 M. Angela Cenci
M. Angela Cenci- Basal Ganglia Pathophysiology Unit, Department of Experimental Medical Science, Lund University, Lund, Sweden
The dopamine (DA) precursor l-DOPA has been the most effective treatment for Parkinson’s disease (PD) for over 40 years. However, the response to this treatment changes with disease progression, and most patients develop dyskinesias (abnormal involuntary movements) and motor fluctuations within a few years of l-DOPA therapy. There is wide consensus that these motor complications depend on both pre- and post-synaptic disturbances of nigrostriatal DA transmission. Several presynaptic mechanisms converge to generate large DA swings in the brain concomitant with the peaks-and-troughs of plasma l-DOPA levels, while post-synaptic changes engender abnormal functional responses in dopaminoceptive neurons. While this general picture is well-accepted, the relative contribution of different factors remains a matter of debate. A particularly animated debate has been growing around putative players on the presynaptic side of the cascade. To what extent do presynaptic disturbances in DA transmission depend on deficiency/dysfunction of the DA transporter, aberrant release of DA from serotonin neurons, or gliovascular mechanisms? And does noradrenaline (which is synthetized from DA) play a role? This review article will summarize key findings, controversies, and pending questions regarding the presynaptic mechanisms of l-DOPA-induced dyskinesia. Intriguingly, the debate around these mechanisms has spurred research into previously unexplored facets of brain plasticity that have far-reaching implications to the treatment of neuropsychiatric disease.
Parkinson’s Disease (PD) is defined by a set of motor signs and symptoms that are caused by dopamine (DA) deficiency and respond well to dopaminergic therapies. Accordingly, functional imaging studies have established a close link between the onset and severity of PD motor features and the loss of dopaminergic markers in the putamen (1, 2). Oral administration of the DA precursor, l-DOPA has provided the backbone of PD treatment for over 40 years [recently reviewed in Ref. (3, 4)]. However, this treatment leads to complications.
After a few years of l-DOPA pharmacotherapy, most PD patients will exhibit a shorter motor response to each medication dose (“wearing-off fluctuation”), often associated with choreiform abnormal involuntary movements (AIMs) that appear when plasma and brain levels of l-DOPA are high (“peak-dose dyskinesias”) (Figure 1). More complex response patterns may also occur, for example, dyskinesias appearing when plasma l-DOPA levels rise or decline after each dose (“diphasic dyskinesia”), or abrupt fluctuations between a good antiparkinsonian response and a severe parkinsonian motor state (“unpredictable on–off fluctuations”) [reviewed in Ref. (3, 5)]. It has recently been established that oral l-DOPA therapy produces non-motor complications too, particularly, fluctuations in mood and cognitive performance (3, 6).
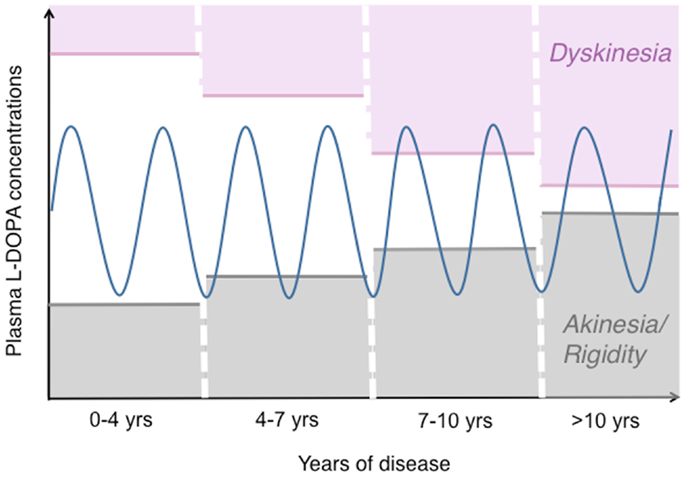
Figure 1. The pattern of motor response to l-DOPA changes during the progression of PD. This drawing illustrates how the therapeutic window of l-DOPA narrows during the progression of PD [based on (172, 173)]. While oral l-DOPA therapy achieves a stable symptomatic control during the first years, it causes motor fluctuations and dyskinesias in more advanced disease stages. Dyskinesias are most commonly associated with high plasma levels of l-DOPA (peak-dose LID), as shown here. The blue sinuous line represents peaks-and-troughs in plasma l-DOPA levels concomitant with oral l-DOPA therapy. The empty area at the centre represents the range of l-DOPA concentrations that induce relief of PD motor features without causing dyskinesia.
Factors associated with a higher incidence and/or early development of l-DOPA-induced dyskinesia (LID) include, l-DOPA dosage, severity and duration of PD (7, 8, 9), and a young age at PD onset [reviewed in Ref. (8, 9)]. Some autosomal recessive forms of PD also entail a high risk of LID (10), possibly because they share many features with young-onset idiopathic PD, in particular, a severe loss of DA neurons with relative preservation of non-dopaminergic systems and slow progression of Lewy-related brain pathology (11, 12). The reasons underlying a high risk for LID in young-onset PD patients have not been resolved, and several valid hypotheses have been put forward, including a faster DA turnover (13) or a larger potential for neuroplasticity in a younger brain (14). Moreover, the relative integrity of non-dopaminergic systems in younger subjects may contribute to a higher risk for LID. These systems may include corticostriatal and/or serotonergic projections, as will be discussed in this article.
“Pre- or Post-Synaptic Mechanisms?” A Brief Historical Perspective
In the most typical cases, dyskinesias and motor fluctuations are temporally related to rises and declines in plasma l-DOPA levels (Figure 1). In advanced stages of PD, the same dosage of l-DOPA that is required to relieve parkinsonian features may also induce AIMs [reviewed in Ref. (3, 15) and schematically illustrated in Figure 1].
Whether this altered response pattern depends on presynaptic or post-synaptic changes in nigrostriatal DA transmission has been a matter of major debate. The presynaptic hypothesis, which prevailed in the 80s, held that the progressive degeneration of nigral neurons causes a loss of DA storage capacity in nigrostriatal nerve terminals (16). Under these conditions, l-DOPA would be immediately converted to DA by a variety of cells in the brain, and rapidly eliminated. Peak-dose LID and wearing-off fluctuations would thus be the clinical counterparts of swift rises and declines in central DA levels, respectively [reviewed in Ref. (17)].
During the 90s, the presynaptic hypothesis appeared to decrease in popularity as many investigators turned one’s attention to the post-synaptic consequences of DA depletion. The attention shift was prompted by studies in 6-hydroxydopamine (6-OHDA)-lesioned rats, which revealed striking effects of chronic l-DOPA treatment on the expression of GABA-biosynthetic enzymes, neuropeptides, and opioid precursors in striatal neurons (18). In addition, studies in PD patients revealed that the therapeutic window of apomorphine, a direct DA agonist, narrowed with the progression from a DOPA-naive to a DOPA-treated dyskinetic state (19). Because apomorphine acts independently of presynaptic nigrostriatal terminals, these results were used to suggest that altered signal-transduction mechanisms in striatal neurons are the main culprit of motor complications to PD therapy (19).
Presynaptic factors were brought back into the limelight by human positron emission tomography (PET) studies using the reversible D2 receptor ligand, [11C] raclopride to estimate DA release. This approach takes advantage of a competition between endogenous DA and [11C] raclopride for binding to D2 receptors. Increased DA levels in the striatum are thus seen as a reduction in [11C] raclopride binding potential compared to baseline values. Using this technique, De la Fuente Fernandez and colleagues showed that standard oral doses of l-DOPA caused larger swings in striatal DA levels in PD patients experiencing motor complications compared to patients with a stable response to treatment (20, 21). Moreover, Piccini and collaborators found a positive linear relationship between putaminal changes in [11C] raclopride binding and AIM scores “on” l-DOPA (22). These human studies provided a strong support to the presynaptic hypothesis of LID, and prompted a new wave of clinical and preclinical research aimed at shedding light on the mechanisms involved.
During the past 10 years, different groups of investigators have continued to debate on whether or not presynaptic factors can by themselves drive the development of LID (23, 24), and experimental evidence has been put forward to either support or reject this standpoint [cf., e.g., Ref. (25, 26)]. Because a disruption of presynaptic DA homeostasis will certainly have post-synaptic consequences (27) (Figure 2), this debate may appear artificially contentious at first glance. However, it is becoming clear that the relative weight of presynaptic versus post-synaptic mechanisms in generating the involuntary movements will condition the response to antidyskinetic interventions (28).
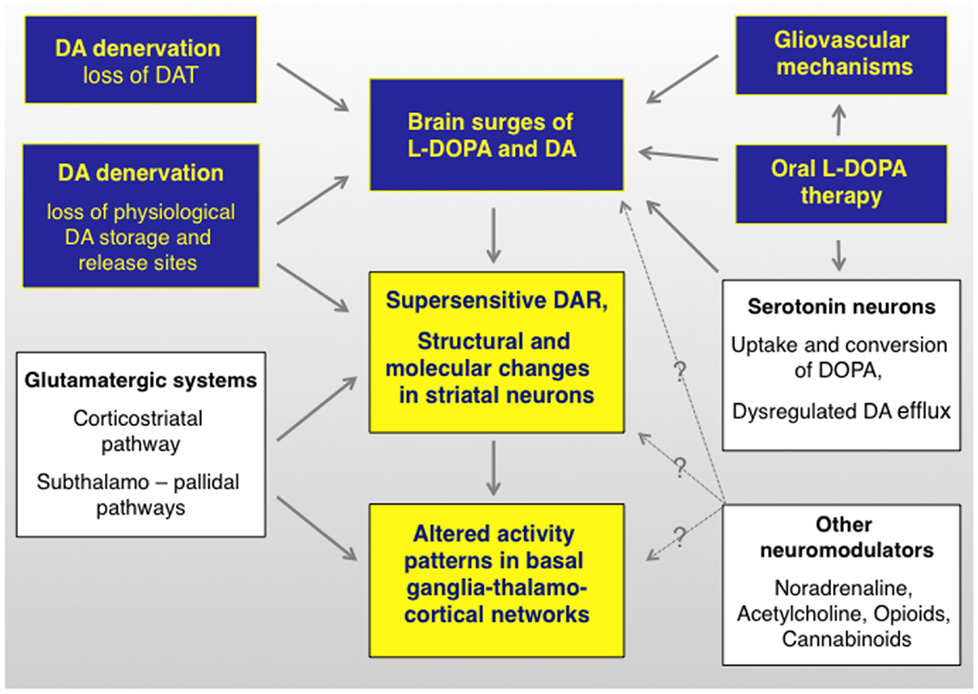
Figure 2. l-DOPA-induced dyskinesia depends on both pre- and postsynaptic disturbances of DA transmission that are modulated by non-dopaminergic transmitter systems. The term “presynaptic” refers to all factors that contribute to generating fluctuating levels of l-DOPA and DA in the brain (blue boxes). The term post-synaptic refers to mechanisms that occur at the level of dopaminoceptive cells (yellow boxes). Non-dopaminergic modulatory systems are shown in white boxes. It is not well understood how these systems modulate different levels of the pathophysiological cascade (hence the question marks). DAR, dopamine receptors. Studies supporting this pathophysiological cascade have been reviewed in Ref. (3, 27, 174, 175). An updated review on the presynaptic factors is presented in this article.
This review article will summarize both the terms of the debate and the valuable research that has stemmed from it. Thanks to this research, conspicuous progress has been made toward understanding specific players on the “presynaptic side” of the cascade (summarized in Figures 2 and 3).
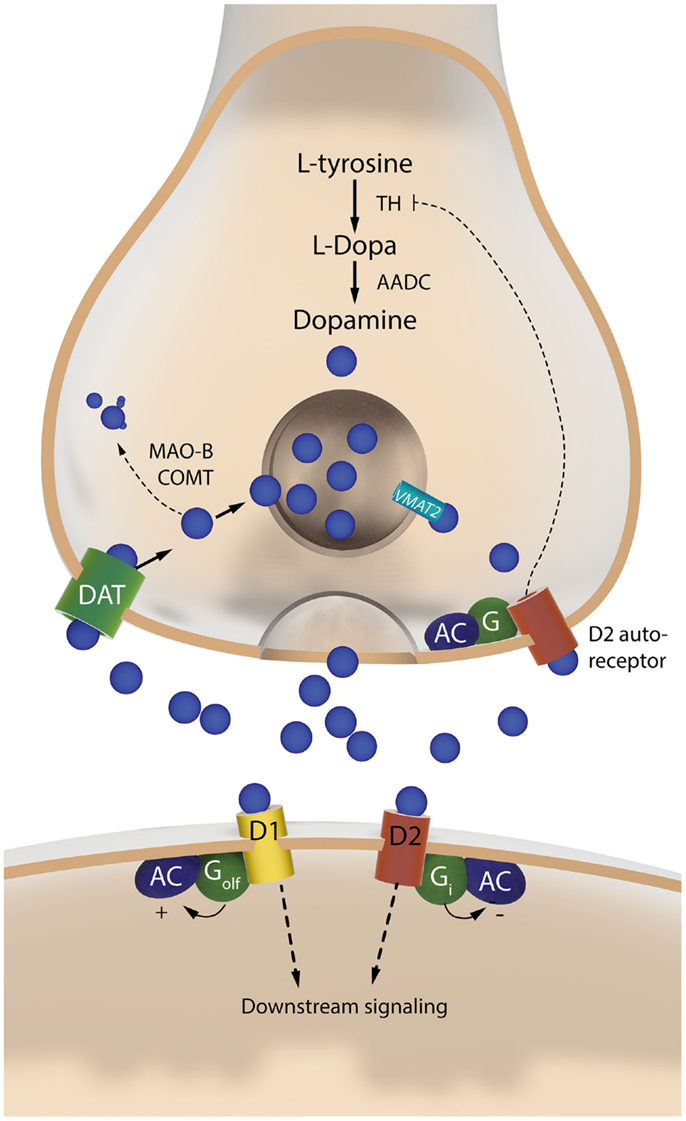
Figure 3. The two sides of a dopaminergic synapse. The drawing illustrates components of the nigrostriatal dopaminergic synapse that are discussed in this article. The presynaptic nigrostriatal terminal releases DA (blue circles), and regulates extracellular DA levels through several mechanisms: DA reuptake from the extracellular fluid (via the DAT), DA transport into synaptic vesicles (via VMAT-2), DA synthesis (which is subjected to autoregulatory control via presynaptic D2 receptors), and DA metabolism (via MAO-B and COMT). The post-synaptic neuron responds to DA via two main types of receptors. The D1 receptor is coupled to Golf and activates c-AMP-dependent intracellular signaling pathways. The D2 receptor is coupled to Gi and inhibits the same pathways. AADC, aromatic L-amino acid decarboxylase; AC, adenylate cyclase; COMT, catechol-O-methyl-transferase; DAT, dopamine transporter; MAO-B, monoamine oxidase B; TH, tyrosine hydroxylase; VMAT-2, vesicular monoamine transporter 2.
Nigrostriatal DA Denervation and l-DOPA Dosage are Critical to LID
Clinical observations suggest that the loss of nigrostriatal DA neurons plays an important role in the development of LID (8, 9, 29). But PD has a complex pathology, and it is difficult to demonstrate the causal link between dopaminergic denervation and LID in human studies. This type of information can however be inferred from experimental models of the movement disorder.
In all the most common animal models of PD–LID, the loss of nigrostriatal neurons is obtained using specific neurotoxins. 6-Hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) have been the most commonly used toxins in rodents and non-human primate species, respectively. In all the current animal models, the AIMs induced by l-DOPA mimic the peak-dose variant of human LID [reviewed in Ref. (30)].
Non-human primate studies examining the relationship between LID and extent of nigrostriatal DA lesion have been sparse and, at first glance, conflicting. A seminal study in MPTP-lesioned macaques reported that therapeutic doses of l-DOPA produced dyskinesia only in monkeys having ≥95% striatal DA loss (31). Accordingly, a study in MPTP-lesioned marmosets reported that only animals with >85% striatal DA loss developed choreoathetoid dyskinesias with therapeutic doses of l-DOPA, and that the most severely parkinsonian animals displayed the most severe LID (32). However, studies in squirrel monkeys reported choreoathetoid dyskinesias in animals with partial striatal DA denervation (33), and even in intact animals treated with a therapeutic l-DOPA regimen (15 mg/kg twice daily for 2 weeks) (34). Furthermore, intact macaque monkeys were reported to develop choreoathetoid dyskinesias if treated with very high doses of l-DOPA (80 mg/kg/day for 13 weeks) (35). Thus, the impact of nigrostriatal DA denervation on the susceptibility to LID differs between non-human primate species, some of which can develop involuntary movements even in the absence of dopaminergic denervation, if given sufficiently high doses of l-DOPA.
The largest rodent study addressing the relationship between nigrostriatal DA lesion and LID severity is the one by Winkler and colleagues (36). In this study, rats sustained partial or complete lesions of the nigrostriatal pathway, and were then treated with l-DOPA at a low therapeutic dose (6 mg/kg/day) for 4 weeks. Only rats with >80% loss of striatal dopamine transporter (DAT) or nigral DA neurons developed dyskinetic behaviors, and involuntary movements of maximal severity occurred only in the subgroup exhibiting >90% loss of dopaminergic markers (36). However, some of the completely DA-denervated animals remained free from dyskinetic behaviors throughout the l-DOPA treatment period (Figure 4). Thus, although a large nigrostriatal DA lesion was necessary for l-DOPA to induce involuntary movements, the severe dopaminergic denervation was not by itself sufficient (36). A similar conclusion was reached by Bezard and collaborators in a study using MPTP-lesioned macaques (37). It should be added, however, that high doses of l-DOPA will induce dyskinesia in all animals exhibiting >90% loss of dopaminergic markers throughout the caudate-putamen, although the actual doses will vary depending on species [c.f. ≥25 mg/kg/day in the rat (38, 39) versus ≥3 mg/kg/day in mice (40, 41)].
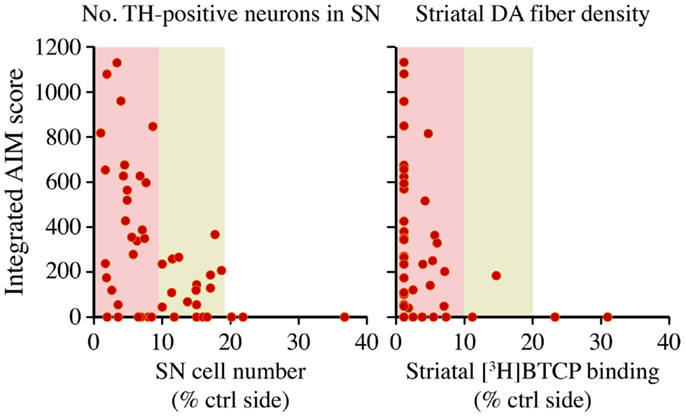
Figure 4. A large nigrostriatal DA lesion is necessary but not sufficient for therapeutic l-DOPA doses to induce dyskinesia. Rats sustained unilateral nigrostriatal DA lesions of varying severity, and were then treated with l-DOPA (6 mg/kg/day) for 4 weeks. Diagrams plot the animals cumulative Abnormal Involuntary Movement (AIM) scores (y axis) on presynaptic markers of DA neuron integrity, that is, tyrosine hydroxylase- positive cells in the substantia nigra (SN) or striatal innervation density, estimated with DAT radioligand binding using [3H]-BTCP. Data collected on the side ipsilateral to the lesion are expressed as a percentage of the values on the contralateral (ctrl) intact side. With either measure, AIM scores were found to occur only in animals that had lost more than 80% of presynaptic dopaminergic markers, and maximally severe AIMs occurred only when this loss exceeded 90%. Note however that some of the completely DA-denervated animals did not develop any dyskinesia. The dataset is derived from Ref. (36).
In summary, the bulk of experimental data indicate that, if l-DOPA is given at a therapeutic dosage, involuntary movements develop only when the loss of DA afferents to the motor striatum exceeds a threshold level of 80–85%. Despite these large lesions, some animals will however remain free from LID during the chronic treatment. Intriguingly, these experimental observations are in keeping with the clinical experience, whereby a proportion of PD patients never develop dyskinesias during their lifetime exposure to l-DOPA (9). Autoradiographic studies of DAT binding in the post-mortem striatum have not detected a difference between dyskinetic and non-dyskinetic PD cases (42, 43), indicating that a severe dopaminergic denervation is not sufficient for some patients to develop LID. Thus, although presynaptic DA depletion predicts the risk of LID (29), the susceptibility to this therapy complication must also depend on additional factors. These factors are likely to include some of the mechanisms discussed in the following sections.
Presynaptic Consequences of Nigrostriatal DA Denervation
The degeneration of nigrostriatal DA neurons in PD implies a severe depletion of the presynaptic compartment that physiologically converts l-DOPA to DA, releases DA in a regulated fashion, and clears DA from the extracellular space via high-affinity reuptake (Figure 3). The nigrostriatal system has a high capacity to mount compensatory mechanisms after partial lesions through, e.g., increased DA turnover, sprouting of residual DA terminals, and downregulation of the DAT [reviewed in Ref. (15, 44)]. Accordingly, parkinsonian motor symptoms have been estimated to appear only after a loss of 50% nigral DA neurons and 70% striatal DA contents [reviewed in Ref. (15)]. Similar phenomena have been observed in 6-OHDA-lesioned rodents, where the compensatory capacity of the nigrostriatal system appears to break down only after a >70% loss of nigral DA neurons (45, 46).
The breakdown of presynaptic DA homeostasis predisposes to large fluctuations in central levels of DA upon treatment with l-DOPA. In a seminal microdialysis study, Abercrombie and collaborators showed that a peripheral injection of l-DOPA results in significantly higher extracellular DA levels in rats with large 6-OHDA lesions compared to intact animals (47). The l-DOPA-induced increase in striatal extracellular DA concentrations (ΔDA) was 30- to 80-fold larger in 6-OHDA-lesioned animals compared to intact controls (the striking difference being partly dependent on the lower baseline DA concentrations in lesioned animals) (47). This study also established a causal relationship between ΔDA and the lesion-induced loss of DAT. Indeed, combined treatment of intact rats with l-DOPA and nomifensine, a DAT inhibitor, produced increases in extracellular DA approaching the magnitude of those in 6-OHDA-lesioned animals (47). More recent studies have confirmed the crucial importance of DAT deficiency in determining large increases in extracellular DA “on” l-DOPA (48). However, these studies have also indicated that, when the nigrostriatal lesion is very severe, the magnitude of such increases depends on factors other than DAT deficiency. Thus, animals with less than 90% DA denervation exhibit a significant negative correlation between ΔDA and striatal DAT binding levels. However, in rats with >90% denervation, DAT levels no longer predict ΔDA (48). What factors may then condition the magnitude of ΔDA in animals with severe nigrostriatal DA lesions?
In addition to the loss of DAT, a severe degeneration of the nigrostriatal pathway inevitably entails a shift in the routes of l-DOPA metabolism from nigrostriatal DA neurons to other sites (15). The conversion of l-DOPA to DA is a one-step enzymatic reaction catalyzed by aromatic L-amino acid decarboxylase (AADC, also called DOPA decarboxylase, DDC) (Figure 3). This enzyme is expressed by catecholaminergic neurons (49), but also by astrocytes (50) and blood vessel-associated cells (51).
A seldom appreciated fact is that AADC and 5-hydroxytry ptophan decarboxylase (which synthetizes 5-hydroxytryptamine, serotonin) are the same enzyme (see, e.g., http://omim.org/entry/107930). Serotonin neurons therefore express relatively high levels of AADC, and they also express vesicular monoamine transporter 2 (VMAT-2), which packages DA into synaptic vesicles and protects it from rapid cytosolic degradation [reviewed in Ref. (15)] (cf. Figure 3). Although AADC and VMAT-2 also are expressed by noradrenergic neurons, these are unlikely to provide a major source of DA upon l-DOPA treatment, because the DA formed from l-DOPA in these neurons is rapidly converted to noradrenaline (NA) by the enzyme, dopamine-beta-hydroxylase.
Thus, serotonin neurons can both synthetize DA from l-DOPA, store the formed DA in synaptic vesicles, and release it in an activity-dependent manner. During the past few years, an abundant literature has documented that serotonin neurons indeed provide a source of DA release in l-DOPA-treated parkinsonian subjects. An intense debate has grown around the extent of this phenomenon and its significance to the occurrence of LID, as will be detailed in the following sections of this review. But before approaching this topic, we need to briefly consider the post-synaptic consequences of DA denervation, which are likely to be crucial to the development of LID.
Post-Synaptic Consequences of Nigrostriatal DA Denervation
Although this article focuses on the presynaptic mechanisms of LID, it is important to keep in mind that a loss of nigrostriatal DA input also entails profound adaptations at the post-synaptic level (Figure 2). In particular, DA-denervating lesions cause pronounced molecular, physiological, and morphological changes in striatal neurons, as demonstrated by a large body of experimental literature, briefly reviewed below.
Already in the 70s, a deafferentation-induced supersensitivity of post-synaptic DA receptors was hypothesized to play a role in the development of LID (52). Today we know that this supersensitivity depends on complex changes in the signal-transduction properties of DA receptors. The changes include, an increased coupling efficiency of both D1 and D2 receptors to their corresponding G proteins, a large activation of downstream intracellular signaling molecules, changes in DA receptor trafficking, and also a striking activation of non-canonical signaling pathways [reviewed in Ref. (52–54)]. Gerfen and collaborators were the first to propose that the denervation-induced supersensitivity of D1 receptors leads to an activation of intracellular pathways that are not recruited under physiological conditions (55). In their seminal study (55), treatment of 6-OHDA-lesioned rats with D1 receptor agonists was found to cause a pronounced striatal activation of extracellular signal regulated kinases 1 and 2 (ERK1/2), a pathway traditionally associated with the stimulation of tyrosine-kinase or glutamate receptors, not Gs/olf-coupled receptors (cf. Figure 2). A link between l-DOPA-induced ERK1/2 activation and the development of dyskinetic behaviors was later demonstrated in both rodent (40, 56–58) and non-human primate models of LID (40).
In addition to altered DA receptor-mediated signaling, an abnormal corticostriatal synaptic plasticity (59) and structural changes of striatal neurons associated with the progression of PD (60) predispose to a dyskinetic response to therapy. Post-mortem investigations of striatal tissue from PD patients have revealed conspicuous loss of spines and dendritic atrophy in medium-sized spiny neurons (61, 62). Similar phenomena have been found to occur in both rodent and non-human primate models of PD (63, 64). The results so far available indicate that treatment with l-DOPA does not normalize the dendritic structure of striatal neurons, but instead superimposes a new layer of changes that are associated with the development of dyskinetic behaviors (65–67).
It has been hypothesized that striatal dendritic atrophy has a major impact on the response to PD treatment favoring the emergence of complications because, “expecting a normal reaction to dopaminergic drugs under these circumstances is like expecting a four-cylinder car engine to turn over normally on three cylinders” (68). Further investigations are however needed to clarify the precise contribution of an altered striatal dendritic morphology to the genesis of LID (69).
l-DOPA-Induced DA Release in the Dyskinetic Brain
PET imaging studies in PD patients have established a link between l-DOPA-induced motor complications and large fluctuations in striatal DA levels (20). In a seminal study using [11C] raclopride PET, De La Fuente Fernandez and coworkers compared the dynamics of striatal DA release between PD patients affected by LID and patients with a stable response to therapy (20). One hour after l-DOPA administration, dyskinetic patients exhibited significantly greater changes in striatal DA levels than did stable l-DOPA responders (21). Similar results were obtained by Piccini’s group, who also established a positive correlation between changes in striatal DA levels and severity of peak-dose LID (22). One limitation of these human studies is that the absolute extracellular concentrations of DA, hence their impact on changes in [11C] raclopride binding, were not accessible to investigation. This concern is relevant because the dyskinetic PD patients in these studies had a longer disease duration than did stable l-DOPA responders (21, 22). A longer disease duration may potentially lead to lower striatal DA levels at baseline.
Microdialysis studies in rodent models of LID have been very useful in clarifying the relationship between dyskinesia and absolute striatal DA concentrations “on” and “off” l-DOPA. In a seminal study, Meissner and colleagues compared striatal extracellular DA levels in 6-OHDA-lesioned rats exposed to a prior course of treatment with l-DOPA or saline (70). l-DOPA was given at a high dose (50 mg/kg/day per 10 days), which induced AIMs in all of the treated animals. A striking result of this study is that the same peripheral dose of l-DOPA elicited a larger increase in striatal extracellular DA levels in l-DOPA-primed animals compared to saline-treated ones (70). Other microdialysis studies were performed in 6-OHDA-lesioned rats that had been chronically treated with a lower dose of l-DOPA (6 mg/kg/day), upon which some of the animals remained free from AIMs. These studies reported larger striatal levels of l-DOPA (71) or DA (72, 73) in dyskinetic animals compared to non-dyskinetic cases. The most pronounced between-group difference in striatal DA levels occurred at the peak of the l-DOPA-induced surge, i.e., 40–60 min after l-DOPA administration. DA concentrations did not however differ between dyskinetic and non-dyskinetic animals either at baseline or at later time points post drug dosing (72, 73). Although dyskinetic animals showed a larger increase above baseline (ΔDA), their absolute DA concentrations never exceeded the values measured in intact control animals (72). Interestingly, a similar pattern of group differences was observed in the substantia nigra, which was monitored simultaneously with the striatum in one study (72).
Taken together, these results show that both ΔDA and absolute DA concentrations at the peak of the l-DOPA effect are larger in animals affected by involuntary movements compared to non-dyskinetic cases, despite similar baseline DA levels. The larger ΔDA values in dyskinetic rats are in keeping with the results of [11C] raclopride-PET studies in dyskinetic PD patients, though apparently at variance with other experimental data. In particular, a recent microdialysis study in the macaque model of LID has failed to detect a significant increase in striatal extracellular DA levels after l-DOPA administration, whereas striatal levels of DOPA showed a robust increase (74). According to the authors interpretation, these data indicate that a low DOPA decarboxylase activity in parkinsonian primates limits the production of DA from exogenous l-DOPA, differently from the situation encountered in 6-OHDA-lesioned rodents (74). These unexpected results prompt the interim reflection that the rat model of LID is more suitable than the macaque one to reproduce the presynaptic disturbances seen in the human condition. Indeed, [11C] raclopride binding is displaced by DA, and not by l-DOPA itself.
Serotonin Neurons as an Aberrant Source of DA Release “on” l-DOPA
The first report implicating serotonin neuron as a source of DA release “on” l-DOPA was provided by Tanaka and colleagues (75). These authors compared extracellular DA levels in the striatum of 6-OHDA-lesioned rats that had sustained or not an additional chemical lesion of serotonin neurons. Rats in the double-lesion group exhibited a dramatic 80% reduction in l-DOPA-induced DA efflux (75). Another important early study used a similar approach to show that a serotonin lesion completely suppressed the induction of both rotational behavior and striatal c-Fos expression by l-DOPA in 6-OHDA-lesioned rats (76). The authors of these studies suggested that the action of l-DOPA in PD critically depends on its conversion to DA in serotonin neurons.
As explained above, serotonin neurons are endowed with the enzymes that convert l-DOPA to DA, and package this DA into synaptic vesicles. A double-labeling immunofluorescence study in rats treated with l-DOPA has indeed revealed immunoreactivity for DA in serotonin-positive dorsal raphe neurons and their striatal projections (77).
It is therefore hardly surprising that serotonin neurons become an important source of l-DOPA-derived DA release in a situation where nigrostriatal neurons are severely damaged. A relationship between LID severity, on one hand, and morphological or autoradiographic measures of striatal serotonin innervation, on the other hand, has been detected in both rat and non-human primate models of PD by several studies (78–81). These results fit well with our observation that chronically l-DOPA-treated rats with larger ΔDA values “on” l-DOPA show higher striatal levels of serotonin and its metabolite at baseline, suggestive of a denser 5-HT innervation (72).
Supporting the notion that 5-HT neurons release DA “on” l-DOPA, several studies in 6-OHDA-lesioned rats have shown that l-DOPA-induced peak DA efflux can be blunted by agonists of the serotonin autoreceptors, 5-HT1a and 5-HT1b (72, 82, 83). Agonists at these receptors dampen the activity of serotonin neurons, measured as either firing rate or neurotransmitter release (84). 5-HT1a and 5-HT1b receptor agonists have marked antidyskinetic effects in both rodent and non-human primate models of LID [reviewed in Ref. (3, 85)]. However, doses of 5-HT1a/b agonists that improve LID do not improve dyskinesias that are induced by apomorphine (86) or D1 receptor agonists (28). This pattern of effects indicates that low-medium doses of 5-HT1a and 5-HT1b agonists [cf. doses in (28, 72, 86)] interfere with presynaptic mechanisms of dyskinesia that are exclusively recruited by l-DOPA, not by dopaminergic agents acting directly on DA receptors. The efficacy of 5-HT1a and 5-HT1b agonists in reducing LID further indicates that DA release from serotonin neurons plays a causal role in LID. A compelling demonstration of this concept was provided by Carta and collaborators using selective lesions of 5-HT neurons (86). These lesions completely suppressed l-DOPA-induced AIMs in previously dyskinetic rats (86). Other studies applied a chemical lesion of 5-HT neurons to 6-OHDA-lesioned rats before treating them with l-DOPA, and demonstrated a positive association between the levels of residual striatal 5-HT innervation and the severity of dyskinetic movements induced by the treatment (87).
Why would DA release from 5-HT neurons be so prone to induce LID? Serotonin neurons lack presynaptic mechanisms that can sense and regulate their DA release, such as DA autoreceptors and DAT [reviewed in Ref. (15)]. Thus, in situations where both baseline DA levels and DAT activity are severely reduced (which is the case in advanced PD), DA release from serotonin neurons is bound to produce large swings in DA levels. Moreover, DA efflux from 5-HT neurons will be ectopic in terms of both subcellular release sites and anatomical distribution. Accordingly, an elegant microdialysis study in 6-OHDA-lesioned rats reported very large increases in DA levels “on” l-DOPA in many brain structures (including hippocampus and prefrontal cortex), and the increases were totally abolished by a complete lesion of serotonin neurons (88). These large extrastriatal DA surges induced by l-DOPA most likely contribute to the development of both motor and non-motor complications to therapy (89). With respect to LID, a recent study in the rat has linked the stimulation of cortical D1 receptors to the expression of involuntary movements through a local generation of high-frequency oscillatory activities (90).
Debate on the Involvement of 5-HT Neurons in LID
Although the studies reviewed above are quite consistent, the concept that 5-HT neurons provide a major source of DA release in LID has met some resistance. Here follows a summary of common objections presented to us in the form of scientific correspondence. First, it is pointed out that the role of 5-HT neurons in LID has been studied in animals with relatively intact serotonin projections, which would be unlike the situation in the advanced stages of PD. Second, it is pointed out that a degree of striatal DA denervation as dramatic as in these animals would occur only in the very terminal stages of PD, implying that there would always be some nigrostriatal fibers ready to release DA in dyskinetic PD patients. Third, the comment has been put forward that astrocytes represent a much more abundant compartment than 5-HT projections to take up l-DOPA and convert it to DA in the striatum. All these objections are warranted, but also quite addressable with data available in the published literature.
As to the first point, post-mortem biochemical studies of 5-HT markers in PD have revealed that the loss of serotonergic innervation is more severe in the caudate than the putamen. In the latter structure, detectable levels of serotonergic markers persist until the terminal stages of PD (91). Accordingly, PET imaging studies in patients with advanced PD have detected only 30% reduction in putaminal serotonin transporter (SERT) binding (92), whereas, dopaminergic markers may be reduced by over 75% in the same structure (93). Post-mortem autoradiographic studies of SERT and DAT binding activities in the PD putamen are in keeping with the PET imaging investigations (42, 43, 80, 94). Furthermore, a post-mortem autoradiographic study has revealed larger SERT binding density in the post-commissural putamen in PD cases with LID compared to non-dyskinetic subjects (80).
Regarding the extent of DA denervation in the human disease, a recent pathological study has reported a virtual absence of DA fiber markers in the posterior putamen already at 4–5 years from PD diagnosis (95). Thus, the levels of DA denervation occurring in the dorsolateral striatum in animal models of LID are comparable to those in the post-commissural putamen (the motor part of the striatum) in mid-advanced stages of PD. And these are the stages where motor complications to therapy start to appear (cf. Figure 1).
As to the role of non-neuronal cells in handling l-DOPA, while this phenomenon certainly deserves further investigation (see below), it should be pointed out that neither glia nor vessel-associated cells have a capacity for vesicular storage and release of neurotransmitters. This is an important point, because microdialysis studies in 6-OHDA-lesioned rats have shown that l-DOPA-induced DA release is significantly reduced by reserpine, a VMAT blocker (96), and also by tetrodotoxin (TTX) (72, 97), a sodium channel blocker inhibiting the generation of action potentials. Thus, the bulk of DA efflux “on” l-DOPA has a neuronal origin even in animals with complete nigrostriatal DA lesions. Some authors have proposed that striatal interneurons expressing TH may provide a source of DA production and l-DOPA conversion in PD (98–100). However, it is as yet unclear whether these neurons can actually release DA [cf. (101)], and the expression of AADC in these cells appears to be very low, at least in rodents (41).
A proof-of-concept that 5-HT neurons release DA in patients affected by LID has been recently provided by Politis and coworkers using PET imaging techniques (102). In this study, dyskinetic PD patients were compared to patients with a stable response to therapy (“stable responders”) using both a SERT ligand ([11C]-DASB PET) and [11C] raclopride. In agreement with previous studies (see above), a standard dose of l-DOPA induced a larger displacement of [11C] raclopride binding in the dyskinetic group. Interestingly, the magnitude of [11C] raclopride displacement was positively correlated with the striatal levels of [11C] DASB binding, suggesting a relationship between peak DA efflux “on” l-DOPA and the density of striatal 5-HT innervation. Further to these observations, the authors evaluated the effects of buspirone, a compound with 5-HT1a agonistic activity, on the change in [11C] raclopride binding induced by l-DOPA administration. Intriguingly, buspirone reduced the magnitude of raclopride displacement only in dyskinetic PD patients, while having no effect at all in the stable responders. Furthermore, dyskinetic patients exhibiting a greater response to buspirone displayed a larger signal on the [11C] DASB PET scans, indicating larger striatal levels of serotonergic terminals. Finally, a strong positive correlation between AIM ratings and [11C] DASB binding density was found in the group of patients with peak-dose LID of mild-moderate severity (102). The authors concluded that striatal serotonergic terminals contribute to LID in human PD via aberrant processing of exogenous l-DOPA and release of DA as false neurotransmitter, quite in agreement with the results obtained in rat studies (102).
Debate on the Plasticity of the Serotonin System in LID and Its Animal Models
The serotonin system is highly vulnerable to age-related degenerative changes, but also highly plastic (103–105). Functional and structural adaptations of the serotonin projections may therefore impact on their role in LID.
In many toxin-based animal models of PD, the neurotoxic lesion induces partial damage of ascending 5-HT projections, followed by a long-term compensatory sprouting of 5-HT axon fibers (81, 106–108). Furthermore, chronic dyskinesiogenic treatment with l-DOPA has a growth-promoting effect on serotonin axon terminals (78, 80, 81), which is likely dependent on the treatment-induced upregulation of BDNF (80). The treatment-induced sprouting of 5-HT axon terminals requires a previous severe DA denervation of the affected region, as well as a partial lesion of 5-HT afferents, as it does not seem to occur when LID is produced in animal models of PD having intact serotonin projections [cf. (109)].
The striking plasticity of the 5-HT system in animal models of PD–LID has raised concerns that the importance of this system may be overestimated in the experimental models relative to the human disease, because serotonin neurons are expected to degenerate, not to grow new axon terminals, in PD. However, in the study by Politis and coworkers (102), the dyskinetic patients with longest disease duration exhibited a remarkably preserved serotonin terminal function. Thus, striatal levels of [11C]-DASB binding did not differ between the severely dyskinetic patients and the subjects with a stable response to therapy, who had a significantly shorter disease duration (102). These results are at variance with the expected loss of [11C]-DASB binding during the progression of PD (92), and may in fact suggest that serotonin axon terminals mount a long-term sprouting response in human LID, analogous to that seen in the animal models. Further support to this interpretation comes from an autoradiographic study of SERT radioligand binding density in the human post-mortem putamen and pallidum, showing larger SERT binding levels in PD patients with clinical records of LID compared to non-dyskinetic cases (80). In this study, a linear correlation was found between SERT binding density and number of SERT-immunoreactive axonal varicosities, at least in the pallidum (80).
At variance with the evidence above, some recent studies in 6-OHDA-lesioned rats have suggested that chronic l-DOPA treatment may have deleterious effects on serotonin neurons. In one study, animals were treated with l-DOPA (12 mg/kg/day) for 28 days, after which tissue levels of DA and serotonin were measured in several brain regions at various intervals following the last l-DOPA dose (89). A reduced ratio between serotonin and DA concentrations occurred for up to 4 h post l-DOPA administration in all the structures examined. The authors concluded that l-DOPA treatment had increased DA levels while reducing 5-HT levels in all brain regions (89). These results may reflect the fact that DA displaces 5-HT from synaptic vesicles within serotonin axon terminals (77, 86). If serotonin is displaced from the vesicles, its degradation will be faster and its tissue contents reduced, at least for a few hours following the administration of l-DOPA. However, Eskow Jaunarajs and colleagues proposed that long-term l-DOPA therapy may be directly detrimental to serotonin neurons through mechanisms involving oxidative stress, an idea supported by some observations in vitro (89). Endorsing the above interpretation, a microdialysis study performed in rats previously treated with l-DOPA (12 mg/kg/day for 10 days) reported a lower magnitude of l-DOPA-induced DA efflux in several brain regions compared to that measured in acutely l-DOPA-treated animals (110). The authors concluded that chronic l-DOPA therapy negatively affects the functionality of serotonin neurons, at least if high drug doses are used (110). These results are, however, at variance with those reported by other studies using high doses of l-DOPA (70).
While the debate on the degeneration and plasticity of 5-HT neurons in PD–LID is still ongoing, there is agreement that 5-HT receptors in the brain show pronounced functional adaptations. In particular, increases in striatal and cortical levels of 5-HT1a and 5-HT1b receptors, as well as their adaptor proteins (111), have been reported by several studies performed in animal models of PD and LID [partially reviewed in Ref. (112)]. Further studies are needed to verify the occurrence of these adaptations in the human disease, and to clarify their functional consequences. For example, it is likely that these receptor adaptations may impact on the responsiveness to antidyskinetic treatments targeting 5-HT1a and 5-HT1b receptors.
Gliovascular Mechanisms
In addition to high DA levels, dyskinetic animals show a large increase in the extracellular levels of l-DOPA following peripheral drug administration (71, 74, 113). A study in non-human primates has even suggested that l-DOPA does not need to be converted to DA in order to elicit AIMs (74).
The concentrations of l-DOPA in the brain extracellular fluid reflect the balance between drug entry and drug uptake/metabolism by brain cells. There are no indications that the uptake of l-DOPA by brain cells is impaired in dyskinetic animals, and it is therefore warranted to ask whether its entry could be increased. l-DOPA enters the brain from the blood stream via the L-type amino acid transporter system present in endothelial cells of the blood–brain barrier (BBB) (114, 115). Thus, the passage of l-DOPA from blood to brain will depend on the same variables that regulate the extraction of any substance, that is: (1) capillary permeability, (2) the capillary surface area, and (3) the regional blood flow (116). In the case of l-DOPA, a fourth variable should be considered, namely, the possibility of an active drug metabolism at the capillary level.
Already in the 60s, studies based on the Falck–Hillarp catecholamine histofluorescence method had indicated that brain capillaries critically regulate the entry of l-DOPA into the brain parenchyma (51). Endothelial cells and pericytes were revealed to be the first site of l-DOPA uptake, conversion, and metabolism in the brain (Figure 5), and were found to express very high levels of both AADC and monoamine oxidase B (51). It was thus proposed that cells lining cerebral microvessels form an enzymatic barrier to the entry of l-DOPA (51). Further to these studies, it was recently reported that l-DOPA accumulates not only in the microvessels, but also in astrocyte cell bodies and astrocytic endfeet surrounding cerebral microvessels (117).
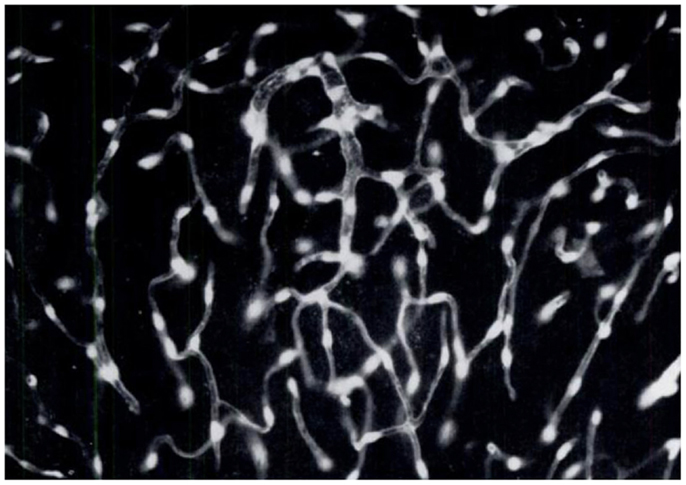
Figure 5. Brain endothelial cells and pericytes produce dopamine following systemic administration of l-DOPA. In the 60s, a group of Swedish pharmacologists led by E. Rosengren discovered that brain endothelial cells and pericytes are a significant site of dopamine production following treatment with l-DOPA. This photomicrograph represents a section of rat cerebellum processed for the Falck–Hillarp catecholamine histofluorescence method to visualize DA-containing cells. The rat had received an injection of l-DOPA (50 mg/kg, combined with the monoamine-B inhibitor nialamide) shortly before being killed. The authors commented, “It was evident that the fluorescent material occurred throughout the capillary walls giving almost a three-dimensional appearance of the capillary tubes. Fluorescence of high intensity (was found) in cytoplasm and nucleus of both endothelial cells and pericytes” [Reproduced with permission from Ref. (51)].
Endothelial cells, pericytes, and perivascular astrocytes form a functional unit that controls both capillary permeability and regional cerebral blood flow (rCBF) [reviewed in Ref. (118, 119)]. Both of these parameters are dynamically regulated in the brain to match the metabolic activity of neurons, and this process (termed “neurovascular coupling”) is modulated by monoaminergic afferents that innervate cerebral arterioles and microvessels (120–124).
Interestingly, while regional glucose metabolism (which is mainly driven by neuronal activity) and rCBF are well-matched in PD patients during the “off” medication state, the administration of l-DOPA greatly increases rCBF without elevating glucose metabolism in a brain network that includes putamen, pallidum, and midbrain–pons (125). In this brain network, the dissociation between flow and metabolism is particularly striking in patients affected by LID (125). These findings suggest that l-DOPA exerts hemodynamic effects that are independent of its modulation of neuronal metabolic activity, thus superseding physiological mechanisms of neurovascular coupling in the affected brain regions. A similar phenomenon occurs in the rat model of LID, which features a large increase in rCBF “on” l-DOPA in many parts of the basal ganglia, often in the absence of large concomitant changes in glucose metabolism (126).
The flow-metabolism dissociation response is a particularly intriguing phenomenon as it may signal a previously overlooked effect of l-DOPA on gliovascular cells (126). Moreover, this phenomenon may result in higher extracellular levels of l-DOPA in dyskinetic subjects (125, 126), impacting on the presynaptic mechanisms of LID. The underlying mechanisms are however unclear. Evidence of flow-metabolism dissociation has thus far been found only in specific regions, and the observed regional pattern cannot be readily explained by regional differences in either DA efflux “on” l-DOPA (88) or gliovascular expression of DA receptors (43, 120, 122, 127). Some interesting mechanistic suggestions have however emerged from studies performed in the rat model of LID. In 6-OHDA-lesioned rats treated with l-DOPA, regions with large increases in blood flow “on medication” exhibit endothelial proliferation and angiogenic activity when the treatment is given chronically (126). Furthermore, some of these regions exhibited an increased microvascular density and upregulation of angiogenesis markers in a post-mortem study of basal ganglia tissue from dyskinetic PD patients (43). These findings suggest that the large increases in rCBF “on” l-DOPA and the angiogenic response to the chronic treatment are interrelated phenomena, which are critically regulated by gliovascular cells in the affected brain regions (126). Investigating this hypothesis is likely to yield important insights into previously overlooked neurovascular effects of l-DOPA, uncovering novel therapeutic targets.
Changes in BBB Permeability: The Findings and the Debate
As mentioned above, capillary permeability is one of the factors determining the central availability of l-DOPA. The BBB is a selective diffusion barrier that relies on specialized properties of the brain’s capillary endothelium, such as the presence of tight cell–cell junctions, low levels of pinocytotic activity, and the expression of selective transporter proteins at the plasma membrane [reviewed in Ref. (128)]. Several independent studies suggest that the functionality of the BBB becomes impaired during the progression of PD (129–131). For example, the ratio between albumin concentrations in cerebrospinal fluid (CSF) and plasma is increased in PD patients with advanced disease compared to age-matched controls (131). Interestingly, higher albumin ratio values were measured in patients receiving DA replacement therapy compared with untreated subjects (131).
It has been suggested that the neuroinflammation associated with neurodegeneration leads to an increased BBB permeability due to the vascular effects of proinflammatory cytokines [see Discussion in Ref. (132), and references therein]. However, while neuroinflammation is a widespread finding in PD (133), the permeability problem appears to depend on focal areas of BBB dysfunction within the striatum and the midbrain. These areas show signs of angiogenic activity (43, 126, 132, 134). Several studies in both parkinsonian animals and human PD have indeed detected endothelial proliferation and other markers of active angiogenesis within the substantia nigra and the striatum (43, 134–137). Because active angiogenesis entails a transient increase in vessel permeability, it will inevitably lead to a localized leakage of the BBB when it occurs in the brain (138). Accordingly, studies in rat models of PD have revealed localized leakage of BBB tracer molecules (132) or downregulation of BBB proteins (139) precisely on vessels having angiogenic features.
When treatment with l-DOPA produces dyskinesias, it may aggravate the BBB dysfunction associated with PD, or even induce a new pattern of dysfunction. In the rat model of LID, dyskinetic animals exhibit endothelial proliferation, increased BBB permeability, and upregulation of vascular endothelial growth factor (VEGF) in the lateral striatum and the basal ganglia output nuclei (the substantia nigra pars reticulata and the entopeduncular nucleus, i.e., rodent equivalent of the GPi) (43, 139–141). These phenomena only occur on the DA-denervated side of the brain, and they are positively associated with the development of LID (139, 141). l-DOPA induces this angiogenic activity via stimulation of D1 receptors and activation of ERK1/2 signaling (140). Treatments that antagonize VEGF attenuate the gradual increase in dyskinesia severity during a chronic course of l-DOPA administration (43, 141), while inhibiting the angiogenic activity and BBB dysfunction induced by l-DOPA in the basal ganglia (43). Along with human pathological observations (43, 137), these findings suggest that a treatment-induced, VEGF-dependent angiogenic activity in the basal ganglia contributes to an aggravation and chronicization of LID in the advanced stages of PD (43).
The pathophysiological implications of these findings are, however, poorly understood. We have proposed that the increased BBB permeability associated with angiogenesis may contribute to an increased entry of l-DOPA in the affected regions (i.e., the motor part of the striatum and the basal ganglia output nuclei) (139). Supporting this proposition, dyskinetic animals were found to exhibit increased striatal and nigral uptake of an intravenous tracer molecule (which normally does not cross the BBB) having a molecular weight similar to l-DOPA (126). Importantly, leakage of this tracer into the striatal parenchyma was detected at significant levels at 60 min, but not 24 h after the administration of l-DOPA (126). This observation is interesting because it suggests an association between increased rCBF “on” l-DOPA and BBB hyperpermeability in dyskinetic subjects (126). In other words, the high rCBF associated with LID (125, 126) would cause BBB leakage at the level of immature microvessels, which form in the striatum and its output nuclei because of the combined effect of DA denervation and chronic l-DOPA treatment (126). In keeping with this suggestion, an increased perfusion has been shown to enhance tight-junction opening between endothelial cells in other models of brain disease involving angiogenesis or microvascular pathology (142). Further investigations are needed to clarify the relative importance of an increased BBB permeability in producing high extracellular levels of l-DOPA in LID.
The suggestion that BBB permeability is enhanced in LID has raised some debate (143). It is often argued that the peripheral DOPA decarboxylase inhibitors included in standard l-DOPA preparations [i.e., carbidopa or benserazide, reviewed in Ref. (3, 4)] are unlikely to enter the brain. If they did, the treatment would not engender an increase in central levels of DA, whereas raclopride–PET studies unequivocally demonstrate striatal DA release after the administration of l-DOPA to PD patients. However, studies in both intact and 6-OHDA-lesioned rats indicate that peripheral DOPA decarboxylase inhibitors significantly reduce central AADC activity only at doses much higher than those given to patients (144, 145). More importantly, doses of benserazide reducing striatal AADC activity by over 50% did not have any effect on either basal DA levels or l-DOPA-induced DA release in the striatum (145, 146). To achieve a significant effect on the above parameters, benserazide had to be administered at the dose of 50 mg/kg, which reduced striatal AADC activity by ≥80% (145, 146). Such a dose is manifold larger than the highest benserazide dosage to which a PD patient will ever be exposed. In a study using 6-OHDA-lesioned rats, not even 50 mg/kg benserazide had any significant effect on the increase in extracellular DA levels induced by l-DOPA, affecting only the time to reach the peak (145).
Role of Noradrenaline Neurons
Dopamine is the immediate precursor of NA along the catecholamine biosynthetic pathway, and extracellular NA levels increase in the DA-denervated striatum after a peripheral injection of l-DOPA. Interestingly, this increase is significantly larger when the treatment induces involuntary movements (73). An elevation in striatal NA levels has been suggested to contribute to LID because local infusions of NA in the DA-denervated striatum induce AIMs in the rat (73, 147). Based on these findings, one would expect LID to be improved by lesions of central NA projections. Quite in contrast with this prediction, most studies addressing the impact of noradrenergic denervation on LID have reported a worsening of dyskinesia, which was due either to an increased peak severity (148, 149) or to an increased duration of the involuntary movements (150). Other studies have not, however, detected a significant worsening of LID, even when the noradrenergic denervation resulted in a worsening of motor and cognitive deficits (151, 152). These apparent discrepancies are likely to depend on technical differences regarding NA lesion procedures and/or types of 6-OHDA models used in different studies. In this regard, it is useful to know that injections of 6-OHDA in the medial forebrain bundle (MFB) damage also ascending NA fibers, an effect that cannot be completely prevented by pretreating animals with blockers of NA uptake, such as desipramine (unpublished data by the Cenci’s lab). Thus, a large 6-OHDA lesion in the MFB may occlude the effect of a subsequent NA lesion, even more so if the latter is applied using toxins that damage NA projections but leave their cell bodies intact (150).
Despite the above discrepancies, a large amount of data point to an involvement of the NA system in the motor complications of PD therapy. This system is highly vulnerable to the neurodegenerative process in PD (153) and to the neurotoxins that are used to create PD models in animals [reviewed in Ref. (30)]. Moreover, treatment with l-DOPA appears to modulate the activity of brain NA neurons, as indicated by changes in NA cell firing in the locus coeruleus region, and by an increased NA efflux in their projection targets (73, 150). That the NA system in causally involved in LID is suggested not only by the results of lesion studies in the rat (148–150), but also by a vast pharmacological literature investigating the effects of NA receptor modulators.
Several studies in rat and primate models of PD have indeed shown that modulators of NA receptors improve LID. Many studies have evaluated antagonists of α2B/C-adrenoceptors, and found that they reduce the severity of l-DOPA-induced AIMs, and that they also can prolong the anti-akinetic effect of single l-DOPA doses (154–158). One potential underlying mechanism may involve a reduction of peak extracellular levels of both DOPA and DA, which the α2C adrenoceptor antagonist idazoxan has been shown to achieve at a dose that significantly reduces the severity of LID (113). The mechanisms by which central NA neurons modulate the effects of l-DOPA remain, however, poorly understood. Given that the NA system has widespread modulatory functions in the brain, these mechanisms are bound to be very complex. Relevant to the presynaptic mechanisms of LID are the modulatory effects of NA on several afferent striatal systems, including 5-HT and DA axon terminals (159–161), and the key role of locus coeruleus neurons in regulating both cerebral blood flow and capillary permeability (124, 162), and in maintaining the integrity of the BBB (163).
Concluding Remarks
l-DOPA remains the most effective treatment for PD and understanding how this drug is handled by, and in turn affects, a parkinsonian brain, is an undisputed research priority, not least for the sake of developing better treatment options.
In the past few years, research on the presynaptic mechanisms of LID has generated results of great translational importance, but also scientific controversy. In this article, I have reviewed both the findings and the controversies, while highlighting important aspects that call for further investigations.
Some of the concepts presented in this article are, however, quite uncontroversial and have already inspired a clinical development of new treatments. Thus, the concept that large swings in striatal DA levels are the culprit behind motor fluctuations and dyskinesia has prompted the development of new methods of continuous l-DOPA delivery, which are now available in several countries [reviewed in Ref. (3)]. While these therapies have a proven efficacy against the motor fluctuations (164), the extent to which they can eliminate already established dyskinesias remains to be demonstrated.
The concept that LID depends on DA release from serotonin neurons has raised both interest and discussion. That 5-HT neurons can produce and release DA “on” l-DOPA is now widely accepted. A debate, however, persists regarding the relative importance of this phenomenon. PD dyskinesias are conceivably more complex than the models of peak-dose LID obtained in animals with “clean” nigrostriatal lesions. For example, in the advanced stages of PD, the involuntary movements may exhibit a variable and unpredictable relationship with the timing of drug administration, and they may be induced by dopaminergic agents that do not release any DA in the brain. A point of recent discussion pertains to the role of DA release from 5-HT neurons in inducing involuntary movements as opposed to “good” anti-akinetic effects. Two recent studies (165, 166) have suggested that DA release from serotonin neurons not only generates dyskinesia but may also mediate the therapeutic benefit of l-DOPA. An implication of these findings is that antidyskinetic treatments based on the stimulation of 5-HT1A/B receptors (dampening transmitter release from 5-HT neurons) may have an unfavorable risk-benefit profile in the advanced stages of PD, when most l-DOPA-derived DA release is likely to come from 5-HT neurons, at least in the motor regions of the striatum. Accordingly, large clinical trials of 5-HT1A receptor agonists in LID appear to have faced some difficulties in defining a suitable therapeutic window for the investigational drugs [reviewed in Ref. (3)]. It should be noted, however, that the 5-HT1A ligands so far evaluated in PD patients had partial agonist activity and many off-target effects. To really appreciate the potential of this strategy, it will therefore be important to test more potent and selective compounds.
During the past few years, we have learned that l-DOPA pharmacotherapy affects not only neurons, but also microvascular (43, 125, 126, 141) and glial compartments (43, 117, 167) within the basal ganglia and the midbrain. Findings obtained in rat models of LID have revealed a previously unappreciated plastic potential of basal ganglia microvessels, sparking a new interest in the effects of dopaminergic medications on the neurovascular unit. This topic clearly deserves further investigation. An emerging research is uncovering orchestrated actions of gliovascular cells, immune cells, and neurons in the maladaptive plasticity associated with brain diseases and their treatments (168–171). Investigating the interactions between neuronal and gliovascular compartments is therefore required to fully understand the long-lasting plasticity at the basis of LID. Such an understanding will make it possible to devise new preventive strategies. Ultimately, preventive interventions may represent the best approach to this medical problem because, once established, LID is probably impossible to completely eliminate with add-on pharmacological treatments.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our work in this area is supported by grants from the Swedish Research Council, The Basal Ganglia Disorders Linnaeus Consortium (BAGADILICO), The Swedish Parkinson Foundation, The Olle Engquist Foundation, The Greta and Johan Kocks Foundation, The Michael J. Fox Foundation, and the NIH Morris K. Udall Centre of Excellence for Parkinson’s Disease Research (NIH P50 NS0716759).
References
1. Leenders KL, Salmon EP, Tyrrell P, Perani D, Brooks DJ, Sager H, et al. The nigrostriatal dopaminergic system assessed in vivo by positron emission tomography in healthy volunteer subjects and patients with Parkinson’s disease. Arch Neurol (1990) 47:1290–8. doi: 10.1001/archneur.1990.00530120034007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Morrish PK, Sawle GV, Brooks DJ. An [18F]dopa-PET and clinical study of the rate of progression in Parkinson’s disease. Brain (1996) 119(Pt 2):585–91. doi:10.1093/brain/119.2.585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Cenci MA, Ohlin KE, Odin P. Current options and future possibilities for the treatment of dyskinesia and motor fluctuations in Parkinson’s disease. CNS Neurol Disord Drug Targets (2011) 10:670–84. doi:10.2174/187152711797247885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Salat D, Tolosa E. Levodopa in the treatment of Parkinson’s disease: current status and new developments. J Parkinsons Dis (2013) 3:255–69. doi:10.3233/JPD-130186
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa-induced dyskinesias. Mov Disord (2007) 22:1379–89. doi:10.1002/mds.21475
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Storch A, Schneider CB, Wolz M, Sturwald Y, Nebe A, Odin P, et al. Nonmotor fluctuations in Parkinson disease: severity and correlation with motor complications. Neurology (2013) 80:800–9. doi:10.1212/WNL.0b013e318285c0ed
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Cilia R, Akpalu A, Sarfo FS, Cham M, Amboni M, Cereda E, et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain (2014) 137:2731–42. doi:10.1093/brain/awu195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Nutt JG. Levodopa-induced dyskinesia: review, observations, and speculations. Neurology (1990) 40:340–5. doi:10.1212/WNL.40.2.340
9. Manson A, Stirpe P, Schrag A. Levodopa-induced-dyskinesias clinical features, incidence, risk factors, management and impact on quality of life. J Parkinsons Dis (2012) 2:189–98. doi:10.3233/JPD-2012-120103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Khan NL, Graham E, Critchley P, Schrag AE, Wood NW, Lees AJ, et al. Parkin disease: a phenotypic study of a large case series. Brain (2003) 126:1279–92. doi:10.1093/brain/awg142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Halliday GM, Holton JL, Revesz T, Dickson DW. Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol (2011) 122:187–204. doi:10.1007/s00401-011-0852-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Selikhova M, Williams DR, Kempster PA, Holton JL, Revesz T, Lees AJ. A clinico-pathological study of subtypes in Parkinson’s disease. Brain (2009) 132:2947–57. doi:10.1093/brain/awp234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Sossi V, de la Fuente-Fernandez R, Schulzer M, Adams J, Stoessl J. Age-related differences in levodopa dynamics in Parkinson’s: implications for motor complications. Brain (2006) 129:1050–8. doi:10.1093/brain/awl028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Linazasoro G. New ideas on the origin of L-dopa-induced dyskinesias: age, genes and neural plasticity. Trends Pharmacol Sci (2005) 26:391–7. doi:10.1016/j.tips.2005.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Cenci MA, Lundblad M. Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J Neurochem (2006) 99:381–92. doi:10.1111/j.1471-4159.2006.04124.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Leenders KL, Palmer AJ, Quinn N, Clark JC, Firnau G, Garnett ES, et al. Brain dopamine metabolism in patients with Parkinson’s disease measured with positron emission tomography. J Neurol Neurosurg Psychiatry (1986) 49:853–60. doi:10.1136/jnnp.49.8.853
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Metman LV, Konitsiotis S, Chase TN. Pathophysiology of motor response complications in Parkinson’s disease: hypotheses on the why, where, and what. Mov Disord (2000) 15:3–8. doi:10.1002/1531-8257(200001)15:1<3::AID-MDS1003>3.0.CO;2-E
18. Engber TM, Susel Z, Kuo S, Gerfen CR, Chase TN. Levodopa replacement therapy alters enzyme activities in striatum and neuropeptide content in striatal output regions of 6-hydroxydopamine lesioned rats. Brain Res (1991) 552:113–8. doi:10.1016/0006-8993(91)90667-K
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Verhagen Metman L, Locatelli ER, Bravi D, Mouradian MM, Chase TN. Apomorphine responses in Parkinson’s disease and the pathogenesis of motor complications. Neurology (1997) 48:369–72. doi:10.1212/WNL.48.2.369
20. de la Fuente-Fernandez R, Schulzer M, Mak E, Calne DB, Stoessl AJ. Presynaptic mechanisms of motor fluctuations in Parkinson’s disease: a probabilistic model. Brain (2004) 127:888–99. doi:10.1093/brain/awh102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. de la Fuente-Fernandez R, Sossi V, Huang Z, Furtado S, Lu JQ, Calne DB, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain (2004) 127:2747–54. doi:10.1093/brain/awh290
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Pavese N, Evans AH, Tai YF, Hotton G, Brooks DJ, Lees AJ, et al. Clinical correlates of levodopa-induced dopamine release in Parkinson disease: a PET study. Neurology (2006) 67:1612–7. doi:10.1212/01.wnl.0000242888.30755.5d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. de la Fuente-Fernandez R. Presynaptic mechanisms of motor complications in Parkinson disease. Arch Neurol (2007) 64:141–3. doi:10.1001/archneur.64.1.141
24. Linazasoro G. Pathophysiology of motor complications in Parkinson disease: postsynaptic mechanisms are crucial. Arch Neurol (2007) 64:137–40. doi:10.1001/archneur.64.1.137
25. Cao X, Yasuda T, Uthayathas S, Watts RL, Mouradian MM, Mochizuki H, et al. Striatal overexpression of DeltaFosB reproduces chronic levodopa-induced involuntary movements. J Neurosci (2010) 30:7335–43. doi:10.1523/JNEUROSCI.0252-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Ulusoy A, Sahin G, Kirik D. Presynaptic dopaminergic compartment determines the susceptibility to L-DOPA-induced dyskinesia in rats. Proc Natl Acad Sci U S A (2010) 107:13159–64. doi:10.1073/pnas.1003432107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Cenci MA. Dopamine dysregulation of movement control in L-DOPA-induced dyskinesia. Trends Neurosci (2007) 30:236–43. doi:10.1016/j.tins.2007.03.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Iderberg H, Rylander D, Bimpisidis Z, Cenci MA. Modulating mGluR5 and 5-HT1A/1B receptors to treat l-DOPA-induced dyskinesia: effects of combined treatment and possible mechanisms of action. Exp Neurol (2013) 250:116–24. doi:10.1016/j.expneurol.2013.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Hong JY, Oh JS, Lee I, Sunwoo MK, Ham JH, Lee JE, et al. Presynaptic dopamine depletion predicts levodopa-induced dyskinesia in de novo Parkinson disease. Neurology (2014) 82:1597–604. doi:10.1212/WNL.0000000000000385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Iderberg H, Francardo V, Pioli EY. Animal models of L-DOPA-induced dyskinesia: an update on the current options. Neuroscience (2012) 211:13–27. doi:10.1016/j.neuroscience.2012.03.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Schneider JS. Levodopa-induced dyskinesias in parkinsonian monkeys: relationship to extent of nigrostriatal damage. Pharmacol Biochem Behav (1989) 34:193–6. doi:10.1016/0091-3057(89)90372-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Pearce RK, Jackson M, Smith L, Jenner P, Marsden CD. Chronic L-DOPA administration induces dyskinesias in the 1-methyl-4- phenyl-1,2,3,6-tetrahydropyridine-treated common marmoset (Callithrix jacchus). Mov Disord (1995) 10:731–40. doi:10.1002/mds.870100606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Di Monte DA, McCormack A, Petzinger G, Janson AM, Quik M, Langston WJ. Relationship among nigrostriatal denervation, parkinsonism, and dyskinesias in the MPTP primate model. Mov Disord (2000) 15:459–66. doi:10.1002/1531-8257(200005)15:3<459::AID-MDS1006>3.0.CO;2-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Togasaki DM, Protell P, Tan LC, Langston JW, Di Monte DA, Quik M. Dyskinesias in normal squirrel monkeys induced by nomifensine and levodopa. Neuropharmacology (2005) 48:398–405. doi:10.1016/j.neuropharm.2004.10.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Pearce RK, Heikkila M, Linden IB, Jenner P. L-dopa induces dyskinesia in normal monkeys: behavioural and pharmacokinetic observations. Psychopharmacology (Berl) (2001) 156:402–9. doi:10.1007/s002130100733
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Winkler C, Kirik D, Bjorklund A, Cenci MA. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of Parkinson’s disease: relation to motor and cellular parameters of nigrostriatal function. Neurobiol Dis (2002) 10:165–86. doi:10.1006/nbdi.2002.0499
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Guigoni C, Dovero S, Aubert I, Li Q, Bioulac BH, Bloch B, et al. Levodopa-induced dyskinesia in MPTP-treated macaques is not dependent on the extent and pattern of nigrostrial lesioning. Eur J Neurosci (2005) 22:283–7. doi:10.1111/j.1460-9568.2005.04196.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Lindgren HS, Rylander D, Ohlin KE, Lundblad M, Cenci MA. The “motor complication syndrome” in rats with 6-OHDA lesions treated chronically with L-DOPA: relation to dose and route of administration. Behav Brain Res (2007) 177:150–9. doi:10.1016/j.bbr.2006.09.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Picconi B, Paille V, Ghiglieri V, Bagetta V, Barone I, Lindgren HS, et al. l-DOPA dosage is critically involved in dyskinesia via loss of synaptic depotentiation. Neurobiol Dis (2008) 29:327–35. doi:10.1016/j.nbd.2007.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Fasano S, Bezard E, D’Antoni A, Francardo V, Indrigo M, Qin L, et al. Inhibition of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) signaling in the striatum reverts motor symptoms associated with L-dopa-induced dyskinesia. Proc Natl Acad Sci U S A (2010) 107:21824–9. doi:10.1073/pnas.1012071107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Francardo V, Recchia A, Popovic N, Andersson D, Nissbrandt H, Cenci MA. Impact of the lesion procedure on the profiles of motor impairment and molecular responsiveness to L-DOPA in the 6-hydroxydopamine mouse model of Parkinson’s disease. Neurobiol Dis (2011) 42:327–40. doi:10.1016/j.nbd.2011.01.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Lindgren HS, Rylander D, Iderberg H, Andersson M, O’Sullivan SS, Williams DR, et al. Putaminal upregulation of FosB/DeltaFosB-like immunoreactivity in Parkinson’s disease patients with dyskinesia. J Parkinsons Dis (2011) 1:347–57. doi:10.3233/JPD-2011-11068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Ohlin KE, Francardo V, Lindgren HS, Sillivan SE, O’Sullivan SS, Luksik AS, et al. Vascular endothelial growth factor is upregulated by L-dopa in the parkinsonian brain: implications for the development of dyskinesia. Brain (2011) 134:2339–57. doi:10.1093/brain/awr165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM. Compensations after lesions of central dopaminergic neurons: some clinical and basic implications. Trends Neurosci (1990) 13:290–6. doi:10.1016/0166-2236(90)90112-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Finkelstein DI, Stanic D, Parish CL, Tomas D, Dickson K, Horne MK. Axonal sprouting following lesions of the rat substantia nigra. Neuroscience (2000) 97:99–112. doi:10.1016/S0306-4522(00)00009-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Stanic D, Finkelstein DI, Bourke DW, Drago J, Horne MK. Timecourse of striatal re-innervation following lesions of dopaminergic SNpc neurons of the rat. Eur J Neurosci (2003) 18:1175–88. doi:10.1046/j.1460-9568.2003.02800.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Abercrombie ED, Bonatz AE, Zigmond MJ. Effects of L-dopa on extracellular dopamine in striatum of normal and 6-hydroxydopamine-treated rats. Brain Res (1990) 525:36–44. doi:10.1016/0006-8993(90)91318-B
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Sossi V, Dinelle K, Topping GJ, Holden JE, Doudet D, Schulzer M, et al. Dopamine transporter relation to levodopa-derived synaptic dopamine in a rat model of Parkinson’s: an in vivo imaging study. J Neurochem (2009) 109:85–92. doi:10.1111/j.1471-4159.2009.05904.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Anden NE, Magnusson T, Rosengren E. On the presence of dihydroxyphenylalanine decarboxylase in nerves. Experientia (1964) 20:328–9. doi:10.1007/BF02171078
50. Tsai MJ, Lee EH. Characterization of L-DOPA transport in cultured rat and mouse astrocytes. J Neurosci Res (1996) 43:490–5. doi:10.1002/(SICI)1097-4547(19960215)43:4<490::AID-JNR10>3.3.CO;2-K
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Bertler A, Falck B, Owman C, Rosengrenn E. The localization of monoaminergic blood-brain barrier mechanisms. Pharmacol Rev (1966) 18:369–85.
52. Klawans HL, Goetz C, Nausieda PA, Weiner WJ. Levodopa-induced dopamine receptor hypersensitivity. Trans Am Neurol Assoc (1977) 102:80–3.
53. Cenci MA, Konradi C. Maladaptive striatal plasticity in L-DOPA-induced dyskinesia. Prog Brain Res (2010) 183:209–33. doi:10.1016/S0079-6123(10)83011-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Murer MG, Moratalla R. Striatal signaling in L-DOPA-induced dyskinesia: common mechanisms with drug abuse and long term memory involving D1 dopamine receptor stimulation. Front Neuroanat (2011) 5:51. doi:10.3389/fnana.2011.00051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Gerfen CR, Miyachi S, Paletzki R, Brown P. D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J Neurosci (2002) 22:5042–54.
56. Ding Y, Won L, Britt JP, Lim SA, McGehee DS, Kang UJ. Enhanced striatal cholinergic neuronal activity mediates L-DOPA-induced dyskinesia in parkinsonian mice. Proc Natl Acad Sci U S A (2011) 108:840–5. doi:10.1073/pnas.1006511108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, et al. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci (2007) 27:6995–7005. doi:10.1523/JNEUROSCI.0852-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Westin JE, Vercammen L, Strome EM, Konradi C, Cenci MA. Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol Psychiatry (2007) 62:800–10. doi:10.1016/j.biopsych.2006.11.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Calabresi P, Di Filippo M, Ghiglieri V, Picconi B. Molecular mechanisms underlying levodopa-induced dyskinesia. Mov Disord (2008) 23(Suppl 3):S570–9. doi:10.1002/mds.22019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Deutch AY. Striatal plasticity in parkinsonism: dystrophic changes in medium spiny neurons and progression in Parkinson’s disease. J Neural Transm Suppl (2006) 70:67–70. doi:10.1007/978-3-211-45295-0_12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. McNeill TH, Brown SA, Rafols JA, Shoulson I. Atrophy of medium spiny I striatal dendrites in advanced Parkinson’s disease. Brain Res (1988) 455:148–52. doi:10.1016/0006-8993(88)90124-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Zaja-Milatovic S, Milatovic D, Schantz AM, Zhang J, Montine KS, Samii A, et al. Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology (2005) 64:545–7. doi:10.1212/01.WNL.0000150591.33787.A4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Ingham CA, Hood SH, van Maldegem B, Weenink A, Arbuthnott GW. Morphological changes in the rat neostriatum after unilateral 6-hydroxydopamine injections into the nigrostriatal pathway. Exp Brain Res (1993) 93:17–27. doi:10.1007/BF00227776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Villalba RM, Lee H, Smith Y. Dopaminergic denervation and spine loss in the striatum of MPTP-treated monkeys. Exp Neurol (2009) 215:220–7. doi:10.1016/j.expneurol.2008.09.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Fieblinger T, Graves SM, Sebel L, Alcacer C, Plotkin JL, Gertler TS, et al. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat Comm (2014) 5:5316. doi:10.1038/ncomms6316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Suarez LM, Solis O, Carames JM, Taravini IR, Solis JM, Murer MG, et al. L-DOPA treatment selectively restores spine density in dopamine receptor D2-expressing projection neurons in dyskinetic mice. Biol Psychiatry (2014) 75:711–22. doi:10.1016/j.biopsych.2013.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Zhang Y, Meredith GE, Mendoza-Elias N, Rademacher DJ, Tseng KY, Steece-Collier K. Aberrant restoration of spines and their synapses in L-DOPA-induced dyskinesia: involvement of corticostriatal but not thalamostriatal synapses. J Neurosci (2013) 33:11655–67. doi:10.1523/JNEUROSCI.0288-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci (2008) 9:665–77. doi:10.1038/nrn2471
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Fieblinger T, Cenci MA. Zooming in on the small: the plasticity of striatal spines in L-DOPA-induced dyskinesia. Mov Disord (2014) (in press).
70. Meissner W, Ravenscroft P, Reese R, Harnack D, Morgenstern R, Kupsch A, et al. Increased slow oscillatory activity in substantia nigra pars reticulata triggers abnormal involuntary movements in the 6-OHDA-lesioned rat in the presence of excessive extracelullar striatal dopamine. Neurobiol Dis (2006) 22:586–98. doi:10.1016/j.nbd.2006.01.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Carta M, Lindgren H, Lundblad M, Stancampiano R, Fadda F, Cenci MA. Role of striatal L-DOPA in the production of dyskinesia in 6-hydroxydopamine lesioned rats. J Neurochem (2006) 96:1718–27. doi:10.1111/j.1471-4159.2006.03696.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Lindgren HS, Andersson DR, Lagerkvist S, Nissbrandt H, Cenci MA. L-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: temporal and quantitative relationship to the expression of dyskinesia. J Neurochem (2010) 112:1465–76. doi:10.1111/j.1471-4159.2009.06556.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Wang Y, Wang HS, Wang T, Huang C, Liu J. L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease is associated with the fluctuational release of norepinephrine in the sensorimotor striatum. J Neurosci Res (2014) 92:1733–45. doi:10.1002/jnr.23439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Porras G, De Deurwaerdere P, Li Q, Marti M, Morgenstern R, Sohr R, et al. L-dopa-induced dyskinesia: beyond an excessive dopamine tone in the striatum. Sci Rep (2014) 4:3730. doi:10.1038/srep03730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Tanaka H, Kannari K, Maeda T, Tomiyama M, Suda T, Matsunaga M. Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport (1999) 10:631–4. doi:10.1097/00001756-199902250-00034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Lopez A, Munoz A, Guerra MJ, Labandeira-Garcia JL. Mechanisms of the effects of exogenous levodopa on the dopamine-denervated striatum. Neuroscience (2001) 103:639–51. doi:10.1016/S0306-4522(00)00588-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Arai R, Karasawa N, Geffard M, Nagatsu T, Nagatsu I. Immunohistochemical evidence that central serotonin neurons produce dopamine from exogenous L-DOPA in the rat, with reference to the involvement of aromatic L-amino acid decarboxylase. Brain Res (1994) 667:295–9. doi:10.1016/0006-8993(94)91511-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Gil S, Park C, Lee J, Koh H. The roles of striatal serotonin and L-amino-acid decarboxylase on L-DOPA-induced Dyskinesia in a Hemiparkinsonian rat model. Cell Mol Neurobiol (2010) 30:817–25. doi:10.1007/s10571-010-9509-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Lundblad M, af Bjerken S, Cenci MA, Pomerleau F, Gerhardt GA, Stromberg I. Chronic intermittent L-DOPA treatment induces changes in dopamine release. J Neurochem (2009) 108:998–1008. doi:10.1111/j.1471-4159.2008.05848.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Rylander D, Parent M, O’Sullivan SS, Dovero S, Lees AJ, Bezard E, et al. Maladaptive plasticity of serotonin axon terminals in levodopa-induced dyskinesia. Ann Neurol (2010) 68:619–28. doi:10.1002/ana.22097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Zeng BY, Iravani MM, Jackson MJ, Rose S, Parent A, Jenner P. Morphological changes in serotoninergic neurites in the striatum and globus pallidus in levodopa primed MPTP treated common marmosets with dyskinesia. Neurobiol Dis (2010) 40:599–607. doi:10.1016/j.nbd.2010.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Kannari K, Yamato H, Shen H, Tomiyama M, Suda T, Matsunaga M. Activation of 5-HT(1A) but not 5-HT(1B) receptors attenuates an increase in extracellular dopamine derived from exogenously administered L-DOPA in the striatum with nigrostriatal denervation. J Neurochem (2001) 76:1346–53. doi:10.1046/j.1471-4159.2001.00184.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Nahimi A, Holtzermann M, Landau AM, Simonsen M, Jakobsen S, Alstrup AK, et al. Serotonergic modulation of receptor occupancy in rats treated with L-DOPA after unilateral 6-OHDA lesioning. J Neurochem (2012) 120:806–17. doi:10.1111/j.1471-4159.2011.07598.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Blier P, Pineyro G, el Mansari M, Bergeron R, de Montigny C. Role of somatodendritic 5-HT autoreceptors in modulating 5-HT neurotransmission. Ann N Y Acad Sci (1998) 861:204–16. doi:10.1111/j.1749-6632.1998.tb10192.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Huot P, Fox SH, Brotchie JM. The serotonergic system in Parkinson’s disease. Prog Neurobiol (2011) 95:163–212. doi:10.1016/j.pneurobio.2011.08.004
86. Carta M, Carlsson T, Kirik D, Bjorklund A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain (2007) 130:1819–33. doi:10.1093/brain/awm082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Eskow KL, Dupre KB, Barnum CJ, Dickinson SO, Park JY, Bishop C. The role of the dorsal raphe nucleus in the development, expression, and treatment of L-dopa-induced dyskinesia in hemiparkinsonian rats. Synapse (2009) 63:610–20. doi:10.1002/syn.20630
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Navailles S, Bioulac B, Gross C, De Deurwaerdere P. Serotonergic neurons mediate ectopic release of dopamine induced by L-DOPA in a rat model of Parkinson’s disease. Neurobiol Dis (2010) 38:136–43. doi:10.1016/j.nbd.2010.01.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Eskow Jaunarajs KL, George JA, Bishop C. L-DOPA-induced dysregulation of extrastriatal dopamine and serotonin and affective symptoms in a bilateral rat model of Parkinson’s disease. Neuroscience (2012) 218:243–56. doi:10.1016/j.neuroscience.2012.05.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Halje P, Tamte M, Richter U, Mohammed M, Cenci MA, Petersson P. Levodopa-induced dyskinesia is strongly associated with resonant cortical oscillations. J Neurosci (2012) 32:16541–51. doi:10.1523/JNEUROSCI.3047-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Kish SJ, Tong J, Hornykiewicz O, Rajput A, Chang LJ, Guttman M, et al. Preferential loss of serotonin markers in caudate versus putamen in Parkinson’s disease. Brain (2008) 131:120–31. doi:10.1093/brain/awm239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Politis M, Wu K, Loane C, Kiferle L, Molloy S, Brooks DJ, et al. Staging of serotonergic dysfunction in Parkinson’s disease: an in vivo 11C-DASB PET study. Neurobiol Dis (2010) 40:216–21. doi:10.1016/j.nbd.2010.05.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Khan NL, Valente EM, Bentivoglio AR, Wood NW, Albanese A, Brooks DJ, et al. Clinical and subclinical dopaminergic dysfunction in PARK6-linked parkinsonism: an 18F-dopa PET study. Ann Neurol (2002) 52:849–53. doi:10.1002/ana.10417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Rylander D, Iderberg H, Li Q, Dekundy A, Zhang J, Li H, et al. A mGluR5 antagonist under clinical development improves L-DOPA-induced dyskinesia in parkinsonian rats and monkeys. Neurobiol Dis (2010) 39:352–61. doi:10.1016/j.nbd.2010.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain (2013) 136:2419–31. doi:10.1093/brain/awt192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Kannari K, Tanaka H, Maeda T, Tomiyama M, Suda T, Matsunaga M. Reserpine pretreatment prevents increases in extracellular striatal dopamine following L-DOPA administration in rats with nigrostriatal denervation. J Neurochem (2000) 74:263–9. doi:10.1046/j.1471-4159.2000.0740263.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Miller DW, Abercrombie ED. Role of high-affinity dopamine uptake and impulse activity in the appearance of extracellular dopamine in striatum after administration of exogenous L-DOPA: studies in intact and 6-hydroxydopamine-treated rats. J Neurochem (1999) 72:1516–22. doi:10.1046/j.1471-4159.1999.721516.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Darmopil S, Muneton-Gomez VC, de Ceballos ML, Bernson M, Moratalla R. Tyrosine hydroxylase cells appearing in the mouse striatum after dopamine denervation are likely to be projection neurones regulated by L-DOPA. Eur J Neurosci (2008) 27:580–92. doi:10.1111/j.1460-9568.2008.06040.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. DiCaudo C, Riverol M, Mundinano IC, Ordonez C, Hernandez M, Marcilla I, et al. Chronic levodopa administration followed by a washout period increased number and induced phenotypic changes in striatal dopaminergic cells in MPTP-monkeys. PLoS One (2012) 7:e50842. doi:10.1371/journal.pone.0050842
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Lopez-Real A, Rodriguez-Pallares J, Guerra MJ, Labandeira-Garcia JL. Localization and functional significance of striatal neurons immunoreactive to aromatic L-amino acid decarboxylase or tyrosine hydroxylase in rat Parkinsonian models. Brain Res (2003) 969:135–46. doi:10.1016/S0006-8993(03)02291-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Unal B, Shah F, Kothari J, Tepper JM. Anatomical and electrophysiological changes in striatal TH interneurons after loss of the nigrostriatal dopaminergic pathway. Brain Struct Funct (2013). doi:10.1007/s00429-013-0658-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Politis M, Wu K, Loane C, Brooks DJ, Kiferle L, Turkheimer FE, et al. Serotonergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J Clin Invest (2014) 124:1340–9. doi:10.1172/JCI71640
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Maeda T, Nagata K, Yoshida Y, Kannari K. Serotonergic hyperinnervation into the dopaminergic denervated striatum compensates for dopamine conversion from exogenously administered l-DOPA. Brain Res (2005) 1046:230–3. doi:10.1016/j.brainres.2005.04.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci (2004) 27:589–94. doi:10.1016/j.tins.2004.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. van Luijtelaar MG, Tonnaer JA, Steinbusch HW. Aging of the serotonergic system in the rat forebrain: an immunocytochemical and neurochemical study. Neurobiol Aging (1992) 13:201–15. doi:10.1016/0197-4580(92)90032-S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Guerra MJ, Liste I, Labandeira-Garcia JL. Effects of lesions of the nigrostriatal pathway and of nigral grafts on striatal serotonergic innervation in adult rats. Neuroreport (1997) 8:3485–8. doi:10.1097/00001756-199711100-00014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Rozas G, Liste I, Guerra MJ, Labandeira-Garcia JL. Sprouting of the serotonergic afferents into striatum after selective lesion of the dopaminergic system by MPTP in adult mice. Neurosci Lett (1998) 245:151–4. doi:10.1016/S0304-3940(98)00198-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Zhou FC, Bledsoe S, Murphy J. Serotonergic sprouting is induced by dopamine-lesion in substantia nigra of adult rat brain. Brain Res (1991) 556:108–16. doi:10.1016/0006-8993(91)90553-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Morin N, Morissette M, Gregoire L, Di Paolo T. Effect of a chronic treatment with an mGlu5 receptor antagonist on brain serotonin markers in parkinsonian monkeys. Prog Neuropsychopharmacol Biol Psychiatry (2014) 56C:27–38. doi:10.1016/j.pnpbp.2014.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Navailles S, Bioulac B, Gross C, De Deurwaerdere P. Chronic L-DOPA therapy alters central serotonergic function and L-DOPA-induced dopamine release in a region-dependent manner in a rat model of Parkinson’s disease. Neurobiol Dis (2011) 41:585–90. doi:10.1016/j.nbd.2010.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Zhang X, Andren PE, Greengard P, Svenningsson P. Evidence for a role of the 5-HT1B receptor and its adaptor protein, p11, in L-DOPA treatment of an animal model of Parkinsonism. Proc Natl Acad Sci U S A (2008) 105:2163–8. doi:10.1073/pnas.0711839105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Huot P, Fox SH. The serotonergic system in motor and non-motor manifestations of Parkinson’s disease. Exp Brain Res (2013) 230:463–76. doi:10.1007/s00221-013-3621-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Buck K, Voehringer P, Ferger B. The alpha(2) adrenoceptor antagonist idazoxan alleviates L-DOPA-induced dyskinesia by reduction of striatal dopamine levels: an in vivo microdialysis study in 6-hydroxydopamine-lesioned rats. J Neurochem (2010) 112:444–52. doi:10.1111/j.1471-4159.2009.06482.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Matsuo H, Tsukada S, Nakata T, Chairoungdua A, Kim DK, Cha SH, et al. Expression of a system L neutral amino acid transporter at the blood-brain barrier. Neuroreport (2000) 11:3507–11. doi:10.1097/00001756-200011090-00021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Wade LA, Katzman R. Synthetic amino acids and the nature of L-DOPA transport at the blood-brain barrier. J Neurochem (1975) 25:837–42. doi:10.1111/j.1471-4159.1975.tb04415.x
116. Renkin EM. B.W. Zweifach award lecture. Regulation of the microcirculation. Microvasc Res (1985) 30:251e63.
117. Inyushin MY, Huertas A, Kucheryavykh YV, Kucheryavykh LY, Tsydzik V, Sanabria P, et al. L-DOPA uptake in astrocytic endfeet enwrapping blood vessels in rat brain. Parkinsons Dis (2012) 2012:321406. doi:10.1155/2012/321406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature (2010) 468:232–43. doi:10.1038/nature09613
119. Itoh Y, Suzuki N. Control of brain capillary blood flow. J Cereb Blood Flow Metab (2012) 32:1167–76. doi:10.1038/jcbfm.2012.5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Bacic F, Uematsu S, McCarron RM, Spatz M. Dopaminergic receptors linked to adenylate cyclase in human cerebromicrovascular endothelium. J Neurochem (1991) 57:1774–80. doi:10.1111/j.1471-4159.1991.tb06380.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Bekar LK, Wei HS, Nedergaard M. The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. J Cereb Blood Flow Metab (2012) 32:2135–45. doi:10.1038/jcbfm.2012.115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Iadecola C. Neurogenic control of the cerebral microcirculation: is dopamine minding the store? Nat Neurosci (1998) 1:263–5. doi:10.1038/1074
123. Krimer LS, Muly EC III, Williams GV, Goldman-Rakic PS. Dopaminergic regulation of cerebral cortical microcirculation. Nat Neurosci (1998) 1:286–9. doi:10.1038/1099
124. Raichle ME, Hartman BK, Eichling JO, Sharpe LG. Central noradrenergic regulation of cerebral blood flow and vascular permeability. Proc Natl Acad Sci U S A (1975) 72:3726–30. doi:10.1073/pnas.72.9.3726
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Hirano S, Asanuma K, Ma Y, Tang C, Feigin A, Dhawan V, et al. Dissociation of metabolic and neurovascular responses to levodopa in the treatment of Parkinson’s disease. J Neurosci (2008) 28:4201–9. doi:10.1523/JNEUROSCI.0582-08.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Ohlin KE, Sebastianutto I, Adkins CE, Lundblad C, Lockman PR, Cenci MA. Impact of L-DOPA treatment on regional cerebral blood flow and metabolism in the basal ganglia in a rat model of Parkinson’s disease. Neuroimage (2012) 61:228–39. doi:10.1016/j.neuroimage.2012.02.066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Choi JK, Chen YI, Hamel E, Jenkins BG. Brain hemodynamic changes mediated by dopamine receptors: role of the cerebral microvasculature in dopamine-mediated neurovascular coupling. Neuroimage (2006) 30:700–12. doi:10.1016/j.neuroimage.2005.10.029
128. Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev (2005) 57:173–85. doi:10.1124/pr.57.2.4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Bartels AL, Willemsen AT, Kortekaas R, de Jong BM, de Vries R, de Klerk O, et al. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm (2008) 115:1001–9. doi:10.1007/s00702-008-0030-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett (1996) 211:13–6. doi:10.1016/0304-3940(96)12706-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Pisani V, Stefani A, Pierantozzi M, Natoli S, Stanzione P, Franciotta D, et al. Increased blood-cerebrospinal fluid transfer of albumin in advanced Parkinson’s disease. J Neuroinflammation (2012) 9:188. doi:10.1186/1742-2094-9-188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Carvey PM, Zhao CH, Hendey B, Lum H, Trachtenberg J, Desai BS, et al. 6-hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur J Neurosci (2005) 22:1158–68. doi:10.1111/j.1460-9568.2005.04281.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis (2006) 21:404–12. doi:10.1016/j.nbd.2005.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Barcia C, Bautista V, Sanchez-Bahillo A, Fernandez-Villalba E, Faucheux B, Poza y Poza M, et al. Changes in vascularization in substantia nigra pars compacta of monkeys rendered parkinsonian. J Neural Transm (2005) 112:1237–48. doi:10.1007/s00702-004-0256-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Desai Bradaric B, Patel A, Schneider JA, Carvey PM, Hendey B. Evidence for angiogenesis in Parkinson’s disease, incidental Lewy body disease, and progressive supranuclear palsy. J Neural Transm (2012) 119:59–71. doi:10.1007/s00702-011-0684-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Faucheux BA, Bonnet AM, Agid Y, Hirsch EC. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet (1999) 353:981–2. doi:10.1016/S0140-6736(99)00641-8
137. Wada K, Arai H, Takanashi M, Fukae J, Oizumi H, Yasuda T, et al. Expression levels of vascular endothelial growth factor and its receptors in Parkinson’s disease. Neuroreport (2006) 17:705–9. doi:10.1097/01.wnr.0000215769.71657.65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Greenberg DA, Jin K. From angiogenesis to neuropathology. Nature (2005) 438:954–9. doi:10.1038/nature04481
139. Westin JE, Lindgren HS, Gardi J, Nyengaard JR, Brundin P, Mohapel P, et al. Endothelial proliferation and increased blood-brain barrier permeability in the basal ganglia in a rat model of 3,4-dihydroxyphenyl-L-alanine-induced dyskinesia. J Neurosci (2006) 26:9448–61. doi:10.1523/JNEUROSCI.0944-06.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Lindgren HS, Ohlin KE, Cenci MA. Differential involvement of D1 and D2 dopamine receptors in L-DOPA-induced angiogenic activity in a rat model of Parkinson’s disease. Neuropsychopharmacology (2009) 34:2477–88. doi:10.1038/npp.2009.74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Munoz A, Garrido-Gil P, Dominguez-Meijide A, Labandeira-Garcia JL. Angiotensin type 1 receptor blockage reduces L-dopa-induced dyskinesia in the 6-OHDA model of Parkinson’s disease. Involvement of vascular endothelial growth factor and interleukin-1beta. Exp Neurol (2014) 261:720–32. doi:10.1016/j.expneurol.2014.08.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis (2008) 32:200–19. doi:10.1016/j.nbd.2008.08.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Astradsson A, Jenkins BG, Choi JK, Hallett PJ, Levesque MA, McDowell JS, et al. The blood-brain barrier is intact after levodopa-induced dyskinesias in parkinsonian primates – evidence from in vivo neuroimaging studies. Neurobiol Dis (2009) 35:348–51. doi:10.1016/j.nbd.2009.05.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Mitala CM, Wang Y, Borland LM, Jung M, Shand S, Watkins S, et al. Impact of microdialysis probes on vasculature and dopamine in the rat striatum: a combined fluorescence and voltammetric study. J Neurosci Methods (2008) 174:177–85. doi:10.1016/j.jneumeth.2008.06.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Shen H, Kannari K, Yamato H, Arai A, Matsunaga M. Effects of benserazide on L-DOPA-derived extracellular dopamine levels and aromatic L-amino acid decarboxylase activity in the striatum of 6-hydroxydopamine-lesioned rats. Tohoku J Exp Med (2003) 199:149–59. doi:10.1620/tjem.199.149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Jonkers N, Sarre S, Ebinger G, Michotte Y. Benserazide decreases central AADC activity, extracellular dopamine levels and levodopa decarboxylation in striatum of the rat. J Neural Transm (2001) 108:559–70. doi:10.1007/s007020170056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Buck K, Ferger B. Comparison of intrastriatal administration of noradrenaline and l-DOPA on dyskinetic movements: a bilateral reverse in vivo microdialysis study in 6-hydroxydopamine-lesioned rats. Neuroscience (2009) 159:16–20. doi:10.1016/j.neuroscience.2008.12.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Fulceri F, Biagioni F, Ferrucci M, Lazzeri G, Bartalucci A, Galli V, et al. Abnormal involuntary movements (AIMs) following pulsatile dopaminergic stimulation: severe deterioration and morphological correlates following the loss of locus coeruleus neurons. Brain Res (2007) 1135:219–29. doi:10.1016/j.brainres.2006.12.030
149. Shin E, Rogers JT, Devoto P, Bjorklund A, Carta M. Noradrenaline neuron degeneration contributes to motor impairments and development of L-DOPA-induced dyskinesia in a rat model of Parkinson’s disease. Exp Neurol (2014) 257:25–38. doi:10.1016/j.expneurol.2014.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Miguelez C, Aristieta A, Cenci MA, Ugedo L. The locus coeruleus is directly implicated in L-DOPA-induced dyskinesia in parkinsonian rats: an electrophysiological and behavioural study. PLoS One (2011) 6:e24679. doi:10.1371/journal.pone.0024679
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Ostock CY, Lindenbach D, Goldenberg AA, Kampton E, Bishop C. Effects of noradrenergic denervation by anti-DBH-saporin on behavioral responsivity to L-DOPA in the hemi-parkinsonian rat. Behav Brain Res (2014) 270:75–85. doi:10.1016/j.bbr.2014.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Perez V, Marin C, Rubio A, Aguilar E, Barbanoj M, Kulisevsky J. Effect of the additional noradrenergic neurodegeneration to 6-OHDA-lesioned rats in levodopa-induced dyskinesias and in cognitive disturbances. J Neural Transm (2009) 116:1257–66. doi:10.1007/s00702-009-0291-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Del Tredici K, Rub U, De Vos RA, Bohl JR, Braak H. Where does Parkinson disease pathology begin in the brain? J Neuropathol Exp Neurol (2002) 61:413–26.
154. Lundblad M, Andersson M, Winkler C, Kirik D, Wierup N, Cenci MA. Pharmacological validation of behavioural measures of akinesia and dyskinesia in a rat model of Parkinson’s disease. Eur J Neurosci (2002) 15:120–32. doi:10.1046/j.0953-816x.2001.01843.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Dekundy A, Lundblad M, Danysz W, Cenci MA. Modulation of L-DOPA-induced abnormal involuntary movements by clinically tested compounds: further validation of the rat dyskinesia model. Behav Brain Res (2007) 179:76–89. doi:10.1016/j.bbr.2007.01.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Gomez-Mancilla B, Bedard PJ. Effect of nondopaminergic drugs on L-dopa-induced dyskinesias in MPTP-treated monkeys. Clin Neuropharmacol (1993) 16:418–27. doi:10.1097/00002826-199310000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Henry B, Fox SH, Peggs D, Crossman AR, Brotchie JM. The alpha2-adrenergic receptor antagonist idazoxan reduces dyskinesia and enhances anti-parkinsonian actions of L-dopa in the MPTP-lesioned primate model of Parkinson’s disease. Mov Disord (1999) 14:744–53. doi:10.1002/1531-8257(199909)14:5<744::AID-MDS1006>3.0.CO;2-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Savola JM, Hill M, Engstrom M, Merivuori H, Wurster S, McGuire SG, et al. Fipamezole (JP-1730) is a potent alpha2 adrenergic receptor antagonist that reduces levodopa-induced dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Mov Disord (2003) 18:872–83. doi:10.1002/mds.10464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Chotibut T, Apple DM, Jefferis R, Salvatore MF. Dopamine transporter loss in 6-OHDA Parkinson’s model is unmet by parallel reduction in dopamine uptake. PLoS One (2012) 7:e52322. doi:10.1371/journal.pone.0052322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Chotibut T, Fields V, Salvatore MF. Norepinephrine transporter inhibition with desipramine exacerbates L-DOPA-induced dyskinesia: role for synaptic dopamine regulation in denervated nigrostriatal terminals. Mol Pharmacol (2014) 86:675–85. doi:10.1124/mol.114.093302
161. Munoz A, Lopez-Real A, Labandeira-Garcia JL, Guerra MJ. Interaction between the noradrenergic and serotonergic systems in locomotor hyperactivity and striatal expression of Fos induced by amphetamine in rats. Exp Brain Res (2003) 153:92–9. doi:10.1007/s00221-003-1582-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Kalaria RN, Stockmeier CA, Harik SI. Brain microvessels are innervated by locus ceruleus noradrenergic neurons. Neurosci Lett (1989) 97:203–8. doi:10.1016/0304-3940(89)90164-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Kalinin S, Feinstein DL, Xu HL, Huesa G, Pelligrino DA, Galea E. Degeneration of noradrenergic fibres from the locus coeruleus causes tight-junction disorganisation in the rat brain. Eur J Neurosci (2006) 24:3393–400. doi:10.1111/j.1460-9568.2006.05223.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Olanow CW, Kieburtz K, Odin P, Espay AJ, Standaert DG, Fernandez HH, et al. Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: a randomised, controlled, double-blind, double-dummy study. Lancet Neurol (2014) 13:141–9. doi:10.1016/S1474-4422(13)70293-X
165. Bezard E, Tronci E, Pioli EY, Li Q, Porras G, Bjorklund A, et al. Study of the antidyskinetic effect of eltoprazine in animal models of levodopa-induced dyskinesia. Mov Disord (2013) 28:1088–96. doi:10.1002/mds.25366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Nevalainen N, Af Bjerken S, Gerhardt GA, Stromberg I. Serotonergic nerve fibers in L-DOPA-derived dopamine release and dyskinesia. Neuroscience (2014) 260:73–86. doi:10.1016/j.neuroscience.2013.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Bortolanza M, Cavalcanti-Kiwiatkoski R, Padovan-Neto FE, da-Silva CA, Mitkoviski M, Raisman-Vozari R, et al. Glial activation is associated with L-DOPA induced dyskinesia and blocked by a nitric oxide synthase inhibitor in a rat model of Parkinson’s disease. Neurobiol Dis (2015) 73:377–87. doi:10.1016/j.nbd.2014.10.017
168. Kousik SM, Napier TC, Carvey PM. The effects of psychostimulant drugs on blood brain barrier function and neuroinflammation. Front Pharmacol (2012) 3:121. doi:10.3389/fphar.2012.00121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Newton SS, Fournier NM, Duman RS. Vascular growth factors in neuropsychiatry. Cell Mol Life Sci (2013) 70:1739–52. doi:10.1007/s00018-013-1281-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Ostergaard L, Aamand R, Gutierrez-Jimenez E, Ho YC, Blicher JU, Madsen SM, et al. The capillary dysfunction hypothesis of Alzheimer’s disease. Neurobiol Aging (2013) 34:1018–31. doi:10.1016/j.neurobiolaging.2012.09.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Xanthos DN, Sandkuhler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci (2014) 15:43–53. doi:10.1038/nrn3617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Mouradian MM, Juncos JL, Fabbrini G, Schlegel J, Bartko JJ, Chase TN. Motor fluctuations in Parkinson’s disease: central pathophysiological mechanisms, Part II. Ann Neurol (1988) 24:372–8. doi:10.1002/ana.410240304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Nutt JG, Holford NH. The response to levodopa in Parkinson’s disease: imposing pharmacological law and order. Ann Neurol (1996) 39:561–73. doi:10.1002/ana.410390504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Cenci MA, Lindgren HS. Advances in understanding L-DOPA-induced dyskinesia. Curr Opin Neurobiol (2007) 17:665–71. doi:10.1016/j.conb.2008.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Sgambato-Faure V, Cenci MA. Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol (2012) 96:69–86. doi:10.1016/j.pneurobio.2011.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: neuroplasticity, neuropharmacology, neuropsychiatry, neurovascular unit, movement disorders, dystonia, basal ganglia, monoamines
Citation: Cenci MA (2014) Presynaptic mechanisms of l-DOPA-induced dyskinesia: the findings, the debate, and the therapeutic implications. Front. Neurol. 5:242. doi: 10.3389/fneur.2014.00242
Received: 08 September 2014; Paper pending published: 18 September 2014;
Accepted: 10 November 2014; Published online: 15 December 2014.
Edited by:
Antonio Cerasa, National Research Council, ItalyReviewed by:
Jose L. Labandeira-Garcia, University of Santiago de Compostela, SpainFilip Bergquist, University of Gothenburg, Sweden
Copyright: © 2014 Cenci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Angela Cenci, Basal Ganglia Pathophysiology Unit, Department of Experimental Medical Science, Lund University, BMC F11, 221 84 Lund, Sweden e-mail:YW5nZWxhLmNlbmNpX25pbHNzb25AbWVkLmx1LnNl