 Jose V. Sanchez-Mut
Jose V. Sanchez-Mut Johannes Gräff
Johannes Gräff- Neuroepigenetics Laboratory – UPGRAEFF, Brain Mind Institute, School of Life Sciences, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland
Alzheimer’s disease (AD) is the major cause of dementia in Western societies. It progresses asymptomatically during decades before being belatedly diagnosed when therapeutic strategies have become unviable. Although several genetic alterations have been associated with AD, the vast majority of AD cases do not show strong genetic underpinnings and are thus considered a consequence of non-genetic factors. Epigenetic mechanisms allow for the integration of long-lasting non-genetic inputs on specific genetic backgrounds, and recently, a growing number of epigenetic alterations in AD have been described. For instance, an accumulation of dysregulated epigenetic mechanisms in aging, the predominant risk factor of AD, might facilitate the onset of the disease. Likewise, mutations in several enzymes of the epigenetic machinery have been associated with neurodegenerative processes that are altered in AD such as impaired learning and memory formation. Genome-wide and locus-specific epigenetic alterations have also been reported, and several epigenetically dysregulated genes validated by independent groups. From these studies, a picture emerges of AD as being associated with DNA hypermethylation and histone deacetylation, suggesting a general repressed chromatin state and epigenetically reduced plasticity in AD. Here we review these recent findings and discuss several technical and methodological considerations that are imperative for their correct interpretation. We also pay particular focus on potential implementations and theoretical frameworks that we expect will help to better direct future studies aimed to unravel the epigenetic participation in AD.
Introduction
The term “epigenetics” was introduced by the developmental biologist Conrad Hal Waddington (1905–1975) in the early 1940s. He defined epigenetics as the branch of biology that studies the causal interactions between genes and their products, which bring the phenotype into being (Waddington, 1942, 2012). This general idea was later narrowed and defined as the science that studies the heritable traits resulting from changes in a chromosome without altering the DNA sequence (Berger et al., 2009). Although nowadays widely accepted, this definition is – strictly speaking – a conception of developmental and cancer researchers, who perceive epigenetics as a way to transmit phenotypic characteristics to daughter cells. Yet, such conception represents a major problem for other disciplines like neurosciences, since neurons do not divide and, accordingly, nothing happening in neurons would be considered epigenetics. As “epigenetic newcomers,” neuroscientists have only recently started to add their viewpoints toward this perception, but due to the emerging importance of epigenetics in the nervous system, this has already stimulated a profound revision of the perception of epigenetics. Currently, epigenetics is rather considered as the study of structural changes of the chromatin that modify the phenotype without altering the genotype (Jaenisch and Bird, 2003), independently of whether the cells divide or not.
Yet, where does this recent interest of neuroscientists in epigenetics or, in other words, in neuroepigenetics originate from? Probably one of the main reasons is because epigenetic mechanisms provide a platform for integrating different inputs and for generating adaptive long-lasting outputs. This capacity of epigenetics touches upon the very fundament of neuroscience, providing a potential substrate for memory allocation, and for articulating the hypothesis of gene × environment interaction associated with many multifactorial diseases such as Parkinson’s and Alzheimer’s disease (AD), amyotrophic lateral and multiple sclerosis, and even epilepsy (Urdinguio et al., 2009). As a matter of fact, it is known that epigenetic mechanisms participate in the processes of learning and memory formation (Levenson and Sweatt, 2005; Gräff and Tsai, 2013a; Zovkic et al., 2013; Guzman-Karlsson et al., 2014; Jarome et al., 2014; Woldemichael et al., 2014), and that – on the other end of the spectrum – life style (Fraga et al., 2005), aging (Heyn et al., 2012), nutrition (Cooney et al., 2002), and environmental toxins (Anway et al., 2005) associated with AD can modify the epigenetic makeup and might thereby contribute to the pathophysiology of AD (Cacabelos and Torrellas, 2014; Coppede, 2014; Bennett et al., 2015).
Epigenetic Mechanisms
At the molecular level, it is generally accepted that epigenetics encompasses two main mechanisms: the direct methylation of the DNA, and the modification of the proteins that package the DNA, the histones. Chromatin remodelers and non-coding RNAs can also participate in the regulation of the chromatin but, because they are not considered purely epigenetic mechanisms, are not included in this review (for a further discussion about these topics see Magistri et al., 2012; Langst and Manelyte, 2015). Here, we first provide a description of the functioning of these two epigenetic modifications in general, before moving to their implication in neuroscience, and in particular, in AD.
DNA Methylation
DNA methylation has thus far been the most studied epigenetic modification. It mainly consists of the addition of a methyl group at cytosines that precede guanines (so-called CpG dinucleotides). These dinucleotides are underrepresented in the genome and tend to accumulate in CpG-dense regions (so-called CpG islands, or CGI) although around 95% of CpGs are scattered through all the genome without showing any type of aggregation. As a general view, CpGs in non-CGI and CGIs tend to be fully methylated and non-methylated, respectively, with a negligible amount of CpGs being partially methylated (Vinson and Chatterjee, 2012).
Historically, DNA methylation has been considered an epigenetic mark of repression, since seminal experiments have shown that the genomic insertion of exogenous DNA results in active transcription only with non-methylated DNA (Pollack et al., 1980; Wigler et al., 1981; Stein et al., 1982), and since CGI hypermethylation has been recurrently associated with the silencing of tissue-specific genes and X inactivation (Straussman et al., 2009). In fact, the existence of CGI together with the bimodal pattern of DNA methylation has evoked the idea that genes can be switch ON an OFF by controlling the DNA methylation of their CGIs. This idea has been predominant in the last years. However, it is now apparent that the reality is much more complex since around 70% of annotated genes contain CGI regions in their promoters (Deaton and Bird, 2011) and most of them are non-methylated. Furthermore, CGI and non-CGI promoter-containing genes show specific particularities, with the former being constitutively expressed and displaying low evolution rates and relaxed use of transcription start sites (TSS), while the latter is characterized by a more restricted pattern of expression, higher evolution rate, and strongly defined TSS usage (Tang and Epstein, 2007). Thus, DNA methylation cannot be the main driver of gene expression in these regions.
Importantly, DNA is not randomly distributed in the nucleus but associated to histones forming the nucleosomes (Schones et al., 2008). The distribution and the compaction of these nucleosomes determines the chromatin structure and thereby the access of the transcriptional machinery to the DNA (Schones et al., 2008). Accordingly, CGIs favors the access of the DNA polymerase and gene expression by constituting a rigid structure that complicates the wrapping of DNA and nucleosome positioning (Ramirez-Carrozzi et al., 2009). Therefore, CGI are not mere platforms for controlling gene expression by DNA methylation, but such effect depends on the nearby sequence and thus, three-dimensional chromatin context. In general, DNA methylation in gene promoters is associated with lower levels of expression (Kelly et al., 2012) whereas in gene bodies, it favors gene expression (Guenther et al., 2007).
The enzymes that carry out the active DNA methylation, the so called DNA methyltransferases (DNMTs), are all associated with the nucleosomes (Jeong et al., 2009), which reinforces the idea that DNA methylation and nucleosome positioning are intimately related. Three different DNMTs have been identified: DNMT1 – necessary for the maintenance of DNA methylation patterns during cell division – and DNMT3A and DNMT3B, both involved in establishing de novo patterns of DNA methylation during development and cell fate determination. Interestingly, DNMTs also show high levels of expression in post-mitotic neurons (Guo et al., 2014a), suggesting that their importance in the adult brain is beyond the classical developmental point of view. A deficit of these enzymes can cause passive DNA demethylation (Rhee et al., 2002), but DNA can also be actively demethylated by the action of several enzymatic reactions. These include the 10–11 translocation proteins (TET), which mediate the oxidation of 5-methylcytosines (5mC) to 5-hydroxymethylcytosine (5hmC), and later on to 5-formilcytosine (5fC) and 5-carboxycytosine (5caC); and the thymine-DNA glycosylases (TDG), which causes the final excision and conversion to cytosines (Kohli and Zhang, 2013).
Newly Identified DNA Methylation Marks
The recently developed techniques of deep-sequencing have documented an unexpected high prevalence of 5hmC and 5fC in brain (Lister et al., 2013; Varley et al., 2013; Guo et al., 2014a,b, Kozlenkov et al., 2014). In spite of that, it is still under discussion whether 5hmC and 5fC constitute new epigenetic marks per se or if they are just intermediate states of the DNA demethylation (Hahn et al., 2014). In the brain, around ∼80% of cytosines in CpG sites are methylated (5mC), whereas ∼8% are hydroxyl-methylated (5hmC), ∼0.8% are formyl-methylated (5fC), and even less are carboxyl-methylated (5caC). These data reflect a high prevalence of the intermediate states, in special for 5hmC, which has been used as an argument to emphasize the specific role of 5hmC in epigenetic signaling (Globisch et al., 2010; Song et al., 2011; Lister et al., 2013; Wen et al., 2014), which together with 5fC/5caC is enriched in enhancers and gene bodies of highly transcribed genes (Song et al., 2011, 2013; Shen et al., 2013; Wen et al., 2014; Raiber et al., 2015).
Also, a certain degree of DNA methylation outside of CpG dinucleotides has recently been reported. The so-called non-CpG DNA methylation mainly occurs in the context of CpA dinucleotides (Lister et al., 2009; Yan et al., 2011; Ziller et al., 2011) and is particularly prevalent in the brain where it accounts for ∼25% of all cytosine modifications (Lister et al., 2013; Guo et al., 2014a). Similarly to 5mC and 5hmC, non-CpG methylation also tends to occur in gene bodies of highly transcribed genes (Lister et al., 2013; Guo et al., 2014a).
Histone Modifications
As aforementioned, nucleosomes are important components of the chromatin structure and their positioning is influenced by DNA methylation and sequence context. Notwithstanding, nucleosomes are primarily regulated by posttranslational modifications that tend to occur in the N-terminal tail of histone proteins (Bowman and Poirier, 2015). The most studied of these are histone acetylation and methylation, which occur as a consequence of the antagonistic activity of histone acetyltransferases (HATs) and deacetylates (HDACs), and of histone methyltransferases (HMTs) and demethylases (HDMTs), respectively, as well as histone phosphorylation, which is mediated by the opposing action of protein kinases and phosphatases. Further, more recently discovered posttranslational modifications include ADP-ribosylation, ubiquitylation, sumoylation, crotonylation, propionylation, deiminiation and O-GlcNAcylation, which are also the consequence of a similar set of enzyme complexes.
These modifications can take place on different amino acids. For instance, lysines can be acetylated; mono-, di-, or trimethylated; and mono- or polyubiquitylated; arginines can be deaminated; mono-, symmetrically or asymmetrically dimethylated, and mono- or poly-ADP-ribosylated; serines and threonines can be phosphorylated and O-GlcNAcylated; glutamates can be mono- or poly-ADP-ribosylated; prolines can be isomerized; and tyrosines can be phosphorylated (Hanes, 2014; Xu et al., 2014; Bowman and Poirier, 2015).
Histone Acetylation
Depending on their identity, posttranslational histone modifications can have different effects on gene expression. In general, histone acetylation is associated with increased gene activity (Kouzarides, 2007), in part because it diminishes the basic charge of histones and thereby reduces the electrostatic interaction with the negatively charged DNA chains. As a result, nucleosome compaction is relaxed facilitating the access of the transcriptional machinery to the DNA (Li and Reinberg, 2011). In line, histone acetylation is enriched in promoter and gene bodies of active genes (Wang et al., 2008b). Some of the most studied lysine acetylation modifications include the acetylation of lysine 9 on histone 3 (H3K9ac) and H3K27ac in gene promoters and H3K4ac and H4K12ac in gene bodies, among others (Wang et al., 2008b).
Three families of HATs – GNAT, MYST, and CBP/p300 – are responsible of the acetylation of these amino acids, and the specificity of these modifications depend on their association with other regulatory proteins (Bannister and Kouzarides, 2011). In line, although many acetylation sites has been individually described, they tend to occur in combinations (Baker, 2011). Zinc-dependent class I, II and IV HDACs as well as the NAD-dependent class III HDACs, the sirtuins, antagonize the activity of HATs. Similarly to the HATs, HDAC proteins show a low level of specificity that is mainly regulated by the interaction with other non-catalytic proteins and complexes (Yang and Seto, 2007).
Histone Methylation
As opposed to acetylation, the effect of histone methylation depends on both the type of modification and the residue on which it occurs. As an example, the mono-methylation of lysine 27 of histone 3 (H3K27me1) is enriched in promoters of active genes, whereas the tri-methylation of the same amino acid (H3K27me3) is mainly found in repressed genes (Wang et al., 2008b). Also in contrast to histone acetylation, the enzymes that regulate histone methylation show a high degree of specificity. Two classes of lysine HMTs are at play: SET domain and non-SET domain-containing HMTs, which install histone methylation at particular lysines to different degrees. For instance, three different members of the SET HMTs perform H4K20 methylation in its mono-, di-, or trimethylated form (Liu et al., 2010; Qi et al., 2010). The activity of the HMTs is antagonized by two classes of HDMTs – LSD1- and Jumonji-related HDMTs – that also show a high degree of specificity. For instance, LSD1 demethylates the monomethylation and dimethylation of H3K4 and H3K9, but not their trimethylation (Bannister and Kouzarides, 2011).
The Histone Code
The sum of different histone modifications, their intricate system of regulation, and the almost infinite number of possible combinations draws a complex landscape of histone modifications that – considering that chromatin is composed by hundreds of millions of nucleosomes – can epigenetically encode an immense amount of information. This idea has inspired the hypothesis of the “histone code,” which suggests that by using different combinations of histone modifications cells can regulate specific and distinct functional chromatin outputs (Strahl and Allis, 2000; Jenuwein and Allis, 2001). This intriguing view has attracted a lot of attention in last years that has undoubtedly helped to push the development of the field. In spite of that, recent chromatin immunoprecipitation (ChIP) sequencing experiments have suggested that the complexity behind such histone code seems smaller than previously estimated. In fact, a high degree of redundancy between different histone marks has been reported (Wang et al., 2008b) and algorithms have recognized recurrent combinations of histone modifications that can be grouped into discrete chromatin states, which account for most of gene transcription variance (Baker, 2011). Obviously, this might be a simplification of the reality, and the numbers of identified chromatin states strongly depends of the level of technical resolution applied, but importantly, it underlines that from all possible combinations only few of them ever really occur together, suggesting that the histone code is simpler than anticipated, which would facilitate the task of epigeneticists considerably.
Epigenetic Crosstalk
Importantly, DNA methylation and histone modifications are not isolated phenomena, and proteins that recognize and regulate both epigenetic marks are orchestrated in multiprotein complexes. For instance, MeCP2 and KAISO proteins, which recognizes methylated DNA, are also associated with HDAC1 and HDAC3, linking DNA methylation and histone deacetylation (Suzuki et al., 2003; Yoon et al., 2003); likewise, other DNA methylation readers such as UHRF1 are known to interact with the H3K9 HMT G9A, associating, in this case, DNA methylation with H3K9 methylation (Meilinger et al., 2009; Rottach et al., 2010). Thus, the concept of a “histone code” should be expanded to one of an epigenetic code, which also applies to the nervous system (Gräff and Mansuy, 2008). Besides, the different histone marks also interact with each other, as it attested by, for instance, the fact that the HAT GCN5 is also able to recognize H3K4me3 (Guillemette et al., 2011), which can help to understand the apparent redundancy of some epigenetic marks (for a more comprehensive view of different epigenetic marks crosstalk see Du and Patel, 2014).
Beyond such crosstalk, the different epigenetic marks are ensembles interpreted by so-called chromatin remodelers – SWI/SNF, NuRD and ISWI families (for a detailed description see Langst and Manelyte, 2015) – which modify the presence, composition, and nucleosome positioning regulating the chromatin accessibility. Therefore, epigenetic modifications should be understood as chromatin states instead of isolated modifications independently associated with particular functions, which seem to have a special relevance in the nervous system.
Neuroepigenetics
The nervous system is a highly specialized system in which millions of cells are organized in different structures with characteristic epigenetic and expression profiles that are associated with particular functions (Xin et al., 2010; Ko et al., 2013; Sanchez-Mut et al., 2013). It is in the nervous system where three out of four genes are expressed (Johnson et al., 2009), where most splicing variants are transcribed (Stamm et al., 2000; Xu et al., 2002; Yeo et al., 2004) and most miRNAs are synthesized (Cao et al., 2006). Also, it is in the nervous system, where the expression patterns of cells show the highest degree of heterogeneity, with more than 70% of genes being expressed in less than 20% of the cells of the entire brain (Lein et al., 2007). Owing to this complexity, the transcription machinery faces a formidable challenge in the nervous system, and, as a consequence, is also highly sensitive to epigenetic perturbations.
Accordingly, the importance of epigenetics in the functioning of the nervous system is underlined by the fact that mutations in epigenetic genes cause severe mental disorders (Berdasco and Esteller, 2013). For example, mutations in genes that establish epigenetic marks, such as DNMT1, NSD1, NSD2, or CBP cause hereditary sensory autonomic neuropathy with dementia (HSAN1), Sotos, Wolf–Hirschhorn and Rubinstein–Taybi syndromes, respectively. Similarly, mutations in genes that remove epigenetic marks, such as KDM5C, recognize them, such as MeCP2, or are in charge of their integration, such as SWI/SNF proteins, are associated with X-linked mental retardation, Rett syndrome and Coffin-Siris syndromes respectively (Urdinguio et al., 2009; Gasser and Li, 2011; Sanchez-Mut et al., 2012; Berdasco and Esteller, 2013).
But one the most important findings that supports the importance of epigenetics in the functioning of the brain has been the discovery that neuronal activity per se modifies DNA methylation and histone modifications patterns, and further, that learning and memory depend on these epigenetic changes (Levenson et al., 2004; Miller and Sweatt, 2007; Guan et al., 2009; Ma et al., 2009; Gupta et al., 2010; Miller et al., 2010; Guo et al., 2011; Gräff et al., 2012; Zovkic et al., 2013). For instance, neuronal activity induces the expression of DNMT3A2, TET1, and TET3 (Guo et al., 2011; Oliveira et al., 2012; Rudenko et al., 2013; Li et al., 2014b) and the shuttling of HDAC4 to the nucleus (Sando et al., 2012), whereas the depletion of DNMTs 1 and 3a, of the HATs KAT2A and KAT2B, of the HDMT neuronal specific isoform LSD1, and of the HMTs GLP and G9A as well as the increased expression of MeCP2, of HDACs 2, 3, 4, and 5 impair learning and memory formation (Guan et al., 2009; Feng et al., 2010; Kramer et al., 2011; McQuown et al., 2011; Na et al., 2012; Sando et al., 2012; Agis-Balboa et al., 2013; Kerimoglu et al., 2013; Morris et al., 2013; Stilling et al., 2014; Wang et al., 2015). Furthermore, several HDAC inhibitors, such as valproic acid, sodium butyrate and others, potentiate learning and memory formation in different paradigms and animal models (Gräff and Tsai, 2013a), as well as in different neurological diseases such as Alzheimer, Parkinson’s, and Huntington diseases (Zhang et al., 2013). Therefore, it is evident that neuronal activity as well as learning and memory engage and to some degree depend on numerous epigenetic players, and that epigenetic perturbations not only impair brain normal functioning but also are associated with many neurological diseases, including AD.
Epigenetic Alterations in AD
Alzheimer’s disease is the main cause of dementia in Western societies, where it affects 17% of people older than 65 years and 50% older than 85 years (Alzheimer’s Association, 2010). AD is a neurodegenerative disorder characterized by a progressive decline in mental abilities, neuronal loss and accumulation of two types of protein aggregates, amyloid plaques and neurofibrillary tangles (NFT; Cummings, 2004). Amyloid plaques are mainly constituted by aggregates of the amyloid-β (Aβ)-peptide, itself being a consequence of the cleavage of the amyloid precursor protein (APP) by β- and γ-secretases, while NFT are aggregates of hyperphosphorylated TAU protein. These two hallmarks are unequivocally associated with AD, but whether they are cause or consequence, and the mechanisms leading to their formation and propagation are poorly understood.
It is known that genetic and non-genetic factors contribute to the development of AD. Rare mutations in three genes – APP, PSEN1, and PSEN2 – are associated with 1% of AD (Chouraki and Seshadri, 2014) and other frequent genetic variants such as APOE-E4 can account for up to 20% of total cases of the disease (Mayeux and Stern, 2012). In total, the heritability for AD is estimated to explain between one half and two thirds of total AD cases (Ertekin-Taner, 2007), the other third/half being attributable to non-genetic risk factors in which epigenetics mechanisms are supposedly involved, namely diabetes mellitus, hypertension, obesity, physical inactivity, depression, smoking and low educational attainment (Figure 1A) (Kivipelto and Mangialasche, 2014).
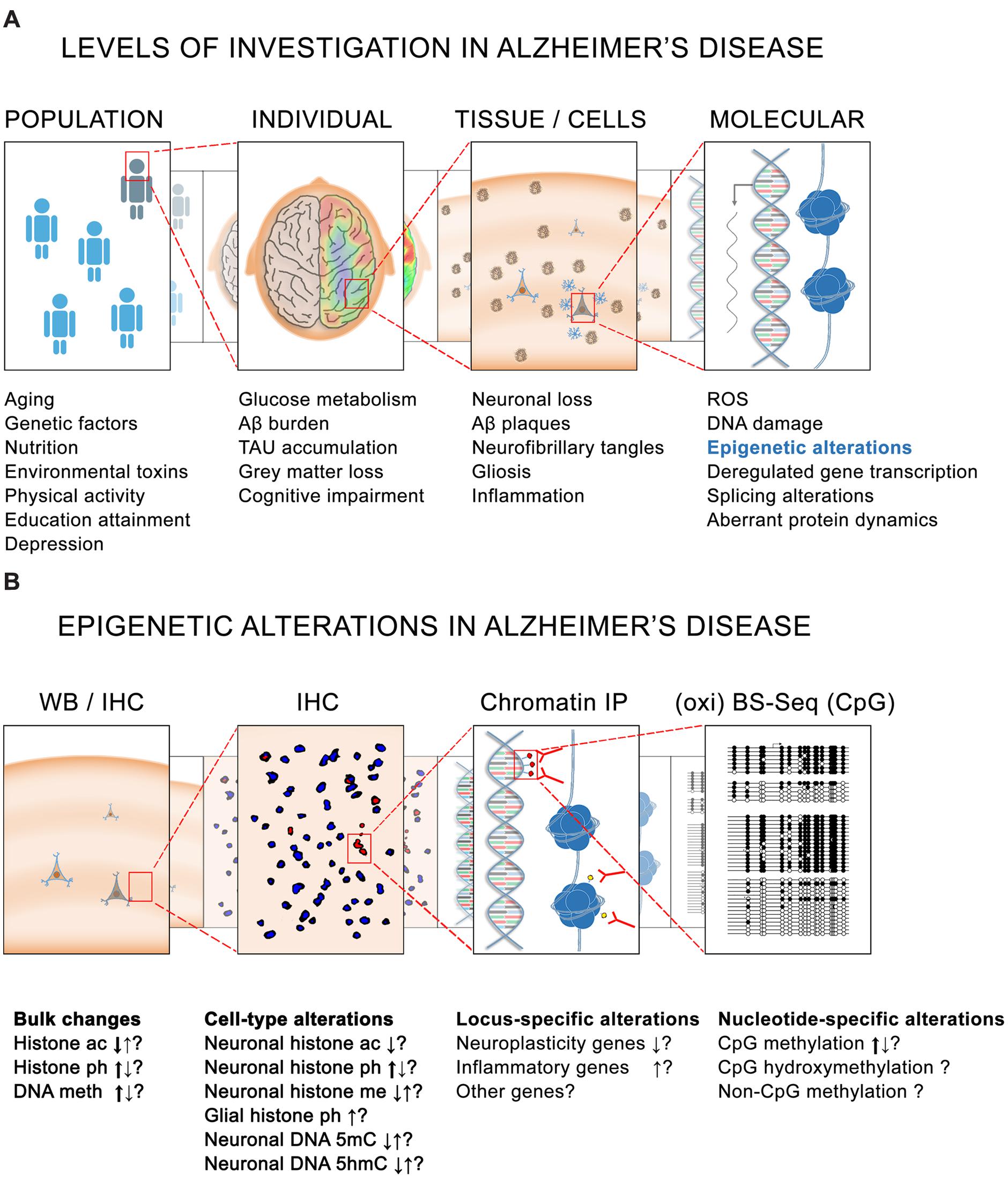
FIGURE 1. Epigenetics in Alzheimer’s disease (AD). (A) Different levels of investigation in AD pathology. At the population level, several genetic and non-genetic factors contribute to the risk for developing the disease. At the level of the individual, several pathophysiological characteristics such as altered glucose metabolism are observed in the brain of cognitively impaired AD patients. Associated with these alterations are – at the tissue and cellular level – yet other pathological hallmarks such as the presence of amyloid plaques and neurofibrillary tangles. Finally, at the intracellular level, higher levels of reactive oxygen species (ROS) and DNA damage, together with dysregulated gene transcription, splicing alterations and aberrant protein dynamics are also believed to be implicated in the onset and development of the pathology. (B) Similarly, epigenetic alterations have been reported in AD at different levels. Bulk histone acetylation (ac), phosphorylation (ph), and methylation (me) changes as well as DNA methylation (5mC) and hydroxymethylation (5hmC) alterations have been reported in AD tissues by IHC and WB. Major tendencies for these changes (as observed by several studies) are indicated by thick arrows. Locus-specific alterations mainly causing a repression of neuroplasticity genes and an activation of inflammatory genes have also been observed by ChIP using antibodies against histone modifications. Greater resolution is also possible for DNA methylation analysis in which nucleotide-specific alterations can be detected by oxi- and BS-sequencing. BS, bisulfite sequencing; ChIP, chromatin immunoprecipitation; CpG, cytosine-guanine dinucleotide; IHC, immunohistochemistry; IP, immunoprecipitation; WB, western blotting.
By far the predominant risk factor for AD is aging itself, since AD only appears in late adulthood, and the risk to develop the disease doubles every 5 years after age 65 (Kawas et al., 2000). Importantly, epigenetic mechanisms have also been suggested to be a major force of aging (Chouliaras et al., 2012, 2013b; Heyn et al., 2012) and similar epigenetic alterations have also been described in AD (Figure 1B) (Cacabelos and Torrellas, 2014; Coppede, 2014; Bennett et al., 2015). But prior to outlining the evidence of an epigenetic implication in AD, it is important to mention that this question has been addressed from different (technical) perspectives, and that the obtained results strongly depend of the experimental approaches, the samples analyzed, and the techniques used. Some of these studies are based on cell lines, others on animal models, and yet others on human post-mortem tissue, sometimes with a limited sample number at their disposal. Equally diverse are the techniques being used for determining the levels of DNA methylation, which range from the use of DNA methylation sensitive restriction enzymes, to antibodies that specifically recognize DNA methylation modifications, and to a direct reading of DNA methylation by bisulfite-sequencing (Figure 1B).
DNA Methylation
Global DNA Methylation Changes
In general, the use of cell lines, independently of the technique used, suggest that AD is associated with lower levels of DNA methylation. For instance, the glioblastoma cell line H4 harboring the Swedish mutation of APP (K670M/N671L double mutation segregating in a Swedish family), which causes an increase in Aβ production (Citron et al., 1992; Mullan et al., 1992; Haass et al., 1995), shows a general tendency toward hypomethylation as measured by DNA microarrays following bisulfite conversion (Sung et al., 2011). Similarly, treatment of the neuronal-like cell line SH-SY5Y with conditioned media obtained from cells harboring the Indiana mutation (V717F mutation identified by a group of Indiana) – associated with higher Aβ levels (Murrell et al., 1991; Suzuki et al., 1994) –induced a general DNA hypomethylation as measured by DNA methylation-sensitive antibodies (Hodgson et al., 2013). In line, brain microvascular endothelial cells subjected to high levels of synthetic Aβ show lower levels of DNA methylation as measured by high-performance liquid chromatography (HPLC) (Chen et al., 2009). Nevertheless, conversely to the previously discussed observations, IMR-32 neuroblastoma cells subjected to high levels of synthetic Aβ do not show significant alterations in DNA methylation as measured by DNA microarrays (Taher et al., 2014).
In mouse models of AD, global levels of DNA methylation have been less studied and, to our knowledge, only one work has addressed this question. Cong et al. (2014) used cortical samples of APPswe/PS1dE9 mice – harboring the Swedish APP mutation in combination with the deletion of the exon 9 of the PSEN1 resulting in increases Aβ formation (Borchelt et al., 1997) – and immunoprecipitation of the DNA using DNA methylation specific antibodies (Methylated DNA immunoprecipitation: MeDIP) followed by the hybridization of the resulting DNA to promoter microarrays. Following this approach, around 10% of analyzed genes (2346 of 20404 promoter genes contained in the array) showed higher levels of DNA methylation in the APPswe/PS1dE9 mice than in the controls, and no hypomethylated genes were reported (Cong et al., 2014). Therefore, conversely to what occurs in cell line models of the disease, at least one AD mouse model displays higher levels of DNA methylation.
The study of human post-mortem samples has not helped to solve this apparent discrepancy. Using antibodies that recognize methylated DNA, a loss of DNA methylation has been observed in the entorhinal cortex (Mastroeni et al., 2010) and the hippocampus of post-mortem samples of AD (Chouliaras et al., 2013a). Conversely, other studies using the same technique have reported no differences in the entorhinal cortex (Lashley et al., 2015) or even gains of DNA methylation in the frontal cortex (Coppieters et al., 2014), the temporal cortex (Coppieters et al., 2014) and the hippocampus of AD samples (Bradley-Whitman and Lovell, 2013). In much the same manner, ELISA 5mC assays of the entorhinal cortex of AD patients (Lashley et al., 2015) as well as DNA methylation microarrays in frontal cortex (Bakulski et al., 2012) have not shown significant DNA methylation differences.
A similarly confusing scenario is also emerging from the study of the DNA hydroxymethylation in AD. Higher levels of DNA hydroxymethylation have been reported in 3xTg-AD mice – harboring the APP Swedish, PSEN1 M146L, and P301L TAU mutations and resulting in Aβ formation and TAU phosphorylation (Oddo et al., 2003) – using specific 5hmC antibodies (Cadena-del-Castillo et al., 2014), but lower levels have been observed using the same technique in the human frontal, entorhinal, and temporal cortex (Condliffe et al., 2014; Coppieters et al., 2014) as well as in the hippocampus of post-mortem AD samples (Chouliaras et al., 2013a), with no significant differences observed in entorhinal cortex using 5hmC specific ELISA assays (Lashley et al., 2015).
It is important to mention that, in all these studies, the magnitude of DNA methylation and hydroxymethylation changes, and the number of samples analyzed, are relatively small, and as a consequence the results can easily be influenced by differences in the analyzed regions (Ladd-Acosta et al., 2007; Xin et al., 2010; Hernandez et al., 2011; Lee et al., 2011; Davies et al., 2012; Sanchez-Mut et al., 2013), interindividual variability (Turan et al., 2010; Heyn et al., 2013) and experimental fluctuations. As a result, if DNA methylation and hydroxymethylation differences are present in AD, these are likely to be either small or to be associated with only discrete regions of the genome. The study of disease-discordant twins has been crucial for unraveling the epigenetic component of common diseases (Bell and Spector, 2011), but unfortunately, only a single couple of monozygotic twins discordant for AD has been studied so far. There, AD was associated with a loss of DNA methylation and an increase of DNA hydroxymethylation (Mastroeni et al., 2009; Chouliaras et al., 2013a).
Gene-Specific DNA Methylation Changes
Attempts to determine whether specific genetic regions or particular genes are altered in AD have initially focused on genes previously associated with the disease – APP, PSEN1, and TAU genes –, and similarly to the global tendencies, no conclusive evidences have emerged from these studies.
In spite of some reports suggesting a hypomethylation in the promoter of APP in the temporal cortex of AD (West et al., 1995) and aging (Tohgi et al., 1999), studies using higher sample numbers have not been able to find differences in frontal cortex, parietal cortex, and hippocampus of AD patients (Yoshikai et al., 1990; Wang et al., 2008a; Barrachina and Ferrer, 2009). Similarly, studies aimed to determine whether PSEN1 might also be epigenetically deregulated in AD have not been conclusive. Methyl groups are directly assimilated from diet, which itself is frequently deficient in aging and AD (Ford and Almeida, 2012; Hinterberger and Fischer, 2013). Indeed, in the case of dietary depletion, the PSEN1 promoter can become hypomethylated in TgCRND8 – harboring the Swedish and V717F Indiana APP mutations (Chishti et al., 2001) – and APPswe/PS1dE9 AD models (Fuso et al., 2011; Li et al., 2015). Similar findings have been obtained in SK-N-BE neuroblastoma cell line using vitamin B6 and B12 deficient media (Fuso et al., 2005). However, although mechanistically possible, PSEN1 hypomethylation has not been observed in frontal cortex and hippocampus of AD samples (Wang et al., 2008a; Barrachina and Ferrer, 2009). And finally, no significant differences in DNA methylation in the frontal cortex or the hippocampus of post-mortem AD samples have been observed in the promoter of TAU (Barrachina and Ferrer, 2009). Therefore, it seems that at least these three classical AD-associated genes are not epigenetically dysregulated in AD at the DNA methylation level, which might indicate that DNA methylation changes do not play a role in AD, or that genetic and non-genetic forms of AD might be the results of alterations in a different subset of genes. As a consequence, unbiased genome wide screening are also starting to be performed.
Genome-Wide DNA Methylation Changes
Unfortunately, genome-wide control-case comparisons have not been more conclusive, with almost every single study reporting a different subset of altered genes which might reflect that current approaches are still immature (Table 1). However, the combination of genome-wide strategies with longitudinal studies of AD patients and mouse models yields more consistent data. Two different genes have been reported to be hypermethylated by two independent groups, namely Sorbin And SH3 Domain Containing 3 (SORBS3) (Siegmund et al., 2007; Sanchez-Mut et al., 2014) and Ankyrin 1 (ANK1) (De Jager et al., 2014; Lunnon et al., 2014). These results were obtained in an age-dependent DNA methylation study and a genome-wide DNA methylation screening in two different AD mouse models – APPswe/dE9 and 3xTg-AD – and later validated in the frontal cortex of human post-mortem AD samples, as well as from two genome-wide DNA methylation screenings in several human brain regions in differentially AD-affected samples respectively. Equally relevant seems to be the hypermethylation of the gene Insulin-Like Growth Factor Binding Protein 7 (IGFBP7), which is sustained by consistent changes in DNA methylation in the APPPS1-21 AD mouse model – harboring the Swedish APP mutation in combination with the L166P PSEN1 mutation (Radde et al., 2006) – and in human frontal cortex samples (Agbemenyah et al., 2014). Lastly, the hypermethylation of Dual Specificity Phosphatase 22 (DUSP22), similarly to ANK1, correlates with the severity of the disease and was demonstrated modify TAU phosphorylation and cell viability in vitro (Sanchez-Mut et al., 2014).
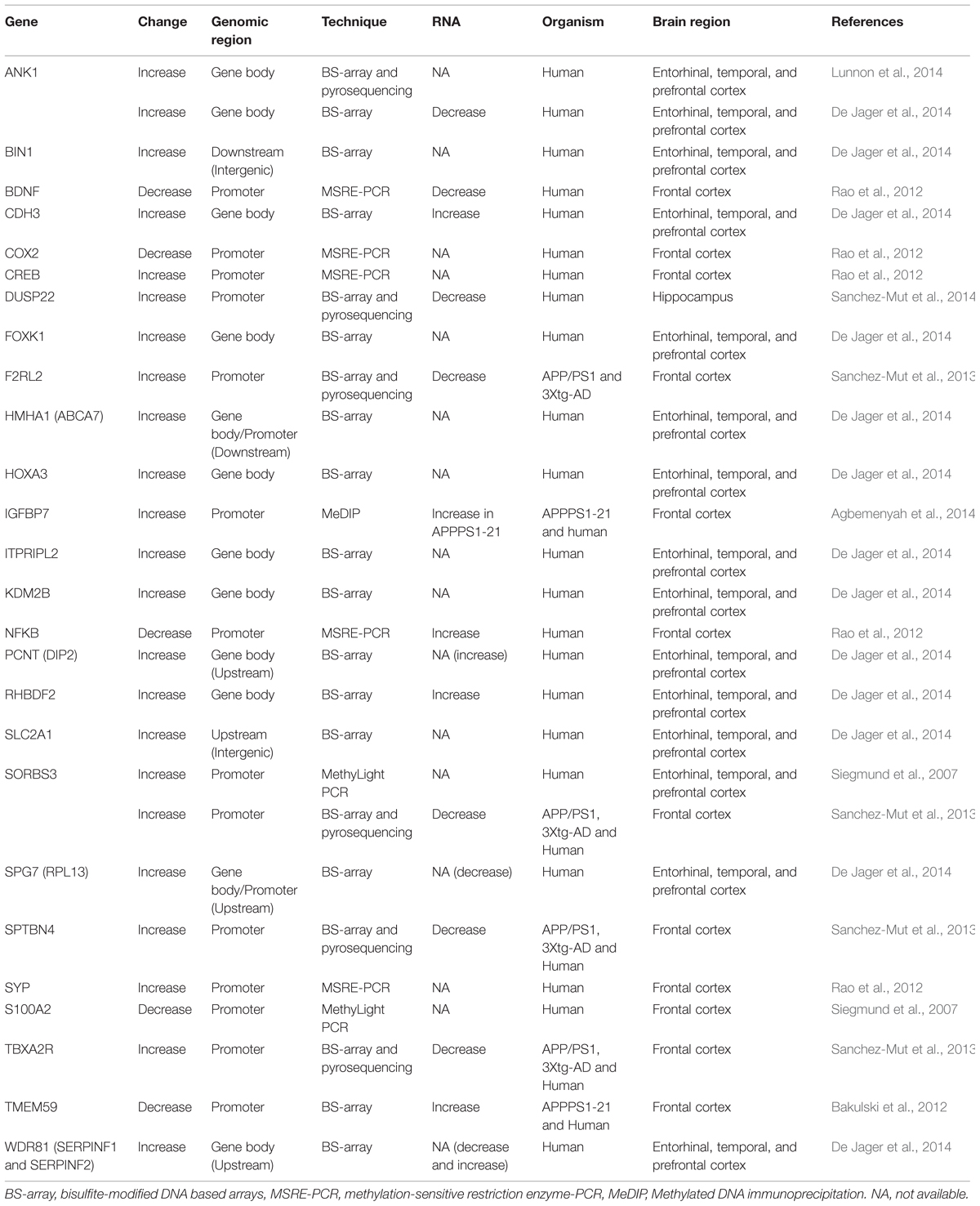
TABLE 1. Differentially DNA methylated genes reported in Alzheimer’s disease (AD).
Nonetheless, it has to be noted that these correlations do not necessarily reflect a causal relation with the disease, and might even be the consequence of secondary alterations. This is particularly important for the ones observed in mouse models, since these models have already a genetic predisposition to develop AD pathology. Also, another limitation of these studies is that they did not distinguish between different cell populations, the proportions of which are already altered in AD (AD being a neurodegenerative disease associated with a prominent gliosis and a specific loss of neurons (Serrano-Pozo et al., 2011), and which present distinct epigenetic profiles (Iwamoto et al., 2011; Labonte et al., 2013; Lister et al., 2013). Therefore, although promising, these results should be considered with caution since they will require further validations using cell-type specific studies.
Histone Modifications
Global Histone Acetylation Changes
Contrary to DNA methylation, histone modifications have been less studied in AD and evidences linking histone modification alterations with AD are mainly indirect. The few studies that, have found that several HDAC inhibitors exert a protective effect in AD, improving dendritic spine density, and facilitating learning and memory formation in different mouse models of the disease (Fischer et al., 2007; Francis et al., 2009; Ricobaraza et al., 2009, 2012; Zhang and Schluesener, 2013; Rumbaugh et al., 2015), although the precise mechanisms by which the HDAC inhibitors work remain to be determined. Furthermore, HDAC2 was found to be elevated with age in mice and humans (Chouliaras et al., 2013b; Singh and Thakur, 2014), in APP/PS1 (Gonzalez-Zuniga et al., 2014), p25/Cdk5 – harboring the Cdk5 activator p25 transgene that induces TAU phosphorylation and neurodegeneration (Cruz et al., 2006) – and 5xFAD AD mouse models – harboring the Swedish, I716V Florida, and V717I London APP mutations in combination with the M146L and L286L PSEN1 mutations with induce Aβ formation and neurodegeneration (Oakley et al., 2006) – as well as in the hippocampus and entorhinal cortex of post-mortem human AD samples (Gräff et al., 2012). In line, it has been shown that HDAC2 is able to differentially bind and regulate the expression of several learning and neuroplasticity-related genes, but that its viral-mediated depletion or its specific pharmacological inhibition is sufficient for restoring the synaptic and cognitive deficits observed in p25/Cdk5 mice (Gräff et al., 2012; Wagner et al., 2015). Therefore, there is compelling evidence that HDAC2 is increased in aging and AD, and probably implicated in the associated cognitive decline, although it should be mentioned that a decrease of HDAC2 in AD patients has been also reported by another study (Mastroeni et al., 2011).
Surprisingly, in spite of these evidences, it is still not clear whether basal histone acetylation is altered in AD. Lower (Zhang et al., 2012), equal (Rao et al., 2012; Lu et al., 2014), and higher (Narayan et al., 2015) levels of histone acetylation have been reported for post-mortem AD human samples, whereas no differences have been observed in two different AD mouse models – namely Tg2576 and 3xTg-AD – (Francis et al., 2009; Cadena-del-Castillo et al., 2014), although an increase of H3 and H4 acetylation in primary cultures of the 3xTg-AD mouse has been described by others (Walker et al., 2013). One possible explanation might be that instead of an alteration of the basal levels of histone acetylation, AD might be more related with the incapacity of modifying the epigenetic patterns in certain conditions, such as learning and memory formation, in which HDAC inhibitors that increase histone acetylation would “prime” the levels of histone acetylation and consequently, of gene activity (Gräff and Tsai, 2013a,b; Gräff et al., 2014). In support of this view, the basal levels of H4K12ac in aging remain constant, but when mice are subjected to learning and memory paradigms only young animals are able to increase these levels and not the aged mice (Peleg et al., 2010). Similarly, in Tg2576 AD mice – harboring the Swedish APP mutation in combination with the M146V PSEN1 mutation which results in higher levels of Aβ formation (Chishti et al., 2001) – the global levels of H4 acetylation are not altered, but when mice are subjected to learning and memory paradigms only wild-type animals are able to increase the levels of histone acetylation and not the Tg2576 mice (Francis et al., 2009). Alternatively, although without excluding the previous hypothesis, it could also be possible that histone acetylation alterations occur just in certain loci, which could be more sensitive to HDAC inhibitors, without reflecting general tendencies in the bulk chromatin. To better understand these scenarios, genome wide screenings of histone modifications are starting to be undertaken.
Global Tendencies in Other Histone Marks
Less attention has been put on posttranslational modifications of other histone marks, despite some results suggesting that histone phosphorylation might be altered in AD. Namely, the linker histone H1 becomes hyperphosphorylated and accumulates in the cytoplasm of astrocytes and neurons of APPswe/PS1dE9 mice (Duce et al., 2006). Interestingly, H1 is a substrate of p25/Cdk5 that accumulates in AD patients (Patrick et al., 1999; Chang et al., 2012) and is associated with neurodegeneration and cellular damage such as false entrance of cell cycle division (Cruz et al., 2006), and H2A.X phosphorylation (Kim et al., 2008), both pathophysiological characteristics of AD (Ogawa et al., 2003; Myung et al., 2008). In addition to H1 phosphorylation, evidence for H3 phosphorylation has been mixed thus far (Rao et al., 2012; Anderson et al., 2015), suggesting that more studies are necessary for elucidating its role in AD.
The first attempts for studying potential alterations on H3K9 methylation in AD has been equally inconclusive since only three studies have address this question with contradictory results: decreased heterochromatin compaction associated with lower H3K9me2 levels in a TAU Drosophila AD model and human samples (Frost et al., 2014), no significant differences in the heterochromatin of p25/cdk5 Ad mouse model measured by H3K9me3 (Gjoneska et al., 2015), and increased compaction in primary cultures of 3xTg-AD mouse measured by H3K9me2 (Walker et al., 2013).
Gene-Specific Histone Alterations
The possibility that in AD specific genes might be posttranslationally modified on their histones has just started to be addressed, and to the extent of our knowledge, only two studies in the p25/Cdk5 AD mouse model addressed this point. In 2012, several neuroplasticity related genes were reported as hypoacetylated and repressed in p25/Cdk5 mice (Table 2; Gräff et al., 2012) and, recently, the catalog of deregulated genes and posttranslational modifications has been enormously enlarged (Gjoneska et al., 2015). There, in general, complementary gains and losses of specific marks at discrete loci were observed, explaining the minor global alterations reported in previous studies (Gjoneska et al., 2015). An interesting finding of this study was further a consistent enrichment of active marks (H3K27ac and H3K4me3) in enhancers and promoters of immune and stimulus-response functions coupled with a specific decrease in synapse and learning-associated functions can be observed (Gjoneska et al., 2015).
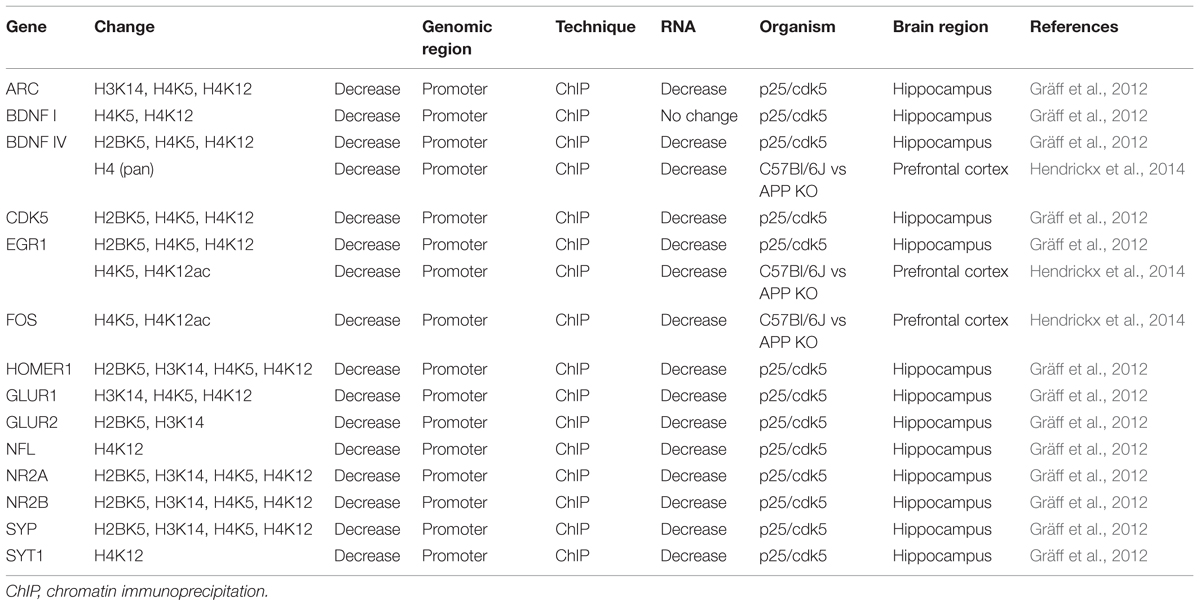
TABLE 2. Differentially histone acetylated genes in AD.
Similarly to reported DNA methylation alterations in AD, these results probably reflect both changes in cell composition and cell-type-specific changes associated with AD pathology, thereby necessitating cell-specific validations for a better evaluation of their significance in AD.
Considerations
There is an increasing interest of neuroscientists in epigenetics that is likely to result in a fruitful synergy, which will undoubtedly push the frontiers of both fields: epigenetic researchers have approached neuroscience, and vice versa, neuroscientists have also approached epigenetics. Nonetheless, the following conceptual, methodological, and biological caveats need to be properly addressed from both the epigenetic and the neuroscience point of view in order draw meaningful conclusions from these studies.
DNA Methylation Marks: Independent or Redundant?
In spite of the abundant literature arguing in favor of the specific roles for the different types of DNA methylation mark, it is still unclear whether they are independent or redundant. 5mC, 5hmC, and 5fC/5caC show similar distribution and association with gene transcription, being mainly enriched in gene bodies and correlated with gene expression (Guenther et al., 2007; Song et al., 2011; Shen et al., 2013; Wu et al., 2014), and when occurring in TSS, associated with gene repression (Guenther et al., 2007; Shen et al., 2013). In line, 5mC and 5hmC are significantly correlated (Lashley et al., 2015). Furthermore, non-CpG methylation tends to occur on gene bodies of highly transcribed genes (Lister et al., 2013; Guo et al., 2014a) and to accumulate in aging (data presented in AD/PD 2015) in the same manner than 5mC and 5hmC (Hernandez et al., 2011; Szulwach et al., 2011). A possible explanation for this might be that CpGs are continuously being methylated and demethylated in promoters of highly transcribed genes questioning their specific effect and explaining the high prevalence and concordance of these DNA methylation marks (Neri et al., 2015). 5mC, 5hmC, and 5fC/5caC are indeed produced in a stepwise manner (Kohli and Zhang, 2013), and it has been suggested that non-CpG methylation could be a consequence of the DNMT3B binding to previously methylated CpG sites (Ramsahoye et al., 2000; Arand et al., 2012, Baubec et al., 2015). Therefore, it is possible that, similarly to the initial high expectations about the histone code, that the complexity behind DNA methylation might be currently overestimated.
Then, it is also worth to mention that different techniques seem to show different scenarios. There is an apparent discrepancy between DNA methylation levels reported by antibody-based immunoprecipitation and by classical bisulfite-dependent modification of DNA, the former usually reporting higher values than the latter (Clark et al., 2012). Furthermore, antibody-based techniques tend to enrich densely modified regions (Pastor et al., 2011) and classical bisulfite DNA modification cannot distinguish between 5mC and 5hmC (Nestor et al., 2010). Therefore, it might be possible that instead of reporting just DNA methylation differences, antibody-based techniques could be reporting a combination of DNA methylation differences and other alterations to the chromatin structure – not necessarily related with the DNA methylation differences – and that the classical bisulfite-dependent modification of DNA might be underestimating the 5mC changes. In line, most of the reported DNA methylation changes based on bisulfite-dependent DNA modifications are gains of methylation, since losses of DNA methylation should be coupled with gains of 5hmC and are consequently masked in this technique. The application of the recently developed oxidative bisulfite-dependent DNA modification could help to resolve these discrepancies since it combines the precision of the classical bisulfite DNA modification with the ability of differentiate between 5mC and 5hmC (Booth et al., 2013).
HDAC Inhibitors in Learning and AD: Only an Epigenetic Effect?
Similarly, it is also important to consider several constraints when analyzing histone modifications. The beneficial effect of HDAC inhibitors in learning and AD can be interpreted as a proof of the involvement of histone acetylation in these processes (Vogel-Ciernia and Wood, 2012; Gräff and Tsai, 2013a) but, instead, it is just proving the involvement of the inhibited enzyme per se, and not necessarily of the acetylation of histones since other non-histones substrates can be also acetylated and deacetylated (Martinez-Redondo and Vaquero, 2013; Li et al., 2014a). For instance, it is known that HDAC6 is elevated in AD patients (Ding et al., 2008), and that its deletion restores the cognitive deficits observed in APPPS1-21 mouse model of AD (Govindarajan et al., 2013), but it is also known that the main effect of this protection is a consequence of the modification of tubulin acetylation (Govindarajan et al., 2013). Similarly, it has been seen that SIRT1 decreases in aging and in AD (Julien et al., 2009; Quintas et al., 2012), and that its restoration protects against neurodegeneration (Kim et al., 2007; Gräff et al., 2013), but again its main effects are associated with non-histone substrates, including PGC-1alpha, p53, and TAU (Kim et al., 2007; Min et al., 2010). Therefore, these studies should be interpreted cautiously not only in light of the particular specificity of the HDAC inhibitor, but also in light of the potential non-histone substrates of the targeted HDAC.
Epigenetics and Gene Expression, Always Coupled?
Also, it is worth to mention that, in most cases, the interpretation of the effect of epigenetic marks on gene transcription is based on genome-wide comparisons in which significant correlations can be seen, but when single genes are being analyzed a strikingly high level of discrepancy is observed (Lopez-Atalaya and Barco, 2014). In fact, changes in gene transcription can occur independently of epigenetic modifications (Zhang et al., 2014), and changes in epigenetic modifications are not necessarily reflected by changes in gene expression (Lopez-Atalaya and Barco, 2014).
Along these lines, it is important to note that epigenetic mechanisms mainly regulate the chromatin structure, which secondarily modifies the accessibility of the genome to impact on gene regulation. Several epigenetic players also interact with the gene transcription machinery, but it is simplistic to assume that epigenetic changes will completely determine the levels of gene transcription since many other factors are also implied. More likely, epigenetics would be one of the variables of the probabilistic model that finally determines the levels and the magnitude of the potential changes in gene expression. Furthermore, most epigenetic changes occur in enhancer and regulatory regions that are not easily assignable to specific genes (Gjoneska et al., 2015; Sun and Yi, 2015). These assignments are frequently based on distance criteria, which do not necessarily reflect real interactions. In fact, chromatin conformation capture experiments have shown that the majority of regulatory elements do not interact with the nearest genes (Sanyal et al., 2012). Therefore, an important part of long-range epigenetic information is still far from being understood.
Cause or Consequence?
Finally, it is important to consider that the majority of studies investigating potential epigenetic alterations in AD are based on correlations, which do not necessarily reflect a causal relationship. Therefore, whether epigenetic modifications are driving the chromatin behavior or whether they are just a consequence of other processes happening nearby is still unknown. These studies also require considerable amounts of chromatin, which is achieved, in the best of the cases, by collecting pools of similar cell types. However, unlike in cell lines, this is a particularly difficult feature to achieve in the heterogeneous central nervous system. In fact, every single cell might have its specific epigenetic and expression profile. As a consequence, the pooling of cells could mix different epigenetic patterns, thereby masking potentially important changes and complicating any analysis. Epigenetic AD studies ought thus to be conducted in particular cell types. Recently, the first report of single-cell DNA methylation profiling has just appeared (Guo et al., 2015), raising hope that cell-type specific epigenetic profiling might in the future also become an option for AD.
In spite of these limitations, it is becoming more and more evident that by modifying the chromatin structure, epigenetic mechanisms can shape genome accessibility and thereby have an impact on gene transcription. And that, by doing so, epigenetic changes might provide a molecular substrate for “chromatin memories” with important implications for learning and memory formation and for diseases such as AD. Indeed, both memory and AD are influenced by non-genetic factors that accumulate over time (Miller and O’Callaghan, 2008). In this sense, epigenetics might store long-lasting information and provide a platform for accumulating hits over time. This idea has inspired the hypothesis of the Latent Early life Associated Regulation (LEARn) model (Lahiri et al., 2008). This model suggests that a series of harmful events throughout lifetime, from gestation to old age, could accumulate epigenetic marks that modify the expression probability of certain genes, which in turn might induce or accelerate the onset and development of AD. Whether this hypothesis will withstand further experimentations remains to be determined but, for the moment, it provides an attractive food for thoughts.
Author Contribution
JVSM and JG conceptualized and wrote the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is funded by the Swiss National Science Foundation (Project Grant 31003A_155898), by the National Center for Competence in Research (NCCR) SYNAPSY, by the Synapsis Foundation for Alzheimer Research, by the Béatrice Ederer-Weber Stiftung, and by an Alzheimer’s Association New Investigator Research Grant to JG.
References
Agbemenyah, H. Y., Agis-Balboa, R. C., Burkhardt, S., Delalle, I., and Fischer, A. (2014). Insulin growth factor binding protein 7 is a novel target to treat dementia. Neurobiol. Dis. 62, 135–143. doi: 10.1016/j.nbd.2013.09.011
Agis-Balboa, R. C., Pavelka, Z., Kerimoglu, C., and Fischer, A. (2013). Loss of HDAC5 impairs memory function: implications for Alzheimer’s disease. J. Alzheimers Dis. 33, 35–44. doi: 10.3233/JAD-2012-121009
Alzheimer’s Association (2010). 2010 Alzheimer’s disease facts and figures. Alzheimers Dement. 6, 158–194. doi: 10.1016/j.jalz.2010.01.009
Anderson, K. W., Mast, N., Pikuleva, I. A., and Turko, I. V. (2015). Histone H3 Ser57 and Thr58 phosphorylation in the brain of 5XFAD mice. FEBS Open Bio 5, 550–556. doi: 10.1016/j.fob.2015.06.009
Anway, M. D., Cupp, A. S., Uzumcu, M., and Skinner, M. K. (2005). Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308, 1466–1469. doi: 10.1126/science.1108190
Arand, J., Spieler, D., Karius, T., Branco, M. R., Meilinger, D., Meissner, A., et al. (2012). In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 8:e1002750. doi: 10.1371/journal.pgen.1002750
Baker, M. (2011). Making sense of chromatin states. Nat. Methods 8, 717–722. doi: 10.1038/nmeth.1673
Bakulski, K. M., Dolinoy, D. C., Sartor, M. A., Paulson, H. L., Konen, J. R., Lieberman, A. P., et al. (2012). Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J. Alzheimers Dis. 29, 571–588. doi: 10.3233/JAD-2012-111223
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Barrachina, M., and Ferrer, I. (2009). DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. J. Neuropathol. Exp. Neurol. 68, 880–891. doi: 10.1097/NEN.0b013e3181af2e46
Baubec, T., Colombo, D. F., Wirbelauer, C., Schmidt, J., Burger, L., Krebs, A. R., et al. (2015). Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243–247. doi: 10.1038/nature14176
Bell, J. T., and Spector, T. D. (2011). A twin approach to unraveling epigenetics. Trends Genet. 27, 116–125. doi: 10.1016/j.tig.2010.12.005
Bennett, D. A., Yu, L., Yang, J., Srivastava, G. P., Aubin, C., and De Jager, P. L. (2015). Epigenomics of Alzheimer’s disease. Transl. Res. 165, 200–220. doi: 10.1016/j.trsl.2014.05.006
Berdasco, M., and Esteller, M. (2013). Genetic syndromes caused by mutations in epigenetic genes. Hum. Genet. 132, 359–383. doi: 10.1007/s00439-013-1271-x
Berger, S. L., Kouzarides, T., Shiekhattar, R., and Shilatifard, A. (2009). An operational definition of epigenetics. Genes Dev. 23, 781–783. doi: 10.1101/gad.1787609
Booth, M. J., Ost, T. W., Beraldi, D., Bell, N. M., Branco, M. R., Reik, W., et al. (2013). Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat. Protoc. 8, 1841–1851. doi: 10.1038/nprot.2013.115
Borchelt, D. R., Ratovitski, T., van Lare, J., Lee, M. K., Gonzales, V., Jenkins, N. A., et al. (1997). Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19, 939–945. doi: 10.1016/S0896-6273(00)80974-5
Bowman, G. D., and Poirier, M. G. (2015). Post-translational modifications of histones that influence nucleosome dynamics. Chem. Rev. 115, 2274–2295. doi: 10.1021/cr500350x
Bradley-Whitman, M. A., and Lovell, M. A. (2013). Epigenetic changes in the progression of Alzheimer’s disease. Mech. Ageing Dev. 134, 486–495. doi: 10.1016/j.mad.2013.08.005
Cacabelos, R., and Torrellas, C. (2014). Epigenetic drug discovery for Alzheimer’s disease. Expert Opin. Drug Discov. 9, 1059–1086. doi: 10.1517/17460441.2014.930124
Cadena-del-Castillo, C., Valdes-Quezada, C., Carmona-Aldana, F., Arias, C., Bermudez-Rattoni, F., and Recillas-Targa, F. (2014). Age-dependent increment of hydroxymethylation in the brain cortex in the triple-transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 41, 845–854. doi: 10.3233/JAD-132285
Cao, X., Yeo, G., Muotri, A. R., Kuwabara, T., and Gage, F. H. (2006). Noncoding RNAs in the mammalian central nervous system. Annu. Rev. Neurosci. 29, 77–103. doi: 10.1146/annurev.neuro.29.051605.112839
Chang, K. H., Vincent, F., and Shah, K. (2012). Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J. Cell Sci. 125, 5124–5137. doi: 10.1242/jcs.108183
Chen, K. L., Wang, S. S., Yang, Y. Y., Yuan, R. Y., Chen, R. M., and Hu, C. J. (2009). The epigenetic effects of amyloid-beta(1-40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem. Biophys. Res. Commun. 378, 57–61. doi: 10.1016/j.bbrc.2008.10.173
Chishti, M. A., Yang, D. S., Janus, C., Phinney, A. L., Horne, P., Pearson, J., et al. (2001). Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570. doi: 10.1074/jbc.M100710200
Chouliaras, L., Mastroeni, D., Delvaux, E., Grover, A., Kenis, G., Hof, P. R., et al. (2013a). Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 34, 2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021
Chouliaras, L., van den Hove, D. L., Kenis, G., Draanen, M., Hof, P. R., van Os, J., et al. (2013b). Histone deacetylase 2 in the mouse hippocampus: attenuation of age-related increase by caloric restriction. Curr. Alzheimer Res. 10, 868–876. doi: 10.2174/1567205011310080009
Chouliaras, L., van den Hove, D. L., Kenis, G., Keitel, S., Hof, P. R., van Os, J., et al. (2012). Prevention of age-related changes in hippocampal levels of 5-methylcytidine by caloric restriction. Neurobiol. Aging 33, 1672–1681. doi: 10.1016/j.neurobiolaging.2011.06.003
Chouraki, V., and Seshadri, S. (2014). Genetics of Alzheimer’s disease. Adv. Genet. 87, 245–294. doi: 10.1016/B978-0-12-800149-3.00005-6
Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y., Seubert, P., et al. (1992). Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 360, 672–674. doi: 10.1038/360672a0
Clark, C., Palta, P., Joyce, C. J., Scott, C., Grundberg, E., Deloukas, P., et al. (2012). A comparison of the whole genome approach of MeDIP-seq to the targeted approach of the Infinium HumanMethylation450 BeadChip((R)) for methylome profiling. PLoS ONE 7:e50233. doi: 10.1371/journal.pone.0050233
Condliffe, D., Wong, A., Troakes, C., Proitsi, P., Patel, Y., Chouliaras, L., et al. (2014). Cross-region reduction in 5-hydroxymethylcytosine in Alzheimer’s disease brain. Neurobiol. Aging 35, 1850–1854. doi: 10.1016/j.neurobiolaging.2014.02.002
Cong, L., Jia, J., Qin, W., Ren, Y., and Sun, Y. (2014). Genome-wide analysis of DNA methylation in an APP/PS1 mouse model of Alzheimer’s disease. Acta Neurol. Belg. 114, 195–206. doi: 10.1007/s13760-013-0267-6
Cooney, C. A., Dave, A. A., and Wolff, G. L. (2002). Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J. Nutr. 132, 2393S–2400S.
Coppede, F. (2014). The potential of epigenetic therapies in neurodegenerative diseases. Front. Genet. 5:220. doi: 10.3389/fgene.2014.00220
Coppieters, N., Dieriks, B. V., Lill, C., Faull, R. L., Curtis, M. A., and Dragunow, M. (2014). Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol. Aging 35, 1334–1344. doi: 10.1016/j.neurobiolaging.2013.11.031
Cruz, J. C., Kim, D., Moy, L. Y., Dobbin, M. M., Sun, X., Bronson, R. T., and Tsai, L. H. (2006). p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J. Neurosci. 26, 10536–10541. doi: 10.1523/JNEUROSCI.3133-06.2006
Davies, M. N., Volta, M., Pidsley, R., Lunnon, K., Dixit, A., Lovestone, S., et al. (2012). Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 13:R43. doi: 10.1186/gb-2012-13-6-r43
De Jager, P. L., Srivastava, G., Lunnon, K., Burgess, J., Schalkwyk, L. C., Yu, L., et al. (2014). Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 17, 1156–1163. doi: 10.1038/nn.3786
Deaton, A. M., and Bird, A. (2011). CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022. doi: 10.1101/gad.2037511
Ding, H., Dolan, P. J., and Johnson, G. V. (2008). Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 106, 2119–2130. doi: 10.1111/j.1471-4159.2008.05564.x
Du, J., and Patel, D. J. (2014). Structural biology-based insights into combinatorial readout and crosstalk among epigenetic marks. Biochim. Biophys. Acta 1839, 719–727. doi: 10.1016/j.bbagrm.2014.04.011
Duce, J. A., Smith, D. P., Blake, R. E., Crouch, P. J., Li, Q. X., Masters, C. L., et al. (2006). Linker histone H1 binds to disease associated amyloid-like fibrils. J. Mol. Biol. 361, 493–505. doi: 10.1016/j.jmb.2006.06.038
Ertekin-Taner, N. (2007). Genetics of Alzheimer’s disease: a centennial review. Neurol. Clin. 25, 611–667, v. doi: 10.1016/j.ncl.2007.03.009
Feng, J., Zhou, Y., Campbell, S. L., Le, T., Li, E., Sweatt, J. D., et al. (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 13, 423–430. doi: 10.1038/nn.2514
Fischer, A., Sananbenesi, F., Wang, X., Dobbin, M., and Tsai, L. H. (2007). Recovery of learning and memory is associated with chromatin remodelling. Nature 447, 178–182. doi: 10.1038/nature05772
Ford, A. H., and Almeida, O. P. (2012). Effect of homocysteine lowering treatment on cognitive function: a systematic review and meta-analysis of randomized controlled trials. J. Alzheimers Dis. 29, 133–149. doi: 10.3233/JAD-2012-111739
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., et al. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609. doi: 10.1073/pnas.0500398102
Francis, Y. I., Fa, M., Ashraf, H., Zhang, H., Staniszewski, A., Latchman, D. S., et al. (2009). Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer’s disease. J. Alzheimers Dis. 18, 131–139. doi: 10.3233/JAD-2009-1134
Frost, B., Hemberg, M., Lewis, J., and Feany, M. B. (2014). Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 17, 357–366. doi: 10.1038/nn.3639
Fuso, A., Nicolia, V., Pasqualato, A., Fiorenza, M. T., Cavallaro, R. A., and Scarpa, S. (2011). Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol. Aging 32, 187–199. doi: 10.1016/j.neurobiolaging.2009.02.013
Fuso, A., Seminara, L., Cavallaro, R. A., D’Anselmi, F., and Scarpa, S. (2005). S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol. Cell. Neurosci. 28, 195–204. doi: 10.1016/j.mcn.2004.09.007
Gasser, S. M., and Li, E. (2011). Epigenetics and disease: pharmaceutical opportunities. Preface. Prog. Drug Res. 67, v–viii. doi: 10.1007/978-3-7643-8989-5
Gjoneska, E., Pfenning, A. R., Mathys, H., Quon, G., Kundaje, A., Tsai, L. H., et al. (2015). Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518, 365–369. doi: 10.1038/nature14252
Globisch, D., Munzel, M., Muller, M., Michalakis, S., Wagner, M., Koch, S., et al. (2010). Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE 5:e15367. doi: 10.1371/journal.pone.0015367
Gonzalez-Zuniga, M., Contreras, P. S., Estrada, L. D., Chamorro, D., Villagra, A., Zanlungo, S., et al. (2014). c-Abl stabilizes HDAC2 levels by tyrosine phosphorylation repressing neuronal gene expression in Alzheimer’s disease. Mol. Cell 56, 163–173. doi: 10.1016/j.molcel.2014.08.013
Govindarajan, N., Rao, P., Burkhardt, S., Sananbenesi, F., Schluter, O. M., Bradke, F., et al. (2013). Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 5, 52–63. doi: 10.1002/emmm.201201923
Gräff, J., Joseph, N. F., Horn, M. E., Samiei, A., Meng, J., Seo, J., et al. (2014). Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell 156, 261–276. doi: 10.1016/j.cell.2013.12.020
Gräff, J., Kahn, M., Samiei, A., Gao, J., Ota, K. T., Rei, D., et al. (2013). A dietary regimen of caloric restriction or pharmacological activation of SIRT1 to delay the onset of neurodegeneration. J. Neurosci. 33, 8951–8960. doi: 10.1523/JNEUROSCI.5657-12.2013
Gräff, J., and Mansuy, I. M. (2008). Epigenetic codes in cognition and behaviour. Behav. Brain Res. 192, 70–87. doi: 10.1016/j.bbr.2008.01.021
Gräff, J., Rei, D., Guan, J. S., Wang, W. Y., Seo, J., Hennig, K. M., et al. (2012). An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226. doi: 10.1038/nature10849
Gräff, J., and Tsai, L. H. (2013a). Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111. doi: 10.1038/nrn3427
Gräff, J., and Tsai, L. H. (2013b). The potential of HDAC inhibitors as cognitive enhancers. Annu. Rev. Pharmacol. Toxicol. 53, 311–330. doi: 10.1146/annurev-pharmtox-011112-140216
Guan, J. S., Haggarty, S. J., Giacometti, E., Dannenberg, J. H., Joseph, N., Gao, J., et al. (2009). HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. doi: 10.1038/nature07925
Guenther, M. G., Levine, S. S., Boyer, L. A., Jaenisch, R., and Young, R. A. (2007). A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77–88. doi: 10.1016/j.cell.2007.05.042
Guillemette, B., Drogaris, P., Lin, H. H., Armstrong, H., Hiragami-Hamada, K., Imhof, A., et al. (2011). H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet. 7:e1001354. doi: 10.1371/journal.pgen.1001354
Guo, H., Zhu, P., Guo, F., Li, X., Wu, X., Fan, X., et al. (2015). Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nat. Protoc. 10, 645–659. doi: 10.1038/nprot.2015.039
Guo, J. U., Su, Y., Shin, J. H., Shin, J., Li, H., Xie, B., et al. (2014a). Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 17, 215–222. doi: 10.1038/nn.3607
Guo, W., Chung, W. Y., Qian, M., Pellegrini, M., and Zhang, M. Q. (2014b). Characterizing the strand-specific distribution of non-CpG methylation in human pluripotent cells. Nucleic Acids Res. 42, 3009–3016. doi: 10.1093/nar/gkt1306
Guo, J. U., Su, Y., Zhong, C., Ming, G. L., and Song, H. (2011). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434. doi: 10.1016/j.cell.2011.03.022
Gupta, S., Kim, S. Y., Artis, S., Molfese, D. L., Schumacher, A., Sweatt, J. D., et al. (2010). Histone methylation regulates memory formation. J. Neurosci. 30, 3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010
Guzman-Karlsson, M. C., Meadows, J. P., Gavin, C. F., Hablitz, J. J., and Sweatt, J. D. (2014). Transcriptional and epigenetic regulation of Hebbian and non-Hebbian plasticity. Neuropharmacology 80, 3–17. doi: 10.1016/j.neuropharm.2014.01.001
Haass, C., Lemere, C. A., Capell, A., Citron, M., Seubert, P., Schenk, D., et al. (1995). The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1, 1291–1296. doi: 10.1038/nm1295-1291
Hahn, M. A., Szabo, P. E., and Pfeifer, G. P. (2014). 5-Hydroxymethylcytosine: a stable or transient DNA modification? Genomics 104, 314–323. doi: 10.1016/j.ygeno.2014.08.015
Hanes, S. D. (2014). Prolyl isomerases in gene transcription. Biochim. Biophys. Acta 1850, 2017–2034. doi: 10.1016/j.bbagen.2014.10.028
Hendrickx, A., Pierrot, N., Tasiaux, B., Schakman, O., Kienlen-Campard, P., De Smet, C., et al. (2014). Epigenetic regulations of immediate early genes expression involved in memory formation by the amyloid precursor protein of Alzheimer disease. PLoS ONE 9:e99467. doi: 10.1371/journal.pone.0099467
Hernandez, D. G., Nalls, M. A., Gibbs, J. R., Arepalli, S., van der Brug, M., Chong, S., et al. (2011). Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 20, 1164–1172. doi: 10.1093/hmg/ddq561
Heyn, H., Li, N., Ferreira, H. J., Moran, S., Pisano, D. G., Gomez, A., et al. (2012). Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. U.S.A. 109, 10522–10527. doi: 10.1073/pnas.1120658109
Heyn, H., Moran, S., Hernando-Herraez, I., Sayols, S., Gomez, A., Sandoval, J., et al. (2013). DNA methylation contributes to natural human variation. Genome Res. 23, 1363–1372. doi: 10.1101/gr.154187.112
Hinterberger, M., and Fischer, P. (2013). Folate and Alzheimer: when time matters. J. Neural Transm. (Vienna) 120, 211–224. doi: 10.1007/s00702-012-0822-y
Hodgson, N., Trivedi, M., Muratore, C., Li, S., and Deth, R. (2013). Soluble oligomers of amyloid-beta cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers Dis. 36, 197–209. doi: 10.3233/JAD-130101
Iwamoto, K., Bundo, M., Ueda, J., Oldham, M. C., Ukai, W., Hashimoto, E., et al. (2011). Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 21, 688–696. doi: 10.1101/gr.112755.110
Jaenisch, R., and Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33(Suppl. ), 245–254. doi: 10.1038/ng1089
Jarome, T. J., Thomas, J. S., and Lubin, F. D. (2014). The epigenetic basis of memory formation and storage. Prog. Mol. Biol. Transl. Sci. 128, 1–27. doi: 10.1016/B978-0-12-800977-2.00001-2
Jenuwein, T., and Allis, C. D. (2001). Translating the histone code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Jeong, S., Liang, G., Sharma, S., Lin, J. C., Choi, S. H., Han, H., et al. (2009). Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol. Cell. Biol. 29, 5366–5376. doi: 10.1128/MCB.00484-09
Johnson, M. B., Kawasawa, Y. I., Mason, C. E., Krsnik, Z., Coppola, G., Bogdanovic, D., et al. (2009). Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron 62, 494–509. doi: 10.1016/j.neuron.2009.03.027
Julien, C., Tremblay, C., Emond, V., Lebbadi, M., and Salem, N. Jr. (2009). Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 68, 48–58. doi: 10.1097/NEN.0b013e3181922348
Kawas, C., Gray, S., Brookmeyer, R., Fozard, J., and Zonderman, A. (2000). Age-specific incidence rates of Alzheimer’s disease: the Baltimore Longitudinal Study of Aging. Neurology 54, 2072–2077. doi: 10.1212/WNL.54.11.2072
Kelly, T. K., Liu, Y., Lay, F. D., Liang, G., Berman, B. P., and Jones, P. A. (2012). Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 22, 2497–2506. doi: 10.1101/gr.143008.112
Kerimoglu, C., Agis-Balboa, R. C., Kranz, A., Stilling, R., Bahari-Javan, S., Benito-Garagorri, E., et al. (2013). Histone-methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J. Neurosci. 33, 3452–3464. doi: 10.1523/JNEUROSCI.3356-12.2013
Kim, D., Frank, C. L., Dobbin, M. M., Tsunemoto, R. K., Tu, W., Peng, P. L., et al. (2008). Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 60, 803–817. doi: 10.1016/j.neuron.2008.10.015
Kim, D., Nguyen, M. D., Dobbin, M. M., Fischer, A., Sananbenesi, F., Rodgers, J. T., et al. (2007). SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179. doi: 10.1038/sj.emboj.7601758
Kivipelto, M., and Mangialasche, F. (2014). Alzheimer disease: to what extent can Alzheimer disease be prevented? Nat. Rev. Neurol. 10, 552–553. doi: 10.1038/nrneurol.2014.170
Ko, Y., Ament, S. A., Eddy, J. A., Caballero, J., Earls, J. C., Hood, L., et al. (2013). Cell type-specific genes show striking and distinct patterns of spatial expression in the mouse brain. Proc. Natl. Acad. Sci. U.S.A. 110, 3095–3100. doi: 10.1073/pnas.1222897110
Kohli, R. M., and Zhang, Y. (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. doi: 10.1038/nature12750
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Kozlenkov, A., Roussos, P., Timashpolsky, A., Barbu, M., Rudchenko, S., Bibikova, M., et al. (2014). Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 42, 109–127. doi: 10.1093/nar/gkt838
Kramer, J. M., Kochinke, K., Oortveld, M. A., Marks, H., Kramer, D., de Jong, E. K., et al. (2011). Epigenetic regulation of learning and memory by Drosophila EHMT/G9a. PLoS Biol. 9:e1000569. doi: 10.1371/journal.pbio.1000569
Labonte, B., Suderman, M., Maussion, G., Lopez, J. P., Navarro-Sanchez, L., Yerko, V., et al. (2013). Genome-wide methylation changes in the brains of suicide completers. Am. J. Psychiatry 170, 511–520. doi: 10.1176/appi.ajp.2012.12050627
Ladd-Acosta, C., Pevsner, J., Sabunciyan, S., Yolken, R. H., Webster, M. J., Dinkins, T., et al. (2007). DNA methylation signatures within the human brain. Am. J. Hum. Genet. 81, 1304–1315. doi: 10.1086/524110
Lahiri, D. K., Zawia, N. H., Greig, N. H., Sambamurti, K., and Maloney, B. (2008). Early-life events may trigger biochemical pathways for Alzheimer’s disease: the “LEARn” model. Biogerontology 9, 375–379. doi: 10.1007/s10522-008-9162-6
Langst, G., and Manelyte, L. (2015). Chromatin remodelers: from function to dysfunction. Genes 6, 299–324. doi: 10.3390/genes6020299
Lashley, T., Gami, P., Valizadeh, N., Li, A., Revesz, T., and Balazs, R. (2015). Alterations in global DNA methylation and hydroxymethylation are not detected in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 41, 497–506. doi: 10.1111/nan.12183
Lee, R. S., Tamashiro, K. L., Aryee, M. J., Murakami, P., Seifuddin, F., Herb, B., et al. (2011). Adaptation of the CHARM DNA methylation platform for the rat genome reveals novel brain region-specific differences. Epigenetics 6, 1378–1390. doi: 10.4161/epi.6.11.18072
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. doi: 10.1038/nature05453
Levenson, J. M., O’Riordan, K. J., Brown, K. D., Trinh, M. A., Molfese, D. L., and Sweatt, J. D. (2004). Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 279, 40545–40559. doi: 10.1074/jbc.M402229200
Levenson, J. M., and Sweatt, J. D. (2005). Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 6, 108–118. doi: 10.1038/nrn1604
Li, G., and Reinberg, D. (2011). Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 21, 175–186. doi: 10.1016/j.gde.2011.01.022
Li, T., Song, B., Wu, Z., Lu, M., and Zhu, W. G. (2014a). Systematic identification of Class I HDAC substrates. Brief. Bioinform. 15, 963–972. doi: 10.1093/bib/bbt060
Li, X., Wei, W., Zhao, Q. Y., Widagdo, J., Baker-Andresen, D., Flavell, C. R., et al. (2014b). Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc. Natl. Acad. Sci. U.S.A. 111, 7120–7125. doi: 10.1073/pnas.1318906111
Li, W., Liu, H., Yu, M., Zhang, X., Zhang, M., Wilson, J. X., et al. (2015). Folic acid administration inhibits amyloid beta-peptide accumulation in APP/PS1 transgenic mice. J. Nutr. Biochem. 26, 883–891. doi: 10.1016/j.jnutbio.2015.03.009
Lister, R., Mukamel, E. A., Nery, J. R., Urich, M., Puddifoot, C. A., Johnson, N. D., et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905. doi: 10.1126/science.1237905
Lister, R., Pelizzola, M., Dowen, R. H., Hawkins, R. D., Hon, G., Tonti-Filippini, J., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. doi: 10.1038/nature08514
Liu, W., Tanasa, B., Tyurina, O. V., Zhou, T. Y., Gassmann, R., Liu, W. T., et al. (2010). PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 466, 508–512. doi: 10.1038/nature09272
Lopez-Atalaya, J. P., and Barco, A. (2014). Can changes in histone acetylation contribute to memory formation? Trends Genet. 30, 529–539. doi: 10.1016/j.tig.2014.09.003
Lu, T., Aron, L., Zullo, J., Pan, Y., Kim, H., Chen, Y., et al. (2014). REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454. doi: 10.1038/nature13163
Lunnon, K., Smith, R., Hannon, E., De Jager, P. L., Srivastava, G., Volta, M., et al. (2014). Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 17, 1164–1170. doi: 10.1038/nn.3782
Ma, D. K., Jang, M. H., Guo, J. U., Kitabatake, Y., Chang, M. L., Pow-Anpongkul, N., et al. (2009). Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science 323, 1074–1077. doi: 10.1126/science.1166859
Magistri, M., Faghihi, M. A., St Laurent, G. III, and Wahlestedt, C. (2012). Regulation of chromatin structure by long noncoding RNAs: focus on natural antisense transcripts. Trends Genet. 28, 389–396. doi: 10.1016/j.tig.2012.03.013
Martinez-Redondo, P., and Vaquero, A. (2013). The diversity of histone versus nonhistone sirtuin substrates. Genes Cancer 4, 148–163. doi: 10.1177/1947601913483767
Mastroeni, D., Grover, A., Delvaux, E., Whiteside, C., Coleman, P. D., and Rogers, J. (2010). Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol. Aging 31, 2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005
Mastroeni, D., Grover, A., Delvaux, E., Whiteside, C., Coleman, P. D., and Rogers, J. (2011). Epigenetic mechanisms in Alzheimer’s disease. Neurobiol. Aging 32, 1161–1180. doi: 10.1016/j.neurobiolaging.2010.08.017
Mastroeni, D., McKee, A., Grover, A., Rogers, J., and Coleman, P. D. (2009). Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS ONE 4:e6617. doi: 10.1371/journal.pone.0006617
Mayeux, R., and Stern, Y. (2012). Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006239. doi: 10.1101/cshperspect.a006239
McQuown, S. C., Barrett, R. M., Matheos, D. P., Post, R. J., Rogge, G. A., Alenghat, T., et al. (2011). HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci. 31, 764–774. doi: 10.1523/JNEUROSCI.5052-10.2011
Miller, C. A., Gavin, C. F., White, J. A., Parrish, R. R., Honasoge, A., Yancey, C. R., et al. (2010). Cortical DNA methylation maintains remote memory. Nat. Neurosci. 13, 664–666. doi: 10.1038/nn.2560
Meilinger, D., Fellinger, K., Bultmann, S., Rothbauer, U., Bonapace, I. M., Klinkert, W. E., et al. (2009). Np95 interacts with de novo DNA methyltransferases, Dnmt3a and Dnmt3b, and mediates epigenetic silencing of the viral CMV promoter in embryonic stem cells. EMBO Rep. 10, 1259–1264. doi: 10.1038/embor.2009.201
Miller, C. A., and Sweatt, J. D. (2007). Covalent modification of DNA regulates memory formation. Neuron 53, 857–869. doi: 10.1016/j.neuron.2007.02.022
Miller, D. B., and O’Callaghan, J. P. (2008). Do early-life insults contribute to the late-life development of Parkinson and Alzheimer diseases? Metab. Clin. Exp. 57(Suppl. 2), S44–S49. doi: 10.1016/j.metabol.2008.07.011
Min, S. W., Cho, S. H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi: 10.1016/j.neuron.2010.08.044
Morris, M. J., Mahgoub, M., Na, E. S., Pranav, H., and Monteggia, L. M. (2013). Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. J. Neurosci. 33, 6401–6411. doi: 10.1523/JNEUROSCI.1001-12.2013
Mullan, M., Crawford, F., Axelman, K., Houlden, H., Lilius, L., Winblad, B., et al. (1992). A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1, 345–347. doi: 10.1038/ng0892-345
Murrell, J., Farlow, M., Ghetti, B., and Benson, M. D. (1991). A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science 254, 97–99. doi: 10.1126/science.1925564
Myung, N. H., Zhu, X., Kruman, I. I., Castellani, R. J., Petersen, R. B., Siedlak, S. L., et al. (2008). Evidence of DNA damage in Alzheimer disease: phosphorylation of histone H2AX in astrocytes. Age 30, 209–215. doi: 10.1007/s11357-008-9050-7
Na, E. S., Nelson, E. D., Adachi, M., Autry, A. E., Mahgoub, M. A., Kavalali, E. T., et al. (2012). A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J. Neurosci. 32, 3109–3117. doi: 10.1523/JNEUROSCI.6000-11.2012
Narayan, P. J., Lill, C., Faull, R., Curtis, M. A., and Dragunow, M. (2015). Increased acetyl and total histone levels in post-mortem Alzheimer’s disease brain. Neurobiol. Dis. 74, 281–294. doi: 10.1016/j.nbd.2014.11.023
Neri, F., Incarnato, D., Krepelova, A., Rapelli, S., Anselmi, F., Parlato, C., et al. (2015). Single-base resolution analysis of 5-Formyl and 5-Carboxyl cytosine reveals promoter DNA methylation dynamics. Cell Rep. doi: 10.1016/j.celrep.2015.01.008 [Epub ahead of print].
Nestor, C., Ruzov, A., Meehan, R., and Dunican, D. (2010). Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. Biotechniques 48, 317–319. doi: 10.2144/000113403
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/S0896-6273(03)00434-3
Ogawa, O., Zhu, X., Lee, H. G., Raina, A., Obrenovich, M. E., Bowser, R., et al. (2003). Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: a mitotic catastrophe? Acta Neuropathol. 105, 524–528.
Oliveira, A. M., Hemstedt, T. J., and Bading, H. (2012). Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat. Neurosci. 15, 1111–1113. doi: 10.1038/nn.3151
Pastor, W. A., Pape, U. J., Huang, Y., Henderson, H. R., Lister, R., Ko, M., et al. (2011). Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature 473, 394–397. doi: 10.1038/nature10102
Patrick, G. N., Zukerberg, L., Nikolic, M., de la Monte, S., Dikkes, P., and Tsai, L. H. (1999). Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622. doi: 10.1038/45159
Peleg, S., Sananbenesi, F., Zovoilis, A., Burkhardt, S., Bahari-Javan, S., Agis-Balboa, R. C., et al. (2010). Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328, 753–756. doi: 10.1126/science.1186088
Pollack, Y., Stein, R., Razin, A., and Cedar, H. (1980). Methylation of foreign DNA sequences in eukaryotic cells. Proc. Natl. Acad. Sci. U.S.A. 77, 6463–6467. doi: 10.1073/pnas.77.11.6463
Qi, H. H., Sarkissian, M., Hu, G. Q., Wang, Z., Bhattacharjee, A., Gordon, D. B., et al. (2010). Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature 466, 503–507. doi: 10.1038/nature09261
Quintas, A., de Solis, A. J., Diez-Guerra, F. J., Carrascosa, J. M., and Bogonez, E. (2012). Age-associated decrease of SIRT1 expression in rat hippocampus: prevention by late onset caloric restriction. Exp. Gerontol. 47, 198–201. doi: 10.1016/j.exger.2011.11.010
Radde, R., Bolmont, T., Kaeser, S. A., Coomaraswamy, J., Lindau, D., Stoltze, L., et al. (2006). Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 7, 940–946. doi: 10.1038/sj.embor.7400784
Raiber, E. A., Murat, P., Chirgadze, D. Y., Beraldi, D., Luisi, B. F., and Balasubramanian, S. (2015). 5-Formylcytosine alters the structure of the DNA double helix. Nat. Struct. Mol. Biol. 22, 44–49. doi: 10.1038/nsmb.2936
Ramirez-Carrozzi, V. R., Braas, D., Bhatt, D. M., Cheng, C. S., Hong, C., Doty, K. R., et al. (2009). A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138, 114–128. doi: 10.1016/j.cell.2009.04.020
Ramsahoye, B. H., Biniszkiewicz, D., Lyko, F., Clark, V., Bird, A. P., and Jaenisch, R. (2000). Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. U.S.A. 97, 5237–5242. doi: 10.1073/pnas.97.10.5237
Rao, J. S., Keleshian, V. L., Klein, S., and Rapoport, S. I. (2012). Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl. Psychiatry 2, e132. doi: 10.1038/tp.2012.55
Rhee, I., Bachman, K. E., Park, B. H., Jair, K. W., Yen, R. W., Schuebel, K. E., et al. (2002). DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416, 552–556. doi: 10.1038/416552a
Ricobaraza, A., Cuadrado-Tejedor, M., Marco, S., Perez-Otano, I., and Garcia-Osta, A. (2012). Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 22, 1040–1050. doi: 10.1002/hipo.20883
Ricobaraza, A., Cuadrado-Tejedor, M., Perez-Mediavilla, A., Frechilla, D., Del Rio, J., and Garcia-Osta, A. (2009). Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology 34, 1721–1732. doi: 10.1038/npp.2008.229
Rottach, A., Frauer, C., Pichler, G., Bonapace, I. M., Spada, F., and Leonhardt, H. (2010). The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res. 38, 1796–1804. doi: 10.1093/nar/gkp1152
Rudenko, A., Dawlaty, M. M., Seo, J., Cheng, A. W., Meng, J., Le, T., et al. (2013). Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 79, 1109–1122. doi: 10.1016/j.neuron.2013.08.003
Rumbaugh, G., Sillivan, S. E., Ozkan, E. D., Rojas, C. S., Hubbs, C. R., Aceti, M., et al. (2015). Pharmacological selectivity within class I histone deacetylases predicts effects on synaptic function and memory rescue. Neuropsychopharmacology 40, 2307–2316. doi: 10.1038/npp.2015.93
Sanchez-Mut, J. V., Aso, E., Heyn, H., Matsuda, T., Bock, C., Ferrer, I., et al. (2014). Promoter hypermethylation of the phosphatase DUSP22 mediates PKA-dependent TAU phosphorylation and CREB activation in Alzheimer’s disease. Hippocampus 24, 363–368. doi: 10.1002/hipo.22245
Sanchez-Mut, J. V., Aso, E., Panayotis, N., Lott, I., Dierssen, M., Rabano, A., et al. (2013). DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 136, 3018–3027. doi: 10.1093/brain/awt237
Sanchez-Mut, J. V., Huertas, D., and Esteller, M. (2012). Aberrant epigenetic landscape in intellectual disability. Prog. Brain Res. 197, 53–71. doi: 10.1016/B978-0-444-54299-1.00004-2
Sando, R. III, Gounko, N., Pieraut, S., Liao, L., Yates, J. III, and Maximov, A. (2012). HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 151, 821–834. doi: 10.1016/j.cell.2012.09.037
Sanyal, A., Lajoie, B. R., Jain, G., and Dekker, J. (2012). The long-range interaction landscape of gene promoters. Nature 489, 109–113. doi: 10.1038/nature11279
Schones, D. E., Cui, K., Cuddapah, S., Roh, T. Y., Barski, A., Wang, Z., et al. (2008). Dynamic regulation of nucleosome positioning in the human genome. Cell 132, 887–898. doi: 10.1016/j.cell.2008.02.022
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Shen, L., Wu, H., Diep, D., Yamaguchi, S., D’Alessio, A. C., Fung, H. L., et al. (2013). Genome-wide analysis reveals TET– and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 153, 692–706. doi: 10.1016/j.cell.2013.04.002
Siegmund, K. D., Connor, C. M., Campan, M., Long, T. I., Weisenberger, D. J., Biniszkiewicz, D., et al. (2007). DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE 2:e895. doi: 10.1371/journal.pone.0000895
Singh, P., and Thakur, M. K. (2014). Reduced recognition memory is correlated with decrease in DNA methyltransferase1 and increase in histone deacetylase2 protein expression in old male mice. Biogerontology 15, 339–346. doi: 10.1007/s10522-014-9504-5
Song, C. X., Szulwach, K. E., Dai, Q., Fu, Y., Mao, S. Q., Lin, L., et al. (2013). Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 153, 678–691. doi: 10.1016/j.cell.2013.04.001
Song, C. X., Szulwach, K. E., Fu, Y., Dai, Q., Yi, C., Li, X., et al. (2011). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29, 68–72. doi: 10.1038/nbt.1732
Stamm, S., Zhu, J., Nakai, K., Stoilov, P., Stoss, O., and Zhang, M. Q. (2000). An alternative-exon database and its statistical analysis. DNA Cell Biol. 19, 739–756. doi: 10.1089/104454900750058107
Stein, R., Razin, A., and Cedar, H. (1982) In vitro methylation of the hamster adenine phosphoribosyltransferase gene inhibits its expression in mouse L cells. Proc. Natl. Acad. Sci. U.S.A. 79, 3418–3422. doi: 10.1073/pnas.79.11.3418
Stilling, R. M., Ronicke, R., Benito, E., Urbanke, H., Capece, V., Burkhardt, S., et al. (2014). K-Lysine acetyltransferase 2a regulates a hippocampal gene expression network linked to memory formation. EMBO J. 33, 1912–1927. doi: 10.15252/embj.201487870
Strahl, B. D., and Allis, C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45. doi: 10.1038/47412
Straussman, R., Nejman, D., Roberts, D., Steinfeld, I., Blum, B., Benvenisty, N., et al. (2009). Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 16, 564–571. doi: 10.1038/nsmb.1594
Sun, D., and Yi, S. V. (2015). Impacts of chromatin states and long-range genomic segments on aging and DNA methylation. PLoS ONE 10:e0128517. doi: 10.1371/journal.pone.0128517
Sung, H. Y., Choi, E. N., Ahn Jo, S., Oh, S., and Ahn, J. H. (2011). Amyloid protein-mediated differential DNA methylation status regulates gene expression in Alzheimer’s disease model cell line. Biochem. Biophys. Res. Commun. 414, 700–705. doi: 10.1016/j.bbrc.2011.09.136
Suzuki, M., Yamada, T., Kihara-Negishi, F., Sakurai, T., and Oikawa, T. (2003). Direct association between PU.1 and MeCP2 that recruits mSin3A-HDAC complex for PU 1-mediated transcriptional repression. Oncogene 22, 8688–8698. doi: 10.1038/sj.onc.1207182
Suzuki, N., Cheung, T. T., Cai, X. D., Odaka, A., Otvos, L. Jr., Eckman, C., et al. (1994). An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 264, 1336–1340. doi: 10.1126/science.8191290
Szulwach, K. E., Li, X., Li, Y., Song, C. X., Wu, H., Dai, Q., et al. (2011). 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616. doi: 10.1038/nn.2959
Taher, N., McKenzie, C., Garrett, R., Baker, M., Fox, N., and Isaacs, G. D. (2014). Amyloid-beta alters the DNA methylation status of cell-fate genes in an Alzheimer’s disease model. J. Alzheimers Dis. 38, 831–844. doi: 10.3233/JAD-131061
Tang, C. S., and Epstein, R. J. (2007). A structural split in the human genome. PLoS ONE 2:e603. doi: 10.1371/journal.pone.0000603
Tohgi, H., Utsugisawa, K., Nagane, Y., Yoshimura, M., Genda, Y., and Ukitsu, M. (1999). Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res. Mol. Brain Res. 70, 288–292. doi: 10.1016/S0169-328X(99)00163-1
Turan, N., Katari, S., Coutifaris, C., and Sapienza, C. (2010). Explaining inter-individual variability in phenotype: is epigenetics up to the challenge? Epigenetics 5, 16–19. doi: 10.4161/epi.5.1.10557
Urdinguio, R. G., Sanchez-Mut, J. V., and Esteller, M. (2009). Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 8, 1056–1072. doi: 10.1016/S1474-4422(09)70262-5
Varley, K. E., Gertz, J., Bowling, K. M., Parker, S. L., Reddy, T. E., Pauli-Behn, F., et al. (2013). Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 23, 555–567. doi: 10.1101/gr.147942.112
Vinson, C., and Chatterjee, R. (2012). CG methylation. Epigenomics 4, 655–663. doi: 10.2217/epi.12.55
Vogel-Ciernia, A., and Wood, M. A. (2012). Molecular brake pad hypothesis: pulling off the brakes for emotional memory. Rev. Neurosci. 23, 607–626. doi: 10.1515/revneuro-2012-0050
Waddington, C. H. (2012). The epigenotype. 1942. Int. J. Epidemiol. 41, 10–13. doi: 10.1093/ije/dyr184
Wagner, F. F., Zhang, Y. L., Fass, D. M., Joseph, N., Gale, J. P., Weiwer, M., et al. (2015). Kinetically selective inhibitors of histone deacetylase 2 (HDAC2) as cognition enhancers. Chem. Sci. 6, 804–815. doi: 10.1039/C4SC02130D
Walker, M. P., LaFerla, F. M., Oddo, S. S., and Brewer, G. J. (2013). Reversible epigenetic histone modifications and Bdnf expression in neurons with aging and from a mouse model of Alzheimer’s disease. Age 35, 519–531. doi: 10.1007/s11357-011-9375-5
Wang, J., Telese, F., Tan, Y., Li, W., Jin, C., He, X., et al. (2015). LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat. Neurosci. 18, 1256–1264. doi: 10.1038/nn.4069
Wang, S. C., Oelze, B., and Schumacher, A. (2008a). Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 3:e2698. doi: 10.1371/journal.pone.0002698
Wang, Z., Zang, C., Rosenfeld, J. A., Schones, D. E., Barski, A., Cuddapah, S., et al. (2008b). Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903. doi: 10.1038/ng.154
Wen, L., Li, X., Yan, L., Tan, Y., Li, R., Zhao, Y., et al. (2014). Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 15:R49. doi: 10.1186/gb-2014-15-3-r49
West, R. L., Lee, J. M., and Maroun, L. E. (1995). Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J. Mol. Neurosci. 6, 141–146. doi: 10.1007/BF02736773
Wigler, M., Levy, D., and Perucho, M. (1981). The somatic replication of DNA methylation. Cell 24, 33–40. doi: 10.1016/0092-8674(81)90498-0
Woldemichael, B. T., Bohacek, J., Gapp, K., and Mansuy, I. M. (2014). Epigenetics of memory and plasticity. Prog. Mol. Biol. Transl. Sci. 122, 305–340. doi: 10.1016/B978-0-12-420170-5.00011-8
Wu, H., Wu, X., Shen, L., and Zhang, Y. (2014). Single-base resolution analysis of active DNA demethylation using methylase-assisted bisulfite sequencing. Nat. Biotechnol. 32, 1231–1240. doi: 10.1038/nbt.3073
Xin, Y., Chanrion, B., Liu, M. M., Galfalvy, H., Costa, R., Ilievski, B., et al. (2010). Genome-wide divergence of DNA methylation marks in cerebral and cerebellar cortices. PLoS ONE 5:e11357. doi: 10.1371/journal.pone.0011357
Xu, Q., Modrek, B., and Lee, C. (2002). Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res. 30, 3754–3766. doi: 10.1093/nar/gkf492
Xu, Y. M., Du, J. Y., and Lau, A. T. (2014). Posttranslational modifications of human histone H3: an update. Proteomics 14, 2047–2060. doi: 10.1002/pmic.201300435
Yan, J., Zierath, J. R., and Barres, R. (2011). Evidence for non-CpG methylation in mammals. Exp. Cell Res. 317, 2555–2561. doi: 10.1016/j.yexcr.2011.08.019
Yang, X. J., and Seto, E. (2007). HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318. doi: 10.1038/sj.onc.1210599
Yeo, G., Holste, D., Kreiman, G., and Burge, C. B. (2004). Variation in alternative splicing across human tissues. Genome Biol. 5:R74. doi: 10.1186/gb-2004-5-10-r74
Yoon, H. G., Chan, D. W., Reynolds, A. B., Qin, J., and Wong, J. (2003). N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol. Cell 12, 723–734. doi: 10.1016/j.molcel.2003.08.008
Yoshikai, S., Sasaki, H., Doh-ura, K., Furuya, H., and Sakaki, Y. (1990). Genomic organization of the human amyloid beta-protein precursor gene. Gene 87, 257–263. doi: 10.1016/0378-1119(90)90310-N
Zhang, H., Gao, L., Anandhakumar, J., and Gross, D. S. (2014). Uncoupling transcription from covalent histone modification. PLoS Genet. 10:e1004202. doi: 10.1371/journal.pgen.1004202
Zhang, K., Schrag, M., Crofton, A., Trivedi, R., Vinters, H., and Kirsch, W. (2012). Targeted proteomics for quantification of histone acetylation in Alzheimer’s disease. Proteomics 12, 1261–1268. doi: 10.1002/pmic.201200010
Zhang, R., Lu, J., Kong, X., Jin, L., and Luo, C. (2013). Targeting epigenetics in nervous system disease. CNS Neurol. Disord. Drug Targets 12, 126–141. doi: 10.2174/1871527311312010018
Zhang, Z. Y., and Schluesener, H. J. (2013). Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model. J. Neuropathol. Exp. Neurol. 72, 178–185. doi: 10.1097/NEN.0b013e318283114a
Ziller, M. J., Muller, F., Liao, J., Zhang, Y., Gu, H., Bock, C., et al. (2011). Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 7:e1002389. doi: 10.1371/journal.pgen.1002389
Keywords: Alzheimer’s disease, epigenetics, neuroepigenetics, DNA methylation, histone acetylation, histone methylation, histone phosphorylation
Citation: Sanchez-Mut JV and Gräff J (2015) Epigenetic Alterations in Alzheimer’s Disease. Front. Behav. Neurosci. 9:347. doi: 10.3389/fnbeh.2015.00347
Received: 07 October 2015; Accepted: 25 November 2015;
Published: 17 December 2015.
Edited by:
Martine Ammassari-Teule, Consiglio Nazionale delle Ricerche, ItalyReviewed by:
Angel Barco, Instituto de Neurociencias de Alicante, SpainTamara Maes, Oryzon Genomics S.A., Spain
Copyright © 2015 Sanchez-Mut and Gräff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johannes Gräff, am9oYW5uZXMuZ3JhZWZmQGVwZmwuY2g=