 Nico Melzer
Nico Melzer Thomas Budde
Thomas Budde Oliver Stork
Oliver Stork Sven G. Meuth
Sven G. Meuth- 1Department of Neurology, University of Münster, Münster, Germany
- 2Institute of Physiology I, University of Münster, Münster, Germany
- 3Department of Genetics and Molecular Neurobiology, Institute of Biology, Otto-von-Guericke University Magdeburg, Magdeburg, Germany
- 4Department of Neuropathophysiology, Institute of Physiology I, University of Münster, Münster, Germany
Limbic encephalitis is characterized by adaptive autoimmune inflammation of the gray matter structures of the limbic system. It has recently been identified as a major cause of temporal lobe epilepsy accompanied by progressive declarative – mainly episodic – memory disturbance as well as a variety of rather poorly defined emotional and behavioral changes. While autoimmune inflammation of the hippocampus is likely to be responsible for declarative memory disturbance, consequences of autoimmune inflammation of the amygdala are largely unknown. The amygdala is central for the generation of adequate homoeostatic behavioral responses to emotionally significant external stimuli following processing in a variety of parallel neuronal circuits. Here, we hypothesize that adaptive cellular and humoral autoimmunity may target and modulate distinct inhibitory or excitatory neuronal networks within the amygdala, and thereby strongly impact processing of emotional stimuli and corresponding behavioral responses. This may explain some of the rather poorly understood neuropsychiatric symptoms in limbic encephalitis.
Introduction – Autoimmune Inflammation of the Amygdala in Limbic Encephalitis
Limbic encephalitis is characterized by adaptive autoimmune inflammation of the gray matter structures of the limbic system (1). It has recently been identified as a major cause of temporal lobe epilepsy accompanied by progressive declarative – mainly episodic – memory disturbance as well as a variety of rather poorly defined emotional and behavioral changes (2–4).
Magnetic resonance imaging exhibits dynamic changes of volume and signal intensity that use to be most prominent in the amygdala and the hippocampus suggesting considerable inflammation and subsequent degeneration (together with structural reorganization) in these brain areas (1, 5–7). While autoimmune inflammation of the hippocampus is likely to be responsible for declarative memory disturbance, consequences of autoimmune inflammation of the amygdala are poorly understood (8).
A role of the amygdala in appropriate human emotional behavior was first reported by Adolphs et al. (9). This finding was supported by studies on individuals with impaired ability to recognize emotions from facial or prosodic expressions after amygdala damage of variable etiology (10–12). However, the recognition of facial (13) and prosodic (14, 15) emotions is apparently normal in some patients with amygdala damage. Two factors related to this discrepancy have been suggested: (i) age at damage onset [early-onset (congenital) vs. late-onset of amygdala damage] and (ii) extent of the damage (selective amygdala damage vs. broad damage to the mesial temporal lobe) (16).
Emotion recognition (happiness, sadness, anger, fear, surprise, disgust) from facial and non-facial stimuli was investigated in two patients with non-herpetic limbic encephalitis 14 weeks after the onset of the disease. One patient who had a lesion relatively restricted to the amygdala and hippocampus experienced difficulty in recognizing fear from facial expressions. In contrast, the other patient who had a lesion that extended beyond the amygdala experienced difficulty in recognizing fear from non-facial (prosodic and written verbal) stimuli (17). Moreover, recognition of emotional stimuli, such as fear, and disgust both from faces and voices has been shown to be impaired in a patient with Ma antibody-associated limbic encephalitis (18).
Furthermore, autonomic responses to such emotional stimuli have been reported to be severely impaired in patients with limbic encephalitis. Sweating on the palms of the hands and soles of the feet, so-called emotional sweating, is considered to be mediated by the limbic system, including the amygdala and anterior cingulate cortex. Hence, sweat and skin vasoconstriction responses to arousal stimuli were recorded on the palms of seven patients with viral (herpes simplex virus and Epstein–Barr virus encephalitis; n = 3) and autoimmune (voltage-gated K+ channel antibody positive, glutamate receptor antibody positive, and antibody-negative limbic encephalitis; n = 4) bilateral limbic encephalitis, which included both the amygdala and hippocampus 3 weeks to 4 months after disease onset. Sweat responses and skin vasoconstriction responses were absent or markedly reduced in patients with limbic encephalitis compared to normal controls following a variety of emotional stimuli (19). The same results were obtained in a patient with autoimmune limbic encephalitis restricted bilaterally to the amygdala (20) indicating that affection of the amygdala rather than the hippocampus seemingly accounts for impaired emotional sweating in these patients.
Interestingly, another case of extensive bilateral limbic damage (including amygdala damage) after an episode of herpes simplex virus encephalitis 30 years ago did not present any emotional impairment besides difficulty to identify anger expression (21), suggesting that some adaptive mechanisms may compensate for the neuropsychological symptoms during the disease course.
The amygdala is an almond-shaped nucleus located deep in the temporal lobe. It is considered as a core region of the limbic system involved in the control of positive and negative affects, as well as the modulation of social and cognitive functions (22). Different amygdala subnuclei exist, of which we will focus here on lateral (LA) and basal amygdala (BA), which are often jointly (together with the accessory-basal nuclei) considered the basolateral complex of the amygdala (BLA). We argue that interference with BLA function through known B cell-derived autoantibodies and T cells may explain, at least in part, some neuropsychiatric features in limbic encephalitis.
The BLA is a main input site for sensory information reaching the amygdala from thalamic and cortical regions and for their convergence with affective information. Sensory input is organized in a topographic manner in that the LA is more concerned with unimodal information, whereas the BA tends to receive more complex multimodal inputs. Through its projections to the central amygdala (CeA), the bed nucleus of the stria terminalis (BNST), and the ventral hippocampus (23, 24), the BLA has been implicated in the control of fear and anxiety [for review see Ref. (25)]. Moreover, the BLA modulates reward and addiction via direct projections to the nucleus accumbens (26) and controls the function of the medial prefrontal cortex (27).
The amygdala receives prominent neuromodulatory input and triggers arousal and stress responding through projections to the septum and locus coeruleus. It is further a key site for the activity of corticosterone in the brain, which increases excitability of BLA projection neurons and reduces the inhibition through γ-aminobutyric acid (GABA) receptors (28). By mediating the effects of these modulatory factors, the BLA is also thought to act as an important modulator of neural plasticity and memory formation in the hippocampus and prefrontal cortex (29, 30).
Glutamatergic principal cells in the BLA are under a tight control by GABAergic interneurons (Figure 1). Approximately 10–20% of neurons in the amygdala are GABAergic and control information flow as well as rhythmic network activity (31). Various types of inhibitory interneurons (32) exist in the BLA that control specific aspects of information flow and behavioral function (33). Disinhibitory local circuits have also been reported (34) and implicated in fear memory formation (35). This internal circuitry of the BLA is particularly well set to process information via rhythmic network activities. BLA neurons show intrinsic low-frequency oscillations (36) and can develop gamma oscillations upon GABAergic blockage (37). GABAergic interneurons are furthermore selectively recruited to theta rhythms originating in the hippocampus (38). Thus, network oscillations in the BLA closely reflect periods of safety and threat exposure (39) and synchronize with the hippocampus and prefrontal cortex during states of fear retrieval and extinction (40, 41).
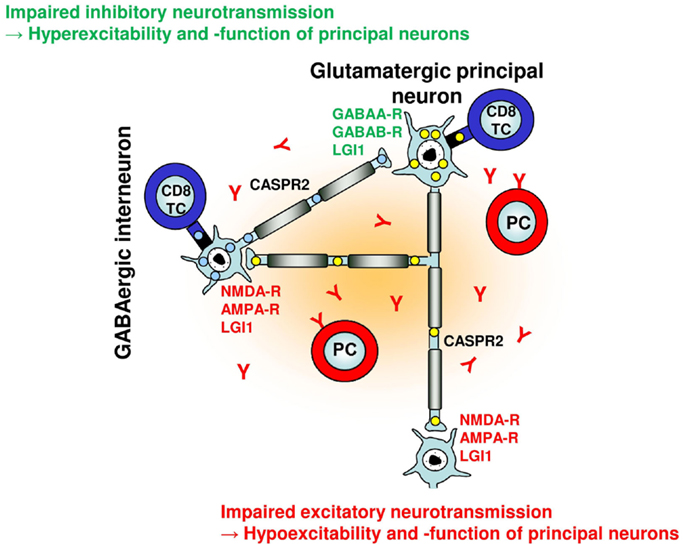
Figure 1. Putative effects of adaptive humoral and cellular autoimmunity on a simplified neuronal network. Glutamatergic principal neurons and GABAergic interneurons can be selectively targeted by neuronal antigen-specific CD8+ T cells based on their differential intracellular antigen expression (and presentation) [e.g., GAD65 in interneurons (blue), Hu in principal neurons (yellow)] with distinct consequences for network function and excitability. With regard to neuronal cell membrane antigens, excitatory glutamatergic synaptic transmission and plasticity can be disturbed by antibodies against NMDA and AMPA receptors, GABAergic synaptic transmission and plasticity can be disturbed by antibodies against GABAA and GABAB receptors. Antibodies against LGI1 and CASPR2 may interfere with both glutamatergic and GABAergic synaptic transmission and intrinsic neuronal excitability within the network, respectively.
Amygdala lesions lead to emotional numbness and fearlessness, whereas hypertrophy of amygdala has been observed in patients with post-traumatic stress disorder and depression (42). Indeed, it has been suggested that the amygdala is a key structure for the long-term behavioral adaptation to stress (43). The amygdala has furthermore been identified as a major epileptic focus in temporal lobe epilepsy, and in rodents it is widely used as a site of stimulation in the kindling model of epilepsy. Importantly, within the amygdala the BLA plays a central role in seizure generation (44).
Considering these findings, we hypothesize that changes in BLA excitability and information processing induced by autoimmune inflammation contribute to seizures, different levels of anxiety, mood disorder, and potentially also memory deficits in limbic encephalitis as – depending on the predominant immune effector mechanism and the neuronal target antigen – autoimmune inflammation of the amygdala may result in decreased or increased excitability and function of principal neurons of the BLA network (Figure 1; Table 1).
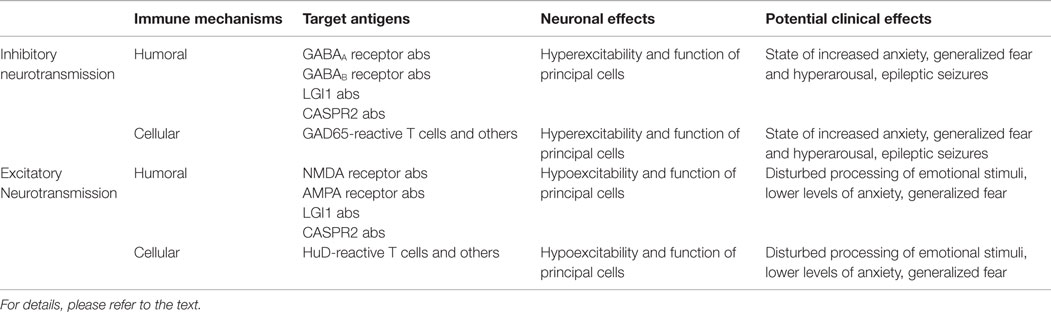
Table 1. Putative effects of adaptive humoral and cellular autoimmunity on inhibitory and excitatory transmission and network activity of the amygdala together with potential clinical consequences.
Putative Pathogenesis of Autoimmune Gray Matter Inflammation and Consequences for Neuronal Function and Integrity
Adaptive neuron-directed autoimmunity underlying limbic encephalitis is illustrated by the presence of specific anti-neuronal antibodies binding to either intracellular or plasma membrane neuronal antigens in sera and cerebrospinal fluid in many cases (45–47). Depending on the cellular localization of their antigens, these antibodies provide some hints on the predominant autoimmune effector mechanisms toward single neurons and neuronal networks.
In a subgroup of patients with limbic encephalitis, autoantibodies are detected that bind to intracellular neuronal antigens. These are molecules with a role in a variety of gene expression and signal transduction processes, which are expressed in distinct neuronal cell populations [reviewed in Ref. (46)]. In these patients, CD8+ T cells usually recognize continuous linear peptide epitopes consisting of 8–10 amino acids that are derived from intracellular neuronal proteins by extensive antigen processing and presented in the context of MHC I molecules on the cell surface membrane (48–51). T cell receptor (TCR)-signaling upon recognition of the appropriate antigen in the context of MHC I molecules during engagement of activated CD8+ T cells with neurons leads to the formation of the immunological synapse (52). Subsequent CD8+ T cell-mediated impairment of neuronal excitability and integrity is predominantly mediated via two largely independent pathways (53, 54). (i) Granule cytotoxicity occurs by liberation of perforin together with a variety of granzymes. Depending on the amounts released, perforin alone can lead to the formation of unselective transmembrane pores with a subsequent impairment of electrical excitability and signaling (50, 55) or neuronal necrosis with immediate swelling and rupture of the neuronal cell membrane (56). Alternatively, perforin mediates the trafficking of granzymes into the target cell promoting apoptosis within few hours (56, 57). (ii) Impairment of neuronal excitability and structural integrity may also occur through the ligation of cell death receptors (50, 58). Moreover, CD8+ T cells are able to liberate potentially neurotoxic cytokines, such as interferon-(IFN)γ and tumor necrosis factor-(TNF)α (59, 60) as well as distinct neurotransmitters like glutamate (61, 62), adding to the repertoire of molecular effector mechanisms directly impacting on neuronal excitability and integrity (63).
Given these molecular effector mechanisms, CD8+ T cells cannot directly impact the function or expression of their cognate neuronal antigens but recognize their expression by the respective neuron. This enables them to contribute to neuronal dysfunction and cell death (50, 55, 64, 65). These effects may be restricted to distinct inhibitory (1) or excitatory (66, 67) neuronal populations and networks due to their differential antigen expression pattern and capability of MHC I-mediated antigen presentation (65, 68).
In a subset of patients with limbic encephalitis, antibodies against a family of intracellular neuron-specific RNA binding proteins, the neuronal “embryonic lethal and abnormal vision-like” RNA binding proteins, HuB, HuC, and HuD, can be detected (69, 70). These Hu proteins are expressed in specific neuronal populations, including large glutamatergic pyramidal-like neurons in layer V of the neocortex, the cornu amonis (CA) 1–4, and dentate gyrus (DG) regions of the hippocampus, as well as principal cells of the amygdala (71). Neuron-specific Hu proteins have recently been shown to maintain neuronal glutamate levels by stabilizing glutaminase mRNA and protein levels (66, 67). Glutamate is the major excitatory neurotransmitter in the mammalian brain and also impacts inhibitory neuronal signaling in two ways: (i) it is the biochemical precursor for the major inhibitory neurotransmitter GABA (72) and (ii) activates inhibitory neuronal feedback loops (73). Consistent with a net inhibitory effect of Hu-expressing neurons on neuronal network excitability, genetic deficiency of neuron-specific Hu proteins leads to spontaneous neuronal hypersynchrony and epileptic seizure activity (66, 67). Moreover, Hu-expressing neurons have been implicated in hippocampus and amygdala-based synaptic plasticity, learning, and memory (74, 75).
In another subset of patients with limbic encephalitis, antibodies against neuronal glutamate decarboxylase (GAD) have been detected (1). GAD is an intracellular enzyme expressed in a subset of interneurons and catalyzes the conversion of glutamate to GABA therein. The brain contains two isoforms, GAD65 and GAD67, which display characteristic differences in localization and activity patterns (76, 77). GAD67 is typically distributed throughout the neuron and almost all of it exists in its active cofactor-bound form, whereas GAD65 is predominantly found in synaptic terminals and much of it is in the form of an inactive apoenzyme (72, 78). In accordance with a net inhibitory effect of GAD65-expressing interneurons on neuronal network excitability, genetic deficiency of GAD65 leads to spontaneous neuronal hypersynchrony and epileptic seizures (79). Moreover, GAD65-expressing neurons have been implicated in hippocampus and amygdala-based synaptic plasticity, learning, and memory (41, 80–82).
In light of these physiological effects of Hu-expressing pyramidal-like neurons and GAD65-expressing interneurons on synaptic transmission, plasticity, learning, and memory, as well as neuronal network excitability and seizure development, it seems conceivable that similar effects observed upon genetic deficiency of these molecules occur if these cell populations become targets of neuronal antigen-specific CD8+ T cells as in limbic encephalitis (51).
In another subgroup of patients with limbic encephalitis, autoantibodies are detected that bind to synaptic and extrasynaptic neuronal cell membrane antigens. These include a variety of ionotropic and metabotropic neurotransmitter receptors and associated molecules [reviewed in Ref. (46)]. In these patients, antibody-mediated disturbance of synaptic transmission and plasticity as well as neuronal (network) excitability occurs together with some neurodegenerative effects (51).
Antibodies recognize and bind to discontinuous conformational epitopes composed of segments of the respective neuronal plasma membrane protein antigen that come in close spatial proximity in their three-dimensional structure and exposed on the neuronal plasma membrane. These are synaptic and extra-synaptic ligand- and voltage-gated ion channels involved in excitatory [mainly N-methyl-d-aspartate (NMDA) (83, 84) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) (85, 86) receptors] and inhibitory [mainly GABAA (87) and GABAB (88) receptors] synaptic transmission and plasticity. Moreover, these antibodies also target neuronal membrane proteins implicated in clustering of voltage- and ligand-gated ion channels inside the synapse [leucine-rich glioma-inactivated 1; LGI1 (89, 90)] or outside the synapse at the juxtaparanodal region of the node of Ranvier [contactin-2 and contactin-associated protein-like 2; CASPR2 (89, 91)], thereby indirectly impacting neuronal excitability.
Depending on the IgG subtype, antibodies may (i) specifically activate or block the function of their target molecules, (ii) crosslink and internalize the receptors, (iii) activate the complement cascade with subsequent formation of the terminal membrane attack complex and target cell lysis, and (iv) activate Fc-receptors with subsequent antibody-dependent cell-mediated cytotoxicity (ADCC) mainly by NK cells (92).
Regarding excitatory synaptic transmission, NMDA receptor antibodies have been described to directly impact the gating behavior of the receptor (93). It has been shown that the N368/G369 region of the GluN1 subunit of NMDA receptors may represent the immunodominant binding region for IgG on the receptor molecule. In single channels recordings, antibody binding to the receptor caused more frequent channel openings and prolonged open time of the receptor as immediate effects (93). Moreover, NMDA receptor antibodies have been shown to cause a selective and reversible decrease in post-synaptic NMDA receptor surface density and synaptic localization in inhibitory as well as excitatory cultured rat hippocampal neurons by selective antibody-mediated cross-linking and internalization (94, 95). Consistently, NMDA receptor antibodies selectively decreased NMDA receptor-mediated miniature excitatory post-synaptic currents (mEPSCs) without affecting AMPA receptor-mediated mEPSCs in cultured rat hippocampal neurons. However, despite these strong effects, NMDA receptor antibodies did not impact the number of synapses, dendritic spines, dendritic complexity, or cell survival in cultured rat hippocampal neurons (94). Consistent with these mechanisms, NMDA receptor antibodies have been shown to suppress induction of long-term potentiation (LTP) at Schaffer collateral-CA1 synapses in mouse hippocampal slices (96). Once internalized, antibody-bound NMDA receptors traffic through both recycling endosomes and lysosomes, but do not induce compensatory changes in glutamate receptor gene expression. This process eventually results in a decrease in inhibitory synapse density onto excitatory cultured rat hippocampal neurons through distinct homeostatic synaptic plasticity mechanisms (95, 97).
Additionally, autoantibodies in limbic encephalitis target GluA1 and GluA2 subunits of the AMPA receptor (85, 98). Application of antibodies to cultures of neurons reversibly decreased the number of GluA1- and GluA2-containing AMPA-receptor clusters at synapses and beyond through increased internalization and degradation of surface AMPA receptors (85, 98). In contrast, antibodies do not alter the density of excitatory synapses or cell viability (98). Consistently, whole-cell patch clamp recordings of cultured hippocampal neurons incubated with antibodies revealed decreased AMPA receptor-mediated currents, but not NMDA receptor-mediated currents. Interestingly, probably by distinct homeostatic plasticity mechanisms like those observed by NMDA receptor antibodies (97), several functional properties of AMPA receptor antibody-targeted neurons are altered. Affected neurons exhibit decreased inhibitory post-synaptic currents (IPSCs) and reduced inhibitory synapse density onto excitatory cultured rat hippocampal neurons, whereas the intrinsic excitability of neurons and short-interval firing increase (98).
Hence, both NMDA and AMPA receptor antibodies eliminate ionotropic glutamate receptors from the post-synaptic (and extra-synaptic) neuronal plasma membrane through cross-linking and internalization, resulting in disturbed excitatory synaptic transmission and a concomitant homeostatic decrease in inhibitory synaptic transmission and increased intrinsic excitability.
Antibodies to LGI1 have been described in limbic encephalitis (89, 90). Extracellularly secreted LGI1 has been reported to link two receptors, ADAM (a disintegrin and metalloproteinase domain-containing protein) 22 and ADAM23, and establish a transsynaptic protein complex that includes presynaptic voltage-gated K+ channels and post-synaptic AMPA receptors. A lack of LGI1 disrupts this synaptic protein connection and selectively reduces AMPA receptor-mediated synaptic transmission in the hippocampus (99, 100). LGI1 antibodies associated with limbic encephalitis specifically inhibit the ligand–receptor interaction between LGI1 and ADAM22/23 by targeting the EPTP repeat domain of LGI1 and reversibly reduce synaptic AMPA receptor clusters in rat hippocampal neurons (101). Interestingly, conditional knockout of LGI1 restricted to glutamatergic pyramidal cells is sufficient to generate seizures, whereas seizure thresholds were shown to be unchanged after depletion of LGI1 in GABAergic interneurons. Hence, LGI1 secreted from excitatory pyramidal neurons, but not inhibitory interneurons, makes a major contribution to LGI1-related epileptogenesis (102). Similar effects may be expected from antibody-mediated disruption of LGI1 function.
Antibodies against contactin-2 and CASPR2 (89, 91) have been implicated to impair clustering of voltage-gated K+ channels at the juxtaparanodal region of the node of Ranvier of myelinated axons, thereby probably interfering with axonal excitability and action potential conduction.
Regarding inhibitory synaptic transmission, antibodies directed against the extracellular epitope of the β3 subunit of the GABAA receptor have been reported (87). Rat hippocampal neuronal cultures exposed to GABAA receptor antibodies specifically decreased both synaptic and surface GABAA receptors, and showed selectively reduced miniature inhibitory post-synaptic currents (mIPSCs) without affecting mEPSCs (103). Concomitant changes in excitatory synaptic transmission have not been reported thus far.
The presence of GABAB receptor autoantibodies constitutes another form of limbic encephalitis (88). GABAB receptors are G-protein-coupled receptors composed of two subunits, GABAB1 and GABAB2 (104). The main antigen recognized by the antibodies, the GABAB1 subunit, is necessary for GABA binding and receptor function, whereas the GABAB2 subunit is required for localization of the receptor to appropriate areas of the cell membrane and G-protein coupling (104). GABAB receptors mediate presynaptic inhibition by activation of G-protein-coupled-inward rectifying K+ channels and inhibition of Ca2+ channels. Post-synaptic GABAB receptors mediate inhibition by similar mechanisms and by inducing a slow inhibitory post-synaptic potential (104). GABAB receptors limit the duration of increased activity states in neuronal networks, preventing excessive neuronal synchronization, and development of epileptic seizures. Hence, although experimental evidence of the molecular consequences of antibody targeting of GABAB receptors is still lacking, it seems conceivable to assume that they will also reduce pre- and post-synaptic GABAergic inhibition and thus promote increased activity states with excessive synchronization in neuronal networks and promotion of epileptic seizures.
Taken together, experimental evidence thus far suggest reduced inhibitory synaptic transmission (and plasticity) – either indirectly via homeostatic plasticity mechanisms following antibody binding to NMDA oder AMPA receptors or directly via antibody binding to GABAA or GABAB receptors – as a major consequence of humoral neuron-directed autoimmunity in different forms of limbic encephalitis. Moreover, cellular immune effects may either directly or indirectly selectively target inhibitory interneuron networks with similar functional consequences (Figure 1; Table 1).
In other forms of limbic encephalitis, humoral immune effects may predominantly affect excitatory synaptic transmission (and plasticity). Furthermore, cellular immune effects may directly target excitatory principal neuron networks with similar consequences (Figure 1; Table 1).
In the following, we speculate on the consequences of humoral and cellular autoimmune impact on either inhibitory or excitatory pathways for amygdala function, i.e., emotion processing and behavioral responses.
Putative Effects of Cellular and Humoral Autoimmunity toward Circuit Function of the Amygdala
The local network in the BLA is organized to integrate sensory and affective information and to control emotional responses and cognitive function through its efferent connections to the brainstem, hypothalamus, and forebrain. Key features of the local network function are the strong GABAergic control of activity and the propensity to develop network oscillations at different frequencies. On the one hand, this allows the BLA to resonate with cortical brain regions during emotional information processing; on the other hand, these characteristics may explain why the BLA is prone to the development of epileptic activity.
T cell responses toward GAD65-expressing GABAergic interneurons and autoantibodies to GABA receptors, by reducing GABAergic tone, can be expected to exert profound effects on the amygdala, leading to hyperexcitability of principal cells and a state of increased anxiety and hyperarousal (Figure 1; Table 1).
Loss of GAD65 in knockout mice and concomitant hyperactivity in the amygdala results in anxiety, hyperactivity, and generalized fear (80, 81, 105, 106). It also increases the susceptibility to seizures (107). In fact, it has also been shown that GAD65 antibodies isolated from a stiff-person-syndrome patient induce anxiety in rats upon binding to the amygdala, hippocampus, and prefrontal cortex (108). However, a considerable ability exists to compensate for reduction in GAD65 expression without emergence of profound fear phenotype (109), likely due to activity-dependent regulation of residual enzyme and the co-expression of its isozyme, GAD67, or other components of the GABA metabolism and synaptic cycling. Thus, a considerable variability can be expected in patients with autoimmunity toward GAD65 in interneurons concerning the development of BLA-related symptoms.
Several other autoantibodies also may have the capacity to disturb the inhibitory control of the BLA. For example, loss of GABAB receptors upon antibody binding would likely disrupt synaptic regulation of GABA release in the BLA, which has been related to fear generalization (110). On the post-synaptic level, endocytosis of β3 subunits of the GABAA receptor has been observed during the reinstatement of extinguished fear memory (111). Thus, autoantibody-induced endocytosis of this receptor subunit in the amygdala could have the potential to reactivate memory for fearful experiences, triggering stress and anxiety.
As discussed above, T cell responses toward glutamatergic Hu-expressing principal neurons and antibody-mediated loss of NMDA and AMPA glutamate receptors may affect both glutamatergic and – by mechanisms of homeostatic plasticity – GABAergic neurons in the BLA. Given the local network architecture, it can be expected that this will lead to a reduced net activation of principal cells in the BLA, potentially resulting in disturbed processing of emotional stimuli. Indeed, loss of Hu in knockout mice is associated with lower levels of anxiety and overall activity compared to wild-type counterparts (67). Moreover, lack of precision in information processing in cortico-amygdala circuits can lead to generalization of fear (112) as mechanisms of synaptic competition in the LA contribute to the specificity of conditioned fear (113). GABAergic neurons in the amygdala can be divided into two major populations: cells, which are scatter distributed within the local neurophil (32), and groups of highly clustered GABAergic neurons, the so-called intercalated cells (ITCs) that are targeted by glutamatergic projections from the medial prefrontal cortex mediating fear extinction (114) have been shown to control the information flow between BLA and CeA. Increased internalization of AMPA receptors has been reported in ITCs to mediate long-term depression (LTD) (115). Reduced excitability and plasticity in this structure is likely relevant for the management of previously acquired fearful memories. Hence, internalization of AMPA receptors, induced by AMPA receptor or LGI1 autoantibodies, may mimic a state of LTD in the amygdala (116).
Evidence suggests that autoantibodies to LGI1 are also associated with neuropsychiatric disturbances and seizures (117, 118). In line, selective genetic ablation of LGI1 in excitatory neurons induces seizures, whereas the conditional knockout in parvalbumin expressing interneurons remains without such effect (102). Interfering with the function of LGI1 released from BLA principle cells, via interaction with the disintegrin ADAM23 could alter neuronal morphology and decrease seizure threshold (119), and via ADAM22 reduce the expression of synaptic AMPA receptors (101). On the other hand, somatostatin-positive interneurons in the amygdaloid complex revealed selective vulnerability in temporal lobe epilepsy and following toxic insults, thereby contributing to hyperexcitability of amygdala synaptic circuits and anxiety-like behavior (120–122). Some neuropsychiatric conditions, like schizophrenia, are characterized by remodeling of the perineuronal nets surrounding somatostatin expressing interneurons in the amygdala, which results in GABAergic dysfunction and immune system dysregulation (123), thereby also pointing to possible localized pathological processes in the amygdala.
Moreover, reduced modulatory input from the BLA impairs synaptic plasticity in the hippocampus- and hippocampus-dependent learning. Thus, in addition to putative direct effects on hippocampal synaptic function, the BLA may be involved in memory deficits observed in limbic encephalitis patients. The amygdala provides a site for the modulation of memory acquisition, storage, recall, and modification via the hippocampus (124) and interaction of the BLA with the nAcc is required for active avoidance learning (125). Patients with autoantibodies to voltage-gated K+ channels display deficits in cognitive and executive function (126). However, these disturbances of memory-related functions are rather likely to involve direct effects of the autoantibodies on the different amygdala target areas, and at this time it is difficult to estimate the contribution of the BLA to the observed deficits.
The findings discussed above open up the possibility to experimentally assess the role of distinct neuronal cell populations using gene targeting and optogenetics. Furthermore, it can be tested whether disruption of the extracellular matrix, which normally can act as a passive diffusion barrier for cell surface molecules (127) and possibly limits access of immune cells and antibodies to the neuronal cell surface, may influence the impact of both immune effector arms on neuronal network structure and function.
Hence, accumulating evidence suggests that adaptive autoimmune amygdala inflammation may be a major determinant of emotional and behavioral disturbance in limbic encephalitis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the German research foundation (DFG) funded excellence cluster “CiM – Cells in Motion.” NM has received honoria for lecturing and travel expenses for attending meetings from Biogen Idec, GlaxoSmith Kline, Teva, and Fresenius Medical Care and has received financial research support from Fresenius Medical Care. TB has received financial research support from Bayer, Biogen Idec, and Novartis. OS has received travel expenses from Boehringer Ingelheim. SM has received honoraria for lecturing and travel expenses for attending meetings and has received financial research support from Bayer, Bayer Schering, Biogen Idec, Genzyme, Merck Serono, MSD, Novartis, Novo Nordisk, Sanofi-Aventis, and Teva.
References
1. Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol (2010) 67(4):470–8. doi: 10.1002/ana.21917
2. Bien CG, Elger CE. Limbic encephalitis: a cause of temporal lobe epilepsy with onset in adult life. Epilepsy Behav (2007) 10(4):529–38. doi:10.1016/j.yebeh.2007.03.011
3. Bien CG, Schulze-Bonhage A, Deckert M, Urbach H, Helmstaedter C, Grunwald T, et al. Limbic encephalitis not associated with neoplasm as a cause of temporal lobe epilepsy. Neurology (2000) 55(12):1823–8. doi:10.1212/WNL.55.12.1823
4. Bien CG, Urbach H, Schramm J, Soeder BM, Becker AJ, Voltz R, et al. Limbic encephalitis as a precipitating event in adult-onset temporal lobe epilepsy. Neurology (2007) 69(12):1236–44. doi:10.1212/01.wnl.0000276946.08412.ef
5. Wagner J, Witt JA, Helmstaedter C, Malter MP, Weber B, Elger CE. Automated volumetry of the mesiotemporal structures in antibody-associated limbic encephalitis. J Neurol Neurosurg Psychiatry (2015) 86(7):735–42. doi:10.1136/jnnp-2014-307875
6. Wagner J, Schoene-Bake JC, Malter MP, Urbach H, Huppertz HJ, Elger CE, et al. Quantitative FLAIR analysis indicates predominant affection of the amygdala in antibody-associated limbic encephalitis. Epilepsia (2013) 54(9):1679–87. doi:10.1111/epi.12320
7. Wagner J, Weber B, Elger CE. Early and chronic gray matter volume changes in limbic encephalitis revealed by voxel-based morphometry. Epilepsia (2015) 56(5):754–61. doi:10.1111/epi.12968
8. Frisch C, Malter MP, Elger CE, Helmstaedter C. Neuropsychological course of voltage-gated potassium channel and glutamic acid decarboxylase antibody related limbic encephalitis. Eur J Neurol (2013) 20(9):1297–304. doi:10.1111/ene.12186
9. Adolphs R, Tranel D, Damasio H, Damasio A. Impaired recognition of emotion in facial expressions following bilateral damage to the human amygdala. Nature (1994) 372(6507):669–72. doi:10.1038/372669a0
10. Broks P, Young AW, Maratos EJ, Coffey PJ, Calder AJ, Isaac CL, et al. Face processing impairments after encephalitis: amygdala damage and recognition of fear. Neuropsychologia (1998) 36(1):59–70. doi:10.1016/S0028-3932(97)00105-X
11. Adolphs R, Tranel D, Hamann S, Young AW, Calder AJ, Phelps EA, et al. Recognition of facial emotion in nine individuals with bilateral amygdala damage. Neuropsychologia (1999) 37(10):1111–7. doi:10.1016/S0028-3932(99)00039-1
12. Scott SK, Young AW, Calder AJ, Hellawell DJ, Aggleton JP, Johnson M. Impaired auditory recognition of fear and anger following bilateral amygdala lesions. Nature (1997) 385(6613):254–7. doi:10.1038/385254a0
13. Hamann SB, Stefanacci L, Squire LR, Adolphs R, Tranel D, Damasio H, et al. Recognizing facial emotion. Nature (1996) 379(6565):497. doi:10.1038/379497a0
14. Anderson AK, Phelps EA. Intact recognition of vocal expressions of fear following bilateral lesions of the human amygdala. Neuroreport (1998) 9(16):3607–13.
15. Adolphs R, Tranel D. Intact recognition of emotional prosody following amygdala damage. Neuropsychologia (1999) 37(11):1285–92. doi:10.1016/S0028-3932(99)00023-8
16. Schmolck H, Squire LR. Impaired perception of facial emotions following bilateral damage to the anterior temporal lobe. Neuropsychology (2001) 15(1):30–8. doi:10.1037/0894-4105.15.1.30
17. Hayakawa Y, Mimura M, Murakami H, Kawamura M. Emotion recognition from stimuli in different sensory modalities in post-encephalitic patients. Neuropsychiatr Dis Treat (2010) 6:99–105. doi:10.2147/NDT.S9215
18. Sprengelmeyer R, Atkinson AP, Sprengelmeyer A, Mair-Walther J, Jacobi C, Wildemann B, et al. Disgust and fear recognition in paraneoplastic limbic encephalitis. Cortex (2010) 46(5):650–7. doi:10.1016/j.cortex.2009.04.007
19. Asahina M, Fujinuma Y, Yamanaka Y, Fukushima T, Katagiri A, Ito S, et al. Diminished emotional sweating in patients with limbic encephalitis. J Neurol Sci (2011) 306(1–2):16–9. doi:10.1016/j.jns.2011.04.007
20. Asahina M, Suzuki A, Mori M, Kanesaka T, Hattori T. Emotional sweating response in a patient with bilateral amygdala damage. Int J Psychophysiol (2003) 47(1):87–93. doi:10.1016/S0167-8760(02)00123-X
21. Feinstein JS, Rudrauf D, Khalsa SS, Cassell MD, Bruss J, Grabowski TJ, et al. Bilateral limbic system destruction in man. J Clin Exp Neuropsychol (2010) 32(1):88–106. doi:10.1080/13803390903066873
22. Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev (2010) 90(2):419–63. doi:10.1152/physrev.00037.2009
23. Felix-Ortiz AC, Beyeler A, Seo C, Leppla CA, Wildes CP, Tye KM. BLA to vHPC inputs modulate anxiety-related behaviors. Neuron (2013) 79(4):658–64. doi:10.1016/j.neuron.2013.06.016
24. Felix-Ortiz AC, Tye KM. Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J Neurosci (2014) 34(2):586–95. doi:10.1523/JNEUROSCI.4257-13.2014
25. Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev (2003) 83(3):803–34. doi:10.1152/physrev.00002.2003
26. Di Ciano P, Everitt BJ. Direct interactions between the basolateral amygdala and nucleus accumbens core underlie cocaine-seeking behavior by rats. J Neurosci (2004) 24(32):7167–73. doi:10.1523/JNEUROSCI.1581-04.2004
27. Likhtik E, Paz R. Amygdala-prefrontal interactions in (mal)adaptive learning. Trends Neurosci (2015) 38(3):158–66. doi:10.1016/j.tins.2014.12.007
28. Duvarci S, Pare D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J Neurosci (2007) 27(16):4482–91. doi:10.1523/JNEUROSCI.0680-07.2007
29. Roozendaal B, McEwen BS, Chattarji S. Stress, memory and the amygdala. Nat Rev Neurosci (2009) 10(6):423–33. doi:10.1038/nrn2651
30. Richter-Levin G, Maroun M. Stress and amygdala suppression of metaplasticity in the medial prefrontal cortex. Cereb Cortex (2010) 20(10):2433–41. doi:10.1093/cercor/bhp311
31. Makkar SR, Zhang SQ, Cranney J. Behavioral and neural analysis of GABA in the acquisition, consolidation, reconsolidation, and extinction of fear memory. Neuropsychopharmacology (2010) 35(8):1625–52. doi:10.1038/npp.2010.53
32. Sosulina L, Graebenitz S, Pape HC. GABAergic interneurons in the mouse lateral amygdala: a classification study. J Neurophysiol (2010) 104(2):617–26. doi:10.1152/jn.00207.2010
33. Spampanato J, Polepalli J, Sah P. Interneurons in the basolateral amygdala. Neuropharmacology (2011) 60(5):765–73. doi:10.1016/j.neuropharm.2010.11.006
34. Woodruff AR, Monyer H, Sah P. GABAergic excitation in the basolateral amygdala. J Neurosci (2006) 26(46):11881–7. doi:10.1523/JNEUROSCI.3389-06.2006
35. Wolff SB, Grundemann J, Tovote P, Krabbe S, Jacobson GA, Muller C, et al. Amygdala interneuron subtypes control fear learning through disinhibition. Nature (2014) 509(7501):453–8. doi:10.1038/nature13258
36. Pape HC, Pare D, Driesang RB. Two types of intrinsic oscillations in neurons of the lateral and basolateral nuclei of the amygdala. J Neurophysiol (1998) 79(1):205–16.
37. Randall FE, Whittington MA, Cunningham MO. Fast oscillatory activity induced by kainate receptor activation in the rat basolateral amygdala in vitro. Eur J Neurosci (2011) 33(5):914–22. doi:10.1111/j.1460-9568.2010.07582.x
38. Bienvenu TC, Busti D, Magill PJ, Ferraguti F, Capogna M. Cell-type-specific recruitment of amygdala interneurons to hippocampal theta rhythm and noxious stimuli in vivo. Neuron (2012) 74(6):1059–74. doi:10.1016/j.neuron.2012.04.022
39. Stujenske JM, Likhtik E, Topiwala MA, Gordon JA. Fear and safety engage competing patterns of theta-gamma coupling in the basolateral amygdala. Neuron (2014) 83(4):919–33. doi:10.1016/j.neuron.2014.07.026
40. Seidenbecher T, Laxmi TR, Stork O, Pape HC. Amygdalar and hippocampal theta rhythm synchronization during fear memory retrieval. Science (2003) 301(5634):846–50. doi:10.1126/science.1085818
41. Sangha S, Narayanan RT, Bergado-Acosta JR, Stork O, Seidenbecher T, Pape HC. Deficiency of the 65 kDa isoform of glutamic acid decarboxylase impairs extinction of cued but not contextual fear memory. J Neurosci (2009) 29(50):15713–20. doi:10.1523/JNEUROSCI.2620-09.2009
42. McEwen BS. Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism (2005) 54(5 Suppl 1):20–3. doi:10.1016/j.metabol.2005.01.008
43. Schmidt MV, Abraham WC, Maroun M, Stork O, Richter-Levin G. Stress-induced metaplasticity: from synapses to behavior. Neuroscience (2013) 250:112–20. doi:10.1016/j.neuroscience.2013.06.059
44. Aroniadou-Anderjaska V, Fritsch B, Qashu F, Braga MF. Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res (2008) 78(2–3):102–16. doi:10.1016/j.eplepsyres.2007.11.011
45. Lancaster E, Dalmau J. Neuronal autoantigens – pathogenesis, associated disorders and antibody testing. Nat Rev Neurol (2012) 8(7):380–90. doi:10.1038/nrneurol.2012.99
46. Melzer N, Meuth SG, Wiendl H. Neuron-directed autoimmunity in the central nervous system: entities, mechanisms, diagnostic clues, and therapeutic options. Curr Opin Neurol (2012) 25(3):341–8. doi:10.1097/WCO.0b013e3283531efb
47. Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol (2011) 10(8):759–72. doi:10.1016/S1474-4422(11)70096-5
48. Neumann H, Cavalie A, Jenne DE, Wekerle H. Induction of MHC class I genes in neurons. Science (1995) 269(5223):549–52. doi:10.1126/science.7624779
49. Neumann H, Schmidt H, Cavalie A, Jenne D, Wekerle H. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. J Exp Med (1997) 185(2):305–16. doi:10.1084/jem.185.2.305
50. Melzer N, Meuth SG, Wiendl H. CD8+ T cells and neuronal damage: direct and collateral mechanisms of cytotoxicity and impaired electrical excitability. FASEB J (2009) 23(11):3659–73. doi:10.1096/fj.09-136200
51. Bien CG, Vincent A, Barnett MH, Becker AJ, Blumcke I, Graus F, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain (2012) 135(Pt 5):1622–38. doi:10.1093/brain/aws082
52. Stinchcombe JC, Griffiths GM. Secretory mechanisms in cell-mediated cytotoxicity. Annu Rev Cell Dev Biol (2007) 23:495–517. doi:10.1146/annurev.cellbio.23.090506.123521
53. Kagi D, Vignaux F, Ledermann B, Burki K, Depraetere V, Nagata S, et al. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science (1994) 265(5171):528–30. doi:10.1126/science.7518614
54. Lowin B, Hahne M, Mattmann C, Tschopp J. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature (1994) 370(6491):650–2. doi:10.1038/370650a0
55. Meuth SG, Herrmann AM, Simon OJ, Siffrin V, Melzer N, Bittner S, et al. Cytotoxic CD8+ T cell-neuron interactions: perforin-dependent electrical silencing precedes but is not causally linked to neuronal cell death. J Neurosci (2009) 29(49):15397–409. doi:10.1523/JNEUROSCI.4339-09.2009
56. Waterhouse NJ, Sutton VR, Sedelies KA, Ciccone A, Jenkins M, Turner SJ, et al. Cytotoxic T lymphocyte-induced killing in the absence of granzymes A and B is unique and distinct from both apoptosis and perforin-dependent lysis. J Cell Biol (2006) 173(1):133–44. doi:10.1083/jcb.200510072
57. Pipkin ME, Lieberman J. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol (2007) 19(3):301–8. doi:10.1016/j.coi.2007.04.011
58. Choi C, Benveniste EN. Fas ligand/Fas system in the brain: regulator of immune and apoptotic responses. Brain Res Brain Res Rev (2004) 44(1):65–81. doi:10.1016/j.brainresrev.2003.08.007
59. Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol (2002) 20:323–70. doi:10.1146/annurev.immunol.20.100201.131730
60. Kreutzfeldt M, Bergthaler A, Fernandez M, Bruck W, Steinbach K, Vorm M, et al. Neuroprotective intervention by interferon-gamma blockade prevents CD8+ T cell-mediated dendrite and synapse loss. J Exp Med (2013) 210(10):2087–103. doi:10.1084/jem.20122143
61. Melzer N, Hicking G, Bittner S, Bobak N, Gobel K, Herrmann AM, et al. Excitotoxic neuronal cell death during an oligodendrocyte-directed CD8+ T cell attack in the CNS gray matter. J Neuroinflammation (2013) 10:121. doi:10.1186/1742-2094-10-121
62. Garg SK, Banerjee R, Kipnis J. Neuroprotective immunity: T cell-derived glutamate endows astrocytes with a neuroprotective phenotype. J Immunol (2008) 180(6):3866–73. doi:10.4049/jimmunol.180.6.3866
63. Liblau RS, Gonzalez-Dunia D, Wiendl H, Zipp F. Neurons as targets for T cells in the nervous system. Trends Neurosci (2013) 36(6):315–24. doi:10.1016/j.tins.2013.01.008
64. Chevalier G, Suberbielle E, Monnet C, Duplan V, Martin-Blondel G, Farrugia F, et al. Neurons are MHC class I-dependent targets for CD8 T cells upon neurotropic viral infection. PLoS Pathog (2011) 7(11):e1002393. doi:10.1371/journal.ppat.1002393
65. Scheikl T, Pignolet B, Dalard C, Desbois S, Raison D, Yamazaki M, et al. Cutting edge: neuronal recognition by CD8 T cells elicits central diabetes insipidus. J Immunol (2012) 188(10):4731–5. doi:10.4049/jimmunol.1102998
66. Ince-Dunn G, Okano HJ, Jensen KB, Park WY, Zhong R, Ule J, et al. Neuronal Elav-like (Hu) proteins regulate RNA splicing and abundance to control glutamate levels and neuronal excitability. Neuron (2012) 75(6):1067–80. doi:10.1016/j.neuron.2012.07.009
67. DeBoer EM, Azevedo R, Vega TA, Brodkin J, Akamatsu W, Okano H, et al. Prenatal deletion of the RNA-binding protein HuD disrupts postnatal cortical circuit maturation and behavior. J Neurosci (2014) 34(10):3674–86. doi:10.1523/JNEUROSCI.3703-13.2014
68. Cebrian C, Zucca FA, Mauri P, Steinbeck JA, Studer L, Scherzer CR, et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat Commun (2014) 5:3633. doi:10.1038/ncomms4633
69. Graus F, Keime-Guibert F, Rene R, Benyahia B, Ribalta T, Ascaso C, et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain (2001) 124(Pt 6):1138–48. doi:10.1093/brain/124.6.1138
70. Alamowitch S, Graus F, Uchuya M, Rene R, Bescansa E, Delattre JY. Limbic encephalitis and small cell lung cancer. Clinical and immunological features. Brain (1997) 120(Pt 6):923–8. doi:10.1093/brain/120.6.923
71. Darnell RB. RNA protein interaction in neurons. Annu Rev Neurosci (2013) 36:243–70. doi:10.1146/annurev-neuro-062912-114322
72. Martin DL, Rimvall K. Regulation of gamma-aminobutyric acid synthesis in the brain. J Neurochem (1993) 60(2):395–407. doi:10.1111/j.1471-4159.1993.tb03165.x
73. McBain CJ, Fisahn A. Interneurons unbound. Nat Rev Neurosci (2001) 2(1):11–23. doi:10.1038/35047544
74. Bolognani F, Qiu S, Tanner DC, Paik J, Perrone-Bizzozero NI, Weeber EJ. Associative and spatial learning and memory deficits in transgenic mice overexpressing the RNA-binding protein HuD. Neurobiol Learn Mem (2007) 87(4):635–43. doi:10.1016/j.nlm.2006.11.004
75. Bolognani F, Tanner DC, Merhege M, Deschenes-Furry J, Jasmin B, Perrone-Bizzozero NI. In vivo post-transcriptional regulation of GAP-43 mRNA by overexpression of the RNA-binding protein HuD. J Neurochem (2006) 96(3):790–801. doi:10.1111/j.1471-4159.2005.03607.x
76. Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ. Two genes encode distinct glutamate decarboxylases. Neuron (1991) 7(1):91–100. doi:10.1016/0896-6273(91)90077-D
77. Bowers G, Cullinan WE, Herman JP. Region-specific regulation of glutamic acid decarboxylase (GAD) mRNA expression in central stress circuits. J Neurosci (1998) 18(15):5938–47.
78. Kaufman DL, Houser CR, Tobin AJ. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J Neurochem (1991) 56(2):720–3. doi:10.1111/j.1471-4159.1991.tb08211.x
79. Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, et al. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A (1997) 94(25):14060–5. doi:10.1073/pnas.94.25.14060
80. Lange MD, Jungling K, Paulukat L, Vieler M, Gaburro S, Sosulina L, et al. Glutamic acid decarboxylase 65: a link between GABAergic synaptic plasticity in the lateral amygdala and conditioned fear generalization. Neuropsychopharmacology (2014) 39(9):2211–20. doi:10.1038/npp.2014.72
81. Bergado-Acosta JR, Sangha S, Narayanan RT, Obata K, Pape HC, Stork O. Critical role of the 65-kDa isoform of glutamic acid decarboxylase in consolidation and generalization of Pavlovian fear memory. Learn Mem (2008) 15(3):163–71. doi:10.1101/lm.705408
82. Sangha S, Ilenseer J, Sosulina L, Lesting J, Pape HC. Differential regulation of glutamic acid decarboxylase gene expression after extinction of a recent memory vs. intermediate memory. Learn Mem (2012) 19(5):194–200. doi:10.1101/lm.025874.112
83. Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol (2008) 7(12):1091–8. doi:10.1016/S1474-4422(08)70224-2
84. Dalmau J, Tuzun E, Wu HY, Masjuan J, Rossi JE, Voloschin A, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol (2007) 61(1):25–36. doi:10.1002/ana.21050
85. Lai M, Hughes EG, Peng X, Zhou L, Gleichman AJ, Shu H, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol (2009) 65(4):424–34. doi:10.1002/ana.21589
86. Bataller L, Galiano R, Garcia-Escrig M, Martinez B, Sevilla T, Blasco R, et al. Reversible paraneoplastic limbic encephalitis associated with antibodies to the AMPA receptor. Neurology (2010) 74(3):265–7. doi:10.1212/WNL.0b013e3181cb3e52
87. Petit-Pedrol M, Armangue T, Peng X, Bataller L, Cellucci T, Davis R, et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: a case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol (2014) 13(3):276–86. doi:10.1016/S1474-4422(13)70299-0
88. Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol (2010) 9(1):67–76. doi:10.1016/S1474-4422(09)70324-2
89. Irani SR, Alexander S, Waters P, Kleopa KA, Pettingill P, Zuliani L, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain (2010) 133(9):2734–48. doi:10.1093/brain/awq213
90. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol (2010) 9(8):776–85. doi:10.1016/S1474-4422(10)70137-X
91. Lancaster E, Huijbers MG, Bar V, Boronat A, Wong A, Martinez-Hernandez E, et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol (2011) 69(2):303–11. doi:10.1002/ana.22297
92. Diamond B, Huerta PT, Mina-Osorio P, Kowal C, Volpe BT. Losing your nerves? Maybe it’s the antibodies. Nat Rev Immunol (2009) 9(6):449–56. doi:10.1038/nri2529
93. Gleichman AJ, Spruce LA, Dalmau J, Seeholzer SH, Lynch DR. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci (2012) 32(32):11082–94. doi:10.1523/JNEUROSCI.0064-12.2012
94. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci (2010) 30(17):5866–75. doi:10.1523/JNEUROSCI.0167-10.2010
95. Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol (2014) 76(1):108–19. doi:10.1002/ana.24195
96. Zhang Q, Tanaka K, Sun P, Nakata M, Yamamoto R, Sakimura K, et al. Suppression of synaptic plasticity by cerebrospinal fluid from anti-NMDA receptor encephalitis patients. Neurobiol Dis (2012) 45(1):610–5. doi:10.1016/j.nbd.2011.09.019
97. Vitureira N, Letellier M, Goda Y. Homeostatic synaptic plasticity: from single synapses to neural circuits. Curr Opin Neurobiol (2012) 22(3):516–21. doi:10.1016/j.conb.2011.09.006
98. Peng X, Hughes EG, Moscato EH, Parsons TD, Dalmau J, Balice-Gordon RJ. Cellular plasticity induced by anti-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann Neurol (2015) 77(3):381–98. doi:10.1002/ana.24293
99. Fukata Y, Lovero KL, Iwanaga T, Watanabe A, Yokoi N, Tabuchi K, et al. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci U S A (2010) 107(8):3799–804. doi:10.1073/pnas.0914537107
100. Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science (2006) 313(5794):1792–5. doi:10.1126/science.1129947
101. Ohkawa T, Fukata Y, Yamasaki M, Miyazaki T, Yokoi N, Takashima H, et al. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J Neurosci (2013) 33(46):18161–74. doi:10.1523/JNEUROSCI.3506-13.2013
102. Boillot M, Huneau C, Marsan E, Lehongre K, Navarro V, Ishida S, et al. Glutamatergic neuron-targeted loss of LGI1 epilepsy gene results in seizures. Brain (2014) 137(Pt 11):2984–96. doi:10.1093/brain/awu259
103. Ohkawa T, Satake S, Yokoi N, Miyazaki Y, Ohshita T, Sobue G, et al. Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis. J Neurosci (2014) 34(24):8151–63. doi:10.1523/JNEUROSCI.4415-13.2014
104. Emson PC. GABA(B) receptors: structure and function. Prog Brain Res (2007) 160:43–57. doi:10.1016/S0079-6123(06)60004-6
105. Stork O, Ji FY, Kaneko K, Stork S, Yoshinobu Y, Moriya T, et al. Postnatal development of a GABA deficit and disturbance of neural functions in mice lacking GAD65. Brain Res (2000) 865(1):45–58. doi:10.1016/S0006-8993(00)02206-X
106. Bergado-Acosta JR, Muller I, Richter-Levin G, Stork O. The GABA-synthetic enzyme GAD65 controls circadian activation of conditioned fear pathways. Behav Brain Res (2014) 260:92–100. doi:10.1016/j.bbr.2013.11.042
107. Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, et al. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun (1996) 229(3):891–5. doi:10.1006/bbrc.1996.1898
108. Geis C, Weishaupt A, Grunewald B, Wultsch T, Reif A, Gerlach M, et al. Human stiff-person syndrome IgG induces anxious behavior in rats. PLoS One (2011) 6(2):e16775. doi:10.1371/journal.pone.0016775
109. Sandhu KV, Yanagawa Y, Stork O. Transcriptional regulation of glutamic Acid decarboxylase in the male mouse amygdala by dietary phyto-oestrogens. J Neuroendocrinol (2015) 27(4):285–92. doi:10.1111/jne.12262
110. Shaban H, Humeau Y, Herry C, Cassasus G, Shigemoto R, Ciocchi S, et al. Generalization of amygdala LTP and conditioned fear in the absence of presynaptic inhibition. Nat Neurosci (2006) 9(8):1028–35. doi:10.1038/nn1732
111. Lin HC, Tseng YC, Mao SC, Chen PS, Gean PW. GABAA receptor endocytosis in the basolateral amygdala is critical to the reinstatement of fear memory measured by fear-potentiated startle. J Neurosci (2011) 31(24):8851–61. doi:10.1523/JNEUROSCI.0979-11.2011
112. Armony JL, Servan-Schreiber D, Romanski LM, Cohen JD, LeDoux JE. Stimulus generalization of fear responses: effects of auditory cortex lesions in a computational model and in rats. Cereb Cortex (1997) 7(2):157–65. doi:10.1093/cercor/7.2.157
113. Kim D, Samarth P, Feng F, Pare D, Nair SS. Synaptic competition in the lateral amygdala and the stimulus specificity of conditioned fear: a biophysical modeling study. Brain Struct Funct (2015). doi:10.1007/s00429-015-1037-4
114. Pare D, Quirk GJ, Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol (2004) 92(1):1–9. doi:10.1152/jn.00153.2004
115. Royer S, Pare D. Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience (2002) 115(2):455–62. doi:10.1016/S0306-4522(02)00455-4
116. Yu SY, Wu DC, Zhan RZ. GluN2B subunits of the NMDA receptor contribute to the AMPA receptor internalization during long-term depression in the lateral amygdala of juvenile rats. Neuroscience (2010) 171(4):1102–8. doi:10.1016/j.neuroscience.2010.09.038
117. Klein CJ, Lennon VA, Aston PA, McKeon A, O’Toole O, Quek A, et al. Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol (2013) 70(2):229–34. doi:10.1001/jamaneurol.2013.592
118. Klein CJ, Lennon VA, Aston PA, McKeon A, Pittock SJ. Chronic pain as a manifestation of potassium channel-complex autoimmunity. Neurology (2012) 79(11):1136–44. doi:10.1212/WNL.0b013e3182698cab
119. Owuor K, Harel NY, Englot DJ, Hisama F, Blumenfeld H, Strittmatter SM. LGI1-associated epilepsy through altered ADAM23-dependent neuronal morphology. Mol Cell Neurosci (2009) 42(4):448–57. doi:10.1016/j.mcn.2009.09.008
120. Tuunanen J, Halonen T, Pitkanen A. Status epilepticus causes selective regional damage and loss of GABAergic neurons in the rat amygdaloid complex. Eur J Neurosci (1996) 8(12):2711–25. doi:10.1111/j.1460-9568.1996.tb01566.x
121. Klueva J, Munsch T, Albrecht D, Pape HC. Synaptic and non-synaptic mechanisms of amygdala recruitment into temporolimbic epileptiform activities. Eur J Neurosci (2003) 18(10):2779–91. doi:10.1111/j.1460-9568.2003.02984.x
122. Prager EM, Pidoplichko VI, Aroniadou-Anderjaska V, Apland JP, Braga MF. Pathophysiological mechanisms underlying increased anxiety after soman exposure: reduced GABAergic inhibition in the basolateral amygdala. Neurotoxicology (2014) 44:335–43. doi:10.1016/j.neuro.2014.08.007
123. Berretta S, Pantazopoulos H, Markota M, Brown C, Batzianouli ET. Losing the sugar coating: potential impact of perineuronal net abnormalities on interneurons in schizophrenia. Schizophr Res (2015). doi:10.1016/j.schres.2014.12.040
124. Roozendaal B, McGaugh JL. Memory modulation. Behav Neurosci (2011) 125(6):797–824. doi:10.1037/a0026187
125. Ramirez F, Moscarello JM, LeDoux JE, Sears RM. Active avoidance requires a serial basal amygdala to nucleus accumbens shell circuit. J Neurosci (2015) 35(8):3470–7. doi:10.1523/JNEUROSCI.1331-14.2015
126. Bettcher BM, Gelfand JM, Irani SR, Neuhaus J, Forner S, Hess CP, et al. More than memory impairment in voltage-gated potassium channel complex encephalopathy. Eur J Neurol (2014) 21(10):1301–10. doi:10.1111/ene.12482
Keywords: amygdala, circuit, autoimmunity, limbic encephalitis, T cells, B cells, antibodies, neurons
Citation: Melzer N, Budde T, Stork O and Meuth SG (2015) Limbic encephalitis: potential impact of adaptive autoimmune inflammation on neuronal circuits of the amygdala. Front. Neurol. 6:171. doi: 10.3389/fneur.2015.00171
Received: 07 May 2015; Accepted: 20 July 2015;
Published: 03 August 2015
Edited by:
Fernando Cendes, University of Campinas, BrazilReviewed by:
João P. Leite, University of São Paulo School of Medicine at Ribeirão Preto, BrazilMarino M. Bianchin, Universidade Federal do Rio Grande do Sul, Brazil
Copyright: © 2015 Melzer, Budde, Stork and Meuth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nico Melzer and Sven G. Meuth, Department of Neurology, University of Münster, Albert-Schweitzer-Campus 1, Münster 48149, Germany,bmljby5tZWx6ZXJAdWttdWVuc3Rlci5kZQ==;c3Zlbi5tZXV0aEB1a211ZW5zdGVyLmRl