



- 1 Department of Psychiatry and Program in Neuroscience, Uniformed Services University, Bethesda, MD, USA
- 2 Center for Neural Science, New York University, NY, USA
Pavlovian auditory fear conditioning involves the integration of information about an acoustic conditioned stimulus (CS) and an aversive unconditioned stimulus in the lateral nucleus of the amygdala (LA). The auditory CS reaches the LA subcortically via a direct connection from the auditory thalamus and also from the auditory association cortex itself. How neural modulators, especially those activated during stress, such as norepinephrine (NE), regulate synaptic transmission and plasticity in this network is poorly understood. Here we show that NE inhibits synaptic transmission in both the subcortical and cortical input pathway but that sensory processing is biased toward the subcortical pathway. In addition binding of NE to β-adrenergic receptors further dissociates sensory processing in the LA. These findings suggest a network mechanism that shifts sensory balance toward the faster but more primitive subcortical input.
Introduction
The neural circuits underlying classical fear conditioning are well characterized (LeDoux, 2000, 2007; Pare et al., 2004; Maren, 2005; Lang and Davis, 2006). In classical fear conditioning a previously neutral stimulus comes to trigger a behavioral fear response. For this to occur, two events become persistently associated through associative memory formation. Fear conditioning requires the coordinated presentation or pairing of two sensory inputs. One input is an initially neutral stimulus, which becomes the conditioned stimulus (CS). The other input is a biologically significant event, the unconditioned stimulus (US). The pairing of these stimuli results in a conditioned response (CR) such that the initially neutral stimulus (CS) now elicits the same behavioral response as the US.
The neurobiology of fear conditioning is best characterized for an auditory fear-conditioning task employing an acoustic CS and a nociceptive US. These stimuli converge in the lateral amygdala (LA; see Romanski et al., 1993; Johnson et al., 2009 for review). The acoustic signal conveying the tone CS enters the LA via the auditory thalamus (subcortical) and the auditory association cortex (Romanski et al., 1993; LeDoux, 2003). Both of these pathways enter the LA, where they converge with US signals. Additionally, both pathways converge onto single LA neurons on adjacent spines (Humeau et al., 2005) and both pathways undergo plasticity during fear conditioning (Mckernan and Shinnick-Gallagher, 1997; Doyere et al., 2003; Shin et al., 2006). The two auditory routes, are generally thought to provide different aspects of the CS to the LA, with the subcortical (thalamus direct to amygdala) input, coined the “low road,” providing a rudimentary version of the CS and the cortical input, coined the “high road,” providing more detail (LeDoux, 1994, 2000).
Stress and arousal are known to activate norepinephrine (NE) release in the brain (Aston-Jones and Bloom, 1981; Mcgaugh, 2000). NE is especially important to conditioned fear because fear learning occurs during times of threat and stress. Thus, NE release may modulate the neural substrates of conditioned fear (Huang et al., 2000; Tully et al., 2007). NE, especially its β receptors, has been implicated in the formation, reconsolidation, and extinction of fear memories, and has been proposed as a potential treatment for PTSD (Berlau and Mcgaugh, 2006; Debiec and LeDoux, 2006; Brunet et al., 2008). Recent data by Tully et al. (2007) shows that NE promotes the formation of long term potentiation (LTP), a cellular model of memory in the thalamo-amygdala pathway. Despite the psychopharmacological data in animals and the therapeutic potential in humans, relatively little is known about the underlying network and cellular mechanism in the LA. A key question is whether the two pathways are differentially regulated by NE. A differential regulation could shift the balance between the “high” and “low” roads of sensory input to the amygdala fear conditioning circuit.
In order to screen for possible network effects of NE and its receptors we used in vitro field potential recordings which would allow us to detect changes in the synaptic activity as well as excitability of the network as a whole. We find NE and its β receptors differentially regulate synaptic transmission between the cortical and subcortical pathways to the LA. Differences in synaptic transmission are dependent upon GABA receptors in the LA. Together these data provide a mechanism by which increased NE signaling in LA, activated by stress and arousal, may critically shift the balance in synaptic transmission toward the faster subcortical afferents and thus promote survival.
Materials and Methods
In order to record the electrophysiological, excitatory, or inhibitory, response of LA neurons to NE and its β receptors, we made extracellular field potential recordings. We measured the evoked field excitatory potentials synaptic potential (fEPSP) in response to evoked activation of the subcortical (or thalamic) and cortical inputs pathways to the LA (Weisskopf and LeDoux, 1997).
Physiological Recordings in vitro: Coronal brain slices containing the LA were prepared from Sprague-Dawley rats aged 4–7 weeks. Once deeply anesthetized with ketamine and xylazine the chest was exposed to reveal the heart. A sharp 26-gage needle was placed in the left ventricle, an incision made in the right atrium, and 5–10 ml of ice cold oxygenated artificial cerebro-spinal fluid (ACSF) was injected in less than 1 min. The animal was then quickly decapitated and the brain removed. Chilled brains were blocked into coronal sections containing the entire amygdala and then sliced in hemi-coronal sections on a vibratome to 400 μm. Brain slices were warmed to 32°C for 30 min and then slowly lowered to room temperature and maintained for several hours. Slices were transferred to a recording chamber, flow rate 2.5 ml/min, containing ACSF (in mM) 115 NaCl, 3.3 KCl, 1 MgSO4, 2 CaCl2, 25.5 NaHCO3, 1.2 NaH2PO4, 5 lactic acid, and 25 glucose, equilibrated with 95% O2/5% CO2.
Slices and neurons were visualized with an upright fixed stage microscope equipped with infrared differential interference contrast optics. Recording electrodes of 5–10 MΩ (Lamprecht et al., 2006) filled with ACSF, were guided to dorsolateral amygdala (LAd) neurons. Two bipolar stimulating electrodes were placed in the brain slice to stimulate auditory subcortical (thalamic) and cortical afferents to the LA (Weisskopf et al., 1999). The subcortical afferents electrode was placed medial to the LA and lateral to stria terminalis, where dorsal to the central nucleus of the amygdala, thalamus to amygdala fibers course. The cortical afferents electrode was placed dorsal to the LA on the external capsule/cortical border (see LeDoux et al., 1990; Romanski et al., 1993 for details of anatomical input to LA). Current was passed at 0–0.2 mA to activate suprathreshold EPSPs and IPSPs. Stimulation was controlled by Axon software (see Weisskopf and LeDoux, 1997 for further description). Voltage recordings were amplified using an AxoClamp 2B amplifier, signals filtered at 3 kHz and digitized at 5 kHz using an Axon analog to digital converter and analyzed off line with pClamp. Drugs (Norepinephrine, Isoproterenol, PTX, Sigma, MO, USA) were added to the superfused ACSF. All drugs were superfused onto slice with a flow rate of 2.5 ml/min. Data was also analysis using Excel (Microsoft, WA, USA) and statistical tests were made with GraphPad, Prism. Data are presented as mean ± SEM. A probability level of <0.05 was considered significant.
Results
We evoked extracellular field potentials in the LA in response to stimulation of both cortical and subcortical pathways (Weisskopf and LeDoux, 1999) in order to directly compare (thalamic) both afferent responses (Figure 1A) to NE in LA. Potentials were sequentially activated, with a suitable separation latency, which allowed for direct comparison of potentials. Extracellular evoked fEPSP amplitudes were measured as previously described (Lamprecht et al., 2006; Johnson et al., 2008, 2009; see also Huang et al., 2000). The fEPSP in both pathways ranged in size from 0.2 to 0.6 mV without picrotoxin (PTX) and from 0.5 to 1.2 mV in the presence of PTX (Figure 1B). Differences in amplitude in fEPSP between the pathways at baseline were not observed (Lamprecht et al., 2006). Stimulation of both pathways was reduced to bring the fEPSP to approximately 50% of maximum amplitude. Recordings were stabilized prior to applications of drugs. We found that NE differentially regulates synaptic transmission in the subcortical and cortical pathways to the LA. Five key findings are described below.
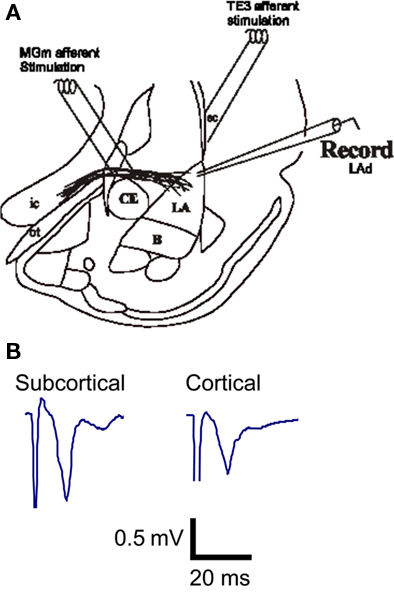
Figure 1. In vitro afferent pathways to the LA established by Weisskopf et al. (1999). Both the cortical afferent pathway to the LA and the direct subcortical (thalamic) pathway to the LA were alternatively stimulated and potential were recorded in the LA (A). Example traces of extracellular excitatory postsynaptic synaptic potentials (fEPSP) recorded from the LA in vitro (B).
Synaptic Transmission is Inhibited More by NE in the Cortical than the Subcortical Pathway
In the first experiment, we tested the effect of NE itself on the fEPSP in both the cortical and subcortical pathways (Figures 2A,B). NE (20 μM) reduced the amplitude of the evoked fEPSP in both pathways. The effect of NE on fEPSP was rapid (Figure 2A). Thus, after addition of the NE to the superfused ACSF the fEPSP began to decrease in amplitude within 2 min of reaching the brain slice. The reduction in the fEPSP amplitude was maximal in both pathways by 10 min (Figure 2A).
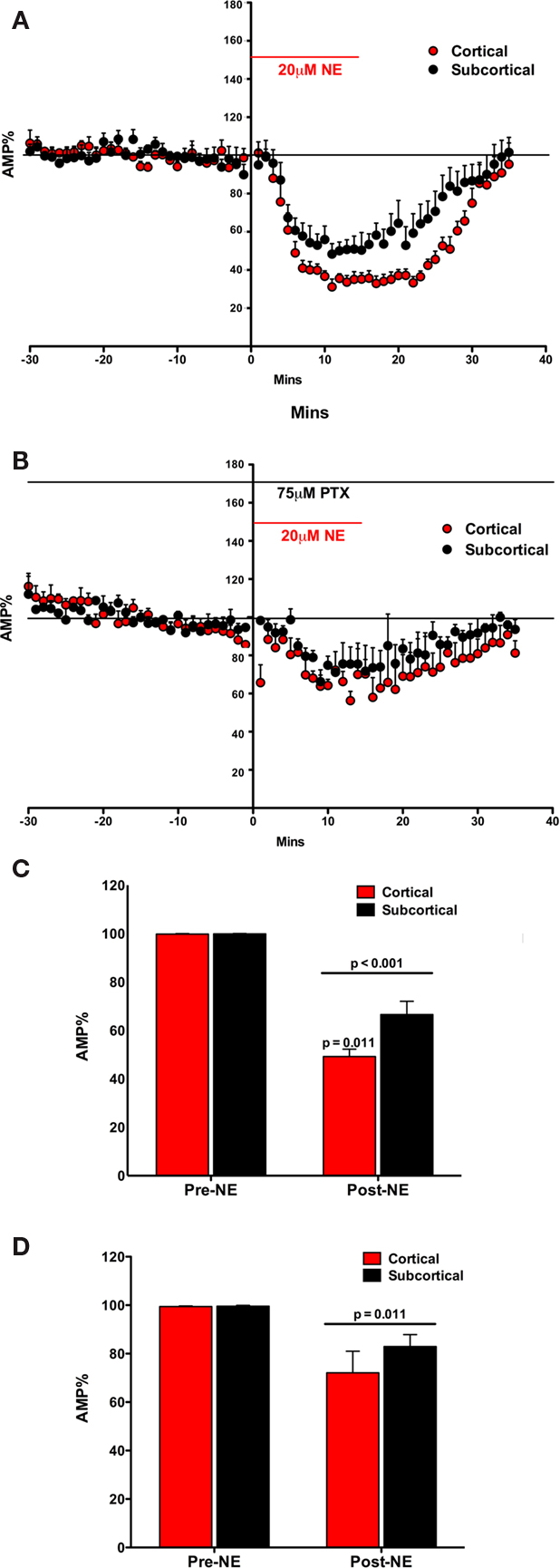
Figure 2. Norepinephrine decreased the amplitude of the fEPSP in both the Subcortical and the Cortical afferents to the LA. The effect is transient on both pathways. Both pathways show a post-NE rebound response (A). In the presence of PTX, NE decreases the amplitude of the fEPSP in both the subcortical and the cortical afferents to the LA. The effect is transient on both pathways. In contrast, a rebound effect is not observed (B). NE significantly decreased the amplitude of the fEPSP in both the subcortical and the cortical pathways. Moreover, NE significantly decreased the cortical pathway more than the subcortical pathway (C). In the presence of PTX, NE significantly decreased the amplitude of the fEPSP in both the subcortical and the cortical pathways (D). However, in contrast, there was no significant difference in NE induced reduction in amplitude between the cortical and the subcortical pathway.
The fEPSP amplitude (mean ± SEM%) was compared to normalized baseline (−30 to −1 min) during NE application (2–30 min). A significant main effect of NE on fEPSP amplitude was found suggesting that NE reduced fEPSP amplitude [F(1, 10) = 175.05; p < 0.001; Figure 2C]. In addition to the main effect, there was a significant interaction between NE and fEPSPs between cortical and subcortical pathways [F(1, 10) = 7.531; p = 0.021]. fEPSPs in the two pathways were not reduced equally. In comparing the amplitude of the fEPSP in response to NE application (2–30 min) the amount of reduction in the cortical and subcortical pathway was significantly different [difference between the means 24.83 ± 7.31; t(7) = 3.397, p < 0.011]. Thus, there is a difference in NE induced reduction in the fEPSP between the subcortical and cortical pathways to the LA.
The effects of NE on the fEPSP were rapidly reversed after wash out of NE. Within 15 min the fEPSP reduction mostly eliminated. However, a very small rebound depolarization lasting about 60 min was qualitatively observed (data not shown; Figure 2A). We hypothesized that this rebound effect may be due to activation of β subtypes of NE receptors (Huang et al., 2000). This was tested below. We tested the potential contribution of GABAA receptors to the differences in the pathways (Figures 2B,D).
NE Receptor Mediated Pathway Differences are Dependent on GABAA Receptors
The fEPSP measures glutamatergic postsynaptic excitability evoked by presynaptic stimulations. This fEPSP postsynaptic excitability is regulated by local GABA neurons (Rainnie et al., 1991; Johnson et al., 2008). Moreover, recent findings report the importance of GABAergic transmission in the NE regulation of LA plasticity (Tully et al., 2007). Thus in order to determine any potential contribution by GABA transmission in the greater inhibition of the cortical pathway than the subcortical pathway by NE, we next repeated the first experiment in the presence of the GABAA antagonist PTX (Figure 2B).
The fEPSP amplitude (mean ± SEM%) was compared to normalized baseline (−30 to −1 min) during NE application (2–30 min). A main effect of NE during PTX administration was found, causing a significant decrease in fEPSP amplitude [F(1, 4) = 19.63; p = 0.011; Figure 2D]. However, no interaction between NE application and pathway was found [F(1, 4) = 0.894; p = 0.398]. Moreover, the two pathways did not significantly differ from each other [t(4) = 0.985; p = 0.381] after NE application. In contrast to experiment one, in the absence of the GABAA antagonist, the inhibition of the two pathways by NE was not significantly different (Figure 2D). In comparing the amplitude of the fEPSP in response to NE application the amount of reduction in the cortical and subcortical pathway was not significantly different (difference between the means 10.11 ± 10.26%). Thus, the difference in NE induced reduction in the fEPSP between the subcortical and cortical pathways to the LA is dependent on intact GABAA receptors.
Synaptic Transmission is Potentiated More by a NE β Agonist (Isoproterenol) in the Cortical Pathway than the Subcortical Pathway
In the third experiment we directly compared the effects of the β adrenergic receptor agonist isoproterenol (ISO) on the fEPSP recorded in LA after stimulation of the cortical and subcortical pathways (Figures 3A,B). Prior to drug treatment, fEPSPs in both pathways were reduced to 50% of maximum and stable recordings established as above. ISO (15 μM) increased the amplitude of the evoked fEPSP in both pathways (Figure 3A). Like NE, the effect of ISO on the fEPSP was rapid. After the addition of the drug, the fEPSPs increased in amplitude within 2 min of reaching the brain slice. The increase in the fEPSP amplitude reach maximum in both pathways in less than 10 min (Figure 3A).
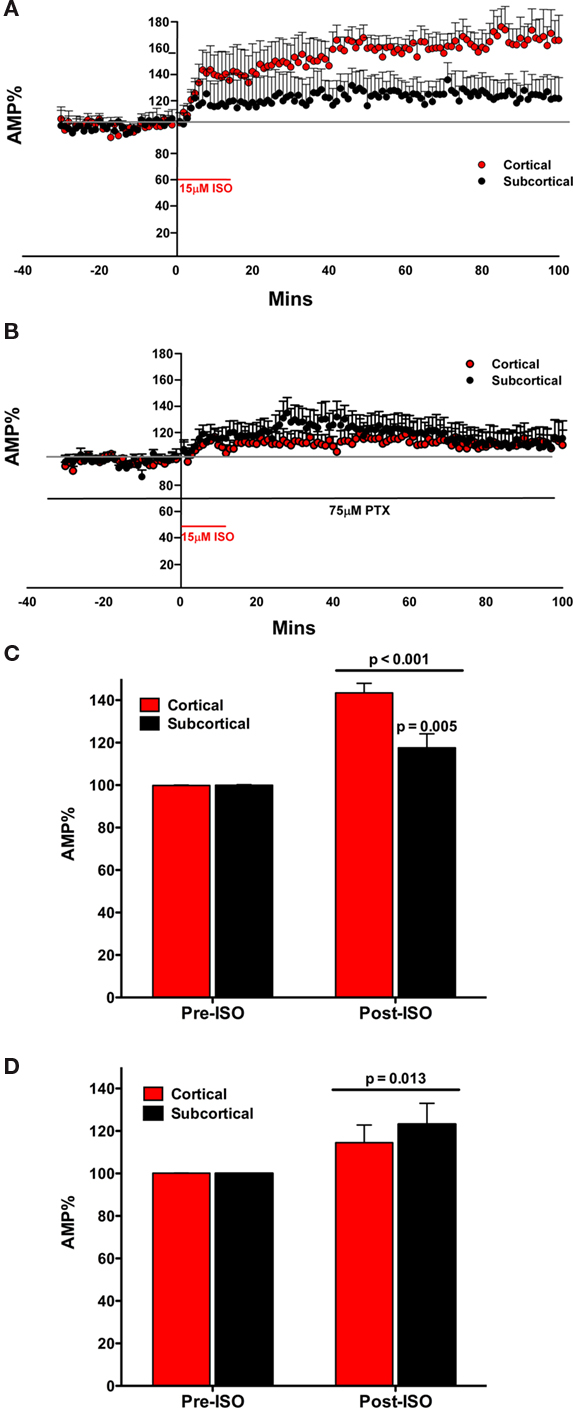
Figure 3. The β agonist isoproterenol (ISO) increased the amplitude of the fEPSP in both the subcortical and the cortical afferents to the LA. The effect is not transient on both pathways, inducing a long lasting response (A). In the presence of PTX, ISO increases the amplitude of the fEPSP in both the subcortical and the cortical afferents to the LA. The effect is not transient on both pathways, inducing a long lasting response (B). ISO significantly increases the amplitude of the fEPSP in both the subcortical and the cortical pathways. Moreover, ISO significantly increased the cortical pathway more than the subcortical pathway (C). In the presence of PTX, ISO significantly increased the amplitude of the fEPSP in both the subcortical and the cortical pathways (D). However, in contrast, there was no significant increase in ISO induced potentiation of amplitude between the cortical and the subcortical pathway.
We tested the effects of ISO on the fEPSP in both the cortical and subcortical pathways. ISO (15 μM) significantly increased the amplitude of the evoked fEPSP in both the subcortical and cortical pathways. This effect occurred within 10 min of application of ISO with the full effect occurring approximately 30 min after the addition of the ISO to the superfused ACSF. Moreover, the effect was very long lasting and remained for the duration of the recording period. Pathways were compared at a long latency. The fEPSP amplitude (mean ± SEM%) was compared to normalized baseline (−30 to −1 min) during ISO application (40–60 min). A significant main effect of ISO on fEPSP amplitude was determined [F(1, 18) = 59.724; p < 0.001; Figure 3C]. Moreover, a significant interaction between ISO application and fEPSP amplitude between cortical and subcortical pathways was found [F(1, 18) = 10.819; p = 0.004]. In comparing the amplitude of the fEPSP in response to ISO application for the two pathways (40–60 min) the increase in the cortical and subcortical pathway was significantly different [difference between means; 25.90 ± 8.01%; t(18) = 3.234, p = 0.005]. The fEPSP amplitude (mean ± SEM%) was compared to normalized baseline (−30 to −1 min) during ISO application (40–60 min). During application of ISO the fEPSPs of both the cortical [43.60 ± 4.487%; t(9) = 9.715; p = 0.042] and subcortical [17.6 ± 6.52%; t(9) = 2.695; p = 0.025] afferents significantly increased compared to the baseline (Figure 3D). These data indicate that the cortical pathway is more potentiated by ISO than the subcortical pathway.
In contrast to the rapid wash out of NE induced inhibition of the fEPSP the ISO effect did not wash out (Figure 3A). The increase in fEPSP remained throughout the recording. Moreover, the cortical path remained more affected than the subcortical pathway (Figure 3D). We next investigated potential mechanisms that may control the different magnitude of the NE and ISO induced changes in the fEPSP in the subcortical and cortical pathways to the LA.
β Receptor Mediated Pathway Differences are Dependent on GABAA Receptors
In the next experiment, as in experiment two, we sought to determine whether inhibitory networks acting via GABAA receptors contribute to the differential effects of ISO in the two input pathways to LA. We repeated experiment three, this time in the presence of the GABAA antagonist PTX (75 μM), when GABAA inhibition is blocked (Figures 3B,D).
We tested the effects of ISO on the fEPSP in both the cortical and subcortical pathways. ISO (15 μM) significantly increased the amplitude of the evoked fEPSP in both the subcortical and cortical pathways. The increase in amplitude of both pathways occurred rapidly. However, in contrast to experiment 3, the two pathways did not appear different (Figure 3B). A significant main effect on fEPSP amplitude was determined when ISO was applied in the presence of PTX [F(1, 12) = 8.580; p = 0.013] but no significant interaction was found between ISO application and fEPSP amplitude between cortical and subcortical pathways [F(1, 12) = 0.464; p = 0.509]. The fEPSP amplitude (mean ± SEM%) was compared to normalized baseline (−30 to −1 min) during ISO application (40–60 min). During application of ISO (in the presence of PTX) the fEPSPs of the subcortical [22.95 ± 8.48%; t(7) = 2.707; p = 0.030] but not the cortical [14.4 ± 8.28; t(6) = 1.741; p = 0.132] afferents increased compared to the baseline (Figure 3D). In comparing the amplitude of the fEPSP in response to ISO application for the two pathways (40–60 min), in the presence of the GABAA antagonist, the increase in the cortical and subcortical pathway was not significantly different [8.62 ± 12.88%; t(12) = 0.669; p = 0.516] from each other. Thus, the difference in ISO induced potentiation of the fEPSP between the subcortical and cortical pathways to the LA is dependent on intact GABAA receptors.
Comparison Across Experiments Confirms the Dependence on GABAA Receptors for the Differential Actions of NE and ISO in the Subcortical and Cortical Pathways
We directly compared the fEPSP responses across experimental groups in order to obtain a comparison of how GABAA affected NE and ISO responses in the cortical and subcortical pathways. To do this we compared the experiments with and without GABAA inhibition (blocked with PTX) as a single experiment and first asked if there was a main effect as a result of PTX being present. Second, we again compared across experiments and asked if there was a difference in the fEPSP between the same pathways with or without PTX. We examined whether inhibiting GABAA transmission altered the reduction of the cortical or subcortical pathways in the presence of NE (Figure 4A); and if inhibiting GABAA transmission altered the potentiation of the cortical or subcortical pathways in the presence of ISO (Figure 4B).
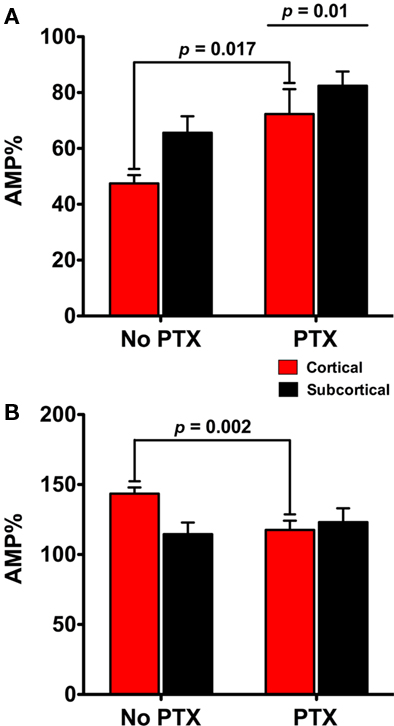
Figure 4. Comparison across experiments confirms the dependence on GABAA receptors for the differential actions of NE and the β agonist isoproterenol (ISO) in the subcortical and cortical pathways. Additional comparison of subcortical and cortical pathways across different experiments supports the finding that the cortical pathway is differently regulated by NE and ISO. The GABAA antagonist (PTX) reduces the reduction on the amplitude of fEPSPs in both the cortical and subcortical pathways in response to NE (A). Second, there was more reduction in the cortical pathway than the subcortical (A). In the presence of PTX the potentiation of the cortical pathway by ISO was reduced (B). Together these data suggest that inhibiting GABAA transmission contributes to a significant change in the cortical fEPSPs response to NE and ISO but not to the subcortical fEPSP.
We found an overall main effect of PTX on the amplitude of fEPSPs between cortical and subcortical pathways [F(1, 17) = 8.546; p = 0.01] in the presence of NE. fEPSPs in the cortical pathway were differently reduced in the presence of PTX [22.82 ± 7.32%; t(7) = 3.119, p = 0.017] but not in the subcortical pathway [16.37 ± 8.66%; t(7) = 1.89; p = 0.101; Figure 4A]. When ISO was applied we found that inhibiting GABAA transmission had no main effect on the increase of fEPSPs in the presence ISO [F(1, 33) = 1.62; p = 0.212]. However, PTX did contribute to a difference in cortical [27.11 ± 7.29%; t(15) = 3.721; p = 0.002] but not subcortical pathway response to ISO [6.87 ± 12.41%; t(15) = 0.554; p = 0.588]. Together these data suggest that inhibiting GABAA transmission contributes to a significant change in the cortical fEPSPs response to NE and ISO but not to the subcortical fEPSP (Figures 4A,B).
This direct comparison provides additional evidence for a difference between the pathways. In summary, when analyzing data both within and between experiments, we find that NE reduces and ISO potentiates the cortical pathway more than the subcortical pathway, and that this effect is dependent upon GABAA receptors. These data suggests a difference between the cortical and subcortical micro network in the LA.
Discussion
The amygdala has been extensively implicated in the neurobiology of conditioned fear (LeDoux, 2000; Sah et al., 2003; Pare et al., 2004). NE, especially its β receptors, has been implicated in formation, reconsolidation, and extinction of fear memories (Debiec and LeDoux, 2004; Berlau and Mcgaugh, 2006; Tully et al., 2007) and is known to be a mechanism of aspects of stress and arousal in the brain (Aston-Jones et al., 1996; Mcgaugh, 2000). The LA receives subcortical and cortical sensory afferents and both have been implicated in conditioned fear (Mckernan and Shinnick-Gallagher, 1997; Doyere et al., 2003; Shin et al., 2006). Here we asked whether NE mediates these effects at afferent input synapses, and if so, what mechanism is involved. We recorded evoked field potential (EFPs) following stimulation of subcortical and cortical afferents in vitro in order to screen for network effects. NE transiently inhibited EFPs in both pathways. However, inhibition was greater in the cortical path. In the presence of the GABAA antagonist PTX, inhibition by NE remained in both pathways but was reduced in amplitude. Importantly the difference in the effects of NE on the two pathways was eliminated, indicating that GABA networks contribute to the NE effects in LA and that NE also regulates the cortical evoked GABA network to a greater extent. Application of the β adrenergic agonist ISO potentiated EFPs in both pathways. ISO produced a greater potentiation of the cortical path. Importantly this difference was removed by PTX, suggesting that an additional potentiation may occur via a separate cortical pathway activated GABA network in LA.
Previous studies have shown that NE has a potent effect on synaptic transmission in amygdala. Consistent with our findings, Braga et al. (2004) found that NE reduces the amplitude of the fEPSP in the external capsule input to basolateral amygdala (BLA). Additionally they found that IPSC frequency was potentiated by NE (Braga et al., 2004). Consistent with an apparent reduction in excitatory transmission, Delaney et al. (2007) found that NE dramatically reduced EPSC amplitude in the parabrachial nucleus inputs to central amygdala pathway. The mechanism involved presynaptic α2 receptors, which inhibits transmitter release. Taken together, these studies in amygdala show multiple mechanisms by which NE can reduce local network excitability, acting at both glutamatergic and GABAergic transmitter systems (Figure 4). The response to NE and the response to the NE β receptor agonist (ISO) are different. NE results in an overall decrease in the field amplitude whereas ISO results in an increase. The NE response occurs only during the drug, whereas the response to ISO continues after the drug. This differential response between NE and the β receptor agonist has been previously described (for example see Huang et al., 2000). The implication from previous data is that NE has a short lasting (during drug) inhibitory effect, whereas the β agonist ISO has a long lasting (beyond drug) effect that is permissive on plasticity (Huang et al., 2000). The difference we observed between the subcortical and cortical pathways suggest that different networks of GABA neurons mediate NE regulation of the two pathways. We propose that an additional local GABA circuit is activated by the cortical pathway, which provides further inhibition to cortically derived sensory input during stress and arousal (Figure 5).
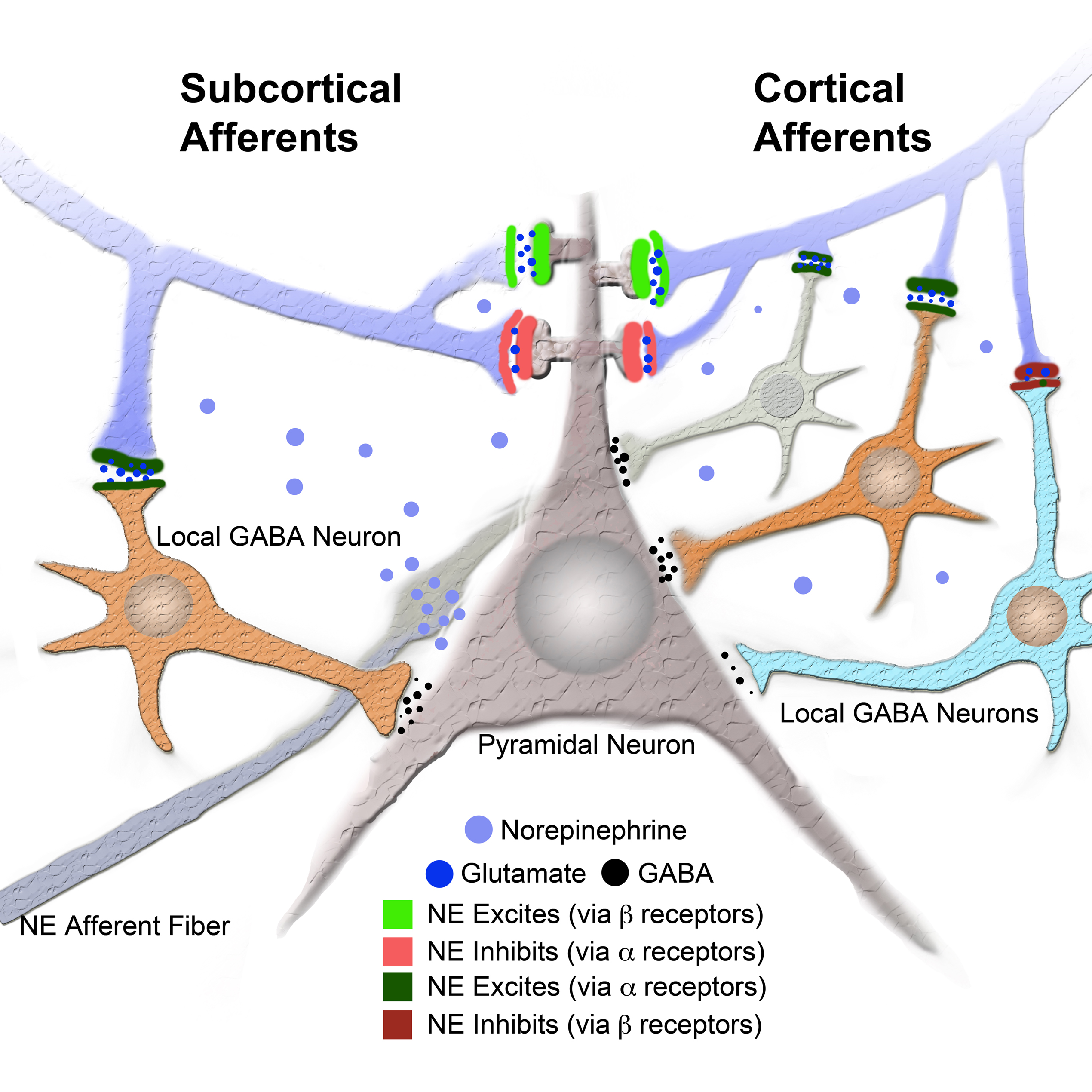
Figure 5. Model of intra LA circuit and its regulation by NE and its receptors. Model provides a network explanation for differential NE modulation of the subcortical and cortical afferents to the LA. Model shows interaction between NE and its receptors and local GABA neurons and LA principal neuron (see legend). Additional local GABA neurons, regulated by α and β receptors, postsynaptic to cortical afferents and not to subcortical afferents could explain the differences in regulation of afferent excitably by NE and β receptors.
In order to directly compare cortical and subcortical afferent responses to NE, we evoked extracellular field potentials in the LA in response to stimulation of both the pathways. These pathways have previously been recognized to contain the subcortical and cortical pathways, including the auditory medial geniculate and auditory TE3 cortical paths to the LA (LeDoux et al., 1990; Doron and LeDoux, 1999; Weisskopf and LeDoux, 1999; Doyere et al., 2003; Humeau et al., 2005; Shin et al., 2006; Johnson et al., 2009). We measured extracellular evoked fEPSP as previously described (Huang et al., 2000; Lamprecht et al., 2006). These pathways have previously been shown to be independent from each other with respect to independently induced monosynaptic plasticity (Weisskopf and LeDoux, 1997; Lamprecht et al., 2006). By using the fEPSP we were able to ascertain overall excitability of the LA network evoked by each of the pathways (Huang and Kandel, 1996; Collins and Pare, 2000; Huang et al., 2000; Shinnick-Gallagher et al., 2003).
In vivo the input to the LA via the cortex, arrives some 30 ms later to provide more sensory detail (Armony et al., 1995, 1998; Quirk et al., 1995; Li et al., 1996; LeDoux, 2000). Either of thalamo-LA or cortico-LA pathways is sufficient for conditioning to simple auditory stimuli. However the cortical pathway appears to be required for learning about more complex stimuli (LeDoux, 2000, 2007). One interpretation of the basis for these two anatomically distinct pathways is that the direct thalamic (subcortical) route to the amygdala can provide a fast, and possibly life saving, sensory signal to warn the amygdala of potential danger (LeDoux, 2000). The two sensory pathways both arrive in the LA, and both synapse onto LA principal neurons where they contact different types of dendritic spines (Humeau et al., 2005). While there is spatial convergence of these sensory inputs, there is also temporal divergence with the two inputs being separated in time. One possible mechanism that may allow for the two temporally segregated sensory inputs to converge in time as well as in space is a recurrent network in the LA. This network may allow for thalamo-LA signals to feedback to the superior parts of the LA during conditioning where they will meet incoming cortical signals (Johnson et al., 2008). A reduction in excitability in the NE pathway could reduce feedback within the LA and create a disconnect with the cortical pathway. These effects may explain why activation of β receptors alone appears to promote plasticity in the cortical pathway.
In studies of the subcortical to LA pathway, Tully et al. (2007; Tully and Bolshakov, 2010) found NE reduced IPSP amplitude. They found that NE increases network excitability, which could then promote the induction of LTP. The NE β antagonist propranolol has previously been shown to inhibit the maintenance of LTP in the cortical pathway (Huang et al., 2000). While NE alone, acting on all its receptors, resulted in a decrease in excitability, activation of β receptors alone resulted in a greater potentiation of the cortical pathway. These β receptor data strongly suggest that plasticity is favored in the cortical pathway compared to the subcortical pathway. This interpretation is consistent with the known, effects of β receptors on synaptic plasticity in the cortical pathway (Huang et al., 2000). Differences in plasticity between the cortical and subcortical pathways is an important question for future studies.
In conclusion, NE may regulate memory formation in the LA with both temporal and synapse precision. These results may indicate that the differential cortical and subcortical regulation helps protect mammals under times of stress. Activation of the subcortical “low road” (LeDoux, 2000) is hypothesized to allow rapid sensory access to the LA in order to initiate defensive amygdala dependent fear responses. These data show that while NE inhibits synaptic transmission in both pathways the balance between the two is shifted toward the subcortical low road. The cortical “high road” (LeDoux, 2000) is hypothesized to allow refined sensory input to the LA in order to better identify threatening stimuli. Our findings that activation of the NE β receptor potentiates the cortical pathway more than the subcortical pathway may allow increase plasticity and hence learning about dangerous stimuli at the cortical pathway under times of stress and arousal, while at the same time preventing learning less accurate non-cortically processed sensory information in the subcortical pathway. Further work on differential plasticity and regulation between the subcortical and cortical pathways is needed to confirm these ideas.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by R01 MH046516 P50 MH058911 K05 MH067048 R37 MH38774 (Joseph E. LeDoux). Luke R. Johnson held a NARSAD Young Investigator Award. We thank Anu Amin for help with a control experiment.
References
Armony, J. L., Quirk, G. J., and LeDoux, J. E. (1998). Differential effects of amygdala lesions on early and late plastic components of auditory cortex spike trains during fear conditioning. J. Neurosci. 18, 2592–2601.
Armony, J. L., Servan-Schreiber, D., Cohen, J. D., and LeDoux, J. E. (1995). An anatomically constrained neural network model of fear conditioning. Behav. Neurosci. 109, 246–257.
Aston-Jones, G., and Bloom, F. E. (1981). Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J. Neurosci. 1, 876–886.
Aston-Jones, G., Rajkowski, J., Kubiak, P., Valentino, R. J., and Shipley, M. T. (1996). Role of the locus coeruleus in emotional activation. Prog. Brain Res. 107, 379–402.
Berlau, D. J., and Mcgaugh, J. L. (2006). Enhancement of extinction memory consolidation: the role of the noradrenergic and GABAergic systems within the basolateral amygdala. Neurobiol. Learn. Mem. 86, 123–132.
Braga, M. F., Aroniadou-Anderjaska, V., Manion, S. T., Hough, C. J., and Li, H. (2004). Stress impairs alpha(1A) adrenoceptor-mediated noradrenergic facilitation of GABAergic transmission in the basolateral amygdala. Neuropsychopharmacology 29, 45–58.
Brunet, A., Orr, S. P., Tremblay, J., Robertson, K., Nader, K., and Pitman, R. K. (2008). Effect of post-retrieval propranolol on psychophysiologic responding during subsequent script-driven traumatic imagery in post-traumatic stress disorder. J. Psychiatr. Res. 42, 503–506.
Collins, D. R., and Pare, D. (2000). Differential fear conditioning induces reciprocal changes in the sensory responses of lateral amygdala neurons to the CS(+) and CS(−). Learn. Mem. 7, 97–103.
Debiec, J., and LeDoux, J. E. (2004). Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience 129, 267–272.
Debiec, J., and LeDoux, J. E. (2006). Noradrenergic signaling in the amygdala contributes to the reconsolidation of fear memory: treatment implications for PTSD. Ann. N. Y. Acad. Sci. 1071, 521–524.
Delaney, A. J., Crane, J. W., and Sah, P. (2007). Noradrenaline modulates transmission at a central synapse by a presynaptic mechanism. Neuron 56, 880–892.
Doron, N. N., and LeDoux, J. E. (1999). Organization of projections to the lateral amygdala from auditory and visual areas of the thalamus in the rat. J. Comp. Neurol. 412, 383–409.
Doyere, V., Schafe, G. E., Sigurdsson, T., and LeDoux, J. E. (2003). Long-term potentiation in freely moving rats reveals asymmetries in thalamic and cortical inputs to the lateral amygdala. Eur. J. Neurosci. 17, 2703–2715.
Huang, Y. Y., and Kandel, E. R. (1996). Modulation of both the early and the late phase of mossy fiber LTP by the activation of beta-adrenergic receptors. Neuron 16, 611–617.
Huang, Y. Y., Martin, K. C., and Kandel, E. R. (2000). Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J. Neurosci. 20, 6317–6325.
Humeau, Y., Herry, C., Kemp, N., Shaban, H., Fourcaudot, E., Bissiere, S., and Luthi, A. (2005). Dendritic spine heterogeneity determines afferent-specific Hebbian plasticity in the amygdala. Neuron 45, 119–131.
Johnson, L. R., Hou, M., Ponce-Alvarez, A., Gribelyuk, L. M., Alphs, H. H., Albert, L., Brown, B. L., LeDoux, J. E., and Doyere, V. (2008). A recurrent network in the lateral amygdala: a mechanism for coincidence detection. Front. Neural Circuits 2:3. doi: 10.3389/neuro.04.003.2008
Johnson, L. R., LeDoux, J. E., and Doyere, V. (2009). Hebbian reverberations in emotional memory micro circuits. Front. Neurosci. 3:2. doi: 10.3389/neuro.01.027.2009
Lamprecht, R., Margulies, D. S., Farb, C. R., Hou, M., Johnson, L. R., and LeDoux, J. E. (2006). Myosin light chain kinase regulates synaptic plasticity and fear learning in the lateral amygdala. Neuroscience 139, 821–829.
Lang, P. J., and Davis, M. (2006). Emotion, motivation, and the brain: reflex foundations in animal and human research. Prog. Brain Res. 156, 3–29.
LeDoux, J. E., Farb, C., and Ruggiero, D. A. (1990). Topographic organization of neurons in the acoustic thalamus that project to the amygdala. J. Neurosci. 10, 1043–1054.
Li, X. F., Stutzmann, G. E., and LeDoux, J. E. (1996). Convergent but temporally separated inputs to lateral amygdala neurons from the auditory thalamus and auditory cortex use different postsynaptic receptors: in vivo intracellular and extracellular recordings in fear conditioning pathways. Learn. Mem. 3, 229–242.
Mckernan, M. G., and Shinnick-Gallagher, P. (1997). Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390, 607–611.
Pare, D., Quirk, G. J., and LeDoux, J. E. (2004). New vistas on amygdala networks in conditioned fear. J. Neurophysiol. 92, 1–9.
Quirk, G. J., Repa, C., and LeDoux, J. E. (1995). Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: parallel recordings in the freely behaving rat. Neuron 15, 1029–1039.
Rainnie, D. G., Asprodini, E. K., and Shinnick-Gallagher, P. (1991). Inhibitory transmission in the basolateral amygdala. J. Neurophysiol. 66, 999–1009.
Romanski, L. M., Clugnet, M. C., Bordi, F., and LeDoux, J. E. (1993). Somatosensory and auditory convergence in the lateral nucleus of the amygdala. Behav. Neurosci. 107, 444–450.
Sah, P., Faber, E. S., Lopez De Armentia, M., and Power, J. (2003). The amygdaloid complex: anatomy and physiology. Physiol. Rev. 83, 803–834.
Shin, R. M., Tsvetkov, E., and Bolshakov, V. Y. (2006). Spatiotemporal asymmetry of associative synaptic plasticity in fear conditioning pathways. Neuron 52, 883–896.
Shinnick-Gallagher, P., Mckernan, M. G., Xie, J., and Zinebi, F. (2003). L-type voltage-gated calcium channels are involved in the in vivo and in vitro expression of fear conditioning. Ann. N. Y. Acad. Sci. 985, 135–149.
Tully, K., and Bolshakov, V. Y. (2010). Emotional enhancement of memory: how norepinephrine enables synaptic plasticity. Mol. Brain 3, 15.
Tully, K., Li, Y., Tsvetkov, E., and Bolshakov, V. Y. (2007). Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc. Natl. Acad. Sci. U.S.A. 104, 14146–14150.
Weisskopf, M. G., Bauer, E. P., and LeDoux, J. E. (1999). L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J. Neurosci. 19, 10512–10519.
Weisskopf, M. G., and LeDoux, J. E. (1997). In-vitro analysis of synaptic input to the dorsal subdivision of the lateral amygdala. Soc. Neurosci. Abstr. 23, 786.
Keywords: stress, PTSD, fear, fear conditioning, high road, low road
Citation: Johnson LR, Hou M, Prager EM and LeDoux JE (2011) Regulation of the fear network by mediators of stress: norepinephrine alters the balance between cortical and subcortical afferent excitation of the lateral amygdala. Front. Behav. Neurosci. 5:23. doi: 10.3389/fnbeh.2011.00023
Received: 27 May 2010;
Accepted: 18 April 2011;
Published online: 23 May 2011.
Edited by:
Carmen Sandi, École Polytechnique Fédérale de Lausanne, SwitzerlandReviewed by:
Constantine Pavlides, The Rockefeller University, USADavid M. Diamond, University of South Florida, USA
Copyright: © 2011 Johnson, Hou, Prager and LeDoux. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Luke R. Johnson, Departments of Psychiatry and Neuroscience, Uniformed Services University, Bethesda, MD 20814, USA. e-mail:bHVrZS5qb2huc29uQHVzdWhzLm1pbA==