains. These OTUs were classified into 193 families, 51 classes, and 24 phyla. Microbial composition and diversity of raptors is still poorly understood. However, studies on other bird species offer some context. For instance, Kursa et al. (2021) identified 144 bacterial genera in the upper respiratory tract of 507 turkeys. Costanzo et al. (2022) screened 1,700-32,000 operational taxonomic units (OTUs) from 19 phyla in the cloaca of cavity-nesting lesser kestrels (Falco naumanni). Similarly, Gao et al. (2021) found 26,000 OTUs, classified into 32 phyla, in the fecal microbiota of six waterbird species. When applied to threatened species, microbiome studies become particularly potent, offering a window into the microbiome that can shed light on critical aspects of host health and survival as the microbiome plays a pivotal role in shaping host fitness. Currently, threats to biodiversity such as climate change, habitat degradation, antimicrobial resistance, and the emergence of infectious diseases, make understanding and managing the microbiome of species of heightened importance (Bahrndorff et al., 2016; West et al., 2019; Akoijam and Joshi, 2022). These risks often induce shifts in microbial communities associated with hosts across all taxonomic groups (Trevelline et al., 2019).
According to our dataset, both the tracheal and cloacal communities showed diversity of bacterial species. The richness of the bacterial community (alpha diversity reflected by the Chao 1 index) suggests variability and complexity of bacterial taxa within the studied environments, which is largely expected given their different physiological functions: the trachea hosts bacterias adapted to respiratory functions, while the cloacal bacterial community is associated with digestive and reproductive processes. Migratory birds harbor a wider variety of microorganisms than resident species, due to adaptations related to long-distance travel between breeding and wintering sites (Zhang et al., 2021; Grond et al., 2023).
Mann-Whitney U test and Kruskal-Wallis test showed significant differences in abundance of bacterial species between tracheal and cloacal body sites. This outcome was expected due to the differences in the studied environment: tracheas oxygen-rich conditions contrary to anaerobic cloaca; trachea’s relatively neutral pH vs. cloaca’s alkaline pH; distinct enzymatic profiles, etc. Besides these environmental differences, 21.5% of the identified bacterial species were shared by tracheal and cloacal samples and were dominated by species from Gammaproteobacteria, Bacilli, Clostridia, and Negativicutes taxa. These four bacterial classes were the most prevalent assigned amplicon sequences and overall, 77.0% of the total 16S rRNA reads were detected across our samples. Although these taxa were abundant in both body parts, their relative proportions varied among the samples. This variability can be influenced by genetic factors, temporal dynamics, and potential sampling bias.
Comparison of tracheal samples apart from cloacal samples by Mantel statistics showed that eaglets living in short geographical distances harbored more similar microbiomes than those who were nested further apart. Interestingly, this trend was not observed in cloacal samples. Besides that nests were located alongside the same Iori river in semi-arid habitat and distance between nests was 3.7-96.2 km. The composition of tracheal bacterial species was shaped by microclimate (two nests were located next to the dumpster and four in the river plains) and at this stage of life the birds might be less dependent on the individual parameters of the host. The relatively stable environment (temperature, pH, oxygen level, etc.) in the cloaca might alter similarities in cloacal bacterial composition by stabilizing selection pressure. These factors create a unique microenvironment that can favor the growth and survival of certain bacterial species over others. Through ongoing interactions between host physiology, microbial communities, and environmental conditions, specific bacterial taxa may thrive, while others are suppressed or eliminated. This process of active selection pressure within the cloaca could lead to similar patterns of bacterial composition among individuals of the same populations. Moreover, studies have highlighted the convergence of microbiota in birds with the same diet preferences (Grond et al., 2018; Oliveira et al., 2020; Bodawatta et al., 2022a). We expected qualitative similarities among the studied individuals, as this trend was also detected by other studies (Crespo et al., 2019; de Jonge et al., 2022), and slight variation in the bacterial species composition in the cloaca of A. heliaca chicks could be explained by interindividual differences.
Negativicutes (16.7%), Clostridia (16.1%), and Actinomycetes (6.2%) were prevalent in the cloaca, while Bacteroidia (9.6%), Fusobacteria (15.4%), and Betaproteobacteria (9.5%) were enriched in the trachea. Notably, the trachea harbored a slightly richer bacterial community, with 440 OTUs, compared to 337 OTUs in the cloaca.
Spearman’s rank correlation coefficient revealed a positive association between the abundance of OTUs in the trachea and cloaca. This suggests that tracheal microbiomes reflects cloacal microbiomes, particularly for abundant taxa such as Gammaproteobacteria, Bacilli, Clostridia, and Negativicutes. Hence, sampling the trachea and cloaca could be a reasonable approach to estimate the overall bacterial composition. However, it is important to recognize that sampling either the trachea or cloaca may not provide a comprehensive understanding of the microbiota, particularly with regard to pathogenic bacterial species and those that could pose zoonotic risks. Furthermore, the correlation with less abundant taxa may be weaker.
The observed similarities in overall bacterial composition (trachea plus cloaca) is more likely due to close geographic locations, similar dietary habits, microclimate of nests than small sample size. Although we recognize the potential limitations of our study, larger, more complex investigations with differing age groups, varied habitats are needed to deepen our understanding and potentially revise our current conclusions. Further research is crucial to fully determine the drivers of A. heliaca microbiome composition.
Bacterial pathogens
Based on our results, commensal and pathogenic bacterial species belong predominantly to the phyla Pseudomonadota, Bacillota, and Actinemicetota. These taxa have also been reported in other bird species (Benskin et al., 2009; Colston and Jackson, 2016; Bahrndorff et al., 2016; Grond et al., 2018; Capunitan et al., 2020). Eastern imperial eagles primarily prey on rodents, which often carry pathogenic bacteria. Additionally, during long breeding periods, the accumulation of feathers, skin remains and pellets in the nest creates a conducive environment for bacterial growth. These factors explain the existence of 382 pathogenic bacterial species in our samples. Blautia, Enterococci, Staphylococci, and Streptococci are common bacterial taxa in the respiratory tract (Bosch et al., 2013; Shabbir et al., 2015; Faldynova et al., 2024) and are widespread among our sampled birds.
Alteromonas australica (represented by the highest number of reads) is associated with seawater and marine sediments (López-Pérez et al., 2014). Unexpectedly, this bacterial species was not detected as part of the tracheal or cloacal microbiome; however, the abundance of the reads could be attributed to the capacity of Alteromonas to rapidly proliferate in the presence of suitable substrates (Chen et al., 2020). The opportunistic pathogens Staphylococcus aureus, Megamonas funiformis, M. hypermegale, and Ornithobacterium rhinotracheale had the most reads in the studied tracheal and cloacal samples. Staphylococcus is a normal flora on the mucous membrane and skin but also has a clumping factor and forms blood clots (Yehia et al., 2023). Staphylococcus aureus can cause skin and respiratory tract infections in birds, especially those under stress or with compromised immune systems (Ferrer and Hiraldo, 1995; Silva et al., 2022). Both Megamonas species are found in the digestive tract and are considered part of the gut microbiome (Bodawatta et al., 2022a,b).
Ornithobacterium rhinotracheale, Mycoplasma gallisepticum, Pasteurella multocida, Riemerella anatipestifer, and Bordetella avium cause respiratory infections with high mortality rates among poultry, particularly during the first month (Barbosa et al., 2019). In addition to these avian respiratory pathogens, we detected the following respiratory disease agents in tracheal samples: five Mycoplasma species (M. californicum, M. gallinaceum, M. iowae, M. maculosum, and M. phocidae), three Bordetella species (B. bronchiseptica, B. parapertussis, and B. pertussis) and two Pasteurella species (P. aerogans and P. dagmatis). The transmission of avian mycoplasma through horizontal (between individuals) and vertical (adults to its progeny) routes has been described (Yehia et al., 2023). However, the prevalence, clinical significance, and transmission dynamics of bacteria in eagles are not well understood. Further research is needed to investigate the frequency of occurrence, their impact on health and welfare, and the mechanisms by which they are transmitted among A. heliaca individuals.
The following opportunistic pathogenic species were detected in the cloaca only: Staphylococcus simulans, Acinetobacter radioresistens, Fastidiosipila sanguinis, and Trueperella bialowiezensis. Moraxella cuniculi, M. ovis, Bacteroides vulgatus, Campylobacter gracilis, Mannheimia varigena, Massilia oculi, Fusobacterium ulcerans, Weeksella virosa, Morganella, Morganorganii, Helicobacter pylori, Neisseria canis, Xanthomonas vasicola, and Yersinia enterocolitica originated from the trachea and could not be detected in cloacal samples.
Various Salmonella species have been detected in both domestic and wild birds (Belo et al., 2023; Smith et al., 2023). We detected S. bongori in all the samples and Salmonella enterica in four eaglets. S. bongori is known in reptiles, and few cases of human infection have been described (Fookes et al., 2011). On the other hand, Salmonella enterica includes a wide range of servers and is responsible for various types of human and animal illnesses and causes of food-borne illness worldwide (Belo et al., 2023; Smith et al., 2023). Understanding the prevalence and diversity of Salmonella species in threatened eagles is important for effective conservation, disease management and public health efforts.
We detected three species: Klebsiella aerogens, K. pneumonia, and K. variicola in our dataset. Among them, pathogenic K. variicola represented approximately 5K reads in each sample. The high level of abundance suggests its potential significance in the sampled population. K. variicola infects various hosts, and people infected with a high titer of K. variicola have a high mortality rate (Maatallah et al., 2014). K. pneumoniae is a component of the normal intestinal flora but has the potential to infect the respiratory tract in humans. K. aerogenes is associated with respiratory and urinary tract infections (Rodríguez-Medina et al., 2019). The presence of these Klebsiella species, particularly pathogenic K. variicola, raises concerns about potential health risks to both humans and animals.
In the present study, some species of bacilli were found to be highly prevalent in B. mycoides in two cloacal and one tracheal sample and in B. coagulans in one tracheal sample. Additionally, B. cereus, B. subtilis, and B. thuringiensis were also identified. However, to the best of our knowledge, B. anthracis has not been previously attributed to the respiratory or digestive system of eagles.
Campylobacteriosis is the most commonly reported zoonotic bacterial food-borne gastroenteritis worldwide (Igwaran and Okoh, 2019). Eleven species were identified in this study (Campylobacter coli, C. concisus, C. curvus, C. fetus, C. gracilis, C. helveticus, C. jejuni, C. lari, C. showae, C. upsaliensis, and C. ureolyticus). Among them, C. concisus was present in all tracheal samples analyzed. Different species of Campylobacter have been isolated from wild and domestic birds worldwide, due to their pathogenicity and antibiotic resistance, this is currently a global challenge (Ahmed and Gulhan, 2022).
Ten Leptospira species with low abundance were detected in our dataset: L. alexanderi, L. borgpetersenii L. fainei, L. inadai L. broomii, L. kirschneri, L. meyeri, L. noguchii, L. santarosai, and L. weilii. Most of the identified species are associated with leptospirosis. Epidemiological studies on leptospirosis in wild birds and its role in transmission to other animals are rare (Balcázar et al., 2024).
Yersinia enterocolitica was detected in only one tracheal sample, while Y. pseudotuberculosis and Y. freundiksenii were detected in all the samples. Vibrio mimicus was identified from only one cloacal sample, but V. cholerae, V. cincinnatiensis, V. mimicus, and V. parahaemolyticus V. vulnificus were identified across all the samples. The detection of Yersinia and Vibrio spps. suggests that A. heliaca might be a carrier, potential reservoir, or source of infection.
To determine whether the abundance of pathogens in the cloaca and trachea is sufficient to cause infections, several factors must be considered, including the type of pathogen, its concentration, and the overall health of the host. The mere presence of certain pathogens in these sites does not automatically indicate active disease. While the existence of a pathogenic agent could potentially lead to infection, it may not always cause development of disease unless factors such as stress, poor nutrition, a compromised immune system, or other environmental conditions are present. Therefore, this balance should be carefully considered, taking into account both the potential risks of infection and the welfare of the host when designing study protocols.
Conclusion
Aquila heliaca can be considered as a reliable source of the presence of pathogenic bacterial species in their respective habitats. However, capturing them for microbiome studies poses a significant challenge, due to elusive nature, low abundance, difficulties in safely handling and vulnerability. Therefore, during conservation activities such as ringing or tagging with telemetry devices, it is imperative to prioritize sample collection for microbiome studies. Efforts aiming at conservation of red-listed birds may not presently focus on microbiome considerations but integrating microbiome research into conservation strategies could yield significant benefits. Understanding bacterial abundance through such studies provides valuable insights into the health of the species, as disease outbreaks have the potential to disrupt ecological interactions, impact reproductive capabilities, alter predator-prey dynamics, decrease population sizes, and even lead to species extinction. Consequently, monitoring efforts are essential for assessing the risk of zoonotic disease transmission to humans and implementing appropriate control measures to prevent the spread of infection among other species. Therefore, integrating microbiome monitoring into ongoing conservation efforts is not only beneficial for the species under study but also for understanding broader ecological health. Ultimately, it provides a comprehensive approach to species conservation that includes the often-overlooked role of microbial ecosystems in the overall wellbeing of ecosystems.
Statements
Data availability statement
The dataset analyzed in this study is available in the NCBI Sequence Read Archive (SRA) under BioProject accession number PRJNA1123231.
Ethics statement
The animal study was approved by Ethics committee at Ilia State University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
MM: Conceptualization, Methodology, Project administration, Visualization, Writing – original draft, Data curation, Formal Analysis, Funding acquisition, Investigation, Validation. LN: Investigation, Writing – review & editing. NB: Data curation, Writing – review & editing. ET: Writing – review & editing. ZG: Writing – review & editing, Data curation. AK: Methodology, Writing – review & editing. NK: Methodology, Writing – review & editing. AG: Methodology, Writing – review & editing. ZJ: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Reagents and consumables and data analysis funded by the European Union, grant GRMP, Sample collection by SABUKO, DNA extraction by Shota Rustaveli National Science Foundation of Georgia, grant # FR-22-2855.
Acknowledgments
We express our gratitude to David Tarkhnishvili for valuable comments and suggestions, and Natia Javakhishvili for her contribution to the sample collection process.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Abellan-SchneyderI.MatchadoM. S.ReitmeierS.SommerA.SewaldZ.BaumbachJ.et al (2021). Primer, pipelines, parameters: Issues in 16S rRNA gene sequencing.mSphere6:e01202-20. 10.1128/msphere.01202-20
2
AhmedN. A.GulhanT. (2022). Campylobacter in wild birds: Is it an animal and public health concern?Front. Microbiol.12:812591. 10.3389/fmicb.2021.812591
3
AkoijamN.JoshiS. R. (2022). “Conservation metagenomics: Understanding microbiomes for biodiversity sustenance and conservation,” in Molecular genetics and genomics tools in biodiversity conservation, edsKumarA.et al (Singapore: Springer), 31–61.
4
AndrewsS. (2010). FastQC: A quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
5
AraghiM.EskandariF.GilasianE. (2015). Avian wound myiasis caused by Calliphora vicina robineau-desvoidy (Diptera: Calliphoridae) in an immature migrating eastern imperial eagle (Aquila heliacal Savigny)(Aves: Accipitridae) in South-western Iran.J. Vet. Sci. Technol.6:212. 10.4172/2157-7579.1000212
6
BahrndorffS.AlemuT.AlemnehT.Lund NielsenJ. (2016). The microbiome of animals: Implications for conservation biology.Int. J. Genom.2016:5304028. 10.1155/2016/5304028
7
BalcázarL.Azócar-AedoL.BarreraV.MeniconiG.MuñozV.Valencia-SotoC. (2024). Detection of antibodies for pathogenic leptospira in wild mammals and birds from southern chile—first record of seropositivity in a guiña (Leopardus guigna).Animals14:601. 10.3390/ani14040601
8
BarbosaE. V.CardosoC. V.SilvaR.deC. F.CerqueiraA.deM. F.et al (2019). Ornithobacterium rhinotracheale: An update review about an emerging poultry pathogen.Vet. Sci.7:3. 10.3390/vetsci7010003
9
BekmansurovR. H.KaryakinI. V.ShnayderE. P. (2015). On Eastern imperial eagle (Aquila heliaca) breeding in atypical habitat under competitive conditions with other eagle species.Slovak Raptor J.995–104. 10.1515/srj-2015-0007
10
BeloN. O.de Angeli DutraD.RodelloD.CrispimN. P. C. C.SilvaA. S. G.CoelhoH. L. G.et al (2023). Detection of Salmonella spp. in wild and domestic birds in an anthropized ecotone between the Cerrado and the Amazon Forest in Brazil.Braz. J. Microbiol.54565–569. 10.1007/s42770-022-00886-z
11
BenskinC. M. H.WilsonK.JonesK.HartleyI. R. (2009). Bacterial pathogens in wild birds: A review of the frequency and effects of infection.Biol. Rev. Camb. Philos. Soc.84349–373. 10.1111/j.1469-185X.2008.00076.x
12
BirdLife (2023). BirdLife international species factsheet: Aquila heliaca.Cambridge: BirdLife
13
BläckbergA.MoreniusC.OlaisonL.BergeA.RasmussenM. (2021). Infective endocarditis caused by HACEK group bacteria—a registry-based comparative study.Eur. J. Clin. Microbiol. Infect. Dis.401919–1924. 10.1007/s10096-021-04240-3
14
BodawattaK. H.HirdS. M.GrondK.PoulsenM.JønssonK. A. (2022a). Avian gut microbiomes taking flight.Trends Microbiol.30268–280. 10.1016/j.tim.2021.07.003
15
BodawattaK. H.KlečkováI.KlečkaJ.PužejováK.KoaneB.PoulsenM.et al (2022b). Specific gut bacterial responses to natural diets of tropical birds.Sci. Rep.12:713. 10.1038/s41598-022-04808-9
16
BolgerA. M.LohseM.UsadelB. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data.Bioinformatics302114–2120. 10.1093/bioinformatics/btu170
17
BordensteinS. R.TheisK. R. (2015). Host biology in light of the microbiome: Ten principles of holobionts and hologenomes.PLoS Biol.13:e1002226. 10.1371/journal.pbio.1002226
18
BoschA. A. T. M.BiesbroekG.TrzcinskiK.SandersE. A. M.BogaertD. (2013). Viral and bacterial interactions in the upper respiratory tract.PLoS Pathogens9:e1003057. 10.1371/journal.ppat.1003057
19
BraginE.HorváthM.PoesselS.KatznerT. (2021). “Eastern imperial eagle (Aquila heliaca),” in Migration strategies of birds of prey in Western Palearctic, edsPanuccioM.MelloneU.AgostiniN. (Boca Raton, FL: CRC Press), 117–124.
20
BrilhanteR. S. N.Castelo BrancoD. S. C. M.DuarteG. P. S.PaivaM. A. N.TeixeiraC. E. C.ZeferinoJ. P. O.et al (2012). Yeast microbiota of raptors: A possible tool for environmental monitoring.Environ. Microbiol. Rep.4189–193. 10.1111/j.1758-2229.2011.00319.x
21
BudagashviliN. (2024). Imperial eagles: Conservation action plan for 2024-2034.Tbilisi: SABUKO.
22
BushnellB.RoodJ.SingerE. (2017). BBMerge – accurate paired shotgun read merging via overlap.PLoS One12:e0185056. 10.1371/journal.pone.0185056
23
CapunitanD. C.JohnsonO.TerrillR. S.HirdS. M. (2020). Evolutionary signal in the gut microbiomes of 74 bird species from equatorial guinea.Mol. Ecol.29829–847. 10.1111/mec.15354
24
CasadevallA.PirofskiL. (2000). Host-Pathogen interactions: Basic concepts of microbial commensalism, colonization, infection, and disease.Infect. Immun.686511–6518. 10.1128/iai.68.12.6511-6518.2000
25
ChenM.SongY.FengX.TangK.JiaoN.TianJ.et al (2020). Genomic characteristics and potential metabolic adaptations of hadal trench roseobacter and alteromonas bacteria based on single-cell genomics analyses.Front. Microbiol.11:1739. 10.3389/fmicb.2020.01739
26
ColstonT. J.JacksonC. R. (2016). Microbiome evolution along divergent branches of the vertebrate tree of life: What is known and unknown.Mol. Ecol.253776–3800. 10.1111/mec.13730
27
CostanzoA.AmbrosiniR.FranzettiA.RomanoA.CecereJ. G.MorgantiM.et al (2022). The cloacal microbiome of a cavity-nesting raptor, the lesser kestrel (Falco naumanni).PeerJ10:e13927. 10.7717/peerj.13927
28
CrespoR.DowdS. E.VarlandD. E.FordS.HamerT. E. (2019). Bacterial diversity in feces of wild bald eagles, turkey vultures and common ravens from the Pacific Northwest Coast, USA.bioRxiv [Preprint].10.1101/511147
29
CunninghamA. A.DaszakP.WoodJ. L. N. (2017). One health, emerging infectious diseases and wildlife: Two decades of progress?Philos. Trans. R. Soc. Lond. B Biol. Sci.372:20160167. 10.1098/rstb.2016.0167
30
DallasJ. W.WarneR. W. (2023). Captivity and animal microbiomes: Potential roles of microbiota for influencing animal conservation.Microb. Ecol.85820–838. 10.1007/s00248-022-01991-0
31
DaszakP.CunninghamA. A.HyattA. D. (2000). Emerging infectious diseases of wildlife–threats to biodiversity and human health.Science287443–449. 10.1126/science.287.5452.443
32
de JongeN.CarlsenB.ChristensenM. H.PertoldiC.NielsenJ. L. (2022). The gut microbiome of 54 mammalian species.Front. Microbiol.13:886252. 10.3389/fmicb.2022.886252
33
DickG. J.AnderssonA. F.BakerB. J.SimmonsS. L.ThomasB. C.YeltonA. P.et al (2009). Community-wide analysis of microbial genome sequence signatures.Genome Biol.10:R85. 10.1186/gb-2009-10-8-r85
34
DiedenhofenB.MuschJ. (2015). cocor: A comprehensive solution for the statistical comparison of correlations.PLoS One10:e0121945. 10.1371/journal.pone.0121945
35
DietzM. W.SallesJ. F.HsuB. Y.DijkstraC.GroothuisT. G.van der VeldeM.et al (2019). Prenatal transfer of gut bacteria in rock pigeon.Microorganisms8:61. 10.3390/microorganisms8010061
36
FaldynovaM.PrikrylovaH.SebkovaA.VolfJ.KarasovaD.CrhanovaM.et al (2024). Contact with adult hens affects the composition of skin and respiratory tract microbiota in newly hatched chicks.Poultry Sci.103:103302. 10.1016/j.psj.2023.103302
37
FerrerM.HiraldoF. (1995). Human-associated staphylococcal infection in Spanish imperial eagles.J. Wildlife Dis.31534–536. 10.7589/0090-3558-31.4.534
38
FookesM.SchroederG. N.LangridgeG. C.BlondelC. J.MamminaC.ConnorT. R.et al (2011). Salmonella bongori provides insights into the evolution of the Salmonellae.PLoS Pathogens7:e1002191. 10.1371/journal.ppat.1002191
39
FridolfssonA.-K.EllegrenH. (1999). A simple and universal method for molecular sexing of non-ratite birds.J. Avian Biol.30116–121. 10.2307/3677252
40
GalvezR. A.GavashelishviliL.JavakhishviliZ. (2005). Raptors and owls of Georgia. Georgian centre for the conservation of wildlife and buneba print publishing.Georgia: Distributed by NHBS.
41
GaoL.LiuL.DuC.HouQ. (2021). Comparative analysis of fecal bacterial microbiota of six bird species.Front. Vet. Sci.8:791287. 10.3389/fvets.2021.791287
42
GrondK.LouyakisA. S.HirdS. M. (2023). Functional and compositional changes in the fecal microbiome of a shorebird during migratory stopover. mSystems 8:e01128-22. 10.1128/msystems.01128-22
43
GrondK.SandercockB. K.JumpponenA.ZeglinL. H. (2018). The avian gut microbiota: Community, physiology and function in wild birds.J. Avian Biol.49:e01788. 10.1111/jav.01788
44
HákF.MisikT.SasváriJ. (2007). Breeding biology of imperial eagle pairs (Aquila heliaca) on the heves-plain.ÉVFOLYAM2641–51. 10.20332/tvk-jnatconserv.2020.26.39
45
HerediaB. O. (1996). International action plan for the Imperial Eagle (Aquila heliaca). globally threatened birds of Europe: Action plans.Strasbourg: Council of Europe Publishing.
46
HohenloheP. A.FunkW. C.RajoraO. P. (2021). Population genomics for wildlife conservation and management.Mol. Ecol.3062–82. 10.1111/mec.15720
47
HorváthM.KovácsA.Gallo-OrsiU. (2006). Action plan for imperial eagle (Aquila heliaca) in the Southern-Caucasus.Wageningen: BirdLife International.
48
HungY.-M.LyuW.-N.TsaiM.-L.LiuC.-L.LaiL.-C.TsaiM.-H.et al (2022). To compare the performance of prokaryotic taxonomy classifiers using curated 16S full-length rRNA sequences.Comp. Biol. Med.145:105416. 10.1016/j.compbiomed.2022.105416
49
IgwaranA.OkohA. I. (2019). Human campylobacteriosis: A public health concern of global importance.Heliyon5:e02814. 10.1016/j.heliyon.2019.e02814
50
IUCN (2016). The IUCN red list of threatened species.Gland: IUCN
51
JavakhishviliZ.AghababianK.SultanovE.TohidifarM.MnatsakanovR.IsfendiyarogluS. (2020). “Status of birds in the caucasus,” in Ecoregional conservation plan for the caucasus, 2020 edition: supplementary reportsedsZazanashviliN.GarforthM.BitsadzeM. (Tbilisi: WWF, KFW).
52
JeongJ.YunK.MunS.ChungW.-H.ChoiS.-Y.NamY.et al (2021). The effect of taxonomic classification by full-length 16S rRNA sequencing with a synthetic long-read technology.Sci. Rep.11:1727. 10.1038/s41598-020-80826-9
53
JohnsonJ. S.SpakowiczD. J.HongB.-Y.PetersenL. M.DemkowiczP.ChenL.et al (2019). Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis.Nat. Commun.10:5029. 10.1038/s41467-019-13036-1
54
Jurado-TarifaE.TorralboA.BorgeC.Cerdà-CuéllarM.AyatsT.CarboneroA.et al (2016). Genetic diversity and antimicrobial resistance of Campylobacter and Salmonella strains isolated from decoys and raptors.Comp. Immunol. Microbiol. Infect. Dis.4814–21. 10.1016/j.cimid.2016.07.003
55
KatznerT. E.JacksonD. S.IvyJ.BraginE. A.DeWoodyA. (2014). Variation in offspring sex ratio of a long-lived sexually dimorphic raptor, the Eastern imperial eagle Aquila heliaca.Ibis156395–403. 10.1111/ibi.12139
56
KhalediM.SameniF.AfkhamiH.HemmatiJ.Asareh Zadegan DezfuliA.SanaeM. J.et al (2022). Infective endocarditis by HACEK: A review.J. Cardiothorac. Surg.17:185. 10.1186/s13019-022-01932-5
57
KovácsA.WilliamsN. P. (2012). Guidelines for Preparing National or Regional Raptor Conservation and Management Strategies.Abu-Dhabi, UAE: CMS Technical Series.
58
KursaO.TomczykG.Sawicka-DurkalecA.GizaA.Słomiany-SzwarcM. (2021). Bacterial communities of the upper respiratory tract of turkeys.Sci. Rep.11:2544. 10.1038/s41598-021-81984-0
59
LiP.-E.LoC.-C.AndersonJ. J.DavenportK. W.Bishop-LillyK. A.XuY.et al (2017). Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform.Nucleic Acids Res.4567–80. 10.1093/nar/gkw1027
60
López-PérezM.GonzagaA.IvanovaE. P.Rodriguez-ValeraF. (2014). Genomes of Alteromonas australica, a world apart.BMC Genom.15:483. 10.1186/1471-2164-15-483
61
MaatallahM.VadingM.KabirM. H.BakhroufA.KalinM.NauclerP.et al (2014). Klebsiella variicola is a frequent cause of bloodstream infection in the Stockholm area, and associated with higher mortality compared to K. pneumoniae.PLoS One9:e113539. 10.1371/journal.pone.0113539
62
McMurdieP. J.HolmesS. (2013). phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data.PLoS One8:e61217. 10.1371/journal.pone.0061217
63
MoellerA. H.SandersJ. G. (2020). Roles of the gut microbiota in the adaptive evolution of mammalian species.Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci.375:20190597. 10.1098/rstb.2019.0597
64
MovalliP.KroneO.OsbornD.PainD. (2018). Monitoring contaminants, emerging infectious diseases and environmental change with raptors, and links to human health.Bird Study65S96–S109. 10.1080/00063657.2018.1506735
65
MueggeB. D.KuczynskiJ.KnightsD.ClementeJ. C.GonzálezA.FontanaL.et al (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans.Science332970–974. 10.1126/science.1198719
66
OksanenJ.BlanchetF. G.KindtR.LegendreP.MinchinP. R.O’HaraR. B.et al (2013). Package ‘vegan’. Community cology package, version, 2, 1–295.
67
OliveiraB. C. M.MurrayM.TsengF.WidmerG. (2020). The fecal microbiota of wild and captive raptors.Anim. Microbiome2:15. 10.1186/s42523-020-00035-7
68
PatelJ. B. (2001). 16S rRNA gene sequencing for bacterial pathogen identification in the clinical laboratory.Mol. Diagn.6313–321. 10.1054/modi.2001.29158
69
Rodríguez-MedinaN.Barrios-CamachoH.Duran-BedollaJ.Garza-RamosU. (2019). Klebsiella variicola: An emerging pathogen in humans.Emerg. Microbes Infect.8973–988. 10.1080/22221751.2019.1634981
70
RossiG.TerraccianoG.GherardiR.GalosiL.PerrucciS. (2021). Parasites, bacteria, and associated pathological changes in the digestive system of diurnal and nocturnal raptors in Central Italy.Pathogens10:1567. 10.3390/pathogens10121567
71
RudnickJ. A.KatznerT. E.BraginE. A.DeWoodyJ. A. (2008). A non-invasive genetic evaluation of population size, natal philopatry, and roosting behavior of non-breeding eastern imperial eagles (Aquila heliaca) in central Asia.Conserv. Genet.9667–676. 10.1007/s10592-007-9397-9
72
SekirovI.RussellS. L.AntunesL. C. M.FinlayB. B. (2010). Gut microbiota in health and disease.Physiol. Rev.90859–904. 10.1152/physrev.00045.2009
73
ShabbirM. Z.MalysT.IvanovY. V.ParkJ.ShabbirM. A. B.RabbaniM.et al (2015). Microbial communities present in the lower respiratory tract of clinically healthy birds in Pakistan.Poultry Sci.94612–620. 10.3382/ps/pev010
74
SilvaV.LopesA. F.SoeiroV.CaniçaM.ManageiroV.PereiraJ. E.et al (2022). Nocturnal birds of prey as carriers of staphylococcus aureus and other staphylococci: Diversity, antimicrobial resistance and clonal lineages.Antibiotics11:240. 10.3390/antibiotics11020240
75
SmithJ. C.VarrianoS.RoachK.SnipesZ.DawsonJ. L.ShealyJ.et al (2023). Prevalence and molecular characterization of Salmonella isolated from wild birds in fresh produce environments.Front. Microbiol.14:1272916. 10.3389/fmicb.2023.1272916
76
SmithK. F.SaxD. F.LaffertyK. D. (2006). Evidence for the role of infectious disease in species extinction and endangerment.Conserv. Biol.201349–1357. 10.1111/j.1523-1739.2006.00524.x
77
SongS. J.SandersJ. G.DelsucF.MetcalfJ.AmatoK.TaylorM. W.et al (2020). Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats.MBio1110–1128. 10.1177/1535370219830075
78
StenkatJ.Krautwald-JunghannsM.-E.Schmitz OrnésA.EilersA.SchmidtV. (2014). Aerobic cloacal and pharyngeal bacterial flora in six species of free-living birds.J. Appl. Microbiol.1171564–1571. 10.1111/jam.12636
79
TrevellineB. K.FontaineS. S.HartupB. K.KohlK. D. (2019). Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices.Proc. R. Soc. B286:20182448. 10.1098/rspb.2018.2448
80
VidalA.BaldomàL.Molina-LópezR. A.MartinM.DarwichL. (2017). Microbiological diagnosis and antimicrobial sensitivity profiles in diseased free-living raptors.Avian Pathol.46442–450. 10.1080/03079457.2017.1304529
81
VogtN. A. (2022). “Wild birds and zoonotic pathogens,” in Zoonoses: Infections affecting humans and animals, ed.SingA. (Berlin: Springer International Publishing), 1–31. 10.1007/978-3-030-85877-3_47-1
82
VogtN. A. (2023). “Wild birds and zoonotic pathogens: Beware of the fowl feces,” in Zoonoses: Infections affecting humans and animals, ed. A. Sing (Cham: Springer International Publishing), 1003–1033.
83
WangS.ChenL.HeM.ShenJ.LiG.TaoZ.et al (2018). Different rearing conditions alter gut microbiota composition and host physiology in Shaoxing ducks. Sci. Rep. 8:7387. 10.1038/s41598-018-25760-7
84
WestA. G.WaiteD. W.DeinesP.BourneD. G.DigbyA.McKenzieV. J.et al (2019). The microbiome in threatened species conservation.Biol. Conserv.22985–98. 10.1016/j.biocon.2018.11.016
85
YehiaN.SalemH. M.MahmmodY.SaidD.SamirM.MawgodS. A.et al (2023). Common viral and bacterial avian respiratory infections: An updated review.Poultry Sci.102:102553. 10.1016/j.psj.2023.102553
86
ZhangZ.YangZ.ZhuL. (2021). Gut microbiome of migratory shorebirds: Current status and future perspectives.Ecol. Evol.113737–3745. 10.1002/ece3.7390
87
ZhaoC.LiuL.GaoL.BaiL. (2022). A comprehensive comparison of fecal microbiota in three ecological bird groups of raptors, waders, and waterfowl.Front. Microbiol.13:919111. 10.3389/fmicb.2022.919111
Summary
Keywords
Eastern Imperial Eagle, tracheal bacterias, cloacal bacterias, 16S rRNA, conservation
Citation
Murtskhvaladze M, Ninua L, Budagashvili N, Tevdoradze E, Gurgenidze Z, Kotorashvili A, Kotaria N, Gavashelishvili A and Javakhishvili Z (2025) Tracheal and cloacal bacterial diversity of red listed Eastern Imperial Eagle (Aquila heliaca). Front. Microbiol. 16:1477032. doi: 10.3389/fmicb.2025.1477032
Received
06 August 2024
Accepted
16 April 2025
Published
09 May 2025
Volume
16 - 2025
Edited by
Flávia Spago, Federal Institute of Espírito Santo (IFES), Brazil
Reviewed by
Vallo Tilgar, University of Tartu, Estonia
Mingzhe Guo, University of Nevada, United States
Updates
Copyright
© 2025 Murtskhvaladze, Ninua, Budagashvili, Tevdoradze, Gurgenidze, Kotorashvili, Kotaria, Gavashelishvili and Javakhishvili.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marine Murtskhvaladze, dna_lab@iliauni.edu.ge
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.