 Jorge Correale
Jorge Correale Mauricio F. Farez
Mauricio F. Farez- Department of Neurology, Institute for Neurological Research Dr. Raúl Carrea, FLENI, Buenos Aires, Argentina
Multiple sclerosis (MS) is an inflammatory disorder causing central nervous system (CNS) demyelination and axonal injury. Although its etiology remains elusive, several lines of evidence support the concept that autoimmunity plays a major role in disease pathogenesis. The course of MS is highly variable; nevertheless, the majority of patients initially present a relapsing–remitting clinical course. After 10–15 years of disease, this pattern becomes progressive in up to 50% of untreated patients, during which time clinical symptoms slowly cause constant deterioration over a period of many years. In about 15% of MS patients, however, disease progression is relentless from disease onset. Published evidence supports the concept that progressive MS reflects a poorly understood mechanism of insidious axonal degeneration and neuronal loss. Recently, the type of microglial cell and of astrocyte activation and proliferation observed has suggested contribution of resident CNS cells may play a critical role in disease progression. Astrocytes could contribute to this process through several mechanisms: (a) as part of the innate immune system, (b) as a source of cytotoxic factors, (c) inhibiting remyelination and axonal regeneration by forming a glial scar, and (d) contributing to axonal mitochondrial dysfunction. Furthermore, regulatory mechanisms mediated by astrocytes can be affected by aging. Notably, astrocytes might also limit the detrimental effects of pro-inflammatory factors, while providing support and protection for oligodendrocytes and neurons. Because of the dichotomy observed in astrocytic effects, the design of therapeutic strategies targeting astrocytes becomes a challenging endeavor. Better knowledge of molecular and functional properties of astrocytes, therefore, should promote understanding of their specific role in MS pathophysiology, and consequently lead to development of novel and more successful therapeutic approaches.
Introduction
Multiple sclerosis (MS) is an inflammatory disorder causing central nervous system (CNS) demyelination and axonal injury. Although its etiology remains elusive, several lines of evidence support the concept that autoimmunity plays a major role in disease pathogenesis (1).
The course of MS is highly variable; nevertheless, most patients initially present a relapsing–remitting clinical course [relapsing–remitting MS (RRMS)]. After 10–15 years of disease, this pattern becomes progressive in up to 50% of untreated patients, during which time clinical symptoms slowly cause constant deterioration over a period of many years [secondary progressive MS (SPMS)]. In about 15% of MS patients, however, disease progression is relentless from disease onset [primary progressive MS (PPMS)] (2).
In recent decades, better understanding of RRMS disease mechanisms has led to development of different disease-modifying therapies, reducing both severity and frequency of new relapses, by modulating or suppressing the immune system (3). By contrast, therapeutic options available for progressive MS are comparatively disappointing, and remain challenging. One possible reason behind this is a lack of understanding of the pathogenic mechanisms driving progressive MS.
The conventional view explaining the sequence of MS events is one in which systemic activation of myelin reactive cells from the periphery migrate into the CNS, leading to inflammation and development of focal demyelinating lesions, which constitute the main pathological substrate for relapses. The progressive phase of MS reflects a poorly understood and insidious form of axonal degeneration with neuronal loss, independent of relapses. Pathological studies have shown that axonal degeneration occurs diffusely throughout normal appearing white matter (4). Although this neurodegenerative component is associated with inflammation (5), there is growing awareness that T cell-mediated inflammatory mechanisms alone, cannot explain the degenerative process. Recently, a number of observations have challenged the concept of an autoimmune attack against myelin mediated only by adaptive immune response to self antigens, as the complete and full explanation behind the disease, particularly during its progressive phases. For example, pathology studies of early lesions show oligodendrocyte and myelin loss, in the absence of T or B cell infiltrates (6). Likewise, large areas of myelin loss are seen in cerebral cortex and deep gray matter nuclei with a paucity of infiltrating immune cells (7–9). Activation and proliferation of microglia and astrocytes observed within demyelinating lesions suggest that innate immune response contribution by resident CNS cells might play a critical role in both oligodendrocyte injury and axonal degeneration (10). Indeed, glial cells and astrocytes, in particular, were found to be highly abnormal, early in the study of MS lesions (11). Large and bizarre astrocytes containing multiple and sometimes fragmented nuclei or engulfing other cells were found in early active lesions (12), and considered by investigators during the late nineteenth and early twentieth century, to be the major cell type targeted in MS (13). However, later identification of oligodendrocytes as the myelinating cell of the CNS, as well as of their depletion from MS lesions, caused the role of astrocytes in MS pathogenesis to be largely ignored after about 1930. Nevertheless, most neuropathologists continue to report astrocyte appearance in MS lesions, and consider it an important indicator of lesional activity and age (14).
This review summarizes current studies on the role of astrocytes in disease progression, and discusses data on some of the mechanisms through which these cells may play a key role in MS pathogenesis.
Overview of Astrocytes
Astrocytes are the most abundant and heterogeneous type of glial cell (15). Two main subtypes exist: fibrous and protoplasmic, based on cell morphology and anatomical location. Fibrous astrocytes of the white matter have small cell bodies, and their processes align with myelinated fibers, giving them an elongated morphology (16). Protoplasmic astrocytes have more primary processes, as well as a higher degree of branching compared to fibrous astrocytes, and are located in the gray matter (17). Additionally, other morphologically distinct and more regional populations of astrocytes have been described, such as Müller cells in the retina, Bergmann glia and velate astrocytes in the cerebellum, radial astrocytes in the spinal cord, among others (18). Consequently, astrocytes can no longer be considered as a homogeneous group of cells. Their morphological diversity, specific density, as well as proliferation rate will be determined by interactions with the microenvironment, particularly during development, reflecting important molecular and functional differences between astrocyte types (19, 20).
Astrocytes have at least two different origins: (1) directly from radial glial cells located in the ventricular zone and (2) from a proliferative and migratory population located in the subventricular zone (SVZ) (21–24). New astrocytes may arise either from the proliferation of mature astrocytes or from differentiation of progenitors. Notably, there is little evidence that mature astrocytes divide in the uninjured brain (19). By contrast, very active proliferation is associated with scar formation following injury (see below).
Astrocyte development is regulated by different molecules and through different intracellular pathways including the IL6/LIF family of cytokines, the TGF-β growth factor family, fibroblast growth factor (FGF), and Notch and Notch ligand pairs (25). Additionally, epigenetic factors also influence astrocyte development. A number of astrocyte genes are methylated early in development, including GFAP, Aldolase C, and Kir4.1, a process that serves to repress astrocyte specific gene transcription (26). By contrast, demethylation occurring during early astrocyte development allows LIF to upregulate genes, through binding to transcription factors present downstream to astrocyte gene promoters in the signaling pathways (26, 27).
Expression of glial fibrillary acidic protein (GFAP) has become the prototypic marker for identifying astrocytes within the CNS; however, expression patterns differ across anatomical regions (28). Moreover, other CNS-resident cells, such as NG2 and pericytes, have also been to shown to be GFAP+ (9). Several other antibodies against intermediate filament proteins, including cytoplasmic or membrane protein markers, such as vimentin, nestin, S100 calcium-binding protein β (S100β), glutamine synthetase (GS), or glutamate/aspartate transporter (GLAST), are also commonly used to label normal and reactive astrocytes (28). A significant drawback of current immunohistochemistry techniques is that no reliable markers exist to identify different astrocyte subtypes, making it hard to establish whether any given behavior observed corresponds to astrocytes in general, or is characteristic of a particular subtype only.
Astrocytes contact blood vessels and are linked to each other via gap junctions, and to oligodendrocytes via heterotypic gap junctions. Adjacent astrocytes present homomeric gap junctions at the cytoplasmic level, formed by connexin (Cx) 43 and Cx 30, through which molecules, such as K+ and glutamate, are dissipated, and intercellular Ca++ waves propagate (29). In addition, astrocytes support several activities essential for neuronal function, including (1) an active role in both formation and pruning of synapses (30); (2) regulation of extracellular concentrations of ions and neurotransmitters (29, 31); (3) synthesis of metabolic substrates for neurons, such as glycogen, sterols, and lipoproteins (23, 32); (4) formation and maintenance of blood–brain barrier (BBB) integrity, thus protecting the brain from influx of toxic substances and ions, as well as maintaining extracellular space volume (33); and (5) removal of neurotransmitters released by active neurons, such as glutamate (34). Central questions remaining include whether astrocytes in general carry out all these functions, and if not, what relevance differences in subpopulations may play in human disease.
Astrocytes in Multiple Sclerosis
Contribution of Astrocytes to MS Lesion Development
Around acute inflammatory lesions, astrocyte reactivity is widespread. A gradient of response is observed, ranging from modestly swollen process-bearing cells in normal adjacent white matter, to hypertrophic astrocytes in the center of a lesion (14, 35). As lesions age not only persist hypertrophic astrocytes but also begin to develop bundles of glial filaments, GFAP immunoreactivity increases, and edema decreases. Relapsing disease activity is associated with recurrent inflammatory activity, astroglial reactivity (particularly along lesion borders), and recent astrocyte mitotic activity (14, 35). Studies in experimental autoimmune encephalomyelitis (EAE) have shown that activation of astrocytes, and loss of their end-feet around small blood vessels represent early events in lesion development, linked to loss of BBB function, subsequent CNS inflammation, as well as perivascular edema (14). It is well recognized that factors produced by astrocytes are required for establishment and maintenance of endothelial cells forming the BBB. For example, astrocyte activation by macrophage produced IL-1, leads to induction of hypoxia inducible factor-1 (HIF-1), and its target, vascular endothelial growth factor A (VEGF-A) in astrocytes, which acting on endothelial cells induces down-regulation or loss of tight proteins claudin-5 (CLN5) and occludin, determining a focal loss of BBB function in injured tissue, a process mediated by eNOS (36, 37). Inactivation of VEGF-A expression, or systemic selective inhibition of eNOS reduces BBB breakdown, decreasing lymphocyte infiltration and tissue damage, protecting against neurological deficit in EAE (38). Besides tight junctions on endothelial cells, astrocyte end-feet forming glia provide an additional barrier against autoreactive cell activity in the CNS. Furthermore, imbalance between upregulation of matrix metalloproteinases (MMPs) in both astrocytes and macrophages, compared to stable expression or reduction of parenchymal basal membrane components aid in encephalitogenic cell dispersion into the CNS (39). However, it should be noted that remodeling of the extracellular matrix (ECM) can be both deleterious and beneficial, depending on the situation and on the type of MMP involved (Table 1) (14, 20, 39).
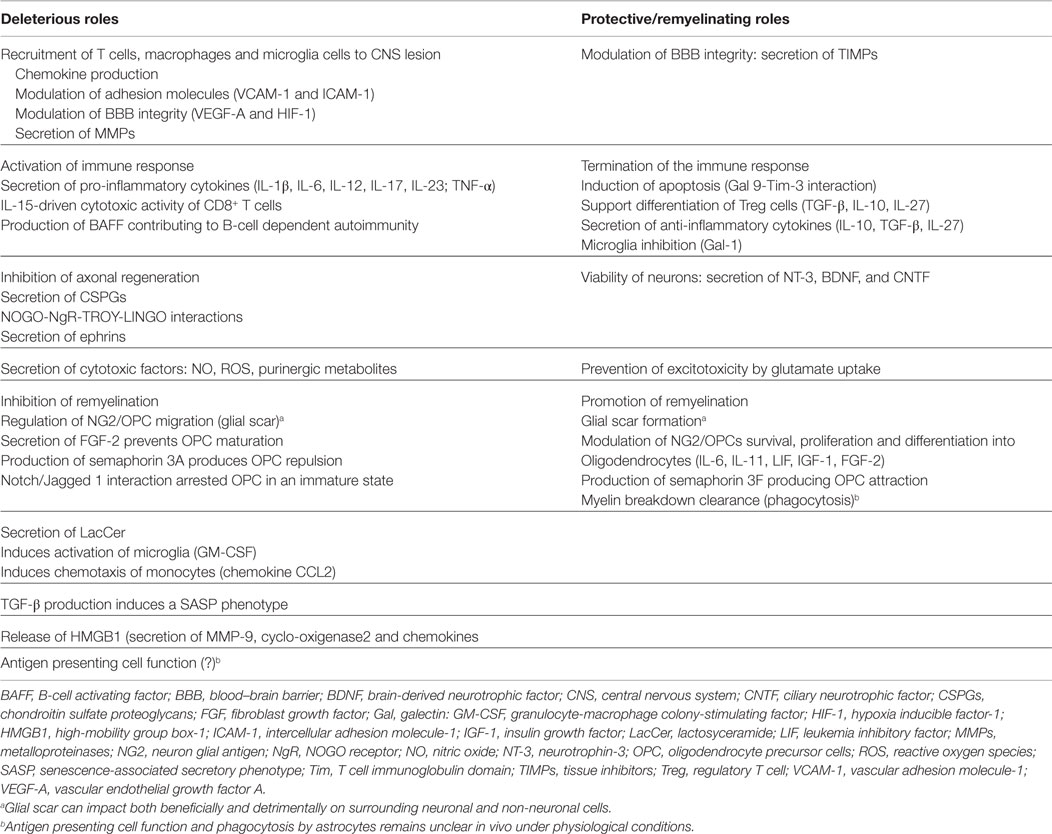
Table 1. The dual role of astrocytes in the pathophysiology of multiple sclerosis.
It is important to point out the dual role of astrocytes, not only aiding in axonal degeneration and demyelination but also creating a permissive environment promoting remyelination (Table 1). The particular impact of astrocytes on pathogenesis and repair of an inflammatory process, therefore, will be dependent on a number of factors, including timing after injury, type of lesion and surrounding microenvironment, as well as interaction with other cell types and factors influencing their activation (39).
Astrocytes and the Innate Immune System
Innate immunity is the initial non-specific response to foreign pathogens. The system includes cellular barriers, such as the BBB and diverse immune cells of myeloid origin, including DCs, macrophages, monocytes, NK cells, NKT cells, mast cells, granulocytes and γδ T cells in the periphery and microglia cells in the CNS. Innate immunity also includes non-myeloid cells, such as astrocytes (9). Cellular innate immune responses to diverse stimuli are accomplished through an array of pattern recognition receptors (PRRs) that bind to diverse pathogen-associated molecular patterns (PAMPs) (40). Notably, PRRs also recognize self-molecules released after cell damage or death. These molecules, known as danger-associated molecular patterns, include diverse ligands, such as heat-shock proteins, double stranded DNA, and purinergic metabolites (9, 41). Responses to endogenous host molecules may trigger inflammatory reactions, and therefore play an important role in autoimmunity.
Astrocytes express diverse PRRs, and can mediate innate immune responses through several mechanisms (10, 42). First, astrocytes directly affect cell entry to the CNS, via the BBB, by regulating expression of adhesion molecules, particularly vascular adhesion molecule-1 (VCAM-1) and intercellular adhesion-molecule-1 (ICAM-1) that bind to lymphocyte receptors, namely, very late antigen-4 (VLA4) and lymphocyte function-associated antigen-1 (LFA-1), respectively (43, 44). In addition, release of IL-6, IL-1β, TNF-α, and TGF-β by astrocytes can control passage of immune cells through the BBB, by acting on endothelial cells and tight junctions (33, 45, 46).
Second, astrocytes secrete different chemokines, such as CCL-2 (MCP-1), CCL5 (RANTES), IP-10 (CXCL10), CXCL12 (SDF-1), and IL-8 (CXCL8), which attract both peripheral immune cells (e.g., T cells, monocytes, and DCs), as well as resident CNS cells (microglia) to lesion sites (47). This could represent the primary mechanism through which astrocytes perpetuate immune-mediated demyelination and neurodegeneration. In vitro studies confirm that human astrocytes secrete IP-10, CCL-2, and CXCL12 in response to inflammatory cytokines IL-1β, TNF-α and IFN-γ, suggesting astrocyte-induced immunopathology may be a consequence of activation by infiltrating T cells (48–50).
Third, astrocytes may affect both the number and the phenotype of T cells in the CNS. Cytokines secreted by astrocytes have the potential of committing T cells to a pro-inflammatory phenotype (Th1 and Th17) or to a regulatory phenotype (Treg, Tr1). Under inflammatory conditions astrocytes express all subunits of IL-12/IL-23, as well as CD24, favoring the development of Th17 and Th1 cells in the CNS during EAE, thereby affecting its severity (51, 52). Additionally, IL-9 receptor complex is constitutively expressed in astrocytes, T cell-derived IL-9 induces astrocytes to produce CCL20, which in turn induces Th17 cell migration in vitro (53). Treatment with anti-IL-9 neutralizing antibodies attenuates EAE, decreasing the number of infiltrating Th17 cells, and reducing CCL-20 expression in astrocytes (53). Furthermore, astrocyte-driven IL-15 production, which has been observed in MS lesions, has been shown to have an important role in encephalitogenic activity of CD8+ T cells (54). By contrast, astrocytes can also terminate T cell responses, either by induction of apoptosis of infiltrating cells through FAS-L, which is highly expressed on astrocyte end-feet (55), or through interaction of galectin-9 and its ligand Tim-3, present in Th1 and CD8+ cytotoxic T cells (56).
Fourth, B-cell-activating factor (BAFF), critical for both B cell development and survival, as well as for the production of immunoglobulins, is constitutively expressed by astrocytes in normal CNS. BAFF expression in astrocytes is upregulated in MS lesions and in EAE affected mice, suggesting astrocytes may contribute to drive B-cell-dependent autoimmunity (57).
Fifth, astrocytes modulate microglial and macrophages activity through two different pathways: (a) inducing their recruitment toward lesion sites by producing chemotactic signals (CXCL-10-CXCR3) (58) and (b) by secreting GM-CSF, M-CSF, or TGF-β, which can regulate Class II expression, and even microglial phagocytosis (59).
Finally, an important function of innate immune cells is to act as antigen presenting cells (APCs). However, although astrocytes express major histocompatibility complex (MHC) class I and class II molecules in vitro capable of presenting myelin antigens, their ability to also express co-stimulatory molecules including CD40, CD80, and CD86 challenges this function, making their final effect unclear (60, 61). Nor is it clear to what degree astrocytes can perform phagocytosis, or process and present antigens, particularly under physiological conditions in vivo (62).
Recent investigations have demonstrated that in chronic phases of EAE, astrocyte depletion ameliorates disease severity. This deleterious effect of astrocytes on EAE is mediated by preferential expression of 4-galactosyltransferase 5 and 6 (B4GALT5 and B4GALT6) (63). Notably, in human MS lesions, B4GALT6 is expressed by reactive astrocytes. These enzymes synthesize the signaling molecule lactosylceramide (LacCer), the expression of which is significantly increased in the CNS during progressive phases of EAE. Furthermore, intraperitoneal administration of LacCer exacerbates existing signs of EAE. LacCer promotes astrocyte activation in an autocrine manner, via the NF-κB and IRF-1 pathways (63, 64), leading to inducing GM-CSF and CCL2 genes, consequently activating microglia and causing infiltration of monocytes from blood, respectively. Remarkably, inhibition or knockout of B4galt6 in mice suppresses disease progression, local CNS innate immunity, and neurodegeneration in EAE, and interferes with human astrocyte activation in vitro (63).
Astrocytes as a Source of Cytotoxic Factors
In most areas of myelin breakdown, it has been documented that activated astrocytes secrete compounds with toxic effects on neurons, axons, and oligodendrocytes/myelin, including reactive oxygen and nitrogen species, glutamate, and ATP (14). In rodents, astrocytes stimulated with IFN-γ, IL-17, or LPS induce nitric oxide synthase (iNOS) (65, 66). Likewise, IL-1β as well as combined treatment with TGF-β plus IFN-γ increases percentage of astrocyte secreted nitric oxide (NO), which is among the most prominent damage-inducing molecules in neurodegeneration (67, 68). Moreover, in situ hybridization and immunohistochemistry of astrocytes in MS, as well as in EAE lesions, demonstrates extensive iNOS reactivity and positive nitrotyrosine presence (69, 70). Furthermore, recent studies suggest a strong relationship between excessive calcium influx mediated by glutamate receptor stimulation (see below), and increased NO synthase activity, as well as amplified formation of reactive oxygen species (ROS), providing a link between excitotoxic insult and NO-mediated damage. Simultaneously, excitotoxicity is further increased by NO, which stimulates glutamate release from astrocytes (71). Remarkably, the predominant contribution of NO to excitotoxicity depends on increased superoxide ion production, which reacts with NO forming peroxinitrite (ONOO−), resulting in neuronal necrosis or apoptosis, depending on its concentration (72). Furthermore, ONOO− inactivates glutamate transporters in astrocytes, directly damaging myelin, oligodendrocytes, and axons (73).
Decreased uptake of glutamate by astrocyte transporters could also contribute to pathologically elevated levels of extracellular glutamate, which are directly toxic to oligodendrocytes, axons, and neurons (74). In mice, TNF-α from cortical astrocytes down-regulates expression of glutamate transporters in astrocytes, thus limiting glutamate uptake (75). Furthermore, knock down of glutamate transporters: GLAS and GLT-1 using antisense oligonucleotide causes neurotoxicity in mice (76). Excitotoxicity is caused mainly by sustained activation of glutamate receptors and massive subsequent influx of Ca++ into viable neurons. Calcium, which is the primary signaling agent involved in excitotoxicity injury, enters cells through various mechanisms, but the most important is entrance through ion channels coupled to NMDA receptors and AMPA/kainate glutamate receptors (77, 78). Calcium overload determined by glutamate receptor activation, in turn, activates several Ca++-dependent enzymes associated with neurodegeneration and cell death by causing membrane breakdown, cytoskeleton alteration, and NO-derived free radical formation. Moreover, intracellular calcium increase results in changes in microtubules and neurofilament phosphorylation, which ultimately leads to axon cytoskeleton breakdown (79, 80). Recent studies have shown glutamate can also be toxic to white matter oligodendrocytes and myelin, via mechanisms triggered by AMPA/kainate receptors (81). Indeed, treatment with glutamate receptor antagonists protects oligodendrocytes from damage, ameliorating EAE (82). Thus, proper function of glutamate uptake in astrocytes is critical to preclude brain cell damage, and strict regulation of extracellular glutamate levels appears to be a very prominent therapeutic strategy preventing neurodegeneration in MS.
Extracellular purine/pyrimidine metabolites are also exogenous signals playing important destructive/protective roles in neuron to glia, or glia to glia communication within normal or injured brain tissue. They activate membrane-bound ionotropic or metabotropic P2 receptors. Astrocytes express various types of metabotropic P2Y metabotropic, and ionotropic P2X purinoreceptors. Studies in MS lesions have shown preferential expression of P2X7 receptor on astrocytes (83). Although expression is low in resting human fetal astrocytes, P2X7 is upregulated in response to IL-1β in vitro and in reactive astrocytes around MS lesions, a putative IL-1β rich environment (84). Functionally, upregulation of P2X7 results in increased responsiveness to ATP, formation of membrane pores, and increased influx of Ca++ (85). Furthermore, purinergic signaling through P2X7 receptors stimulates IL-1β-induced upregulation of NO synthase (84). Thus, activation of the P2X7 receptor in EAE can trigger toxic effects on oligodendrocytes, axons, and neurons through different mechanisms, producing in vivo lesions reminiscent of MS plaques, displaying oligodendrocyte death, demyelination, and axonal damage.
Astrocytes Inhibit Remyelination and Axon Regeneration by Forming a Glial Scar
Astrocytes respond to injuries through a process commonly referred to as reactive astrogliosis, which involves changes in cell morphology and molecular expression. It is important to remember that although some aspects of glial reactivity are likely to be protective, others may contribute to disease progression. Establishing the molecular basis of such differences may therefore help identify novel therapeutic strategies. Although the best known aspect of reactive astrogliosis is scar formation the concept of reactive astrogliosis is still incomplete, we are only just starting to understand its molecular and cellular characteristics, as well as its multifaceted functions in disease pathogenesis and in CNS recovery from injury. The scar is composed primarily of astrocytes, however, in severe lesions, interaction with other cell types including oligodendrocyte progenitor cells (OPCs) and fibromeningeal cells also occurs (86, 87). Several specific molecular and morphological features have been observed in astrocytes during reactive astrogliosis in both human pathology and animal models (88, 89), of which upregulation of GFAP, vimentin, nestin, and the less investigated synemin are hallmarks. A number of other molecules, such as TGF-α, ciliary neurotrophic factor (CNTF), LIF, and oncostatin M, trigger astrocyte activation in the rodent brain (90). Interestingly, levels of IL-6, LIF, and oncostatin M mRNA, all ligands in the gp130/activator of transcription 3 (STAT3) signaling pathway, are elevated prior phosphorylation and nuclear transcription of STAT 3, both in astrocytes and during astroglyosis induction (91). Nevertheless, it is also conceivable that at least some of these molecules exert effects on astrocytes through other cell types, such as microglia, neurons, or endothelial cells. By contrast, signaling mediated by β1-integrin has the opposite effect on astrocyte activation and is required to promote development of a mature non-reactive astrocyte (92). Other mechanisms may also contribute to astrogliosis. It has been shown that inositol 1,4,5-triphophate (IP3)-dependent Ca2+ signaling and the downstream functions of N-cadherin in astrocytes are required for normal reactive astrogliosis (93). Likewise, epidermal growth factor receptor (EGFR) has been implicated in astrocyte transition from a non-reactive to a reactive state (94). Moreover, astrocytes react to endogenous or exogenous ATP with hypertrophy, swelling cell body and main processes, and generating proliferation, ultimately resulting in an astrogliosis phenotype, which subsequently forms a glial scar (83). ATP per se can trigger these biological effects through activation of P2 receptors (P2R), or through its metabolites ADP, activating some P2R and adenosine through P1R activation (95). Another factor that might contribute to astrocyte ability to react to injury is cell polarity and migration: astrocytes depleted of RhoGTPase Cdc42, a key regulator of polarization, impaired recruitment to lesions despite GFAP upregulation and hypertrophic response (96). It is important to note that reactive astrogliosis is, at least partially, disease specific. For example, reactive astrocytes profoundly affect post-ischemic stages by secreting VEGF, which in turn stimulates formation of new blood vessels and synaptogenesis (97, 98), this beneficial effect contrasts sharply with induction of BBB breakdown and lymphocyte infiltration observed in autoimmune CNS inflammation, which worsen disease (38).
Several experimental approaches have been used to either eliminate reactive astrocytes, or prevent them from becoming fully reactive. Thus, infiltration of CD11b+ microglia/monocytes in a retinal detachment model was blocked in GFAP−/− Vim−/− mice, suggesting activated glial cells are critical for recruitment of microglia/monocytes to injured areas (99). Similarly, GFAP or nestin promoter ablation of STAT3 in astrocytes attenuated upregulation of GFAP, reduced astrocytes hypertrophy, limited astrocyte migration, and led to more widespread infiltration of CD11b+ inflammatory cells, associated with larger lesions and more prominent impairment (100, 101). Conversely, mice with nestin promoter-driven ablation of SOCS3, which inhibits STAT3 signaling, showed increased astrocyte migration, and enhanced contraction of lesions as well as improvement of functional recovery after spinal cord injury (101). Also, ATP released from damaged cells after injury, acting via P2Y receptors enhanced the proliferative effects of FGF2, whereas P2X receptor stimulation inhibited the ability of FGF2 to stimulate DNA synthesis in astrocyte cultures (102). These variable effects of ATP and of other purinergic ligands are mediated by phosphorylation of different STAT3 residues (103). Therefore, pharmacological antagonists of P2X/P2Y receptors might ameliorate long-lasting consequences of different CNS injuries. Overall, these results point to an important role of STAT3 signaling in CNS injury, which may limit development of a potential toxic environment by the rest of the CNS (100, 101), although this might also restrict regenerative responses at a later stage [Ref. (104); see below]. Consequently, there is urgent need for better understanding of the molecular pathways regulating distinct aspects of reactive astrogliosis, in order to allow selective blockade of molecules inhibiting axonal outgrowth, but still permit reactive astrocytes to form a protective scar.
Glial scars are evident in tissue from MS patients and mice with EAE and surround areas of demyelination (105). The purpose of scar formation would appear to be isolation of damaged CNS areas, to prevent spread of tissue destruction. However, glial scar rigidity results in inhibition of both remyelination and axonal regeneration, both negative effects mediated through different mechanisms. First, astrocytes may be detrimental for remyelination by over secreting FGF-2, which in turn promotes OPC proliferation and survival, but prevents maturation (106). Another molecule that appears to play an important role in preventing OPC maturation is the glycosaminoglycan (GAG) hyaluronan, which is found throughout the ECM and in CNS white matter (107). Hyaluronan is produced by astrocytes, and interacts with CD44, a receptor present on OPCs, astrocytes, and T cells in both MS and EAE CNS tissue (19, 108). Oligodendrocytes that co-localize with hyaluronan express an immature phenotype, and treatment of OPCs with hyaluronan in vitro prevents maturation (109).
Second, astrocytes release inhibitory ECM molecules known as chondroitin sulfate proteoglycans (CSPGs) in injured areas (110). CSPGs are a family of molecules characterized by a protein core to which highly sulfated GAG chains are attached. Three types of CSPGs are preferentially localized to astrocytes in vivo: neurocan, brevican, and NG2. Neurocan (secreted) and brevican (cell bound) are the major proteoglycans produced by astrocytes in vitro and both have been shown to inhibit axon growth, following CNS damage (104). There is clear evidence that CSPGs are produced in excess by astrocytes when they become reactive and that inhibitory activity of CSPGs depends on the GAG component, as removal of GAG chains from the protein core eliminates inhibition (104, 111). After injury, CSPGs expression is rapidly upregulated by reactive astrocytes, forming an inhibitory gradient that is highest at the center of lesions and diminishes gradually toward the periphery (112). Meanwhile, NG2 is most often regarded as a marker of OPCs in adult CNS tissue. Along the borders of glial scars, NG2+ cells are found in great numbers. While many of these cells are regarded as OPCs, evidence indicates that NG2+ cells are also able to become astrocytes in vivo (113). Therefore, astrocyte-derived NG2 cells may provide inhibitory signals, suppressing axon regeneration. In vitro studies have demonstrated that NG2 inhibits axonal growth, and that this inhibition can be overcome by anti-NG2 antibody treatment (114). CSPG-mediated inhibition could severely affect both cytoskeleton and membrane components of growth cone architecture. In addition, many signaling pathways that mediate inhibition, such as those involving the GTPase RhoA, share similarities with those triggered by myelin-associated inhibitors (MAIs) (see below).
Aside from CSPGs, there are other less studied inhibitory molecules expressed by astrocytes that suppress axonal growth. Ephrins (EPH) and their receptors, for example, are secreted by normal astrocytes and increased in MS lesions (115). Evidence indicates that astrocyte-derived ephrins create a basal lamina around areas of injury, contributing to scar formation. Additionally, ephrins induce collapse of the axonal growth cone through activation of axon-bound EPH tyrosine-receptor kinase (116).
Finally, MAIs, such as Nogo-A, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein, can also inhibit axonal growth (112). These three proteins share two common neuronal receptors NgR1, together with its co-receptors (p75, TROY, and LINGO-1), and the recently described paired immunoglobulin receptor-B (PirB) (117, 118). In addition, new ligands binding to the NOGO receptor complex have been reported: glioma-inactivated gene product (LGI), B lymphocyte stimulator (BLyS), and FGF (119–121). Moreover, new receptors for MAIs have been recently described, such as NgR1 isoform, NgR2, and NgR3 (122). CNS regeneration inhibitors target the actin cytoskeleton thus regulating dendritic spine maturation, as well as long-term synaptic stability and plasticity. Although most evidence shows that many CNS inhibitors and their receptors are present or near synapses, astrocytes express p75, TROY, and BLyS, therefore, interaction with these ligands suggests astrocytes may also inhibit remyelination and axonal regeneration through these pathways (123).
Astrocytes Contribute to Axon Mitochondrial Dysfunction
There is emerging evidence that mitochondrial dysfunction actively contributes to neurodegeneration and axonal damage. Mitochondria are also key for ATP production and calcium signaling regulation (71).
Astrocytes may reduce mitochondrial energy metabolism in axons through different mechanisms. One related to increase NO production. Nitric oxide synthase (NOS2) expression is increased in both active focal lesions and normal appearing white matter (124). Interestingly, immunostaining shows NOS2+ cells are predominantly astrocytes. A loss of astrocytic β2 adrenergic receptors might explain increased NOS2 expression (125). Indeed, noradrenergic stimulation leads to increased cAMP levels and consequently inhibits NOS2 expression in astrocytes. Elevated levels of NO can compete with oxygen for binding on complex IV of the mitochondrial respiratory chain, reducing electron flow and subsequent ATP synthesis (126); a second mechanism is excitoxicity triggered by increased glutamate levels (see above), and intracellular calcium overload. Increased Ca++ influx into axons mediated by overstimulation of glutamate receptors may damage mitochondria by promoting Ca++ entry into the matrix, leading to inhibition of respiratory chain complex I, and release of cytochrome c into the cytosol (77, 78, 127). Furthermore, excess of intra-axonal Ca++ may stimulate a variety of Ca++-dependent catabolic enzyme systems, including proteases, phospholipases, and calpains, ultimately leading to progressive cytoskeletal degeneration within axons (128). These observations have been confirmed not only in animal models but also in post-mortem studies of MS patients. A third possible mechanism is impaired glycogenolysis and lactate formation secondary to β2 adrenergic receptor deficiency in astrocytes, leading to decreased axonal mitochondrial metabolism and reduced N-acetyl aspartate (NAA) synthesis, as well as impaired GS activity (129–131). Reduced NAA may alter myelin membrane turnover, leading to myelin loss. Damage of the myelin sheath may contribute to axonal degeneration by reducing trophic support and impairing axonal transport. Overall, evidence is accumulating that defective axonal energy metabolism may cause diffuse axon degeneration observed in MS. A number of findings suggest that, at least in part, this metabolism defect might be secondary to astrocyte dysfunction.
Effects of Aging on Astrocytes
Aging affects many functional brain characteristics regulated by astrocytes, e.g., synaptic plasticity, metabolic balance, and BBB permeability. Increased expression of GFAP and vimentin has been the most common change observed in astrocytes with aging (132, 133). Interestingly, TGFβ1 signaling increases in the aging brain and can not only inhibit astrocyte proliferation but also stimulate GFAP expression. Furthermore, TGFβ1 is considered one of the main inducers of the senescence-associated secretory phenotype (SASP) observed in other cell populations (134, 135). Notably, senescent astrocytes can repress their capacity to support neuronal survival and neurite outgrowth, causing changes resembling those observed in the SASP, namely (1) increased expression of GFAP and vimentin filaments (132, 133); (2) accumulation of membrane-bound inclusion material in cytoplasm that appears to be lipofucsin, and ultraestructural changes in nuclei (136); and (3) increased expression of pro-inflammatory cytokines such as IL-6, TNF-α, IL-1β, and prostaglandins, which can enhance BBB permeability (134, 137). This age-related dysfunction can alter Ca++ homeostasis, and induce purinergic signaling in the gliovascular interface (138). In addition, astrocytes can release high-mobility group box-1 (HMGB1) protein, which promotes secretion of a specific subset of inflammatory factors, such as MMP-9, cyclo-oxygenase-2, and other chemokines facilitating monocyte infiltration (139). Indeed, during EAE progression, total and extracellular HMGB1 in the spinal cord is increased, and more positive astrocytes, neurons, and microglia are observed. Local block of CNS HMGB1 significantly attenuates EAE severity, suggesting HMGB1 expression in the spinal cord is associated with EAE progression (140).
Overall, inflammatory mediators appear to generate a vicious age-dependent cycle, where cellular senescence induces a low level of chronic inflammation, enhancing acute pathological conditions, and aggravating age-related neurodegenerative processes. This occurs through triggering of NO-induced pathways, and ROS-mediated dysfunction in mitochondria and endoplasmic reticulum.
Concluding Remarks
Astrocytes are a diverse cell population, differing across the CNS in their morphology, physiology, and function. In recent years, growing evidence indicates that astrocytes are more than simple bystander cells providing an optimal physical and metabolic environment for neuronal activity. In MS lesions, they exert active, dual, and paradoxical roles during disease development (39). Some experimental data implicate astrocytes as actual mediators of inflammation, as observed in sites of injury, ultimately limiting neuronal repair and remyelination (Figure 1). By contrast, other evidence suggests astrocytes curtail detrimental effects of pro-inflammatory factors, thus providing support and protection for oligodendrocytes and neurons. This dichotomy in astrocyte effects makes designing new therapeutic strategies targeting astrocytes a challenging endeavor. In this context, a better definition of astroglial subtypes based on their molecular, functional, and structural properties, should greatly promote our understanding of their specific roles in MS pathophysiology, and consequently lead to the development of novel cell-targeting therapies for the disease.
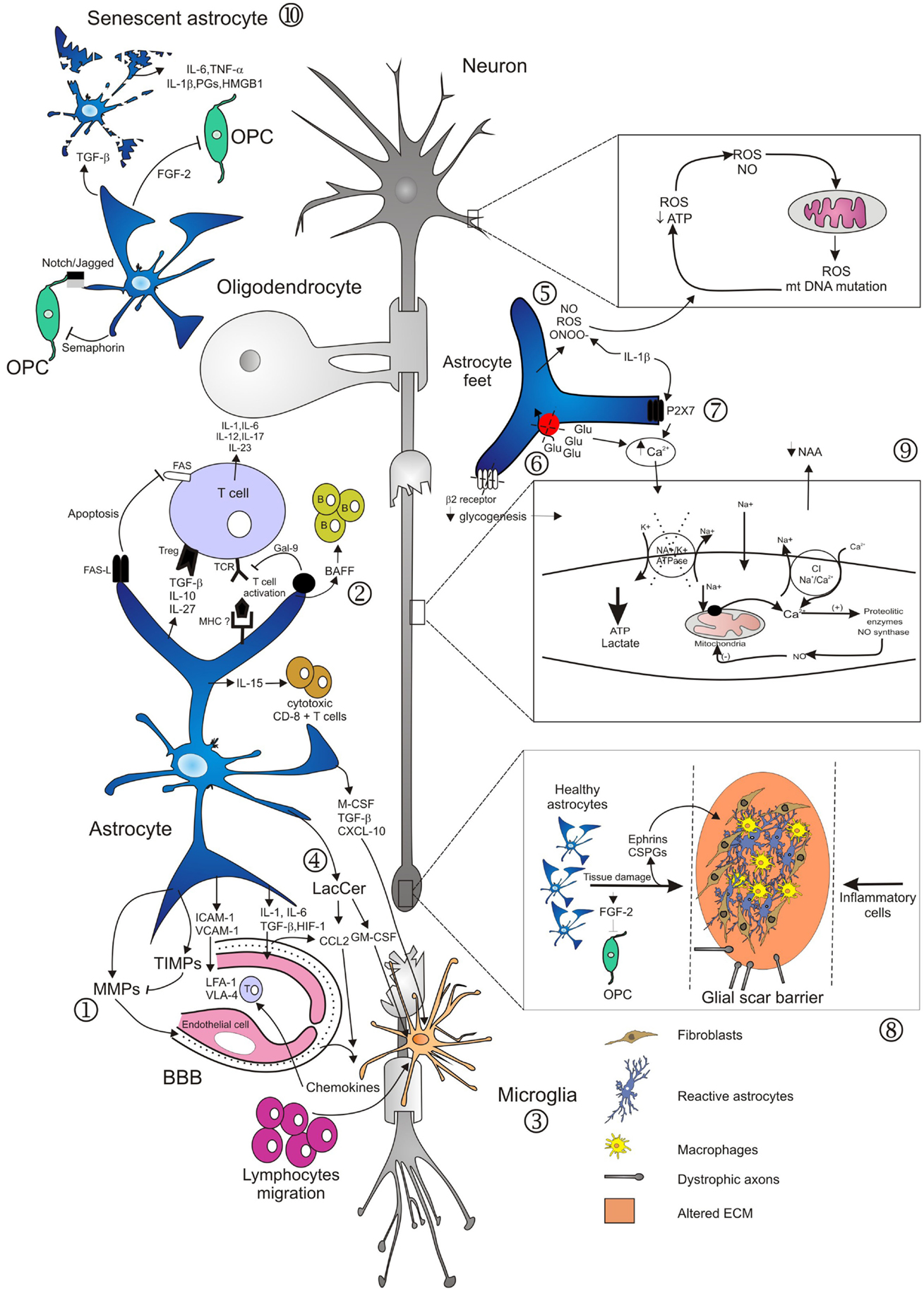
Figure 1. Main mechanisms involved in neurodegeneration driven by astrocytes. Several studies have demonstrated diverse roles of astrocytes in lesion development during the course of MS. Activation of astrocytes and loss of end-feet around small vessels are early events in lesion development, associated to loss of BBB function and consequently to CNS inflammation (1). Astrocytes mediate innate immune responses through several mechanisms. They modulate cell entry into the CNS by regulating adhesion molecule expression profiles, particularly of VCAM-1 and ICAM-1 (1). Astrocytes may also affect the number and phenotype of T cells in the CNS, committing T cells to a pro-inflammatory or regulatory phenotype. By contrast, astrocytes may also terminate T cell response, either by induction of apoptosis, or induction of Galectin-9. Furthermore, production of IL-15 or of BAFF drives immune responses mediated by cytotoxic CD8+ T cells or by B cells (2). Activated astrocytes secrete different chemokines, which attract both peripheral immune cells and microglia to MS lesions (2, 3). In the EAE model, astrocytes produce LacCer during the chronic phase, leading to induction of GM-CSF and CCL2 genes, and to subsequent microglial activation and monocyte infiltration (4). In areas of myelin breakdown, it has been documented that astrocytes secrete compounds with toxic effects for neurons, axons, and oligodendrocytes (5), reduce glutamate uptake by astrocyte transporters (6), and increase expression of purinergic receptors (7). These factors contribute to loss of glutamate buffering capacity mediated by astrocytes, mitochondrial dysfunction, energy deficiency, accumulation of intra-axonal Ca2+, and subsequent activation of proteolitic enzymes (9). Astrocytes respond to injuries by forming a glial scar that inhibits remyelination and axonal regeneration. These effects are mediated through secretion of fibroblast growth factor-2 (FGF-2) and of inhibitory extracellular matrix (ECM) molecules, such as condroitin sulfate proteoglycans (CSPGs) and Ephrins (8). Old age adversely affects astrocyte viability and self-renewal capacity, resulting in the generation of senescent and/or dysfunctional cells, evidenced in the form of cell fragmentation (10). Senescent astrocytes appear to be in a state of chronic activation, associated with pro-inflammatory cytokine and prostaglandins secretion.
Author Contributions
JC conceived the manuscript, outlined the subject of the review, searched for, analyzed and interpreted the literature, wrote the manuscript, edited and revised it for important intellectual content, gave final approval of the version for publication, and accepts full accountability for all aspects of the work. MF edited and revised the manuscript for important intellectual content, gave final approval of the version for publication, and accepts accountability for all aspects of the work.
Conflict of Interest Statement
Jorge Correale is a board member of Merck-Serono Argentina, Novartis Argentina, Genzyme LATAM, Genzyem global, Biogen-Idec LATAM, and Merck-Serono LATAM. Dr. Jorge Correale has received reimbursement for developing educational presentations for Merck-Serono Argentina, Merck-Serono LATAM, Biogen-Idec Argentina, Genzyme Argentina, Novartis Argentian, Novartis LATAM, and TEVA Argentina as well as professional travel/accommodations stipends. Mauricio F. Farez has received professional travel/accommodations stipends from Merck-Serono Argentina and Novartis Argentina.
Acknowledgments
This study was supported by an unrestricted grant from Novartis Argentina. The authors thank Adriana Zufriategui for preparation of the Figure.
References
1. McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol (2007) 8(9):913–9. doi: 10.1038/ni1507
2. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on clinical trials of new agents in multiple sclerosis. Neurology (1996) 46(4):907–11. doi:10.1212/WNL.46.4.907
3. Cohen JA, Rudick RA. Multiple Sclerosis Therapeutics. New York, NY: Cambridge UniveristyPress (2011). 752 p.
4. Mahad DH, Trapp BD, Lassman H. Progressive multiple sclerosis 1. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol (2015) 14(2):183–93. doi:10.1016/S1474-4422(14)70256-X
5. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med (1998) 338(5):278–85. doi:10.1056/NEJM199801293380502
6. Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the new forming lesions. Ann Neurol (2004) 55(4):458–68. doi:10.1002/ana.20016
7. Petterson JW, Bo L, Mork S, Chang A, Trapp BD. Transected neuritis, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol (2001) 50(3):389–400. doi:10.1002/ana.1123
8. Trapp BD, Nave KA. Multiple sclerosis: an immune and neurodegenerative disorder? Annu Rev Neurosci (2008) 31:247–69. doi:10.1146/annurev.neuro.30.051606.094313
9. Bø L. The histopathology of grey matter demyelination in multiple sclerosis. Acta Neurol Scand (2009) 189:51–7. doi:10.1111/j.1600-0404.2009.01216.x
10. Mayo L, Quintana FJ, Weiner HL. The innate immune system in demyelinating disease. Immunol Rev (2012) 248(1):170–87. doi:10.1111/j.1600-0404.2009.01216.x
11. Rindfleish E. Histologisches detail zur grauen degeneration von gehirn und rückermark. Arch Pathol Anat Physiol Klin Med (1863) 26:474–83. doi:10.1007/BF01878008
12. Müller E. Pathologishes anatomie and pathogenese. In: Fisher G, editor. Die Multiple Skleroze des Gehirms und Rückenmarks. Jena: Verlag (1904). p. 300–44.
13. Anton G, Wohlwill F. Multiple nicht eitirge encephalomyelitis und multiple skleroze. Z Ges Neurol Psych (1912) 12:31–99. doi:10.1007/BF02866371
14. Brosnan CF, Raine CS. The astrocyte in multiple sclerosis revisited. Glia (2013) 61(4):453–65. doi:10.1002/glia.22443
15. He F, Sun YE. Glial cells more tan support cells? Int J Biochem Cell Biol (2007) 39(4):661–5. doi:10.1016/j.biocel.2006.10.022
16. Butt AM, Duncan A, Berry M. Astrocyte associations with nodes of ranvier: ultrastructural analysis of HRP-filled astrocytes in the mouse optic nerve. J Neurocytol (1994) 23(8):486–99. doi:10.1007/BF01184072
17. Chao TI, Rickman M, Wolff JR. The synapse-astrocyte boundary: anatomical basis for an integrative role of glia in synaptic transmission. In: Volterra A, Magistretti P, Haydon P, editors. Tripartate Synapses: Synaptic Transmission with Glia. New York, NY: Oxford University Press (2002). p. 3–23.
18. Reinchenbach A, Wolburg H. Astrocytes and ependymal glia. In: Kettenmann H, Ranson BR, editors. Neuroglia. New York, NY: Oxford University Press (2013). p. 35–49.
19. Lundgaard I, Osório MJ, Krees BT, Sangaard S, Nedergaard M. White matter astrocytes in health and disease. Neuroscience (2014) 276:161–73. doi:10.1016/j.neuroscience.2013.10.050
20. Nair A, Frederick TJ, Miller D. Astrocytes in multiple sclerosis: a product of their environment. Cell Mol Life Sci (2008) 65(17):2702–20. doi:10.1007/s00018-008-8059-5
21. Tsai HH, Li H, Fuentealba LC, Molofsky AV, Taveira-Marques R, Zhuang H, et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science (2012) 337(6092):358–62. doi:10.1126/science.1222381
22. Molofsky AV, Krencik R, Ulian EM, Tsai HH, Denereen B, Richardson WD, et al. Astrocytes and disease: a neurodevelopment perspective. Genes Dev (2012) 26(9):891–907. doi:10.1101/gad.188326.112
23. Wang DD, Bordey A. The astrcoyte odyssey. Prog Neurobiol (2008) 86(4):342–67. doi:10.1016/j.pneurobio.2008.09.015
24. Emsley JG, Macklis JD. Astroglial heterogeneity closely reflects the neuronal-defined anatomy of the adult murine CNS. Neuron Glia Biol (2006) 2(3):175–86. doi:10.1017/S1740925X06000202
25. Goldan JE. Astrocyte development. In: Kettenmann H, Ranson BR, editors. Neuroglia. New York, NY: Oxford Univeristy Press (2013). p. 137–47.
26. Hatada I, Namihira M, Morita S, Kimura M, Horii T, Nakashima K, et al. Astrocyte-specific genes are geenrally demethylated in neural precursor cells prior to astrocytic differentiation. PLoS One (2008) 3:3189. doi:10.1371/journal.pone.0003189
27. Fan G, Martinowich K, Chin MH, He F, Fouse SD, Hutnick L, et al. DNA methylation controls the timing of astroglio-genesis through regulation of JAK-STAT signaling. Development (2005) 132(15):3345–56. doi:10.1242/dev.01912
28. Sun D, Jakobs TC. Structural remodeling of astrocytes in the injured CNS. Neuroscientist (2012) 18(6):567–88. doi:10.1177/1073858411423441
29. Seifert G, Schilling K. Steinhäuser. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci (2006) 7(3):194–206. doi:10.1038/nrn1870
30. Ulian EM, Sapperstein SK, Christopherson KS, Barres A. Control of synapses number by glia. Science (2001) 291(5504):657–61. doi:10.1126/science.291.5504.657
31. MacVicar BA, Feighan D, Brown A, Ramson B. Intrinsic optical signals in the rat optic nerve role for K+ uptake via NKCC1 and swelling of astrocytes. Glia (2002) 37(2):114–23. doi:10.1002/glia.10023
32. Göritz C, Mauch DH, Nägler K, Pfrieger FW. Role of glia-derived cholesterol in synaptogenesis: new revelations in the synapse-glia affair. J Physiol Paris (2002) 96(3–4):257–63. doi:10.1016/S0928-4257(02)00014-1
33. Abbot NJ, Ronnback L, Hansson E. Astrocye-endothelial interactions at the blood brain barrier. Nat Rev Neurosci (2006) 7(1):41–53. doi:10.1038/nrn1824
34. Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocye-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci (1998) 1(8):683–92. doi:10.1038/3684
35. Ludwin SK, Raine CS. The neuropathology of MS. In: Raine CS, McFarland HF, Hohlfeld R, editors. Multiple Sclerosis: A Comprehensive Text. New York, NY: Sanders/Elsevier (2008). p. 151–77.
36. Argaw AT, Zang Y, Snyder BJ, Zhao ML, Kopp N, Lee SC, et al. IL-1beta regulates blood-brain-barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol (2006) 177(8):5574–84. doi:10.4049/jimmunol.177.8.5574
37. Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain-barrier breakdown. Proc Natl Acad Sci USA (2009) 106(6):1977–82. doi:10.1073/pnas.0808698106
38. Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Marianin JN, et al. Astrocyte derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest (2012) 122(7):2454–68. doi:10.1172/JCI60842
39. Williams A, Piaton G, Lubetzki C. Astrocytes-friends or foes in multiple sclerosis? Glia (2007) 55(13):1300–12. doi:10.1002/glia.20546
40. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol (2012) 4(3):a006049. doi:10.1101/cshperspect.a006049
41. Miyake Y, Yamasaki S. Sensing necrotic cells. Adv Exp Med Biol (2012) 738:144–52. doi:10.1007/978-1-4614-1680-7_9
42. Farina C, Aloisi F, Mainl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol (2007) 28(3):138–45. doi:10.1016/j.it.2007.01.005
43. Gimenez MAT, SIM JE, Rusell JH. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol (2004) 151(1–2):116–25. doi:10.1016/j.jneuroim.2004.02.012
44. Sobel RA, Mitchell ME, Fondren G. Intracellular adhesion molecule-1 (ICAM-1) in cellular immune reactions in the human central nervous system. Am J Pathol (1990) 136(6):1309–16.
45. Minagar A, Alexander JS. Blood-brain barrier disruption in multiple sclerosis. Mult Scler (2003) 9(6):540–9. doi:10.1191/1352458503ms965oa
46. Didier N, Romero JA, Creminon C, Wijkhuisen A, Grassi J, Mabondzo A. Secretion of interleukin-1 beta by astrocytes mediates endothelin-1 and tumor necrosis factor-alpha effects on human brain microvascualr endothelial cell permeability. J Neurochem (2003) 86(1):246–54. doi:10.1046/j.1471-4159.2003.01829.x
47. Dong Y, Benveniste EN. Immune functions of astrocytes. Glia (2001) 36(2):180–90. doi:10.1002/glia.1107
48. Calderon TM, Eugenin EA, Lopez L, Kumar SS, Hesselgesser J, Raine CS, et al. A role of CXCL12 (SDF-1alpha) in the pathogenesis of multiple sclerosis: regulation of CXCL12 expression in astrocytes by soluble myelin basic protein. J Neuroimmunol (2006) 177(1–2):27–39. doi:10.1016/j.jneuroim.2006.05.003
49. Salmaggi A, Gelati M, Dufour A, Corsini E, Pagano S, Baccalini R, et al. Expression and modulation of IFN-gamma inducible chemokines (IP-10, Mig, and I-TAC) in human brain endothelium and astrocytes: possible relevance for the immune invasion of the central nervous system and the pathogenesis of multiple sclerosis. J Interferon Cytokine Res (2002) 22(6):631–40. doi:10.1089/10799900260100114
50. Karpus WJ, Ransohoff RM. Chemokine regulation of experimental autoimmune encephalomyelitis: temporal and spatial expression patterns govern disease pathogenesis. J Immunol (1998) 161(6):2667–71.
51. Milijkovic D, Moncilovic M, Stojanovic I, Stosio-Grujicic S, Ramiz Z, Mostarica-Stojovic M. Astrocytes stimulate interleukin-17 and interferon-gamma production in vitro. J Neurosci Res (2007) 85(16):3598–606. doi:10.1002/jnr.21453
52. Liu JQ, Carl JW Jr, Joshi PS, RayCahudhury A, Pu XA, Shi FD, et al. CD24 on the resident cells of the central nervous system enhances experimental autoimmune encephalomyelitis. J Immunol (2007) 178(10):6227–35. doi:10.4049/jimmunol.178.10.6227
53. Zhou Y, Sonobe Y, Akahori T, Jin S, Kawanokuchi J, Noda M, et al. IL-9 promotes Th17 cell migration into the central nervoous system via CC chemokine ligand-20 produced by astrocytes. J Immunol (2011) 186(7):4415–21. doi:10.4049/jimmunol.1003307
54. Saikali P, Antel JP, Pittet CL, Newcombe J, Arbour N. Contribution of astrocyte-derived IL-15 to CD8 T cell effector functions in multiple sclerosis. J Immunol (2010) 185(10):5693–703. doi:10.4049/jimmunol.1002188
55. Bechmann I, Steiner B, Gimsa U, Mor G, Wolf S, Beyer M, et al. Astrocyte-induced T cell elimination is CD95 ligand dependent. J Neuroimmunol (2002) 132(1–2):60–5. doi:10.1016/S0165-5728(02)00311-9
56. Zhu C, Anderson AC, Kuchroo VK. Tim-3 and its regulatory role in immune responses. Curr Top Microbiol Immunol (2011) 350:1–15. doi:10.1007/82_2010_84
57. Krumbholz M, Theil D, Derfuss T, Rosenwald A, Schrader F, Monoranu CM, et al. Baff is produced by astrocytes and upregualted in multiple sclerosis lesions and primary centrol nervous system lymphoma. J Exp Med (2005) 201(2):195–200. doi:10.1084/jem.20041674
58. Toft-Hansen H, Füchtbauer L, Owens T. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia (2011) 59(1):166–76. doi:10.1002/glia.21088
59. DeWitt DA, Perry G, Cohen M, Doller C, Silver J. Astrocytes regulate microglial phagocytosis of senile plaque core of Alzheimer’s disease. Exp Neurol (1998) 149(2):329–40. doi:10.1006/exnr.1997.6738
60. Zeinstra E, Wilczak N, Streefland C, Keyser JD. Astrocytes in chronic active multiple sclerosis plaques express MHC Class II molecules. Neuroreport (2000) 11(1):89–91. doi:10.1097/00001756-200001170-00018
61. Chastain EML, Duncan DS, Rodgers JM, Miller DS. The role of antigen presenting cells in multiple sclerosis. Biochem Biophys Acta (2011) 1812(2):265–74. doi:10.1016/j.bbadis.2010.07.008
62. Kort JJ, Kawamura K, Fugger L, Weissert R, Forsthuber TG. Efficient presentation of myelin oligodendrocyte glycoprotein peptides but not proteins by astrocytes from HLA-DR2 and HLA-DR4 transgenic mice. J Neuroimmunol (2006) 173(1–2):23–34. doi:10.1016/j.jneuroim.2005.11.014
63. Mayo L, Trauger SA, Balin M, Nadeu M, Patel B, Alvarez JI, et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med (2014) 20(10):1147–56. doi:10.1038/nm.3681
64. Pannu R, Won JS, Khan M, Singh AK, Singh I. A novel role of lactosylceramida in the regulation of lipopolysaccharide interferon-gamma mediated inducible nitric oxide syntahse gene expression: implications for neuroinflammatory diseases. J Neurosci (2004) 24(26):5942–54. doi:10.1523/JNEUROSCI.1271-04.2004
65. Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci (2001) 21(17):6480–91.
66. Trajkovic V, Stosic-Grujicic S, Samardzic T, Markovic M, Milijovic D, Ramicz Z, et al. Interleukin-17 stimulates inducible nitric oxide synthase activation in rodent astrocytes. J Neuroimmunol (2001) 119(2):183–91. doi:10.1016/S0165-5728(01)00391-5
67. Hamby ME, Hewet JA, Hewet SJ. TGF-beta 1 potentiates astrocytic nitric oxide production by expanding the population of astrocytes that express NOS-2. Glia (2006) 54(6):566–77. doi:10.1002/glia.20411
68. Lee SC, Dickson DW, Liu W, Brosnan CF. Induction of nitric oxide synthase activity in human astrocytes by interleukin-1 beta and interferon-gamma. J Neuroimmunol (1993) 46(1–2):19–24. doi:10.1016/0165-5728(93)90229-R
69. Brosnan CF, Battistini L, Raine CS, Dickson DW, Casadevall A, Lee SC. Reactive nitrogen intermediates in human neuropathology: an overview. Dev Neurosci (1994) 16(3–4):152–61. doi:10.1159/000112102
70. Liu JS, Zhao ML, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am J Pathol (2001) 158(6):2057–66. doi:10.1016/S0002-9440(10)64677-9
71. Stojanovic IR, Kostic M, Ljubisavljevic S. The role of glutamate and its recpeors in multiple sclerosis. J Neural Transm (2014) 121(8):945–55. doi:10.1007/s00702-014-1188-0
72. Kumar P, Kalonia H, Kumar A. Possible GBAERGIC mechanism in the neurorptective effect of gabapentin and lamotirgine against 3-nitropropionic acid induced neurotoxicity. Eur J Pharmacol (2012) 674(2–3):265–74. doi:10.1016/j.ejphar.2011.11.030
73. Rossi S, Motta C, Studer V, Barbieri F, Butari F, Bergami A, et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult Scler (2014) 20(3):304–12. doi:10.1177/1352458513498128
74. Matute C, Sanchez-Gomez MV, Martinez-Millan L, Miledi R. Glutamate receptor-mediated toxicity in optic nerve oligodendrocytes. Proc Natl Acad Sci U S A (1997) 94(16):8830–5. doi:10.1073/pnas.94.16.8830
75. Korn T, Magnus T, Jung S. Autoantigen specific T cells inhibit glutamate uptake in astrocytes by decreasing expression of astrocytic glutamate transporter GLAST: a mechanism mediated by tumor necrosis-alpha. FASEB J (2005) 19(13):1878–80.
76. Rothstein JD, Dykes-Hoberg M, PArdo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron (2006) 16(3):675–86. doi:10.1016/S0896-6273(00)80086-0
77. Ouardouz M, Coderre E, Basak A, Chen A, Zamponi GW, Hameed S, et al. Glutamate receptors on myelinated spinal cord axons. I. GluR6 kainate receptors. Ann Neurol (2009) 65(2):151–9. doi:10.1002/ana.21533
78. Ouardouz M, Coderre E, Zamponi GW, Hameed S, Yin X, Trapp BD, et al. Glutamate receptors on myelinated spinal cord axons. II. AMPA and GluR5 receptors. Ann Neurol (2009) 65(2):160–6. doi:10.1002/ana.21539
79. Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med (2004) 4(2):149–77. doi:10.2174/1566524043479239
80. Norenberg MD, Rao KV. The mitochondrial permeability transition in neurological disease. Neurochem Int (2007) 50(7–8):983–97. doi:10.1016/j.neuint.2007.02.008
81. Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature (2005) 438(7071):1167–71. doi:10.1038/nature04301
82. Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med (2000) 6(1):67–70. doi:10.1038/71555
83. Franke H, Verkhratsky A, Burnstock G, Illes P. Pathophysiology of astroglial purinergic signaling. Purinergic Signal (2012) 8(3):629–57. doi:10.1007/s11302-012-9300-0
84. Narcisse L, Scemes E, Zhao Y, Lee SC, Brosnan CF. The cytokine IL-1beta transiently enhances P2X7 receptor expression and function in human astrocytes. Glia (2005) 49(2):245–58. doi:10.1002/glia.20110
85. Matute C, Torre I, Pérez-Cerdá F, Pérez-Samartín A, Alberdi E, Etxebarria E, et al. P2X(7) recpetor blockade prevents ATP excitotoxicity in oligodendrocyes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci (2007) 27(35):9525–37. doi:10.1523/JNEUROSCI.0579-07.2007
86. Chen ZJ, Negra M, Levine A, Ughrin Y, Levine JM. Oligodendrocyte precursor cells: reactive cells that inhibit axon growth and regeneration. J Neurocytol (2002) 31(6–7):481–95. doi:10.1023/A:1025791614468
87. Bundesen LQ, Scheel TA, Bregman BS, Kromer LF. EPhrin-B2 and EPhB2 regulation of astrocyte-meningeal fibroblast interactions in response to spinal cord lesions in adult rats. J Neurosci (2003) 23(21):7789–800.
88. Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty one years (1969-2000). Neurochem Res (2000) 25(9–10):1439–51. doi:10.1023/A:1007677003387
89. Robel S, Berninger B, Gôtz M. The stem cell potential of glia: lessons from reactive gliosis. Nat Rev Neurosci (2013) 12(2):88–104. doi:10.1038/nrn2978
90. Balasingam V, Tejada-Borges T, Wright E, Bouckova R, Yong VW. Reactive astrogliosis in the neonatal mouse brain and its modulation by cytokines. J Neurosci (1994) 14(2):846–56.
91. Sriram K, Benkovic SA, Hebert MA, Miller DB, O’Callaghan JP. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J Biol Chem (2004) 279(19):19936–47.
92. Robel S, Mori T, Zoubaa S, Schlegel J, Sirko S, Faissner A, et al. Conditional deletion of beta-1 integrin in astroglia causes partial reactive gliosis. Glia (2009) 57(15):1630–47. doi:10.1002/glia.20876
93. Kanemaru K, Kubota J, Sekiya H, Hirose K, Okubo Y, Iino M. Calcium-dependent N-cadherin up-regulation mediates reactive astrogliosis and neuroprotection after brain injury. Proc Natl Acad Sci U S A (2013) 110(28):11612–7. doi:10.1073/pnas.1300378110
94. Erschbamer M, Pernold K, Olson L. Inhibiting epidermal growth factor receptor improves structural, locomotor, sensory, and bladder recovery from experimental spinal cord injury. J Neurosci (2007) 27(24):6428–35. doi:10.1523/JNEUROSCI.1037-07.2007
95. Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev (1998) 50(3):413–92.
96. Robel S, Bardehle S, Lepier A, Brakebusch C, Götz M. Genetic deletion of cdc42 reveals a crucial role for astrocyte recruitment to the injury site in vitro and in vivo. J Neurosci (2011) 31(35):12471–82. doi:10.1523/JNEUROSCI.2696-11.2011
97. Beck H, Plate KH. Angiogenesis after cerebral ischemia. Acta Neuropathol (2009) 117(5):481–96. doi:10.1007/s00401-009-0483-6
98. Zhang ZG, Chopp M. Neurorestorative therapies for stroke; underlying mechanisms and translation to the clinic. Lancet Neurol (2009) 8(5):491–500. doi:10.1016/S1474-4422(09)70061-4
99. Nakazawa T, Takeda M, Lewis GP, Cho KS, Jiao J, Wilhelmsson U, et al. Attenuated glial recations and photoreceptor degeneration after retinal detachment in mice deficient of glial fibrillary acidic protein and vimentin. Invest Ophthalmol Vis Sci (2007) 48(6):2760–8. doi:10.1167/iovs.06-1398
100. Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci (2008) 28(28):7231–43. doi:10.1523/JNEUROSCI.1709-08.2008
101. Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med (2006) 12(7):829–34. doi:10.1038/nm1425
102. Neary JT, Kang Y. Signaling from P2 nucleotide receptors to protein kinase cascades induced by CNS injury: implications for reactive gliosis and neurodegeneration. Mol Neurobiol (2005) 31(1–3):95–105. doi:10.1385/MN:31:1-3:095
103. Washburn KB, Neary JT. P2 purinergic receptors signal to STAT3 in astrocytes: Difference in STAT3 responses to P2Y and P2X receptor activation. Neuroscience (2006) 142(2):411–25. doi:10.1016/j.neuroscience.2006.06.034
104. Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci (2004) 5(2):146–56. doi:10.1038/nrn1326
105. Holley JE, Gveric D, Newcombe J, Cuzner ML, Gutowski NJ. Astrocyte characterization in the multiple sclerosis glial scar. Neuropathol Appl Neurobiol (2003) 29(5):434–44. doi:10.1046/j.1365-2990.2003.00491.x
106. Goddard DR, Berry M, Butt AM. In vivo actions of fibroblast growth factor-2 and insulin-like growth factor-I on oligodendrocyte development and myelination in the central nervous system. J Neurosci Res (1999) 57(1):74–85. doi:10.1002/(SICI)1097-4547(19990701)57:1<74::AID-JNR8>3.0.CO;2-O
107. Sherman LS, Struve JN, Rangwala R, Wallingford NM, Tuohy TM, Kuntz CT. Hyaluronate-based extracellular matrix: keeping glia in their place. Glia (2002) 38(2):93–102. doi:10.1002/glia.10053
108. Soilu-Hanninen M, Laaksonen M, Hanninen A. Hyaluronate receptor (CD44) and integrin alpha4 (CD49d) are upregulated on T cells during MS relapses. J Neuroimmunol (2005) 166(1–2):189–92. doi:10.1016/j.jneuroim.2005.05.008
109. Back SA, Tuohy TM, Chen H, Wallingford N, Craig A, Struve J, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med (2005) 11(9):966–72.
110. Johnson-Green PC, Dow KE, Riopelle RJ. Characterization of glycosaminoglycans produced by primary astrocytes in vitro. Glia (1991) 4(3):314–21. doi:10.1002/glia.440040309
111. Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature (2002) 416(6881):636–40. doi:10.1038/416636a
112. Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci (2006) 7(8):617–27. doi:10.1038/nrn1956
113. Zhu X, Bergles DE, Nishiyama A. NG2 cells generate both oligodendrocytes and grey matter astrocytes. Development (2008) 135(1):145–57. doi:10.1242/dev.004895
114. Fidler PS, Schuette K, Asher RA, Dobbertim A, Thornton SR, Calle-Pantino Y, et al. Comparing astrocytic cell lines that are inhibitory or permissive for axon growth: the major axon-inhibitory proteoglycan is NG2. J Neurosci (1999) 19(20):8778–88.
115. Sobel RA. Ephrin A receptors and ligands in lesions and normal-appearing white matter in multiple sclerosis. Brain Pathol (2005) 15(1):35–45. doi:10.1111/j.1750-3639.2005.tb00098.x
116. Wahl S, Barth H, Ciossek T, Aktories K, Mueller BK. Ephrin-A5 induces collapse of growth cones by activating Rho and Rho kinase. J Cell Biol (2000) 149(2):263–70. doi:10.1083/jcb.149.2.263
117. Satoh J, Tabunoki H, Yamamura T, Arima K, Konno H. TROY and LINGO-1 expression in astrocytes and macrophages/microglia in multiple sclerosis lesions. Neuropathol Appl Neurobiol (2007) 33(1):99–107. doi:10.1111/j.1365-2990.2006.00787.x
118. Fujita Y, Takashima R, Endo S, Takai T, Yamashita T. The p75 receptor mediates axon growth inhibition through an association with PIR-B. Cell Death Dis (2011) 2:e198. doi:10.1038/cddis.2011.85
119. Thomas R, Favell K, Morante-Redolat J, Pool M, Kent C, Wright M, et al. LGl1 is a Nogo receptor 1 ligand that antagonizes myelin-based growth inhibition. J Neurosci (2010) 30(19):6607–12. doi:10.1523/JNEUROSCI.5147-09.2010
120. Zhang L, Zheng S, Wu H, Wu Y, Liu S, Fan M, et al. Identification of BLyS (B lymphocyte stimulator) a non-myelin-associated protein as a functional ligand for Nogo-66 receptor. J Neurosci (2009) 29(19):6348–52. doi:10.1523/JNEUROSCI.5040-08.2009
121. Lee H, Raiker SJ, Venkatesh K, Geary R, Robak LA, Zhang Y, et al. Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J Neurosci (2008) 28(11):2753–65. doi:10.1523/JNEUROSCI.5586-07.2008
122. Borrie SC, Baeumer BE, Bandtlow CE. The Nogo-66 receptor family in the intact and disease CNS. Cell Tissue Res (2012) 349(1):105–17. doi:10.1007/s00441-012-1332-9
123. Schwab ME, Strittmatter SM. Nogo limits neural plasticity and recovery from injury. Curr Opin Neurobiol (2014) 27:53–60. doi:10.1016/j.conb.2014.02.011
124. Broholm H, Andersen B, Wanscher B, Frederiksen JL, Rubin I, Pakkenberg B, et al. Nitric oxide synthase expression and enzymatic activity in multiple sclerosis. Acta Neurol Scand (2004) 109(4):261–9. doi:10.1111/j.1600-0404.2004.00207.x
125. De Keyser J, Zeinstra E, Wilczak N. Astrcoytic beta-2 adrenergic receptors and multiple sclerosis. Neurobiol Dis (2004) 15(2):331–9. doi:10.1016/j.nbd.2003.10.012
126. Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, et al. Mitochondrial changes within axons in multiple sclerosis. Brain (2009) 132(Pt 5):1161–74. doi:10.1093/brain/awp046
127. Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD. Calcium and mitochondria. FEBSS Lett (2004) 567(1):96–102. doi:10.1016/j.febslet.2004.03.071
128. Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci (2005) 233(1–2):3–13. doi:10.1016/j.jns.2005.03.031
129. De Keyser J, Wilczak N, Leta R, Streetland C. Astrocytes in multiple sclerosis lack beta-2 adrenergic recpetors. Neurology (1999) 53(8):1628–33. doi:10.1212/WNL.53.8.1628
130. Patel TB, Clark JB. Synthesis of N-Acetyl-L-aspartate by rat brain mitochondria and its involvement in mitochondrial/cytosolic carbon transport. Biochem J (1979) 184(3):539–46.
131. Brookes N. Regualtion of the glutamine content of astrocytes by cAMP and hydrocortisone: effect of pH. Neurosci Lett (1992) 147(2):139–42. doi:10.1016/0304-3940(92)90579-V
132. Sloane JA, Hollander W, Rosene DL, Moss MB, Kemper T, Abraham CR. Astrcoytic hypertrophy and altered GFAP degradation with age in subcortical white matter of the rhesus monkey. Brain Res (2000) 862(1–2):1–10. doi:10.1016/S0006-8993(00)02059-X
133. Porchet R, Probst A, Bouras C, Drabevora E, Draber P, Riederer BM. Analysis of glial fibrillary protein protein in the human enthorinal cortex during aging and Alzheimer’s disease. Proteomics (2003) 3(8):1476–85. doi:10.1002/pmic.200300456
134. Salminen A, Ojala J, Kaarmiranta K, Haapasalo A, Hiltunen M, Soininen H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur J Neurosci (2011) 34(1):3–11. doi:10.1111/j.1460-9568.2011.07738.x
135. de Sampaio e Spohr TC, Martinez R, da Silva EF, Neto VM, Gomes FC. Neuro-glia interactions effects on GFAP gene: a novel role for transforming growth factor-beta 1. Eur J Neurosci (2002) 16(11):2059–69. doi:10.1046/j.1460-9568.2002.02283.x
136. Landfield PW, Braun LD, Pitler TA, Lindsey JD, Lynch G. Hippocampal aging in rats: a morphometric study of multiple variables in semithin sections. Neurobiol Aging (1981) 2(4):265–75. doi:10.1016/0197-4580(81)90034-8
137. Li C, Zhao R, Gao K, Wei Z, Yin MY, Lau LT, et al. Astrocytes implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res (2011) 8(1):67–80. doi:10.2174/156720511794604543
138. Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascualr interface. J Neurosci (2003) 23(27):9254–62.
139. Pedrazzi M, Patrone M, Passalacqua M, Ranzato E, Colamassaro D, Sparatore B, et al. Selective proinflammatory activation of astrcoytes by high-mobility group box 1 protein signaling. J Immunol (2007) 179(12):8525–32. doi:10.4049/jimmunol.179.12.8525
Keywords: multiple sclerosis, astrocytes, multiple sclerosis progression, microglia, myelin, axon, glial scar, mitochondria
Citation: Correale J and Farez MF (2015) The role of astrocytes in multiple sclerosis progression. Front. Neurol. 6:180. doi: 10.3389/fneur.2015.00180
Received: 23 May 2015; Accepted: 03 August 2015;
Published: 18 August 2015
Edited by:
V. Wee Yong, University of Calgary, CanadaReviewed by:
Yuhong Yang, The Ohio State University Medical Center, USAAstrid E. Cardona, The University of Texas at San Antonio, USA
Copyright: © 2015 Correale and Farez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorge Correale, Department of Neurology, Institute for Neurological Research Dr. Raúl Carrea, FLENI, Montañeses 2325, Buenos Aires 1428, Argentina,amNvcnJlYWxlQGZsZW5pLm9yZy5hciw=am9yZ2UuY29ycmVhbGVAZ21haWwuY29t