 Sook-Kyung C. Kwon1
Sook-Kyung C. Kwon1 Erzsebet Kovesdi1
Erzsebet Kovesdi1 Andrea B. Gyorgy1 Daniel Wingo1
Andrea B. Gyorgy1 Daniel Wingo1 Alaa Kamnaksh1 John Walker1 Joseph B. Long2
Alaa Kamnaksh1 John Walker1 Joseph B. Long2 Denes V. Agoston1*
Denes V. Agoston1*
- 1 Department of Anatomy, Physiology and Genetics, School of Medicine, Uniformed Services University, Bethesda, MD, USA
- 2 Division of Military Casualty Research, Walter Reed Army Institute of Research, Silver Spring, MD, USA
Psychological stress and traumatic brain injury (TBI) can both result in lasting neurobehavioral abnormalities. Post-traumatic stress disorder and blast induced TBI (bTBI) have become the most significant health issues in current military conflicts. Importantly, military bTBI virtually never occurs without stress. In this experiment, we assessed anxiety and spatial memory of rats at different time points after repeated exposure to stress alone or in combination with a single mild blast. At 2 months after injury or sham we analyzed the serum, prefrontal cortex (PFC), and hippocampus (HC) of all animals by proteomics and immunohistochemistry. Stressed sham animals showed an early increase in anxiety but no memory impairment at any measured time point. They had elevated levels of serum corticosterone (CORT) and hippocampal IL-6 but no other cellular or protein changes. Stressed injured animals had increased anxiety that returned to normal at 2 months and significant spatial memory impairment that lasted up to 2 months. They had elevated serum levels of CORT, CK-BB, NF-H, NSE, GFAP, and VEGF. Moreover, all of the measured protein markers were elevated in the HC and the PFC; rats had an increased number of TUNEL-positive cells in the HC and elevated GFAP and Iba1 immunoreactivity in the HC and the PFC. Our findings suggest that exposure to repeated stress alone causes a transient increase in anxiety and no significant memory impairment or cellular and molecular changes. In contrast, repeated stress and blast results in lasting behavioral, molecular, and cellular abnormalities characterized by memory impairment, neuronal and glial cell loss, inflammation, and gliosis. These findings may have implications in the development of diagnostic and therapeutic measures for conditions caused by stress or a combination of stress and bTBI.
Introduction
Traumatic brain injury (TBI) is one of the leading causes of death and chronic disability worldwide (Bruns and Hauser, 2003; Tagliaferri et al., 2006). Blast induced TBI (bTBI) caused by explosive devices is especially frequent in recent conflicts in Iraq and in Afghanistan (Warden and French, 2005; Taber et al., 2006; Warden, 2006). Epidemiological studies have shown that mild bTBI (mbTBI) can result in chronic neurobehavioral changes such as increased anxiety and memory impairment (Ryan and Warden, 2003; Okie, 2005). Importantly, virtually no bTBI occurs on the battlefield without the exposure to psychological stress. Exposure to stress alone (i.e., traumatic and/or life-threatening events) without physical injury can lead to a chronic condition called post-traumatic stress disorder (PTSD) in some but not all affected individuals (Breslau and Kessler, 2001). PTSD is especially frequent among soldiers; about 14% of soldiers suffer from PTSD-like symptoms compared to 4% of the US adult population (Keane et al., 2006; Richardson et al., 2010).
Although bTBI shares some of the clinical features of the closed head and the penetrating TBIs, bTBI appears to be a fundamentally different form of neurotrauma (Ling et al., 2009). Several factors are responsible for the uniqueness of bTBI; explosive blast rapidly dissipates very high levels of energy in the form of supersonic pressure waves (Mayorga, 1997; Okie, 2005). Within the brain parenchyma, the high-energy high-velocity blast waves can cause substantial damage to blood vessels, neuronal and glial cell bodies and their processes (Kaur et al., 1997; Cernak et al., 2001). Clinical hallmarks of severe bTBI include an unusually early onset (hours after injury) and rapid development of diffuse malignant cerebral edema, and delayed (10–14 days post-injury) vasoconstriction (Armonda et al., 2006; Ling et al., 2009; Ling and Ecklund, 2011). Given that blast almost always affects the whole body, subclinical thoracic or abdominal injuries can also contribute to the pathophysiology of bTBI (Cernak et al., 2010). It has been thought that the secondary injury process in bTBI includes vascular changes, neuroinflammation, and gliosis (Kaur et al., 1997; Mayorga, 1997; Cernak et al., 2001; Taber et al., 2006).
Exposure to mild blast poses especially difficult challenges. Even though there are no life-threatening injuries in mbTBI and soldiers do not lose consciousness, 6–9 months later many soldiers develop neurobehavioral abnormalities that include memory impairment, anxiety, and mood disorders (Belanger et al., 2007; Brenner et al., 2009). These symptoms indicate damage to the hippocampus (HC) and the prefrontal cortex (PFC), brain structures that are also indicated as the neuroanatomical substrates of PTSD (Jaffee and Meyer, 2009). Stress is a constant factor on the battlefield (Warden et al., 1997; Brenner et al., 2009); soldiers are repeatedly exposed to various life-threatening situations and to the visual and audible cues of blasts without necessarily suffering from any visible physical injury.
In order to better understand the long-term consequences of stress with and without the exposure to blast, we used a rodent model of stress and mbTBI and assessed the behavior of animals at various time points after sham or injury and analyzed their sera and brains for cellular and molecular changes. Due to our experimental setup, we were unable to determine the effect of blast injury alone. During our pilot studies we learned that handling and transporting animals (associated with the exposure to blast) resulted in a significant amount of stress as indicated by substantially elevated serum CORT levels of animals.
Materials and Methods
Animals, Housing Conditions, and Experimental Schedule
Sprague-Dawley male rats (245–265 g; Charles River Laboratories, Wilmington, MA, USA) were housed in cages with free access to food and water in a reverse 12/12 h light/dark cycle. The experimental schedule is depicted in Figure 1. After 7 days of acclimation and handling in our animal facility, control animals (C) were housed two per cage; stressed sham injured (SS) and stressed injured animals (SI) were housed individually. All animals were handled according to protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the Uniformed Services University of the Health Sciences. All behavioral tests were conducted during animals’ dark cycle.
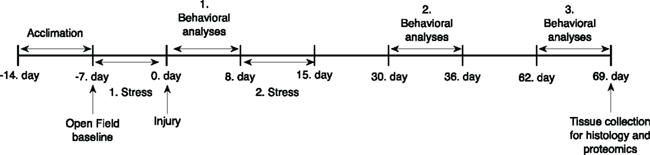
Figure 1. Outline of the experimental schedule.
Injury
On the day of the exposure, animals in the SS and SI groups were transported from USU (Bethesda, MD, USA) to Walter Reed Army Institute of Research (Silver Spring, MD, USA) for injury. Blast TBI was generated using a compression-driven shock tube as described earlier (Long et al., 2009). Before injury, rats in the SI group were anesthetized with 4% isoflurane (Forane, Baxter Healthcare Corporation, Deerfield, IL, USA), placed in an animal holder in a transverse prone position and then transferred to the shock tube where they were exposed to whole body blast overpressure (20.6 ± 3 psi). Immediately after exposure, the duration of apnea was measured and rats were moved back to their home cages. Animals in the SS group received the same amount of anesthesia and underwent the same procedure but were not exposed to blast overpressure. However, SS as well as SI animals were exposed to the sounds of the blast, which is likely an additional stressor. Following exposures (or sham), animals were transported back to USU.
Chronic Stress
Rats in the SI and SS groups were exposed to a combination of predator and unpredictable stressors for 1 week prior to and 1 week after the first behavioral testing session (Figure 1). Unpredictable stress is a face-valid model of human stress that has been shown to reliably elevate stress hormone levels in rodents (Fride et al., 1986; Weinstock et al., 1992).
A combination of fox urine and unpredictable stress (Campbell et al., 2003) was performed with modifications (Berger and Grunberg, in preparation). Rats were exposed to fox urine for 10 min/day (Red fox urine, Buck Stop Lure Company, Stanton, MI, USA) as a predator stress, and loud noises and sudden cage movements at irregular times as an unpredictable stressor. During the stress routine, rats were moved to the animal facility’s procedure room. Each rat was transferred to a clean empty cage with a cotton ball containing 15 ml of fox urine; each day the position of the cotton ball was changed. Within the 10 min stress period, rats were exposed to loud noises and irregular cage movement. After stress, the rats were immediately moved back to their home cages and transferred back to the animal housing room. Control rats were neither transferred to the procedure room nor exposed to any of the stressors.
Schedule of Behavioral Tests
Before chronic stress and injury all animals underwent a baseline open field (OF) measurement (Figure 1). Horizontal activity results were used to create three groups with no statistical significance among them. After the first stress phase and blast (or sham) injury, rats underwent a series of behavioral evaluations. In each behavioral session OF was conducted first to ascertain whether animals had any motor problems, which can confound other behavioral tests. One day after OF, elevated plus maze (EPM) was performed to measure anxiety levels. Barnes maze (BM) was performed last to measure spatial learning and memory. The three behavioral assessments were performed on separate days starting at 24 h, 1, and 2 months after injury (Figure 1).
Open field
The OF test is an indicator of potential motor deficits but can also reflect fluctuations in anxiety levels based on changes in exploratory behavior (Heath and Vink, 1999). Several studies have shown this test to reliably measure anxiety and depression in rodents (von Horsten et al., 1998) including after TBI (Vink et al., 2003). OF tests were performed using Omnitech Electronics’ Digiscan infrared photocell system (test box model RXYZCM, Omnitech Electronics, Columbus, OH; Elliott and Grunberg, 2005). The OF system is a 40 × 40 × 30 (L × W × H) cm clear Plexiglas arena with a perforated lid. During the 60 min testing period, we measured horizontal activity (locomotor activity), time spent in the margins (anxiety), and in the center. Data were automatically gathered and transmitted to a computer via an Omnitech Model DCM-BBU analyzer.
Elevated plus maze
The EPM is a widely used, ethologically relevant test that assesses anxiety states in rodents (Carobrez and Bertoglio, 2005; Salzberg et al., 2007; Walf and Frye, 2007). The maze is an elevated structure (1 m above ground) consisting of four intersecting arms. The arms of the maze are 50 cm long and 10 cm wide; the closed arms have walls on three sides that are 40 cm high while the open arms have none. The lighting in the middle of the maze was set at 90 lux. On testing days rats were placed one by one in the center of the maze facing one of the open arms; each animal was allowed to explore freely for 5 min while its movement was video-tracked. Total distance traveled, number of entries made, and time spent (duration) in each arm was recorded using ANY-maze 4.2 Software (Stoelting Company, Wood Dale, IL, USA).
Barnes maze
Barnes maze was used to assess spatial learning and memory (Barnes, 1979; Maegele et al., 2005; Doll et al., 2009). BM represents a widely used, validated, and less stressful alternative to the commonly used water maze test (Harrison et al., 2009). The maze is a circular platform (1.2 m in diameter) with 18 evenly spaced holes around the periphery. One of the holes is the entrance to a darkened escape box that is not visible from the surface of the board. Each rat was tested twice per day for six consecutive days to find the escape box (only day 1 of the first BM session had three trials). In each trial, latency to locate and enter the escape box was measured (ANY-maze 4.2 Software, Stoelting Company, Wood Dale, IL, USA).
During the first (teaching) trial of the first BM session, all animals were trained to locate the escape chamber. Each animal was placed in the escape box and covered for 30 s. The escape box was then removed with the animal inside and moved to the center of the maze. The rat was removed from the box and allowed to explore the maze for a few seconds, after which the rat was returned to its home cage. No latency times were recorded for the teaching trials. The escape box and the maze were cleaned with 30% ethanol solution between each trial. In the second trial the same rat was placed under a start box in the center of the maze for 30 s; the start box was then removed and the rat was allowed to explore freely to find the escape chamber. Training sessions ended after the animal had entered the escape box or when a pre-determined time (240 s) had elapsed. If the animal had not found the escape box during the given time period, it was placed in the escape box for 1 min at the end of the trial.
Tissue Collection and Processing
On day 67 post-injury (or sham; Figure 1) all animals were deeply anesthetized with isoflurane until a tail pinch produced no reflex movement. Anesthesia was maintained using a mask/nose cone attached to the anesthetic vaporizer, and blood was collected (1.5 ml) from a tail vein. For proteomics and ELISA assays, rats were decapitated under deep anesthesia. Brains were immediately removed and placed on ice. The PFC and HC were dissected, frozen, and stored at −80°C. For histology, rats were transcardially perfused with cold phosphate-buffered saline (PBS) followed by 4% buffered paraformaldehyde solution under deep isoflurane anesthesia. Following overnight post-fixation in 4% buffered paraformaldehyde solution brains were consecutively immersed in cold 15 and 30% sucrose solutions in 1× PBS for cryoprotection and then frozen on dry ice. Frozen brains were sectioned coronally at a 20-μm thickness using a cryostat (Cryocut 1800; Leica Microsystems, Bannockburn, IL, USA) and sections were kept at −80°C until use.
Proteomics
Preparation of samples
Sample preparation, printing, scanning, and data analysis were performed as described later in detail (Gyorgy et al., 2010). Flash-frozen tissues were briefly pulverized in liquid nitrogen; 200 mg of the frozen powder was transferred into 1 ml of T-per lysis buffer (Thermo Fisher, Waltham, MA, USA) with protease and phosphatase inhibitors (Thermo Fisher) and then sonicated. Samples were centrifuged for 15 min at 4°C; the supernatants were aliquoted and stored at −80°C. Protein concentrations were measured by using a BCA assay (Thermo Fisher). Blood samples were promptly centrifuged after removal at 10,000×g for 15 min at 4°C; the supernatants were aliquoted, flash-frozen, and stored at −80°C. Tissue samples were diluted in print buffer (10% glycerol, 0.05% SDS, 50 mM DTT in 1× TBS) to a final protein concentration of 1 mg/ml, while serum samples were diluted 1:10. Samples were then subjected to an 11-point serial 1:2 dilution and transferred into Genetix 384-well plates (X7022, Fisher Scientific, Pittsburgh, PA, USA) as described (Gyorgy et al., 2010). Plates were transferred into an Aushon 2470 Arrayer (Aushon Biosystems, Billerica, MA, USA) and samples were printed on ONCYTE Avid (tissue samples) or ONCYTE Nova (serum samples) single-pad nitrocellulose coated glass slides (Grace Bio-Labs, Bend, OR, USA).
Printing parameters
The Aushon Arrayer was programmed to use 16 pins (4 × 4 pattern). Each sample was printed in 12 dilutions (12 rows) and in triplicate (3 columns), resulting in a block of 3 × 12 dots per sample. The Spot Diameter was set to 250 nm with a spacing of 500 nm between dots on the x-axis and 375 nm on the y-axis. Wash time was set at 2 s without delays. The printer was programmed for a single deposition per dot for printing serum and tissue extracts.
Immunochemical detection
Primary antibodies were diluted to 10× the optimal Western analysis concentration in antibody incubation buffer (0.1% bovine serum albumin (BSA), protease inhibitors (EDTA-free Halt protease and phosphatase inhibitor cocktail, Thermo Fisher, Waltham, MA, USA; 1× TBS, 0.5% Tween 20) as described (Gyorgy et al., 2010). Primary antibodies were used in the following dilutions: VEGF 1:100 (Abcam ab-53465), s100b 1:50 (Abcam ab-41548), GFAP 1:500 (Abcam ab-7260), Tau-protein 1:20 (Santa Cruz sc-1995), CK-BB 1:20 (Santa Cruz sc-15157), NSE 1: 100 (Abcam, Cat# ab53025), NF-H 1:20 (Sigma N4142). Slides were incubated with the primary antibody solution overnight at 4°C covered by a cover slip (Nunc? mSeries LifterSlips, Fisher Scientific, Pittsburg, PA). The following day slides were washed and then incubated with an Alexa Fluor® 635 goat anti-mouse (Cat# A-31574), goat anti-rabbit (Cat# A-31576), or rabbit anti-goat IgG (H + L; Cat# A-21086) secondary antibodies from Invitrogen at 1:6000 dilution in antibody incubation buffer for 1 h at room temperature (RT). After washing and drying, the fluorescent signals were measured in a Scan Array Express HT microarray scanner (Perkin Elmer, Waltham, MA, USA) using a 633 nm wavelength laser and a 647 nm filter. Data from the scanned images were imported into a Microsoft Excel-based bioinformatics program developed in house for analysis (Gyorgy et al., 2010).
Data analysis and bioinformatics
The program calculates total net intensity after local background subtraction for each spot. The intensity data from the dilution series of each sample are then plotted against dilution on a log–log graph. The linear regression of the log–log data was calculated after the removal of flagged data, which include signal to noise ratios of less than 2, spot intensities in the saturation range or noise range, or high variability between duplicate spots (>10–15%). The total amount of antigen is determined by the y-axis intercept (Gyorgy et al., 2010).
Corticosterone Assay
Serum corticosterone (CORT) levels were measured using Cayman’s Corticosterone EIA Kit according to the manufacturer’s instructions (Cayman Chemical, Ann Arbor, MI, USA). Each sample was diluted 1:500 and measured in triplicate.
IL-6 and INFγ Assays
INFγ and IL-6 levels were measured from brain tissues using the rat Interferon gamma ELISA and the Rat IL-6 ELISA kits (both are from Thermo Fisher, Waltham, MA, USA). The IL-6 ELISA kit required a 1:5 dilution using the supplied dilution buffer in order to avoid saturation in the wells. After the dilution of brain samples, the assay was performed according to the manufacturer’s instructions.
Histology
Immunohistochemistry
Every first and tenth coronal section containing either the PFC, the dorsal HC (DHC), or the ventral HC (VHC) were mounted on positively charged glass slides with two sections per slide. Three slides per animal, containing sections with identical z-axes, were selected per brain region for each immunostaining. Slides containing the frozen sections were equilibrated at RT and hydrated with 1× PBS for 30 min. Antigen retrieval was performed by incubating the sections in 10 mM citrate buffer (pH 6.0) at 80°C for 30 min followed by cooling down to RT. After rehydration with 1× PBS, sections were permeabilized with 0.5% Triton X-100 in PBS for 1 h and blocked in 1× PBS containing 5% normal goat serum (NGS), 5% BSA, 0.1% sodium azide, and 0.5% Triton X-100 for 1 h. The same solution minus NGS was used to dilute primary antibodies. The following primary antibodies were used: mouse anti-GFAP (Millipore, Temecula, CA, USA; 1:400) rabbit anti-doublecortin (DCX; Cell Signaling Technology, Beverly, MA, USA; 1:1000), and goat anti-Iba1 (Abcam, Cambridge, MA, USA; 1:1000). Sections were incubated with the primary antibodies overnight at 4°C. After washing with 1× PBS, they were incubated with appropriate secondary antibodies (Alexa Fluor 555 goat anti-mouse IgG, 488 goat anti-rabbit IgG, or 488 donkey anti-goat IgG (Invitrogen, Carlsbad, CA) was applied for 1 h at RT 1:100), Hoechst 33342 (Molecular Probes, Eugene, OR, USA) at 1 μg/ml was applied for 2 min, sections washed and coverslipped using anti-fading media (Vectashield, Vector Laboratories, Burlingame, CA, USA).
TUNEL assay
Apoptotic cell death marked by DNA fragmentation was determined using a TUNEL in situ cell death detection kit, POD (Roche, Indianapolis, IN, USA) according to the manufacturer’s instructions. Sections were hydrated with PBS and endogenous peroxidase activity was quenched by 3% H2O2 in methanol for 10 min at RT. Sections were permeabilized by 0.1 M sodium citrate buffer (pH 6.0) at 70°C for 30 min followed by PBST for 30 min at RT. TUNEL reaction was performed for 1 h at 37°C and the signal was converted using converter-POD. TUNEL-positive cells were visualized by DAB substrate. Dark brown TUNEL-positive cells were counted from four sections per animal.
Histological data acquisition
Histological sections were visualized in an Olympus IX-71 microscope using the appropriate filters and images were collected using a SPOT digital camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA). The collected images were colored using TIFFany Caffeine Software.
Statistical Analysis and Comparison of Data
Behavioral test results were analyzed with ANOVA and Tukey post hoc tests. Differences with a p-value of <0.05 were considered significant. Statistical analyses were performed using GraphPad InStat software. Proteomics and ELISA results were analyzed with Student’s t-test. Data are reported as the average ± standard error of the mean. Proteomics data results were followed up with a one-way ANOVA. For each of our numerical measurements, we determined statistical significance among experimental groups by (p < 0.05 depicted by one star; p < 0.01 by two and p < 0.001 by three).
Results
Behavioral Effects
Locomotor activity
Twenty-four hours after exposure to blast (or sham), rats in the SI group showed a decrease in horizontal activity compared to rats in the SS and C groups (Figure 2A) but the difference was statistically insignificant. Animals in the SI group spent significantly more time in the periphery and significantly less time in the center (Figures 2B,C) compared to C and SS animals. SS animals showed no significant difference compared to C animals.
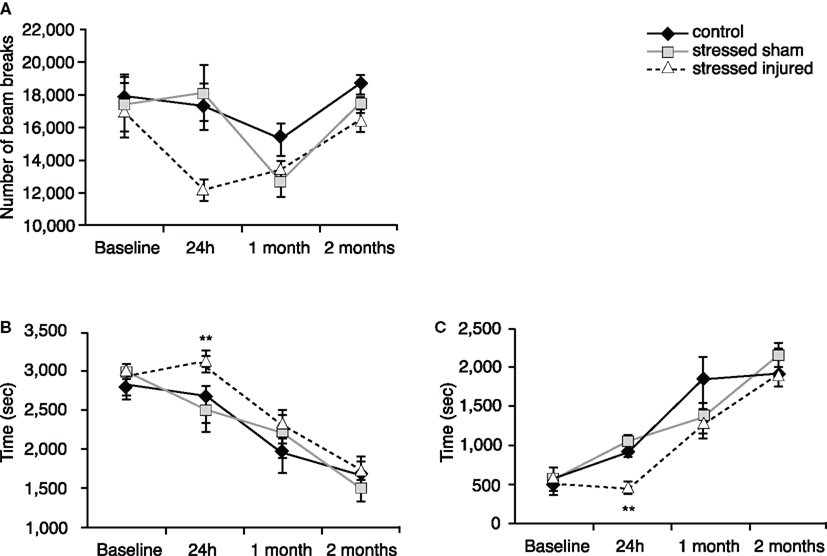
Figure 2. Locomotor activity of animals in the various experimental groups. Open field was used to assess (A) horizontal activity (number of beam breaks), (B) time spent in the peripheral zone (seconds) and (C) time spent in the central zone (seconds). **p < 0.01 SI compared to C rats. Data are presented in mean ± SEM. (C: n = 4; SS: n = 6; and SI: n = 6).
At 1 and 2 months after injury (or sham), we found no significant differences in any of the measured parameters between animals in all experimental groups (Figures 2A–C).
Anxiety
Forty-eight hours after blast (or sham) injury, we found that SI rats traveled significantly shorter distances than C animals (Figure 3A). Animals in the SS group also traveled shorter distances but the differences were not statistically significant. Animals in both SS and SI groups spent significantly less time in the open arms and more time in the closed arms (Figures 3B,C).
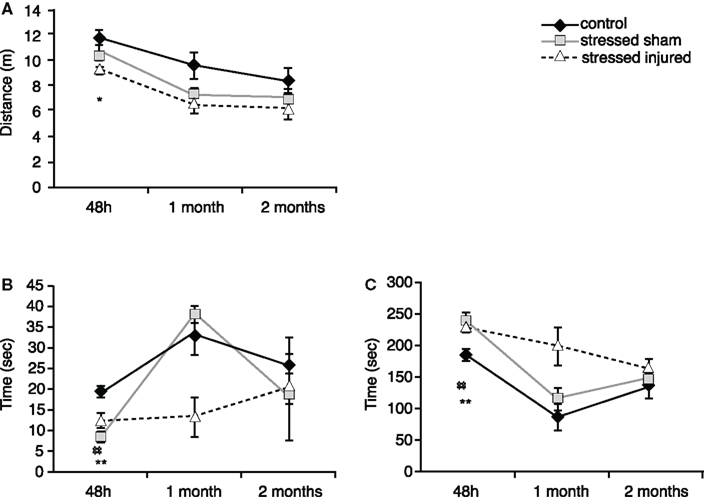
Figure 3. Anxiety of animals in the various experimental groups. Elevated plus maze was used to assess (A) total distance traveled (meter), (B) time spent in the open arms (seconds) and (C) time spent in the closed arms (seconds). *p < 0.05 and **p < 0.01 SI compared to C rats, ##p < 0.01 SS to C rats. Data are presented in mean ± SEM. (C: n = 4; SS: n = 6; and SI: n = 6).
At 1 month both SS and SI groups traveled shorter distances compared to C animals (Figure 3A). Among the stressed groups only SI animals exhibited raised anxiety by spending less time in the open arms and more time in the closed arms (Figures 3B,C). Animals in the SS group did not show significant differences in the time spent in the open arms vs. closed arms compared to the controls.
Two months after blast or sham injury, all animals performed similarly with no significant differences in total distance traveled or time spent in the open and the closed arms.
Spatial learning and memory
During the first BM session, performed between days 3 and 8 post-blast (or sham; see Figure 1 for schedule) we observed significant differences in the performance of animals on the last testing day only (day 8). SS rats needed significantly more time to find the escape box compared to C rats. SI rats spent less time finding the escape box than SS rats, but more time compared to C rats; the difference was not statistically significant (Figure 4A).
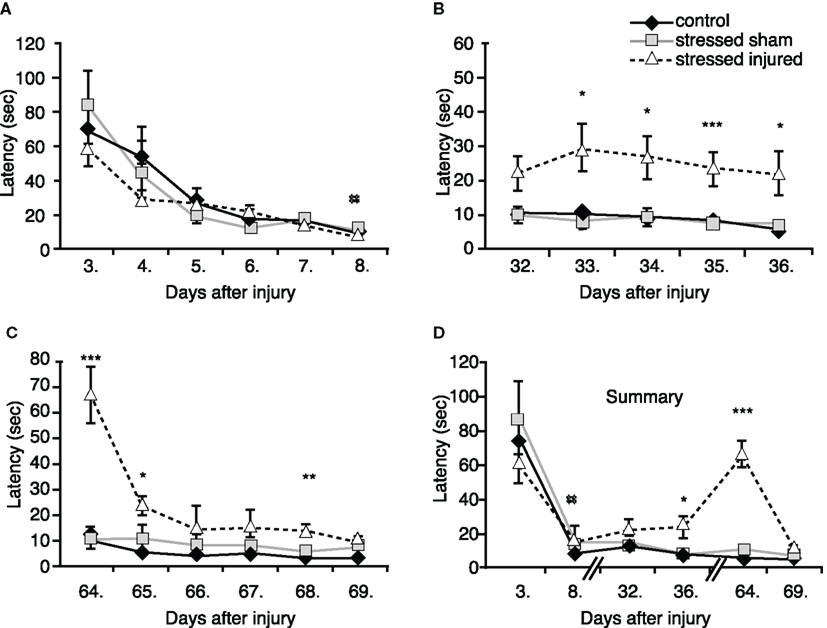
Figure 4. Spatial learning and memory of animals in the various experimental groups. Barnes maze was used to determine latencies to find the escape box (A) 3–8 days post-injury, (B) 32–36 days post-injury, (C) 64–69 days post-injury. (D) Summarized time-line of latencies of the three sets of Barnes maze test. *p < 0.05, **p < 0.01, and ***p < 0.01 SI compared to C rats, ##p < 0.01 SS to C rats. Data are presented in mean ± SEM (C: n = 4; SS: n = 6; and SI: n = 6).
During the second BM session, performed between days 32 and 36 post-blast (or sham), we found no difference in the latency times of animals in the SS and the C groups. However, SI animals required significantly longer times to find the escape box on day 33 through 36 (Figure 4B).
The last BM session was performed between days 64 and 69 post-injury (or sham). Again, the performance of SS animals was not statistically different from animal in the control group (Figure 4C). In contrast, SI rats performed very poorly in the BM on day 64. These animals had increased latency times similar to those measured during the first BM session (Figures 4C,D) showing significant memory problems on days 65 and 68, indicating lasting memory impairment caused by blast.
Effects on Protein Markers
Serum changes
At the end of the last behavioral session (69 days post-injury or sham), we obtained serum from each animal and compared NF-H, CK-BB, GFAP, VEGF, and NSE levels across all experimental groups. We found that serum levels of all protein markers listed above were significantly elevated at this late time point in SI animals (Figure 5). As expected, serum CORT levels were significantly elevated in both SS and SI groups. However, the difference in serum CORT levels between the two groups was statistically insignificant.
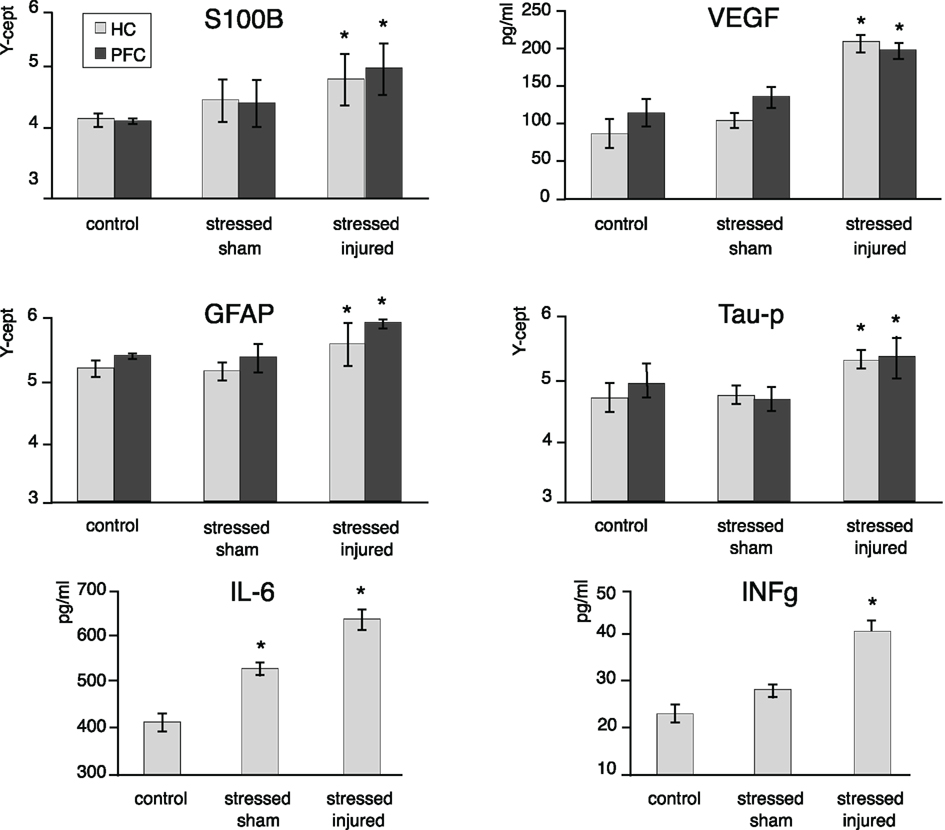
Figure 5. Protein markers in the PFC and the HC of animals in the various experimental groups. Tissue extracts were prepared from dissected PFC and HC regions of C, SS, and SI rat brains. The tissue levels of the selected marker proteins were assayed using either RPPM or ELISA. The y-axis intercept (Y-cept) and pg/ml (IL-6 and IFNg) indicate the measured protein levels. *p < 0.05, **p < 0.01 compared to C rats, error bars are ± SEM. (C: n = 2; SS: n = 3; and SI: n = 3).
Changes in the brain
At the end of the last behavioral session we analyzed changes in the expression of S100β, VEGF, GFAP, Tau-protein, IL-6, and INFγ in the PFC and the HC of all animals. We found significantly elevated levels of all markers (except IL-6) in the PFC and the HC of SI animals only (Figure 6). Interestingly, we found that IL-6 levels were significantly increased in the HC of SS animals while no such effect of stress alone was seen on hippocampal INFγ levels.
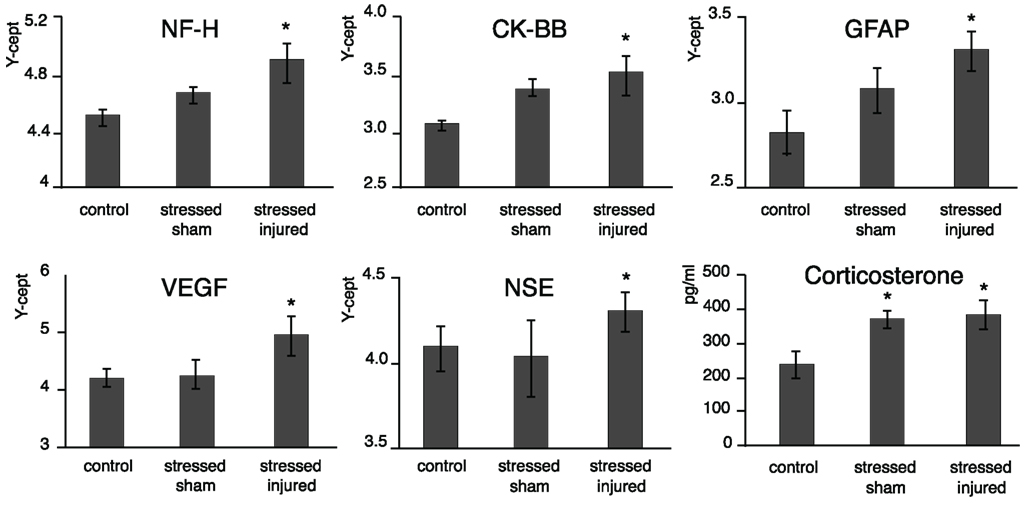
Figure 6. Serum markers of animals in the various experimental groups. Blood was drawn and sera were prepared from the C, SS, and SI rats. The levels of the selected marker proteins were assayed using either RPPM or ELISA, and corticosterone levels were assayed using ELISA. The y-axis intercept (Y-cept) and pg/ml (corticosterone) indicate the measured protein levels. *p < 0.05, **p < 0.01 compared to C rats, error bars are ± SEM (C: n = 4; SS: n = 6; and SI: n = 6).
Cellular Changes
Astrogliosis
To identify the cellular changes underlying the observed behavioral abnormalities, we analyzed the HC and the PFC for GFAP expression at 2 months post-blast (or sham). Consistent with our proteomics data, we found that exposure to stress alone (SS) had no observable effect on GFAP expression in the PFC or the HC (Figure 7). However, in SI animals we detected a noticeable increase in GFAP immunoreactivity in the PFC and the VHC.
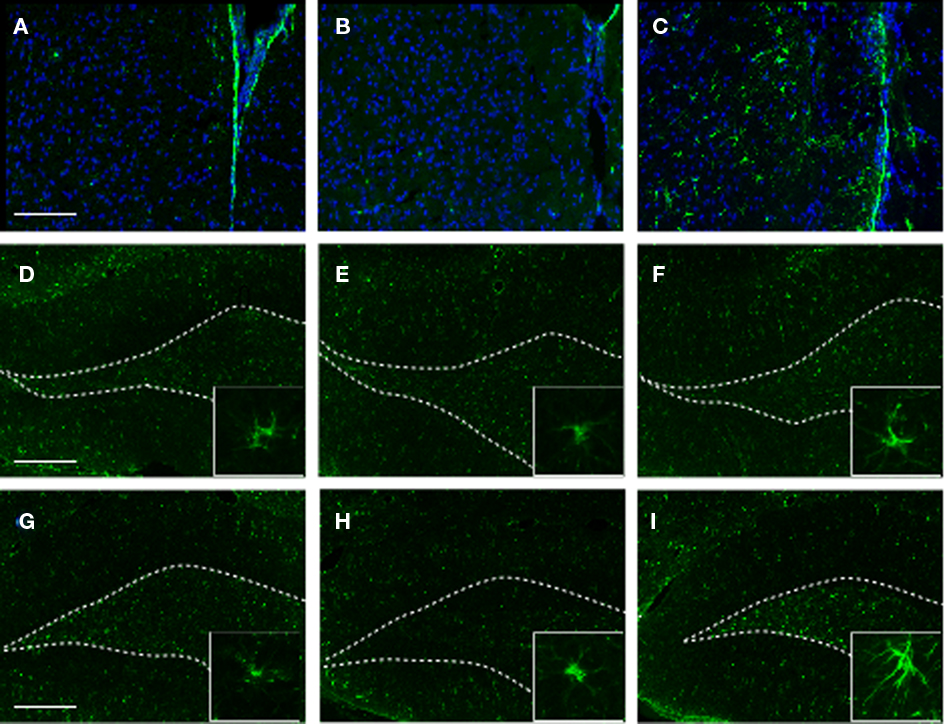
Figure 7. GFAP immunoreactivity in selected brain regions of animals in the various experimental groups. Frozen coronal sections containing the PFC, DHC, and VHC were cut and processed for immunohistochemistry using a GFAP antibody. Sections were stained with Hoechst 33342 to mark cellular nuclei. PFC of (A) C, (B) SS, and (C) SI rats; DHC of (D) C, (E) SS, and (F) SI rats; VHC of (G) C, (H) SS, and (I) SI rats. Inlays display morphological extension of stellar-like processes in VHC of SI rats. White-dotted line outlines DG layer of hippocampus.
Importantly, an increase in GFAP+ cells displaying a stellar morphology characteristic of reactive astrocytes was observed in the PFC and the VHC of SI animals (Figure 7C, and insert).
Inflammation
Similar to the gliotic response above, exposure to stress alone had no significant effect on Iba1 immunoreactivity in the PFC or the HC (Figure 8). While increased Iba1 immunoreactivity was observed in the SI group, there was a noticeable increase in Iba1 immunoreactivity (albeit to a lesser degree) in the PFC of SS animals. No significant differences were observed in Iba1 immunoreactivity in the DHC of any of the experimental groups.
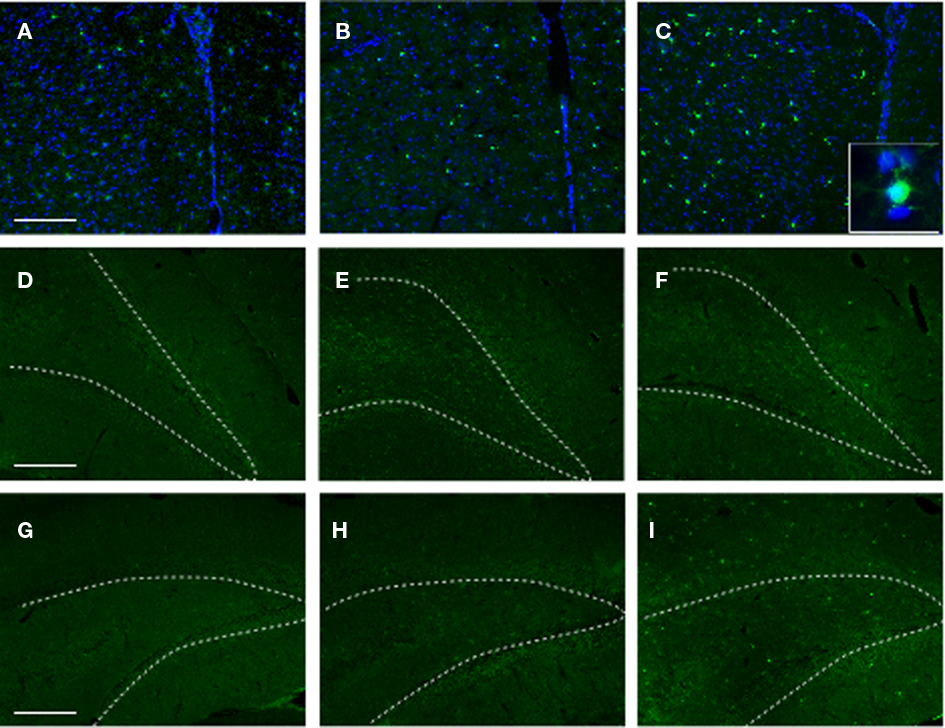
Figure 8. Iba1 immunoreactivity in selected brain regions of animals in the various experimental groups. Frozen coronal sections containing the PFC, DHC, and VHC were cut and processed for immunohistochemistry using an Iba1 antibody. Sections were stained with Hoechst 33342 to mark cellular nuclei. PFC of (A) C, (B) SS, and (C) SI rats; DHC of (D) C, (E) SS, and (F) SI rats; VHC of (G) C, (H) SS, and (I) SI rats. White-dotted line outlines DG layer of hippocampus.
Apoptotic cell death
TUNEL histology was used to determine the extent of apoptotic cell death in the PFC, VHC, and DHC across all experimental groups. The number of TUNEL-positive cells was significantly increased in the hilus of the VHC and the DHC of the SI group (Figure 9). Interestingly, there was no increase in the number of TUNEL-positive cells in the PFC of animals in any of the experimental groups (Figure A1 in Appendix). The exposure to stress alone resulted in no increase in the number of TUNEL-positive cells in any of the SS brain regions investigated.
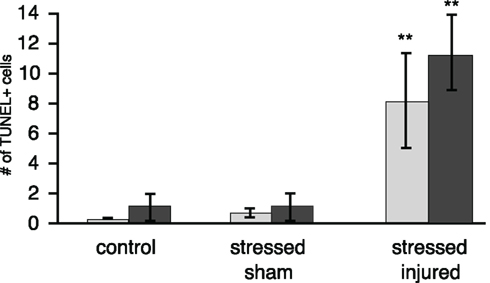
Figure 9. TUNEL-positive cells in selected brain regions of animals in the various experimental groups. Frozen coronal sections containing the hilus of the DHC and VHC of C, SS, and SI rats were cut and processed for TUNEL histology. The number of TUNEL-positive cells were counted and expressed as positive cells per unit area ± SEM. *p < 0.05 and **p < 0.01 compared to C rats.
Neurogenesis
To gain insight into the potential effects of stress (with or without injury) on hippocampal de novo neurogenesis, we performed DCX immunohistochemistry. We observed a noticeable increase in DCX expression in the VHC of SI animals; DCX immunoreactive cells displayed specific morphologies with elaborate processes reaching well into the DG (Figure 10F and insert). Stress alone caused an apparent increase in DCX positive cells in the VHC compared to the controls (Figure 10). However, the DCX+ cells in SS animals lacked the elaborate processes seen in SI animals.
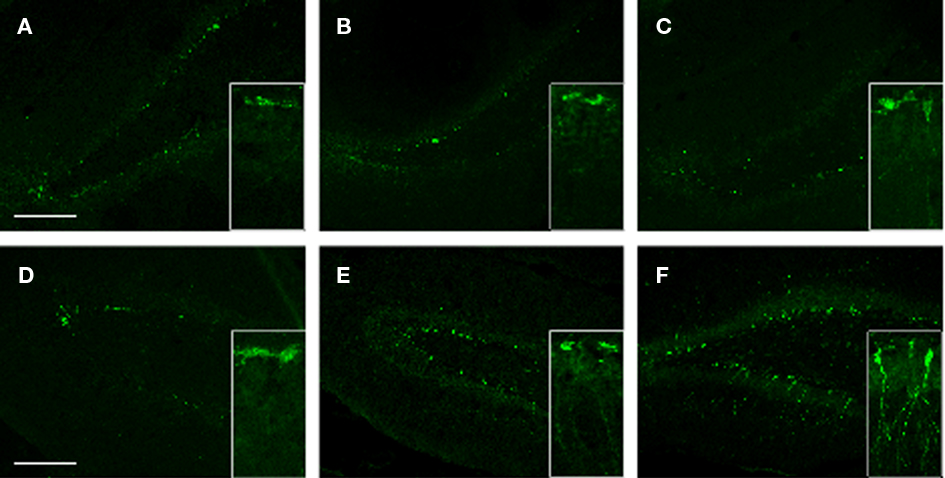
Figure 10. DCX immunoreactivity in selected brain regions of animals in the various experimental groups. Frozen coronal sections containing the PFC, DHC, and VHC were cut. Immunohistochemistry was performed using a DCX antibody. Sections were stained with Hoechst 33342 to mark cellular nuclei. DHC of (A) C, (B) SS, and (C) SI rats; VHC of (D) C, (E) SS, and (F) SI rats. Inlays show the finer details of the cell bodies and processes of DCX cells within the DG of the VHC.
Discussion
The main finding of this study is that repeated stress alone caused a transient increase in anxiety and no major cellular and molecular abnormalities, while the exposure to stress and a single mild blast resulted in a transient (but longer lasting) increase in anxiety and chronic memory impairment. These behavioral changes are associated with neuroinflammation, vascular changes, and neuronal and glial cell loss.
Repetitive stress alone resulted in an early (48 h) increase in anxiety that dissipated at later time points. Our finding is consistent with previous studies where the exposure of rats to a fear-provoking environment resulted in a short-term increase in anxiety (Pynoos et al., 1996). Other studies also reported increased anxiety that lasted up to 7 days after daily exposure to predator odor (Adamec and Shallow, 1993; Cohen et al., 1996, 2000, 2003; Adamec et al., 1997, 1999). On the other hand, exposure of stressed animals to blast appears to prolong the period of increased anxiety, as these animals had elevated anxiety levels up to 1 month after injury. Importantly, anxiety levels of even these animals returned to normal at 2 months post-injury (or sham).
These findings suggest that the long-term effects of stress and injury can dissipate. However, epidemiological studies show that traumatic event(s) or repeated stress can result in PTSD, a chronic condition (Woon et al., 2010), although not all individuals exposed to traumatic events develop PTSD (Gross and Hen, 2004; Yehuda and LeDoux, 2007). The neuroanatomical substrates mediating symptom formation in PTSD include the medial PFC, amygdala, and HC (Bremner, 2007; Liberzon and Sripada, 2008). Within the HC, the VHC is predominantly involved in mediating anxiety-related functions while the DHC is involved in learning and memory-associated functions (Henke, 1990; Moser et al., 1995). In vivo imaging studies have found correlation between hippocampal volume and susceptibility to PTSD development (Karl et al., 2006). A recent meta-analytic study concluded that hippocampal volume reduction is associated with the exposure to trauma independent of PTSD diagnosis, but additional hippocampal reduction is associated with the development of PTSD compared to the trauma-exposed group without PTSD (Woon et al., 2010). A recent comprehensive in vivo imaging study showed that the volume of CA3/DG fields of the HC was significantly reduced in veterans with combat-related PTSD (Wang et al., 2010).
Both genetic and epigenetic factors are suspected in individuals’ susceptibility to developing PTSD. These include an abnormal serotonin system and an altered response to CORT (Gross and Hen, 2004). Stress can alter the function of the hypothalamus–pituitary–adrenal axis (HPA), which in turn leads to abnormally elevated levels of CORT (Yehuda, 2006). Chronically elevated levels of CORT, as we observed, can adversely affect the architecture of the HC. However, a recent study found that chronically elevated CORT levels did not reduce cell number but caused a pronounced loss of synapses, suggesting that volume measures can substantially underestimate the effects of CORT on hippocampal structure and importantly on function (Tata et al., 2006; Tata and Anderson, 2010).
The HC, specifically the DHC, is also a critical neuroanatomical substrate of learning and memory (Henke, 1990; Moser and Humpel, 2005); elevated CORT levels can adversely affect memory (Bannerman et al., 2004). Although we found that CORT levels remained elevated even after 2 months in both SS and SI groups, we found that SS animals did not display memory deficits. Previous studies showed that following daily (foot-shock) stress for 14 days, CORT levels were elevated during the first 7 days post-stress, but returned to control levels by day 14 (Kant et al., 1987). However, there have been no studies to our knowledge that measure the long-term (e.g., 2 months) effects of stress on CORT levels.
In contrast to the transient increase in anxiety, we found that memory impairment was both specific to blast injury and also appeared to be a “chronic” condition. In BM, the performance of SS animals was not significantly different from C animals at any given time point, except on day 8 post-injury. Previous behavioral paradigms, in which rats were exposed to another form of predator stress (Diamond et al., 2006) have indicated that stress can have different effects on memory formation and consolidation depending on the time of exposure. Importantly, our data showed that memory impairment was at its highest at 2 months post-injury. These findings suggest that blast injury predominantly affects the DHC as evidenced by impaired spatial memory.
We found an increased number of TUNEL-positive cells in the hilus of the HC but not in the PFC of animals in the SI group at 2 months post-injury. This apparent lasting apoptotic processes after TBI is quite unusual. The limited information available indicates that in other models of TBI apoptotic activity returned to control levels 2 months after injury (Luo et al., 2002). There were no significant differences in the number of TUNEL-positive cells of animals that were exposed to chronic stress only. As discussed above, SS animals had elevated serum CORT levels but showed no increase in the number of TUNEL-positive cells in their HC or PFC. These findings suggest that in addition to elevated CORT levels, other factors like inflammation may be required to sustain the increase in apoptotic cell death.
Our immunohistochemical data showed an increase in Iba1 immunoreactivity in the PFC as well as the HC of SI animals. Within the HC of SI animals, Iba1 immunoreactivity appeared to be higher in the VHC than in the DHC. However, we also found that exposure to stress alone resulted in an increase in hippocampal IL-6 (but not INFγ) levels 2 month after injury (or sham). IL-6 and INFγ are inflammatory cytokines produced by activated microglia and astroglia and are involved in mediating various responses to injury (Morganti-Kossmann et al., 2001, 2002; Nimmo et al., 2004). Depending on the cellular and molecular context, IL-6 can be neurotoxic or can act as a neuroprotectant (Toulmond et al., 1992).
IL-6 can act as a potent inhibitor of de novo hippocampal neurogenesis; one of several innate regenerative processes triggered by TBI (Vallieres et al., 2002). After a latency period following TBI, the rate of de novo hippocampal neurogenesis increases (Dash et al., 2001; Chirumamilla et al., 2002; Lee and Agoston, 2010). Newborn neurons, marked by DCX expression, migrate from the SGL to the granule cell layer (GCL) where many of the surviving neurons differentiate into granule cells (Altman and Das, 1965; Cameron et al., 1993). We found an apparent increase in DCX positive cells at 2 months after injury in the VHC but not in the DHC of SI rats. Additionally, these cells showed elaborate processes extending well into the DG. Animals in the SS group also showed an increase in DCX immunoreactivity in the VHC but the cells lacked the elaborate processes observed in SI rats. These findings suggest an increase in de novo neurogenesis in the VHC in response to stress, but particularly to the combination of stress and blast. This may indicate that increased de novo neurogenesis in the VHC is partly responsible for the normalization of anxiety observed at 2 months.
The apparent increase in DCX+ cells in the VHC of SI animals contradict what the Iba1 immunohistochemistry and the IL-6 ELISA data would imply. In addition to chronically elevated CORT levels, elevated hippocampal IL-6 levels and the presence of Iba1+ cells in the HC would imply a decrease or repression in neurogenetic activity as neuroinflammation and elevated CORT levels are known inhibitors of hippocampal de novo neurogenesis (Cameron and Gould, 1994; Yu et al., 2004; Montaron et al., 2006); with males showing a greater vulnerability to elevated CORT (Brummelte and Galea, 2010). However, the regulation of de novo hippocampal neurogenesis is rather complex; large numbers of molecules are involved and the exact nature of the regulatory process is currently not fully understood (Kempermann and Gage, 2000; Kempermann, 2002). Even though it has been accepted that an increase in DCX+ cells indicates increased de novo neurogenesis, DCX is only transiently expressed by de novo neurons. Thus, a more detailed BrdU/Prox1 double immunohistochemical and stereological quantification of the histology results is required to determine changes in de novo neurogenesis after stress and blast.
One of the positive regulators of de novo neurogenesis is VEGF; we and others have found that VEGF is significantly upregulated in various forms of TBI (Jin et al., 2002; Lee and Agoston, 2010) increased VEGF level promotes survival of de novo hippocampal neurons by blocking apoptotic cell death (Lee and Agoston, 2010). Consistent with the previous observation, we found that injury and stress, but not stress alone, increases VEGF levels in the HC and also in the PFC. Consistent with the previous observation, we found that injury and stress, but not stress alone, increases VEGF levels in the HC and also in the PFC. Previous studies have demonstrated that an increase in VEGF concentration can also increase vascular permeability as indicated by the breach of the BBB (Dvorak et al., 1995). Interestingly, a recent clinical study has shown that increased serum level of VEGF is indicative of good outcome after ischemic stroke (Sobrino et al., 2009).
We found increased serum levels of several neuronal and glia-specific proteins including NF-H, NSE, CK-BB, and GFAP 2 months post-injury. These molecules have previously been used to assess the extent and the outcome of TBI (Berger, 2006; Korfias et al., 2009). For example, NF-H has been used as a biomarker of neuronal loss and BBB damage and predicting outcome (Anderson et al., 2008). That serum levels of these proteins remain elevated at 2 month after injury suggest ongoing neuronal and glial cell loss as well as a chronically increased BBB permeability in which elevated levels of VEGF may play a role (Ay et al., 2008; Gerstner et al., 2009).
We also found significantly elevated levels of S100β and GFAP in the HC and in the PFC of SI animals. Elevated tissue levels of S100β may indicate astroglial proliferation and overall glial response to injury (Kleindienst et al., 2005). Elevated expression of GFAP by astrocytes, combined with morphological changes (reactive astrogliosis), is a hallmark of CNS neurotrauma (Eng and Ghirnikar, 1994; Fitch and Silver, 2008). We found that the VHC and the DHC appear to have differential expression of stellar GFAP astroglia following exposure to stress only and to the combination stress and injury. The role of increased GFAP immunoreactivity and stellar astroglia in CNS injury is complex. Astrocytic responses can lead to either reparative or detrimental outcomes depending on the type and time after injury (Eng and Ghirnikar, 1994; Fitch and Silver, 2008). However, increasing evidence also indicates a protective role of astrogliosis in reducing the toxic effects of extracellular glutamate and enabling barrier reconstruction after TBI (Buffo et al., 2010). In light of our behavioral findings, showing normalized anxiety levels of SI animals at 2 months post-injury, we speculate that the apparent increase in GFAP positive cells in the VHC may contribute to reparative pathomechanisms and the restoration of normal anxiety levels. Chronic stress has been shown to significantly reduce both the number and somal volume of astroglia in the HC (Czeh et al., 2006). Interestingly, we did see a differential effect of stress alone and stress and injury in the DHC and VHC. While the VHC displayed an apparent increase in GFAP+ cells with stellar processes, the same was not observed in the DHC and memory impairment remained significant 2 months post-injury. A possible explanation for the lasting memory deficits occurring in SI animals is that stress may selectively impair the restorative and/or regenerative action of reactive astrocytes in the DHC. While compelling, the results obtained require further exploration.
In summary, we found that when stressed animals are exposed to a single mild blast overpressure, there are lasting behavioral, molecular, and cellular abnormalities characterized by memory impairment, neuronal and glial cell losses, inflammation, and gliosis. In contrast, stress alone resulted only in a transient increase in anxiety, no memory deficit and no detectable tissue damage. If our findings are independently verified, the potential ramifications can be significant in developing tools to assess the severity and to predict the outcome of psychological and physical traumas.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Neurotrauma Team (WRAIR) for their technical help during the exposures; Ms. Cara Olsen (USU) for her help in statistical analysis; Drs. Grunberg and Wu (USU) for their input in designing and interpreting the behavioral experiments. The work was supported by CDMRP, grant# W81XWH-08-2-0176.
References
Adamec, R. E., Burton, P., Shallow, T., and Budgell, J. (1999). NMDA receptors mediate lasting increases in anxiety-like behavior produced by the stress of predator exposure – implications for anxiety associated with posttraumatic stress disorder. Physiol. Behav. 65, 723–737.
Adamec, R. E., and Shallow, T. (1993). Lasting effects on rodent anxiety of a single exposure to a cat. Physiol. Behav. 54, 101–109.
Adamec, R. E., Shallow, T., and Budgell, J. (1997). Blockade of CCK(B) but not CCK(A) receptors before and after the stress of predator exposure prevents lasting increases in anxiety-like behavior: implications for anxiety associated with posttraumatic stress disorder. Behav. Neurosci. 111, 435–449.
Altman, J., and Das, G. D. (1965). Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 124, 319–335.
Anderson, K. J., Scheff, S. W., Miller, K. M., Roberts, K. N., Gilmer, L. K., Yang, C., and Shaw, G. (2008). The phosphorylated axonal form of the neurofilament subunit NF-H (pNF-H) as a blood biomarker of traumatic brain injury. J. Neurotrauma 25, 1079–1085.
Armonda, R. A., Bell, R. S., Vo, A. H., Ling, G., Degraba, T. J., Crandall, B., Ecklund, J., and Campbell, W. W. (2006). Wartime traumatic cerebral vasospasm: recent review of combat casualties. Neurosurgery 59, 1215–1225; discussion 1225.
Ay, I., Francis, J. W., and Brown, R. H. Jr. (2008). VEGF increases blood–brain barrier permeability to Evans blue dye and tetanus toxin fragment C but not adeno-associated virus in ALS mice. Brain Res. 1234, 198–205.
Bannerman, D. M., Rawlins, J. N., Mchugh, S. B., Deacon, R. M., Yee, B. K., Bast, T., Zhang, W. N., Pothuizen, H. H., and Feldon, J. (2004). Regional dissociations within the hippocampus – memory and anxiety. Neurosci. Biobehav. Rev. 28, 273–283.
Barnes, C. A. (1979). Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J. Comp. Physiol. Psychol. 93, 74–104.
Belanger, H. G., Vanderploeg, R. D., Curtiss, G., and Warden, D. L. (2007). Recent neuroimaging techniques in mild traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 19, 5–20.
Berger, R. P. (2006). The use of serum biomarkers to predict outcome after traumatic brain injury in adults and children. J. Head Trauma Rehabil. 21, 315–333.
Bremner, J. D. (2007). Functional neuroimaging in post-traumatic stress disorder. Expert Rev. Neurother. 7, 393–405.
Brenner, L. A., Vanderploeg, R. D., and Terrio, H. (2009). Assessment and diagnosis of mild traumatic brain injury, posttraumatic stress disorder, and other polytrauma conditions: burden of adversity hypothesis. Rehabil. Psychol. 54, 239–246.
Breslau, N., and Kessler, R. C. (2001). The stressor criterion in DSM-IV posttraumatic stress disorder: an empirical investigation. Biol. Psychiatry 50, 699–704.
Brummelte, S., and Galea, L. A. (2010). Chronic high corticosterone reduces neurogenesis in the dentate gyrus of adult male and female rats. Neuroscience 168, 680–690.
Bruns, J., and Hauser, W. A. (2003). The epidemiology of traumatic brain injury: a review. Epilepsia 44(Suppl.), 2.
Buffo, A., Rolando, C., and Ceruti, S. (2010). Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 79, 77–89.
Cameron, H. A., and Gould, E. (1994). Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience 61, 203–209.
Cameron, H. A., Woolley, C. S., Mcewen, B. S., and Gould, E. (1993). Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience 56, 337–344.
Campbell, T., Lin, S., Devries, C., and Lambert, K. (2003). Coping strategies in male and female rats exposed to multiple stressors. Physiol. Behav. 78, 495–504.
Carobrez, A. P., and Bertoglio, L. J. (2005). Ethological and temporal analyses of anxiety-like behavior: the elevated plus-maze model 20 years on. Neurosci. Biobehav. Rev. 29, 1193–1205.
Cernak, I., Merkle, A. C., Koliatsos, V. E., Bilik, J. M., Luong, Q. T., Mahota, T. M., Xu, L., Slack, N., Windle, D., and Ahmed, F. A. (2010). The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol. Dis. 41, 538–551.
Cernak, I., Wang, Z., Jiang, J., Bian, X., and Savic, J. (2001). Ultrastructural and functional characteristics of blast injury-induced neurotrauma. J. Trauma. 50, 695–706.
Chirumamilla, S., Sun, D., Bullock, M. R., and Colello, R. J. (2002). Traumatic brain injury induced cell proliferation in the adult mammalian central nervous system. J. Neurotrauma 19, 693–703.
Cohen, H., Benjamin, J., Kaplan, Z., and Kotler, M. (2000). Administration of high-dose ketoconazole, an inhibitor of steroid synthesis, prevents posttraumatic anxiety in an animal model. Eur. Neuropsychopharmacol. 10, 429–435.
Cohen, H., Friedberg, S., Michael, M., Kotler, M., and Zeev, K. (1996). Interaction of CCK-4 induced anxiety and post-cat exposure anxiety in rats. Depress. Anxiety 4, 144–145.
Cohen, H., Zohar, J., and Matar, M. (2003). The relevance of differential response to trauma in an animal model of posttraumatic stress disorder. Biol. Psychiatry 53, 463–473.
Czeh, B., Simon, M., Schmelting, B., Hiemke, C., and Fuchs, E. (2006). Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology 31, 1616–1626.
Dash, P. K., Mach, S. A., and Moore, A. N. (2001). Enhanced neurogenesis in the rodent hippocampus following traumatic brain injury. J. Neurosci. Res. 63, 313–319.
Diamond, D. M., Campbell, A. M., Park, C. R., Woodson, J. C., Conrad, C. D., Bachstetter, A. D., and Mervis, R. F. (2006). Influence of predator stress on the consolidation versus retrieval of long-term spatial memory and hippocampal spinogenesis. Hippocampus 16, 571–576.
Doll, H., Truebel, H., Kipfmueller, F., Schaefer, U., Neugebauer, E. A., Wirth, S., and Maegele, M. (2009). Pharyngeal selective brain cooling improves neurofunctional and neurocognitive outcome after fluid percussion brain injury in rats. J. Neurotrauma 26, 235–242.
Dvorak, H. F., Brown, L. F., Detmar, M., and Dvorak, A. M. (1995). Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 146, 1029–1039.
Elliott, B. M., and Grunberg, N. E. (2005). Effects of social and physical enrichment on open field activity differ in male and female Sprague-Dawley rats. Behav. Brain Res. 165, 187–196.
Fitch, M. T., and Silver, J. (2008). CNS injury, glial scars, and inflammation: inhibitory extracellular matrices and regeneration failure. Exp. Neurol. 209, 294–301.
Fride, E., Dan, Y., Feldon, J., Halevy, G., and Weinstock, M. (1986). Effects of prenatal stress on vulnerability to stress in prepubertal and adult rats. Physiol. Behav. 37, 681–687.
Gerstner, E. R., Duda, D. G., Di Tomaso, E., Ryg, P. A., Loeffler, J. S., Sorensen, A. G., Ivy, P., Jain, R. K., and Batchelor, T. T. (2009). VEGF inhibitors in the treatment of cerebral edema in patients with brain cancer. Nat. Rev. Clin. Oncol. 6, 229–236.
Gross, C., and Hen, R. (2004). Genetic and environmental factors interact to influence anxiety. Neurotox. Res. 6, 493–501.
Gyorgy, A. B., Walker, J., Wingo, D., Eidelman, O., Pollard, H. B., Molnar, A., and Agoston, D. V. (2010). Reverse phase protein microarray technology in traumatic brain injury. J. Neurosci. Methods. 192, 96–101.
Harrison, F. E., Hosseini, A. H., and Mcdonald, M. P. (2009). Endogenous anxiety and stress responses in water maze and Barnes maze spatial memory tasks. Behav. Brain Res. 198, 247–251.
Heath, D. L., and Vink, R. (1999). Optimization of magnesium therapy after severe diffuse axonal brain injury in rats. J. Pharmacol. Exp. Ther. 288, 1311–1316.
Henke, P. G. (1990). Hippocampal pathway to the amygdala and stress ulcer development. Brain Res. Bull. 25, 691–695.
Jaffee, M. S., and Meyer, K. S. (2009). A brief overview of traumatic brain injury (TBI) and post-traumatic stress disorder (PTSD) within the Department of Defense. Clin. Neuropsychol. 23, 1291–1298.
Jin, K., Zhu, Y., Sun, Y., Mao, X. O., Xie, L., and Greenberg, D. A. (2002). Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 99, 11946–11950.
Kant, G. J., Leu, J. R., Anderson, S. M., and Mougey, E. H. (1987). Effects of chronic stress on plasma corticosterone, ACTH and prolactin. Physiol. Behav. 40, 775–779.
Karl, A., Schaefer, M., Malta, L. S., Dorfel, D., Rohleder, N., and Werner, A. (2006). A meta-analysis of structural brain abnormalities in PTSD. Neurosci. Biobehav. Rev. 30, 1004–1031.
Kaur, C., Singh, J., Lim, M. K., Ng, B. L., Yap, E. P., and Ling, E. A. (1997). Ultrastructural changes of macroglial cells in the rat brain following an exposure to a non-penetrative blast. Ann. Acad. Med. Singap. 26, 27–29.
Keane, T. M., Marshall, A. D., and Taft, C. T. (2006). Posttraumatic stress disorder: etiology, epidemiology, and treatment outcome. Annu. Rev. Clin. Psychol. 2, 161–197.
Kempermann, G. (2002). Regulation of adult hippocampal neurogenesis – implications for novel theories of major depression. Bipolar Disord. 4, 17–33.
Kempermann, G., and Gage, F. H. (2000). Neurogenesis in the adult hippocampus. Novartis Found. Symp. 231, 220–235; discussion 235–241, 302–226.
Kleindienst, A., Mcginn, M. J., Harvey, H. B., Colello, R. J., Hamm, R. J., and Bullock, M. R. (2005). Enhanced hippocampal neurogenesis by intraventricular S100B infusion is associated with improved cognitive recovery after traumatic brain injury. J. Neurotrauma 22, 645–655.
Korfias, S., Papadimitriou, A., Stranjalis, G., Bakoula, C., Daskalakis, G., Antsaklis, A., and Sakas, D. E. (2009). Serum biochemical markers of brain injury. Mini Rev. Med. Chem. 9, 227–234.
Lee, C., and Agoston, D. V. (2010). Vascular endothelial growth factor is involved in mediating increased de novo hippocampal neurogenesis in response to traumatic brain injury. J. Neurotrauma 27, 541–553.
Liberzon, I., and Sripada, C. S. (2008). The functional neuroanatomy of PTSD: a critical review. Prog. Brain Res. 167, 151–169.
Ling, G., Bandak, F., Armonda, R., Grant, G., and Ecklund, J. (2009). Explosive blast neurotrauma. J. Neurotrauma 26, 815–825.
Ling, G. S., and Ecklund, J. M. (2011). Traumatic brain injury in modern war. Curr. Opin. Anaesthesiol. doi: 10.1097/ACO.0b013e32834458da. [Epub ahead of print].
Long, J. B., Bentley, T. L., Wessner, K. A., Cerone, C., Sweeney, S., and Bauman, R. A. (2009). Blast overpressure in rats: recreating a battlefield injury in the laboratory. J. Neurotrauma 26, 827–840.
Luo, C., Jiang, J., Lu, Y., and Zhu, C. (2002). Spatial and temporal profile of apoptosis following lateral fluid percussion brain injury. Chin. J. Traumatol. 5, 24–27.
Maegele, M., Lippert-Gruener, M., Ester-Bode, T., Sauerland, S., Schafer, U., Molcany, M., Lefering, R., Bouillon, B., Neiss, W. F., Angelov, D. N., Klug, N., Mcintosh, T. K., and Neugebauer, E. A. (2005). Reversal of neuromotor and cognitive dysfunction in an enriched environment combined with multimodal early onset stimulation after traumatic brain injury in rats. J. Neurotrauma 22, 772–782.
Montaron, M. F., Drapeau, E., Dupret, D., Kitchener, P., Aurousseau, C., Le Moal, M., Piazza, P. V., and Abrous, D. N. (2006). Lifelong corticosterone level determines age-related decline in neurogenesis and memory. Neurobiol. Aging 27, 645–654.
Morganti-Kossmann, M. C., Rancan, M., Otto, V. I., Stahel, P. F., and Kossmann, T. (2001). Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock 16, 165–177.
Morganti-Kossmann, M. C., Rancan, M., Stahel, P. F., and Kossmann, T. (2002). Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr. Opin. Crit. Care 8, 101–105.
Moser, K. V., and Humpel, C. (2005). Vascular endothelial growth factor counteracts NMDA-induced cell death of adult cholinergic neurons in rat basal nucleus of Meynert. Brain Res. Bull. 65, 125–131.
Moser, M. B., Moser, E. I., Forrest, E., Andersen, P., and Morris, R. G. (1995). Spatial learning with a minislab in the dorsal hippocampus. Proc. Natl. Acad. Sci. U.S.A. 92, 9697–9701.
Nimmo, A. J., Cernak, I., Heath, D. L., Hu, X., Bennett, C. J., and Vink, R. (2004). Neurogenic inflammation is associated with development of edema and functional deficits following traumatic brain injury in rats. Neuropeptides 38, 40–47.
Pynoos, R. S., Ritzmann, R. F., Steinberg, A. M., Goenjian, A., and Prisecaru, I. (1996). A behavioral animal model of posttraumatic stress disorder featuring repeated exposure to situational reminders. Biol. Psychiatry 39, 129–134.
Richardson, L. K., Frueh, B. C., and Acierno, R. (2010). Prevalence estimates of combat-related post- traumatic stress disorder: critical review. Aust. N. Z. J. Psychiatry 44, 4–19.
Salzberg, M., Kumar, G., Supit, L., Jones, N. C., Morris, M. J., Rees, S., and O’brien, T. J. (2007). Early postnatal stress confers enduring vulnerability to limbic epileptogenesis. Epilepsia 48, 2079–2085.
Sobrino, T., Arias, S., Rodriguez-Gonzalez, R., Brea, D., Silva, Y., De La Ossa, N. P., Agulla, J., Blanco, M., Pumar, J. M., Serena, J., Davalos, A., and Castillo, J. (2009). High serum levels of growth factors are associated with good outcome in intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 29, 1968–1974.
Taber, K. H., Warden, D. L., and Hurley, R. A. (2006). Blast-related traumatic brain injury: what is known? J. Neuropsychiatry Clin. Neurosci. 18, 141–145.
Tagliaferri, F., Compagnone, C., Korsic, M., Servadei, F., and Kraus, J. (2006). A systematic review of brain injury epidemiology in Europe. Acta Neurochir. (Wien) 148, 255–268.
Tata, D. A., and Anderson, B. J. (2010). The effects of chronic glucocorticoid exposure on dendritic length, synapse numbers and glial volume in animal models: implications for hippocampal volume reductions in depression. Physiol. Behav. 99, 186–193.
Tata, D. A., Marciano, V. A., and Anderson, B. J. (2006). Synapse loss from chronically elevated glucocorticoids: relationship to neuropil volume and cell number in hippocampal area CA3. J. Comp. Neurol. 498, 363–374.
Toulmond, S., Vige, X., Fage, D., and Benavides, J. (1992). Local infusion of interleukin-6 attenuates the neurotoxic effects of NMDA on rat striatal cholinergic neurons. Neurosci. Lett. 144, 49–52.
Vallieres, L., Campbell, I. L., Gage, F. H., and Sawchenko, P. E. (2002). Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J. Neurosci. 22, 486–492.
Vink, R., O’Connor, C. A., Nimmo, A. J., and Heath, D. L. (2003). Magnesium attenuates persistent functional deficits following diffuse traumatic brain injury in rats. Neurosci. Lett. 336, 41–44.
von Horsten, S., Exton, M. S., Voge, J., Schult, M., Nagel, E., Schmidt, R. E., Westermann, J., and Schedlowski, M. (1998). Cyclosporine A affects open field behavior in DA rats. Pharmacol. Biochem. Behav. 60, 71–76.
Walf, A. A., and Frye, C. A. (2007). The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat. Protoc. 2, 322–328.
Wang, Z., Neylan, T. C., Mueller, S. G., Lenoci, M., Truran, D., Marmar, C. R., Weiner, M. W., and Schuff, N. (2010). Magnetic resonance imaging of hippocampal subfields in posttraumatic stress disorder. Arch. Gen. Psychiatry 67, 296–303.
Warden, D. (2006). Military TBI during the Iraq and Afghanistan wars. J. Head Trauma Rehabil. 21, 398–402.
Warden, D. L., and French, L. (2005). Traumatic brain injury in the war zone. N. Engl. J. Med. 353, 633–634.
Warden, D. L., Labbate, L. A., Salazar, A. M., Nelson, R., Sheley, E., Staudenmeier, J., and Martin, E. (1997). Posttraumatic stress disorder in patients with traumatic brain injury and amnesia for the event? J. Neuropsychiatry Clin. Neurosci. 9, 18–22.
Weinstock, M., Matlina, E., Maor, G. I., Rosen, H., and Mcewen, B. S. (1992). Prenatal stress selectively alters the reactivity of the hypothalamic-pituitary adrenal system in the female rat. Brain Res. 595, 195–200.
Woon, F. L., Sood, S., and Hedges, D. W. (2010). Hippocampal volume deficits associated with exposure to psychological trauma and posttraumatic stress disorder in adults: a meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 34, 1181–1188.
Yehuda, R. (2006). Advances in understanding neuroendocrine alterations in PTSD and their therapeutic implications. Ann. N. Y. Acad. Sci. 1071, 137–166.
Yehuda, R., and LeDoux, J. (2007). Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron 56, 19–32.
Keywords: stress, blast traumatic brain injury, anxiety, memory, gliosis, inflammation, neurogenesis
Citation: Kwon S-KC, Kovesdi E, Gyorgy AB, Wingo D, Kamnaksh A, Walker J, Long JB and Agoston DV (2011) Stress and traumatic brain injury: a behavioral, proteomics, and histological study. Front. Neur. 2:12. doi: 10.3389/fneur.2011.00012
Received: 11 December 2010;
Accepted: 19 February 2011;
Published online: 07 March 2011.
Edited by:
Marten Risling, Karolinska Institutet, SwedenReviewed by:
Linda Noble, University of California at San Francisco, USARobert Vink, University of Adelaide, Australia
Copyright: © 2011 Kwon, Kovesdi, Gyorgy, Wingo, Kamnaksh, Walker, Long and Agoston. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Denes V. Agoston, Department of Anatomy, Physiology and Genetics, School of Medicine, Uniformed Services University, 4301 Jones Bridge Road, Bethesda, MD 20814, USA. e-mail:dmFnb3N0b25AdXN1aHMuZWR1