 Suzanne Elizabeth Schindler
Suzanne Elizabeth Schindler Anne M. Fagan
Anne M. Fagan- Department of Neurology, Knight Alzheimer’s Disease Research Center, Hope Center for Neurological Disorders, Washington University School of Medicine, St. Louis, MO, USA
Our understanding of the pathogenesis of Alzheimer disease (AD) has been greatly influenced by investigation of rare families with autosomal dominant mutations that cause early onset AD. Mutations in the genes coding for amyloid precursor protein (APP), presenilin 1 (PSEN-1), and presenilin 2 (PSEN-2) cause over-production of the amyloid-β peptide (Aβ) leading to early deposition of Aβ in the brain, which in turn is hypothesized to initiate a cascade of processes, resulting in neuronal death, cognitive decline, and eventual dementia. Studies of cerebrospinal fluid (CSF) from individuals with the common form of AD, late-onset AD (LOAD), have revealed that low CSF Aβ42 and high CSF tau are associated with AD brain pathology. Herein, we review the literature on CSF biomarkers in autosomal dominant AD (ADAD), which has contributed to a detailed road map of AD pathogenesis, especially during the preclinical period, prior to the appearance of any cognitive symptoms. Current drug trials are also taking advantage of the unique characteristics of ADAD and utilizing CSF biomarkers to accelerate development of effective therapies for AD.
Introduction
In 1901, Dr. Alois Alzheimer began treating Auguste D., a 51-year-old woman with memory loss and hallucinations. Ms. D’s dementia progressed and she died at the age of 56. Upon histopathological examination, Alzheimer found two types of abnormalities in the brain that were later termed amyloid plaques and neurofibrillary tangles (1). Over a century later, when patients die with a characteristic history of progressive cognitive decline and upon autopsy are found to have significant quantities of amyloid plaques and neurofibrillary tangles, they are assigned the neuropathological diagnosis of Alzheimer disease (AD). The vast majority of patients with AD develop dementia at age 65 or older. Genetic studies of patients like Ms. D, who develop cognitive decline before age 65, have revealed rare autosomal dominant mutations that cause AD (2). Recently, surviving samples from Ms. D were subjected to genetic analysis and found to have a genetic mutation in presenilin 1 (PSEN-1) (3), although there has been some controversy about this finding (4).
There is some concern that the pathogenesis of autosomal dominant AD (ADAD) may vary from the common late-onset AD (LOAD). However, while there are certainly some differences between ADAD and LOAD in terms of disease etiology, clinical features, and neuropathology, they share many characteristics including an abnormal pattern of cerebrospinal fluid (CSF) biomarkers (Table S1 in Supplementary Material). Although we cannot completely dismiss the notions that the pathogenesis of ADAD and LOAD are distinct and that findings from ADAD do not apply to LOAD, investigation of families with ADAD have contributed enormously to our understanding of AD. Finding mutations that cause ADAD identified key molecules in the disease process (5–9). Transgenic mice expressing human ADAD mutations revolutionized the field and have been used to examine almost every aspect of the disease (10). Recently, studies of CSF and brain imaging biomarkers have helped establish the time course of AD-related brain changes in individuals affected by ADAD, especially during the preclinical stage, prior to the appearance of cognitive symptoms (11, 12). Furthermore, after the failure of numerous drug trials to halt, slow, or reverse cognitive decline in symptomatic individuals with LOAD, clinical trials are now utilizing the unique nature of ADAD and the data derived from these families to design prevention trials for AD dementia in both ADAD mutation carriers (MCs) and individuals at risk for LOAD, while they are still asymptomatic (13, 14). Just as Ms. D’s genetic misfortune benefited the entire field of AD research, it is likely that ADAD patients will lead us to better treatments for all people afflicted by this disease.
Epidemiology
Alzheimer disease is the most common cause of dementia and, in the United States, affects ~4.7 million individuals aged 65 and older (15). Less than five percent of AD patients develop symptoms before age 65 and are classified as having early onset Alzheimer disease (EOAD) (16). Even rarer are the <1% of AD patients who carry mutations that cause ADAD with 100% penetrance who are distributed world-wide. Carriers of ADAD mutations typically develop symptoms of dementia in their 30s to 60s, depending on their specific gene mutation and the age of onset within their family (17, 18). Much of our current knowledge about ADAD and biomarkers of ADAD comes from two large studies: the multi-center, international Dominantly Inherited Alzheimer Network (DIAN) cohort, and the Alzheimer’s Prevention Initiative (API) cohort that studies a large pedigree living in the state of Antioquia in Colombia, South America. The DIAN cohort includes carriers and non-carrier (NC) family members with many different ADAD mutations, while the Colombian kindred is likely descended from a single individual (19) and carries the E280A mutation in the PSEN-1 gene.
Clinical Features
Regardless of whether patients develop symptoms of AD before age 65 (EOAD) or after age 65 (LOAD), the typical first symptom of brain dysfunction is progressive episodic memory loss that slowly worsens over years (20). However, about 30–40% of patients with early symptom onset either from non-familial EOAD or ADAD have an increased frequency of atypical presentations, such as impairments in non-memory domains, including executive, behavioral, language, and visuospatial (21–23). PSEN-1 MCs have been reported to be more likely to have headaches, myoclonus, gait abnormalities, pseudobulbar affect, and spastic paraparesis (24–26). Some mutations in the gene for amyloid precursor protein (APP) cause severe cerebral amyloid angiopathy (CAA), with resultant strokes and brain hemorrhages (27). These clinical features are rarely observed in LOAD.
Neuropathology
The hallmarks of AD, regardless of the age at dementia onset and its underlying cause (ADAD versus LOAD), are aggregation of the amyloid-β (Aβ) peptide into amyloid plaques and region-specific development of intraneuronal neurofibrillary tangles composed of hyperphosphorylated forms of the microtubule-associated protein, tau (28). AD-affected brains also demonstrate significant neuronal loss and associated neuroinflammation (29–31), although these features are not specific to AD.
In addition to these classic pathologies, some ADAD mutations have been associated with neuropathological abnormalities not typically seen in LOAD. For example, amyloid deposition has been observed in the cerebellum of PSEN-1 E280A carriers (32), an area not typically affected in LOAD. “Cotton-wool” type plaques that are larger than typical plaques, lack congophilic cores and have few associated dystrophic neurites (33) are often seen in individuals carrying certain PSEN-1 mutations (34). Some ADAD mutations (notably in APP) result in severe CAA, which appears histologically as deposition of Aβ40 in the blood vessel wall. The specific pattern of CAA distribution in the brain depends on the mutation (e.g., Dutch, Flemish, Arctic, Iowa, and Italian) (34).
Genetics and Pathogenesis
The genetics of ADAD have provided key insights into the molecular pathogenesis of AD. The observation in 1984 that older adults with Trisomy 21, also known as Down syndrome, develop the brain changes of AD suggested that a genetic locus on chromosome 21 might be involved in AD (35). Indeed, the first ADAD mutations were identified in the APP gene that resides on chromosome 21, thus implicating amyloid as a key player in AD pathogenesis (5–7, 36). We also now know that duplication of the APP locus results in ADAD (37, 38), likely because of amyloid over-production. Following the discovery of APP mutations, mutations in PSEN-1 (8) and the gene for presenilin 2 (PSEN-2) (9) were identified and found to increase the amount of the more aggregation-prone Aβ42 compared to Aβ40 (39). Later, it was discovered that presenilin 1 is a critical component of the γ-secretase enzyme complex that cleaves APP to form Aβ (40). To date, 40 mutations in APP, 197 mutations in PSEN-1, and 25 mutations in PSEN-2 have been identified that cause ADAD (2).
Since ADAD mutations either increase total Aβ or increase the ratio of Aβ42:Aβ42, amyloid has been hypothesized to be the initiator of AD, an idea described as the “Amyloid Hypothesis” (41). In further support of this hypothesis, a mutation was recently discovered in APP that decreases Aβ production and lowers the risk for AD (42). According to this hypothesis, initial deposition of Aβ into amyloid plaques leads to downstream tau-related neuronal pathology (tangles), neuronal injury, and subsequent neuronal death, which is then manifested as cognitive impairment, ultimately culminating in dementia at the end stage of the disease. Data from neuropathological, brain imaging, and CSF biomarker studies in LOAD are consistent with this hypothesis (43–49), but it has only been through study of ADAD that we have a more precise knowledge of the timing of these changes during the early, preclinical (presymptomatic) stage.
CSF Biomarkers in ADAD
Due to its high prevalence, the majority of AD biomarker studies to date have evaluated individuals with LOAD. CSF levels of Aβ42, tau, and phosphotau181 (ptau) (markers of amyloid, neuronal injury, and tangles, respectively) have stood the test of time in exhibiting both diagnostic and prognostic utility (50). Individuals diagnosed with very mild or mild AD dementia have low levels of CSF Aβ42 (51–54) that inversely correlate with the presence of amyloid as visualized by positron emission tomography (PET) (55–59). Concentrations of CSF tau and ptau are increased in AD and have been shown to positively correlate (albeit to differing degrees) with tangle load at autopsy (52, 53, 60) and regional brain atrophy as defined by magnetic resonance imaging (MRI) (61–64). When paired, the combination of low CSF Aβ42 and high tau/ptau has been shown to be a strong predictor of future cognitive decline in both early symptomatic (very mild dementia or mild cognitive impairment, MCI) and asymptomatic individuals (55, 65–68). However, while such analyses in individuals at risk for LOAD can estimate the risk for decline, they cannot provide the information that is most useful for clinical care – where an individual falls along the pathologic disease cascade or when an individual can expect to develop symptoms of dementia.
In contrast, ADAD provides a unique resource for characterizing changes in CSF biomarkers, especially those that occur long before the onset of dementia. With ADAD families, investigators know if and when an individual will develop dementia. Mutations have 100% penetrance, allowing investigators to know with certainty that an individual will develop AD. Furthermore, within a given family, the age of dementia onset remains fairly consistent, allowing researchers to calculate an estimated number of years until symptom onset (EYO). The EYO construct permits evaluation of biomarker concentrations as a function of where along the disease trajectory an individual falls, independent of the actual age of dementia onset of their parent (17). Using ADAD families, studies can examine biomarker levels in MCs and NCs at distinct time points throughout the course of the disease, including the preclinical AD interval many years prior to dementia onset. However, the low prevalence of ADAD has historically created difficulties in evaluating CSF biomarkers in these families. Most early studies analyzed CSF from fewer than 10 MCs (69–71) (Table 1), and with the exception of those evaluating the large Columbia kindred (PSEN-1 E280A) (12, 72), most have pooled together carriers of different mutations. Despite the relatively small sample sizes and potential heterogeneity caused by pooling together individuals with different mutations, the pattern of CSF biomarker changes seen in ADAD MCs is remarkably similar to that observed in LOAD, namely, reduced levels of CSF Aβ42 and elevated levels of tau and ptau (Table 1; Table S1 in Supplementary Material). The one exception is very young MCs (in their 20s, about 25 years prior to AD symptom onset), who have elevated CSF Aβ42 (72). This was hypothesized to reflect over-production of CSF Aβ42 in ADAD MCs, which has more recently been confirmed directly in kinetic studies (73).
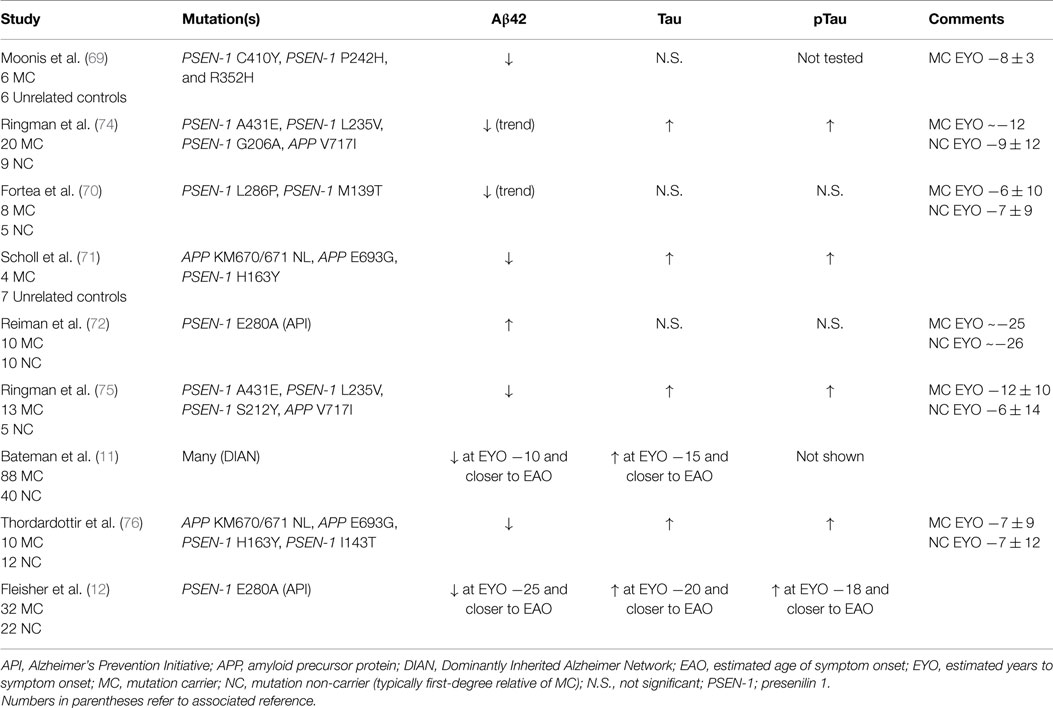
Table 1. Studies examining CSF biomarkers in participants with autosomal dominant Alzheimer disease.
The larger DIAN and API studies have permitted analysis of CSF and imaging biomarkers in greater numbers of both asymptomatic and symptomatic individuals that span a wide range of EYOs, thus allowing conclusions to be drawn regarding the timing of such biomarker changes during the preclinical period (Figure 1). Results from cross-sectional analyses demonstrate higher levels of CSF Aβ42 in MCs compared to NCs very early in the disease process (~20–30 years prior to estimated symptom onset, EYO −20 to −30), which then drop with disease progression, becoming significantly lower than NCs ~10–20 years prior to symptom onset (~EYO −10 to −20) (11, 12, 72, 77). These low levels then begin to plateau with the development of cognitive symptoms. After Aβ42 levels begin to drop, levels of tau and ptau in MCs become significantly higher than NCs (~EYO −15), and then continue to increase with disease progression. However, a recent study of within-person change in biomarkers in a small sub-cohort of DIAN participants with longitudinal biomarker data has shown that although levels of CSF tau and ptau increase in MCs during the preclinical (asymptomatic) phase, levels stabilize or decline over time in individuals who are symptomatic (77). Similar patterns were observed in levels of visinin-like protein 1 (VILIP-1) (77), a neuronal calcium sensor protein that is a marker of neuronal injury/death (78). Consistent with this pattern, a previous report of a single asymptomatic ADAD (APP V717I) MC showed substantial increases in tau and ptau over a 4- to 5-year period very early in the disease process (~EYO −19 to −14) (79), whereas a longitudinal decrease (or a lack of increase) in ptau was reported in a small Japanese cohort (n = 4) of symptomatic PSEN1 MCs (80). Although not often discussed, results consistent with these changes in the trajectories of neuronal injury-related markers have been reported in LOAD (81–83).
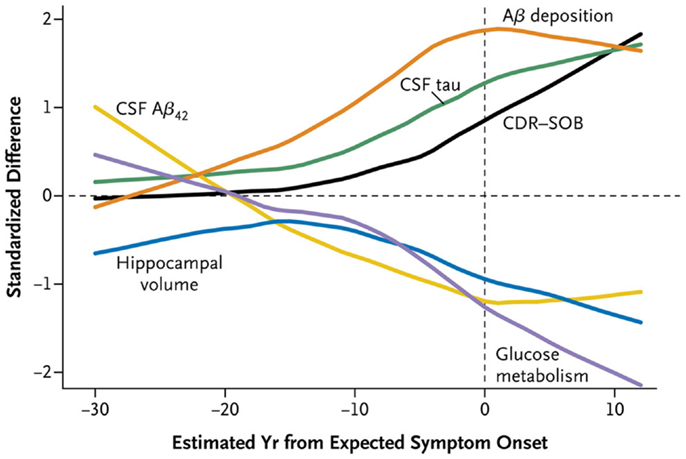
Figure 1. A time course of changes in ADAD mutation carriers versus non-carriers. Cross-sectional data obtained in the DIAN cohort demonstrates that CSF Aβ42 (yellow) declines as Aβ deposition increases as shown by amyloid PET imaging (orange). CSF tau (green) increases as hippocampal volume (blue) and glucose metabolism as shown by FDG PET (purple) decreases. CDR-SOB (Clinical Dementia Rating-Sum of Boxes) (black), which quantifies clinical symptoms of dementia, increases (indicating worse performance) relatively late in the disease course. Reproduced with permission from Bateman et al. (11).
Although this general model is consistent with data obtained from cross-sectional studies in LOAD (49, 84–86) and suggests a common pathophysiology for AD due to mutations and the much more common “sporadic” form, the longitudinal data from DIAN supports a model that incorporates an eventual slowing down of the rate of neuronal injury and death as may be indicated by reductions in these markers. It is also possible that the later decreases during the symptomatic phase may reflect fewer neurons left to contribute to the pool of CSF tau/ptau/VILIP-1. If corroborated in additional cohorts, this reversing pattern of marker change will likely have an impact on the definition of a positive neurodegenerative biomarker outcome in clinical trials, especially during the symptomatic phase. For example, an effective therapy may only slow the rate of increase in injury markers in individuals who are in the preclinical phase, but stabilize or decrease the rate of change in injury markers later in the disease. Confirmation of such patterns awaits evaluation of biomarker trajectories in clinical trials.
Use of ADAD in Clinical Trial Design
Many clinical trials in symptomatic individuals with LOAD have failed to meet their clinical endpoints of delaying, halting, or reversing cognitive decline. One possibility proposed to explain this failure is that therapies must be delivered earlier, in individuals known to have underlying AD pathology, but before significant symptoms are manifest (87). However, there are several challenges associated with the design and implementation of such “prevention trials,” including identifying asymptomatic participants with known underlying AD pathology and who are at a point in their disease trajectory when they are close to becoming symptomatic. Although CSF and imaging biomarkers are currently being used in clinical trials to confirm underlying amyloid pathology in individuals at risk for developing LOAD (http://www.nia.nih.gov/alzheimers/clinical-trials/), the onset of dementia in LOAD is characteristically difficult to predict, even in individuals who are biomarker-positive. As a result, large numbers of participants are required in order to provide adequate statistical power to show a potential drug effect. In contrast, since ADAD is fully penetrant and the time until onset of dementia symptoms in MCs can be predicted with relatively high precision, fewer trial participants are required to demonstrate treatment efficacy within a suitable timeframe. Two such prevention trials in ADAD are currently underway; the DIAN-Trials Unit (DIAN-TU) and API, both of which are testing monoclonal antibodies directed against various forms of Aβ (13, 14).
Another possibility to explain the failure of previous clinical trials in LOAD is that the drug did not engage its purported target. Given the compelling data from observational biomarker studies of ADAD (Table 1), biomarkers can serve as meaningful endpoints to verify target engagement even before the possible appearance of significant cognitive effects. To this end, the DIAN-TU has defined biomarkers as the primary endpoint [amyloid PET or CSF Aβ, with CSF tau(s) as downstream targets], with the trial design transitioning to a cognitive endpoint only for those drugs shown to have properly engaged their pathologic targets (14, 88, 89). CSF biomarkers are also being used as exploratory measures in the API trial (13) and the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s (A4) prevention trial in LOAD (90).
Conclusion
Although there are some differences in the pathology and clinical expression in ADAD compared to LOAD (Table S1 in Supplementary Material), studies of ADAD have provided critical insight that has propelled our knowledge and investigation of all forms of AD. Investigators have proposed the relative timing of biomarker changes in LOAD (48, 49), but these hypotheses cannot yet be empirically verified because we do not know a priori when individuals with LOAD will develop symptoms. Because the EYO is known in ADAD cases, data-based models of AD can be generated (Figure 1) (11, 12). Curves representing changes in CSF and imaging biomarkers over the disease course in ADAD can be superimposed on curves of cognitive function, resulting in a detailed road map of AD pathologic processes. These analyses confirm that AD brain changes begin to develop over two decades before the onset of dementia. Now, as researchers work to develop drugs that prevent dementia associated with AD pathology, they are using ADAD to accelerate clinical trials (13, 14). It would be appropriate if ADAD, which represents <1% of all AD but has provided so much insight into the disease, leads to a drug that ultimately prevents all forms of AD.
Author Contributions
SS and AF were involved in all aspects of preparing, writing, and editing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
SS is supported by a NIH K career development award (5K12 HD001459-15). AF is supported by grants from the NIH (P01AG026276, PO1AG003991, 2UF1AG032438).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fneur.2015.00142
References
1. Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR. An English translation of Alzheimer’s 1907 paper, “uber eine eigenartige Erkankung der hirnrinde”. Clin Anat (1995) 8(6):429–31. doi:10.1002/ca.980080612
2. Chouraki V, Seshadri S. Genetics of Alzheimer’s disease. Adv Genet (2014) 87:245–94. doi:10.1016/B978-0-12-800149-3.00005-6
3. Muller U, Winter P, Graeber MB. A presenilin 1 mutation in the first case of Alzheimer’s disease. Lancet Neurol (2013) 12(2):129–30. doi:10.1016/S1474-4422(12)70307-1
4. Rupp C, Beyreuther K, Maurer K, Kins S. A presenilin 1 mutation in the first case of Alzheimer’s disease: revisited. Alzheimer Dement (2014) 10(6):869–72. doi:10.1016/j.jalz.2014.06.005
5. Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature (1991) 349(6311):704–6. doi:10.1038/349704a0
6. Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature (1991) 353(6347):844–6. doi:10.1038/353844a0
7. Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science (1991) 254(5028):97–9. doi:10.1126/science.1925564
8. Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature (1995) 375(6534):754–60. doi:10.1038/375754a0
9. Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, et al. A familial Alzheimer’s disease locus on chromosome 1. Science (1995) 269(5226):970–3. doi:10.1126/science.7638621
10. Hall AM, Roberson ED. Mouse models of Alzheimer’s disease. Brain Res Bull (2012) 88(1):3–12. doi:10.1016/j.brainresbull.2011.11.017
11. Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med (2012) 367(9):795–804. doi:10.1056/NEJMoa1202753
12. Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gutierrez Gomez M, Langois CM, et al. Associations between biomarkers and age in the presenilin 1 E280a autosomal dominant Alzheimer disease kindred: a cross-sectional study. JAMA Neurol (2015) 72(3):316–24. doi:10.1001/jamaneurol.2014.3314
13. Reiman EM, Langbaum JB, Fleisher AS, Caselli RJ, Chen K, Ayutyanont N, et al. Alzheimer’s prevention initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimer Dis (2011) 26(Suppl 3):321–9. doi:10.3233/JAD-2011-0059
14. Mills SM, Mallmann J, Santacruz AM, Fuqua A, Carril M, Aisen PS, et al. Preclinical trials in autosomal dominant AD: implementation of the DIAN-TU trial. Rev Neurol (2013) 169(10):737–43. doi:10.1016/j.neurol.2013.07.017
15. Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology (2013) 80(19):1778–83. doi:10.1212/WNL.0b013e31828726f5
16. Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, et al. Global prevalence of dementia: a Delphi consensus study. Lancet (2005) 366(9503):2112–7. doi:10.1016/S0140-6736(05)67889-0
17. Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology (2014) 83(3):253–60. doi:10.1212/WNL.0000000000000596
18. Wallon D, Rousseau S, Rovelet-Lecrux A, Quillard-Muraine M, Guyant-Marechal L, Martinaud O, et al. The French series of autosomal dominant early onset Alzheimer’s disease cases: mutation spectrum and cerebrospinal fluid biomarkers. J Alzheimers Dis (2012) 30(4):847–56. doi:10.3233/JAD-2012-120172
19. Lalli MA, Cox HC, Arcila ML, Cadavid L, Moreno S, Garcia G, et al. Origin of the PSEN1 E280A mutation causing early-onset Alzheimer’s disease. Alzheimer Dement (2014) 10(5 Suppl):S277–83. doi:10.1016/j.jalz.2013.09.005
20. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer Dement (2011) 7(3):263–9. doi:10.1016/j.jalz.2011.03.005
21. Balasa M, Gelpi E, Antonell A, Rey MJ, Sanchez-Valle R, Molinuevo JL, et al. Clinical features and APOE genotype of pathologically proven early-onset Alzheimer disease. Neurology (2011) 76(20):1720–5. doi:10.1212/WNL.0b013e31821a44dd
22. Koedam EL, Lauffer V, van der Vlies AE, van der Flier WM, Scheltens P, Pijnenburg YA. Early-versus late-onset Alzheimer’s disease: more than age alone. J Alzheimers Dis (2010) 19(4):1401–8. doi:10.3233/JAD-2010-1337
23. Mendez MF. Early-onset Alzheimer’s disease: nonamnestic subtypes and type 2 AD. Arch Med Res (2012) 43(8):677–85. doi:10.1016/j.arcmed.2012.11.009
24. Joshi A, Ringman JM, Lee AS, Juarez KO, Mendez MF. Comparison of clinical characteristics between familial and non-familial early onset Alzheimer’s disease. J Neurol (2012) 259(10):2182–8. doi:10.1007/s00415-012-6481-y
25. Houlden H, Baker M, McGowan E, Lewis P, Hutton M, Crook R, et al. Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann Neurol (2000) 48(5):806–8. doi:10.1002/1531-8249(200011)48:5<806::AID-ANA18>3.3.CO;2-6
26. Lopera F, Ardilla A, Martinez A, Madrigal L, Arango-Viana JC, Lemere CA, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA (1997) 277(10):793–9. doi:10.1001/jama.277.10.793
27. Natte R, Maat-Schieman ML, Haan J, Bornebroek M, Roos RA, van Duinen SG. Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann Neurol (2001) 50(6):765–72. doi:10.1002/ana.10040
28. Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med (2011) 3(77):77sr1. doi:10.1126/scitranslmed.3002369
29. Mouton PR, Martin LJ, Calhoun ME, Dal Forno G, Price DL. Cognitive decline strongly correlates with cortical atrophy in Alzheimer’s dementia. Neurobiol Aging (1998) 19(5):371–7. doi:10.1016/S0197-4580(98)00080-3
30. Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol (2001) 58(9):1395–402. doi:10.1001/archneur.58.9.1395
31. Heneka MT, Carson MJ, Khoury JE, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol (2015) 14(4):388–405. doi:10.1016/S1474-4422(15)70016-5
32. Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, et al. The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat Med (1996) 2(10):1146–50. doi:10.1038/nm1096-1146
33. Crook R, Verkkoniemi A, Perez-Tur J, Mehta N, Baker M, Houlden H, et al. A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med (1998) 4(4):452–5. doi:10.1038/nm0498-452
34. Shepherd C, McCann H, Halliday GM. Variations in the neuropathology of familial Alzheimer’s disease. Acta Neuropathol (2009) 118(1):37–52. doi:10.1007/s00401-009-0521-4
35. Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun (1984) 122(3):1131–5. doi:10.1016/0006-291X(84)91209-9
36. Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L Jr, Eckman C, et al. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science (1994) 264(5163):1336–40. doi:10.1126/science.8191290
37. Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet (2006) 38(1):24–6. doi:10.1038/ng1718
38. Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, et al. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain (2006) 129(Pt 11):2977–83. doi:10.1093/brain/awl203
39. Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med (1996) 2(8):864–70. doi:10.1038/nm0896-864
40. De Strooper B. Aph-1, Pen-2, and nicastrin with presenilin generate an active gamma-secretase complex. Neuron (2003) 38(1):9–12. doi:10.1016/S0896-6273(03)00205-8
41. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science (2002) 297(5580):353–6. doi:10.1126/science.1072994
42. Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature (2012) 488(7409):96–9. doi:10.1038/nature11283
43. Gomez-Isla T, Price J, McKeel D, Morris J, Growdon J, Hyman B. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neursci (1996) 16:4491–500.
44. Morris J, Price J. Pathologic correlates of nondemented aging, mild cognitive impairment, and early stage Alzheimer’s disease. J Mol Neurosci (2001) 17:101–18. doi:10.1385/JMN:17:2:101
45. Hulette CM, Welsh-Bohmer KA, Murray MG, Saunders AM, Mash DC, McIntyre LM. Neuropathological and neuropsychological changes in “normal” aging: evidence for preclinical Alzheimer disease in cognitively normal individuals. J Neuropathol Exp Neurol (1998) 57:1168–74. doi:10.1097/00005072-199812000-00009
46. Markesbery W, Schmitt F, Kryscio R, Davis D, Smith C, Wekstein D. Neuropathologic substrate of mild cognitive impairment. Arch Neurol (2006) 63:38–46. doi:10.1001/archneur.63.1.38
47. Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging (1997) 18:351–7. doi:10.1016/S0197-4580(97)00056-0
48. Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol (2013) 12(2):207–16. doi:10.1016/S1474-4422(12)70291-0
49. Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol (2010) 9(1):119–28. doi:10.1016/S1474-4422(09)70299-6
50. Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol (2010) 6(3):131–44. doi:10.1038/nrneurol.2010.4
51. Jagust WJ, Landau SM, Shaw LM, Trojanowski JQ, Koeppe RA, Reiman EM, et al. Relationships between biomarkers in aging and dementia. Neurology (2009) 73(15):1193–9. doi:10.1212/WNL.0b013e3181bc010c
52. Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, et al. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol (2009) 66(3):382–9. doi:10.1001/archneurol.2008.596
53. Clark CM, Xie S, Chittams J, Ewbank D, Peskind E, Galasko D, et al. Cerebrospinal fluid tau and beta-amyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol (2003) 60(12):1696–702. doi:10.1001/archneur.60.12.1696
54. Strozyk D, Blennow K, White LR, Launer LJ. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology (2003) 60(4):652–6. doi:10.1212/01.WNL.0000046581.81650.D0
55. Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol (2007) 64(3):343–9. doi:10.1001/archneur.64.3.noc60123
56. Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, et al. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging (2008) 29(10):1456–65. doi:10.1016/j.neurobiolaging.2007.03.029
57. Tolboom N, van der Flier WM, Yaqub M, Boellaard R, Verwey NA, Blankenstein MA, et al. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J Nucl Med (2009) 50(9):1464–70. doi:10.2967/jnumed.109.064360
58. Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol (2006) 59(3):512–9. doi:10.1002/ana.20730
59. Grimmer T, Riemenschneider M, Forstl H, Henriksen G, Klunk WE, Mathis CA, et al. Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry (2009) 65(11):927–34. doi:10.1016/j.biopsych.2009.01.027
60. Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain (2006) 129(Pt 11):3035–41. doi:10.1093/brain/awl269
61. de Souza LC, Chupin M, Lamari F, Jardel C, Leclercq D, Colliot O, et al. CSF tau markers are correlated with hippocampal volume in Alzheimer’s disease. Neurobiol Aging (2012) 33(7):1253–7. doi:10.1016/j.neurobiolaging.2011.02.022
62. Stricker NH, Dodge HH, Dowling NM, Han SD, Erosheva EA, Jagust WJ, et al. CSF biomarker associations with change in hippocampal volume and precuneus thickness: implications for the Alzheimer’s pathological cascade. Brain Imaging Behav (2012) 6(4):599–609. doi:10.1007/s11682-012-9171-6
63. Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol (2009) 65(2):176–83. doi:10.1002/ana.21559
64. Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, et al. CSF biomarkers in prediction of cerebral and clinical change in mild cognitive impairment and Alzheimer’s disease. J Neursci (2010) 30(6):2088–101. doi:10.1523/JNEUROSCI.3785-09.2010
65. Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol (2006) 5(3):228–34. doi:10.1016/S1474-4422(06)70355-6
66. Li G, Sokal I, Quinn JF, Leverenz JB, Brodey M, Schellenberg GD, et al. CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology (2007) 69(7):631–9. doi:10.1212/01.wnl.0000267428.62582.aa
67. Roe CM, Fagan AM, Grant EA, Hassenstab J, Moulder KL, Maue Dreyfus D, et al. Amyloid imaging and CSF biomarkers in predicting cognitive impairment up to 7.5 years later. Neurology (2013) 80(19):1784–91. doi:10.1212/WNL.0b013e3182918ca6
68. Snider BJ, Fagan AM, Roe C, Shah AR, Grant EA, Xiong C, et al. Cerebrospinal fluid biomarkers and rate of cognitive decline in very mild dementia of the Alzheimer type. Arch Neurol (2009) 66(5):638–45. doi:10.1001/archneurol.2009.55
69. Moonis M, Swearer JM, Dayaw MP, St George-Hyslop P, Rogaeva E, Kawarai T, et al. Familial Alzheimer disease: decreases in CSF Abeta42 levels precede cognitive decline. Neurology (2005) 65(2):323–5. doi:10.1212/01.wnl.0000171397.32851.bc
70. Fortea J, Llado A, Bosch B, Antonell A, Oliva R, Molinuevo JL, et al. Cerebrospinal fluid biomarkers in Alzheimer’s disease families with PSEN1 mutations. Neurodegener Dis (2011) 8(4):202–7. doi:10.1159/000322229
71. Scholl M, Wall A, Thordardottir S, Ferreira D, Bogdanovic N, Langstrom B, et al. Low PiB PET retention in presence of pathologic CSF biomarkers in arctic APP mutation carriers. Neurology (2012) 79(3):229–36. doi:10.1212/WNL.0b013e31825fdf18
72. Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol (2012) 11(12):1048–56. doi:10.1016/S1474-4422(12)70228-4
73. Potter R, Patterson BW, Elbert DL, Ovod V, Kasten T, Sigurdson W, et al. Increased in vivo amyloid-beta42 production, exchange, and loss in presenilin mutation carriers. Sci Transl Med (2013) 5(189):189ra77. doi:10.1126/scitranslmed.3005615
74. Ringman JM, Younkin SG, Pratico D, Seltzer W, Cole GM, Geschwind DH, et al. Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology (2008) 71(2):85–92. doi:10.1212/01.wnl.0000303973.71803.81
75. Ringman JM, Coppola G, Elashoff D, Rodriguez-Agudelo Y, Medina LD, Gylys K, et al. Cerebrospinal fluid biomarkers and proximity to diagnosis in preclinical familial Alzheimer’s disease. Dement Geriatr Cogn Disord (2012) 33(1):1–5. doi:10.1159/000335729
76. Thordardottir S, Stahlbom AK, Ferreira D, Almkvist O, Westman E, Zetterberg H, et al. Preclinical cerebrospinal fluid and volumetric magnetic resonance imaging biomarkers in Swedish familial Alzheimer’s disease. J Alzheimer Dis (2015) 43(4):1393–402. doi:10.3233/JAD-140339
77. Fagan AM, Xiong C, Jasielec MS, Bateman RJ, Goate AM, Benzinger TL, et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci Transl Med (2014) 6(226):226ra30. doi:10.1126/scitranslmed.3007901
78. Schnurra I, Bernstein HG, Riederer P, Braunewell KH. The neuronal calcium sensor protein VILIP-1 is associated with amyloid plaques and extracellular tangles in Alzheimer’s disease and promotes cell death and tau phosphorylation in vitro: a link between calcium sensors and Alzheimer’s disease? Neurobiol Dis (2001) 8(5):900–9. doi:10.1006/nbdi.2001.0432
79. Ringman JM, Taylor K, Teng E, Coppola G, Gylys K. Longitudinal change in CSF biomarkers in a presymptomatic carrier of an APP mutation. Neurology (2011) 76(24):2124–5. doi:10.1212/WNL.0b013e31821f4488
80. Ikeda M, Yonemura K, Kakuda S, Tashiro Y, Fujita Y, Takai E, et al. Cerebrospinal fluid levels of phosphorylated tau and Abeta1-38/Abeta1-40/Abeta1-42 in Alzheimer’s disease with PS1 mutations. Amyloid (2013) 20(2):107–12. doi:10.3109/13506129.2013.790810
81. Kanai M, Matsubara E, Isoe K, Urakami K, Nakashima K, Arai H, et al. Longitudinal study of cerebrospinal fluid levels of tau, Ab1-40, and Ab1-42(43) in Alzheimer’s disease: a study in Japan. Ann Neurol (1998) 44:17–26. doi:10.1002/ana.410440108
82. Sunderland T, Wolozin B, Galasko D, Levy J, Dukoff R, Bahro M, et al. Longitudinal stability of CSF tau levels in Alzheimer patients. Biol Psychiatry (1999) 46(6):750–5. doi:10.1016/S0006-3223(99)00143-2
83. Tapiola T, Pirttila T, Mikkonen M, Mehta PD, Alafuzoff I, Koivisto K, et al. Three-year follow-up of cerebrospinal fluid tau, beta-amyloid 42 and 40 concentrations in Alzheimer’s disease. Neurosci Lett (2000) 280(2):119–22. doi:10.1016/S0304-3940(00)00767-9
84. Perrin R, Fagan A, Holtzman D. Multi-modal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature (2009) 461:916–22. doi:10.1016/j.neurobiolaging.2008.08.013
85. Jack C, Vemuri P, Wiste H, Weigand S, Aisen P, Trojanowski J, et al. Evidence for ordering of Alzheimer disease biomarkers. Arch Neurol (2011) 68:1526–35. doi:10.1001/archneurol.2011.183
86. Sperling R, Aisen P, Beckett L, Bennett D, Craft S, Fagan A, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer Dement (2011) 7:280–92. doi:10.1016/j.jalz.2011.03.003
87. Kozauer N, Katz R. Regulatory innovation and drug development for early-stage Alzheimer’s disease. N Engl J Med (2013) 368(13):1169–71. doi:10.1056/NEJMp1302513
88. Bateman RJ, Aisen PS, De Strooper B, Fox NC, Lemere CA, Ringman JM, et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res Ther (2011) 3(1):1. doi:10.1186/alzrt59
89. Moulder KL, Snider BJ, Mills SL, Buckles VD, Santacruz AM, Bateman RJ, et al. Dominantly inherited Alzheimer network: facilitating research and clinical trials. Alzheimers Res Ther (2013) 5(5):48. doi:10.1186/alzrt213
Keywords: cerebrospinal fluid, biomarkers, Alzheimer disease, autosomal dominant, familial
Citation: Schindler SE and Fagan AM (2015) Autosomal dominant Alzheimer disease: a unique resource to study CSF biomarker changes in preclinical AD. Front. Neurol. 6:142. doi: 10.3389/fneur.2015.00142
Received: 24 April 2015; Accepted: 12 June 2015;
Published: 29 June 2015
Edited by:
Sylvain Lehmann, Montpellier University Hospital, FranceCopyright: © 2015 Schindler and Fagan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne M. Fagan, Department of Neurology, Washington University School of Medicine, Campus Box 8111, 660 South Euclid Avenue, St. Louis, MO 63110, USA,ZmFnYW5hQG5ldXJvLnd1c3RsLmVkdQ==