 Cunyao Pan1,2,3†
Cunyao Pan1,2,3† Chongyi Ai1†Lanlan Liang1,2Baoyi Zhang1Qionglin Li1,2Lingling Pu1Zirou Wang1
Chongyi Ai1†Lanlan Liang1,2Baoyi Zhang1Qionglin Li1,2Lingling Pu1Zirou Wang1 Weili Liu1*Zhaoli Chen1*Hui Liu2*
Weili Liu1*Zhaoli Chen1*Hui Liu2* Xinxing Wang1*
Xinxing Wang1*- 1Tianjin Institute of Environmental and Operational Medicine, Tianjin, China
- 2School of Public Health, Lanzhou University, Lanzhou, China
- 3Chengdu Center for Disease Prevention and Control, Chengdu, China
Hypoxia induced by high altitude can lead to severe neurological dysfunction. Mitophagy is known to play a crucial role in hypoxic nerve injury. However, the regulatory mechanism of mitophagy during this injury remains unclear. Recent studies have highlighted the role of Sestrin2 (SESN2), an evolutionarily conserved stress-inducible protein against acute hypoxia. Our study demonstrated that hypoxia treatment increased SESN2 expression and activated mitophagy in PC12 cells. Furthermore, the knock-out of Sesn2 gene led to a significant increase in mitochondrial membrane potential and ATP concentrations, which protected the PC12 cells from hypoxic injury. Although the AMPK/mTOR pathway was significantly altered under hypoxia, it does not seem to participate in mitophagy regulation. Instead, our data suggest that the mitophagy receptor FUNDC1 plays a vital role in hypoxia-induced mitophagy. Moreover, SESN2 may function through synergistic regulation with other pathways, such as SESN2/AMPK, to mediate cellular adaptation to hypoxia, including the regulation of mitophagy in neuron cells. Therefore, SESN2 plays a critical role in regulating neural cell response to hypoxia. These findings offer valuable insights into the underlying molecular mechanisms governing the regulation of mitophagy under hypoxia and further highlight the potential of SESN2 as a promising therapeutic target for hypoxic nerve injury.
1 Introduction
In recent years, increasing numbers of people, including Europeans and Americans, are traveling to high altitude regions for various purposes including recreation, religion and adventure sports such as mountain climbing in the Everest, Qogir or other high altitude locations around the world (Burtscher, et al., 2021; Mikołajczak, et al., 2021). Despite the inherent risks associated with exposure to high altitude hypoxia, mountaineers are drawn to the breathtaking scenery and the physical and mental challenges presented by these environments. Climbing to high altitudes can lead to exposure to hypoxia, which may result in severe neurological dysfunction such as cognitive impairment, memory loss, and even life-threatening conditions such as high-altitude cerebral edema (HACE) and high-altitude pulmonary edema (HAPE) (Singh and Ansari, 2022). Epidemiological studies and laboratory studies have confirmed that acute exposure to high altitude can contribute to neurological decline (Limmer and Platen, 2018). Mechanistically, declined working memory capacity is closely related to insufficient oxygen supply in the central nervous system. As the “power plant” of cells, mitochondria have a vital function in maintaining neuronal homeostasis in neurons.
Mitophagy is important to remove fragmented or damaged mitochondria and in their quality control (Onishi, et al., 2021). Mitophagy induced by hypoxia mainly exerts a protective function, and is activated principally by autophagy-related proteins (Sulkshane, et al., 2021). Beclin1, LC3, P62 and other conserved proteins participate in the autophagy process and are regarded as autophagy-related proteins (Lv, et al., 2021). Studies have proven that BCL2 interacting protein 3 (BNIP3) and its homolog BCL2 interacting protein 3 like (BNIP3L) belong to the BH3-only protein family and interact directly with microtubule associated protein 1 light chain 3 alpha (LC3) to promote mitophagy (Zhang, et al., 2008). NOD-like receptor (NLR) family member X1 (NLRX1) is suggested to participate in a variety of pathophysiological processes, such as cell death, mitochondrial dynamics, and oxidative damage (Imbeault, et al., 2014; Stokman, et al., 2017; Killackey, et al., 2019). Moreover, recent evidence suggested that FUN14 domain containing 1 (FUNDC1) could serve as a receptor for LC3 and is involved in autophagy (Liu, et al., 2012). The mitophagy receptors harbor LC3-interacting region (LIR) consensus sequences (Ploumi, et al., 2017; Killackey, et al., 2022). Therefore, we hypothesized that BNIP3L, BNIP3, NLRX1, and FUNDC1 function as receptors for selective mitophagy in response to hypoxia via interaction with LC3.
Sestrin2 (SESN2), also known as hypoxia-induced 95 (HI95), is a member of the Sestrin family that has been conserved throughout evolution (Budanov, et al., 2002). SESN2 has been shown to activate adenosine monophosphate-activated protein kinase (AMPK) signaling and downregulate mechanistic target of rapamycin (mTOR) signaling to regulate autophagy (Hou, et al., 2015). In addition, studies have suggested that, as an adaptive response to ischemia-reperfusion (IR), SESN2 plays an essential role in maintaining the function and integrity of mitochondria, thus regulating substrate metabolism (Ren, et al., 2021). However, despite considerable research on SESN2 focusing on hypoxia-ischemia injury and IR injury, the extent to which it regulates hypoxia-induced mitophagy remains unclear (Shi, et al., 2016; Yang, et al., 2017; Quan, et al., 2018). The pathophysiological causes of hypoxic nerve injury vary considerably between IR injury and simple hypoxic injury. Therefore, it is important to investigate the specific role of SESN2 in hypoxia-induced brain injury.
Our study investigated the role of SESN2 in regulating hypoxia-induced mitophagy and neuronal cell adaptation. To this end, we used PC12 cells to investigate the role of SESN2 in the AMPK/mTOR/mitophagy downstream signaling pathway in response to hypoxia-induced injury. We found that hypoxia treatment increased SESN2 expression and activated the mitophagy in PC12 cells. Additionally, our findings suggest that SESN2 may function synergistically with other pathways, such as SESN2/AMPK, to mediate cellular adaptation to hypoxia, potentially making it a therapeutic target for hypoxic nerve injury.
2 Materials and methods
2.1 Culture of cells and modeling of hypoxia
PC12 cells (BFN60070191) were grown in a 5% CO2 atmosphere at 37°C in Dulbecco’s modified Eagle’s medium (DMEM [Sigma-Aldrich Corporation, D6429]) containing 10% fetal bovine serum [FBS (Atlanta Biologicals, S12450)], 2 mmol L-glutamine (GIBCO, 25,030-081), and 1% penicillin/streptomycin (Life Technologies, 15140163). Cells to be treated with hypoxia were placed in a tri-gas incubator (InvivO2, I400) at 37°C under 5% CO2 and 0.5% O2. The incubator could control the oxygen conc from 0.1% to 23.0% by regulating the ratio of N2/O2. The Subsequent steps would continue in this incubator if needed.
2.2 Knockout of Sesn2 using a lentivirus-delivered CRISPR/cas9 system
A Sesn2-knockout cell line was generated using the CRISPER/Cas9 system (Shanghai Jikai Gene Chemical Technology Co., Ltd). Green fluorescent protein (GFP)-LC3 (Shanghai Jikai Gene Chemical Technology Co., Ltd.) was used to assess autophagic flux by an inverted light microscope (Leica DMi8; Leica Microsystems, Wetzlar, Germany). Stable cell lines were produced by selection with 2.5 μg/mL puromycin (Sigma).
2.3 Cell viability detection
1A Cell Counting Kit-8 [CCK-8 (Biosharp, BS350C)] was used to test cell viability. Briefly, cells were seeded at 8,000 cells per well into 96-well plates. Then, the cells were exposed to hypoxia, followed by the addition of 100 μL fresh medium with 10 μL of CCK-8 reagent and incubated for 30 min at 37°C. The absorbance of the cells at 450 nm was assessed using a Microplate Reader (M5; Molecular Devices, San Jose, CA, United States).
2.4 Determination of cell reactive oxygen species (ROS)
A ROS assay kit (Beyotime, S0033M) was applied to measure ROS levels. Briefly, PC12 cells were added with 10 μM 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) and incubated for 30 min. Thereafter, the DCFH-DA was removed by rinsing the cells two times using Phosphate-buffered saline [PBS (Procell, PB180327)]. The cells were then digested using pancreatin, resuspended in PBS, and subjected to flow cytometry (BD Biosciences, San Jose, CA, United States) to detect the ROS levels.
2.5 Cell apoptosis assay
Flow cytometry was used to determine cell apoptosis. Collected cells were rinsed using PBS and incubated in the presence of propidium iodide (PI) and annexin V-fluorescein isothiocyanate (FITC) (BD Biosciences, 556,547) for 15 min at room temperature in the dark. Flow cytometry was then used to determine cell apoptosis following the supplier’s protocols.
2.6 Determination of the mitochondrial membrane potential (MMP)
The MMP was detected using the fluorescent probe JC-1 (Beyotime, C2005). Briefly, 1 mL of JC-1 working solution was added to PC12 cells and incubated for 20 min at 37°C, washed two times using JC-1 buffer, and then subjected to flow cytometry analysis.
2.7 ATP content determination
Cells were collected into pre-cooled PBS, frozen quickly as aliquots, and stored in liquid nitrogen. For use, aliquots were allowed to melt slowly in an ice water bath and vortexed for 10 s. The ATP content was assayed via Luciferase driven bioluminescence using an ATP Bioluminescence Assay Kit HS II (Sigma-Aldrich, 11699709001). The relative light units of each sample were detected with Microplate Reader (M5; Molecular Devices, San Jose, CA, United States).
2.8 Western blotting
Western blotting was performed according to a standard protocol. The cell lysates (20 μg/lane) were separated using 4–12% or 15% SDS-PAGE gel and then transferred to nitrocellulose membranes. The membrane was blocked with 5% skim milk diluted in TBST, and further incubated with primary antibodies overnight at 4°C. Primary antibodies to the following proteins were used: SESN2 (1:1,000, Cell Signaling Technology, 8,487), AMPK (1:1,000, Cell Signaling Technology, 5,832), phosphorylated (p)-AMPK (1:1,000, Cell Signaling Technology, 2,535), mTOR (1:1,000, Cell Signaling Technology, 2,983), p-mTOR (1:1,000, Cell Signaling Technology, 2,974), Beclin1 (1:1,000, Cell Signaling Technology, 3,495), P62 (1:1,000, Cell Signaling Technology, 23,214), LC3B (1:1,000, Cell Signaling Technology, 2,775), translocase of outer mitochondrial membrane 20 (Tomm20) (1:1,000, Cell Signaling Technology, 42,406), BNIP3L (1:1,000, Cell Signaling Technology, 12,396), BNIP3 (1:1,000, Cell Signaling Technology, 3,769), FUNDC1 (1:1,000, Cell Signaling Technology, 49,240), NLRX1 (1:1,000, Cell Signaling Technology, 13,829), and β-actin (1:50,000, Abclonal, AC038). Secondary antibodies to the following proteins were used: Goat Anti-Rabbit HRP (1:10,000, Abclonal, AS014). Membrane stripping was performed by incubating the membrane in Stripping Buffer (CWBIO, Stripping Buffer, CW0056M) according to the manufacturer’s instructions. The immunoreactive protein bands were exposed by the enhanced chemiluminescence (ECL) method (General Electric, Boston, MA, United States) and normalized by densitometry using ImageJ (NIH, Bethesda, MD, United States).
2.9 Quantitative real-time reverse transcription PCR (qRT-PCR)
The Trizol reagent (Invitrogen, 15596026) was used to extract total RNA following the manufacturer’s protocol. The mRNA was reverse transcribed to cDNA. The cDNA was then used as the template in the quantitative real-time PCR (qPCR) step, which was carried out using a SYBR Green Real-time PCR Master Mix (Thermo Fisher Scientific, A46109), and Actb (encoding β-actin) as the internal control. The 2−ΔΔCT method was used to analyze the results (Livak and Schmittgen, 2001). The primers used had the following sequences:
Bnip3l forward, 5′-TCTCACTTAGTCGAGCCGC-3′ and reverse 5′-CTCCACCCAGGAACTGTTGA-3’; Bnip3 forward, 5′-TCTCACTTAGTCGAGCCGC-3′ and reverse 5′-CTCCACCCAGGAACTGTTGA-3’; Fundc1 forward, 5′-TCTCACTTAGTCGAGCCGC-3′ and reverse 5′-CTCCACCCAGGAACTGTTGA-3’; Nlrx1 forward, 5′-TCTCACTTAGTCGAGCCGC-3′ and reverse 5′-CTCCACCCAGGAACTGTTGA-3′
2.10 Statistical analysis
The statistical analyses were carried out using GraphPad Prism (GraphPad Inc., V5.0.1). Student’s t-test was used to calculate the difference between two groups, whereas analysis of variance (ANOVA) followed by the Tukey-Kramer multiple comparisons test or an unpaired two-tailed Student-t-test were used to analyze three and more groups of data. All data are shown as mean ± SD for each group. p < 0.05 was considered statistically significant.
3 Results
3.1 Hypoxia induced PC12 cell injury and increased expression of SESN2
Firstly, a hypoxia-induced model of PC12 cell damage was constructed. In the model, hypoxia induced injury to PC12 cells in a time-dependent manner. Hypoxic (0.5% O2) conditions for 12 h resulted in significant injury to PC12 cells, with the OD450 value in the CCK8 assay (representing cell viability) decreasing by about 50% after 48 h (Figure 1A). Hypoxia increased ROS activity in PC12 cells, and the ROS level correlated with increased hypoxia time (Figures 1B, C). The apoptotic rate of PC12 cells increased with hypoxia time in culture (Figures 1D, E). Furthermore, western blotting showed that the level of SESN2 increased at first and then decreased, with the highest levels being observed at 24 h (Figures 1F, G). Thus, we hypothesized that SESN2 might be involved in response to the duration of hypoxia.
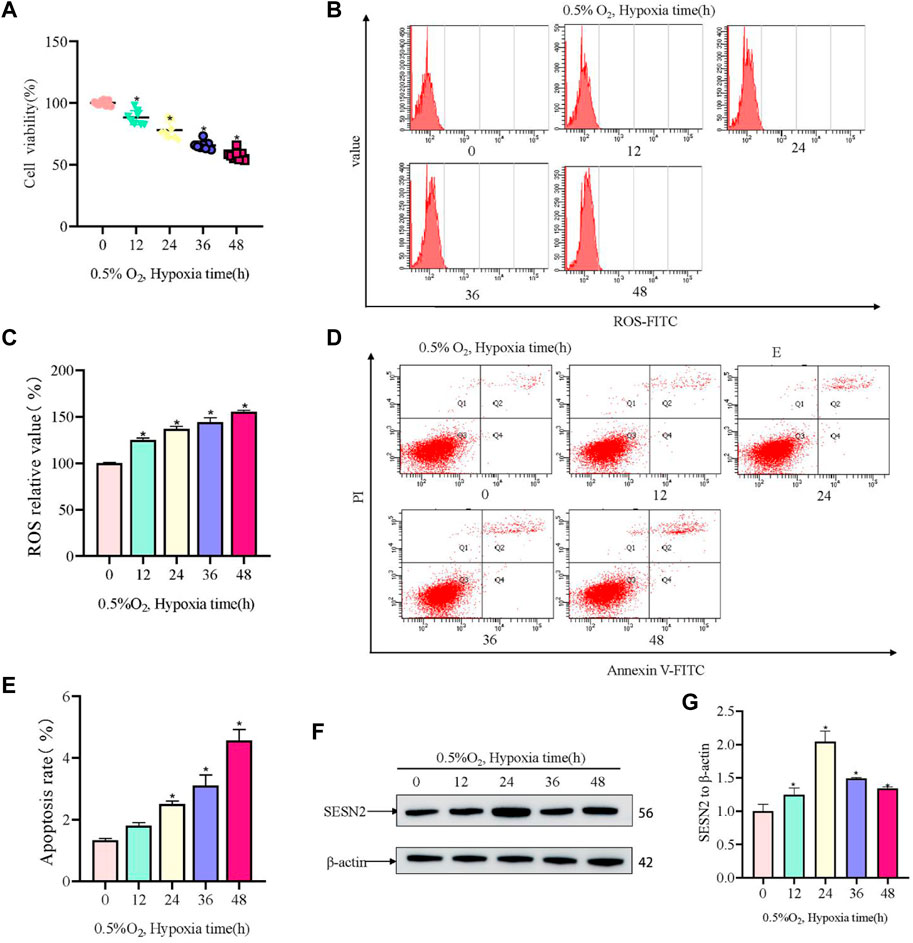
Figure 1. Hypoxia led to decreased cellular activity, elevated ROS levels, and elevated apoptosis rates in PC12 cells. (A) Cellular activity over hypoxia time in PC12 cells (0, 12, 24, 36, and 48 h at 0.5% O2). (B) ROS levels over hypoxia time in PC12 cells. (C) Quantitative analysis of (B). (D) Apoptosis rate over hypoxia time in PC12 cells. (E) Quantitative analysis of (D). (F) Protein levels of SESN2 over hypoxia time in PC12 cells (0, 12, 24, 36, and 48 h at 0.5% O2). (G) Quantitative analysis of (F). Data are presented as mean ± SD. * indicates a significant increase compared with the control value (p < 0.05, n = 3).
3.2 Sesn2 knockout could protect mitochondrial function in hypoxia-damaged PC12 cells
Based on above results, to investigate the role of SESN2 in hypoxic metabolism, the Sesn2 gene was knocked out using the CRISPR/cas9 system, and a stable Sesn2−/− PC12 cell line was constructed, which was screened using puromycin for 5–7 days. Western blotting of proteins extracted from Sesn2−/− PC12 cells showed the almost complete absence of SESN2 (Figure 2A). To determine the effect of Sesn2 knockout on mitochondrial function, we determined the ROS and ATP levels, and the incorporation of JC-1 to assess the mitochondrial membrane potential (MMP), in response to hypoxia 24 h. The results showed knockout of Sesn2 reduced ROS levels under hypoxic conditions compared with those under normal conditions (Figure 2B, C). The mitochondrial membrane potential and mitochondrial ATP levels were increased after Sesn2 knockout under hypoxic conditions compared with those under normal conditions (Figures 2D–F).
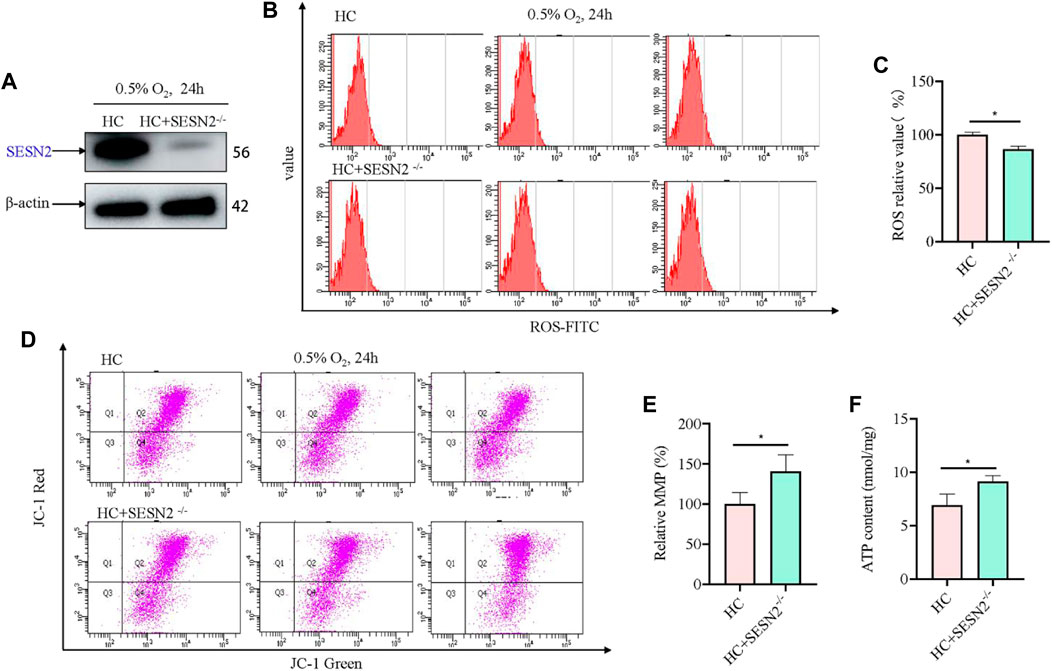
Figure 2. Sesn2 knockout improved mitochondrial function of PC12 cells under hypoxia. (A) Effect of Sesn2 knockout. (B) Knockout of Sesn2 decreased intracellular ROS levels of PC12 cells under hypoxic conditions for 24 h. 3 replicate data of HC and HC + SESN2−/− group was shown respectively. (C) Quantitative analysis of (B). (D) Knockout of Sesn2 increased mitochondria membrane potential in PC12 cells under hypoxic conditions for 24 h. (E) Quantitative analysis of (D). (F) Knockout of Sesn2 upregulated the mitochondrial ATP content in PC12 cells under in hypoxic conditions for 24 h. HC: Hypoxia Control. Data are presented as mean ± SD. * indicates a significant increase compared with the control value (p < 0.05, n = 3).
3.3 Mitophagy is involved in the regulation of hypoxic metabolism in PC12 cells
Hypoxic injury involves damage to mitochondria; therefore, to assess to changes to mitochondria and autophagosomes, the levels of autophagosome-associated proteins and mitophagy receptors were assessed using western blotting. Hypoxia increased the levels of the autophagy proteins Beclin1 and LC3II/I, but reduced the expression of P62 under hypoxic conditions compared with those under normal conditions (Figures 3A–D). The Sestrin2/AMPK/mTOR pathway has been well-established as a critical regulator of autophagy; therefore, the levels of AMPK, p-AMPK, mTOR, and p-mTOR were determined. Somewhat to our surprise, there was no change in the level of p-AMPK and p-mTOR in response to hypoxia (Figures 3E–G). Moreover, we evaluated changes in mitophagy protein markers. Hypoxia increased the levels of BNIP3L, BNIP3, and FUNDC1, but decreased the levels of NLRX1 (Figure 3H–L). TOMM20, which served as a mitochondrial marker, decreased in a time dependent manner in response to hypoxia. Concomitantly, the expression levels of mitophagy-related genes were investigated using qRT-PCR, the results of which were consistent with those of western blotting (Figure 3M–Q).
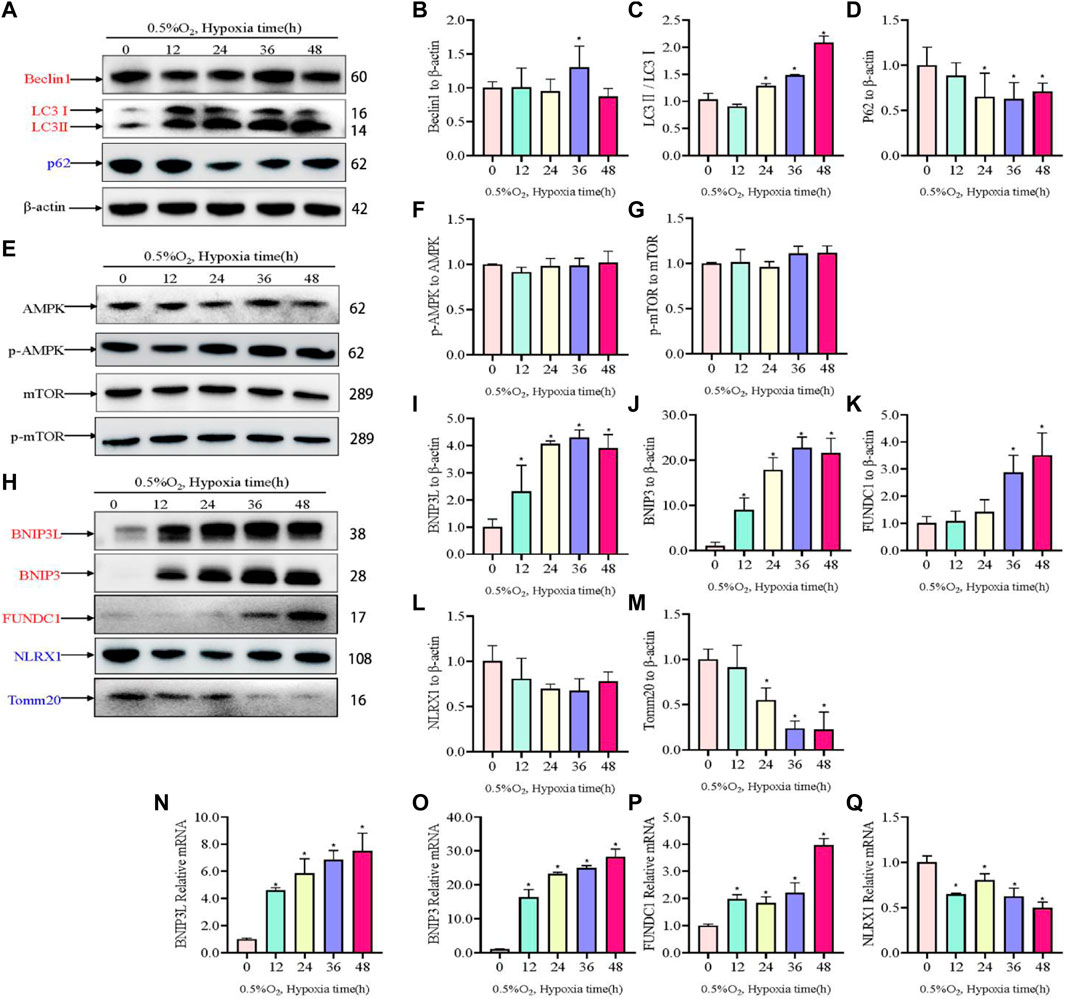
Figure 3. Levels of autophagy-related proteins under hypoxia in PC12 cells. (A) Protein levels of Beclin1, LC3 I, LC3 II, and p62 over hypoxia time in PC12 cells (0, 12, 24, 36, and 48 h at 0.5% O2). (B–D) Quantitative analysis of (A). (E) AMPK, p-AMPK, mTOR and p-mTOR levels over hypoxia time in PC12 cells. (F–G) Quantitative analysis of p-AMPK-AMPK and p-mTOR-mTOR. (H) Protein levels of BNIP3L, BNIP3, FUNDC1, NLRX1, and Tomm20 over hypoxia time in PC12 cells. (I–M) Quantitative analysis of (H). (N–Q) mRNA expression of Bnip3l, Bnip3, Fundc1, and Nlrx1 over hypoxia time. Blue indicates a decrease in the protein level and red indicates an increase in the protein level. Data are presented as mean ± SD. * indicates a significant increase compared with the control value (p < 0.05, n = 3).
3.4 Inhibition of autophagy by 3-methyladenine (3 MA) also improved mitochondrial function after hypoxia
To further validate the role of mitophagy in hypoxia-injured neural cells, the autophagy inhibitor 3 MA was used to pretreat cells at 5 mM for 12 h. Compared with Sesn2-knockout cells, the analysis of ROS showed the opposite results, i.e., 3 MA pre-treatment caused elevation of ROS levels (Figure 4A, B). The results from the JC-1 and ATP experiments showed the same trend to those obtained in the Sesn2-knockout cells, indicating the protective effect of autophagy inhibition on mitochondrial repair in response to hypoxia (Figures 4C–E).
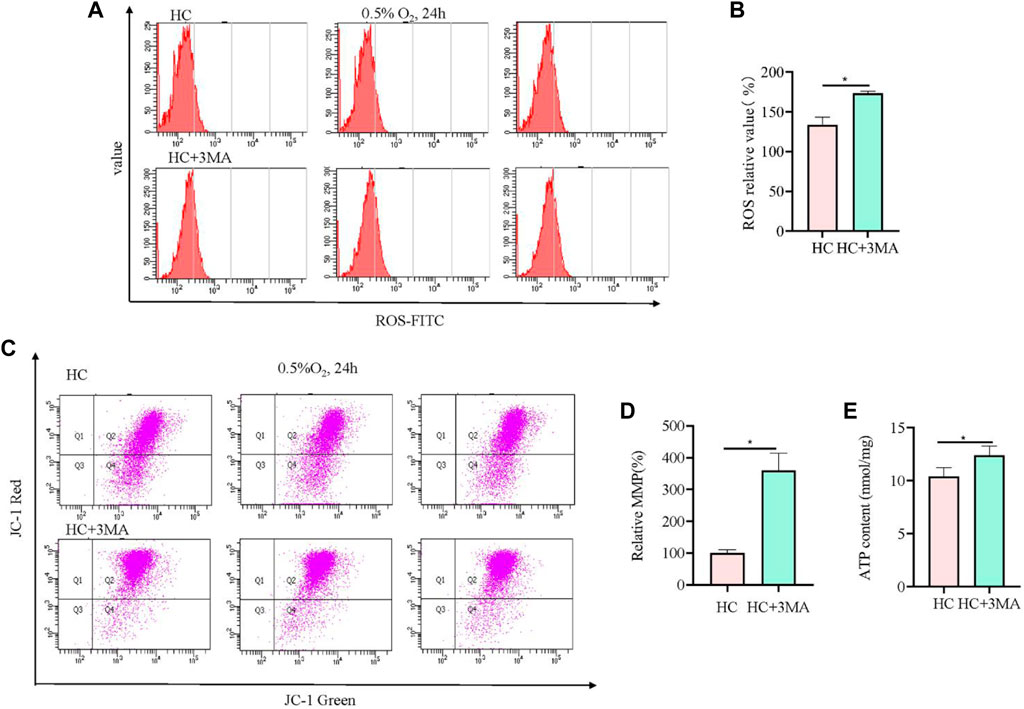
Figure 4. 3MA led to elevated ROS levels, increased mitochondrial membrane potential, and upregulated mitochondrial ATP content. (A) In PC12 cells subjected to hypoxia (0.5% O2, 24 h), 3MA increased intracellular ROS. 3 replicate data of HC and HC + 3MA group was shown respectively. (B) Quantitative analysis of (A). (C) In PC12 cells subjected to hypoxia (0.5% O2, 24 h), 3 MA increased the mitochondrial membrane potential. (D) Quantitative analysis of (C). (E) In PC12 cells subjected to hypoxia (0.5% O2, 24 h), 3 MA upregulated the mitochondrial ATP content. HC: Hypoxia Control. Data are presented as mean ± SD. * indicates a significant increase compared with the control value (p < 0.05, n = 3).
3.5 Sesn2 knockout significantly inhibited p-AMPK levels and mitophagy processes after hypoxia
Next, in view of its role in hypoxia regulation, AMPK-mediated mitophagy was investigated to further study the mechanism by which Sesn2 knockout protected mitochondria against hypoxic injury. Sesn2 knockout did not affect Beclin1 and P62 levels significantly, whereas the level of LC3II/I decreased (Figure 5A). Moreover, Sesn2 knockout decreased the p-AMPK:AMPK ratio significantly, but had no effect on the p-mTOR: mTOR ratio (Figure 5B). The expression of mitophagy proteins and mitochondrial outer membrane proteins were measured using western blotting and qRT-PCR. We found that the mRNA and protein levels of BNIP3 and FUNDC1 decreased significantly, and NLRX1 and TOMM20 mRNA and protein levels increased significantly after Sesn2-knockout (Figure 5C, D). Concomitantly, transfection with GFP-LC3 was employed to assess autophagic flux, which confirmed the above results: Hypoxia caused mitophagy and Sesn2-knockout inhibited autophagy activation (Figure 5E).
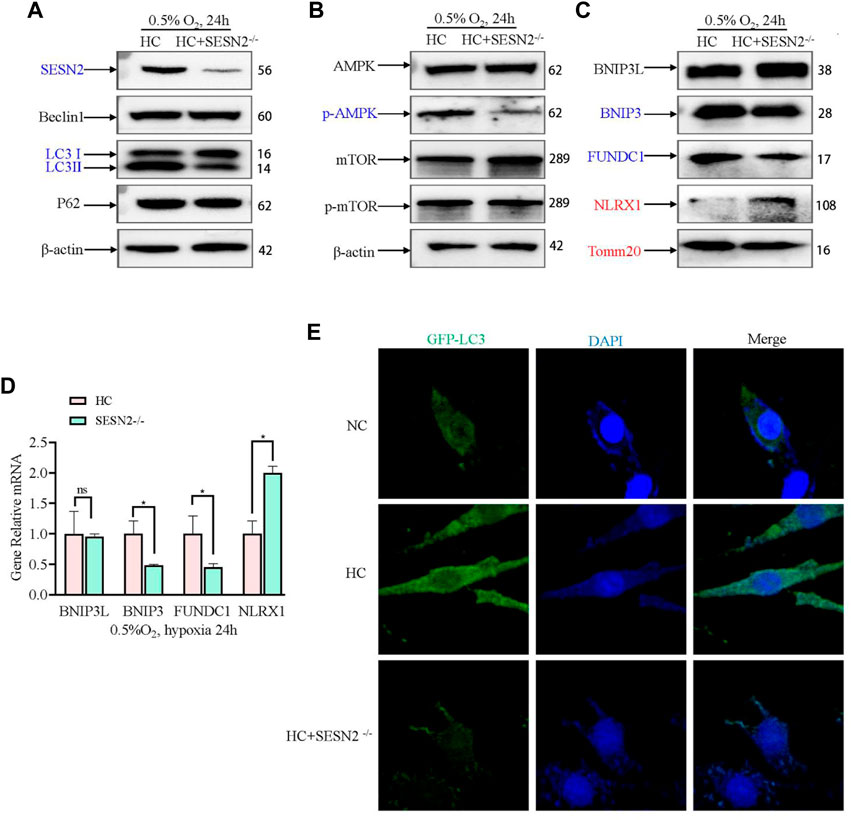
Figure 5. Effect of SESN2 knockout on mitophagy-related proteins under hypoxia in PC12 cells. (A) Levels of Beclin1, LC3 I, LC3 II, and p62 proteins were determined after Sesn2 knockout using western blotting in PC12 cells subjected to hypoxia (0.5% O2, 24 h). (B) AMPK, p-AMPK, mTOR and p-mTOR levels after Sesn2 knockout in PC12 cells subjected to hypoxia (0.5% O2, 24 h). (C) Protein levels of BNIP3L, BNIP3, FUNDC1, NLRX1, and Tomm20 after Sesn2 knockout in PC12 cells (0.5% O2, 24 h). (D) mRNA expression of Bnip3l, Bnip3, Fundc1, and Nlrx1 after knockout in PC12 cells subjected to hypoxia (0.5% O2, 24 h). Data are presented as mean ± SD. * indicates a significant increase compared with the control value (p < 0.05, n = 3). (E) Detection of GFP-LC3 autophagosomes after Sesn2 knockout in PC12 cells subjected to hypoxia (0.5% O2, 24 h, magnification: ×400). HC: Hypoxia Control. NC: Normoxia Control. Blue indicates a decrease in the protein level and red indicates an increase in the protein level.
3.6 Mitophagy was inhibited by 3 MA but did not alter SESN2 expression
To confirm that the expression of SESN2 mediates mitophagy by autophagy activation, we used an autophagy inhibitor (3 MA) to block the initiation of autophagy. The AMPK signaling pathway and mitophagy proteins were then detected. 3MA reduced the level of Beclin1 and LC3B, but exhibited no appreciable effect on the levels of SESN2 and P62 (Figure 6A). 3MA activated mTOR by inhibiting AMPK (Figure 6B). Furthermore, 3 MA treatment significantly decreased the BNIP3L, BNIP3, and FUNDC1 levels, increased the level of TOMM20, and had no effect on NLRX1 (Figure 6C, D). Consistent with Sesn2 knockdown, the addition of 3 MA also decreased the numbers of GFP-LC3–positive cells after hypoxia treatment (Figure 6E).
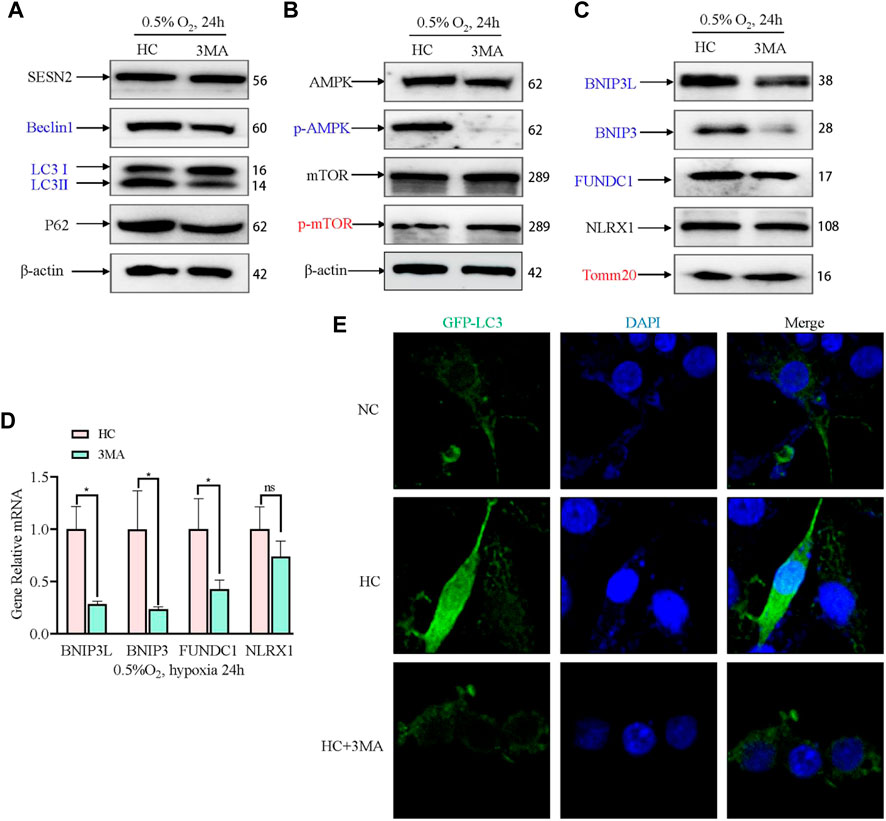
Figure 6. Effect of 3 MA on mitophagy-related proteins under hypoxia in PC12 cells. (A) Effects of 3 MA on the protein expression of SESN2, Beclin1, LC3B, and P62 proteins by western blotting in PC12 cells subjected to hypoxia (0.5% O2, 24 h). (B) Effects of 3 MA on p-mTOR, mTOR, p-AMPK, and AMPK levels in PC12 cells subjected to hypoxia (0.5% O2, 24 h). (C) Effects of 3 MA on the protein levels of BNIP3L, BNIP3, FUNDC1, NLRX1, and Tomm20 in PC12 cells subjected to hypoxia (0.5% O2, 24 h). (D) mRNA expression of Bnip3l, Bnip3, Fundc1, and Nlrx1 after 3 MA intervention in PC 12 cells subjected to hypoxia (0.5% O2, 24 h). Data are presented as mean ± SD. * indicates a significant increase compared with the control value (p < 0.05, n = 3). (E) Detection of GFP-LC3 autophagosomes after 3 MA intervention in PC12 cells subjected to hypoxia (0.5% O2, 24 h, magnification: ×400). HC: Hypoxia Control. NC: Normoxia Control. Blue indicates a decrease in the protein level and red indicates an increase in the protein level.
4 Discussion
SESN2 is a stress-responsive protein that has been shown to respond to various insults, such as oxidative stress, genotoxic stress, energy deficiency, and hypoxia (Budanov, et al., 2002; Pan, et al., 2021). While the role of SESN2 has been extensively studied in I/R-related diseases, its effect on high altitude hypoxia has been rarely reported (Shi, et al., 2016; Wang, et al., 2019a; Wang, et al., 2019b). The present study aimed to investigate whether SESN2 plays a role in the hypoxic response and to elucidate its possible mechanisms. Using in vitro studies, we found that SESN2 promotes mitophagy and reduces the mitochondrial quantity of hypoxic neuroblasts. Our results suggest that the SESN2/AMPK/mTOR/FUNDC1 signaling pathway regulates the process of mitophagy, as shown in Figure 7. These findings provide new insights into the molecular mechanisms underlying hypoxia-induced neuronal injury and highlight SESN2 as a promising therapeutic target for treating cognitive impairment caused by high altitude. In our experiments, we observed significant upregulation of SESN2 expression following hypoxia treatment, consistent with previous reports investigating hypoxia-ischemia (Shi, et al., 2016) and ischemia-reperfusion (Liu, et al., 2020).
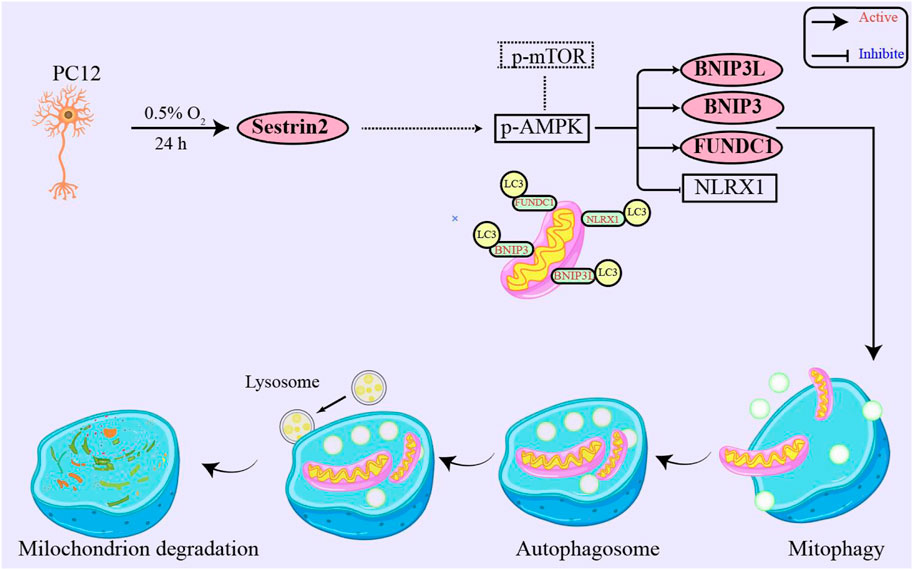
Figure 7. SESN2 upregulation activated mitophagy in PC12 cells by hypoxia treatment. Mitophagy receptor FUNDC1 plays a vital role in hypoxia-induced mitophagy. Moreover, SESN2 upregulation may function through synergistic regulation with other pathways, such as SESN2/AMPK, to mediate cellular adaptation to hypoxia. Therefore, SESN2 plays a critical role in regulating neural cell response to hypoxia.
To assess autophagic flux, we measured the levels of LC3-II, Beclin1, and p62 proteins, which are indicators of autophagy. Our results indicated that hypoxia treatment activated autophagy and increased autophagic flux. Autophagy can be triggered by either mTOR downregulation or AMPK activation (Egan, et al., 2011). However, our findings suggested that hypoxia-induced autophagy flux is not dependent on the AMPK/mTOR pathway. Degradation of damaged or dysfunctional mitochondria involves mitophagy, a specific form of autophagy (Ashrafi and Schwarz, 2013). Mitophagy receptors such as BNIP3L, BNIP3, FUNDC1, and NLRX1 recruit LC3 to initiate mitophagy in response to mitochondrial stress (Rouschop and Wouters, 2009; Fang, et al., 2015), including oxidative stress, bioenergetic stress, and IR stress (Liu, et al., 2012; Ney, 2015). The different mitophagy mechanisms that sense these stresses are still unclear. Our results suggest that BNIP3L, BNIP3, FUNDC1, and NLRX1 are involved in hypoxia-induced mitophagy.
To determine the role of SESN2 in neurocytes under hypoxia, we knocked out Sesn2 in PC12 cells. We observed that Sesn2 knockout increased ROS and ATP levels and mitochondrial membrane potential. Mechanistically, Sesn2 knockout significantly reduced AMPK phosphorylation, inhibited mitophagy, and protected PC12 cells. Sesn2 knockout also suppressed BNIP3 and FUNDC1 expression, while increasing NLRX1 protein. Treatment with 3MA, an autophagy inhibitor, effectively suppressed hypoxia-induced mitophagy, resulting in increased mitochondrial membrane potential and ATP production without any notable effect on SESN2 protein levels. Therefore, we conclude that SESN2 acts upstream of the autophagic cascade. Our experiments further suggest that Sesn2 knockout repairs mitochondrial function by inhibiting AMPK-mediated mitophagy.
Previous studies have indicated that the SESN2/AMPK/mTOR pathway plays a dominant role in mediating autophagy in hypoxia-related diseases (Hou, et al., 2015; Hwang, et al., 2017). To test whether SESN2 regulates AMPK and its downstream mitophagy receptor, we carried out a series of experiments. Sesn2 knockout suppressed AMPK activation, while upregulation of SESN2 contributed to AMPK phosphorylation. Further, Sesn2 knockout significantly reduced BNIP3 and FUNDC1 expression, but NLRX1 protein was decreased too. Zhang et al. (2019a) identified NLRX1 as a novel mitophagy receptor that induces mitophagy, and Li et al. (2021) demonstrated that intestinal I/R injury downregulates NLRX1 levels, consistent with our findings. The pathophysiological role of BNIP3 in our experiments remains unknown, though most studies have reported that it can induce apoptosis (Prabhakaran, et al., 2007; Jia, et al., 2020) while Zhang et al. revealed that BNIP3 could promote mitophagy (Zhang, et al., 2019b).
In summary, our study provides valuable insights into the role of SESN2 in hypoxia-induced nerve injury and its potential as a therapeutic target. Hypoxia treatment increases SESN2 expression, while Sesn2 knockout leads to reduced AMPK activation and mitophagy signaling. These observations highlight the critical role of the SESN2/AMPK pathway in regulating hypoxia-induced autophagy to exert neuroprotective effects and provide a deeper understanding of the role of SESN2 in regulating hypoxia-induced autophagy. Hence, our study reveals a previously unexplored role for SESN2 as a therapeutic intervention for hypoxic nerve injury. Furthermore, targeting SESN2 may hold promise in regulating the response to high altitude hypoxia. However, further research is required to confirm these findings and fully understand the underlying pathways involved.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
CP: Data-curation, Methodology, Writing–original draft. CA: Data-curation, Methodology, Writing–review and editing. LL: Writing–review and editing. BZ: Writing–review and editing. QL: Writing–review and editing. LP: Writing–review and editing. ZW: Writing–review and editing. WL: Conceptualization, Investigation, Writing–review and editing. ZC: Project administration, Resources, Visualization, Writing–review and editing. HL: Software, Writing–review and editing. XW: Formal Analysis, Funding acquisition, Supervision, Validation, Writing–review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Natural Science Foundation of China (31971106) and Foundation of Tianjin Institute of Environmental and Operational Medicine (2022ZZKY07).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2023.1266243/full#supplementary-material
References
Ashrafi, G., and Schwarz, T. L. (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20 (1), 31–42. doi:10.1038/cdd.2012.81
Budanov, A. V., Shoshani, T., Faerman, A., Zelin, E., Kamer, I., Kalinski, H., et al. (2002). Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 21 (39), 6017–6031. doi:10.1038/sj.onc.1205877
Burtscher, M., Hefti, U., and Hefti, J. P. (2021). High-altitude illnesses: old stories and new insights into the pathophysiology, treatment and prevention. Sports Med. Health Sci. 3 (2), 59–69. doi:10.1016/j.smhs.2021.04.001
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331 (6016), 456–461. doi:10.1126/science.1196371
Fang, Y., Tan, J., and Zhang, Q. (2015). Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol. Int. 39 (8), 891–898. doi:10.1002/cbin.10463
Hou, Y. S., Guan, J. J., Xu, H. D., Wu, F., Sheng, R., and Qin, Z. H. (2015). Sestrin2 protects dopaminergic cells against rotenone toxicity through AMPK-dependent autophagy activation. Mol. Cell. Biol. 35 (16), 2740–2751. doi:10.1128/mcb.00285-15
Hwang, H. J., Jung, T. W., Choi, J. H., Lee, H. J., Chung, H. S., Seo, J. A., et al. (2017). Knockdown of sestrin2 increases pro-inflammatory reactions and ER stress in the endothelium via an AMPK dependent mechanism. Biochimica Biophysica Acta-Molecular Basis Dis. 1863 (6), 1436–1444. doi:10.1016/j.bbadis.2017.02.018
Imbeault, E., Mahvelati, T. M., Braun, R., Gris, P., and Gris, D. (2014). Nlrx1 regulates neuronal cell death. Mol. Brain 7, 90. doi:10.1186/s13041-014-0090-x
Jia, J., Yang, X., Zhao, Q., Ying, F., Cai, E., Sun, S., et al. (2020). BNIP3 contributes to cisplatin-induced apoptosis in ovarian cancer cells. FEBS open bio 10 (8), 1463–1473. doi:10.1002/2211-5463.12881
Killackey, S. A., Bi, Y., Soares, F., Hammi, I., Winsor, N. J., Abdul-Sater, A. A., et al. (2022). Mitochondrial protein import stress regulates the LC3 lipidation step of mitophagy through NLRX1 and RRBP1. Mol. Cell 82 (15), 2815–2831.e5. doi:10.1016/j.molcel.2022.06.004
Killackey, S. A., Rahman, M. A., Soares, F., Zhang, A. B., Abdel-Nour, M., Philpott, D. J., et al. (2019). The mitochondrial Nod-like receptor NLRX1 modifies apoptosis through SARM1. Mol. Cell Biochem. 453 (1-2), 187–196. doi:10.1007/s11010-018-3444-3
Li, S., Zhou, Y., Gu, X., Zhang, X., and Jia, Z. (2021). NLRX1/FUNDC1/NIPSNAP1-2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 54 (3), e12986. doi:10.1111/cpr.12986
Limmer, M., and Platen, P. (2018). The influence of hypoxia and prolonged exercise on attentional performance at high and extreme altitudes: A pilot study. PloS one 13 (10), e0205285. doi:10.1371/journal.pone.0205285
Liu, J., Li, Y., Mei, C. L., Ning, X. B., Pang, J. F., Gu, L., et al. (2020). Phytic acid exerts protective effects in cerebral ischemia-reperfusion injury by activating the anti-oxidative protein sestrin2. Biosci. Biotechnol. Biochem. 84, 1401–1408. doi:10.1080/09168451.2020.1754158
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14 (2), 177–185. doi:10.1038/ncb2422
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 (4), 402–408. doi:10.1006/meth.2001.1262
Lv, S., Liu, H., and Wang, H. (2021). Exogenous hydrogen sulfide plays an important role by regulating autophagy in diabetic-related diseases. Journal 22, 6715. doi:10.3390/ijms22136715
Mikołajczak, K., Czerwińska, K., Pilecki, W., Poręba, R., Gać, P., and Poręba, M. (2021). The impact of temporary stay at high altitude on the circulatory system. Journal 10, 1622. doi:10.3390/jcm10081622
Ney, P. A. (2015). Mitochondrial autophagy: origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 1853 (10), 2775–2783. doi:10.1016/j.bbamcr.2015.02.022
Onishi, M., Yamano, K., Sato, M., Matsuda, N., and Okamoto, K. (2021). Molecular mechanisms and physiological functions of mitophagy. Embo J. 40 (3), e104705. doi:10.15252/embj.2020104705
Pan, C., Chen, Z., Li, C., Han, T., Liu, H., and Wang, X. (2021). Sestrin2 as a gatekeeper of cellular homeostasis: physiological effects for the regulation of hypoxia-related diseases. J. Cell. Mol. Med. 25 (12), 5341–5350. doi:10.1111/jcmm.16540
Ploumi, C., Daskalaki, I., and Tavernarakis, N. (2017). Mitochondrial biogenesis and clearance: a balancing act. Febs J. 284 (2), 183–195. doi:10.1111/febs.13820
Prabhakaran, K., Li, L., Zhang, L., Borowitz, J. L., and Isom, G. E. (2007). Upregulation of BNIP3 and translocation to mitochondria mediates cyanide-induced apoptosis in cortical cells. Neuroscience 150 (1), 159–167. doi:10.1016/j.neuroscience.2007.07.033
Quan, N. H., Wang, L., Chen, X., Luckett, C., Cates, C., Rousselle, T., et al. (2018). Sestrin2 prevents age-related intolerance to post myocardial infarction via AMPK/PGC-1 alpha pathway. J. Mol. Cell. Cardiol. 115, 170–178. doi:10.1016/j.yjmcc.2018.01.005
Ren, D., He, Z., Fedorova, J., Zhang, J., Wood, E., Zhang, X., et al. (2021). Sestrin2 maintains OXPHOS integrity to modulate cardiac substrate metabolism during ischemia and reperfusion. Redox Biol. 38, 101824. doi:10.1016/j.redox.2020.101824
Rouschop, K. M., and Wouters, B. G. (2009). Regulation of autophagy through multiple independent hypoxic signaling pathways. Curr. Mol. Med. 9 (4), 417–424. doi:10.2174/156652409788167131
Shi, X. D., Doycheva, D. M., Xu, L., Tang, J. P., Yan, M., and Zhang, J. H. (2016). Sestrin2 induced by hypoxia inducible factor1 alpha protects the blood-brain barrier via inhibiting VEGF after severe hypoxic-ischemic injury in neonatal rats. Neurobiol. Dis. 95, 111–121. doi:10.1016/j.nbd.2016.07.016
Singh, S., and Ansari, M. A. (2022). High altitude related diseases: milder effects, HACE, HAPE, and effect on various organ systems. Berlin, Germany: Springer, 37–49.
Stokman, G., Kors, L., Bakker, P. J., Rampanelli, E., Claessen, N., Teske, G. J. D., et al. (2017). NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J. Exp. Med. 214 (8), 2405–2420. doi:10.1084/jem.20161031
Sulkshane, P., Ram, J., Thakur, A., Reis, N., Kleifeld, O., and Glickman, M. H. (2021). Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol. 45, 102047. doi:10.1016/j.redox.2021.102047
Wang, H. X., Li, N., Shao, X., Li, J., Guo, L. P., Yu, X. Y., et al. (2019a). Increased plasma sestrin2 concentrations in patients with chronic heart failure and predicted the occurrence of major adverse cardiac events: A 36-month follow-up cohort study. Clin. Chim. Acta 495, 338–344. doi:10.1016/j.cca.2019.04.084
Wang, H. X., Zhao, Y. P., Li, Y. X., Wu, J. X., Yu, S. S., Zhu, J., et al. (2019b). Sestrin2 overexpression attenuates focal cerebral ischemic injury in rat by increasing Nrf2/HO-1 pathway-mediated angiogenesis. Neuroscience 410, 140–149. doi:10.1016/j.neuroscience.2019.05.005
Yang, K. P., Xu, C. H., Zhang, Y. F., He, S. L., and Li, D. Z. (2017). Sestrin2 suppresses classically activated macrophages-mediated inflammatory response in myocardial infarction through inhibition of mTORC1 signaling. Front. Immunol. 8, 728. doi:10.3389/fimmu.2017.00728
Zhang, H., Bosch-Marce, M., Shimoda, L. A., Tan, Y. S., Baek, J. H., Wesley, J. B., et al. (2008). Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283 (16), 10892–10903. doi:10.1074/jbc.M800102200
Zhang, Y., Liu, D., Hu, H., Zhang, P., Xie, R., and Cui, W. (2019a). HIF-1α/BNIP3 signaling pathway-induced-autophagy plays protective role during myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 120, 109464. doi:10.1016/j.biopha.2019.109464
Keywords: hypoxia, mitophagy, SESN2, AMPK, FUNDC1, neurological dysfunction
Citation: Pan C, Ai C, Liang L, Zhang B, Li Q, Pu L, Wang Z, Liu W, Chen Z, Liu H and Wang X (2023) Sestrin2 protects against hypoxic nerve injury by regulating mitophagy through SESN2/AMPK pathway. Front. Mol. Biosci. 10:1266243. doi: 10.3389/fmolb.2023.1266243
Received: 24 July 2023; Accepted: 11 September 2023;
Published: 21 September 2023.
Edited by:
Guanghong Jia, University of Missouri, United StatesReviewed by:
Alejandro Silva-Palacios, National Institute of Cardiology Ignacio Chavez, MexicoShane Austin, The University of the West Indies, Cave Hill, Barbados
Copyright © 2023 Pan, Ai, Liang, Zhang, Li, Pu, Wang, Liu, Chen, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weili Liu, bGl1d2VpbGktMDAyQDE2My5jb20=; Zhaoli Chen, emhhb2xpY2hlbkAxMjYuY29t; Hui Liu, bGl1aHVpQGx6dS5lZHUuY24=; Xinxing Wang, d3h4ZW1haWxAc2luYS5jbg==
†These authors have contributed equally to this work