 Firas Kobeissy1,2*†
Firas Kobeissy1,2*† Stefania Mondello3*†
Stefania Mondello3*† Nihal Tümer4,5
Nihal Tümer4,5 Hale Z. Toklu4,5,6
Hale Z. Toklu4,5,6 Melissa A. Whidden7
Melissa A. Whidden7 Nataliya Kirichenko4,5
Nataliya Kirichenko4,5 Zhiqun Zhang1
Zhiqun Zhang1 Victor Prima8
Victor Prima8 Walid Yassin9
Walid Yassin9 John Anagli8
John Anagli8 Namas Chandra10
Namas Chandra10 Stan Svetlov8
Stan Svetlov8 Kevin K. W. Wang1*
Kevin K. W. Wang1*- 1Department of Psychiatry, Center of Neuroproteomics & Biomarker Research, University of Florida, Gainesville, FL, USA
- 2Department of Biochemistry and Molecular Genetics, American University of Beirut Medical Center, Beirut, Lebanon
- 3Department of Neurosciences, University of Messina, Messina, Italy
- 4Geriatric Research, Education and Clinical Center, Department of Veterans Affairs Medical Center, University of Florida, Gainesville, FL, USA
- 5Department of Pharmacology and Therapeutics, University of Florida, Gainesville, FL, USA
- 6Department of Pharmacology, Marmara University, Istanbul, Turkey
- 7Department of Kinesiology, West Chester University, West Chester, PA, USA
- 8Banyan Laboratory, Banyan Biomarkers, Inc., Alachua, FL, USA
- 9Department of Neuropsychiatry, Kyoto University, Kyoto, Japan
- 10Department of Biomedical Engineering, New Jersey Institute of Technology, Newark, NJ, USA
Among the U.S. military personnel, blast injury is among the leading causes of brain injury. During the past decade, it has become apparent that even blast injury as a form of mild traumatic brain injury (mTBI) may lead to multiple different adverse outcomes, such as neuropsychiatric symptoms and long-term cognitive disability. Blast injury is characterized by blast overpressure, blast duration, and blast impulse. While the blast injuries of a victim close to the explosion will be severe, majority of victims are usually at a distance leading to milder form described as mild blast TBI (mbTBI). A major feature of mbTBI is its complex manifestation occurring in concert at different organ levels involving systemic, cerebral, neuronal, and neuropsychiatric responses; some of which are shared with other forms of brain trauma such as acute brain injury and other neuropsychiatric disorders such as post-traumatic stress disorder. The pathophysiology of blast injury exposure involves complex cascades of chronic psychological stress, autonomic dysfunction, and neuro/systemic inflammation. These factors render blast injury as an arduous challenge in terms of diagnosis and treatment as well as identification of sensitive and specific biomarkers distinguishing mTBI from other non-TBI pathologies and from neuropsychiatric disorders with similar symptoms. This is due to the “distinct” but shared and partially identified biochemical pathways and neuro-histopathological changes that might be linked to behavioral deficits observed. Taken together, this article aims to provide an overview of the current status of the cellular and pathological mechanisms involved in blast overpressure injury and argues for the urgent need to identify potential biomarkers that can hint at the different mechanisms involved.
Introduction
Traumatic Brain Injury (TBI) represents a major public health problem with an over 150,000 military personnel diagnosed with form of mild traumatic brain injury (mTBI), due to the exposure to blast resulting in a wide range of neurological and psychological symptoms (1, 2). Blast-related brain injuries can be provocatively described as “a silent epidemic of an invisible wound.” Current Explosive mechanisms [improvised explosive devices (IEDs), landmines, and rocket-propelled grenades (RPGs)] are believed to account for 56–78% of Operation Enduring Freedom (OEF), Operation Iraqi Freedom (OIF), and Operation New Dawn (OND) related injuries (3, 4). This has led to labeling the blast-induced TBI (bTBI) as the signature brain injury for combat troops in today’s military (5, 6).
Between 2000 and 2010, the Department of Defense (DoD) reported ~200,000 head injuries as a consequence of combat-related incidents as well as events occurred in a non-deployed environment (civilian injuries) (7). However, even this number may be an underestimate due to the fact that the majority of blast-related mTBIs go misdiagnosed and untreated as a consequence of in-appropriate approaches of screening, invalidated diagnostic criteria or specific detectable abnormalities, and lack of diagnostic tools. Acute blunt penetrating injuries comprised 2.8% of this total, the rest were classified as mTBI (7).
Out of more than 8,000 cases of TBI reviewed by the Defense and Veterans Brain Injury Center, ~50% were related to blast-related barotrauma (8). The clinical features observed in mTBI resulting from blast exposure vary, these include: headache, fatigue, tinnitus, and irritability which have been highly recognized in recent conflicts. Blast overpressure (BOP) injury has been considered the main cause of both morbidity and mortality in neurotrauma (9, 10). Furthermore, blast TBI has been the center for military medical concern in the context of polytrauma, since blast-induced injury, due to its complex components (primary, secondary, tertiary, and quaternary injuries) is often accompanied by hemorrhagic blood loss, multiple fractures, burns, and systemic injury coupled with TBI (11–13).
The recognition of the high incidence and impact of bTBI; in addition, to the need for a more accurate diagnosis and effective therapeutic interventions, led to an impressive number of experimental and human blast injury studies aiming at investigating the complex interconnected pathways involved in the blast-induced neuropathological/behavioral changes.
This review will focus on three major questions: (i) What is the experimental and human evidence that blast is associated with progressive alterations in the brain and via what mechanism(s) they are mediated? (ii) What is the relation between blast-induced brain injury and the development of neuropsychological disorders such as post-traumatic stress disorder (PTSD)? (iii) What are the biochemical markers that can identify, track and predict the injury and symptoms observed in patients exposed to blast injury?
Biomechanics of Blast Injury
Blast overpressure-induced injury results from an explosion characterized by an abrupt release of energy in such a short period of time within a small volume creating a non-linear shock and pressure wave (14). The blast shock wave of the primary blast is solitary supersonic pressure wave (peak overpressure) characterized with a rapid (sub-milliseconds–milliseconds) increase in pressure followed by sharp fall in pressure, often to sub-atmospheric levels before returning to ambient pressure (15, 16). This is coupled with the “blast wind” (forced super-heated air flow) that gives rise to a very large volume of gas that may throw victim’s body against other objects. Blast wind, along with the shock wave are the main components of the “blast wave” (17, 18). Blast waves comprise the shock front followed by the blast wind (19). Blast waves impinge on the head-brain complex while mechanical pressure pulses in the brain; the severity of the injury is dependent upon the magnitude and duration of the pressure cycle (20). The net loading at a material point in the brain comprised of a direct transmissive load and deflection-induced indirect loads. The pressure pulse in the brain is governed by the acoustic impedance mismatches between the head and the brain, and the flexural rigidity of the skull (20).
Blast can cause four different types of insults: (i) the primary injury resulting from the BOP waves due to the shock-wave overpressure or/and under pressure. This event is usually associated with contusion, edema, hemorrhage, and diffuse axonal injury (DAI) (11, 17, 21, 22). (ii) The secondary injury that is due to shrapnel or hard objects propelled at the body. (iii) The tertiary insult involves head translation/rotation coupled with acceleration/deceleration due to blunt impact arising from blast wind and finally (iv) the quaternary insult resulting from thermal burns or the probable use of toxic gases or chemicals.
Compared to previous past conflicts, the majority of war zone wounds have been attributed to secondary blast injury (shrapnel propelled by explosions), while tertiary and quaternary blast injuries were related to terrorist-linked acts involving structural collapse and the use of toxic material. Previous studies on primary injury (BOP) have traditionally focused on gas-containing hollow organs such as the lungs and gastrointestinal tract (14, 23).
In one study by Clemedson discussing blast injury, the term “blast injury” has been used to describe the biophysical and pathophysiological events post exposure to high explosion or the shock wave associated with it (24). The greatest interest was devoted to study the peak pressure, as well as the impulse relevant to pulmonary injuries produced (25–28). Interestingly, on the pathophysiology focused on the sudden alteration in the body ambient pressure, primarily in gas-air filled organs including the lungs, intestines, or in tissues with different specific weight such as the ear and intestines; this occurred at the interface between media with very large differences in density (16, 24, 29, 30).
Furthermore, BOP can induce a mild form of brain injury with significant neurological conditions involving cerebral edema, neuroinflammation, and vasospasm along with DAI and neuronal death. This neuronal injury phase is followed by a series of complex neuropsychiatric symptoms which may include memory loss and behavioral changes (5, 13, 31–33). As such, exposure to complex blast waves can be viewed as the inducer of multitude of injuries or even polytrauma involving several organ injuries interaction that exacerbates blast insult outcome (13). Finally, blast wave propagation to the brain parenchyma is another controversial mechanism which may involves both direct propagation through the skull or in an indirect propagation via blood vessels which has a direct implication on vascular disturbance (31, 34).
Blast wind passage to the skull causes acceleration/rotation to the brain comprising the direct injury. Indirect injury involves the compression of the abdomen and chest transferring kinetic energy to the body’s biofluid. This rippling effect generates oscillating waves from blood to the brain distant from the contact point. In turn, this kinetic energy transfer will induce functional and morphological changes in brain structures which represent a distinct complex feature of blast-induced brain injury not present in other traditional brain injury models (21, 31, 35). The complex mechanism of blast injury involves consequences of primary blast effects on autonomous nervous system. Taken together, it should be comprehended that the mechanics of neurotrauma due to blast injuries are quite different from that of other types of injuries arising from motor vehicle accidents (blunt) or penetrating injuries (ballistics).
Neuropathological Alteration in Blast Injury
Experimental studies of primary blast brain injuries (though limited) have shown evidence of altered cellular, molecular and biochemical processes, and behavioral outcomes. For instance, different studies have shown a heterogeneous profile of brain-associated cellular impairments including: elevation in β-amyloid precursor protein, altered expression of protooncogenes c-Myc, c-Fos, and c-Jun and impaired axonal transport along with oxidative stress with elevated nitric oxide generation (8, 33, 36–44). In addition, neuronal injury and glial activation (discussed later) coupled with elevation of biochemical markers such as, neuron specific enolase (NSE), ubiquitin C-terminal hydrolase 1 (UCH-L1), and glial fibrillary acidic protein (GFAP) have been also reported. Other studies have shown evidence of axonopathy, edema, and hypertrophic astrogliosis with pronounced altered gene expression post-injury event (40, 44–46). However, there were a lot of ambiguity in the overpressure and duration utilized and the methods used to measure these parameters which were often unclear and not standardized (33, 43, 47).
Furthermore, such heterogeneous neural profile has been attributed to several factors including the suitable experimental model systems that can closely mimic “composite” primary, secondary, tertiary, and quaternary components of blast exposure, the lack of standardized blast wave instruments, different body localization and body armor, and the use of different animal species (31, 32, 41, 48) (see Table 1).
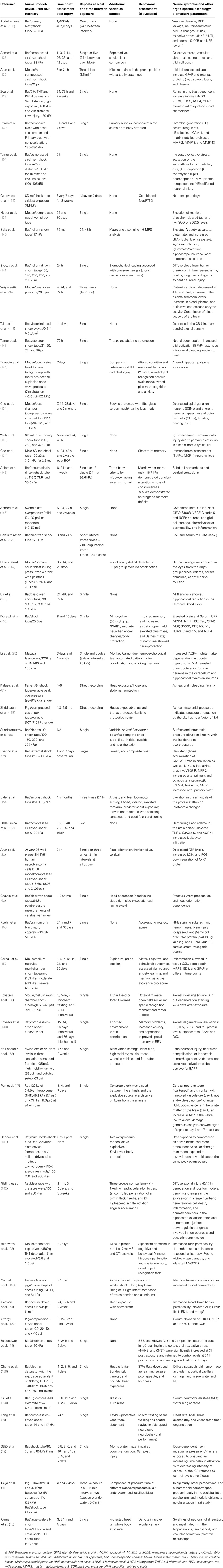
Table 1. Recent major studies on experimental blast injury with different parameters assessed (behavioral, neuropathological, and biomarker changes).
Several studies have been performed to assess neuropathological effect of BOP coupled with other comorbid factors (17, 29, 47–51). In these studies, several parameters were varied (different blast injury models, intensity, animal species used) or other modifications were included (protective vests, stressors, and animal localization).
One representative study is that of Kamnaksh et al. where they assessed different stressors and their contribution to blast injury. These stressors included transportation and blast sound with or without blast injury. Of interest, all groups exhibited increased anxiety, while injured and blast noise-exposed rats showed elevated corticosterone, interferon-c (IFN-c), and interleukin-6 (IL-6) in the amygdala and hippocampus. Injured animals showed elevated Iba1, GFAP, and apoptotic immunoreactivity (52). These data demonstrate that exposure to biological stressors can lead to behavioral changes and trigger specific neuropathological alteration even in the absence of detectable injury.
Pun et al. using a rat model, assessed the effects of a single sublethal blast over pressure (BOP) exposure (48.9–77.3 kPa) in an open-field set up. Histopathological analysis of inflicted brains revealed “darkened” and shrunken cortical neurons with narrowed vasculature at day 1 post-injury. Signs of recovery were demonstrated at days 4 and 7 post-blast exposure. Oligodendrocytes and astrocytes showed TUNEL-positivity in the white matter at day 1. Acute axonal damage was observed in the white matter as indicated by elevated amyloid precursor protein immunoreactivity with no sign of macrophages/microglia change. Major gene changes were observed at day 1 and 4 post-blast pointing toward signs of repair at day 4 and 7. These findings suggest that the BOP levels in the study resulted in mild cellular injury and white matter perturbations (47). In another study by Koliatsos et al. primary (BOP) wave effect of mild BOP (68, 103, and 183 kPag) was compared to secondary and tertiary effects. Using a shock tube generating shock waves, the effects of blast on parenchymatous organs including brain, were evaluated. The main injuries in non-brain organs included hemorrhages in the lung interstitium, hemorrhagic infarcts in liver, spleen, and kidney. Neuropathological changes and behavioral outcomes were evaluated at mild blast intensity showing signs of multifocal axonal injury in the cerebellum, the corticospinal system, and optic tract. These findings were accompanied with prolonged behavioral and motor abnormalities (deficits in social recognition, spatial memory, and in motor coordination). Interestingly, shielding of the torso ameliorated axonal injury and behavioral deficits (50).
In a different study, de Lanerolle et al. used a swine model to assess different scenarios of blast exposure including: simulated free field (blast tube), high-mobility multipurpose wheeled vehicle surrogate, and building 4-walled structure. Of interest, histological changes in the three blast scenarios showed minimal neuronal injury with fiber tract demyelination and intra-cranial hemorrhage. Neuropathological changes involving increased astrocyte activation coupled with proliferation and periventricular axonal injury detected were observed with β-amyloid precursor protein (53).
Long et al. assessed blast-induced physiological, neuropathological, and neurobehavioral changes coupled with Kevlar protective vest encasing the thorax and part of the abdomen using a compression-driven shock tube (at 126- and 147-kPa). Kevlar vest effect reduced air blast mortality and also ameliorated the widespread fiber degeneration in rat brains. BOP was shown to induce abnormal neurologic and neurobehavioral performance along with cardiovascular disruptions involving hemorrhagic hypotension with disruption in cardio-compensatory resilience (reduced peak shed blood volume, etc.) (10). Similarly, Rafaels et al. using a male ferrets with protected thorax and abdomen, evaluated intra-cranial hemorrhage and cardiorespiratory coupling at different ranges of blast exposures. Increasing severity of blast exposure demonstrated increasing apnea immediately after blast accompanied by hemorrhages in proximity to the brain stem (51).
In an interesting study, Garman et al. characterized the neuropathological changes produced by a single blast exposure in rats with body shielding using a helium-driven shock tube (exposure of 35 Psi with left side-head-only exposure) (54). Neuropathological analysis was conducted at various time points (24 h, 72 h, or 2 weeks post-blast). Multifocal axonal degeneration was present in all blast-exposed rats at all-time points coupled with diffused axonal injury in the cerebellar and brainstem white matter tracts. In addition, reactive microglial activation was also identified despite subtle GFAP, ED1, and Iba1 staining. Finally, increased blood–brain barrier (BBB) permeability was seen at 24 h. Findings from this study indicated axonal, dendritic, neuronal, and synaptic degeneration in the initial 2 weeks post exposure with body shielding. Over time, there was also evidence of progression of the axonal degenerative process characterized by increased axonal fragmentation similar to the process of DAI that follows TBI which is suggestive of a therapeutic window in the immediate post-blast period (54).
In conclusion, these different blast studies presented distinguished heterogeneous results (summarized in Table 1); and provided different insights into the associated neuropathological changes occurring post-blast exposure. These findings highlight the challenges encountered in modeling experimental blast injury and translating the findings into preclinical brain injury studies to be evaluated and verified clinically (discussed in different sections).
Neuronal Injury Mechanisms
The exact mechanism by which BOP mediates neuronal injury has not been fully elucidated (47). The neuropathological changes evoked by BOP are different than those described following acute models of brain injury (i.e., acceleration–deceleration injury or direct impact) (10, 55–58) highlighting at the complex pathways involved. Elegant work with experimental data by Cernak et al. has shown that primary closed non-impact blast injury-induced neurotrauma involves the interaction of cerebral, local, and systemic responses (31, 32, 45, 48). These experimental data seem to highlight the fact that blood vessels vasculature (venous as well as arterial) may be acting as a conduit for blast energy transfer to the brain contributing to blast pressure-induced fiber degeneration.
In non-blast brain injury, the primary injury occurs as a consequence of mechanical force due to direct contusion of the brain against skull’s rough interior or due to shearing and stretching forces against the brain tissue (31, 59). This may also involve vascular injury including subdural hematoma from ruptured blood, brain edema from elevated permeability of cerebral vasculature along with reduced blood flow due to intra-cranial pressure or infarction (59). Taken together, these complications represent the secondary and tertiary phases of blast injury.
Cernak et al. assessed the contribution of body-central nervous system (CNS) cross talk involved in blast-induced trauma related to the activation of autonomous nervous system and the neuroendocrine–immune system which contributes significantly to the mechanism of blast injury. Inflammation has been proposed to play an important role in the pathogenesis of long-term neurological deficits due to blast (31). Experiments using rigid body- or head-protection in animals subjected to blast showed that head protection failed to prevent inflammation in the brain while body protection was able to alleviate blast-induced brain functional impairments highlighting the role of body-CNS interaction (31).
Cernak et al. studies have demonstrated that blast exposure (mild-to-moderate) induces the activation of autonomous nervous system in rabbit exposed to BOP. Distinct pathological components in the brain including impaired energy metabolism, and increase in the sodium–potassium ATPase measured in the brainstem and erythrocyte membranes were coupled with edema formation (48, 60). In addition, to link systemic alteration and cerebral inflammation to long-term neurological deficits caused by blast, migration, and accumulation of polymorphonuclear leukocytes as key inflammatory markers of host response were assessed after helium-driven shock tube delivering mild blast injury (103 kPa). In vivo real time imaging of myeloperoxidase (MPO) inflammatory enzyme activity of activated phagocytes was conducted on three groups of rats: (1) whole-body blast; (2) blast with “body armor,” (chest and abdomen) with the head exposed; or (3) blast with “helmet” as head protection (neck and skull) while the rest of the body exposed. One day post-blast exposure, MPO activity was observed in the gastrointestinal tract and the diaphragmal mediastinal parts of the lungs (61).
In the brain, this activity was observed at 7, 14, and 30 days post-blast injury. Of interest, MPO increase in the brain was independent of head protection at 14 and 30 days post-injury suggesting chronic inflammation and highlighting the role of systemic origin of the inflammatory activation mediating brain injury which highly reflects on the role of the vagal afferent neurons mediating gut–brain communication. Taken together, the results of this study clearly demonstrate the importance of the indirect, i.e., blast–body interaction as well as the decisive role of autonomous nervous–neuroendocrine–immune systems interaction in the pathogenesis of blast-induced brain trauma (31).
Similarly, Chavko et al. assessed the theory of the indirect effect of kinetic energy transfer via the blood vessels and the surrounding cerebrospinal fluid (CSF) to the CNS (62). In their work, they evaluated the contribution of direct versus indirect transfer and its correlation to the head orientation and the surface area exposed. Brain biomechanical responses involving pressure inside the brains were assessed in rats exposed to low blast exposure (35 kPa) and positioned in three different orientations with respect to primary blast wave. These positions included: frontal exposure (i.e., head facing blast) right side exposed and head positioned away from blast. Frontal exposures showed higher traces of pressure amplitude and longer duration, suggestive of dynamic pressure transfer (62). On the other hand, the pressure wave inside the brain in the head facing away was similar to hydrodynamic pressure within the brain. It has become more evident that the primary pressure wave can induce functional, biochemical, and morphological alterations in different ways than those observed in other types of traumatic injuries (penetrating head injury).
Mild TBI and Neuropsychiatric Impairments in Blast Injury and PTSD Comorbidity
Another significant aspect of blast injury is psychological health which is highly affected. Many injured troops returning from war zones are afflicted with blast-induced BI experiencing post concussive symptoms (PCS), characterized by memory and cognitive disruption, irritability, anxiety, and fatigue (63). Among these with mTBI, PCS can persist long after exposure leading to major functional impairments (64). Unlike casualties suffered from moderate to severe TBI patients diagnosed with mTBI present with no apparent structural injury and are conscious with typical symptoms including headache, confusion, dizziness, memory impairment, and behavioral changes.
The nomenclature of mTBI has been a challenge for both civilian and military settings as described by Rosenfeld et al. (65). mTBI, according to the DoD, involves head trauma with loss of consciousness for <30 min or exhibiting post-traumatic amnesia for <24 h (66). Patients with mTBI have a Glasgow coma score of 13–15 usually experiencing poor unspecific diagnostic symptoms involving headaches, cognitive dysfunction, etc. independent whether mTBI is blast related or not. It is of high interest to deliver accurate diagnosis for such condition due to the overlapping symptoms mistaken with neuropsychiatric disorders. This in contrary to the moderate and severe blast-related TBI which have 9–12 and 3–8 Glasgow coma score respectively and require special treatment as they exhibit intra-cranial hemorrhage and brain edema (2, 67, 68). Patients with blast-related severe TBI are characterized with delayed vasospasm, and pseudoaneurysm formation requiring early intervention (2, 67). Severe blast-related TBI cases are usually due to the primary and secondary (penetrating injury) phases of blast and would require strict clinical guidelines that are similar to those in non-blast-related severe TBI cases (65).
Mild traumatic brain injury is the most frequent form of brain trauma among deployed military populations (69). It has been shown that repeated exposure to multiple low levels of blast injury account for the majority of mTBIs cases. These victims remain conscious and often are redeployed without proper diagnosis and treatment while they undergo severe mental stress (70, 71). The heterogeneous presentation of BOB injuries among mTBI patients depends on several factors (similar to what is observed in experimental blast injury studies) including: device composition, environment (e.g., presence of intervening protective barriers), distance from blast, and the use of protective shields, etc. (11, 72).
Primary blast component of blast injury is among the main contributors in developing neuropsychiatric impairments associated with the primary phase profile (30, 73). There had been an urgent quest to for future research examining the impact of blast concussion (particularly recurrent concussion) on neuropsychological performance. Neuropsychological evaluation of cognitive status post-blast exposure can be challenging for a variety of reasons. In particular, clinicians may have difficulty assessing: true concussion severity due to limited knowledge of the blast events which may be reflective of self-report months or years post the event(s) occurrence. In addition, the lack of several features of the blast environment may complicate the accuracy of the “blast self-report” involving distance from the blast, concussion severity which these are often unavailable from primary records (74). Thus, the lack of reliable information pertaining to injury characteristics makes it challenging to determine the course of cognitive recovery and rehabilitation. Usually, concussion severity is usually determined based on current PCS on screening instruments which are not necessarily specific to concussion and can be shared with depression or PTSD or even these PCS may be reflective of PTSD itself as elegantly discussed by Nelson et al. (74). Of interest, Hoge et al. reported that more than 40% of soldiers who experienced symptoms associated with mTBI (loss of consciousness) met the criteria for PTSD (1). This same study suggested that increased rates of health problems reported by soldiers exposed to mTBI are mediated mainly via neuropsychiatric disorders such as PTSD or depression, rather than mTBI (1).
Post-traumatic stress disorder, a psychiatric condition that arises after exposure to a life threatening experience such as conditions experienced in combat war zone with or without blast exposure as a form of mTBI (75). This, by itself, poses a challenge in the clinical diagnosis in veterans who are exposed to mTBI since the symptoms may overlap between these conditions exacerbated by other comorbid conditions such as drug abuse or other neuropsychiatric complications (75, 76). A Rand Corporation study indicated that ~20% of returning service personnel (~300,000) have had a TBI and that there was substantial overlap of TBI with the occurrence of PTSD (77).
Psychological stress resulting from exposure to blast wave leads to an altered psychological health status which contribute significantly to the development of PTSD (52, 70). However, a major recurring question arises-due to the similarity of blast injury clinical symptoms and those of PTSD, is how do we clinically differentiate between these two conditions and other neuropsychiatric conditions?
Post-traumatic stress disorder is deemed an effect of psychological and emotional determinants/trauma (i.e., event associated with threat of harm or loss of life to which the individual responds with extreme fear or horror), while mild bTBI is a result of destructive biomechanical forces acting on the brain (78). There is substantial overlap in symptom profile associated with these two conditions (1). For instance, impaired concentration, increased irritability, insomnia, and lack of interest are among the symptoms shared in the diagnosis for mTBI and PTSD (79). Additionally, blast TBI is a well-documented risk factor for the development of PTSD (80–82). The association between the two conditions is further supported by structural and functional neuroimaging studies showing similar abnormalities in patients with blast-related mTBI as well as in those with PTSD (83–86).Such overlap and link determines and contributes to several ambiguities emphasizing the urgent need for finding reliable objective test to make an accurate diagnosis and to improve the understanding of the nature of the interaction and pathophysiology of PTSD and mild bTBI.
Clinical evaluation of a blast-exposed personnel can be challenging as symptoms may range from neurologic problems, psychiatric, or emotional difficulties which may be attributed to blast or due to other psychiatric disorder where in several instances the occurrence of TBI and PTSD may be suggested (81, 87). For neurological assessment in TBI, similar criterion-based methodology to that in PTSD has been used rendering a specific diagnosis to either condition or even to those with both conditions (PTSD or TBI-exposed) uncertain (87–89). Thus, in many cases, clinical diagnosis may result in high rate of inaccurate PTSD diagnosis in persons exposed to TBI (87).
Based on the above, it is of high interest that an accurate detailed knowledge of blast injury biophysics and injury threshold may assist clinicians in better diagnosis (87). This includes expanded neuropsychological studies of blast injury (both experimental and clinical) to identify accurate, specific and sensitive anatomic, pathophysiologic, and behavioral responses to blast injury as discussed by Bass et al. (87). This is complicated by the complex nature of blast injury involving several combinations of primary or other phases of blast injury (secondary, tertiary, and/or quaternary blast).
Animal Models of Blast Injury
Over the last several decades, a number of experimental animal models have been implemented to study the mechanisms of blast wave impact which included rats, mice, ferrets, rabbits, and larger animals involving sheep and swine (33, 90–97). These experimental models exhibited heterogeneous outcomes and even contradictory findings which have been attributed to several factors. A summary of the recent and major blast injury studies (2001, 2009–2013) is summarized in Table 1. In addition, there is a lack in the reproducibility of blast injury models and a need to develop blast injury generators that precisely control blast injury parameters similar to other well-defined acute brain injury models such as (controlled cortical impact (CCI) and the fluid percussion (FP) which have been well characterized with predictable neurological, histological, physiological, and behavioral outcomes. Thus, the need of establishing well characterized reproducible models (animal and blast framework) is vital to identify relevant pathogenic pathways involved that can assist in the development of effective diagnostic, prognostic blast specific-biomarkers (panel of biomarkers) (98). Several blast injury instrumentations are available which include: compressed gas-driven shock tubes which are driven by air, helium, or nitrogen gas which may result in unrealistic duration of the overpressure wave leading to an in-appropriate scaling between species (humans and animal models; Table 1) (99).
Challenges in Animal Models of Blast Injury
There are limited available basic and translational studies relevant to the mechanisms of primary blast-induced brain injury. A better understanding of injury mechanisms is required for the development of protection and treatment options and biomarker identification for prognosis.
Several animal models have been proposed at translating intra-cranial biophysics and pathophysiology experienced in human blast exposure (87). These models have a number of limitations including: neuronal tissue biomechanical properties, anatomical differences as well as physiological differences (87). In addition, other factors that are challenging for proper scaling between experimental and human blast injury are associated with neuroanatomy and physiology involving: size of different brain structures, neural mass (brain size, head, body, position, and architecture), as well as body fluid composition (thickness, volume, and components) (87). Other key factors that need to be considered are the potential for exposure scaling, consistency in experimental protocols, frequency of exposure, and overpressure levels, which should be mimicking real life exposure or at least translate equally to human exposure (Figure 1). Other external factors include: distance from the blast, the use of protective shields and the presence or absence of noise stressors, etc. (12) (Figure 1). In real life situation, soldiers are often deployed several times and exposed to numerous psychological stressors such as blast noise with or without blast injury (87). Such conditions can induce adverse physiological changes leading to post-traumatic symptoms without sustaining any prior physical injury (discussed previously). Taken together, these challenging factors contribute to the difficulty of truly modeling blast injury in animals resulting in an in-appropriate neuropathological and neurobehavioral assessment.
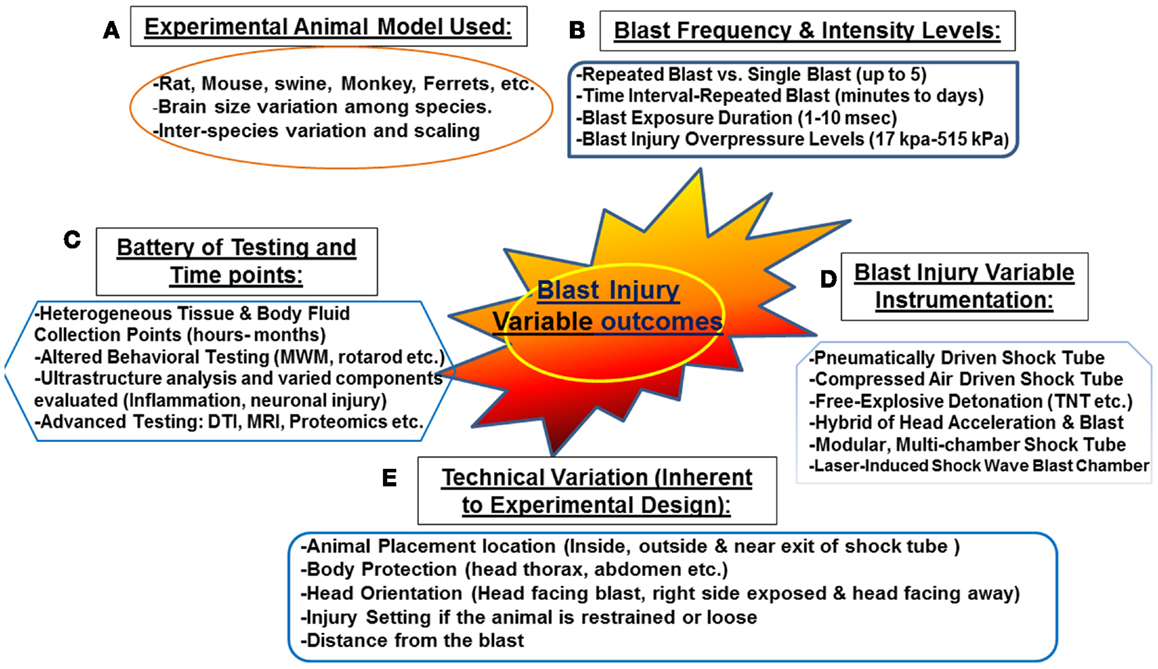
Figure 1. Challenges associated with “experimental blast injury” modeling real life blast exposure. Several factors contribute to the heterogeneous behavioral, neuropathological, and systemic profile observed in the several experimental blast injury models. Even with models using the same injury parameters (animal model, blast shock tube, and intensity levels, etc.); reproducing the same results is rather challenging (refer to Table 1). These challenging variables are summarized in the following: (A) various animal models and interspecies variation, (B) blast injury frequency and intensity levels ranging from single blast up to five blast with some overpressure intensities reaching 515 kPa (C) the heterogeneous selection of biochemical/behavioral testing conducted and the several time points selected (hours to few months) (D) the non-standardized blast and not well characterized blast injury instrumentation (E) technical variation inherent to experimental design related to animal setting, body armor, head protection, and the distance from the blast. These factors contribute to the variable outcome observed in published work in blast injury field.
Blood–Brain Barrier and Secondary Injury in Blast Overpressure
Traumatic brain injury leads to progressive pathophysiological changes resulting in a reduction in cerebral blood flow and a decrease in tissue oxygen levels leading to ischemia, BBB disruption with brain edema (100). Death of resident cells of the CNS has traditionally been accepted to take place in two phases: an early necrotic and an on-going long-term apoptotic phase. Secondary brain injury develops in minutes to months following the original insult, progressively contributing to the worsened neurological impairment. This complex phenomenon is defined by the activation of various neurochemical cascades and the systemic physiological responses which manifest following the traumatic event (101).
At the cellular level, the biphasic nature of secondary injury is mediated by numerous disturbed pathways which include: (a) excitotoxicity caused by an excess of the neurotransmitter glutamate; (b) free radical generation by mitochondrial dysfunction, causing damage to proteins and phospholipid membranes of neurons and glia; and (c) the neuroinflammatory response which takes place due to both CNS and systemic immunoactivation. Thus, diffuse brain injury mediated immune responses, BBB alterations, and neuroinflammation seem to play an important role in the pathology of BOP. The increase in BBB permeability was shown to recover by the third day after the blast exposure (44, 102). Following blast injury, loosening of the vasculature and perivascular unit is mediated by the activation of matrix metalloproteinases and water channel aquaporin-4, promoting edema, enhanced leakiness of the BBB, and progression of neuroinflammation and neuronal degeneration (102). Although many studies demonstrate a similar pathophysiologic progression as the conventional TBI, a recent study reported that cerebrovascular injury due to primary blast is distinct from it; suggesting that BBB disruption in blast injury was an acute one, not resulting from a delayed inflammation as it is in the conventional ones (103).
Recent work from our laboratory has shown that blast injury leads to oxidative stress and autonomic dysfunction (104). Generation of free radicals and hypoxia leads to the failure of the Na+, K+-ATPase, a membrane-bound enzyme required for cellular transport. Dysfunction of this pump is a common feature in CNS pathologies related to ischemic conditions and TBI. The activity of Na+, K+-ATPase pump is very sensitive to free radical reactions and lipid peroxidation. Reductions in this activity can indicate membrane damage indirectly. Thus, Na+, K+-ATPase is clearly down regulated under low O2 conditions which in turn triggers brain edema, enhances the loosening of tight junctions and causes BBB breakdown. MPO activity, an index for neutrophil infiltration, also increases as an evidence of inflammation (105). In summary, failure of pumps, cerebral edema, BBB permeability, neuroinflammation, and oxidative damage are among the major mechanisms that play important roles in the development of secondary brain injury following TBI.
Traumatic Brain Injury and Autonomic Dysfunction
One deleterious consequence of brain injury is autonomic nervous system dysregulation and/or dysautonomia. Autonomic nervous system dysfunction has been documented after TBI but is not well understood. Ninety percent of TBI patients demonstrate signs of autonomic dysfunction during the first week after injury, with about one third of the patients developing longer lasting autonomic dysfunction. Autonomic dysregulation is characterized by distinct changes in cardiovascular hyperactivity, sleep function, and specific biomarkers of neural damage. System dysregulation might lead to a range of comorbidities such as hypertension, endothelial dysfunction, and end-organ perfusion abnormalities. Specifically, TBI disruption of autonomic function most often results in sustained sympatho-activation. This sympathetic hyperactivity after TBI remains poorly understood, although sympathetic hyperactivity likely contributes to the high morbidity and mortality associated with TBI. Sympathetic hyperactivity contributes to systemic stress, including neuroinflammation and oxidative stress in the autonomic nervous system. Eventually these disturbances lead to cardiovascular dysfunction (31, 32, 106) and sleep complications (107). Systemic stress is associated with activation of the hypothalamic-pituitary-adrenal (HPA) axis (108) and the hypothalamic sympatho-adrenal medullary axis (109). It is known that TBI activates the HPA, however little is known regarding the TBI-induced activation of the sympatho-adrenal medullary axis, and there are limited therapeutic options to treat this sympatho-activation.
We recently demonstrated selective biochemical markers of autonomic function and oxidative stress in male Sprague Dawley rats subjected to head-directed overpressure insult (104). There were increased levels of tyrosine hydroxylase (TH), dopamine-β hydroxylase (DβH), Neuropeptide Y (NPY) along with plasma norepinephrine (NE). In addition, blast-induced injury significantly elevated TH in the nucleus tractus solitarius (NTS) of the brain stem while AT1 receptor expression and NADPH oxidase activity, a marker of oxidative stress, was elevated in the hypothalamus suggesting that single BOP exposure results in increased sympatho-excitation. The mechanism may involve the elevated AT1 receptor expression and NADPH oxidase levels in the hypothalamus. Taken together, such effects may be important factors contributing to pathology of brain injury and autonomic dysfunction associated with the clinical profile of patients following BOP exposure (104).
Blast Brain Injury and Oxidative Stress
The primary effects of BOP have been generally attributed to its external physical impact on the body, causing internal mechanical damage. The pathophysiological effects on hollow organs have been extensively studied, but little attention has been given to the biochemical manifestations and molecular mechanism(s) of injury occurring in the brain after BOP exposure. Due to the biochemical nature of BOP compared to physical nature of TBI (impact or penetrating injury), subtle molecular changes such as free radical-mediated oxidative stress occur and contribute to the manifestation of BOP-induced brain injury (40, 44, 110). Previous studies have demonstrated that reactive oxygen species such as the superoxide radicals and nitric oxide can form peroxynitrite, a powerful oxidant that impairs cerebral vascular function following blast-induced brain injury (46, 111). Cernak et al. reported that bilateral vagotomy successfully mitigated bradycardia, hypotension, and apnea caused by blast; prevented extreme metabolic alterations and brain edema; but failed to eliminate oxidative stress in the brain due to blast (48). More recently, it was reported that the induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mTBI induced by primary blast (102). Brain-specific oxidatively modified protein markers that are indicative of biochemical/proteomic and functional changes occurring post-BOP need to be considered. Insufficient published data are available to describe the long-term effects of TBI on central noradrenergic systems, particularly on neuroplastic adaptations within numerous targets of central noradrenergic projections. In addition, understanding the etiology of these changes may shed new light on the molecular mechanism(s) of injury, potentially offering new strategies for treatment.
Blast Injury Biomarkers Identification and Limitations
The widespread recognition of the brain vulnerability to blast exposure and inadequate approaches to diagnose blast-related TBI led to design an mTBI Diagnostics Workshop (66) and the foundation of the Demographics and Clinical Assessment Working Group of the International and Interagency Initiative (112) to assess the current diagnostics technologies that can be used to detect brain injury following mTBI and BOP. One of the major recommendations was the use of biomarkers to supplement functional and imaging-based assessments for significant improvements in the diagnosis and characterization of the effects of blast exposure on brain and for distinguishing bTBI from other neuropsychiatric disorders including PTSD.
Current available imaging modalities, such as computed tomography (CT) and magnetic resonance imaging (MRI), primarily detect major structural changes in the brain (113); however, their utility has not been fully optimized following blast-related mTBI. More advanced neuroimaging techniques such as DTI, while have shown abnormalities post-blast-related TBI (114), have not been able to show consistent relationship to mild bTBI diagnosis (115). Additionally, there is no consensus on the ideal scan method or timing. Therefore, multiple studies have been conducted to identify ideal sensitive, inexpensive, non-invasive biochemical markers that can offer diagnostic and prognostic information, and reflect bTBI pathogenic mechanisms and pathology (116, 117).
To date, several biomarkers such as GFAP (118), UCH-L1 (119), and S-100ß(120) have been identified as potential excellent “candidates” of blast TBI. However, a limited number of studies did specifically evaluate biochemical brain damage markers in the setting of blast-induced brain injury (43, 121). In one study by Svetlov et al. they assessed temporal pattern of serum putative biomarkers that have been characterized in acute TBI including GFAP, NSE, and UCH-L1 in brain tissue, CSF, and blood. Serum biomarkers levels distinctively increased 24 h post-blast, followed by a decline thereafter, indicating a potential use to assess blast-induced brain damage acutely after injury (33). Supporting these observations, Gyorgy and colleagues, using reverse phase protein microarray (RPPM) technology to determine serum protein levels, showed a rise in S-100B, MBP, NF-H, and NSE protein levels in serum after injury in a large-animal model of bTBI. Remarkably, serum NF-H was reported to increase in an overpressure dose-dependent manner reflecting the extent of the damage caused by bTBI (122).
More recently, Balakathiresan et al. proposed microRNAs as novel serum diagnostic biomarkers of bTBI. They investigated microRNA signatures in CSF and serum of rats exposed to BOP injury. Specifically, microRNA let-7i was elevated in both CSF and serum post-blast wave exposure and was considered as an ideal candidate biomarker of brain injury (123). Importantly, microRNAs can be considered the third generation molecular signature after proteomics and genomics studies (123). Elevated concentrations of serum vascular endothelial growth factor, associated with neuroinflammation and vascular pathology in blast-related TBI have also been reported (124).
Studies investigating biomarkers of mTBI in humans continue to be limited as illustrated in one study by Ingebrigtsen and Romner (125). In their research paper, MEDLINE database was surveyed for biochemical serum markers specific to mild head injuries. Three serum markers including creatine kinase isoenzyme BB (CKBB), NSE, and S-100B were evaluated. Of these markers, S-100B protein was proposed as the most promising marker for mTBI while the other two lacked specificity, sensitivity, or injury correlation (125). In an another study by Blennow et al. military personnel exposed to explosions or repeated firing of heavy weapons did not show any evidence of brain damage as assessed by CSF biomarkers. (126). Conversely, the New Zealand Breacher Study demonstrated a degree of brain perturbation as assessed by serum biomarker levels, neurocognitive performance, and self-reported symptoms in members of the New Zealand Defense Force exposed to repeated low-level blast (127). Taken the controversial results of these different studies, these findings, in fact, stimulate the need for further research to evaluate the usefulness of biochemical markers after repeated exposure of different blast levels.
Interestingly, recent experimental and human studies are suggesting a link between blast exposure and chronic traumatic encephalopathy (CTE), a tau protein-linked neurodegenerative disease (128–131). To date, no biofluid marker has been shown to assist with diagnosis of CTE. However, future studies to identify biomarkers tracking chronic processes and on-going degeneration and able to predict the development of neurodegenerative diseases of bTBI are of a critical need.
Future Recommendations
For long, TBI has been considered one of the “signature injuries” of current conflicts in Iraq and Afghanistan which attracted concern from the DoD, Department of Veteran Affairs, and National Institutes of Health, encouraging combined efforts to understand brain injury pathophysiology and identify therapeutics and assess different approaches for rehabilitation platforms as well as deciphering novel blast specific biomarkers (7, 11). Better understanding of the biophysics of blast shock injury and its body propagation to the neural tissue may enhance the development body armor protection. Given the complexity of blast TBI pathobiology, the development of an objective, specific, and quantifiable panel of biomarkers is highly needed for the purpose of providing better monitoring of the real time injury mechanism and progression post-blast exposure (121, 122, 132, 133). An important consideration is that a panel combining different biomarkers be assembled that can establish the nature and severity of the head injury and reflect the contributing pathogenic mechanism(s) of the acute phase as well as the neurodegeneration and recovery (rehabilitative stages). Additionally, the integration of such bTBI diagnostic markers into routine clinical care will require a thorough validation and extensive standardization protocols coupled with well-defined recommendations for immunoassay and different measurement technologies.
A non-trivial and urgent issue in biomarker-panel design will be determining an appropriate instrument platform that is suited to measure these biomarker changes. At present, biomarkers are analyzed in clinical laboratories using closed, high throughput immunoassay analyzers allowing for high performance in terms of accuracy and precision which are suitable for major hospitals. Future recommendation is to focus research on the development of a miniaturized point-of-care (POC) system, which can be transported in the “field” (military and civilian) providing accurate measurements at a reasonable cost with short turnaround time (116).
Conflict of Interest Statement
Drs. Prima and Svetlov are employees and receive salaries from Banyan Biomarkers, Inc. The other co-authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hoge CW, Mcgurk D, Thomas JL, Cox AL, Engel CC, Castro CA. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N Engl J Med (2008) 358:453–63. doi: 10.1056/NEJMoa072972
2. Ling G, Bandak F, Armonda R, Grant G, Ecklund J. Explosive blast neurotrauma. J Neurotrauma (2009) 26:815–25. doi:10.1089/neu.2007.0484
3. Owens BD, Kragh JF Jr, Wenke JC, Macaitis J, Wade CE, Holcomb JB. Combat wounds in operation Iraqi freedom and operation enduring freedom. J Trauma (2008) 64:295–9. doi:10.1097/TA.0b013e318163b875
4. Sayer NA, Chiros CE, Sigford B, Scott S, Clothier B, Pickett T. Characteristics and rehabilitation outcomes among patients with blast and other injuries sustained during the global war on terror. Arch Phys Med Rehabil (2008) 89:163–70. doi:10.1016/j.apmr.2007.05.025
5. Okie S. Reconstructing lives – a tale of two soldiers. N Engl J Med (2006) 355:2609–15. doi:10.1056/NEJMp068235
6. Bhattacharjee Y. Neuroscience. Shell shock revisited: solving the puzzle of blast trauma. Science (2008) 319:406–8. doi:10.1126/science.319.5862.406
7. DePalma RG, Cross GM, Beck LB, Chandler DW. Epidemiology of mTBI (mild traumatic brain injury) due to blast: history, DOD/VA data bases: challenges and opportunities. Proceedings of the NATO RTO-MP-HFM-207 Symposium on A Survey of Blast Injury across the Full Landscape of Military Science. Halifax (2011). p. 1–8.
8. Benzinger TL, Brody D, Cardin S, Curley KC, Mintun MA, Mun SK, et al. Blast-related brain injury: imaging for clinical and research applications: report of the 2008 St. Louis workshop. J Neurotrauma (2009) 26:2127–44. doi:10.1089/neu.2009-0885
10. Long JB, Bentley TL, Wessner KA, Cerone C, Sweeney S, Bauman RA. Blast overpressure in rats: recreating a battlefield injury in the laboratory. J Neurotrauma (2009) 26:827–40. doi:10.1089/neu.2008.0748
11. DePalma RG, Burris DG, Champion HR, Hodgson MJ. Blast injuries. N Engl J Med (2005) 352:1335–42. doi:10.1056/NEJMra042083
12. Belanger HG, Proctor-Weber Z, Kretzmer T, Kim M, French LM, Vanderploeg RD. Symptom complaints following reports of blast versus non-blast mild TBI: does mechanism of injury matter? Clin Neuropsychol (2011) 25:702–15. doi:10.1080/13854046.2011.566892
13. Schultz BA, Cifu DX, Mcnamee S, Nichols M, Carne W. Assessment and treatment of common persistent sequelae following blast induced mild traumatic brain injury. NeuroRehabilitation (2011) 28:309–20. doi:10.3233/NRE-2011-0659
14. Moore DF, Jaffee MS. Military traumatic brain injury and blast. NeuroRehabilitation (2010) 26:179–81. doi:10.3233/NRE-2010-0553
16. Elsayed NM. Toxicology of blast overpressure. Toxicology (1997) 121:1–15. doi:10.1016/S0300-483X(97)03651-2
17. Kirkman E, Watts S, Cooper G. Blast injury research models. Philos Trans R Soc Lond B Biol Sci (2011) 366:144–59. doi:10.1098/rstb.2010.0240
18. Lemonick DM. Bombings and blast injuries: a primer for physicians. Am J Clin Med (2011) 8:134–40.
19. Chandra N, Ganpule S, Kleinschmit NN, Feng R, Holmberg AD, Sundaramurthy A, et al. Evolution of blast wave profiles in simulated air blasts: experiment and computational modeling. Shock Waves (2012) 22:403–15. doi:10.1007/s00193-012-0399-2
20. Selvan V, Ganpule S, Kleinschmit N, Chandra N. Blast wave loading pathways in heterogeneous material systems-experimental and numerical approaches. J Biomech Eng (2013) 135:61002–14. doi:10.1115/1.4024132
21. Warden DL, French LM, Shupenko L, Fargus J, Riedy G, Erickson ME, et al. Case report of a soldier with primary blast brain injury. Neuroimage (2009) 47(Suppl 2):T152–3. doi:10.1016/j.neuroimage.2009.01.060
22. Arun P, Spadaro J, John J, Gharavi RB, Bentley TB, Nambiar MP. Studies on blast traumatic brain injury using in-vitro model with shock tube. Neuroreport (2011) 22:379–84. doi:10.1097/WNR.0b013e328346b138
23. Baker AJ, Topolovec-Vranic J, Michalak A, Pollmann-Mudryj MA, Ouchterlony D, Cheung B, et al. Controlled blast exposure during forced explosive entry training and mild traumatic brain injury. J Trauma (2011) 71:S472–7. doi:10.1097/TA.0b013e318232e7da
25. Clemedson CJ, Pettersson H. Genesis of respiratory and circulatory changes in blast injury. Am J Physiol (1953) 174:316–20.
26. Clemedson CJ. Correlation between respiratory phase and extent of lung damage in air blast injury. J Appl Physiol (1954) 7:38–42.
27. Celander H, Clemedson CJ, Ericsson UA, Hultman HI. A study on the relation between the duration of a shock wave and the severity of the blast injury produced by it. Acta Physiol Scand (1955) 33:14–8. doi:10.1111/j.1748-1716.1955.tb01189.x
28. Clemedson CJ, Hartelius H, Holmberg G. The effect of high explosive blast on the cerebral vascular permeability. Acta Pathol Microbiol Scand (1957) 40:89–95.
29. Mayorga MA. The pathology of primary blast overpressure injury. Toxicology (1997) 121:17–28. doi:10.1016/S0300-483X(97)03652-4
30. Guy RJ, Glover MA, Cripps NP. Primary blast injury: pathophysiology and implications for treatment. Part III: injury to the central nervous system and the limbs. J R Nav Med Serv (2000) 86:27–31.
31. Cernak I. The importance of systemic response in the pathobiology of blast-induced neurotrauma. Front Neurol (2010) 1:151. doi:10.3389/fneur.2010.00151
32. Cernak I, Noble-Haeusslein LJ. Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J Cereb Blood Flow Metab (2010) 30:255–66. doi:10.1038/jcbfm.2009.203
33. Svetlov SI, Prima V, Kirk DR, Gutierrez H, Curley KC, Hayes RL, et al. Morphologic and biochemical characterization of brain injury in a model of controlled blast overpressure exposure. J Trauma (2010) 69:795–804. doi:10.1097/TA.0b013e3181bbd885
34. Valiyaveettil M, Alamneh Y, Wang Y, Arun P, Oguntayo S, Wei Y, et al. Contribution of systemic factors in the pathophysiology of repeated blast-induced neurotrauma. Neurosci Lett (2013) 539:1–6. doi:10.1016/j.neulet.2013.01.028
35. Cernak I, Savic J, Ignjatovic D, Jevtic M. Blast injury from explosive munitions. J Trauma (1999) 47:96–103. doi:10.1097/00005373-199907000-00021 discussion 103-104,
36. Saljo A, Bao F, Haglid KG, Hansson HA. Blast exposure causes redistribution of phosphorylated neurofilament subunits in neurons of the adult rat brain. J Neurotrauma (2000) 17:719–26. doi:10.1089/089771500415454
37. Saljo A, Bao F, Hamberger A, Haglid KG, Hansson HA. Exposure to short-lasting impulse noise causes microglial and astroglial cell activation in the adult rat brain. Pathophysiology (2001) 8:105–11. doi:10.1016/S0928-4680(01)00067-0
38. Saljo A, Bao F, Jingshan S, Hamberger A, Hansson HA, Haglid KG. Exposure to short-lasting impulse noise causes neuronal c-Jun expression and induction of apoptosis in the adult rat brain. J Neurotrauma (2002) 19:985–91. doi:10.1089/089771502753594945
39. Saljo A, Huang YL, Hansson HA. Impulse noise transiently increased the permeability of nerve and glial cell membranes, an effect accentuated by a recent brain injury. J Neurotrauma (2003) 20:787–94. doi:10.1089/089771503767870014
40. Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med (2008) 45:443–52. doi:10.1016/j.freeradbiomed.2008.04.038
41. Saljo A, Arrhen F, Bolouri H, Mayorga M, Hamberger A. Neuropathology and pressure in the pig brain resulting from low-impulse noise exposure. J Neurotrauma (2008) 25:1397–406. doi:10.1089/neu.2008.0602
42. Saljo A, Bolouri H, Mayorga M, Svensson B, Hamberger A. Low-level blast raises intracranial pressure and impairs cognitive function in rats: prophylaxis with processed cereal feed. J Neurotrauma (2009) 27:383–9. doi:10.1089/neu.2009.1053
43. Svetlov SI, Larner SF, Kirk DR, Atkinson J, Hayes RL, Wang KK. Biomarkers of blast-induced neurotrauma: profiling molecular and cellular mechanisms of blast brain injury. J Neurotrauma (2009) 26:913–21. doi:10.1089/neu.2008.0609
44. Readnower RD, Chavko M, Adeeb S, Conroy MD, Pauly JR, McCarron RM, et al. Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J Neurosci Res (2010) 88:3530–9. doi:10.1002/jnr.22510
45. Cernak I, Wang Z, Jiang J, Bian X, Savic J. Ultrastructural and functional characteristics of blast injury-induced neurotrauma. J Trauma (2001) 50:695–706. doi:10.1097/00005373-200104000-00017
46. DeWitt DS, Prough DS. Blast-induced brain injury and posttraumatic hypotension and hypoxemia. J Neurotrauma (2009) 26:877–87. doi:10.1089/neu.2007.0439
47. Pun PB, Kan EM, Salim A, Li Z, Ng KC, Moochhala SM, et al. Low level primary blast injury in rodent brain. Front Neurol (2011) 2:19. doi:10.3389/fneur.2011.00019
48. Cernak I, Savic J, Malicevic Z, Zunic G, Radosevic P, Ivanovic I, et al. Involvement of the central nervous system in the general response to pulmonary blast injury. J Trauma (1996) 40:S100–4. doi:10.1097/00005373-199603001-00023
49. Davenport ND, Lim KO, Armstrong MT, Sponheim SR. Diffuse and spatially variable white matter disruptions are associated with blast-related mild traumatic brain injury. Neuroimage (2011) 59:2017–24. doi:10.1016/j.neuroimage.2011.10.050
50. Koliatsos VE, Cernak I, Xu L, Song Y, Savonenko A, Crain BJ, et al. A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J Neuropathol Exp Neurol (2011) 70:399–416. doi:10.1097/NEN.0b013e3182189f06
51. Rafaels KA, Bass CR, Panzer MB, Salzar RS, Woods WA, Feldman SH, et al. Brain injury risk from primary blast. J Trauma Acute Care Surg (2012) 73:895–901. doi:10.1097/TA.0b013e31825a760e
52. Kamnaksh A, Kovesdi E, Kwon SK, Wingo D, Ahmed F, Grunberg NE, et al. Factors affecting blast traumatic brain injury. J Neurotrauma (2011) 28:2145–53. doi:10.1089/neu.2011.1983
53. de Lanerolle NC, Bandak F, Kang D, Li AY, Du F, Swauger P, et al. Characteristics of an explosive blast-induced brain injury in an experimental model. J Neuropathol Exp Neurol (2011) 70:1046–57. doi:10.1097/NEN.0b013e318235bef2
54. Garman RH, Jenkins LW, Switzer RC III, Bauman RA, Tong LC, Swauger PV, et al. Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J Neurotrauma (2011) 28:947–59. doi:10.1089/neu.2010.1540
55. Lighthall JW. Controlled cortical impact: a new experimental brain injury model. J Neurotrauma (1988) 5:1–15. doi:10.1089/neu.1988.5.1
56. McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, et al. Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience (1989) 28:233–44. doi:10.1016/0306-4522(89)90247-9
57. Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods (1991) 39:253–62. doi:10.1016/0165-0270(91)90104-8
58. Hall ED, Bryant YD, Cho W, Sullivan PG. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J Neurotrauma (2008) 25:235–47. doi:10.1089/neu.2007.0383
59. Greve MW, Zink BJ. Pathophysiology of traumatic brain injury. Mt Sinai J Med (2009) 76:97–104. doi:10.1002/msj.20104
60. Cernak I, Radosevic P, Malicevic Z, Savic J. Experimental magnesium depletion in adult rabbits caused by blast overpressure. Magnes Res (1995) 8:249–59.
61. Cernak I, Merkle AC, Koliatsos VE, Bilik JM, Luong QT, Mahota TM, et al. The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol Dis (2010) 41:538–51. doi:10.1016/j.nbd.2010.10.025
62. Chavko M, Watanabe T, Adeeb S, Lankasky J, Ahlers ST, McCarron RM. Relationship between orientation to a blast and pressure wave propagation inside the rat brain. J Neurosci Methods (2011) 195:61–6. doi:10.1016/j.jneumeth.2010.11.019
63. Okie S. Traumatic brain injury in the war zone. N Engl J Med (2005) 352:2043–7. doi:10.1056/NEJMp058102
64. Carroll LJ, Cassidy JD, Peloso PM, Borg J, Von Holst H, Holm L, et al. Prognosis for mild traumatic brain injury: results of the WHO collaborating centre task force on mild traumatic brain injury. J Rehabil Med (2004) 43:84–105. doi:10.1080/16501960410023859
65. Rosenfeld JV, Mcfarlane AC, Bragge P, Armonda RA, Grimes JB, Ling GS. Blast-related traumatic brain injury. Lancet Neurol (2013) 12:882–93. doi:10.1016/S1474-4422(13)70161-3
66. Marion DW, Curley KC, Schwab K, Hicks RR, mTBI Diagnostics Workgroup. Proceedings of the military mTBI diagnostics workshop, St. Pete Beach, August 2010. J Neurotrauma (2011) 28:517–26. doi:10.1089/neu.2010.1638
67. Armonda RA, Bell RS, Vo AH, Ling G, Degraba TJ, Crandall B, et al. Wartime traumatic cerebral vasospasm: recent review of combat casualties. Neurosurgery (2006) 59:1215–25. doi:10.1227/01.NEU.0000249190.46033.94 discussion 1225,
68. Management of patients with severe head trauma: joint theater trauma system clinical practice guideline. (2012). Available from: http://www.usaisr.amedd.army.mil/assets/cpgs/Mgmt_of_Patients_with_%20Severe_Head_Trauma_7_Mar_12.pdf
69. Vanderploeg RD, Belanger HG, Horner RD, Spehar AM, Powell-Cope G, Luther SL, et al. Health outcomes associated with military deployment: mild traumatic brain injury, blast, trauma, and combat associations in the Florida national guard. Arch Phys Med Rehabil (2012) 93:1887–95. doi:10.1016/j.apmr.2012.05.024
70. Trudeau DL, Anderson J, Hansen LM, Shagalov DN, Schmoller J, Nugent S, et al. Findings of mild traumatic brain injury in combat veterans with PTSD and a history of blast concussion. J Neuropsychiatry Clin Neurosci (1998) 10:308–13.
71. Santiago PN, Wilk JE, Milliken CS, Castro CA, Engel CC, Hoge CW. Screening for alcohol misuse and alcohol-related behaviors among combat veterans. Psychiatr Serv (2010) 61:575–81. doi:10.1176/appi.ps.61.6.575
72. Taber KH, Warden DL, Hurley RA. Blast-related traumatic brain injury: what is known? J Neuropsychiatry Clin Neurosci (2006) 18:141–5. doi:10.1176/appi.neuropsych.18.2.141
73. Kocsis JD, Tessler A. Pathology of blast-related brain injury. J Rehabil Res Dev (2009) 46:667–72. doi:10.1682/JRRD.2008.08.0100
74. Nelson NW, Hoelzle JB, Mcguire KA, Ferrier-Auerbach AG, Charlesworth MJ, Sponheim SR. Neuropsychological evaluation of blast-related concussion: illustrating the challenges and complexities through OEF/OIF case studies. Brain Inj (2011) 25:511–25. doi:10.3109/02699052.2011.558040
75. Zatzick DF, Rivara FP, Jurkovich GJ, Hoge CW, Wang J, Fan MY, et al. Multisite investigation of traumatic brain injuries, posttraumatic stress disorder, and self-reported health and cognitive impairments. Arch Gen Psychiatry (2010) 67:1291–300. doi:10.1001/archgenpsychiatry.2010.158
76. Seal KH, Cohen G, Waldrop A, Cohen BE, Maguen S, Ren L. Substance use disorders in Iraq and Afghanistan veterans in VA healthcare, 2001-2010: implications for screening, diagnosis and treatment. Drug Alcohol Depend (2011) 116:93–101. doi:10.1016/j.drugalcdep.2010.11.027
77. Tanielian E, Jayco LH. Invisible Wounds of War: Psychological and Cognitive Injuries, Their Consequences, and Services to Assist Recovery. Los Angeles: Rand Corporation (2008).
78. Stein MB, McAllister TW. Exploring the convergence of posttraumatic stress disorder and mild traumatic brain injury. Am J Psychiatry (2009) 166:768–76. doi:10.1176/appi.ajp.2009.08101604
79. APA. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association (2000).
80. Vasterling JJ, Verfaellie M, Sullivan KD. Mild traumatic brain injury and posttraumatic stress disorder in returning veterans: perspectives from cognitive neuroscience. Clin Psychol Rev (2009) 29:674–84. doi:10.1016/j.cpr.2009.08.004
81. Rosenfeld JV, Ford NL. Bomb blast, mild traumatic brain injury and psychiatric morbidity: a review. Injury (2010) 41:437–43. doi:10.1016/j.injury.2009.11.018
82. Bryant R. Post-traumatic stress disorder vs traumatic brain injury. Dialogues Clin Neurosci (2011) 13:251–62.
83. Di Stefano G, Bachevalier J, Levin HS, Song JX, Scheibel RS, Fletcher JM. Volume of focal brain lesions and hippocampal formation in relation to memory function after closed head injury in children. J Neurol Neurosurg Psychiatry (2000) 69:210–6. doi:10.1136/jnnp.69.2.210
84. Geuze E, Vermetten E, Bremner JD. MR-based in vivo hippocampal volumetrics: 2. Findings in neuropsychiatric disorders. Mol Psychiatry (2005) 10:160–84. doi:10.1038/sj.mp.4001580
85. Francati V, Vermetten E, Bremner JD. Functional neuroimaging studies in posttraumatic stress disorder: review of current methods and findings. Depress Anxiety (2007) 24:202–18. doi:10.1002/da.20208
86. Scheibel RS, Newsome MR, Troyanskaya M, Lin X, Steinberg JL, Radaideh M, et al. Altered brain activation in military personnel with one or more traumatic brain injuries following blast. J Int Neuropsychol Soc (2012) 18:89–100. doi:10.1017/S1355617711001433
87. Bass CR, Panzer MB, Rafaels KA, Wood G, Shridharani J, Capehart B. Brain injuries from blast. Ann Biomed Eng (2011) 40:185–202. doi:10.1007/s10439-011-0424-0
88. First MB, Frances A, Pincus HA. DSM-IV Handbook of Differential Diagnosis. Washington, DC: American Psychiatric Press (1995).
89. Bombardier CH, Fann JR, Temkin N, Esselman PC, Pelzer E, Keough M, et al. Posttraumatic stress disorder symptoms during the first six months after traumatic brain injury. J Neuropsychiatry Clin Neurosci (2006) 18:501–8. doi:10.1176/appi.neuropsych.18.4.501
90. Irwin RJ, Lerner MR, Bealer JF, Lightfoot SA, Brackett DJ, Tuggle DW. Global primary blast injury: a rat model. J Okla State Med Assoc (1998) 91:387–92.
91. Garner JP, Watts S, Parry C, Bird J, Kirkman E. Development of a large animal model for investigating resuscitation after blast and hemorrhage. World J Surg (2009) 33:2194–202. doi:10.1007/s00268-009-0105-4
92. Svetlov SI, Prima V, Glushakova O, Svetlov A, Kirk DR, Gutierrez H, et al. Neuro-glial and systemic mechanisms of pathological responses in rat models of primary blast overpressure compared to “composite” blast. Front Neurol (2012) 3:15. doi:10.3389/fneur.2012.00015
93. Rubovitch V, Ten-Bosch M, Zohar O, Harrison CR, Tempel-Brami C, Stein E, et al. A mouse model of blast-induced mild traumatic brain injury. Exp Neurol (2011) 232:280–9. doi:10.1016/j.expneurol.2011.09.018
94. Lei T, Xie L, Tu W, Chen Y, Tan Y. Development of a finite element model for blast injuries to the pig mandible and a preliminary biomechanical analysis. J Trauma Acute Care Surg (2012) 73:902–7. doi:10.1097/TA.0b013e3182515cb1
95. Li J, Topaz M, Xun W, Li W, Wang X, Liu H, et al. New swine model of infected soft tissue blast injury. J Trauma Acute Care Surg (2012) 73:908–13. doi:10.1097/TA.0b013e318253b592
96. Sundaramurthy A, Alai A, Ganpule S, Holmberg A, Plougonven E, Chandra N. Blast-induced biomechanical loading of the rat: an experimental and anatomically accurate computational blast injury model. J Neurotrauma (2012) 29:2352–64. doi:10.1089/neu.2012.2413
97. Yarnell AM, Shaughness MC, Barry ES, Ahlers ST, McCarron RM, Grunberg NE. Blast traumatic brain injury in the rat using a blast overpressure model. Curr Protoc Neurosci (2013). Chapter 9, Unit 9.41. doi:10.1002/0471142301.ns0941s62
98. Kochanek PM, Bauman RA, Long JB, Dixon CR, Jenkins LW. A critical problem begging for new insight and new therapies. J Neurotrauma (2009) 26:813–4. doi:10.1089/neu.2008.0893
99. Pervin F, Chen WW. Effect of inter-species, gender, and breeding on the mechanical behavior of brain tissue. Neuroimage (2011) 54(Suppl 1):S98–102. doi:10.1016/j.neuroimage.2010.03.077
100. Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience (2004) 129:1021–9. doi:10.1016/j.neuroscience.2004.06.046
101. Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury (2007) 38:1392–400. doi:10.1016/j.injury.2007.10.005
102. Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, et al. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med (2013) 60:282–91. doi:10.1016/j.freeradbiomed.2013.02.029
103. Yeoh S, Bell ED, Monson KL. Distribution of blood-brain barrier disruption in primary blast injury. Ann Biomed Eng (2013) 41(10):2206–14. doi:10.1007/s10439-013-0805-7
104. Tumer N, Svetlov S, Whidden M, Kirichenko N, Prima V, Erdos B, et al. Overpressure blast-wave induced brain injury elevates oxidative stress in the hypothalamus and catecholamine biosynthesis in the rat adrenal medulla. Neurosci Lett (2013) 544:62–7. doi:10.1016/j.neulet.2013.03.042
105. Biber N, Toklu HZ, Solakoglu S, Gultomruk M, Hakan T, Berkman Z, et al. Cysteinyl-leukotriene receptor antagonist montelukast decreases blood-brain barrier permeability but does not prevent oedema formation in traumatic brain injury. Brain Inj (2009) 23:577–84. doi:10.1080/02699050902926317
106. Hinson HE, Sheth KN. Manifestations of the hyperadrenergic state after acute brain injury. Curr Opin Crit Care (2012) 18:139–45. doi:10.1097/MCC.0b013e3283513290
107. Viola-Saltzman M, Watson NF. Traumatic brain injury and sleep disorders. Neurol Clin (2012) 30:1299–312. doi:10.1016/j.ncl.2012.08.008
108. Griesbach GS. Exercise after traumatic brain injury: is it a double-edged sword? PM R (2011) 3:S64–72. doi:10.1016/j.pmrj.2011.02.008
109. Kvetnansky R, Sabban EL, Palkovits M. Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol Rev (2009) 89:535–606. doi:10.1152/physrev.00042.2006
110. Kochanek PM, Dixon CE, Shellington DK, Shin SS, Bayir H, Jackson E, et al. Screening of biochemical and molecular mechanisms of secondary injury and repair in the brain after experimental blast-induced traumatic brain injury in rats. J Neurotrauma (2012) 30(11):920–37. doi:10.1089/neu.2013.2862
111. Vuceljic M, Zunic G, Romic P, Jevtic M. Relation between both oxidative and metabolic-osmotic cell damages and initial injury severity in bombing casualties. Vojnosanit Pregl (2006) 63:545–51. doi:10.2298/VSP0606545V
112. Menon DK, Schwab K, Wright DW, Maas AI, Demographics and Clinical Assessment Working Group of the International and Interagency Initiative toward Common Data Elements for Research on Traumatic Brain Injury and Psychological Health. Position statement: definition of traumatic brain injury. Arch Phys Med Rehabil (2010) 91:1637–40. doi:10.1016/j.apmr.2010.05.017
113. Bazarian JJ, Blyth B, Cimpello L. Bench to bedside: evidence for brain injury after concussion – looking beyond the computed tomography scan. Acad Emerg Med (2006) 13:199–214. doi:10.1197/j.aem.2005.07.031
114. Mac Donald CL, Johnson AM, Cooper D, Nelson EC, Werner NJ, Shimony JS, et al. Detection of blast-related traumatic brain injury in U.S. military personnel. N Engl J Med (2011) 364:2091–100. doi:10.1056/NEJMoa1008069
115. Levin HS, Wilde E, Troyanskaya M, Petersen NJ, Scheibel R, Newsome M, et al. Diffusion tensor imaging of mild to moderate blast-related traumatic brain injury and its sequelae. J Neurotrauma (2010) 27:683–94. doi:10.1089/neu.2009.1073
116. Mondello S, Muller U, Jeromin A, Streeter J, Hayes RL, Wang KK. Blood-based diagnostics of traumatic brain injuries. Expert Rev Mol Diagn (2011) 11:65–78. doi:10.1586/erm.10.104
117. Mondello S, Schmid K, Berger RP, Kobeissy F, Italiano D, Jeromin A, et al. The challenge of mild traumatic brain injury: role of biochemical markers in diagnosis of brain damage. Med Res Rev (2013). doi:10.1002/med.21295
118. Papa L, Lewis LM, Falk JL, Zhang Z, Silvestri S, Giordano P, et al. Elevated levels of serum glial fibrillary acidic protein breakdown products in mild and moderate traumatic brain injury are associated with intracranial lesions and neurosurgical intervention. Ann Emerg Med (2012) 59:471–83. doi:10.1016/j.annemergmed.2011.08.021
119. Papa L, Lewis LM, Silvestri S, Falk JL, Giordano P, Brophy GM, et al. Serum levels of ubiquitin C-terminal hydrolase distinguish mild traumatic brain injury from trauma controls and are elevated in mild and moderate traumatic brain injury patients with intracranial lesions and neurosurgical intervention. J Trauma Acute Care Surg (2012) 72:1335–44. doi:10.1097/TA.0b013e3182491e3d
120. Unden J, Romner B. Can low serum levels of S100B predict normal CT findings after minor head injury in adults? an evidence-based review and meta-analysis. J Head Trauma Rehabil (2010) 25:228–40. doi:10.1097/HTR.0b013e3181e57e22
121. Agoston DV, Gyorgy A, Eidelman O, Pollard HB. Proteomic biomarkers for blast neurotrauma: targeting cerebral edema, inflammation, and neuronal death cascades. J Neurotrauma (2009) 26:901–11. doi:10.1089/neu.2008.0724
122. Gyorgy A, Ling G, Wingo D, Walker J, Tong L, Parks S, et al. Time-dependent changes in serum biomarker levels after blast traumatic brain injury. J Neurotrauma (2011) 28:1121–6. doi:10.1089/neu.2010.1561
123. Balakathiresan N, Bhomia M, Chandran R, Chavko M, McCarron RM, Maheshwari RK. MicroRNA let-7i is a promising serum biomarker for blast-induced traumatic brain injury. J Neurotrauma (2012) 29:1379–87. doi:10.1089/neu.2011.2146
124. Agoston DV, Elsayed M. Serum-based protein biomarkers in blast-induced traumatic brain injury spectrum disorder. Front Neurol (2012) 3:107. doi:10.3389/fneur.2012.00107
125. Ingebrigtsen T, Romner B. Biochemical serum markers for brain damage: a short review with emphasis on clinical utility in mild head injury. Restor Neurol Neurosci (2003) 21:171–6.
126. Blennow K, Jonsson M, Andreasen N, Rosengren L, Wallin A, Hellstrom PA, et al. No neurochemical evidence of brain injury after blast overpressure by repeated explosions or firing heavy weapons. Acta Neurol Scand (2011) 123:245–51. doi:10.1111/j.1600-0404.2010.01408.x
127. Tate CM, Wang KK, Eonta S, Zhang Y, Carr W, Tortella FC, et al. Serum brain biomarker level, neurocognitive performance, and self-reported symptom changes in soldiers repeatedly exposed to low-level blast: a breacher pilot study. J Neurotrauma (2013) 30(19):1620–30. doi:10.1089/neu.2012.2683
128. Omalu B, Hammers JL, Bailes J, Hamilton RL, Kamboh MI, Webster G, et al. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus (2011) 31:E3. doi:10.3171/2011.9.FOCUS11178
129. Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med (2012) 4:134ra60. doi:10.1126/scitranslmed.3003716
130. Miller G. Neuropathology. Blast injuries linked to neurodegeneration in veterans. Science (2012) 336:790–1. doi:10.1126/science.336.6083.790
131. Huber BR, Meabon JS, Martin TJ, Mourad PD, Bennett R, Kraemer BC, et al. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J Alzheimers Dis (2013) 37(2):309–23. doi:10.3233/JAD-130182
132. Berger RP. The use of serum biomarkers to predict outcome after traumatic brain injury in adults and children. J Head Trauma Rehabil (2006) 21:315–33. doi:10.1097/00001199-200607000-00004
133. Beers SR, Berger RP, Adelson PD. Neurocognitive outcome and serum biomarkers in inflicted versus non-inflicted traumatic brain injury in young children. J Neurotrauma (2007) 24:97–105. doi:10.1089/neu.2006.0055
134. Cho HJ, Sajja VS, Vandevord PJ, Lee YW. Blast induces oxidative stress, inflammation, neuronal loss and subsequent short-term memory impairment in rats. Neuroscience (2013) 253C:9–20. doi:10.1016/j.neuroscience.2013.08.037
135. Genovese RF, Simmons LP, Ahlers ST, Maudlin-Jeronimo E, Dave JR, Boutte AM. Effects of mild TBI from repeated blast overpressure on the expression and extinction of conditioned fear in rats. Neuroscience (2013) 254C:120–9. doi:10.1016/j.neuroscience.2013.09.021
136. Ahmed FA, Kamnaksh A, Kovesdi E, Long JB, Agoston DV. Long-term consequences of single and multiple mild blast exposure on select physiological parameters and blood-based biomarkers. Electrophoresis (2013) 34:2229–33. doi:10.1002/elps.201300077
137. Arun P, Abu-Taleb R, Oguntayo S, Tanaka M, Wang Y, Valiyaveettil M, et al. Distinct patterns of expression of traumatic brain injury biomarkers after blast exposure: role of compromised cell membrane integrity. Neurosci Lett (2013) 552:87–91. doi:10.1016/j.neulet.2013.07.047
138. Zou YY, Kan EM, Lu J, Ng KC, Tan MH, Yao L, et al. Primary blast injury-induced lesions in the retina of adult rats. J Neuroinflammation (2013) 10:79. doi:10.1186/1742-2094-10-79
139. Prima V, Serebruany V, Svetlov A, Hayes RL, Svetlov S. Impact of moderate blast exposures on thrombin biomarkers assessed by Calibrated Automated Thrombography (CAT) in rats. J Neurotrauma (2013) 30(22):1881–7. doi:10.1089/neu.2012.2758
140. Sajja VS, Galloway M, Ghoddoussi F, Kepsel A, Vandevord P. Effects of blast-induced neurotrauma on the nucleus accumbens. J Neurosci Res (2013) 91:593–601. doi:10.1002/jnr.23179
141. Skotak M, Wang F, Alai A, Holmberg A, Harris S, Switzer RC, et al. Rat injury model under controlled field-relevant primary blast conditions: acute response to a wide range of peak overpressures. J Neurotrauma (2013) 30:1147–60. doi:10.1089/neu.2012.2652
142. Takeuchi S, Nawashiro H, Sato S, Kawauchi S, Nagatani K, Kobayashi H, et al. A better mild traumatic brain injury model in the rat. Acta Neurochir Suppl (2013) 118:99–101. doi:10.1007/978-3-7091-1434-6_17
143. Turner RC, Naser ZJ, Logsdon AF, Dipasquale KH, Jackson GJ, Robson MJ, et al. Modeling clinically relevant blast parameters based on scaling principles produces functional & histological deficits in rats. Exp Neurol (2013) 248:520–9. doi:10.1016/j.expneurol.2013.07.008
144. Tweedie D, Rachmany L, Rubovitch V, Zhang Y, Becker KG, Perez E, et al. Changes in mouse cognition and hippocampal gene expression observed in a mild physical- and blast-traumatic brain injury. Neurobiol Dis (2013) 54:1–11. doi:10.1016/j.nbd.2013.02.006
145. Ahlers ST, Vasserman-Stokes E, Shaughness MC, Hall AA, Shear DA, Chavko M, et al. Assessment of the effects of acute and repeated exposure to blast overpressure in rodents: toward a greater understanding of blast and the potential ramifications for injury in humans exposed to blast. Front Neurol (2012) 3:32. doi:10.3389/fneur.2012.00032
146. Ahmed F, Gyorgy A, Kamnaksh A, Ling G, Tong L, Parks S, et al. Time-dependent changes of protein biomarker levels in the cerebrospinal fluid after blast traumatic brain injury. Electrophoresis (2012) 33:3705–11. doi:10.1002/elps.201200299
147. Hines-Beard J, Marchetta J, Gordon S, Chaum E, Geisert EE, Rex TS. A mouse model of ocular blast injury that induces closed globe anterior and posterior pole damage. Exp Eye Res (2012) 99:63–70. doi:10.1016/j.exer.2012.03.013
148. Bir C, Vandevord P, Shen Y, Raza W, Haacke EM. Effects of variable blast pressures on blood flow and oxygen saturation in rat brain as evidenced using MRI. Magn Reson Imaging (2012) 30:527–34. doi:10.1016/j.mri.2011.12.003
149. Kovesdi E, Gyorgy AB, Kwon SK, Wingo DL, Kamnaksh A, Long JB, et al. The effect of enriched environment on the outcome of traumatic brain injury; a behavioral, proteomics, and histological study. Front Neurosci (2011) 5:42. doi:10.3389/fnins.2011.00042
150. Kovesdi E, Kamnaksh A, Wingo D, Ahmed F, Grunberg NE, Long JB, et al. Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front Neurol (2012) 3:111. doi:10.3389/fneur.2012.00111
151. Reneer DV, Hisel RD, Hoffman JM, Kryscio RJ, Lusk BT, Geddes JW. A multi-mode shock tube for investigation of blast-induced traumatic brain injury. J Neurotrauma (2011) 28:95–104. doi:10.1089/neu.2010.1513
152. Risling M, Plantman S, Angeria M, Rostami E, Bellander BM, Kirkegaard M, et al. Mechanisms of blast induced brain injuries, experimental studies in rats. Neuroimage (2011) 54(Suppl 1):S89–97. doi:10.1016/j.neuroimage.2010.05.031
153. Shridharani JK, Wood GW, Panzer MB, Capehart BP, Nyein MK, Radovitzky RA, et al. Porcine head response to blast. Front Neurol (2012) 3:70. doi:10.3389/fneur.2012.00070
154. Elder GA, Dorr NP, De Gasperi R, Gama Sosa MA, Shaughness MC, Maudlin-Jeronimo E, et al. Blast exposure induces post-traumatic stress disorder-related traits in a rat model of mild traumatic brain injury. J Neurotrauma (2012) 29:2564–75. doi:10.1089/neu.2012.2510
155. Dalle Lucca JJ, Chavko M, Dubick MA, Adeeb S, Falabella MJ, Slack JL, et al. Blast-induced moderate neurotrauma (BINT) elicits early complement activation and tumor necrosis factor alpha (TNFalpha) release in a rat brain. J Neurol Sci (2012) 318:146–54. doi:10.1016/j.jns.2012.02.002
156. Kuehn R, Simard PF, Driscoll I, Keledjian K, Ivanova S, Tosun C, et al. Rodent model of direct cranial blast injury. J Neurotrauma (2011) 28:2155–69. doi:10.1089/neu.2010.1532
157. Cernak I, Merkle AC, Koliatsos VE, Bilik JM, Luong QT, Mahota TM, et al. The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol Dis (2011) 41:538–51. doi:10.1016/j.nbd.2010.10.025
158. Connell S, Gao J, Chen J, Shi R. Novel model to investigate blast injury in the central nervous system. J Neurotrauma (2011) 28:1229–36. doi:10.1089/neu.2011.1832
159. Cheng J, Gu J, Ma Y, Yang T, Kuang Y, Li B, et al. Development of a rat model for studying blast-induced traumatic brain injury. J Neurol Sci (2010) 294:23–8. doi:10.1016/j.jns.2010.04.010
Keywords: biomarkers, blast injury, brain injury, neurotrauma, blast overpressure, mild TBI, PTSD, neuropsychiatry
Citation: Kobeissy F, Mondello S, Tümer N, Toklu HZ, Whidden MA, Kirichenko N, Zhang Z, Prima V, Yassin W, Anagli J, Chandra N, Svetlov S and Wang KKW (2013) Assessing neuro-systemic & behavioral components in the pathophysiology of blast-related brain injury. Front. Neurol. 4:186. doi: 10.3389/fneur.2013.00186
Received: 31 October 2012; Accepted: 02 November 2013;
Published online: 21 November 2013.
Edited by:
Mårten Risling, Karolinska Institutet, SwedenReviewed by:
Karin A. Rafaels, Army Research Laboratory, USADenes V. Agoston, Uniformed Services University, USA
Copyright: © 2013 Kobeissy, Mondello, Tümer, Toklu, Whidden, Kirichenko, Zhang, Prima, Yassin, Anagli, Chandra, Svetlov and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Firas Kobeissy and Kevin K. W. Wang, Department of Psychiatry, University of Florida, 100 S Newell Drive Room L4-100, Gainesville, FL 32611, USA e-mail:ZmlyYXNrb0BnbWFpbC5jb20sa3dhbmdAdWZsLmVkdQ==;
Stefania Mondello, Department of Neurosciences, University of Messina, Via Consolare Valeria, Messina 98125, Italy e-mail:c3RtX21vbmRlbGxvQGhvdG1haWwuY29t
†Firas Kobeissy and Stefania Mondello have contributed equally to this work.