 Carlos Sindreu and Daniel R. Storm*
Carlos Sindreu and Daniel R. Storm*- Department of Pharmacology, University of Washington, Seattle, WA, USA
The physiological role of synaptic zinc has remained largely enigmatic since its initial detection in hippocampal mossy fibers over 50 years ago. The past few years have witnessed a number of studies highlighting the ability of zinc ions to regulate ion channels and intracellular signaling pathways implicated in neuroplasticity, and others that shed some light on the elusive role of synaptic zinc in learning and memory. Recent behavioral studies using knock-out mice for the synapse-specific zinc transporter ZnT-3 indicate that vesicular zinc is required for the formation of memories dependent on the hippocampus and the amygdala, two brain centers that are prominently innervated by zinc-rich fibers. A common theme emerging from this research is the activity-dependent regulation of the Erk1/2 mitogen-activated-protein kinase pathway by synaptic zinc through diverse mechanisms in neurons. Here we discuss current knowledge on how synaptic zinc may play a role in cognition through its impact on neuronal signaling.
Initial Identification of Vesicular Zinc in Synaptic Terminals of the Brain
The first indication of the accumulation of zinc in nerve fibers came from Maske (1955). He found that intravital injection of the heavy metal chelator dithizone results in a band of bright red staining – corresponding to metal: dithizonate – in the mossy fiber region of the hippocampus. Subsequently, histochemical methods were developed (i.e., Timm’s stain) to precipitate metal cations in situ and visualize them by silver amplification (Timm, 1958; Danscher, 1981). When combined with electron microscopy, these methods revealed the precise localization of metal ion staining within synaptic vesicles of the telencephalon (Figure 1A; Haug, 1967; Perez-Clausell and Danscher, 1985). Suggesting the involvement of zinc in the vesicular staining, autoradiographic and analytical studies had shown high levels of zinc in the mossy fiber region (Dencker and Tjalve, 1979; Frederickson et al., 1983). Indeed, proton-induced X-ray emission spectroscopy identified zinc as the main ion detected by the above histochemical techniques in brain samples (Danscher et al., 1985). More recently, membrane-permeable fluorophores with good selectivity for zinc ions (such as TSQ, ZP-1, ZnAF-2, or ZnIC, to name a few) have confirmed the abundance of zinc in synaptic terminal fields (Figure 1B; Frederickson et al., 1987; Ueno et al., 2002; Woodroofe et al., 2004; Komatsu et al., 2007).
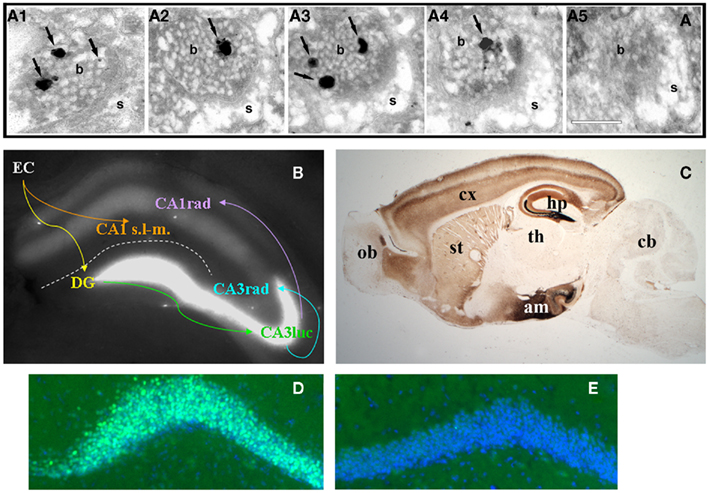
Figure 1. (A)Consecutive serial sections (A1–5) of an axospinous synapse in CA1 stratum radiatum stained for vesicular zinc using the Neo-Timm method and observed by electron microscopy. Arrows point to silver granules of varying size detecting reactive zinc in vesicles. Note the presence of zinc staining in four of five profiles. b, bouton; s, spine. Scale bar, 200 nm. (B) Zinpyr-1 fluorescence in a fresh hippocampal slice with major hippocampal connections indicated by arrows. Reactive zinc is highest in stratum lucidum (luc) of CA3, moderate in stratum radiatum (rad), and absent in stratum lacunosum-moleculare (s.l-m.) of CA1. (C) Bright field image of a sagittal brain section from a 5 month-old C57/BL6J mouse stained with the Neo-Timm method. ob, olfactory bulb; cx, cortex; st, striatum; th, thalamus; hp, hippocampus; am; amygdala; cb, cerebellum. (D,E) Beta-galactosidase immunofluorescence (green) in dentate gyrus granule cells of a ZnT-3 KO (D) and control (E) mouse, counterstained with Hoescht (blue). C. Sindreu, unpublished observations.
Pre-synaptic zinc is observed in all the vertebrates that have been examined (Frederickson, 1989). Also common between species is the remarkable neuroanatomical distribution of pre-synaptic zinc, which is greatly restricted to cortical and limbic regions of the brain in a distinct laminar pattern (Figure 1C). Vesicular zinc is also present in boutons of the olfactory bulb and spinal cord (Friedman and Price, 1984; Birinyi et al., 2001; Danscher et al., 2001). In contrast with the distribution of vesicular zinc, absolute zinc levels are relatively even across the gray matter (60–80 ppm; Frederickson et al., 1983). The apparent disparity between absolute and pre-synaptic zinc levels stems from the fact that about 80% of total zinc in brain is not amenable to histochemical detection (Cole et al., 1999). This “invisible” pool of zinc is found in all six classes of enzymes and in intracellular receptors, where it is normally coordinated by four to six ligands, frequently supplied by histidine and cysteine, in conserved binding motifs (Vallee and Falchuk, 1993; Auld, 2001).
Zinc uptake into synaptic vesicles requires the zinc transporter protein ZnT-3, which is exclusively sorted to clear synaptic vesicles (Palmiter et al., 1996; Cole et al., 1999). Accordingly, zinc staining is excluded from dense core vesicles in pre-synaptic boutons (Perez-Clausell and Danscher, 1985). Zinc staining of synaptic vesicles is evenly distributed at various distances from the active zone (Lavoie et al., 2007), suggesting that both ready-releasable and reserve vesicle pools may contain vesicular zinc. In keeping with this, all synaptic vesicles appear to stain for ZnT-3 in labeled boutons (Wenzel et al., 1997). The fact that only a fraction of these vesicles stains for zinc may reflect a sensitivity issue or a requirement for additional proteins to uptake zinc into vesicles (see next section). In the telencephalon, all boutons stocked with vesicular zinc (i.e., zincergic) establish asymmetric synapses that typically involve dendritic spines (Perez-Clausell and Danscher, 1985). As predicted from their ultrastructure, these zincergic boutons are glutamatergic (Martinez-Guijarro et al., 1991; Beaulieu et al., 1992; Sindreu et al., 2003) and are apposed to post-synaptic membranes enriched in glutamate receptor subunits (Sindreu et al., 2003). In contrast, zincergic boutons in the spinal cord often form symmetric synapses immunoreactive to γ-aminobutyric acid (Danscher et al., 2001). The zincergic MF terminals of the hippocampus represent a special case, as they are both glutamatergic and GABAergic (Ruiz et al., 2004). A common observation in electron microscopy studies is that only a fraction of excitatory synapses show vesicular zinc staining. Studies quantifying the incidence of vesicular zinc or ZnT-3 among axospinous synapses are scarce, but measurements in the CA1 region suggest that about half of Schaffer-collateral synapses may be zincergic (Sindreu et al., 2003). This raises the question as to whether zincergic boutons arise from a subset of glutamatergic neurons or instead only a subset of boutons from any given neuron may be zincergic. Retrograde tracing studies strongly support the former possibility, as parent neurons of zincergic boutons constitute a subpopulation of neurons in the brain, and they are often interspersed with non-zincergic neurons of similar morphology (Slomianka et al., 1990, 1997; Slomianka, 1992; Brown and Dyck, 2004; Cunningham et al., 2007). The hippocampal MFs are a notable exception to this rule: all giant terminals are zincergic and hence all mature dentate gyrus granule cells may give rise to zincergic terminals. This can be appreciated by the expression of a β-galactosidase reporter in virtually all granule cells when driven by the ZnT-3 promoter (Figures 1D,E). Double-tracing studies have further confirmed that the vast majority of zincergic boutons emanate from pyramidal cells in the telencephalon (Miro-Bernie et al., 2003). Interestingly, neuronal projections from or to subcortical structures are largely devoid of pre-synaptic zinc. Instead, zincergic projections selectively (and often reciprocally) inter-connect cortical and limbic structures (Mandava et al., 1993; Long et al., 1995; Christensen and Frederickson, 1998; Casanovas-Aguilar et al., 2002). This suggests that the zincergic connectome represents a subnetwork of intrinsic projections embedded in the general cortical system.
From a cognitive perspective, the abundance of vesicular zinc in synapses of the cortex, hippocampus, and amygdala suggests a possible role in learning and memory. Indeed, these three structures have been strongly implicated in memory formation, storage, and retrieval (Martin et al., 2000). Because activity-dependent changes in synaptic strength are widely assumed to support memory (Neves et al., 2008), several studies have also examined the effects of zinc on synaptic ion channels, receptors, and plasticity. Comparatively less is known as to the role that zinc plays on neuronal signal transduction, which controls the expression of long-term synaptic plasticity and underlies memory consolidation. At any level, a full understanding of the role that synaptic zinc plays in learning and memory requires identification of the different zinc reservoirs and mechanisms of zinc homeostasis operating at synapses.
Homeostasis of Synaptic Zinc
Several lines of evidence indicate the existence of multiple transmembrane zinc gradients among the main constituents the synapse (Figure 2).
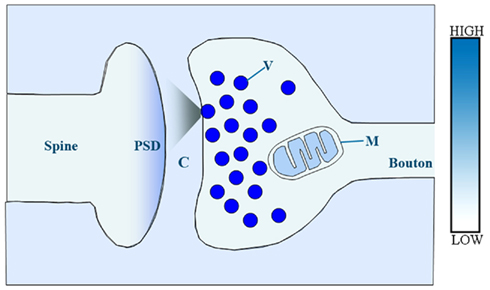
Figure 2. Schematic illustration of presumed relative concentrations of reactive zinc in a central glutamatergic synapse. In this model, relative zinc levels are orientatively coded by color intensity. V, synaptic vesicle; M, mitochondrion; C, synaptic cleft; PSD, post-synaptic density.
Plasma Membrane
Using a carbonic anhydrase-based biosensor, the concentration of loosely bound or free zinc (i.e., reactive zinc) was estimated to be 5–20 nM in the extracellular brain fluid (Frederickson et al., 2006b). Given that total zinc in the cerebrospinal fluid is about 150 nM (Hershey et al., 1983), this value suggests that most extracellular zinc also is tightly bound. Establishing the concentration of cytosolic reactive zinc in neurons has been hampered by the technical limitations of using fluorescent indicators for intracellular zinc measurements, as discussed elsewhere (Nolan and Lippard, 2009; Vinkenborg et al., 2010). Attempts in cortical neuron cultures using the dye mag-fura-5 suggested that cytosolic zinc may be found at subnanomolar levels (Sensi et al., 1997). Similar estimates have been obtained in non-neuronal cells with different indicators: ∼0.6 nM Zn2+ in HT293 cells by using FluoZin-3 (Krezel and Maret, 2006), and ∼0.4 nM Zn2+ in INS-1(832/13) β-cells by using eCALWYs FRET-based sensors (Vinkenborg et al., 2009). Together, the data suggest that the concentration of reactive zinc may be an order of magnitude lower in the cytosol compared with the extracellular brain space. This is noteworthy in view of the fact that the total intracellular zinc concentration largely exceeds that in the extracellular space (∼150 μM; Ehmann et al., 1984). Thus, reactive zinc in the cytosol may represent less than 0.001% of total cellular zinc. Attesting to the importance of controlling the levels of cytosolic zinc, prolonged increases or reductions in intra- or extracellular zinc can compromise neuronal viability (Canzoniero et al., 1999; Bozym et al., 2010).
How circulating zinc, previously absorbed in the gastrointestinal tract, enters the brain or CSF is poorly understood, although it appears to involve active transport mechanisms (Takeda, 2000). Likewise, the homeostatic mechanisms maintaining the zinc gradient across the plasma membrane in neurons are poorly defined, and currently none of the candidate proteins have been directly localized to synapses. The principal mechanism for zinc extrusion in neurons appears to be a secondary active transporter dependent on the Na+ and Ca2+ electrochemical gradients (Ohana et al., 2004; Qin et al., 2008), but the identity of this exchanger remains elusive. Given that neuronal activity elicits voltage-gated ion fluxes across the plasma membrane, such a zinc extrusion mechanism could potentially be subject to activity-dependent regulation. The zinc transporter ZnT-1 of the Slc30a family was the first plasma membrane protein found to regulate zinc extrusion (Palmiter and Findley, 1995). ZnT-1 is expressed in neurons and glia in several regions of the brain, including some that lack vesicular zinc (Sekler et al., 2002), and its genetic deletion is embryonic lethal (Andrews et al., 2004). Knock-down of ZnT-1 in cultured neurons decreases the rate of zinc efflux (Qin et al., 2009), but it is unclear if ZnT-1 affects the resting level of cytosolic zinc in the presence of an inwardly directed zinc gradient. ZnT-1-dependent zinc efflux is not affected by extracellular cations or ATP depletion, and it may involve oligomerization or association with other proteins (Palmiter and Findley, 1995). In addition, ZnT-1 suppresses the influx of other divalent cations via voltage-gated calcium channels (Ohana et al., 2006; Levy et al., 2009), suggesting additional roles besides zinc transport.
On the other hand, the ZIP-1 and -3 members of the Slc39a protein family have been implicated in the tonic import of extracellular zinc into neurons. ZIP-1 and -3 are highly expressed in CA1 hippocampus, and pyramidal neurons from double-knock-out mice show ∼ 50% reduction in the amount of extracellular zinc uptake (Qian et al., 2011). Assuming that zinc extrusion mechanisms are intact in this mutant mouse, the data indicate a major contribution of ZIP-1 and -3 to neuronal zinc import. The mechanism of zinc transport by ZIP proteins is not fully understood, although it was found to be energy-independent (Gaither and Eide, 2000). Evidence from the bacterial homolog ZIPB indicates that ZIP proteins may act as non-saturable zinc channels allowing passive zinc entry down its concentration gradient (Lin et al., 2010). ZIPB-dependent zinc entry increases with the proton concentration, in keeping with the effect of intracellular pH on neuronal zinc import (Colvin, 2002). Intriguingly, earlier studies found that uptake of Zn65 into the neuropil was energy-dependent (Howell et al., 1984), implying that alternative or additional mechanisms may import zinc at synapses.
There is also an interest in the role that the small Zn/Cu-binding protein metallothionein-3 (i.e., MT-3) may play on cytosolic zinc homeostasis. MT-3 has many remarkable biochemical properties with regards to the binding and release of zinc (Maret, 2011), and MT-3 over-expression confers resistance to zinc toxicity in vitro (Palmiter, 1995). An elegant study showed that MT-3 may confer neuroprotection from excitotoxicity by releasing zinc following phosphorylation by PKC, leading to downstream gene expression (Aras et al., 2009). However, MT-3 KO mice thrive under normal conditions and seem to respond normally to systemic zinc overload (Erickson et al., 1997). Hence, the physiological role of neuronal MT-3 in zinc homeostasis is unclear. A safety mechanism that may help maintain the zinc gradient across the plasma membrane is the sequestration of excess zinc into lysosomes. Thus, in CA3 neurons, kainate-induced seizures trigger the accumulation of zinc into lysosomes (Hwang et al., 2008) as well as the up-regulation of the zinc importer ZIP-4 (Emmetsberger et al., 2010). Suggesting a possible link between the two, endocytosis inhibitors prevent the lysosomal degradation of surface ZIP-4 that occurs during zinc overload (Mao et al., 2007).
Post-Synaptic Density
Biochemically isolated post-synaptic densities (PSD) contain a high amount of zinc (∼4 nmol/mg protein) that supports the structural organization of the PSD (Jan et al., 2002; Baron et al., 2006). Recent data indicate that at least a fraction of this zinc might be reactive or exchangeable, as it can be detected in association with Shank2/3 scaffold protein aggregates in dendritic spines with fluorescent zinc indicators (Grabrucker et al., 2011). In line with this, the same study also showed that zinc chelators can quickly alter the ultrastructural appearance of the PSD in cultured neurons.
Synaptic Vesicles
Direct measurements of the concentration of reactive zinc in synaptic vesicles are lacking, but simple calculations indicate that it could easily reach high micromolar levels. A single zinc atom would result in a concentration of ∼40 μM within a 0.64 × 10−5 μm3 synaptic vesicle, which is the average vesicle volume in CA1 boutons (Schikorski and Stevens, 1997). The Neo-Timm’s staining method has been claimed to be able to detect as little as 3–4 zinc atoms (Danscher and Stoltenberg, 2006), which would translate into a minimal concentration of ≥150 μM vesicular zinc. Along these lines, a concentration of ∼1 mM vesicular zinc was inferred by extrapolating from the amount of total zinc found in the hilar region of the hippocampus (Frederickson et al., 1983). On balance, it appears that reactive zinc is several-fold more concentrated within synaptic vesicles than in the cytosol or the extracellular space. The different concentrations likely account for the preferential labeling of synaptic vesicles with zinc probes. Intra-vesicular pH may also affect zinc speciation, releasing it from protein complexes therein and making it more reactive to zinc probes (Vinkenborg et al., 2009). By comparison, the concentration of glutamate in synaptic vesicles ranges from 60 to 210 mM (Clements, 1996). Interestingly, studies in PC12 cells indicate a functional coupling between the vesicular uptake of zinc and glutamate; recombinant murine VGLUT-1 and ZnT-3 were targeted to the same population of synaptic-like microvesicles and reciprocally facilitated their transport activities, perhaps through effects on the transvesicular electrochemical gradient (Salazar et al., 2005). In keeping with current estimates of the cytosolic zinc concentration, ZnT-3 enhanced VGLUT-1-mediated glutamate uptake at low nanomolar zinc levels. Whether ZnT-3 and VGLUT-1 cooperate in a similar manner in synaptic vesicles of the brain has not been examined. However, destroying the vesicular proton gradient with the H+-ATPase inhibitor concanamycin A, which prevents glutamate uptake, also reduces pre-synaptic fluorescence of the zinc indicator Zinpyr-1 at putative single MFs (Ketterman and Li, 2008). Intriguingly, functional differences between human and mouse ZnT-3 have been reported. Thus, human, but not mouse, ZnT-3 tends to form dimmers that promote vesicular targeting and stimulate zinc uptake (Salazar et al., 2009).
The concentration of zinc in synaptic vesicles has led to the natural hypothesis that zinc may be co-released with glutamate into the synaptic cleft following vesicular exocytosis. Evidence that neuronal depolarization increases the extracellular level of elemental zinc, either in vivo or in vitro, and in parallel with glutamate, is overwhelming (Howell et al., 1984; Charton et al., 1985; Aniksztejn et al., 1987; Minami et al., 2002). However, no study has yet confirmed the synaptic vesicle origin of this zinc by performing similar experiments in ZnT-3 knock-out mice (which lack any reactive zinc within synaptic vesicles; Cole et al., 1999; Linkous et al., 2008). Other studies have addressed the issue of vesicular zinc release by stimulating brain slices in the presence of fluorescent zinc indicators. Thus, action potential-evoked zinc exocytosis has been reported in CA3 and CA1 synapses by detecting changes in FluoZin-3 fluorescence, an extracellular zinc dye (Qian and Noebels, 2005, 2006; Carter et al., 2011). Importantly, zinc release was found to be highly sensitive to changes in release probability, dependent on vesicular exocytosis, and it was ablated in ZnT-3 KO mice. These are remarkable observations considering the potential limitations of the approach (e.g., ex vivo zinc depletion, inadequate metal contamination or temperature, small fractional volume of the synaptic cleft, optical sensitivity, perturbation of zinc kinetics by the indicator, etc.). Indeed, mixed results have been obtained when using maximal stimulation to compensate for low optical resolution (Li et al., 2001b; Kay, 2003; Datki et al., 2007). The extracellular zinc increase associated with exocytosis should be paralleled by a corresponding decrease in vesicular zinc, and there are some zinc fluorescence measurements from MFs suggesting that this is also the case (Ketterman and Li, 2008). The results suggest a model where released zinc may diffuse into the synaptic cleft, transiently increasing its extracellular concentration in an activity-dependent manner. In keeping with this, vesicular zinc release has been shown to modulate post-synaptic receptors at the MF synapse (Vogt et al., 2000; Pan et al., 2011, see section below). Even more strikingly, intracellular zinc increases in post-synaptic neurons were recently reported following intense axonal depolarization (Suh, 2009; Carter et al., 2011). Again, ZnT-3 deletion abolished the post-synaptic zinc transient, suggesting that vesicular zinc can be trans-synaptically mobilized under some conditions. One intriguing observation from these experiments is the slow decay of the extracellular zinc transient that is triggered by exocytosis. Whether this is a property of individual synapses is still unknown, as zinc transients were not induced with minimal stimulation protocols to activate single MF boutons. The factors governing the synaptic diffusion and clearance of released zinc are not understood. It has been proposed that vesicular zinc may remain bound to synaptic proteins after exocytosis, although the putative synaptic transporters or proteins that may act as fixed buffers are undefined (Vogt et al., 2000; Kay, 2003). It has also been proposed that the high tortuosity of the MF synapse may delay the clearance of zinc (Qian and Noebels, 2005). Direct comparisons of the fluorescence kinetics shown by extracellular indicators for zinc and glutamate (Hires et al., 2008) after release have not been reported.
Mitochondria
Another potential source of synaptic zinc is that originating from mitochondria. Virtually all MF terminals and about half of small pre-synaptic boutons contain at least one mitochondrion (Ventura and Harris, 1999; Chang et al., 2006), where it can modulate synaptic plasticity by handling Ca2+ and supplying ATP (Cai et al., 2011). Recent evidence in neurons indicates that resting mitochondria may harbor two to threefold higher concentrations of reactive zinc relative to the cytosol (Dittmer et al., 2009). Zinc appears to access the mitochondrial matrix mainly via the Ca2+ uniporter, a process dependent on the mitochondrial membrane potential (Malaiyandi et al., 2005; Caporale et al., 2009). The mechanism whereby mitochondrial zinc is extruded is unknown, as no zinc transporters have been localized to mitochondria. In vitro data suggest that large intracellular Ca2+ increases, often associated with neuronal injury, may possibly couple to mitochondrial zinc release into the cytosol (Sensi et al., 2003; Dineley et al., 2008; Dittmer et al., 2009).
Synaptic Zinc and Memory
The subsynaptic distribution of reactive zinc suggests three possible modes whereby synaptic zinc could affect cognitive processes: (i) tonic binding of zinc to extracellular protein domains with nanomolar affinity; (ii) phasic modulation of surface proteins following exocytosis of vesicular zinc; (iii) tonic or phasic modulation of cytosolic proteins in pre- or post-synaptic compartments.
One way to test the cognitive role of synaptic zinc has been to lower its physiological concentration with the use of zinc chelators. As such, local zinc chelation allows assessing the net effect of zinc at the expense of not differentiating between potentially overlapping facilitatory and inhibitory effects (Timofeeva and Nadler, 2006), or between the synaptic and extra-synaptic actions of zinc. A number of studies have found zinc chelators to modify hippocampus- or amygdala-dependent behaviors. Thus, intra-hippocampal infusion of the membrane-permeable chelator diethyldithiocarbamate (DEDTC) was shown to impair a delayed matching-to-place version of the water maze task (Frederickson et al., 1990). In this task, rats must remember the last location of a hidden platform submerged in a pool of opaque water. The platform location remains constant between two consecutive runs in a trial, whereas it is changed to a different location between trials separated by 10 min. The shorter latency to find the platform on the second run of a given trial (i.e., savings) indicates a memory for the last location (Steele and Morris, 1999). Because the animal has to update its memory in every trial, a high cognitive load is necessary to perform the task. DEDTC injection (i.e., spreading across all hippocampal subregions) shortly before training selectively suppresses the savings in time normally observed on the second run. Moreover, the memory impairment is transitory and parallels the time course of vesicular zinc chelation, arguing against the possibility of hippocampal damage from the injection. Another study assessed the effect of intra-hippocampal DEDTC injections on the standard version of the Morris water maze, which measures incremental, spatial reference memory (Lassalle et al., 2000). DEDTC during training prevents mice from learning the location of a fixed hidden platform, as they show no spatial bias during the probe test. In contrast, post-training DEDTC injections do not affect memory consolidation or recall. Interestingly, these two studies found that injection of the cell-impermeable zinc chelator CaEDTA fails to replicate the effects of DEDTC. Although initially interpreted as a lack of effect of extracellular zinc on spatial memory, subsequent reports suggested that the high concentrations of CaEDTA that were used (i.e., 200 mM) can also deplete intra-vesicular zinc, perhaps by creating an outward gradient (Frederickson et al., 2002). Hence, the basis for the memory impairment induced by DEDTC, but not CaEDTA, remains unclear. On the other hand, both DEDTC and CaEDTA were found to impair contextual fear conditioning (Daumas et al., 2004). This rapid, one-trial form of associative learning allows better temporal separation between the different phases of memory. Acute zinc chelation impairs the consolidation of contextual memory, but not its recall once the memory has been established. Of note, hippocampal zinc chelation in the same mice does not impair acoustic conditioning, which does not depend on the hippocampus. Together, these studies suggest that reactive zinc, perhaps of synaptic origin, is required for the acquisition or consolidation of hippocampus-dependent memory, depending on the task involved.
A recent study analyzed the role of reactive zinc in the rat amygdala (Takeda et al., 2010). Continuous perfusion of the amygdala with 1 mM CaEDTA during fear conditioning or during recall was found to increase, rather than decrease, the expression of memory. It should be noted that the intense conditioning protocol used here (i.e., >100 foot shocks over 1 h) is more akin to inescapable shock stress models of depression than to associative learning paradigms. Interestingly, this low concentration of CaEDTA elevates the basal extracellular levels of zinc and glutamate by two to fourfold, as well as the probability of vesicular release, suggesting that its potentiating effect may be due to disinhibition of amygdala synapses. The notion that zinc could indirectly modulate memory through effects on excitability is consistent with the predominantly anti-convulsant effects of zinc (Mitchell and Barnes, 1993; Dominguez et al., 2003).
Dietary zinc deficiency has also been used to assess the role of brain zinc in memory. A number of reports have demonstrated behavioral alterations in animals fed a zinc-restricted diet for several weeks, including increased anxiety or depression, and impaired spatial memory and extinction learning (Keller et al., 2000; Takeda et al., 2007; Tassabehji et al., 2008; Whittle et al., 2010). One caveat with dietary zinc deficiency models is that they can lead to multisystemic dysfunction, particularly in peripheral tissues with rapid turnover, and compromise many zinc-dependent reactions (Vallee and Falchuk, 1993). In the hippocampus, zinc deficiency has been shown to reduce the total number of synaptic vesicles in boutons (Lu et al., 2000). Hence, the extent to which behavioral changes associated with zinc deficiency are directly attributable to a reduction in synaptic zinc or even to reactive zinc in neurons is controversial.
To circumvent the lack of subcellular specificity attained by zinc chelation or dietary zinc deficiency, recent studies have analyzed the behavioral phenotype of ZnT-3 mutant (KO) mice. These mice are anatomically normal, but show no zinc staining or enrichment of total zinc in synaptic terminals (Cole et al., 1999; Linkous et al., 2008). In addition, total zinc levels in brain regions normally devoid of vesicular zinc, or seizure-induced increases in somatic (i.e., extra-synaptic) zinc are intact in ZnT-3 KO mice (Lee et al., 2000), indicating that zinc depletion is restricted to synaptic vesicles. These data are consistent with the selective expression of ZnT-3 in synaptic vesicles (Wenzel et al., 1997), and imply that extra-synaptic zinc pools can be regulated independently of vesicular zinc. Behaviorally, ZnT-3 KO mice are characterized by normal sensorimotor function and open field activity (Cole et al., 2001). Anxiety and depressive-like behaviors are also normal in the ZnT-3 KO mice. Interestingly, most of the cognitive deficits identified in ZnT-3 KO mice have been observed in tasks that require disambiguation, detailed attention or reverse learning. Thus, ZnT3 KO mice show normal fear conditioning after repeated delivery of tone-shock pairs (Cole et al., 2001), but display reduced memory when the tones are made discontinuous or are explicitly unpaired with the shocks (Martel et al., 2010, 2011). One possible explanation for this result is that the latter paradigms are thought to recruit cerebrocortical areas (i.e., zincergic), whereas simple cued conditioning is largely supported by thalamo-amygdala synapses devoid of vesicular zinc (Kodirov et al., 2006). Spatial reference memory following intense training in the Morris water maze task is also preserved in ZnT-3 KO mice (Cole et al., 2001; Martel et al., 2011). However, mutants show a transient learning delay in the reversal phase (i.e., when the platform was relocated to the opposite quadrant). Another study found that ZnT-3 KO mice have a profound deficit in a T-maze non-matching-to-place working memory task, in which mice are rewarded for alternating in quick succession in a two-choice maze (Sindreu et al., 2011). Spatial working memory has been shown to be highly sensitive to hippocampal dysfunction (Deacon and Rawlins, 2006). Because the hippocampus also is indispensable for the formation of detailed contextual memories (Wiltgen et al., 2010), the same group assessed the performance of ZnT-3 KO mice in a contextual discrimination task. Strikingly, ZnT-3 KO mice are unable to differentiate between the conditioning context and a similar context where they have never been shocked (Sindreu et al., 2011). The fact that ZnT-3 KO mice display normal levels of contextual memory in non-discriminative conditioning rules out an impairment of performance that prevented the expression of otherwise intact memory. Moreover, the contextual discrimination deficit in ZnT-3 KO mice can be reproduced by injecting adult wild type mice with the zinc chelator TPEN into the MF-rich CA3 subregion. This latter result points to an acute effect of vesicular zinc in CA3 that was not compensated for in ZnT-3 KO mice. A marginal deficit in social discrimination, which is another form of hippocampus-dependent memory, has also been reported in ZnT-3 KO mice (Martel et al., 2011). Together, the evidence suggests that vesicular zinc is required for some forms of hippocampus- and amygdala-dependent memories, but it does not affect normal general performance.
The neuronal circuits where ZnT-3 is acting to support these different forms of memory are poorly defined. The contribution of specific synaptic pathways to learning and memory is just starting to emerge, and such understanding will be instrumental to fully account for the complex mnemonic phenotype of ZnT-3 KO mice. A useful example comes from the effect of genetically inactivating CA3 neurons (Nakashiba et al., 2008). Although Schaffer-collateral inputs to CA1 are greatly silenced, mice are still capable of acquiring spatial reference memory in the Morris water maze task. This finding not only implies that direct cortical projections to distal CA1 can support incremental learning, but it may also explain why ZnT-3 KO mice perform this task normally, as the entorhinal projection to CA1 is entirely non-zincergic (Figure 1). In contrast, the deficits in contextual discrimination and complex tone conditioning in ZnT-3 KO mice may reflect the recruitment of zincergic mossy fibers and cortico-amygdaloid projections during performance of these tasks, respectively.
Another lingering question is why zinc chelators appear to cause stronger behavioral deficits than the genetic removal of vesicular zinc. Given the absence of reactive zinc in vesicles of ZnT-3 KO mice, molecular redundancy in vesicular zinc transport seems unlikely. As discussed earlier, reactive zinc also may be found in other subsynaptic compartments. One possibility, then, is that zinc chelators may remove additional pools of zinc important for behavior that are spared in ZnT-3 KO mice. In keeping with this view, age-related decreases in non-vesicular zinc are observed in mouse hippocampus and correlate with late-onset deficits in spatial reference memory (Adlard et al., 2010). If non-vesicular zinc has any behavioral relevance, one would predict that zinc chelators aggravate the memory deficits of ZnT-3 KO mice. We know of no such experiments from the literature. One pool of non-vesicular zinc that potentially may participate in memory is the cytosolic zinc bound to MT-3. However, neither deletion nor over-expression of MT-3 alter passive avoidance, spatial reference memory or reversal learning (Erickson et al., 1997). An alternative view, not necessarily incompatible with the above, is that mice compensate, at least in part, for the absence of vesicular zinc during development. Although no compensatory mechanism has been reported in ZnT-3 KO mice so far, rescue experiments in adult subjects or conditional deletion of ZnT-3 will be necessary before compensation can be formally ruled out. Finally, the development of cell-impermeable chelators with faster rates of zinc chelation and improved ion selectivity would help draw more direct comparisons between genetic and pharmacological manipulations of zinc on behavior.
Synaptic Zinc and Neuronal Signaling
Synaptic Transmission and Plasticity
The synaptic accumulation of reactive zinc and its involvement in several forms of memory suggest a role(s) for zinc in synaptic physiology. Zinc chelators or ZnT-3 deletion do not alter fast excitatory, AMPAR-mediated synaptic transmission evoked by low-frequency stimulation in brain slices (Li et al., 2001a; Lopantsev et al., 2003; Kodirov et al., 2006; Mott et al., 2008; Pan et al., 2011). However, a number of synaptic receptors are known to contain binding sites with nanomolar affinity for extracellular zinc, raising the possibility that zinc modulates their activity in a tonic or phasic manner. In keeping with this, chelation of extracellular zinc increases GABAAR-, NMDAR-, and kainate receptor-mediated currents at MF synapses (Vogt et al., 2000; Molnar and Nadler, 2001; Ruiz et al., 2004; Mott et al., 2008), potentiates ASIC-mediated currents in cortical neurons (Chu et al., 2004), and reduces glycinergic currents in hypoglossal neurons of the brainstem (Hirzel et al., 2006). Zinc inhibition of NMDARs has been greatly examined in heterologous expression systems and found to operate via two mechanisms: one that is low-affinity and voltage-dependent, and another one that is high-affinity and voltage independent (Paoletti et al., 1997; Traynelis et al., 1998; Choi and Lipton, 1999). Importantly, knock-in mice with point mutations previously implicated in zinc binding to the α1 glycine receptor 1 subunit (Hirzel et al., 2006) or the GluN2A NMDAR subunit (Nozaki et al., 2011) display sensorimotor deficits and impaired pain processing, respectively. Likewise, high-affinity zinc inhibition of Cav3.2 channels may limit hyperalgesia in peripheral neurons (Nelson et al., 2007). These findings highlight the importance of zinc modulation of ion channels in clinically relevant responses. However, few of the above studies have examined the origin of the zinc that binds to these ion channels. Using glutamate iontophoresis in CA3 neurons, it has been shown that the increase in NMDAR currents caused by zinc chelators is abolished in ZnT-3 KO mice and is occluded by an increase of similar magnitude triggered by briefly blocking pre-synaptic exocytosis (Vogt et al., 2000). Analogously, zinc chelation no longer enhances kainate receptors in mocha mice with reduced vesicular zinc (Mott et al., 2008). These data indicate that, at least at MFs, tonic vesicular zinc release modulates post-synaptic receptors. At variance with this conclusion, post-synaptic NMDAR currents were found to be of similar amplitude in ZnT-3 KO and control CA3 neurons, although in this case the differential effect of zinc chelation between genotypes or the presence of contaminant zinc was not considered (Lopantsev et al., 2003). Perhaps more importantly, little is known as to the impact of extracellular zinc binding to synaptic ion channels on mnemonic processes or neuronal plasticity. The recent development of GluN2A knock-in mice (Nozaki et al., 2011), which express NMDARs insensitive to nanomolar zinc (H128S substitution) but are presumably still inhibited by higher zinc concentrations, will help establish the role of high-affinity zinc binding on NMDAR-dependent forms of learning and synaptic plasticity.
Ideally, one would like to know the effect of synaptic zinc on synaptic responses elicited by the behavior of interest or to the sort of spike patterns associated with that behavior. This information is not yet available at the electrophysiological level. Converging data from three types of zincergic synapses (the MF-to-CA3, the CA3-to-CA1, and the auditory cortex-to-lateral amygdala synapse) indicate that endogenous zinc is required for the induction of long-term potentiation (i.e., LTP) of AMPA currents, a candidate cellular correlate of learning. Whereas post-synaptic mechanisms have been strongly implicated in the expression of LTP at CA3-to-CA1 synapses, MF-to-CA3 plasticity appears to largely rely on pre-synaptic increases in release probability (Nicoll and Schmitz, 2005). Thus, Lu et al. (2000) showed that the cell-permeable zinc chelators dithizone and DEDTC reversibly block MF LTP. Along these lines, CaEDTA (7.5–10 mM) was also shown to block the induction of MF LTP by two other laboratories (Li et al., 2001a; Huang et al., 2008). Notably, lower CaEDTA concentrations (1–2 mM) or partial (∼50%) removal of vesicular zinc by mutation of the AP-3 δ subunit (Seong et al., 2005) fail to inhibit MF LTP (Lu et al., 2000; Vogt et al., 2000; Li et al., 2001a). In contrast, low concentrations of CaEDTA (1–2 mM) or TPEN (0.1 μM) suffice to inhibit LTP at CA1 and lateral amygdala synapses (Izumi et al., 2006; Kodirov et al., 2006; Takeda et al., 2009). The surprising observation here is that chelator-induced LTP block appears to be complete in CA1 and amygdala, even though only a fraction of their synapses react for vesicular zinc. Paralleling the level of zinc chelation required to affect LTP, application of a high zinc concentration (50–100 μM) is needed to potentiate MF synapses (Li et al., 2001a; Huang et al., 2008), whereas a lower zinc concentration (1–10 μM) enhances LTP in CA1 (Izumi et al., 2006; Takeda et al., 2009; Lorca et al., 2011). Importantly, none of the above studies observed an effect of zinc chelation on basal transmission, paired-pulse modulation, or LTP maintenance, suggesting a role for synaptic zinc in the induction of long-term plasticity. Despite this emerging consensus, not all data are concordant, and others have failed to block MF LTP when using TPEN (20–100 μM; Matias et al., 2006). Collectively, it could be argued that the differential sensitivity to zinc manipulation among synapses grossly correlates with the amount of vesicular zinc that they accumulate. So what, then, is the effect of completely (and selectively) removing vesicular zinc on synaptic plasticity? Surprisingly, only one recent study examined long-term synaptic plasticity in ZnT-3 KO mice, and obtained both confirmatory and unexpected results (Pan et al., 2011). On one hand, the increase in release probability normally observed following tetanus-induced MF LTP was absent in ZnT3 KO mice, confirming a role for vesicular zinc in pre-synaptic plasticity. However, MF LTP was still observed in ZnT3 KO mice, and this was found to be due to disinhibiton of a separate, post-synaptic calcium-dependent mechanism of MF LTP that is normally blocked by zinc in wild type mice (Pan et al., 2011). Thus, in the presence of vesicular zinc, MF LTP is expressed via a pre-synaptic mechanism, but when vesicular zinc is removed, a post-synaptic mechanism is recruited instead, indicating a dual role for zinc. Similar conclusions were reached when using a new, fast-acting extracellular zinc chelator, ZX1. Importantly, both pre- and post-synaptic components of MF LTP were observed in the presence of NMDAR antagonists, differentiating them from a recently described form of MF LTP affecting NMDAR currents (Kwon and Castillo, 2008; Rebola et al., 2008). LTP at other synapses in ZnT3 KO mice remains to be examined. Regarding synaptic plasticity in ZnT3 KO mice, two reported observations require clarification: first, and in contrast to ZX1-perfused wild type mice, post-synaptic calcium chelation does not fully block MF LTP in ZnT3 KO mice; secondly, a basal increase in the frequency of spontaneous MF glutamate release in ZnT3 KO mice has been shown (Lopantsev et al., 2003; Pan et al., 2011). Inducible downregulation of ZnT3 in adult brain will help determine whether these differences reflect homeostatic compensations to gene deletion.
Conclusions on the effects of CaEDTA in LTP are somewhat confounded by its properties and should be confirmed in the future with more selective extracellular zinc chelators, such as Tricine (Paoletti et al., 2009) or ZX1. Given the high-affinity constant of CaEDTA for zinc (KD = 10−16), its concentration-dependent effects have been attributed to the slow kinetics of zinc chelation, which are largely dictated by the slow off-rate for calcium. Thus, low CaEDTA would remove extracellular zinc at equilibrium but it would fail to intercept much of synaptically released zinc. On the other hand, concentrations of CaEDTA higher than 5 mM (i.e., those that block MF LTP) have been shown to reduce the vesicular zinc content under steady-state conditions (Lavoie et al., 2007; Ketterman and Li, 2008). As CaEDTA is negatively charged at physiological pH and hence unlikely to cross the plasma membrane, the intracellular zinc reduction may reflect a homeostatic response to extracellular chelation of zinc or other cations. It is therefore unclear if high CaEDTA blocks MF LTP by chelating zinc before or following its actual release. The extracellular specificity of alternative zinc chelators may be assessed by quantifying the extent of vesicular zinc depletion when blocking pre-synaptic exocytosis in the presence of the chelator. Future studies should also address the importance of intracellular zinc chelation in pre- and post-synaptic compartments on LTP.
Synaptic Signal Transduction
Both learning and repetitive synaptic activation stimulate intracellular signal transduction cascades necessary for memory formation and synaptic plasticity. The involvement of synaptic zinc in some forms of memory and LTP hence suggests that zinc may participate in the mechanisms of neuronal signal transduction. Indeed, three independent groups have recently implicated endogenous zinc in the activation of intracellular signal cascades at MFs (Figure 3; Huang et al., 2008; Besser et al., 2009; Chorin et al., 2011; Sindreu et al., 2011). MF tetanization or direct zinc application (100 μM) in the presence of NMDAR antagonists induces a form of LTP that requires TrkB activity, and is abrogated by CaEDTA (Huang et al., 2008). Whether this type of LTP requires a cAMP-dependent increase in the probability of glutamate release as in classic MF LTPAMPA has not been shown yet. Providing a possible mechanism, zinc application to cultured neurons activates the cytosolic tyrosine kinase Src, increasing phospho-activation of the TrkB receptor in the absence of its ligand BDNF. It had been previously suggested that zinc activation of TrkB operates through the conversion of pro-BDNF to mature BDNF (Hwang et al., 2005). However, the fact that zinc still potentiates MFs in BDNF KO mice suggests that such mechanism might not be essential. Downstream signals, including the Erk1/2 MAP kinase and PLCγ1 pathways, are also activated by zinc in cultures. Although it remains to be shown that synaptic zinc activates Src–TrkB signaling during induction of this form of MF LTP, the data provide circumstantial evidence that this may be the case. TrkB activation is most prominent in axon terminals (Spencer-Segal et al., 2011), whereas the Src kinase accumulates in NMDAR complexes (Salter and Kalia, 2004). Hence, the pre- or post-synaptic MF localization of this pathway and the route(s) of zinc entry remain to be clarified. Since primary neuron cultures typically fail to stain for vesicular zinc (Li et al., 2003; Love et al., 2005; Grabrucker et al., 2011), these issues may need to be addressed in more intact preparations.
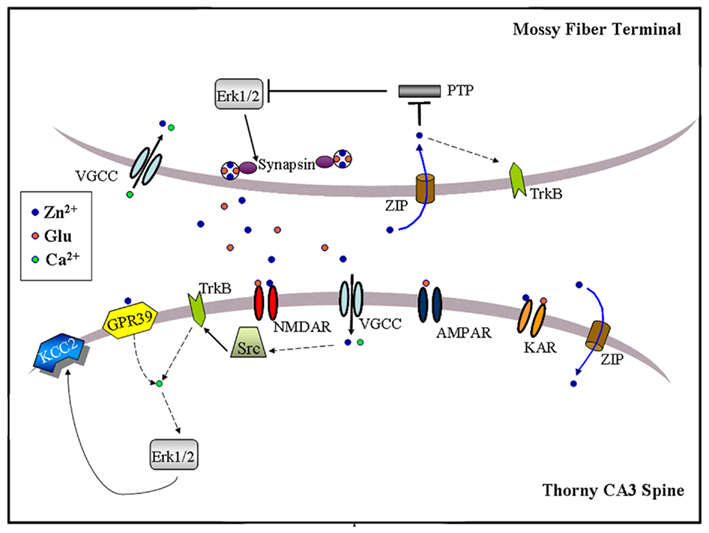
Figure 3. Schematic illustration of zinc-activated signal transduction pathways at the MF-to-CA3 pyramid synapse. Synaptic zinc may activate three signaling pathways: (1) Trans-activation of TrkB receptors at either pre- or post-synaptic sites, perhaps through Src kinase and leading to downstream activation of Erk1/2, Akt or PLCγ; (2) Post-synaptic activation of a GPR39, leading to IP3-dependent intracellular calcium release and KCC2 up-regulation, possibly via Erk1/2 activation; (3) Pre-synaptic Erk1/2 activation, perhaps via inhibition of a MAPK tyrosine phosphatase, leading to synapsin I phosphorylation. Synaptically released zinc may cross the plasma membrane via calcium-permeable channels (VGCC) or dedicated zinc transporter mechanisms (ZIP). Extracellular zinc may also inhibit surface NMDAR and kainate receptors (KAR). Dashed arrows indicate the presence of intermediate steps between connected elements.
Exogenous zinc application (100 μM) has also been reported to increase phosphorylation of Erk1/2 in CA3 somata of young (P8-P16) mice, an effect that is prevented by the Gαq inhibitor YM-254890 (Besser et al., 2009). Interestingly, brief MF tetanization (10 pulses at 66 Hz) triggers a Gq–dependent post-synaptic calcium increase that is reduced by half in ZnT-3 KO mice, suggesting that synaptic zinc can contribute to G protein-coupled receptor (GPCR) signaling even at a developmental age when synaptic vesicles are only partially stocked with zinc (Frederickson et al., 2006a). Furthermore, zinc-induced Gq signaling leads to rapid Erk-dependent up-regulation of KCC2, a K+/Cl− co-transporter, increasing the transmembrane Cl− gradient in CA3 neurons (Chorin et al., 2011). Because KCC2 up-regulation is absent in GPR39 KO mice, this orphan receptor might mediate the metabotropic actions of synaptic zinc. Consistent with this, GPR39 contains extracellular domains that coordinate zinc (Popovics and Stewart, 2011). Interestingly, Erk1/2 activation via TrkB also up-regulates KCC2 as long as PLCγ1 is not co-activated (Rivera et al., 2004). Future studies may clarify the significance of these converging signals to adult synaptic plasticity or memory formation. Intriguingly, similar tetanization regimes and molecular determinants have been associated with the induction of MF LTPNMDA (Kwon and Castillo, 2008; Rebola et al., 2008). In this regard, another study showed that MF stimulation at 50 Hz triggers a form of short-term plasticity (∼10 min-long) that activates, and is modulated by, the pre-synaptic Erk1/2-Synapsin pathway (Vara et al., 2009). Synapsin is a protein that tethers synaptic vesicles to the actin cytoskeleton to modulate transmitter release (Fdez and Hilfiker, 2006), and shown to interact with the plasticity-related protein Rab3A (Giovedi et al., 2004). Suggesting some behavioral relevance for the above observations, dentate gyrus granule cells can fire in bursts of up to 50 Hz when the animal visits a location corresponding to the place field center of the cell (Jung and Mcnaughton, 1993). More recently, it was found that contextual conditioning induces the pre-synaptic activation of the Erk1/2-Synapsin I pathway at MFs (Sindreu et al., 2011). Furthermore, training-induced activation of Erk1/2 is selectively ablated in MFs of ZnT-3 KO mice; partial inhibition of Erk1/2 restricted to granule cells causes a similar deficit in contextual discrimination as that observed in ZnT-3 KO mice. These data represent the first direct evidence that vesicular zinc regulates neuronal signal transduction associated with memory formation. Mechanistically, ZnT-3 KO mice show disinhibition of MAPK tyrosine phosphatases, suggesting that it may underlie impaired Erk activation. Consistent with this, the authors showed that activity-dependent zinc release couples to phosphatase inhibition and Erk1/2 stimulation at MF terminals. The inhibition of MAPK phosphatases by zinc is in keeping with previous observations in neuron cultures (Ho et al., 2008). Because direct Erk1/2 inhibition has been shown to completely block glutamate release (Jovanovic et al., 2000), zinc inhibition of MAPK phosphatases may regulate other pre-synaptic proteins in addition to Synapsin I. At present, it is unclear how zinc may be recycled back into the pre-synaptic terminal to inhibit phosphatases. The identity of the zinc-sensitive MAPK phosphatase also remains to be shown. Suggesting an important role for tyrosine phosphatases in Erk1/2 regulation, catalyzed dephosphorylation of the tyrosine residue (Y185) appears to be the initial and rate-limiting step in Erk1/2 inactivation (Zhou et al., 2002). Previous studies indicated that zinc can inhibit tyrosine phosphatases with high-affinity by binding to their conserved catalytic domain (Brautigan et al., 1981; Haase and Maret, 2003; Redman et al., 2009). How, then, synaptic zinc regulates select signaling pathways via phosphatase inhibition is an intriguing question.
The aggregate evidence indicates that synaptic zinc is a positive regulator of Erk1/2 at MF synapses, activating it through one or more mechanisms (Figure 3). Whereas zinc-dependent Gq signaling may not require zinc influx, released zinc would need to gain access into the cytosol regulate Src or MAPK phosphatases. At MFs, voltage-gated calcium channels represent the main route for pre- and post-synaptic entry of divalent cations (Reid et al., 2001), and calcium channel inhibition suppresses pre-synaptic Erk1/2 at MFs (Sindreu et al., 2011). In addition, currently undefined zinc importers or co-transporters could promote (re-)uptake of extracellular zinc. Whether, similarly to Erk1/2, activation of TrkB or Gq signaling specifically in granule cells or CA3 neurons supports hippocampus-dependent memory is still not known. Also, the possibility of cross-talk between these zinc-regulated pathways has not been examined. For instance, although basal TrkB activation is normal in ZnT3 KO mice (Sindreu et al., 2011), it is unclear if TrkB further stimulates Erk1/2 during learning. Conclusively establishing the pre- or post-synaptic localization of these various pathways will help draw connections among them. Mechanistically, zinc-activated intracellular cascades will need to be put in context with the previous molecules implicated in MF plasticity, such as the adenylyl cyclases and their downstream targets (Nicoll and Schmitz, 2005). A large body of literature indicates that the Erk1/2 pathway (i.e., Ras–b–Raf–MEK–Erk) acts as a node integrating several upstream signals to control memory and neuronal plasticity, mainly through actions on dendritic proteins and gene transcription (Impey et al., 1999; Thomas and Huganir, 2004; Ye and Carew, 2010). However, there is also evidence that Erk1/2 affects cognition via pre-synaptic mechanisms (Kushner et al., 2005; Cui et al., 2008). Circumstantially, zinc can have opposite effects on MAPK in other cellular models. For instance, zinc can decrease Erk signaling in oocytes by inhibiting Raf, an effect that may be counteracted by ZnT-1 (Jirakulaporn and Muslin, 2004). It remains to be explored if reactive zinc can bidirectionally modulate Erk signaling by engaging various mechanisms in neurons.
A pertinent question is to what extent the effects of synaptic zinc on ion channels and signal transduction cascades are functionally related, and whether they co-occur at individual synapses. At MFs, the effects of zinc on Gq-, TrkB-, and Erk-dependent signaling are all spared by NMDAR blockers, in line with the ability of this synapse to express NMDAR-independent forms of plasticity. Moreover, the effects of synaptic zinc on Erk1/2 activation or TrkB-dependent plasticity are not reproduced at CA1 synapses (Huang et al., 2008; Sindreu et al., 2011), possibly as a result of molecular heterogeneity between synapses. In contrast, blockade of CA1 LTP by zinc chelators has been attributed to the untimely activation (due to disinhibition) of NMDARs upstream of calcineurin, nitric oxide, and p38 (Izumi et al., 2006, 2008). Whether NMDAR-dependent forms of plasticity activate other downstream signals that regulate, or are regulated by, reactive zinc is unclear. CaMKII, PKA, PKC, or Akt are normally activated in ZnT-3 KO hippocampus (Sindreu et al., 2011). However, in vitro studies have shown that zinc unbinding from PKC can trigger the activation of this kinase (Knapp and Klann, 2000; Korichneva et al., 2002), and that constitutively active PKC is, in turn, sufficient to increase the expression of genes containing metal regulatory elements (Aras et al., 2009). Thus, PKC activation during synaptic plasticity could potentially trigger intracellular zinc signaling. The effect of acutely chelating reactive zinc from brain tissue on these pathways remains to be shown. On the other hand, block of LTP at cortico-amygdala synapses by zinc chelators can be rescued by GABAAR antagonists (Kodirov et al., 2006), implying that zinc can gate LTP via heterosynaptic inhibition of feed-forward interneurons. The authors proposed that such a mechanism may provide spatial control of amygdala LTP between cortical and thalamic afferents.
Future experiments examining how these, and other, mechanisms of zinc action affect cognition are warranted. The deficits in synaptic plasticity and biochemical activation induced by zinc blockade are in agreement with the memory impairments observed in ZnT-3 KO mice or after zinc chelation. While the ZnT-3 KO mouse model has now provided strong evidence for a role of vesicular zinc in cognition, circumstantial evidence suggests that other sources of reactive zinc may also be involved. Altogether, the data we have discussed here lend further support to the hypothesis that synaptic zinc may promote learning and memory through actions on neuronal signal transduction. Traditionally viewed as an inhibitor of glutamatergic transmission, synaptic zinc may turn out to play complementary roles with glutamate in the service of memory and neuronal plasticity: zinc may recruit additional plasticity mechanisms during increased neuronal firing associated with learning, prevent tonic interference from other signaling pathways, or select between converging synaptic contacts to promote memory formation. Many of the recent findings on synaptic zinc had not been anticipated only 5 years ago. Should this trajectory continue, the next few years will reveal interesting new facets of zinc signaling in the brain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. Victor Faundez, James McNamara, and Jeffrey Noebels for discussing some topics during preparation of this manuscript. This work was funded by NIH grant NS020498.
References
Adlard, P. A., Parncutt, J. M., Finkelstein, D. I., and Bush, A. I. (2010). Cognitive loss in zinc transporter-3 knock-out mice: a phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 30, 1631–1636.
Andrews, G. K., Wang, H., Dey, S. K., and Palmiter, R. D. (2004). Mouse zinc transporter 1 gene provides an essential function during early embryonic development. Genesis 40, 74–81.
Aniksztejn, L., Charton, G., and Ben-Ari, Y. (1987). Selective release of endogenous zinc from the hippocampal mossy fibers in situ. Brain Res. 404, 58–64.
Aras, M. A., Hara, H., Hartnett, K. A., Kandler, K., and Aizenman, E. (2009). Protein kinase C regulation of neuronal zinc signaling mediates survival during preconditioning. J. Neurochem. 110, 106–117.
Baron, M. K., Boeckers, T. M., Vaida, B., Faham, S., Gingery, M., Sawaya, M. R., Salyer, D., Gundelfinger, E. D., and Bowie, J. U. (2006). An architectural framework that may lie at the core of the postsynaptic density. Science 311, 531–535.
Beaulieu, C., Dyck, R., and Cynader, M. (1992). Enrichment of glutamate in zinc-containing terminals of the cat visual cortex. Neuroreport 3, 861–864.
Besser, L., Chorin, E., Sekler, I., Silverman, W. F., Atkin, S., Russell, J. T., and Hershfinkel, M. (2009). Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 29, 2890–2901.
Birinyi, A., Parker, D., Antal, M., and Shupliakov, O. (2001). Zinc co-localizes with GABA and glycine in synapses in the lamprey spinal cord. J. Comp. Neurol. 433, 208–221.
Bozym, R. A., Chimienti, F., Giblin, L. J., Gross, G. W., Korichneva, I., Li, Y., Libert, S., Maret, W., Parviz, M., Frederickson, C. J., and Thompson, R. B. (2010). Free zinc ions outside a narrow concentration range are toxic to a variety of cells in vitro. Exp. Biol. Med. (Maywood) 235, 741–750.
Brautigan, D. L., Bornstein, P., and Gallis, B. (1981). Phosphotyrosyl-protein phosphatase. Specific inhibition by Zn . J. Biol. Chem. 256, 6519–6522.
Brown, C. E., and Dyck, R. H. (2004). Distribution of zincergic neurons in the mouse forebrain. J. Comp. Neurol. 479, 156–167.
Cai, Q., Davis, M. L., and Sheng, Z. H. (2011). Regulation of axonal mitochondrial transport and its impact on synaptic transmission. Neurosci. Res. 70, 9–15.
Canzoniero, L. M., Turetsky, D. M., and Choi, D. W. (1999). Measurement of intracellular free zinc concentrations accompanying zinc-induced neuronal death. J. Neurosci. 19, RC31.
Caporale, T., Ciavardelli, D., Di Ilio, C., Lanuti, P., Drago, D., and Sensi, S. L. (2009). Ratiometric-pericam-mt, a novel tool to evaluate intramitochondrial zinc. Exp. Neurol. 218, 228–234.
Carter, R. E., Aiba, I., Dietz, R. M., Sheline, C. T., and Shuttleworth, C. W. (2011). Spreading depression and related events are significant sources of neuronal Zn2+ release and accumulation. J. Cereb. Blood Flow Metab. 31, 1073–1084.
Casanovas-Aguilar, C., Miro-Bernie, N., and Perez-Clausell, J. (2002). Zinc-rich neurones in the rat visual cortex give rise to two laminar segregated systems of connections. Neuroscience 110, 445–458.
Chang, D. T., Honick, A. S., and Reynolds, I. J. (2006). Mitochondrial trafficking to synapses in cultured primary cortical neurons. J. Neurosci. 26, 7035–7045.
Charton, G., Rovira, C., Ben-Ari, Y., and Leviel, V. (1985). Spontaneous and evoked release of endogenous Zn2+ in the hippocampal mossy fiber zone of the rat in situ. Exp. Brain Res. 58, 202–205.
Choi, Y. B., and Lipton, S. A. (1999). Identification and mechanism of action of two histidine residues underlying high-affinity Zn2+ inhibition of the NMDA receptor. Neuron 23, 171–180.
Chorin, E., Vinograd, O., Fleidervish, I., Gilad, D., Herrmann, S., Sekler, I., Aizenman, E., and Hershfinkel, M. (2011). Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J. Neurosci. 31, 12916–12926.
Christensen, M. K., and Frederickson, C. J. (1998). Zinc-containing afferent projections to the rat corticomedial amygdaloid complex: a retrograde tracing study. J. Comp. Neurol. 400, 375–390.
Chu, X. P., Wemmie, J. A., Wang, W. Z., Zhu, X. M., Saugstad, J. A., Price, M. P., Simon, R. P., and Xiong, Z. G. (2004). Subunit-dependent high-affinity zinc inhibition of acid-sensing ion channels. J. Neurosci. 24, 8678–8689.
Clements, J. D. (1996). Transmitter time course in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 19, 163–171.
Cole, T. B., Martyanova, A., and Palmiter, R. D. (2001). Removing zinc from synaptic vesicles does not impair spatial learning, memory, or sensorimotor functions in the mouse. Brain Res. 891, 253–265.
Cole, T. B., Wenzel, H. J., Kafer, K. E., Schwartzkroin, P. A., and Palmiter, R. D. (1999). Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. U.S.A. 96, 1716–1721.
Colvin, R. A. (2002). pH dependence and compartmentalization of zinc transported across plasma membrane of rat cortical neurons. Am. J. Physiol. Cell Physiol. 282, C317–C329.
Cui, Y., Costa, R. M., Murphy, G. G., Elgersma, Y., Zhu, Y., Gutmann, D. H., Parada, L. F., Mody, I., and Silva, A. J. (2008). Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 135, 549–560.
Cunningham, M. G., Ames, H. M., Christensen, M. K., and Sorensen, J. C. (2007). Zincergic innervation of medial prefrontal cortex by basolateral projection neurons. Neuroreport 18, 531–535.
Danscher, G. (1981). Histochemical demonstration of heavy metals. A revised version of the sulphide silver method suitable for both light and electron microscopy. Histochemistry 71, 1–16.
Danscher, G., Howell, G., Perez-Clausell, J., and Hertel, N. (1985). The dithizone, Timm’s sulphide silver and the selenium methods demonstrate a chelatable pool of zinc in CNS. A proton activation (PIXE) analysis of carbon tetrachloride extracts from rat brains and spinal cords intravitally treated with dithizone. Histochemistry 83, 419–422.
Danscher, G., Jo, S. M., Varea, E., Wang, Z., Cole, T. B., and Schroder, H. D. (2001). Inhibitory zinc-enriched terminals in mouse spinal cord. Neuroscience 105, 941–947.
Danscher, G., and Stoltenberg, M. (2006). Silver enhancement of quantum dots resulting from (1) metabolism of toxic metals in animals and humans, (2) in vivo, in vitro and immersion created zinc-sulphur/zinc-selenium nanocrystals, (3) metal ions liberated from metal implants and particles. Prog. Histochem. Cytochem. 41, 57–139.
Datki, Z. L., Hunya, A., and Penke, B. (2007). A novel and simple fluorescence method for the measurement of presynaptic vesicular zinc release in acute hippocampal slices with a fluorescence plate reader. Brain Res. Bull. 74, 183–187.
Daumas, S., Halley, H., and Lassalle, J. M. (2004). Disruption of hippocampal CA3 network: effects on episodic-like memory processing in C57BL/6J mice. Eur. J. Neurosci. 20, 597–600.
Dencker, L., and Tjalve, H. (1979). An autoradiographic study on the fate of 65Zn in zinc-rich tissues in some rodents. Med. Biol. 57, 391–397.
Dineley, K. E., Devinney, M. J. II, Zeak, J. A., Rintoul, G. L., and Reynolds, I. J. (2008). Glutamate mobilizes [Zn2+] through Ca2+-dependent reactive oxygen species accumulation. J. Neurochem. 106, 2184–2193.
Dittmer, P. J., Miranda, J. G., Gorski, J. A., and Palmer, A. E. (2009). Genetically encoded sensors to elucidate spatial distribution of cellular zinc. J. Biol. Chem. 284, 16289–16297.
Dominguez, M. I., Blasco-Ibanez, J. M., Crespo, C., Marques-Mari, A. I., and Martinez-Guijarro, F. J. (2003). Zinc chelation during non-lesioning overexcitation results in neuronal death in the mouse hippocampus. Neuroscience 116, 791–806.
Ehmann, W. D., Alauddin, M., Hossain, T. I., and Markesbery, W. R. (1984). Brain trace elements in Pick’s disease. Ann. Neurol. 15, 102–104.
Emmetsberger, J., Mirrione, M. M., Zhou, C., Fernandez-Monreal, M., Siddiq, M. M., Ji, K., and Tsirka, S. E. (2010). Tissue plasminogen activator alters intracellular sequestration of zinc through interaction with the transporter ZIP4. J. Neurosci. 30, 6538–6547.
Erickson, J. C., Hollopeter, G., Thomas, S. A., Froelick, G. J., and Palmiter, R. D. (1997). Disruption of the metallothionein-III gene in mice: analysis of brain zinc, behavior, and neuron vulnerability to metals, aging, and seizures. J. Neurosci. 17, 1271–1281.
Fdez, E., and Hilfiker, S. (2006). Vesicle pools and synapsins: new insights into old enigmas. Brain Cell Biol. 35, 107–115.
Frederickson, C. J. (1989). Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 31, 145–238.
Frederickson, C. J., Giblin, L. J. III., Balaji, R. V., Masalha, R., Frederickson, C. J., Zeng, Y., Lopez, E. V., Koh, J. Y., Chorin, U., Besser, L., Hershfinkel, M., Li, Y., Thompson, R. B., and Krezel, A. (2006a). Synaptic release of zinc from brain slices: factors governing release, imaging, and accurate calculation of concentration. J. Neurosci. Methods 154, 19–29.
Frederickson, C. J., Giblin, L. J., Krezel, A., Mcadoo, D. J., Mueller, R. N., Zeng, Y., Balaji, R. V., Masalha, R., Thompson, R. B., Fierke, C. A., Sarvey, J. M., De Valdenebro, M., Prough, D. S., and Zornow, M. H. (2006b). Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp. Neurol. 198, 285–293.
Frederickson, C. J., Kasarskis, E. J., Ringo, D., and Frederickson, R. E. (1987). A quinoline fluorescence method for visualizing and assaying the histochemically reactive zinc (bouton zinc) in the brain. J. Neurosci. Methods 20, 91–103.
Frederickson, C. J., Klitenick, M. A., Manton, W. I., and Kirkpatrick, J. B. (1983). Cytoarchitectonic distribution of zinc in the hippocampus of man and the rat. Brain Res. 273, 335–339.
Frederickson, C. J., Suh, S. W., Koh, J. Y., Cha, Y. K., Thompson, R. B., Labuda, C. J., Balaji, R. V., and Cuajungco, M. P. (2002). Depletion of intracellular zinc from neurons by use of an extracellular chelator in vivo and in vitro. J. Histochem. Cytochem. 50, 1659–1662.
Frederickson, R. E., Frederickson, C. J., and Danscher, G. (1990). In situ binding of bouton zinc reversibly disrupts performance on a spatial memory task. Behav. Brain Res. 38, 25–33.
Friedman, B., and Price, J. L. (1984). Fiber systems in the olfactory bulb and cortex: a study in adult and developing rats, using the Timm method with the light and electron microscope. J. Comp. Neurol. 223, 88–109.
Gaither, L. A., and Eide, D. J. (2000). Functional expression of the human hZIP2 zinc transporter. J. Biol. Chem. 275, 5560–5564.
Giovedi, S., Vaccaro, P., Valtorta, F., Darchen, F., Greengard, P., Cesareni, G., and Benfenati, F. (2004). Synapsin is a novel Rab3 effector protein on small synaptic vesicles. I. Identification and characterization of the synapsin I-Rab3 interactions in vitro and in intact nerve terminals. J. Biol. Chem. 279, 43760–43768.
Grabrucker, A. M., Knight, M. J., Proepper, C., Bockmann, J., Joubert, M., Rowan, M., Nienhaus, G. U., Garner, C. C., Bowie, J. U., Kreutz, M. R., Gundelfinger, E. D., and Boeckers, T. M. (2011). Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 30, 569–581.
Haase, H., and Maret, W. (2003). Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Exp. Cell Res. 291, 289–298.
Haug, F. M. (1967). Electron microscopical localization of the zinc in hippocampal mossy fibre synapses by a modified sulfide silver procedure. Histochemie 8, 355–368.
Hershey, C. O., Hershey, L. A., Varnes, A., Vibhakar, S. D., Lavin, P., and Strain, W. H. (1983). Cerebrospinal fluid trace element content in dementia: clinical, radiologic, and pathologic correlations. Neurology 33, 1350–1353.
Hires, S. A., Zhu, Y., and Tsien, R. Y. (2008). Optical measurement of synaptic glutamate spillover and reuptake by linker optimized glutamate-sensitive fluorescent reporters. Proc. Natl. Acad. Sci. U.S.A. 105, 4411–4416.
Hirzel, K., Muller, U., Latal, A. T., Hulsmann, S., Grudzinska, J., Seeliger, M. W., Betz, H., and Laube, B. (2006). Hyperekplexia phenotype of glycine receptor alpha1 subunit mutant mice identifies Zn(2+) as an essential endogenous modulator of glycinergic neurotransmission. Neuron 52, 679–690.
Ho, Y., Samarasinghe, R., Knoch, M. E., Lewis, M., Aizenman, E., and Defranco, D. B. (2008). Selective inhibition of mitogen-activated protein kinase phosphatases by zinc accounts for extracellular signal-regulated kinase 1/2-dependent oxidative neuronal cell death. Mol. Pharmacol. 74, 1141–1151.
Howell, G. A., Welch, M. G., and Frederickson, C. J. (1984). Stimulation-induced uptake and release of zinc in hippocampal slices. Nature 308, 736–738.
Huang, Y. Z., Pan, E., Xiong, Z. Q., and Mcnamara, J. O. (2008). Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 57, 546–558.
Hwang, J. J., Lee, S. J., Kim, T. Y., Cho, J. H., and Koh, J. Y. (2008). Zinc and 4-hydroxy-2-nonenal mediate lysosomal membrane permeabilization induced by H2O2 in cultured hippocampal neurons. J. Neurosci. 28, 3114–3122.
Hwang, J. J., Park, M. H., Choi, S. Y., and Koh, J. Y. (2005). Activation of the Trk signaling pathway by extracellular zinc. Role of metalloproteinases. J. Biol. Chem. 280, 11995–12001.
Impey, S., Obrietan, K., and Storm, D. R. (1999). Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 23, 11–14.
Izumi, Y., Auberson, Y. P., and Zorumski, C. F. (2006). Zinc modulates bidirectional hippocampal plasticity by effects on NMDA receptors. J. Neurosci. 26, 7181–7188.
Izumi, Y., Tokuda, K., and Zorumski, C. F. (2008). Long-term potentiation inhibition by low-level N-methyl-D-aspartate receptor activation involves calcineurin, nitric oxide, and p38 mitogen-activated protein kinase. Hippocampus 18, 258–265.
Jan, H. H., Chen, I. T., Tsai, Y. Y., and Chang, Y. C. (2002). Structural role of zinc ions bound to postsynaptic densities. J. Neurochem. 83, 525–534.
Jirakulaporn, T., and Muslin, A. J. (2004). Cation diffusion facilitator proteins modulate Raf-1 activity. J. Biol. Chem. 279, 27807–27815.
Jovanovic, J. N., Czernik, A. J., Fienberg, A. A., Greengard, P., and Sihra, T. S. (2000). Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat. Neurosci. 3, 323–329.
Jung, M. W., and Mcnaughton, B. L. (1993). Spatial selectivity of unit activity in the hippocampal granular layer. Hippocampus 3, 165–182.
Kay, A. R. (2003). Evidence for chelatable zinc in the extracellular space of the hippocampus, but little evidence for synaptic release of Zn. J. Neurosci. 23, 6847–6855.
Keller, K. A., Chu, Y., Grider, A., and Coffield, J. A. (2000). Supplementation with L-histidine during dietary zinc repletion improves short-term memory in zinc-restricted young adult male rats. J. Nutr. 130, 1633–1640.
Ketterman, J. K., and Li, Y. V. (2008). Presynaptic evidence for zinc release at the mossy fiber synapse of rat hippocampus. J. Neurosci. Res. 86, 422–434.
Knapp, L. T., and Klann, E. (2000). Superoxide-induced stimulation of protein kinase C via thiol modification and modulation of zinc content. J. Biol. Chem. 275, 24136–24145.
Kodirov, S. A., Takizawa, S., Joseph, J., Kandel, E. R., Shumyatsky, G. P., and Bolshakov, V. Y. (2006). Synaptically released zinc gates long-term potentiation in fear conditioning pathways. Proc. Natl. Acad. Sci. U.S.A. 103, 15218–15223.
Komatsu, K., Urano, Y., Kojima, H., and Nagano, T. (2007). Development of an iminocoumarin-based zinc sensor suitable for ratiometric fluorescence imaging of neuronal zinc. J. Am. Chem. Soc. 129, 13447–13454.
Korichneva, I., Hoyos, B., Chua, R., Levi, E., and Hammerling, U. (2002). Zinc release from protein kinase C as the common event during activation by lipid second messenger or reactive oxygen. J. Biol. Chem. 277, 44327–44331.
Krezel, A., and Maret, W. (2006). Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J. Biol. Inorg. Chem. 11, 1049–1062.
Kushner, S. A., Elgersma, Y., Murphy, G. G., Jaarsma, D., Van Woerden, G. M., Hojjati, M. R., Cui, Y., Leboutillier, J. C., Marrone, D. F., Choi, E. S., De Zeeuw, C. I., Petit, T. L., Pozzo-Miller, L., and Silva, A. J. (2005). Modulation of presynaptic plasticity and learning by the H-ras/extracellular signal-regulated kinase/synapsin I signaling pathway. J. Neurosci. 25, 9721–9734.
Kwon, H. B., and Castillo, P. E. (2008). Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron 57, 108–120.
Lassalle, J. M., Bataille, T., and Halley, H. (2000). Reversible inactivation of the hippocampal mossy fiber synapses in mice impairs spatial learning, but neither consolidation nor memory retrieval, in the Morris navigation task. Neurobiol. Learn. Mem. 73, 243–257.
Lavoie, N., Peralta, M. R. III., Chiasson, M., Lafortune, K., Pellegrini, L., Seress, L., and Toth, K. (2007). Extracellular chelation of zinc does not affect hippocampal excitability and seizure-induced cell death in rats. J. Physiol. (Lond.) 578, 275–289.
Lee, J. Y., Cole, T. B., Palmiter, R. D., and Koh, J. Y. (2000). Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J. Neurosci. 20, RC79.
Levy, S., Beharier, O., Etzion, Y., Mor, M., Buzaglo, L., Shaltiel, L., Gheber, L. A., Kahn, J., Muslin, A. J., Katz, A., Gitler, D., and Moran, A. (2009). Molecular basis for zinc transporter 1 action as an endogenous inhibitor of L-type calcium channels. J. Biol. Chem. 284, 32434–32443.
Li, Y., Hough, C. J., Frederickson, C. J., and Sarvey, J. M. (2001a). Induction of mossy fiber – >Ca3 long-term potentiation requires translocation of synaptically released Zn2+. J. Neurosci. 21, 8015–8025.
Li, Y., Hough, C. J., Suh, S. W., Sarvey, J. M., and Frederickson, C. J. (2001b). Rapid translocation of Zn(2+) from presynaptic terminals into postsynaptic hippocampal neurons after physiological stimulation. J. Neurophysiol. 86, 2597–2604.
Lin, W., Chai, J., Love, J., and Fu, D. (2010). Selective electrodiffusion of zinc ions in a Zrt-, Irt-like protein, ZIPB. J. Biol. Chem. 285, 39013–39020.
Linkous, D. H., Flinn, J. M., Koh, J. Y., Lanzirotti, A., Bertsch, P. M., Jones, B. F., Giblin, L. J., and Frederickson, C. J. (2008). Evidence that the ZNT3 protein controls the total amount of elemental zinc in synaptic vesicles. J. Histochem. Cytochem. 56, 3–6.
Long, Y., Hardwick, A. L., and Frederickson, C. J. (1995). Zinc-containing innervation of the subicular region in the rat. Neurochem. Int. 27, 95–103.
Lopantsev, V., Wenzel, H. J., Cole, T. B., Palmiter, R. D., and Schwartzkroin, P. A. (2003). Lack of vesicular zinc in mossy fibers does not affect synaptic excitability of CA3 pyramidal cells in zinc transporter 3 knockout mice. Neuroscience 116, 237–248.
Lorca, R. A., Rozas, C., Loyola, S., Moreira-Ramos, S., Zeise, M. L., Kirkwood, A., Huidobro-Toro, J. P., and Morales, B. (2011). Zinc enhances long-term potentiation through P2X receptor modulation in the hippocampal CA1 region. Eur. J. Neurosci. 33, 1175–1185.
Love, R., Salazar, G., and Faundez, V. (2005). Neuronal zinc stores are modulated by non-steroidal anti-inflammatory drugs: an optical analysis in cultured hippocampal neurons. Brain Res. 1061, 1–12.
Lu, Y. M., Taverna, F. A., Tu, R., Ackerley, C. A., Wang, Y. T., and Roder, J. (2000). Endogenous Zn(2+) is required for the induction of long-term potentiation at rat hippocampal mossy fiber-CA3 synapses. Synapse 38, 187–197.
Malaiyandi, L. M., Vergun, O., Dineley, K. E., and Reynolds, I. J. (2005). Direct visualization of mitochondrial zinc accumulation reveals uniporter-dependent and -independent transport mechanisms. J. Neurochem. 93, 1242–1250.
Mandava, P., Howell, G. A., and Frederickson, C. J. (1993). Zinc-containing neuronal innervation of the septal nuclei. Brain Res. 608, 115–122.
Mao, X., Kim, B. E., Wang, F., Eide, D. J., and Petris, M. J. (2007). A histidine-rich cluster mediates the ubiquitination and degradation of the human zinc transporter, hZIP4, and protects against zinc cytotoxicity. J. Biol. Chem. 282, 6992–7000.
Maret, W. (2011). Redox biochemistry of mammalian metallothioneins. J. Biol. Inorg. Chem. 16, 1079–1086.
Martel, G., Hevi, C., Friebely, O., Baybutt, T., and Shumyatsky, G. P. (2010). Zinc transporter 3 is involved in learned fear and extinction, but not in innate fear. Learn. Mem. 17, 582–590.
Martel, G., Hevi, C., Kane-Goldsmith, N., and Shumyatsky, G. P. (2011). Zinc transporter ZnT3 is involved in memory dependent on the hippocampus and perirhinal cortex. Behav. Brain Res. 223, 233–238.
Martin, S. J., Grimwood, P. D., and Morris, R. G. (2000). Synaptic plasticity and memory: an evaluation of the hypothesis. Annu. Rev. Neurosci. 23, 649–711.
Martinez-Guijarro, F. J., Soriano, E., Del Rio, J. A., and Lopez-Garcia, C. (1991). Zinc-positive boutons in the cerebral cortex of lizards show glutamate immunoreactivity. J. Neurocytol. 20, 834–843.
Maske, H. (1955). A new method for demonstrating A and B cells in the islands of Langerhans. Klin. Wochenschr. 33, 1058.
Matias, C. M., Matos, N. C., Arif, M., Dionisio, J. C., and Quinta-Ferreira, M. E. (2006). Effect of the zinc chelator N,N,N′,N′-tetrakis (2-pyridylmethyl)ethylenediamine (TPEN) on hippocampal mossy fiber calcium signals and on synaptic transmission. Biol. Res. 39, 521–530.
Minami, A., Takeda, A., Yamaide, R., and Oku, N. (2002). Relationship between zinc and neurotransmitters released into the amygdalar extracellular space. Brain Res. 936, 91–94.
Miro-Bernie, N., Sancho-Bielsa, F. J., Lopez-Garcia, C., and Perez-Clausell, J. (2003). Retrograde transport of sodium selenite and intracellular injection of micro-ruby: a combined method to describe the morphology of zinc-rich neurones. J. Neurosci. Methods 127, 199–209.
Mitchell, C. L., and Barnes, M. I. (1993). Proconvulsant action of diethyldithiocarbamate in stimulation of the perforant path. Neurotoxicol. Teratol. 15, 165–171.
Molnar, P., and Nadler, J. V. (2001). Synaptically-released zinc inhibits N-methyl-D-aspartate receptor activation at recurrent mossy fiber synapses. Brain Res. 910, 205–207.
Mott, D. D., Benveniste, M., and Dingledine, R. J. (2008). pH-dependent inhibition of kainate receptors by zinc. J. Neurosci. 28, 1659–1671.
Nakashiba, T., Young, J. Z., Mchugh, T. J., Buhl, D. L., and Tonegawa, S. (2008). Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science 319, 1260–1264.
Nelson, M. T., Woo, J., Kang, H. W., Vitko, I., Barrett, P. Q., Perez-Reyes, E., Lee, J. H., Shin, H. S., and Todorovic, S. M. (2007). Reducing agents sensitize C-type nociceptors by relieving high-affinity zinc inhibition of T-type calcium channels. J. Neurosci. 27, 8250–8260.
Neves, G., Cooke, S. F., and Bliss, T. V. (2008). Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci. 9, 65–75.
Nicoll, R. A., and Schmitz, D. (2005). Synaptic plasticity at hippocampal mossy fibre synapses. Nat. Rev. Neurosci. 6, 863–876.
Nolan, E. M., and Lippard, S. J. (2009). Small-molecule fluorescent sensors for investigating zinc metalloneurochemistry. Acc. Chem. Res. 42, 193–203.
Nozaki, C., Vergnano, A. M., Filliol, D., Ouagazzal, A. M., Le Goff, A., Carvalho, S., Reiss, D., Gaveriaux-Ruff, C., Neyton, J., Paoletti, P., and Kieffer, B. L. (2011). Zinc alleviates pain through high-affinity binding to the NMDA receptor NR2A subunit. Nat. Neurosci. 14, 1017–1022.
Ohana, E., Segal, D., Palty, R., Ton-That, D., Moran, A., Sensi, S. L., Weiss, J. H., Hershfinkel, M., and Sekler, I. (2004). A sodium zinc exchange mechanism is mediating extrusion of zinc in mammalian cells. J. Biol. Chem. 279, 4278–4284.
Ohana, E., Sekler, I., Kaisman, T., Kahn, N., Cove, J., Silverman, W. F., Amsterdam, A., and Hershfinkel, M. (2006). Silencing of ZnT-1 expression enhances heavy metal influx and toxicity. J. Mol. Med. (Berl) 84, 753–763.
Palmiter, R. D. (1995). Constitutive expression of metallothionein-III (MT-III), but not MT-I, inhibits growth when cells become zinc deficient. Toxicol. Appl. Pharmacol. 135, 139–146.
Palmiter, R. D., Cole, T. B., Quaife, C. J., and Findley, S. D. (1996). ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc. Natl. Acad. Sci. U.S.A. 93, 14934–14939.
Palmiter, R. D., and Findley, S. D. (1995). Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 14, 639–649.
Pan, E., Zhang, X. A., Huang, Z., Krezel, A., Zhao, M., Tinberg, C. E., Lippard, S. J., and Mcnamara, J. O. (2011). Vesicular zinc promotes presynaptic and inhibits postsynaptic long-term potentiation of Mossy fiber-CA3 synapse. Neuron 71, 1116–1126.
Paoletti, P., Ascher, P., and Neyton, J. (1997). High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J. Neurosci. 17, 5711–5725.
Paoletti, P., Vergnano, A. M., Barbour, B., and Casado, M. (2009). Zinc at glutamatergic synapses. Neuroscience 158, 126–136.
Perez-Clausell, J., and Danscher, G. (1985). Intravesicular localization of zinc in rat telencephalic boutons. A histochemical study. Brain Res. 337, 91–98.
Popovics, P., and Stewart, A. J. (2011). GPR39: a Zn(2+)-activated G protein-coupled receptor that regulates pancreatic, gastrointestinal and neuronal functions. Cell. Mol. Life Sci. 68, 85–95.
Qian, J., and Noebels, J. L. (2005). Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J. Physiol. (Lond.) 566, 747–758.
Qian, J., and Noebels, J. L. (2006). Exocytosis of vesicular zinc reveals persistent depression of neurotransmitter release during metabotropic glutamate receptor long-term depression at the hippocampal CA3-CA1 synapse. J. Neurosci. 26, 6089–6095.
Qian, J., Xu, K., Yoo, J., Chen, T. T., Andrews, G., and Noebels, J. L. (2011). Knockout of Zn transporters Zip-1 and Zip-3 attenuates seizure-induced CA1 neurodegeneration. J. Neurosci. 31, 97–104.
Qin, Y., Thomas, D., Fontaine, C. P., and Colvin, R. A. (2008). Mechanisms of Zn2+ efflux in cultured cortical neurons. J. Neurochem. 107, 1304–1313.
Qin, Y., Thomas, D., Fontaine, C. P., and Colvin, R. A. (2009). Silencing of ZnT1 reduces Zn2+efflux in cultured cortical neurons. Neurosci. Lett. 450, 206–210.
Rebola, N., Lujan, R., Cunha, R. A., and Mulle, C. (2008). Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 57, 121–134.
Redman, P. T., Hartnett, K. A., Aras, M. A., Levitan, E. S., and Aizenman, E. (2009). Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. J. Physiol. (Lond.) 587, 4393–4404.
Reid, C. A., Fabian-Fine, R., and Fine, A. (2001). Postsynaptic calcium transients evoked by activation of individual hippocampal mossy fiber synapses. J. Neurosci. 21, 2206–2214.
Rivera, C., Voipio, J., Thomas-Crusells, J., Li, H., Emri, Z., Sipila, S., Payne, J. A., Minichiello, L., Saarma, M., and Kaila, K. (2004). Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J. Neurosci. 24, 4683–4691.
Ruiz, A., Walker, M. C., Fabian-Fine, R., and Kullmann, D. M. (2004). Endogenous zinc inhibits GABA(A) receptors in a hippocampal pathway. J. Neurophysiol. 91, 1091–1096.
Salazar, G., Craige, B., Love, R., Kalman, D., and Faundez, V. (2005). Vglut1 and ZnT3 co-targeting mechanisms regulate vesicular zinc stores in PC12 cells. J. Cell. Sci. 118, 1911–1921.
Salazar, G., Falcon-Perez, J. M., Harrison, R., and Faundez, V. (2009). SLC30A3 (ZnT3) oligomerization by dityrosine bonds regulates its subcellular localization and metal transport capacity. PLoS ONE 4, e5896. doi:10.1371/journal.pone.0005896
Salter, M. W., and Kalia, L. V. (2004). Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 5, 317–328.
Schikorski, T., and Stevens, C. F. (1997). Quantitative ultrastructural analysis of hippocampal excitatory synapses. J. Neurosci. 17, 5858–5867.
Sekler, I., Moran, A., Hershfinkel, M., Dori, A., Margulis, A., Birenzweig, N., Nitzan, Y., and Silverman, W. F. (2002). Distribution of the zinc transporter ZnT-1 in comparison with chelatable zinc in the mouse brain. J. Comp. Neurol. 447, 201–209.
Sensi, S. L., Canzoniero, L. M., Yu, S. P., Ying, H. S., Koh, J. Y., Kerchner, G. A., and Choi, D. W. (1997). Measurement of intracellular free zinc in living cortical neurons: routes of entry. J. Neurosci. 17, 9554–9564.
Sensi, S. L., Ton-That, D., Sullivan, P. G., Jonas, E. A., Gee, K. R., Kaczmarek, L. K., and Weiss, J. H. (2003). Modulation of mitochondrial function by endogenous Zn2+ pools. Proc. Natl. Acad. Sci. U.S.A. 100, 6157–6162.
Seong, E., Wainer, B. H., Hughes, E. D., Saunders, T. L., Burmeister, M., and Faundez, V. (2005). Genetic analysis of the neuronal and ubiquitous AP-3 adaptor complexes reveals divergent functions in brain. Mol. Biol. Cell 16, 128–140.
Sindreu, C., Palmiter, R. D., and Storm, D. R. (2011). Zinc transporter ZnT-3 regulates presynaptic Erk1/2 signaling and hippocampus-dependent memory. Proc. Natl. Acad. Sci. U.S.A. 108, 3366–3370.
Sindreu, C. B., Varoqui, H., Erickson, J. D., and Perez-Clausell, J. (2003). Boutons containing vesicular zinc define a subpopulation of synapses with low AMPAR content in rat hippocampus. Cereb. Cortex 13, 823–829.
Slomianka, L. (1992). Neurons of origin of zinc-containing pathways and the distribution of zinc-containing boutons in the hippocampal region of the rat. Neuroscience 48, 325–352.
Slomianka, L., Danscher, G., and Frederickson, C. J. (1990). Labeling of the neurons of origin of zinc-containing pathways by intraperitoneal injections of sodium selenite. Neuroscience 38, 843–854.
Slomianka, L., Ernst, E., and Ostergaard, K. (1997). Zinc-containing neurons are distinct from GABAergic neurons in the telencephalon of the rat. Anat. Embryol. 195, 165–174.
Spencer-Segal, J. L., Waters, E. M., Bath, K. G., Chao, M. V., Mcewen, B. S., and Milner, T. A. (2011) Distribution of phosphorylated TrkB receptor in the mouse hippocampal formation depends on sex and estrous cycle stage. J Neurosci 31, 6780–6790.
Steele, R. J., and Morris, R. G. (1999). Delay-dependent impairment of a matching-to-place task with chronic and intrahippocampal infusion of the NMDA-antagonist D-AP5. Hippocampus 9, 118–136.
Suh, S. W. (2009). Detection of zinc translocation into apical dendrite of CA1 pyramidal neuron after electrical stimulation. J. Neurosci. Methods 177, 1–13.
Takeda, A. (2000). Movement of zinc and its functional significance in the brain. Brain Res. Brain Res. Rev. 34, 137–148.
Takeda, A., Fuke, S., Ando, M., and Oku, N. (2009). Positive modulation of long-term potentiation at hippocampal CA1 synapses by low micromolar concentrations of zinc. Neuroscience 158, 585–591.
Takeda, A., Tamano, H., Imano, S., and Oku, N. (2010). Increases in extracellular zinc in the amygdala in acquisition and recall of fear experience and their roles in response to fear. Neuroscience 168, 715–722.
Takeda, A., Tamano, H., Kan, F., Itoh, H., and Oku, N. (2007). Anxiety-like behavior of young rats after 2-week zinc deprivation. Behav. Brain Res. 177, 1–6.
Tassabehji, N. M., Corniola, R. S., Alshingiti, A., and Levenson, C. W. (2008). Zinc deficiency induces depression-like symptoms in adult rats. Physiol. Behav. 95, 365–369.
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183.
Timm, F. (1958). Histochemistry of the region of Ammon’s horn. Z. Zellforsch. Mikrosk. Anat. 48, 548–555.
Timofeeva, O., and Nadler, J. V. (2006). Facilitation of granule cell epileptiform activity by mossy fiber-released zinc in the pilocarpine model of temporal lobe epilepsy. Brain Res. 1078, 227–234.
Traynelis, S. F., Burgess, M. F., Zheng, F., Lyuboslavsky, P., and Powers, J. L. (1998). Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J. Neurosci. 18, 6163–6175.
Ueno, S., Tsukamoto, M., Hirano, T., Kikuchi, K., Yamada, M. K., Nishiyama, N., Nagano, T., Matsuki, N., and Ikegaya, Y. (2002). Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-D-aspartate receptor activity in hippocampal CA3 circuits. J. Cell Biol. 158, 215–220.
Vallee, B. L., and Falchuk, K. H. (1993). The biochemical basis of zinc physiology. Physiol. Rev. 73, 79–118.
Vara, H., Onofri, F., Benfenati, F., Sassoe-Pognetto, M., and Giustetto, M. (2009). ERK activation in axonal varicosities modulates presynaptic plasticity in the CA3 region of the hippocampus through synapsin I. Proc. Natl. Acad. Sci. U.S.A. 106, 9872–9877.
Ventura, R., and Harris, K. M. (1999). Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 19, 6897–6906.
Vinkenborg, J. L., Koay, M. S., and Merkx, M. (2010). Fluorescent imaging of transition metal homeostasis using genetically encoded sensors. Curr. Opin. Chem. Biol. 14, 231–237.
Vinkenborg, J. L., Nicolson, T. J., Bellomo, E. A., Koay, M. S., Rutter, G. A., and Merkx, M. (2009). Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat. Methods 6, 737–740.
Vogt, K., Mellor, J., Tong, G., and Nicoll, R. (2000). The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 26, 187–196.
Wenzel, H. J., Cole, T. B., Born, D. E., Schwartzkroin, P. A., and Palmiter, R. D. (1997). Ultrastructural localization of zinc transporter-3 (ZnT-3) to synaptic vesicle membranes within mossy fiber boutons in the hippocampus of mouse and monkey. Proc. Natl. Acad. Sci. U.S.A. 94, 12676–12681.
Whittle, N., Hauschild, M., Lubec, G., Holmes, A., and Singewald, N. (2010). Rescue of impaired fear extinction and normalization of cortico-amygdala circuit dysfunction in a genetic mouse model by dietary zinc restriction. J. Neurosci. 30, 13586–13596.
Wiltgen, B. J., Zhou, M., Cai, Y., Balaji, J., Karlsson, M. G., Parivash, S. N., Li, W., and Silva, A. J. (2010). The hippocampus plays a selective role in the retrieval of detailed contextual memories. Curr. Biol. 20, 1336–1344.
Woodroofe, C. C., Masalha, R., Barnes, K. R., Frederickson, C. J., and Lippard, S. J. (2004). Membrane-permeable and -impermeable sensors of the Zinpyr family and their application to imaging of hippocampal zinc in vivo. Chem. Biol. 11, 1659–1666.
Ye, X., and Carew, T. J. (2010). Small G protein signaling in neuronal plasticity and memory formation: the specific role of ras family proteins. Neuron 68, 340–361.
Keywords: synaptic zinc, memory, neuronal plasticity, signal transduction
Citation: Sindreu C and Storm DR (2011) Modulation of neuronal signal transduction and memory formation by synaptic zinc. Front. Behav. Neurosci. 5:68. doi: 10.3389/fnbeh.2011.00068
Received: 19 August 2011;
Accepted: 03 October 2011;
Published online: 09 November 2011.
Edited by:
Riccardo Brambilla, San Raffaele Scientific Institute and University, ItalyCopyright: © 2011 Sindreu and Storm. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Daniel R. Storm, Department of Pharmacology, University of Washington, 1959 NE Pacific Street, Seattle, WA 98103, USA. e-mail:ZHN0b3JtQHV3LmVkdQ==