 Sverre E. Aune
Sverre E. Aune Daniel J. Herr
Daniel J. Herr Donald R. Menick
Donald R. Menick- Gazes Cardiac Research Institute, Medical University of South Carolina, Charleston, SC, USA
Ischemia-reperfusion (IR) injury comprises a significant portion of morbidity and mortality from heart and brain diseases worldwide. This enduring clinical problem has inspired myriad reports in the scientific literature of experimental interventions seeking to elucidate the pathology of IR injury. Elective cardiac surgery presents perhaps the most viable scenario for protecting the heart and brain from IR injury due to the opportunity to condition the organs prior to insult. The physiological parameters for the preconditioning of vital organs prior to insult through mechanical and pharmacological maneuvers have been heavily examined. These investigations have revealed new insights into how preconditioning alters cellular responses to IR injury. However, the promise of preconditioning remains unfulfilled at the clinical level, and research seeking to implicate cell signals essential to this protection continues. Recent discoveries in molecular biology have revealed that gene expression can be controlled through posttranslational modifications, without altering the chemical structure of the genetic code. In this scenario, gene expression is repressed by enzymes that cause chromatin compaction through catalytic removal of acetyl moieties from lysine residues on histones. These enzymes, called histone deacetylases (HDACs), can be inhibited pharmacologically, leading to the de-repression of protective genes. The discovery that HDACs can also alter the function of non-histone proteins through posttranslational deacetylation has expanded the potential impact of HDAC inhibitors for the treatment of human disease. HDAC inhibitors have been applied in a very small number of experimental models of IR. However, the scientific literature contains an increasing number of reports demonstrating that HDACs converge on preconditioning signals in the cell. This review will describe the influence of HDACs on major preconditioning signaling pathways in the heart and brain.
Introduction
Worldwide, 33 million people suffer a stroke each year (1). Ischemia, which can occur in all tissues, is defined as the stress that a tissue experiences when both oxygen and substrate are reduced (2). Stroke, defined as insufficient blood flow to the brain, is one major type of cerebral ischemia and may cause transient to permanent loss of brain function or death (3). In a subset of patients (<15%), stroke occurs as a result of cardiac arrest and cardiac surgery; secondary stroke is, by definition, the result of embolic events originating in the heart (4). In spite of intense research effort directed toward reducing the impact of these conditions, world rates of morbidity and mortality as a result of brain and heart disease continue to rise, most strikingly in developing countries (5). The lack of broadly effective treatment continues to fuel the search for new molecular targets in ischemia-reperfusion injury (IRI). Intriguingly, studies of molecular pathology in the brain and heart are often informed by cancer research. Recent clinical advances have revealed the efficacy of using small molecule inhibitors of histone deacetylases (HDACs) to target malignancy (6). HDACs are enzymes which control signal transduction and gene expression in all cell types (7). Here we review the experimental evidence in support of applying HDAC inhibitors in settings of cerebral and cardiac ischemia, with emphasis on the roles that HDACs play in signaling events that occur as a result of IRI. Importantly, this nascent experimental work indicates that HDAC inhibitors show great promise for treating patients at high risk for stroke or cardiac arrest and for patients electing to receive brain and heart surgeries.
Endogenous tolerance to ischemia can be evoked in the heart and brain (4, 8). Classical ischemic conditioning in a tissue requires the mechanical application of several momentary, sub-lethal reductions in oxygen and substrate delivery, which reduce the injury caused by a more severe ischemic insult (9). Organs subjected to classical ischemic conditioning experience only transient stimulation of endogenous protective mechanisms, which limits application of the conditioning stimulus to shortly before prolonged insult, as in preconditioning a patient of elective surgery, or to shortly afterward, as in postconditioning a patient of out-of-hospital stroke or cardiac arrest. The idea that similar magnitudes of endogenous protection can be evoked pharmacologically is a considerable advancement over the barrier of time-dependence instituted by mechanical ischemic conditioning (10). It follows that drug-evoked conditioning is a promising treatment strategy for patients undergoing elective brain or heart surgery, or those at high risk of stroke or cardiac arrest (11).
Prevention of cell death is the primary effect of ischemic conditioning (12, 13). Many forms of ischemic conditioning (including classical, pharmacological, and remote conditioning) converge on signaling pathways that either inhibit cell death or activate endogenous cell survival maintenance programs. Once active, cell survival programs improve endoplasmic reticulum stabilization (14), increase expression of antioxidant enzymes (SOD2; catalase) (15), inhibit endogenous cell death programs (Bax/Bad/Bcl2; c-jun) (16), activate transcription of genes for repair enzymes (Hsp70; HIF-1-alpha) (17, 18), and stimulate autophagic flux (HDAC6) (19).
Acute adaptation to ischemia through conditioning stimuli is mediated by protein posttranslational modifications (PTMs) (20, 21). Historically, protein phosphorylation has been the most rigorously characterized of the PTMs in settings of experimental ischemic conditioning. These include the reperfusion injury salvage kinases (RISK) pathways of Raf/MEK/ERK1/2 and PI3K/Akt/eNOS, the JAK/STAT transcriptional pathways and the calcium-responsive PKC pathways (22–25). However, accumulating evidence shows that protein acetylation also plays major roles in regulating cell survival through ischemic conditioning (7).
Histone Deacetylases Regulate Cell Fate in Cerebral Ischemia
Histone deacetylases are a class of epigenetic enzymes that have come under recent intense scrutiny as pharmacological targets for patients suffering stroke. HDACs remove acetyl moieties from ε-amino groups of lysine residues on histones and non-histone proteins (25). Deacetylation of histones enhances chromatin compaction, which renders DNA less available for binding by regulatory factors leading to repression of gene expression (26, 27). In this function, HDACs exert classical transcriptional control. This process is reversible via enzymatic histone acetyltransferase (HAT) activity. The zinc-dependent HDACs have been divided into classes based on homology to yeast transcriptional repressors. Class I comprises HDACs 1, 2, 3, and 8; class IIa comprises 4, 5, 7, and 9; class IIb comprises 6 and 10; and class IV comprises HDAC11 (7). Class III, called sirtiuns, are NAD+-dependent and will not be discussed in this review, but have been expertly reviewed elsewhere (28). HDAC enzymes are widely expressed in rodent brains, and are localized to specific cellular compartments in isoform-specific patterns (29, 30). Class I HDACs are generally restricted to the nucleus where they impose transcriptional control, whereas class IIa HDACs transit the nuclear membranes and enter the cytoplasm in a process mediated by phosphorylation (31). Furthermore, HDAC6 is primarily, though not constitutively, cytoplasmic (19).
As mentioned above, HDACs deacetylate both histone and non-histone proteins. When HDACs deacetylate histones and repress gene transcription, cell survival is impacted after several hours and days on time scales necessary for protein expression (7). Non-histone deacetylation is another element of this complex code of enzymatic crosstalk, which is distinct from direct inhibition of gene expression by histone deacetylation. Importantly, the acetylation state of a given metabolic signaling factor may mediate its phosphorylation, methylation, and ubiquitination state, thereby determining its subcellular location, activation, or degradation, with immediate implications for cell survival in the seconds to minutes following the insult (32). The acetylation state of transcription factors, co-activators, and co-repressors can regulate their activity (33). Decoding the complex patterns of HDAC enzymatic crosstalk will enhance our understanding of histone and non-histone protein lysine deacetylation and its impact on the survival of cells under ischemic stress (34).
Importantly, experimental cerebral ischemia causes upregulation of class I/IIb HDAC expression, which possibly implicates them in ischemic pathology (29). For example, HDAC1 must exist in complex with HDAC3 to promote apoptosis in cerebellar granule neurons; the toxic effects of HDAC1:HDAC3 association were mitigated by activation of PI3K/Akt signaling (35). Cortical neurons transfected with HDAC3 or HDAC6 shRNA each exhibited decreased apoptosis when exposed to prolonged oxygen–glucose deprivation (30). Furthermore, HDAC2 mutant mice exhibited reduced retinal degeneration following ischemia (36). In response to these findings and many others, a variety of pharmacological HDAC inhibitors have been developed and applied in animal models of cerebral ischemia. Results from these studies have revealed that HDAC inhibitors can pharmacologically condition the neuron against ischemic injury through de-repression of transcription (37, 38). While these studies suggest that pharmacological inhibition of class I/IIb HDAC isoforms (so-called “pan-inhibitors”) promote survival of stressed neurons, the evidence reviewed below indicates that the catalytically inactive IIa isoforms may be beneficial to cell survival from IRI.
Small Molecule HDAC Inhibitors Condition the Brain Against Ischemic Injury
Reduction in infarct volume and prevention of apoptosis are the most obvious physiological effects of class I/IIb HDAC inhibitors in experimental cerebral ischemia (see Table 1). The molecular mediators are numerous but consensus is forming around certain cellular processes: modulation of apoptotic intermediates (caspases, Bcl-2), stabilization of the cellular stress response (Hsp-70, EIF-2α, CHOP), transcription of oxygen-responsive enzymes (HIF-1α, Nrf2), regulation of calcium handling (BDNF, CREB), and activation of survival kinase cascades (Akt, ERK, AMPK, p21).
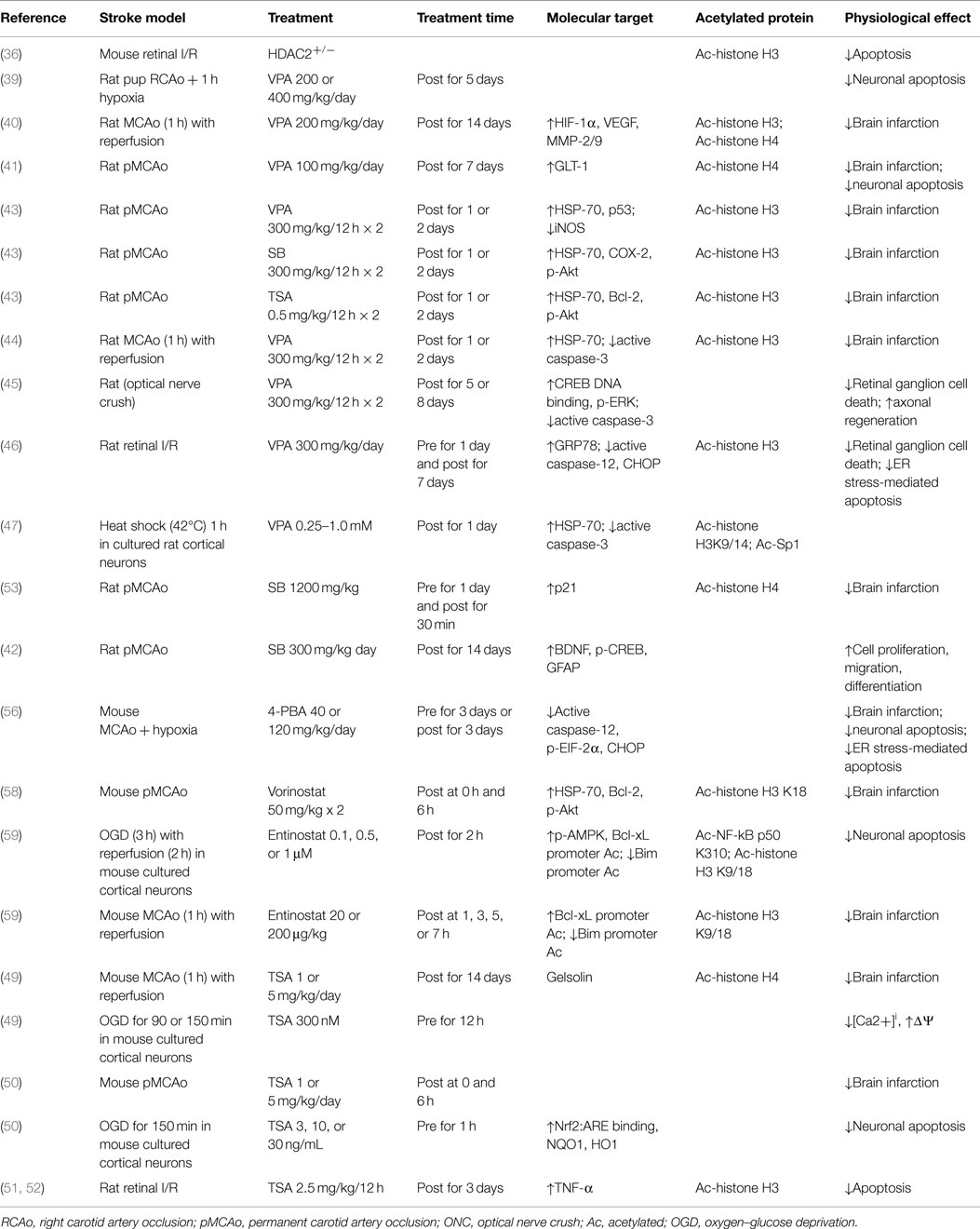
Table 1. Physiological effects of HDAC inhibitors in experimental models of stroke.
Administration of the mood stabilizer and weak HDAC inhibitor valproic acid (VPA) after permanent right carotid artery occlusion prevented neuronal apoptosis in a dose-dependent manner in neonatal rats (39). Rats treated with VPA during middle cerebral artery occlusion (MCAo) exhibited reduced infarct volume, enhanced angiogenesis (40), neurogenesis (41, 42), and reduction of monocyte infiltration (43). These effects were correlated with increased transcription of Hsp70 (43, 44), HIF1-alpha and MMP-2/9 (40), or GLT-1, a transporter protein, which accelerates clearance of glutamate in damaged gray and white matter (41). VPA protected retinal ganglion cells from ischemia-reperfusion (IR) through reduction of mitochondria-mediated apoptosis (45) and endoplasmic reticulum stress-induced apoptosis (46), and enhanced Hsp70 promoter acetylation in cortical neurons through inhibition of a class I HDAC (47). The potent hydroxamate trichostatin A (TSA) decreased infarct volume in rodents given MCAo, which depended on induction of Hsp70, Bcl-2, and Akt phosphorylation, or on gelsolin, an essential regulator of actin homeostasis (43, 48, 49). Mice treated with TSA at the onset of permanent MCAo exhibited reduced infarct volume through increased Nrf2-dependent transcription of antioxidant enzymes (50). TSA also reduced transcription of inflammatory proteins MMP-1 and MMP-3, and reduced caspase-3 activation up to 24 h after the onset of ischemia in the retina (51, 52). Sodium butyrate (a potent analog of VPA) treatment before permanent MCAo evoked a 50% reduction in infarct volume through increased expression of p21, a cyclin-dependent kinase inhibitor, which prevents pro-apoptotic gene transcription (53–55). Furthermore, mice treated with 4-phenylbutyrate before or after ischemia–hypoxia exhibited reduced ER-stress-mediated apoptosis through reduction of EIF2-alpha phosphorylation (56). Mice treated with the potent hydroxamate Vorinostat (FDA approved for treatment of T-cell lymphoma) at the onset of MCAo exhibited reduced infarct size and increased transcription of Hsp70 and Bcl2 (57, 58). Furthermore, neurons exposed to oxygen–glucose deprivation in vitro and mice subjected to MCAo in vivo exhibited increased acetylation at the Bcl-xL promoter when treated with Entinostat, a class I selective HDAC inhibitor; the effect was mediated by enhanced NF-kB p50 acetylation and decreased activation of the Bim promoter (59).
While class I HDACs seem to play pathological roles in cerebral ischemia, there is evidence that class IIa HDACs are required for cell survival following neuronal stress. Genetic heterogeneity surrounding the HDAC9 gene is associated with large vessel ischemic stroke (60). By directly inhibiting the c-jun promoter, HDAC4 (61) and HDAC7 prevented neuronal cell death induced by low potassium (62). HDAC4 is required for the normal development of retinal neurons through the stabilization of HIF-1-alpha (63). HDAC4 and HDAC5 knock-in protected neuron-like pheochromocytoma cells from apoptosis induced by OGD, which was partly dependent on HMGB1 activity (64). Conversely, nuclear export of HDAC5 was required for regeneration after acute axonal injury, a condition that promotes rapid influx of calcium (65). In fact, nuclear calcium levels regulate the association of class IIa HDACs with a MEF2-SMRT corepressor complex (66–68). Given this, it is possible that class IIa HDACs may correct calcium-induced pathological gene expression in neuronal ischemia.
HDAC Enzymatic Crosstalk in Cerebral Ischemia
Evidence is accumulating that HDAC signal transduction pathways communicate in crosstalk with kinase signal cascades in cerebral ischemia. The ability of HDAC inhibitors to condition the neuron in the seconds to minutes following acute ischemic stress may be dependent on the concurrent activity of certain cell survival kinases. As mentioned above, TSA prevented oxidative cell death in cortical neurons through increased transcription of p21, which inactivates pro-apoptotic c-jun transcription by inhibiting the kinase ASK-1 (53–55). HDAC3 was phosphorylated by GSK3-beta and was required for cell death induced by low potassium in cultured cortical neurons; neuronal death was prevented by pharmacological inhibition of GSK3-beta, and with constitutively active Akt, a known inhibitor of GSK3-beta (69). Conversely, the class IIa HDAC4 protects neurons from cell death induced by low potassium by direct inhibition of cyclin-dependent kinase-1 activity, independent of PI3K/AKT, c-jun, or RAF/MEK/ERK signaling (61).
PI3K and AKT activities are both required for the neuronal conditioning achieved with VPA (47). Interestingly, induction of Hsp70 by VPA and other Class I HDAC inhibitors resulted in increased histone methylation in primary neurons and astrocytes (70). In particular, as confirmed by chromatin immunoprecipitation, HDAC inhibition caused increased methylation at the Hsp70 promoter, a histone landscape favoring transcriptional activation. This suggests an intricate interplay between histone acetylation and histone methylation. In fact, this phenomenon of functional and structural cooperation between HDACs and lysine-specific demethylase (LSD) enzymes is well established, as in the multifaceted corepressor CoREST/REST/HDAC/LSD complex (71). However, complex crosstalk between lysine “readers” (enzymes that recruit PTM enzymes to acetyl-lysine residues) and “writers” (enzymes that catalyze acetylation of lysine residues) results in combinations of histone modifications that form a hierarchal landscape, which dictates the transition between silencing and activation of a certain transcription domain (72). Clearly, HDAC enzymatic crosstalk with other PTM enzymes occurs on both histones and non-histone proteins.
HDAC Inhibitors Mitigate Cardiac Infarction Following IRI
Histone deacetylase inhibitors have also shown potential in mitigating cardiac IRI (73). Importantly, HDAC activity is also upregulated in hearts after IR. Mice treated with TSA following in vivo IRI exhibited marked reduction of infarct area which correlated with stabilization of HIF-1a. This effect was abrogated in HDAC4 knockout cardiomycytes, in another example of the putative protective nature of a class IIa HDAC (74).
Multiple kinase pathways have been implicated in promoting myocyte survival in response to ischemic injury, including p38 MAPK (75–77), the RISK PI3K/AKT/eNOS (78–83) and RAF/MEK/ERK1/2 (84), and the survivor activating factor enhancement pathway (SAFE) (85, 86). Evidence for enzymatic crosstalk between HDACs and these pathways is growing. The cardioprotective action of HDAC inhibitors in IRI is also apparently dependent on the gp-91 subunit of NADPH oxidase (87), p38 (88), Akt1, and Mkk3 (89, 90). The transcription factor NF-kB has been suggested as a common target of multiple types of preconditioning stimuli (91). Interestingly, NF-kB is a common target of the p38 MAPK (92), Akt (93), and Erk1/2 (94) signaling pathways, and is required for p38-mediated adaptation to mechanical preconditioning in isolated hearts (92). TSA-induced pharmacological preconditioning was recently correlated with nuclear translocation, activation, and hyperacetylation of NF-kB, while specific deletion of NF-kB p50 abrogated the TSA protective effect (95). The effects of HDAC inhibition on ERK1/2 signaling have not been directly assessed in a model of IR. Intriguingly, however, HDAC inhibition prevents cell death in multiple cell lines by activating ERK1/2, while this effect is not present in several cancer cell lines (96). The absence of HDAC-mediated ERK1/2 activation in cancer cells is likely the result of suppressed HDAC1 activity (97).
Recent work in mouse tissues demonstrated that the endogenous HDAC1 inhibitor d-β-hydroxybutarate (β-OHB) caused increased acetylation at the promoter of the FOXO3a transcription factor. This led to upregulation of FOXO3a and its targets SOD2 and catalase, free radical scavengers, which further prevented paraquat-induced renal oxidative stress in mouse kidneys (98–100). Additionally, we recently reported preserved cardiac contractile function and reduced infarction following IR by pre-treating rats with Entinostat (101). This was associated with dramatic nuclear FOXO3a enrichment, along with increased transcript and protein levels of SOD2 and catalase. Nuclear enrichment of FOXO3a was likely due to decreased Akt-mediated phosphorylation at the key nuclear exclusion site S318/321 (102), suggesting that class I HDAC inhibition also influences the nuclear trafficking of transcription factors which upregulate these enzymes. HDAC4/5 are excluded from the nucleus by phosphorylation by AMPK, but localize to the nucleus in situations of low glucose, where they recruit HDAC3. Intriguingly, in this case, HDAC3 deacetylates and activates FOXO-mediated transcription of anti-apoptotic genes (103).
Conclusion
Histone deacetylases evidently form signaling hubs for cellular communication in cerebral and cardiac IRI. Class I HDACs appear to play mainly pathologic roles in IRI, by repressing transcription of genes required for cell survival, while Class IIa HDACs appear necessary for cell survival. The roles of the class IIb HDAC6 in preconditioning are not full understood, though HDAC6 is a major regulator of autophagic flux in neurodegenerative diseases (104) and a contributor to pathological responses in the heart (105). Recent studies demonstrate that the modulation of endogenous antioxidant transcription is a significant mechanism by which the inhibition of HDACs confers preconditioning protection against IRI. Neurons and cardiomyocytes may share epigenetic signaling mechanisms for activation of endogenous protection from ischemia by HDAC inhibitors.
Lines of evidence have begun to accumulate that support a role for communication between HDAC and kinase signaling networks in ischemia of the heart–brain axis. While the mechanisms behind the protective preconditioning effect of HDAC inhibition remain to be fully elucidated, it is evident that the RISK pathway and related kinases are integral components. More research into the details of the specific interactions between HDACs and other PTMs will advance our understanding of the role of HDAC inhibition in ischemic preconditioning. Dissecting the dual roles of HDACs as transcriptional repressors and as effectors of enzymatic crosstalk is needed to dissect the chronic and acute phases of preconditioning protection. Given that several small molecule inhibitors of HDAC activity are currently used in patients or in clinical trials, HDAC inhibitors represent promising treatment modalities for patients undergoing elective brain or heart surgery, or patients at high risk of stroke or cardiac arrest.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This publication was supported by the South Carolina Clinical and Translational Research (SCTR) Institute, with an academic home at the Medical University of South Carolina, NIH/NCATS Grant Number UL1 TR000062. SA was supported by NIH Grant Number T32 HL07260. DH was supported by NIH/NCATS Grant Number TL1 TR000061 and by NIH Grant Number T32 GM008716. Further support was provided by VA merit award BX002327-01 to DM.
References
1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics-2015 update. Circulation (2014) 131:e29–322. doi:10.1161/cir.0000000000000152
2. Schweizer S, Meisel A, Marschenz S. Epigenetic mechanisms in cerebral ischemia. J Cereb Blood Flow Metab (2013) 33:1335–46. doi:10.1038/jcbfm.2013.93
3. Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol (2009) 8(4):398–412. doi:10.1016/S1474-4422(09)70054-7
4. Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci (2006) 7(6):437–48. doi:10.1038/nrn1927
5. Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJL. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet (2006) 367(9524):1747–57. doi:10.1016/S0140-6736(06)68770-9
6. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest (2014) 124(1):30–9. doi:10.1172/JCI69738
7. Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene (2007) 26:5310–8. doi:10.1038/sj.onc.1210599
8. Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, et al. “Ischemic tolerance” phenomenon found in the brain. Brain Res (1990) 528(1):21–4. doi:10.1016/0006-8993(90)90189-I
9. Dezfulian C, Garrett M, Gonzalez NR. Clinical application of preconditioning and postconditioning to achieve neuroprotection. Transl. Stroke Res (2013) 4(1):19–24. doi:10.1007/s12975-012-0224-3
10. Moskowitz MA, Lo EH, Iadecola C. The science of stroke-mechanisms in search of treatments. Neuron (2010) 67(2):181–98. doi:10.1016/j.neuron.2010.07.002
11. Ginsberg MD. Neuroprotection for Ischemic stroke: past, present and future. Neuropharmacology (2008) 55(3):363–89. doi:10.1016/j.neuropharm.2007.12.007
12. Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci (2003) 26(5):248–54. doi:10.1016/S0166-2236(03)00071-7
13. Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis (2009) 14(4):469–77. doi:10.1007/s10495-008-0304-8
14. Bodalia A, Li H, Jackson MF. Loss of endoplasmic reticulum Ca2+ homeostasis: contribution to neuronal cell death during cerebral ischemia. Acta Pharmacol Sin (2013) 34(1):49–59. doi:10.1038/aps.2012.139
15. Sugawara T, Noshita N, Lewén A, Gasche Y, Ferrand-Drake M, Fujimura M, et al. Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci (2002) 22(1):209–17.
16. Gao Y, Signore AP, Yin W, Cao G, Yin X-M, Sun F, et al. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab (2005) 25(6):694–712. doi:10.1038/sj.jcbfm.9600062
17. Sharp FR, Kinouchi H, Koistinaho J, Chan PH, Sagar SM. HSP70 heat shock gene regulation during ischemia. Stroke (1993) 24(12 Suppl):I72–5.
18. Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci (2007) 27(23):6320–32. doi:10.1523/JNEUROSCI.0449-07.2007
19. Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao T-P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell (2003) 115(6):727–38. doi:10.1016/S0092-8674(03)00939-5
20. Porter K, Medford HM, McIntosh CM, Marsh SA. Cardioprotection requires flipping the “posttranslational modification” switch. Life Sci (2012) 90(3–4):89–98. doi:10.1016/j.lfs.2011.10.026
21. Hausenloy DJ, Yellon DM. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res (2006) 70:240–53. doi:10.1016/j.cardiores.2006.01.017
22. Hashiguchi A, Yano S, Morioka M, Hamada J, Ushio Y, Takeuchi Y, et al. Up-regulation of endothelial nitric oxide synthase via phosphatidylinositol 3-kinase pathway contributes to ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab (2004) 24(3):271–9. doi:10.1097/01.WCB.0000110539.96047.FC
23. Zhao H. Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J Cereb Blood Flow Metab (2009) 29(5):873–85. doi:10.1038/jcbfm.2009.13
24. Lai EW, Toledo-Pereyra LH, Walsh J, Lopez-Neblina F, Anaya-Prado R. The role of MAP kinases in trauma and ischemia-reperfusion. J Invest Surg (2004) 17(1):45–53. doi:10.1080/08941930490269646
25. Gershey EL, Vidali G, Allfrey VG. Chemical studies of histone acetylation. the occurrence of epsilon-N-acetyllysine in the f2a1 histone. J Biol Chem (1968) 243(19):5018–22.
26. Rundlett SE, Carmen AA, Kobayashi R, Bavykin S, Turner BM, Grunstein M. HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci U S A (1996) 93(25):14503–8. doi:10.1073/pnas.93.25.14503
27. Taunton J, Hassiq CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science (1996) 272(5260):408–11. doi:10.1126/science.272.5260.408
28. Zhang F, Wang S, Gan L, Vosler PS, Gao Y, Zigmond MJ, et al. Protective effects and mechanisms of sirtuins in the nervous system. Prog Neurobiol (2011) 95(3):373–95. doi:10.1016/j.pneurobio.2011.09.001
29. Baltan S, Bachleda A, Morrison RS, Murphy SP. Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. Transl Stroke Res (2011) 2(3):411–23. doi:10.1007/s12975-011-0087-z
30. Chen YT, Zang XF, Pan J, Zhu XL, Chen F, Chen ZB, et al. Expression patterns of histone deacetylases in experimental stroke and potential targets for neuroprotection. Clin Exp Pharmacol Physiol (2012) 39(9):751–8. doi:10.1111/j.1440-1681.2012.05729.x
31. Yang X-J, Grégoire S. Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol Cell Biol (2005) 25(8):2873–84. doi:10.1128/MCB.25.8.2873-2884.2005
32. Yang XJ, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell (2008) 31(4):449–61. doi:10.1016/j.molcel.2008.07.002
33. Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene (2005) 363:15–23. doi:10.1016/j.gene.2005.09.010
34. Thompson JW, Dave KR, Young JI, Perez-Pinzon MA. Ischemic preconditioning alters the epigenetic profile of the brain from ischemic intolerance to ischemic tolerance. Neurotherapeutics (2013) 10(4):789–97. doi:10.1007/s13311-013-0202-9
35. Bardai FH, Price V, Zaayman M, Wang L, D’Mello SR. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J Biol Chem (2012) 287(42):35444–53. doi:10.1074/jbc.M112.394544
36. Fan J, Alsarraf O, Dahrouj M, Platt KA, Chou CJ, Rice CJ, et al. Inhibition of HDAC2 protects the retina from ischemic injury. Invest Ophthalmol Vis Sci (2013) 54(6):4072–80. doi:10.1167/iovs.12-11529
37. Gibson CL, Murphy SP. Benefits of histone deacetylase inhibitors for acute brain injury-a systematic review of animal studies. J Neurochem (2010) 115(4):806–13. doi:10.1111/j.1471-4159.2010.06993.x
38. Langley B, Brochier C, Rivieccio MA. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke (2009) 40:2899–905. doi:10.1161/STROKEAHA.108.540229
39. Kabukas N, Ay I, Aysun S, Söylemezoglu F, Ozcan A, Celasun B. Protective effects of valproic acid against hypoxic-ischemic brain injury in neonatal rats. J Child Neurol (2005) 20(7):582–7. doi:10.1177/08830738050200070801
40. Wang Z, Tsai LK, Munasinghe J, Leng Y, Fessler EB, Chibane F, et al. Chronic valproate treatment enhances post-ischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke (2012) 43(9):2430–6. doi:10.1161/STROKEAHA.112.652545
41. Liu XS, Chopp M, Kassis H, Jia LF, Hozeska-Solgot A, Zhang RL, et al. Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience (2012) 220:313–21. doi:10.1016/j.neuroscience.2012.06.012
42. Kim HJ, Leeds P, Chuang MW. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J Neurochem (2009) 110(4):1226–40. doi:10.1111/j.1471-4159.2009.06212.x
43. Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke-multiple mechanisms of action. J Pharmacol Exp Ther (2007) 321(3):892–901. doi:10.1124/jpet.107.120188
44. Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem (2004) 89(6):1358–67. doi:10.1111/j.1471-4159.2004.02406.x
45. Biermann J, Grieshaber P, Goebel U, Martin G, Thanos S, Di Giovanni S, et al. Valproic-acid mediated neuroprotection and regeneration in injured retinal ganglion cells. Invest Opthalmol Vis Sci (2010) 51(1):526–34. doi:10.1167/iovs.09-3903
46. Zhang Z, Tong N, Gong Y, Qiu Q, Yin L, Lv X, et al. Valproate protects the retina from endoplasmic reticulum stress-induced apoptosis after ischemia-reperfusion injury. Neurosci Lett (2011) 504(2):88–92. doi:10.1016/j.neulet.2011.09.003
47. Marinova Z, Ren M, Wendland JR, Leng Y, Liang MH, Yasuda S, et al. Valproic acid induces functional heat-shock protein 70 via class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J Neurochem (2009) 111(4):976–87. doi:10.1111/j.1471-4159.2009.06385.x
48. Endres M, Meisel A, Biniszkiewicz D, Namura S, Prass K, Ruscher K, et al. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci (2000) 20(9):3175–81.
49. Yildirim F, Gertz K, Kronenberg G, Harms C, Fink KB, Meisel A, et al. Inhibition of histone deacetylation protects wildtype but not gelsolin-deficient mice from ischemic brain injury. Exp Neurol (2008) 210(2):531–42. doi:10.1016/j.expneurol.2007.11.031
50. Wang B, Zhu X, Kim Y, Li J, Huang S, Saleem S, et al. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med (2012) 52(5):928–36. doi:10.1016/j.freeradbiomed.2011.12.006
51. Crosson CE, Mani SK, Husain S, Alsarraf O, Menick DR. Inhibition of histone deacetylase protects the retina from ischemic injury. Invest Ophthalmol Vis Sci (2010) 51(7):3639–45. doi:10.1167/iovs.09-4538
52. Alsarraf O, Fan J, Dahrouj M, Chou CJ, Menick DR, Crosson CE. Acetylation: a lysine modification with neuroprotective effects in ischemic retinal degeneration. Exp Eye Res (2014) 127:124–31. doi:10.1016/j.exer.2014.07.012
53. Langley B, D’Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, et al. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-dependent neuroprotection. J Neurosci (2008) 28(1):163–76. doi:10.1523/JNEUROSCI.3200-07.2008
54. Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, et al. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci U S A (2000) 97(1):436–41. doi:10.1073/pnas.97.1.436
55. Shim J, Lee H, Park J, Kim H, Choi EJ. A non-enzymatic p21 protein inhibitor of stress-activated protein kinases. Nature (1996) 381(6585):804–6. doi:10.1038/381804a0
56. Qi X, Hosoi T, Okuma Y, Kaneko M, Nomura Y. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol (2004) 66:899–908. doi:10.1124/mol.104.001339
57. Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol (2007) 25(1):84–90. doi:10.1038/nbt1272
58. Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, et al. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol (2006) 70(6):1876–84. doi:10.1124/mol.106.027912
59. Lanzillota A, Pignataro G, Branca C, Cuomo O, Sarnico I, Benarese M, et al. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol Dis (2013) 49:177–89. doi:10.1016/j.nbd.2012.08.018
60. Bellenguez C, Bevan S, Gschwendtner A, Spence CCA, Burgess AI, Pirinen M, et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet (2012) 44(3):328–33. doi:10.1038/ng.1081
61. Majdzadeh N, Wang L, Morrison BE, Bassel-Duby R, Olson EN, D’Mello SR. HDAC4 inhibits cell cycle progression and protects neurons from cell death. Dev Neurobiol (2008) 68(8):1076–92. doi:10.1002/dneu.20637
62. Ma C, D’Mello SR. Neuroprotection by histone deacetylase-7 (HDAC7) occurs by inhibition of c-jun expression through a deacetylase-independent mechanism. J Biol Chem (2011) 286(6):4819–28. doi:10.1074/jbc.M110.146860
63. Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science (2009) 323(5911):256–9. doi:10.1126/science.1166226
64. He M, Zhang B, Wei X, Wang Z, Fan B, Du P, et al. HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J Cell Mol Med (2013) 17(4):531–42. doi:10.1111/jcmm.12040
65. Cho Y, Sloutsky R, Naegle KM, Cavalli V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell (2013) 155(4):894–908. doi:10.1016/j.cell.2013.10.004
66. Schlumm F, Mauceri D, Freitag HE, Bading H. Nuclear calcium signaling regulates nuclear export of a subset of class IIa histone deacetylases following synaptic activity. J Biol Chem (2013) 288(12):8074–84. doi:10.1074/jbc.M112.432773
67. Soriano FX, Chawla S, Skehel P, Hardingham GE. SMRT-mediated co-shuttling enables export of class IIa HDACs independent of their CaM kinase phosphorylation sites. J Neurochem (2013) 124(1):26–35. doi:10.1111/jnc.12058
68. McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature (2000) 408(6808):106–11. doi:10.1038/35040593
69. Bardai FH, D’Mello SR. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3beta. J Neurosci (2011) 31(5):1746–51. doi:10.1523/JNEUROSCI.5704-10.2011
70. Marinova Z, Leng Y, Leeds P, Chuang DM. Histone deacetylase inhibition alters histone methylation associated with heat shock protein 70 promoter modifications in astrocytes and neurons. Neuropharmacology (2011) 60(7–8):1109–15. doi:10.1016/j.neuropharm.2010.09.022
71. Vasilatos SN, Katz TA, Oesterreich S, Wan Y, Davidson NE, Huang Y. Crosstalk between lysine-specific demethylase 1 (LSD1) and histone deacetylases mediates antineoplastic efficacy of HDAC inhibitors in human breast cancer cells. Carcinogenesis (2013) 34(6):1196–207. doi:10.1093/carcin/bgt033
72. Jenuwein TL, Allis CD. Translating the histone code. Science (2001) 293(5532):1074–80. doi:10.1126/science.1063127
73. McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol (2012) 52:303–16. doi:10.1146/annurev-pharmtox-010611-134712
74. Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, et al. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J (2008) 22:3549–60. doi:10.1096/fj.08-108548
75. Dana A, Skarli M, Papakrivopoulou J, Yellon DM. Adenosine A(1) receptor induced delayed preconditioning in rabbits: induction of p38 mitogen-activated protein kinase activation and Hsp27 phosphorylation via a tyrosine kinase- and protein kinase C-dependent mechanism. Circ Res (2000) 86(9):989–97. doi:10.1161/01.RES.86.9.989
76. Martindale JJ, Wall JA, Martinez-Longoria DM, Aryal P, Rockman HA, Guo Y, et al. Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J Biol Chem (2005) 280(1):669–76. doi:10.1074/jbc.M406690200
77. Zhao TC, Taher MM, Valerie KC, Kukreja RC. p38 triggers late preconditioning elicited by anisomycin in heart: involvement of NF-kappaB and iNOS. Circ Res (2001) 89(10):915–22. doi:10.1161/hh2201.099452
78. Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the reperfusion injury salvage kinase (RISK)-pathway. Cardiovasc Res (2004) 61(3):448–60. doi:10.1016/j.cardiores.2003.09.024
79. Gao F, Gao E, Yue TL, Ohlstein EH, Lopez BL, Christopher TA, et al. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: the roles of PI3-kinase, akt, and endothelial nitric oxide synthase phosphorylation. Circulation (2002) 105(12):1497–502. doi:10.1161/01.CIR.0000012529.00367.0F
80. Jonassen AK, Sack MN, Mjøs OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via akt and p70s6 kinase cell-survival signaling. Circ Res (2001) 89(12):1191–8. doi:10.1161/hh2401.101385
81. Matsui T, Li L, del Monte F, Fukui Y, Franke TF, Hajjar RJ, et al. Adenoviral gene transfer of activated phosphatidylinositol 3’-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation (1999) 100(23):2373–9. doi:10.1161/01.CIR.100.23.2373
82. Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation (2000) 101(6):660–7. doi:10.1161/01.CIR.101.6.660
83. Vondriska TM, Zhang J, Song C, Tang X-L, Cao X, Baines CP, et al. Protein kinase C ε-Src modules direct signal transduction in nitric oxide-induced cardioprotection: complex formation as a means for cardioprotective signaling. Circ Res (2001) 88:1306–13. doi:10.1161/hh1201.092994
84. Párrizas M, Saltiel AR, LeRoith D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3’-kinase and mitogen-activated protein kinase pathways. J Biol Chem (1997) 272(1):154–61. doi:10.1074/jbc.272.1.154
85. Hausenloy DJ, Lecour S, Yellon DM. Reperfusion injury salvage kinase and survivor activating factor enhancement prosurvival signaling pathways in ischemic postconditioning: two sides of the same coin. Antioxid Redox Signal (2011) 14(5):893–907. doi:10.1089/ars.2010.3360
86. Tsang A, Hausenloy DJ, Mocanu MM, Yellon DM. Postconditioning: a form of “modified reperfusion” protects the myocardium by activating the phosphatidylinositol 3-Kinase-Akt pathway. Circ Res (2004) 95(3):230–2. doi:10.1161/01.RES.0000138303.76488.fe
87. Zhao TC, Zhang LX, Cheng G, Liu JT. gp-91 mediates histone deacetylase inhbition-induced cardioprotection. Biochim Biophys Acta (2010) 1803(7):872–80. doi:10.1016/j.bbamcr.2010.04.007
88. Zhao TC, Cheng G, Zhang LX, Tseng YT, Padbury JF. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc Res (2007) 76(3):473–81. doi:10.1016/j.cardiores.2007.08.010
89. Zhao TC, Du J, Zhuang S, Liu P, Zhang LX. HDAC inhibition elicits myocardial protective effect through modulation of MKK3/Akt-1. PLoS One (2013) 8(6):e65474. doi:10.1371/journal.pone.0065474
90. Zhang L, Qin X, Zhao Y, Fast L, Zhuang S, Liu P, et al. Inhibition of histone deacetylases preserves myocardial performance and prevents cardiac remodeling through stimulation of endogenous angiomyogenesis. J Pharmacol Exp Ther (2012) 341(1):285–93. doi:10.1124/jpet.111.189910
91. Xuan YT, Tang XL, Banerjee S, Takano H, Li RC, Han H, et al. Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ Res (1999) 84(9):1095–109. doi:10.1161/01.RES.84.9.1095
92. Maulik N, Sato M, Price BD, Das DK. An essential role of NFkappaB in tyrosine kinase signaling of p38 MAP kinase regulation of myocardial adaptation to ischemia. FEBS Lett (1998) 429(3):365–9. doi:10.1016/S0014-5793(98)00632-2
93. Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer (2009) 125(12):2863–70. doi:10.1002/ijc.24748
94. Li RC, Ping P, Zhang J, Wead WB, Cao X, Gao J, et al. PKCepsilon modulates NF-kappaB and AP-1 via mitogen-activated protein kinases in adult rabbit cardiomyocytes. Am J Physiol Heart Circ Physiol (2000) 279(4):H1679–89.
95. Zhang LX, Zhao Y, Cheng G, Guo TL, Chin YE, Liu PY, Zhao TC. Targeted deletion of NF-kappaB p50 diminishes the cardioprotection of histone deacetylase inhibition. Am J Physiol Heart Circ Physiol (2010) 298(6):H2154–63. doi:10.1152/ajpheart.01015.2009
96. Zhang Y, Yu G, Wang D, Hu Y, Lei W. ERK1/2 activation plays important roles in the opposite effects of trichostatin A in non-cancer and cancer cells. Toxicon (2011) 57(6):932–7. doi:10.1016/j.toxicon.2011.03.008
97. Lei WW, Zhang KH, Pan XC, Wang DM, Hu Y, Yang YN, et al. Histone deacetylase 1 and 2 differentially regulate apoptosis by opposing effects on extracellular signal-regulated kinase 1/2. Cell Death Dis (2010) 1:e44. doi:10.1038/cddis.2010.21
98. Bolli R. Oxygen-derived free radicals and postischemic myocardial dysfunction (“stunned myocardium”). J Am Coll Cardiol (1988) 12(1):239–49. doi:10.1016/0735-1097(88)90381-6
99. Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature (2002) 419(6904):316–21. doi:10.1038/nature01036
100. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science (2013) 339(6116):211–4. doi:10.1126/science.1227166
101. Aune SE, Herr DJ, Mani SK, Menick DR. Selective inhibition of class I but not class IIb histone deacetylases exerts cardiac protection from ischemia reperfusion. J Mol Cell Cardiol (2014) 72:138–45. doi:10.1016/j.yjmcc.2014.03.005
103. Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud P-D, Yu RT, Alvarez JG, et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homoestasis. Cell (2011) 145(4):607–21. doi:10.1016/j.cell.2011.03.043
104. Yan J. Interplay between HDAC6 and its interacting partners: essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol (2014) 33(9):567–80. doi:10.1089/dna.2013.2300
Keywords: ischemia-reperfusion injury, histone deacetylase inhibitors, stroke, posttranslational modification, enzymatic crosstalk, preconditioning, postconditioning, reperfusion injury salvage kinase
Citation: Aune SE, Herr DJ, Kutz CJ and Menick DR (2015) Histone deacetylases exert class-specific roles in conditioning the brain and heart against acute ischemic injury. Front. Neurol. 6:145. doi: 10.3389/fneur.2015.00145
Received: 14 February 2015; Accepted: 15 June 2015;
Published: 30 June 2015
Edited by:
Giuseppe Pignataro, University of Naples Federico II, ItalyReviewed by:
Akihiko Urayama, University of Texas Houston Medical School, USAMaria Jose Sisalli, University of Naples Federico II, Italy
Copyright: © 2015 Aune, Herr, Kutz and Menick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Donald R. Menick, Gazes Cardiac Research Institute, Medical University of South Carolina, 114 Doughty Street, Room 331, Charleston, SC 29425, USA,bWVuaWNrZEBtdXNjLmVkdQ==