 Ryan C. Turner1,2†
Ryan C. Turner1,2† Brandon P. Lucke-Wold1,2†
Brandon P. Lucke-Wold1,2† Aric F. Logsdon2,3†
Aric F. Logsdon2,3† Matthew J. Robson4
Matthew J. Robson4 John M. Lee5
John M. Lee5 Julian E. Bailes6Matthew L. Dashnaw7Jason D. Huber2,3
Julian E. Bailes6Matthew L. Dashnaw7Jason D. Huber2,3 Anthony L. Petraglia8‡Charles L. Rosen1,2*‡
Anthony L. Petraglia8‡Charles L. Rosen1,2*‡
- 1Department of Neurosurgery, West Virginia University School of Medicine, Morgantown, WV, USA
- 2Center for Neuroscience, West Virginia University School of Medicine, Morgantown, WV, USA
- 3Department of Basic Pharmaceutical Sciences, West Virginia University School of Pharmacy, Morgantown, WV, USA
- 4Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN, USA
- 5Department of Pathology and Laboratory Medicine, NorthShore University Health System, University of Chicago Pritzker School of Medicine, Evanston, IL, USA
- 6Department of Neurosurgery, NorthShore University Health System, University of Chicago Pritzker School of Medicine, Evanston, IL, USA
- 7Department of Neurosurgery, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
- 8Division of Neurosurgery, Rochester Regional Health, Rochester, NY, USA
Despite the extensive media coverage associated with the diagnosis of chronic traumatic encephalopathy (CTE), our fundamental understanding of the disease pathophysiology remains in its infancy. Only recently have scientific laboratories and personnel begun to explore CTE pathophysiology through the use of preclinical models of neurotrauma. Some studies have shown the ability to recapitulate some aspects of CTE in rodent models, through the use of various neuropathological, biochemical, and/or behavioral assays. Many questions related to CTE development, however, remain unanswered. These include the role of impact severity, the time interval between impacts, the age at which impacts occur, and the total number of impacts sustained. Other important variables such as the location of impacts, character of impacts, and effect of environment/lifestyle and genetics also warrant further study. In this work, we attempt to address some of these questions by exploring work previously completed using single- and repetitive-injury paradigms. Despite some models producing some deficits similar to CTE symptoms, it is clear that further studies are required to understand the development of neuropathological and neurobehavioral features consistent with CTE-like features in rodents. Specifically, acute and chronic studies are needed that characterize the development of tau-based pathology.
Introduction
Corsellis described the original case series of chronic traumatic encephalopathy (CTE) in boxers (1). The disease consisted of brain atrophy, dilated ventricles, a cavum septum pellucidum, and pallor of the substantia nigra. CTE was reintroduced into the medical lexicon by Dr. Bennet Omalu in 2005 (2). Omalu described a progressive tauopathy that was seen in the brains of deceased football players. In recent years, McKee, Goldstein, and Stern have defined clinical and pathological features of the disease. These include behavioral disturbances such as impulsivity, depression, and lack of oversight (3). Pathological criteria include neurofibrillary tangles (NFTs) in a perivascular distribution and within superficial cortical areas with occasional amyloid and TDP-43 protein aggregations (4). Stern recently expanded the CTE criteria further by describing young versus old onset based on symptom manifestation (5). Interestingly, blast traumatic brain injury has been linked to CTE following a single exposure, where athletes develop the disease following repetitive head injury (6).
Chronic traumatic encephalopathy has been defined as a slowly progressive disease that takes years to decades to develop, often providing a significant latent period between when the neurotrauma occurs and when symptoms develop. A few cases involve athletes/soldiers as early as late teen’s to early 20s (7, 8). The reason for the discrepancies in age of presentation observed is currently unknown but is likely due to the age at which impacts were sustained and the severity of the injury. Prior studies have shown that children with TBI have inadequate development of social cognition (9) and that adolescents can develop post-traumatic headaches. Very rarely, however, do either of these groups experience symptoms of CTE (10). Early behavioral symptoms of CTE usually do not appear until the mid-30s, and cognitive impairment does not begin until the early sixties (4). Recent evidence suggests that neurotrauma may be linked to other neurodegenerative diseases such as Alzheimer’s disease (AD) as well (11).
Chronic traumatic encephalopathy and AD, despite both being tauopathies, have generally been viewed as separate diseases. Each disease has distinct clinical presentations and unique clinical risk values. This view has gradually been changing with some reports showing that CTE can develop within the context of AD. The relationship between these two conditions, however, is poorly understood. Prior studies have indicated that TBI is a risk factor for development of cognitive impairment and AD (12), but whether the conditions are additive or synergistic remains unclear (13). Recent evidence suggests that neurotrauma may both increase the likelihood of disease development and accelerate the development of AD (14). Notably, the wealth of AD pathology observed in preclinical neurotrauma models supports the idea of disease acceleration (15). In addition to AD, some groups have presented a possible link between CTE and a variant of amyotrophic lateral sclerosis that has been termed “chronic traumatic encephalomyelopathy” (CTEM) (16). Therefore, it is clear that neurotrauma may have many lasting deleterious consequences, including the potential for increased risk and accelerated development of neurodegenerative diseases such as AD, CTE, and CTEM. While the data on these studies are preliminary in nature and not prospective, these findings demonstrate the need for further investigation with both preclinical models and clinical trials.
The Quest for the Ideal CTE Model
The search for ideal preclinical models to study CTE remains an area of ongoing investigation with a relative paucity of prior studies. Of those published previously, the majority of studies fail to recapitulate the extensive neuropathological and neurobehavioral aspects of injury (17, 18). Post-mortem identification of NFTs are a key diagnostic marker used clinically (19). Therefore, the development of hyperphosphorylated tau in animals following injury must be an important component in establishing a CTE model. Tau hyperphosphorylation appears to correlate with the emergence of neuropathological and neuropsychiatric deficits representative of neurotrauma-related neurodegeneration (20). By modeling CTE in rodents, we can better understand disease development and discover potential therapeutic avenues. Similarly, little justification is given to injury severity, number of impacts, interval between impacts, and age at which the impacts occur. Few studies have also evaluated secondary mechanisms of injury. In the following sections, we discuss the advancements and shortcomings of prior research while highlighting areas in need of further investigation.
Proper Controls to Use When Studying CTE
One of the challenges in creating a preclinical model for CTE is the establishment of proper controls comparing sham to TBI animals (13). Specifically, sham animals must undergo the exact same procedure each day of the injury paradigm. They must be anesthetized for the same duration as injured animals. The age of the animal must also be considered. Control animals should be the same age as experimental animals at time of sacrifice. This is an important consideration in designing extended studies with behavioral assays. Biochemical time courses also require control animals for each time point. Numerous questions must be addressed in planning experiments while considering the implications of each decision in terms of experimental question addressed and potential complications (Figure 1).
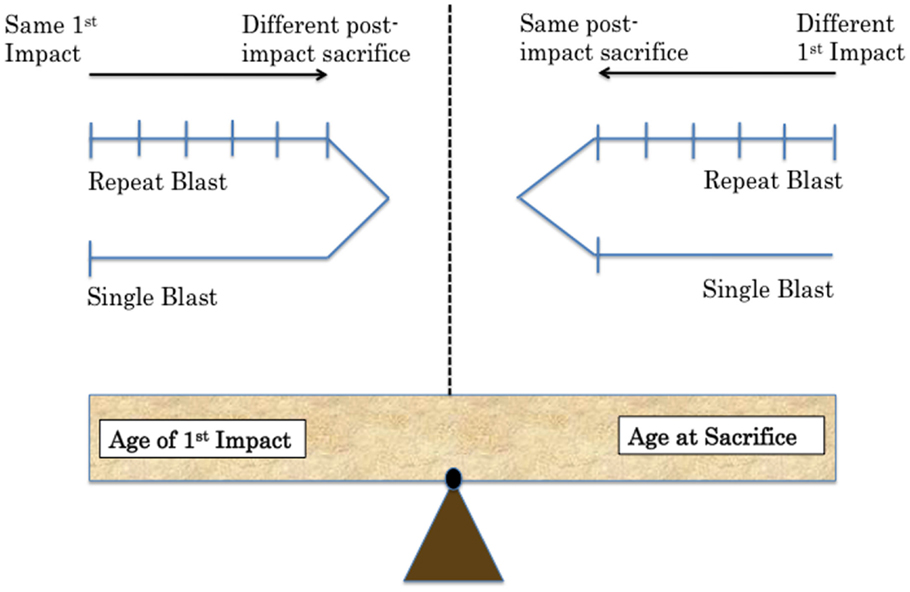
Figure 1. Methodological challenges associated with repeat injury in comparison to single-injury paradigms include balancing equal age at time of exposure versus age at sacrifice. Two possibilities are shown depending on the variable the experimenter wants to control in future work.
Available Tools to Study CTE
One of the current weaknesses of clinical investigation of CTE is that the disease remains a neuropathological diagnosis. Some advancement has been made in ligand-based PET imaging. The imaging was used in a cohort of patients with an extensive history of neurotrauma, but the modality is not readily available (21). Consequently, clinical observations are largely retrospective in nature and it is nearly impossible to investigate disease onset and progression. These clinical shortcomings can be readily addressed through the use of adequately designed preclinical models.
The first step in creating a preclinical model of CTE is to choose a model that can be used to generate a combination of biochemical and behavioral changes post-injury consistent with the CTE-like phenotype. Specifically, tauopathy or a precursor of tauopathy (tau hyperphosphorylation), must be present. The changes in tau must be inducible in genetically unaltered animals and then must be verified with transgenic rodents. Behavioral changes must be induced by neurotrauma. These changes should persist or worsen at chronic time points post-injury. Assessment of behavior should include tests capable of evaluating behavioral symptoms reported in clinical CTE cases. In particular, tests for cognition, depressive-like behavior, and impulsive-like behavior should be used. Cognition can be measured using Morris water maze (MWM), whereas depressive-like behavior can be assessed with the forced swim test or tail suspension test. Impulsivity can be elucidated with the elevated-plus maze (EPM).
Once a model is established, biochemical, electrophysiological, and advanced imaging techniques can be employed to assess neural injury. Biochemical mechanisms of potential interest include those related to cell survival/death, regulation of tau phosphorylation/dephosphorylation (kinases/phosphatases), bioenergetics, and propagation of tau-based changes. Electrophysiological studies can be performed to identify effects of injury and CTE development both on individual synapses and larger tracts such as the Schaffer collaterals within the hippocampus. Imaging modalities, such as magnetic resonance imaging (MRI), functional MRI (fMRI), spectroscopy (MRS), and PET studies, have the unique advantage of being able to be performed longitudinally with multiple assessments of the same animal at different time points. Taken together, integration of biochemical, behavioral, electrophysiological, and imaging modalities may provide insight into the mechanisms and time course of CTE development. This approach will also allow for more definitive evidence to be gathered. This evidence will provide a stepping-stone in addressing key questions about the effect of inter-injury interval, injury severity, and number of cumulative injuries necessary for the development of CTE. The answers garnered from this potential work may then influence the design of clinical trials that dictate return-to-play decisions. Baseline monitoring may become required in sports arenas and battlefields with high incidences of neurotrauma. The monitoring may assist in detecting cumulative subthreshold injury levels and be used to decrease the overall level of concussions.
Implications for Modeling CTE: Results from Prior CTE Modeling Studies
Perhaps one of the most promising studies, with regards to demonstrating CTE-like disease in a rodent model, was performed by Luo et al. In this study, the authors developed a closed-head model of neurotrauma utilizing an electromagnetic stereotaxic impact device. The authors showed that enhanced force of injury or using repeat injury increased GFAP-tagged luciferase. Intriguingly, when this same repeat-injury paradigm was applied to wild-type mice, spatial learning, and memory deficits were observed 2–6 months after injury and were accompanied by increased hyperphosphorylated tau and astrogliosis (22). The gliosis response in human CTE has not been well characterized, but the findings by Luo and colleagues show that GFAP was increased near areas of tauopathy. Similar findings have been seen by Petraglia and colleagues who performed a rigorous investigation of the behavioral effects of both single and repetitive closed-head injury in wild-type mice (23). The authors found that a single injury, in un-anesthetized animals, produced notable short-term abnormalities in behavior similar to a post-concussive state. Repetitive injury (42 impacts total: 6 impacts per day over 7 days) produced chronic deficits, particularly with regard to depressive-like and risk-taking behaviors as well as spatial learning and memory (23). This same group published a recent follow-up study demonstrating the presence of hyperphosphorylated tau, a precursor of NFT development, in repetitively injured animals (20). The model produces several of the same behavioral symptoms reported by patients suspected of having CTE. We have recently shown that endoplasmic reticulum stress contributes to tauopathy and CTE-like behavioral deficits following repeat blast injury (24).
Other studies, conducted by Mouzon and colleagues report both behavioral and neuropathological changes following repetitive neurotrauma in mice. Specifically, five injuries administered 48 h apart, produced durable cognitive deficits, learning disabilities, diminished rotarod performance, and changes in anxiety-like behavior on EPM. Notably, these behavioral changes occurred in conjunction with persistent neuroinflammatory changes and disruption of white matter integrity. No changes in Aβ and tau phosphorylation were seen at the chronic time points of 6 and 12 months post-neurotrauma, likely because rodents do not naturally develop NFTs (25). Rodents do, however, demonstrate acute tau changes due to phosphorylation and cleavage following injury (26). Liu and colleagues found that tau hyperphosphorylation was increased in rats acutely post-injury and triggers caspase activation in rat cortices (27). The activation of cell death can lead to circuit dysfunction and behavioral deficits (28). Goldstein and colleagues show that tauopathy contributes to mitochondrial dysfunction and microtubule injury that ultimately leads to apoptosis. In this study, they found that tau modulation is a potential avenue for therapeutic intervention (29). We have recently reported that caspase activation is increased in human CTE brains near sites of NFT formation (30).
Other repeat-injury studies have investigated changes in tau and amyloid post-injury with the goal being to more clearly elucidate the relationship between TBI- and neurotrauma-related neurodegeneration. One report showed an increase in neuronal tau immunoreactivity (31) and another showed elevated amyloid precursor protein (APP) (13) at a variety of time points post-injury. A final study by Zhang and colleagues showed that monoacylglycerol lipase can lead to behavioral deficits and tauopathy characteristic of a CTE-like phenotype (32). These findings were further verified in other studies using transgenic models of amyloidosis and tauopathy in which repetitive injury paradigms produced elevated amyloid and tau levels with increased deposition. Single injury, however, failed to produce changes above control levels (33, 34). Using a T44 tau Tg mouse line, Yoshiyama and colleagues sought to study dementia pugilistica (DP), a condition sharing many features with modern day CTE, but with several key differences. DP by definition occurs only in boxers and has more severe gross anatomical changes that are not always present in CTE. The Yoshiyama group found that four impacts per day given once a week for 4 weeks produced only modest neuropathology with only one mouse demonstrating CTE or DP characteristics out of a total of 18 Tg mice and 24 wild-type mice (13, 35). The affected mouse displayed neuropathological changes that included heightened tau burden, the presence of NFTs, and cognitive deficits (35). While it remains unclear why only one mouse developed such pathology, neuropathological findings from this one animal demonstrated that iron deposition was increased and associated with blood–brain barrier disruption. Iron deposition, associated with degradation of heme, activates oxidative stress-related pathways. Importantly, this oxidative stress is then associated with accelerated NFT formation in these perivascular locales (35).
Notably, even single-injury paradigms have been shown to produce tauopathy in a variety of injury models, including blast (17), fluid percussion (36), and controlled cortical impact, findings consistent with prior clinical reports (37, 38). Goldstein and colleagues demonstrated that tauopathy following single blast injury was associated with hippocampal-dependent learning and memory deficits at subacute and chronic time points. These changes were also associated with electrophysiological alterations in long-term potentiation (LTP) (17). Therefore, even a single mild to moderate injury may induce neurodegeneration and neurological deficits leading to impaired cognition and disrupted synaptic transmission (17). Single injury has also been shown to contribute to blood–brain barrier disruption (39). This is a significant finding considering the numerous concussive and subconcussive injuries occurring in athletics and on the battlefield. Other studies using single-injury models have also demonstrated activation of numerous pathological processes and behavioral changes associated with neurodegeneration. Modeling neurotrauma-related neurodegeneration is a key component in the search for a model of CTE (40–42). While the importance of repeat injury in CTE modeling cannot be overstated, some single-injury studies have led to advances in the ability to detect phospho-tau in serum at weeks to months post-injury. These advances indicate the potential role of biomarkers in monitoring and understanding disease pathophysiology (38). The development of animal-based models that exhibit similar characteristics of CTE will afford researchers the opportunity to characterize the acute and chronic effects of injury on the phosphorylation of tau in controlled experimental conditions. In vitro models may even be beneficial in elucidating changes at the cellular level (43). Evaluating imaging modalities, potential biomarkers, such as phosphorylated tau, and proposed therapeutics in a controlled context will promote advancement toward clinical applications and could be instrumental for monitoring and understanding disease pathophysiology in the future.
Implications for Modeling CTE: Learning from Past Shortcomings with TBI Models
Despite these notable advancements in CTE modeling as outlined above, the vast majority of repeat-injury studies fail to address the role of injury severity, inter-injury interval, and the total number of impacts needed to reproduce a CTE-like state (44, 45). Most studies simply describe features of TBI without relating the findings to neurodegeneration (46). Furthermore, because CTE is a neuropathological diagnosis, any study claiming to serve as a model of CTE must demonstrate the hallmark neuropathological changes (tau hyperphosphorylation and NFTs) and show that these changes persist at delayed time points and coexist with behavioral deficits. Few studies have looked at amyloid, tau, or TDP-43 accumulation following injury.
One of the most common omissions from repetitive injury paradigms is the lack of consideration for inter-injury interval. Work by Longhi and colleagues directly explored this issue, demonstrating that in a mouse model of closed-head injury (CHI), mice had a period of vulnerability estimated between 3 and 5 days where the effect of injuries was additive and produced deficits in cognition. When the inter-injury interval was lengthened to 7 days, these deficits were not present (47). While the focus of that study was elucidating differences in the inter-injury interval, other groups employing a repeat-injury paradigm should consider how the inter-injury interval may relate to their findings. Inter-interval design will be an important consideration in developing CTE models. Attempting to provide a clinical context for these decisions would be desirable, with the interval between severe concussive-type impacts ideally being longer than that between mild subconcussive-type impacts. This would expand scientific insight for “return-to-play” guidelines that require players to be asymptomatic following a diagnosed concussion prior to returning to contact sports. These types of questions have generally not been considered in preclinical work, with most studies using brief inter-injury intervals, often ranging from minutes to 24 h with poor justification for timing (23, 48–52).
Another important limitation, related to interpretation of injury severity and neuropathological outcome, is the sensitivity of detecting neuropathology using current scientific approaches. Shitaka and colleagues demonstrated previously that silver staining was more reliable for detecting axonal injury and pathology in comparison to routine histological analysis, assessment of neuronal cell loss, and APP immunohistochemistry (49). It may be increasingly important, particularly in studies of repetitive subconcussive impacts, to utilize measures of high sensitivity for injury detection. Silver staining, which has been shown repeatedly to exhibit a higher degree of sensitivity in detecting axonal injury than many immunohistochemical techniques, electron microscopy, or other markers with these characteristics may prove promising (49). It has not yet been determined if NFTs accumulate around sites of axonal shearing.
Key Questions Going Forward
Improving experimental models will enhance the quest for developing therapeutic agents that can be used to prevent and treat CTE. The search for a model of CTE raises a number of questions that are important clinically. These questions include issues such as length of the inter-injury interval, the number and severity of impacts, and the age at time of impacts, as well as the mechanism of impact, the gender of the patient, and what role genetic predisposition may play in the development of neurodegenerative disease following neurotrauma. Another important question that must be addressed is does a history of neurotrauma and potential presence of CTE accelerate the development of AD? Furthermore, how can the period of susceptibility following neurotrauma be identified most readily? In the following sections, we attempt to address these questions based on available evidence. We also provide suggestions for handling shortcomings going forward. Studies on which these sections are based are referenced in Tables 1–7 for quick reference.
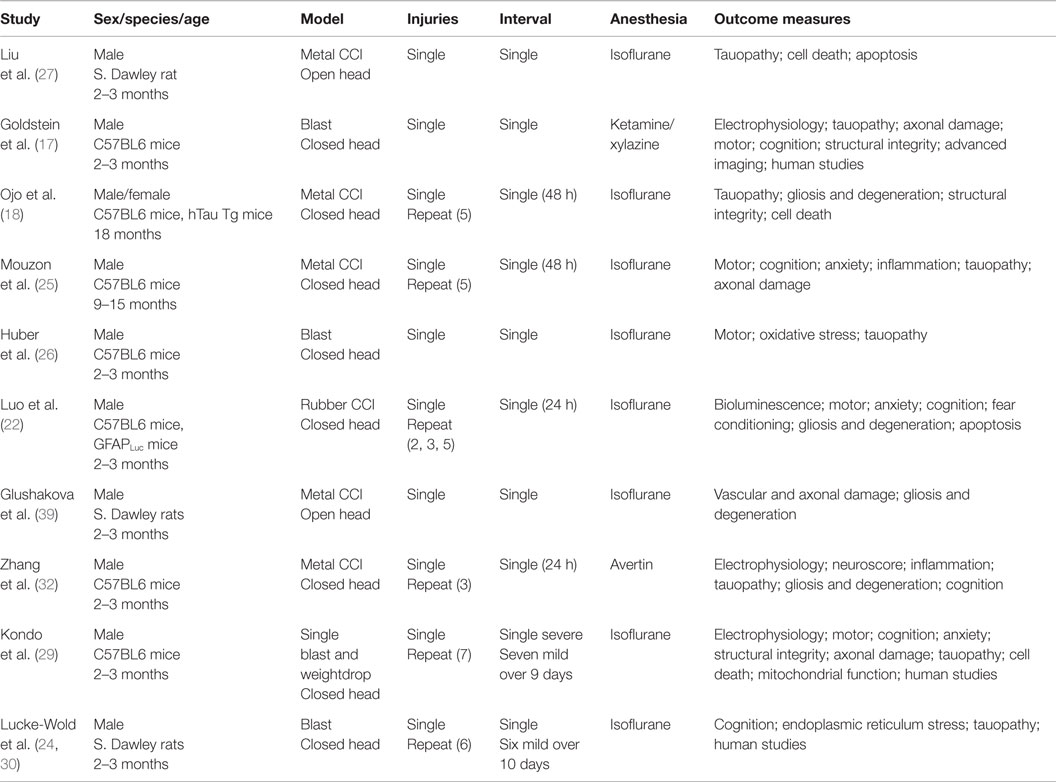
Table 1. Chronic traumatic encephalopathy TBI models.
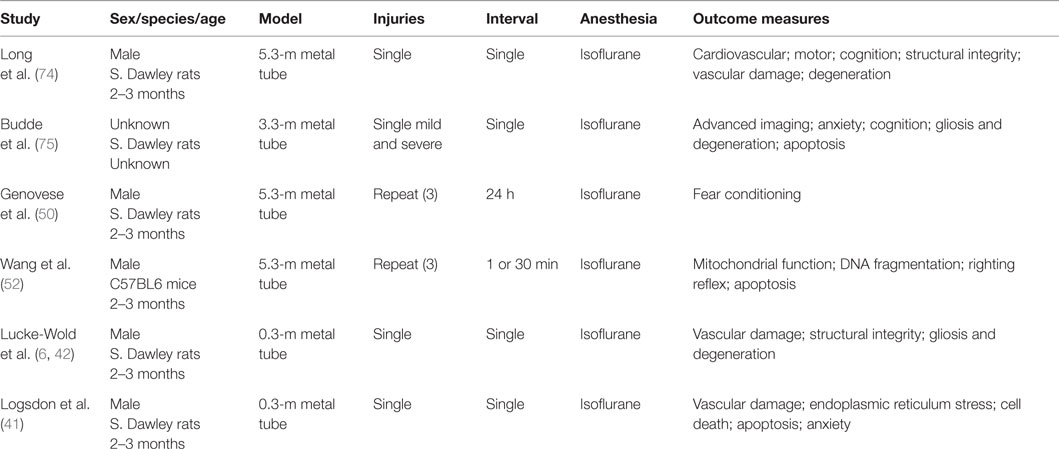
Table 2. Shock tube TBI models.
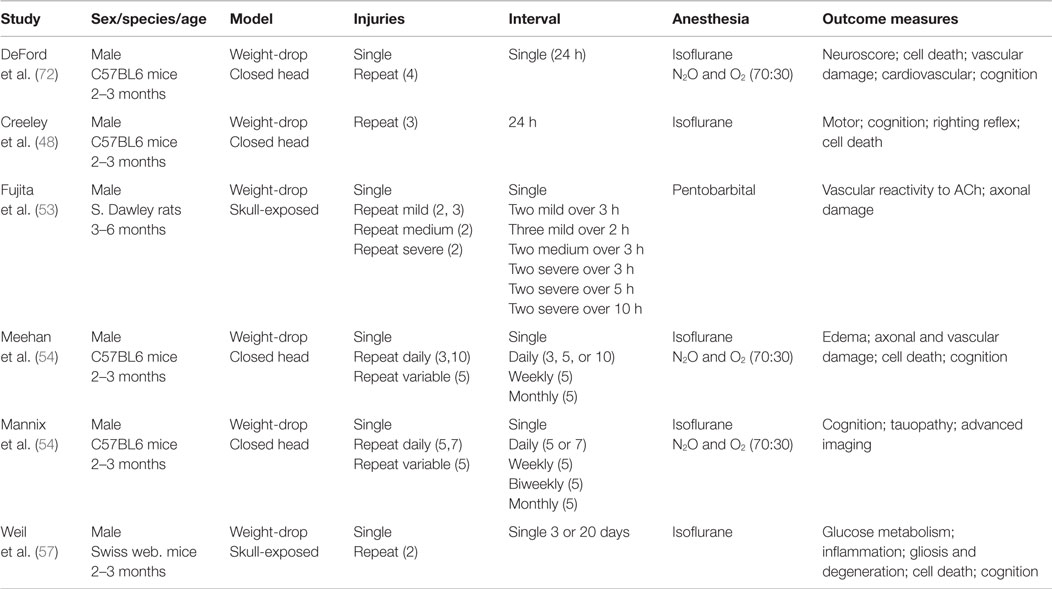
Table 3. Weight-drop TBI models.
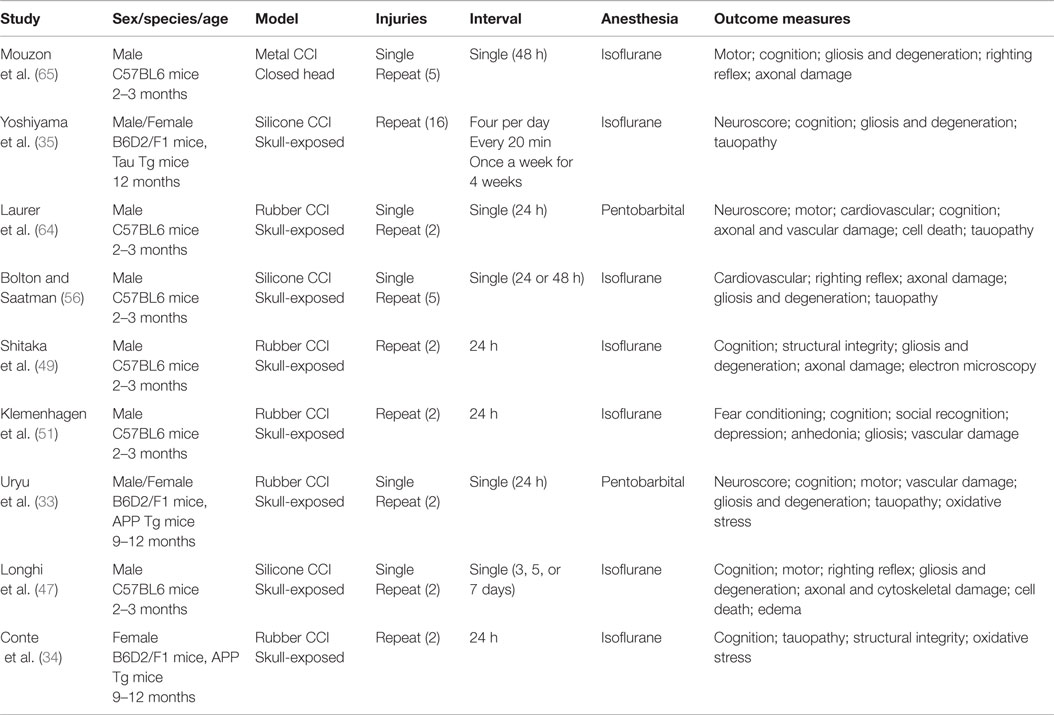
Table 4. TBI models not using craniotomy.
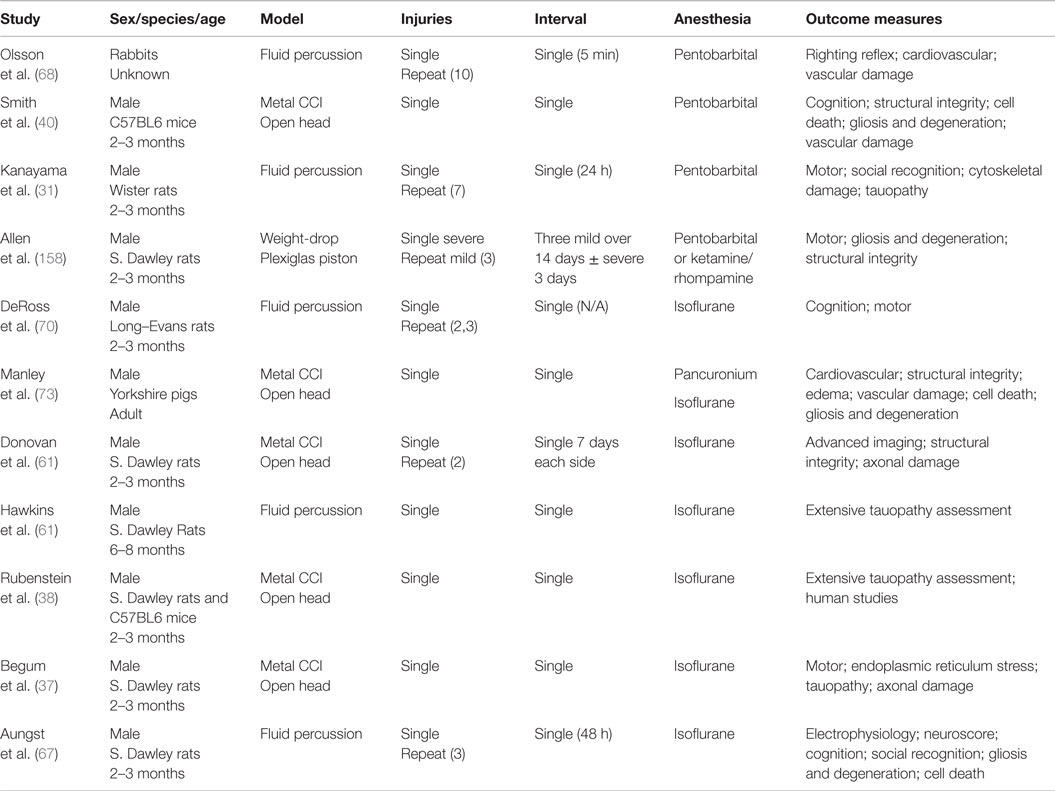
Table 5. TBI models using craniotomy.
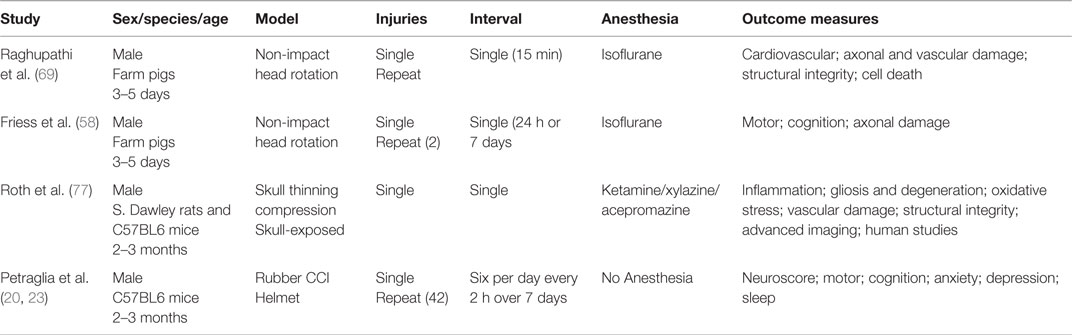
Table 6. Other in vivo TBI models.
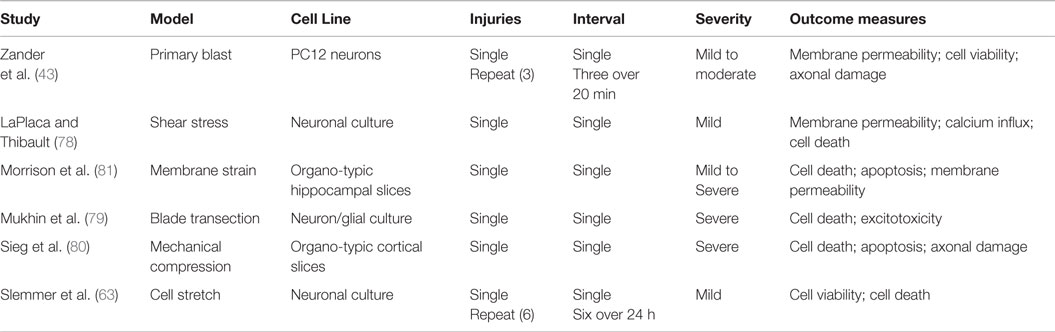
Table 7. In vitro and ex vivo TBI models.
Inter-Injury Interval
The effect of inter-injury interval on outcome following TBI has only recently been investigated. Studies have explored a variety of intervals ranging from 2 min apart (in vitro) to a few hours (in vivo) to as long as 30 days apart (in vivo). Using a novel approach assessing vasoreactivity in TBI, Fujita and colleagues demonstrated that administration of seemingly mild injuries at brief intervals (3 h apart) produced dramatic declines in vasoreactivity and axonal pathology. When the inter-injury interval was lengthened to 5 h, the magnitude of these changes was diminished substantially with complete dissolution of changes in both pathology and vasoreactivity at 10 h (53).
Other studies that have investigated longer inter-injury intervals have identified periods of susceptibility following an initial impact at periods ranging from 24 h to a few weeks (54–57). Bolton and colleagues demonstrated with a CHI model that a single impact produced extensive gliosis bilaterally in the hippocampi and entorhinal cortices. Repeat injury after 24 h produced a more severe injury consisting of hemorrhage in the entorhinal cortices as well as heightened measures of neurodegeneration, gliosis, and neuroinflammation (56). When the experimental paradigm was changed such that impacts were given with a 48 h inter-injury interval, the histopathology resembled that of a single impact suggesting enhanced susceptibility when a second impact was administered within 24 h (56).
Mannix and colleagues performed one of the most rigorous investigations of the effect of inter-injury interval on outcomes associated with TBI. These studies included measures of both cognition and neuropathology related to the development of neurodegenerative diseases such as CTE and AD. In this study, the investigators showed that animals that received daily or weekly injuries with weight-drop had persistent cognitive deficits up to 1 year post-injury (55). This was in contrast to when animals were injured biweekly or monthly, which failed to produce deficits at such a chronic time point (55). Notably, the cognitive deficits seen in the daily- and weekly injured animals did not correlate with elevations in tau phosphorylation or amyloid-β when measured by ELISA nor brain volume loss when measured by MRI (55). This finding may indicate that in addition to inter-interval time, injury severity must be considered. Tauopathy is essential for modeling CTE therefore an appropriate inter-injury interval might be best characterized in transgenic animals. Weight drop produces variable injury based on the height of the drop. A more severe TBI can produce cognitive deficits but may not be representative of the concussive and subconcussive injuries associated with CTE. A mild TBI with a transgenic animal will likely produce the most relevant deficits.
Meehan and colleagues performed a similar study with primarily behavioral assays. The investigators subjected mice to a CHI via weight-drop for a total of five impacts at various intervals. These intervals included daily, weekly, and monthly intervals. Mice receiving five impacts total at daily or weekly intervals were impaired in the MWM compared to sham animals (54). This was not the case when injuries were delivered at monthly intervals, as these animals exhibited no impairment in the MWM (54). Interestingly, at 1 month post-injury, the daily- or weekly injured animals still exhibited deficits in the MWM and this deficit persisted in daily-injured animals out to 1 year (54). This finding may represent why football lineman who experience daily subconcussive injuries appear more likely to develop CTE based on the clinical cases reported. It is still necessary to establish if tauopathy is the driving mechanism behind behavior. The findings confirm and expand upon the inter-interval studies completed by Longhi and colleagues (47). Longhi reported that shorter inter-interval injuries produce worse outcomes, which is in agreement with the Mannix and Meehan findings. These studies were consistent with findings in higher phylogenetic species as well, specifically piglets. Friess and colleagues showed that a 24-h inter-injury interval produced more severe deficits and higher mortality rates than when the interval was extended to 7 days (58). Finally, Kanayama and colleagues demonstrated a graded response in locomotor activity. Both shorter inter-injury intervals and greater number of total injuries were associated with worse outcome (31).
Weil and colleagues explored the effect of altering the inter-injury interval in relation to recovery from TBI. They used a clear clinical-minded approach and utilization of metabolic imaging (PET). This group showed that injuries separated by only 3 days were associated with worse neuropathology and an inability to mount the typical hypermetabolic response with regard to glucose utilization following TBI. This worse outcome was not seen following either a single injury or repeat injuries with an extended inter-injury interval of 20 days (57). Similarly, a brief inter-injury interval of 3 days was associated with elevated IL-1β and TNFα gene expression when compared with other experimental groups (57).
The longest interval between injuries used in preclinical studies, to the best knowledge of the authors, was 30 days. The additional injury had no additive effect on anxiety (EPM), depression (FST), and cognitive function (MWM) when compared to animals receiving only one injury (59). These findings indicate that either the window of vulnerability following the first injury was avoided or that the response to the first injury may protect the animal from subsequent injuries, a concept known as preconditioning (59). The progression toward tauopathy was not well characterized in this work.
In vitro studies have shown similar findings to the in vivo studies described above. Shorter inter-injury intervals between mechanical stretching resulted in an elevation in S-100β protein release and increased cellular permeability identified with propidium iodide staining (13). Similarly, a “subthreshold” level of stretch did not produce any overt cellular damage or death when repeated at 1 h intervals but did cause neuronal loss and neuron-specific enolase (NSE) release when performed at incredibly short intervals (every 2 min) (13, 60). Remarkably, this rapid and repetitive “subthreshold” stretch that produced changes in neurons, failed to produce an increase in S-100β protein release, indicating a differential response between neurons and glia to neurotrauma severity and interval (13, 60).
In contrast to the above studies, one group showed that repeat injury, when administered in different anatomical locations within the brain, failed to result in heightened damage when an inter-injury interval was 3 days. It did, however, increase tissue vulnerability with a 7-day interval as evident by increased hemorrhage (61, 62). The authors of the study therefore argue that the period of susceptibility likely depends on not only the time interval between injuries but also the anatomic location of injury (61, 62). Importantly, this study utilized an open injury model (controlled cortical impact), a scenario that is only seen in a subset of clinical neurotraumas.
In summary, these studies demonstrate a period of vulnerability following initial injury in which sustaining a second brain injury may result in an additive effect. Additive injury is not clearly apparent when the brain is allowed a more extensive recovery period. Interestingly, this vulnerability may not solely be due to initial axonal pathology but may also be the result of cerebrovascular reactivity and the inability to utilize glucose effectively (53, 57). This concept of a varied cellular response to TBI is consistent with findings from in vitro studies that demonstrate a varied response amongst glia and neurons (13, 60, 63).
Number of Impacts
The prevailing theory for “mild” neurotrauma is that repetitive injuries are associated with more short- and long-term detrimental effects than a single injury alone (56, 64, 65). This concept has been applied regardless of injury severity (concussive versus subconcussive) with emerging evidence indicating that even subthreshold impacts are cumulatively detrimental (53, 66). In this section, we explore the effect of repeat injury through analysis of studies employing both repeat and single-injury paradigms.
One of these studies, conducted by Mouzon and colleagues, showed that when rodents are exposed to five total injuries with an interval of 48 h, these animals exhibit both impaired learning and memory at extended time points (25). These findings are in contrast to single-injured animals that display only learning deficits but no retention impairment at the same time points (25). This study closely parallels the clinical findings documented by Guskiewicz and colleagues in which former athletes with a history of repetitive concussions experience memory-related issues at a rate of five times higher than those without a history of concussion (12, 25).
Other work that investigated various iterations of impacts (0, 1, 3, 5, and 10) demonstrated that while a single injury does not produce deficits in MWM performance in comparison to sham-injured animals, repetitive injury does in fact produce deficits and these deficits may exhibit a dose-dependent relationship. When mice were given 10 concussive weight-drop injuries, those in which the weight was dropped from a height of 42″ performed worse than those injured from a height of 38″. Therefore, this work demonstrates the potential for both injury number and injury severity in contributing to neurological dysfunction (54). Luo and colleagues utilized a GFAP-driven luciferase mouse line and a repetitive closed-head injury model to investigate cumulative decline. The investigators showed that there appeared to be a linear increase in GFAP luminescence from 1 to 3 injuries but that this response appeared to reach a plateau by five injuries (22). In addition to the increase in GFAP fluorescence with repetitive injuries, mice receiving three injuries demonstrated less freezing time than sham animals in both cued and contextual fear conditioning (22). This was in contrast to single-injury animals that did not differ from sham-injured animals in cued or contextual memory (22). Consequently, these data demonstrate that an increase in injury number is associated with an increasing severity of injury markers (based on protein expression of GFAP) as well as functional deficits (fear conditioning). Others employing both single- and repeat-injury paradigms have shown that while single injury may not induce pathological findings, repeat injury, of the same severity, does. This was particularly notable in work by Uryu and colleagues in which repetitive injury produced an increase in Aβ deposition in Tg2576 animals, whereas no increase was observed in single-injury paradigms in these same animals (33). Likewise, Kanayama and colleagues demonstrated that repeat-injury paradigms induced tau hyperphosphorylation, a precursor to NFT formation, in conditions such as CTE and AD (31).
The observed findings in closed-head, repeat-injury models, were also consistent with those found in open-head injury using a craniectomy and controlled cortical impact. In work by Donovan and colleagues, the investigators showed that repeat injury induces progressive and evolving changes that are not observed in single-injury paradigms (62). The basis of many of the memory and cognitive changes following repeat TBI may be explained by electrophysiological alterations in synaptic transmission. In work by Aungst and colleagues, repetitive TBI was found to prevent the induction of LTP 28-days post-injury due to alterations in the NMDA receptor. This is in stark contrast to single-injury paradigms that revealed the ability to induce LTP in both hemispheres, with the contralateral hemisphere exhibiting less LTP than the ipsilateral hemisphere. The impairments in excitatory neurotransmission following repeat injury were accompanied by extensive neuroinflammation and neurodegeneration as well as behavioral/functional impairments (67). Specifically, repeat injury produced deficits more severe than single injury when measured at 1-week intervals out to a month. Importantly, even single injury produced deficits at chronic time points post-injury when compared to sham animals, indicating long-term effects of TBI (67).
Povlishock and colleagues expanded the concept of subthreshold injury. In these studies, the investigators showed that administering a single weight-drop injury from 1.0 m resulted in neither axonal nor microvascular change. With repeat injury of short inter-injury intervals (hours), significant axonal and microvascular pathology was observed (53). This work was the first to assess microvascular reactivity to acetylcholine (ACh) following repetitive subthreshold brain injury. This work demonstrates the clear danger of subthreshold impacts when sustained in a repeated and rapid fashion. It also illustrates the role of the microvasculature in neuronal injury, showing that the neurovascular unit is essential for neuronal homeostasis.
Importantly, work regarding the number of impacts has been extended higher up the phylogenetic tree to rabbits and piglets (68, 69). In rabbits, repeated loading with loads of up to 1.5 atm failed to produce an additive concussive response over a single load. A multi-loading paradigm at higher loads, however, caused respiratory arrest (68). In piglets, multiple less severe injuries induced neuropathological findings similar to a severe load based on the density of injured axons as well as number and distribution of foci (69).
In vitro studies have reported similar findings to the in vivo studies. Weber and colleagues applied a mild stretch to hippocampal neuronal cultures that produced low-grade injury when applied at a single time. When this injury was repeated, the cells exhibited cumulative damage with two injuries inducing an increase in NSE (13, 60, 63).
Notably, the group led by Mychasiuk and colleagues provides evidence contrary to the widely held belief that an increased number of impacts are associated with detrimental findings on behavioral or histological measures. With a 30-day inter-injury interval, rodents receiving multiple injuries performed similar to single-injury animals on measures of anxiety, depression, and cognitive ability. Therefore, they propose that receiving head injury at an early age may prime the brain to be less susceptible to the effects of a later neurotrauma, a theory known as “preconditioning” (59). DeRoss et al. also showed that while one concussive impact resulted in diminished performance in 85% of animals, less deviation was seen with subsequent impacts. The number of impacts has an inverse relationship with animal performance in the water maze (70). Again, the mechanism behind these findings is not entirely clear as those animals sustaining multiple injuries also received additional exposure to the water maze, allowing for enhanced training/learning of the maze (70). Therefore, while repetitively injured animals did better than single-injured animals in the maze, this is likely a product of increased training rather than a protective response but this cannot be said with absolute certainty.
Severity of Impacts
The effect of injury severity on likelihood of neurodegenerative disease development is not entirely clear, although some clinical reports indicate that more severe injury results in a greater predisposition for AD development (33, 71). What is known from preclinical studies using an array of animal models is that there is a dose-dependent increase in neural injury markers and cognitive deficits with more severe injury (22, 72–76). Similarly, repetitive “mild” injuries may produce a phenotype more consistent with a single more severe injury (72).
How “mild” TBI contributes to the likelihood of developing CTE remains unclear. Emerging evidence from preclinical studies raises concern about lasting effects of subthreshold injuries when sustained in a rapid and repetitive fashion. These repetitive injuries contribute to vascular reactivity and subsequent axonal degeneration in vivo (53). Some studies suggest that severity of injury may dictate the rest period required to minimize cumulative cognitive deficits, although further studies are required to validate these findings (54).
Similarly, preclinical studies, even those in which only a mild force is imparted to a thinned cranium, indicate that a substantive inflammatory response is produced quickly after injury (77). This response is associated with heightened vascular permeability, also seen clinically, as well as microglial response (77). It is these mechanisms, both primary and secondary, that may contribute to neurodegenerative disease post-neurotrauma.
In vitro studies may be of further use in addressing the role of injury severity, particularly in subconcussive/subthreshold-type studies, as levels of injury and subsequent cellular responses can be monitored rapidly and performed over a greater number of iterations at a lower cost in comparison to in vivo studies. In fact, a number of injury paradigms and mechanisms have been investigated successfully in this manner, including fluid pulse-induced shear stress, repetitive stretching, and other mechanical deformation procedures, both in vitro and ex vivo (60, 63, 78–81). The in vitro shearing studies found a significant amount of axonal beading and glial death (82).
Age at Time of Impacts
Age is the biggest risk factor for the development of neurodegenerative disease and has been associated with poor outcomes following TBI in a variety of clinical and preclinical reports (83–91). Similarly, neurotrauma has been associated with an increased risk of neurodegenerative disease development with regard to AD (71, 92–94), PD (95), and CTE (2, 5, 7, 16, 17, 96–103). One of the primary questions currently in the field is how the age at which the patient sustains the neurotrauma pertains to the development, or lack thereof, of CTE.
Similar findings have been observed in preclinical studies. Mychasiuk and colleagues showed that TBI during brain development leads to worse outcomes than TBI affecting the mature brain (59). Preclinical work has also shown that young animals experience less edema than middle-aged animals following TBI (104). The increased edema is associated with an increase in lesion size in aged rodents experiencing TBI (105). TBI in aged rodents is also more likely to increase sensorimotor and cognitive decline (85). TBI in youth may ultimately be more detrimental for social development, whereas severe injury in the elderly results in rapid cognitive decline due to increased edema and therefore, lesion size. In regard to human TBI, it is unclear if the elderly would have a more progressive form of the disease similar to rodent studies or if the disease would develop in the normal manner. It is also important to consider that males <35 years old are the most likely to have repetitive TBIs, a group associated with heavy participation in sports and now the military (106). Future preclinical studies should therefore continue to investigate TBI secondary mechanisms in both young and aged animals with particular attention paid to addressing repetitive injury paradigms and the development of CTE-like features, both behaviorally and biochemically.
Mechanism of Impact
Chronic traumatic encephalopathy has been diagnosed in athletes sustaining direct impacts as a result of participation in warfare, football, wrestling, and soccer (2, 5, 7, 16, 17, 96–103). The mechanism of injury is different between blast and athletic concussions, but how these mechanisms relate to injury progression remains to be elucidated. For blast, primary to quaternary injury must be considered (107). Other important questions include what is the influence of linear versus rotational impacts? What is the effect of direct impacts such as a football tackle versus indirect impacts such as primary blast exposure? Furthermore, how can comparisons most accurately be made across these various impacts? Are accelerometers and recording systems (such as the HITS system) the best method for understanding and comparing these impacts? What role does high-speed videography and subsequent kinematic analysis play? Each of these questions remains to be answered and may provide further insight into understanding the role that impact type plays in CTE development.
Biochemical Mechanisms
Interestingly, glymphatic clearance has recently been shown to play a role in injury progression. Iliff and colleagues showed increased tauopathy accumulation in aquaporin knock-out mice following traumatic brain injury due to disrupted glymphatic clearance (108). It has yet to be determined how the primary injury mechanism causes the disruption to glymphatic channels. Cernak proposed an interesting theory about low-frequency stress waves transmitting kinetic energy through tissue (109). This mechanism may account for the dysfunction of the aquaporin channels. The energy transfer may also injure other cellular components such as axons or vessels. Chodobski and colleagues have shown that kinetic transfer of energy can account for blood–brain barrier disruption (110). Blood–brain barrier disruption post-injury can trigger increased neuroinflammation. Agoston and colleagues showed that blast traumatic brain injury, in particular, causes persistent neuroinflammation, which leads to behavioral deficits (111). The neuroinflammation can also contribute to post-traumatic epilepsy and tauopathy (112). Investigating how tauopathy spreads is a topic warranting further investigation (113). Furthermore, Kobeissy and colleagues highlight in their recent review that underlying neuronal damage can cause lasting neuropsychiatric deficits such as post-concussion syndrome and post-traumatic stress disorder (107).
Role of Genetics
Genetics and lifestyle choices may play a role in likelihood of sustaining a TBI and also the outcome following a TBI. Little to no evidence currently exists regarding lifestyle choices associated with TBI and only recently has genetic contribution to CTE been addressed. Specifically, it is well known in the human literature that the APOϵ4 allele is associated with worse outcome following TBI (114). APOϵ4 and APOϵ3 have also been implicated in the development of both CTE and AD (13, 115). The mechanism by which APOϵ4 worsens outcome following TBI is poorly defined (55), but targeted replacement of the allele in mice has allowed focused research into cholesterol metabolism and may lead to insights in the field of traumatic brain injury and subsequent neurodegeneration (116). Notably, a study by Maroon and colleagues recently demonstrated that there was no significant difference between ApoE4 carriers in a population of patients afflicted with CTE when compared to the general population, suggesting that perhaps ApoE may not represent a significant risk factor for CTE development, even in individuals exposed to neurotrauma (117).
Other genetic influences have also been observed but are currently limited to preclinical evidence. Rare genetic alterations such as mutation of the CACNA1A calcium subunit gene have also been shown to lead to poor TBI outcomes in human patients (118). Recent preclinical studies have shed light on other potential genetic factors that may influence TBI outcomes. Decreases in micro RNA 23a and 27a increase apoptosis following TBI in rodents (119). Deficient caveolin expression can exacerbate neuroinflammation post-TBI (120). The knock-in mouse APP696swe has accelerated deposition of Aβ following TBI (35). Emerging evidence also suggests dysfunctional mitochondrial genes following TBI such as Fas, Apaf1, and Chp. Interestingly, these genes become more dysfunctional and mutated with time after mild TBI (121). TBI can also induce DNA fragmentation leading to an upregulation of p53, a critical regulator of cell cycle (122). On the other hand, upregulation of insulin growth factor expression prior to TBI is associated with neuroprotection (123). Similarly, genetic regulation of aquaporin 4 channels can reduce edema formation following TBI in rodent models (124). Surprisingly, disruption or the PARP1 gene offers protection against TBI hypoxia (125). Studying genetic risk factors for TBI is an area of growing importance and requires further investigation. Understanding factors that lead to increased or decreased TBI severity may also allow the development of novel pharmaceutics for the prevention of neurodegenerative disease.
An emerging area is the role of epigenetic modulation following TBI. Epigenetic markers are now being used preliminarily to predict recovery following injury (126). VandeVord and colleagues show enhanced methylation of DNA in the rat hippocampus following blast traumatic brain injury (127). These epigenetic changes are mediated by HDACs and DNMTs (128). Interestingly, the methylation changes are cumulative with repetitive injury (129). HDAC has been shown to contribute to GSK3β activation, which is a known tau kinase. When HDAC is inhibited, white matter damage is reduced (130). Epigenetic regulation has been tied to the development of post-traumatic stress disorder clinically (131). Targeting epigenetic regulation may therefore be a viable target in preventing tauopathy and behavioral deficits following traumatic brain injury.
Influence of Gender
The influence of gender on outcome after TBI remains controversial, particularly in light of the few cases of CTE diagnosed in women. Some studies claim that females have better outcomes following neural injury (132), while others report no change (133, 134), or worse outcome (135). Estrogen treatment has shown improved outcomes in rodent models of neural injury (136–139), including TBI (140, 141). Female rodents exhibit better outcomes after neural injury as evidenced by increased neurotrophin production (140), decreased neuroinflammation (142), and better performance on motor tasks (143). Clinical evidence shows that female patients exhibit lower oxidative damage after TBI (144), which could be the result of a higher estrogen circulation following injury. Gender differences should be considered when conducting clinical trials for TBI therapy.
Interestingly, Dixon and colleagues found that TBI in female humans reduces estradiol in the CSF (145). The reduction of estradiol may have unique long-term effects because it is not yet known if this is transient or permanent. The limited amount of data about women with TBI has restricted the comparisons of injury between genders (146). The majority of traumatic brain injuries in women are subdural hematomas from falls. Elderly women with subdural hematomas tend to fair worse than the general TBI population as a whole (147). Menopause may therefore dampen the neuroprotective physiological properties mediated by estrogen post-injury. TBI in elderly women is also linked with earlier onset AD (148). Hormonal changes may also be a contributing factor to CTE, but this has yet to be verified. It is clearly based on the limited preclinical and clinical data that further investigation into the gender differences surrounding TBI outcome is warranted.
Effect of Environment
Another area of investigation required for elucidation of factors influencing the likelihood and/or severity of CTE development following neurotrauma includes the effect of environment. Areas of particular interest include social support, diet, use of supplements, and use of drugs or anabolic steroids.
Good social support has been shown to decrease the likelihood of developing a postconcussional disorder following acute head injury (149). Strong family support is linked to better outcomes (150). Another important feature of environment is diet. While the effect of diet is well understood with regard to general health, there are limited studies relating diet and mental health, particularly in the context of traumatic brain injury. A preclinical study conducted by Mychasiuk and colleagues showed that high-fat diet in conjunction with TBI resulted in cumulative deficits on assessments of motor function, short-term working memory, and produced depressive-like effects compared to animals with normal diet and TBI (59). Similarly, administration of dietary supplements such as Vitamin E and DHA has been shown to improve outcomes from TBI in numerous studies (34, 151–154). The suggestion has been made previously, based on evidence that some cases of diagnosed CTE occurred in former athletes with a history of anabolic steroid use, that anabolic steroid use may predispose these athletes to CTE development. While this question has not been completely resolved in terms of studying markers of CTE, preliminary studies found no difference in APP expression post-TBI regardless of when anabolic steroids were used (155).
Preconditioning
Any neural injury model has the potential to be complicated by the concept of preconditioning. Preconditioning at the most basic levels refers to neuroprotection for a given injury induced by a prior stimulus/injury. The concept of preconditioning has been well documented in a variety of neural injury models ranging from ischemic stroke to TBI (13). Initial stimuli that serve a protective effect in a subsequent injury include but are not limited to brief periods of ischemia, chronic exposure to moderate heat or heat acclimation, and subthreshold or mild injury (13). The proposed mechanism is that neuronal antioxidant machinery is upregulated with subthreshold injury thereby increasing the cells ability to respond to free radical production during subsequent injuries (156). Whether preconditioning serves a protective phenomenon in the development of CTE is unclear, but the general clinical consensus is that any brain injury no matter how small may be detrimental long term, minimizing any potential benefit of the preconditioning phenomenon (66, 157). This is in contrast to preclinical literature demonstrating that sustaining repetitive mild injury prior to a single severe injury protects the animal from the most deleterious effects of the severe injury (158). In other words, animals receiving repetitive mild injury prior to a severe injury do better than animals receiving a single severe injury (158). Similar findings have been observed in vitro in which subthreshold stretch prevented more deleterious injury when a threshold stimulus was given (60). The concept of preconditioning versus additive injury is captured pictorially in Figure 2.
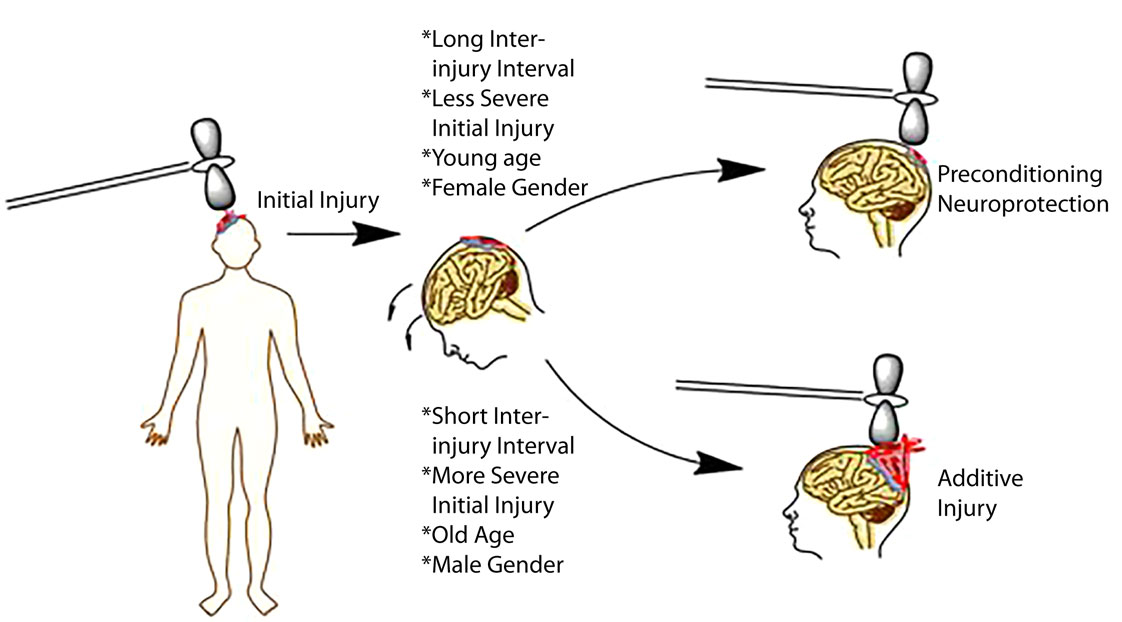
Figure 2. Schematic representation of factors influencing injury outcomes, particularly in repetitive injury paradigms. Initial evidence indicates that longer inter-injury intervals, less severe initial injury, a younger age, and female gender may serve as protective effects in repeat paradigms. By contrast, shorter intervals, increased severity, older age, and male gender may be associated with worse outcomes.
Summary
In conclusion, understanding CTE as a disease remains in its infancy and current studies remain largely speculative in nature without prospective clinical investigation. The required clinical studies to advance the field mandate extensive financial resources and time. Preclinical studies represent the most promising mechanism for studying many of the basic biologic questions about CTE, as discussed above. While these studies are continuing to evolve, numerous groups have reported exciting findings. Better modeling has allowed more extensive biochemical and behavioral characteristics to be defined. Now that our laboratory and others have established CTE models, options for translational investigation of CTE pathophysiology abound. In this work, we discussed numerous avenues for addressing translational questions, namely the role of (1) inter-injury interval, (2) number of impacts, (3) impact severity, (4) age at time of impacts, (5) mechanism of impact, (6) genetics, (7) gender, and (8) effect of environment on the development of CTE. We also highlighted some of the challenges of CTE modeling and specific requirements for successful models. By improving our understanding about CTE mechanisms, we believe that significant strides can be made not only in understanding CTE but also potentially developing prevention and therapeutic-related approaches. A companion manuscript describes our collective experience in modeling CTE, both neuropathologically and behaviorally.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
A Research Funding and Development Grant from West Virginia University supported the project. An American Medical Association Foundation Seed-Grant, a Neurosurgery Research and Education Foundation Medical Student Summer Research Fellowship, and an American Foundation of Pharmaceutical Education Pre-doctoral Fellowship supported BL-W. An American Foundation of Pharmaceutical Education Pre-doctoral Fellowship also supported AL. MR was supported by a PhRMA Foundation award and NIH grant NS007491.
References
1. Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med (1973) 3:270–303. doi:10.1017/S0033291700049588
2. Omalu BI, Dekosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery (2005) 57:128–34. doi:10.1227/01.NEU.0000163407.92769.ED
3. Mez J, Stern RA, McKee AC. Chronic traumatic encephalopathy: where are we and where are we going? Curr Neurol Neurosci Rep (2013) 13:407. doi:10.1007/s11910-013-0407-7
4. Baugh CM, Robbins CA, Stern RA, McKee AC. Current understanding of chronic traumatic encephalopathy. Curr Treat Options Neurol (2014) 16:306. doi:10.1007/s11940-014-0306-5
5. Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology (2013) 81:1122–9. doi:10.1212/WNL.0b013e3182a55f7f
6. Lucke-Wold BP, Turner RC, Logsdon AF, Bailes JE, Huber JD, Rosen CL. Linking traumatic brain injury to chronic traumatic encephalopathy: identification of potential mechanisms leading to neurofibrillary tangle development. J Neurotrauma (2014) 31:1129–38. doi:10.1089/neu.2013.3303
7. Omalu B, Bailes J, Hamilton RL, Kamboh MI, Hammers J, Case M, et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery (2011) 69:173–83. doi:10.1227/NEU.0b013e318212bc7b
8. Tartaglia MC, Hazrati LN, Davis KD, Green RE, Wennberg R, Mikulis D, et al. Chronic traumatic encephalopathy and other neurodegenerative proteinopathies. Front Hum Neurosci (2014) 8:30. doi:10.3389/fnhum.2014.00030
9. Ryan NP, Catroppa C, Cooper JM, Beare R, Ditchfield M, Coleman L, et al. Relationships between acute imaging biomarkers and theory of mind impairment in post-acute pediatric traumatic brain injury: a prospective analysis using susceptibility weighted imaging (SWI). Neuropsychologia (2015) 66:32–8. doi:10.1016/j.neuropsychologia.2014.10.040
10. Dubrovsky AS, Friedman D, Kocilowicz H. Pediatric post-traumatic headaches and peripheral nerve blocks of the scalp: a case series and patient satisfaction survey. Headache (2014) 54:878–87. doi:10.1111/head.12334
11. Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci (2010) 11:361–70. doi:10.1038/nrn2808
12. Guskiewicz KM, Marshall SW, Bailes J, McCrea M, Cantu RC, Randolph C, et al. Association between recurrent concussion and late-life cognitive impairment in retired professional football players. Neurosurgery (2005) 57:719–26. doi:10.1227/01.NEU.0000175725.75780.DD
13. Weber JT. Experimental models of repetitive brain injuries. Prog Brain Res (2007) 161:253–61. doi:10.1016/S0079-6123(06)61018-2
14. Barnes DE, Kaup A, Kirby KA, Byers AL, Diaz-Arrastia R, Yaffe K. Traumatic brain injury and risk of dementia in older veterans. Neurology (2014) 83:312–9. doi:10.1212/WNL.0000000000000616
15. Tran HT, Laferla FM, Holtzman DM, Brody DL. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci (2011) 31:9513–25. doi:10.1523/JNEUROSCI.0858-11.2011
16. McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol (2010) 69:918–29. doi:10.1097/NEN.0b013e3181ee7d85
17. Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med (2012) 4:134ra160. doi:10.1126/scitranslmed.3004862
18. Ojo JO, Mouzon BC, Crawford F. Repetitive head trauma, chronic traumatic encephalopathy and tau: challenges in translating from mice to men. Exp Neurol (2015). doi:10.1016/j.expneurol.2015.06.003
19. Baugh CM, Stamm JM, Riley DO, Gavett BE, Shenton ME, Lin A, et al. Chronic traumatic encephalopathy: neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imaging Behav (2012) 6:244–54. doi:10.1007/s11682-012-9164-5
20. Petraglia AL, Plog BA, Dayawansa S, Dashnaw ML, Czerniecka K, Walker CT, et al. The pathophysiology underlying repetitive mild traumatic brain injury in a novel mouse model of chronic traumatic encephalopathy. Surg Neurol Int (2014) 5:184. doi:10.4103/2152-7806.147566
21. Small GW, Kepe V, Siddarth P, Ercoli LM, Merrill DA, Donoghue N, et al. PET scanning of brain tau in retired national football league players: preliminary findings. Am J Geriatr Psychiatry (2013) 21:138–44. doi:10.1016/j.jagp.2012.11.019
22. Luo J, Nguyen A, Villeda S, Zhang H, Ding Z, Lindsey D, et al. Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front Neurol (2014) 5:12. doi:10.3389/fneur.2014.00012
23. Petraglia AL, Plog BA, Dayawansa S, Chen M, Dashnaw ML, Czerniecka K, et al. The spectrum of neurobehavioral sequelae after repetitive mild traumatic brain injury: a novel mouse model of chronic traumatic encephalopathy. J Neurotrauma (2014) 31:1211–24. doi:10.1089/neu.2013.3255
24. Lucke-Wold BP, Turner RC, Logsdon AF, Nguyen L, Bailes JE, Lee JM, et al. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J Neurosurg (2015). doi:10.3171/2015.3.JNS141802
25. Mouzon BC, Bachmeier C, Ferro A, Ojo JO, Crynen G, Acker CM, et al. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol (2014) 75:241–54. doi:10.1002/ana.24064
26. Huber BR, Meabon JS, Martin TJ, Mourad PD, Bennett R, Kraemer BC, et al. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J Alzheimers Dis (2013) 37:309–23. doi:10.3233/JAD-130182
27. Liu MC, Kobeissy F, Zheng W, Zhang Z, Hayes RL, Wang KK. Dual vulnerability of tau to calpains and caspase-3 proteolysis under neurotoxic and neurodegenerative conditions. ASN Neuro (2011) 3:e00051. doi:10.1042/AN20100012
28. Abisambra JF, Scheff S. Brain injury in the context of tauopathies. J Alzheimers Dis (2014) 40:495–518. doi:10.3233/JAD-131019
29. Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature (2015) 523:431–6. doi:10.1038/nature14658
30. Lucke-Wold BP, Naser ZJ, Logsdon AF, Turner RC, Smith KE, Robson MJ, et al. Amelioration of NADPH-mediated stress reduces cell death following blast-induced traumatic brain injury. Transl Res (2015). doi:10.1016/j.trsl.2015.08.005
31. Kanayama G, Takeda M, Niigawa H, Ikura Y, Tamii H, Taniguchi N, et al. The effects of repetitive mild brain injury on cytoskeletal protein and behavior. Methods Find Exp Clin Pharmacol (1996) 18:105–15.
32. Zhang J, Teng Z, Song Y, Hu M, Chen C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J Cereb Blood Flow Metab (2015) 35:706. doi:10.1038/jcbfm.2014.247
33. Uryu K, Laurer H, McIntosh T, Pratico D, Martinez D, Leight S, et al. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci (2002) 22:446–54.
34. Conte V, Uryu K, Fujimoto S, Yao Y, Rokach J, Longhi L, et al. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J Neurochem (2004) 90:758–64. doi:10.1111/j.1471-4159.2004.02807.x
35. Yoshiyama Y, Uryu K, Higuchi M, Longhi L, Hoover R, Fujimoto S, et al. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J Neurotrauma (2005) 22:1134–41. doi:10.1089/neu.2005.22.1134
36. Hawkins BE, Krishnamurthy S, Castillo-Carranza DL, Sengupta U, Prough DS, Jackson GR, et al. Rapid accumulation of endogenous tau oligomers in a rat model of traumatic brain injury: possible link between traumatic brain injury and sporadic tauopathies. J Biol Chem (2013) 288:17042–50. doi:10.1074/jbc.M113.472746
37. Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci (2014) 34:3743–55. doi:10.1523/JNEUROSCI.2872-13.2014
38. Rubenstein R, Chang B, Davies P, Wagner AMD, Robertson CS, Wang KK. A novel, ultrasensitive assay for tau: potential for assessing traumatic brain injury in tissues and biofluids. J Neurotrauma (2015) 32:342–52. doi:10.1084/neu.2014.3548
39. Glushakova OY, Johnson D, Hayes RL. Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J Neurotrauma (2014) 31(13):1180–93. doi:10.1089/neu.2013.3080
40. Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, et al. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma (1995) 12:169–78. doi:10.1089/neu.1995.12.169
41. Logsdon AF, Turner RC, Lucke-Wold BP, Robson MJ, Naser ZJ, Smith KE, et al. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci (2014) 8:421. doi:10.3389/fncel.2014.00421
42. Lucke-Wold BP, Logsdon AF, Smith KE, Turner RC, Alkon DL, Tan Z, et al. Bryostatin-1 restores blood brain barrier integrity following blast-induced traumatic brain injury. Mol Neurobiol (2015) 52:1119–34. doi:10.1007/s12035-014-8902-7
43. Zander NE, Piehler T, Boggs ME, Banton R, Benjamin R. In vitro studies of primary explosive blast loading on neurons. J Neurosci Res (2015) 93:1353–63. doi:10.1002/jnr.23594
44. Prins ML, Hales A, Reger M, Giza CC, Hovda DA. Repeat traumatic brain injury in the juvenile rat is associated with increased axonal injury and cognitive impairments. Dev Neurosci (2010) 32:510–8. doi:10.1159/000316800
45. Goddeyne C, Nichols J, Wu C, Anderson T. Repetitive mild traumatic brain injury induces ventriculomegaly and cortical thinning in juvenile rats. J Neurophysiol (2015) 113:3268–80. doi:10.1152/jn.00970.2014
46. Loane DJ, Kumar A. Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol (2015). doi:10.1016/j.expneurol.2015.08.018
47. Longhi L, Saatman KE, Fujimoto S, Raghupathi R, Meaney DF, Davis J, et al. Temporal window of vulnerability to repetitive experimental concussive brain injury. Neurosurgery (2005) 56:364–74. doi:10.1227/01.NEU.0000149008.73513.44
48. Creeley CE, Wozniak DF, Bayly PV, Olney JW, Lewis LM. Multiple episodes of mild traumatic brain injury result in impaired cognitive performance in mice. Acad Emerg Med (2004) 11:809–19. doi:10.1111/j.1553-2712.2004.tb00761.x
49. Shitaka Y, Tran HT, Bennett RE, Sanchez L, Levy MA, Dikranian K, et al. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol (2011) 70:551–67. doi:10.1097/NEN.0b013e31821f891f
50. Genovese RF, Simmons LP, Ahlers ST, Maudlin-Jeronimo E, Dave JR, Boutte AM. Effects of mild TBI from repeated blast overpressure on the expression and extinction of conditioned fear in rats. Neuroscience (2013) 254:120–9. doi:10.1016/j.neuroscience.2013.09.021
51. Klemenhagen KC, O’Brien SP, Brody DL. Repetitive concussive traumatic brain injury interacts with post-injury foot shock stress to worsen social and depression-like behavior in mice. PLoS One (2013) 8:e74510. doi:10.1371/journal.pone.0074510
52. Wang Y, Arun P, Wei Y, Oguntayo S, Gharavi R, Valiyaveettil M, et al. Repeated blast exposures cause brain DNA fragmentation in mice. J Neurotrauma (2014) 31:498–504. doi:10.1089/neu.2013.3074
53. Fujita M, Wei EP, Povlishock JT. Intensity- and interval-specific repetitive traumatic brain injury can evoke both axonal and microvascular damage. J Neurotrauma (2012) 29:2172–80. doi:10.1089/neu.2012.2357
54. Meehan WP III, Zhang J, Mannix R, Whalen MJ. Increasing recovery time between injuries improves cognitive outcome after repetitive mild concussive brain injuries in mice. Neurosurgery (2012) 71:885–91. doi:10.1227/NEU.0b013e318265a439
55. Mannix R, Meehan WP, Mandeville J, Grant PE, Gray T, Berglass J, et al. Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol (2013) 74:65–75. doi:10.1002/ana.23858
56. Bolton AN, Saatman KE. Regional neurodegeneration and gliosis are amplified by mild traumatic brain injury repeated at 24-hour intervals. J Neuropathol Exp Neurol (2014) 73:933–47. doi:10.1097/NEN.0000000000000115
57. Weil ZM, Gaier KR, Karelina K. Injury timing alters metabolic, inflammatory and functional outcomes following repeated mild traumatic brain injury. Neurobiol Dis (2014) 70:108–16. doi:10.1016/j.nbd.2014.06.016
58. Friess SH, Ichord RN, Ralston J, Ryall K, Helfaer MA, Smith C, et al. Repeated traumatic brain injury affects composite cognitive function in piglets. J Neurotrauma (2009) 26:1111–21. doi:10.1089/neu.2008-0845
59. Mychasiuk R, Hehar H, Van Waes L, Esser MJ. Diet, age, and prior injury status differentially alter behavioral outcomes following concussion in rats. Neurobiol Dis (2015) 73:1–11. doi:10.1016/j.nbd.2014.09.003
60. Slemmer JE, Weber JT. The extent of damage following repeated injury to cultured hippocampal cells is dependent on the severity of insult and inter-injury interval. Neurobiol Dis (2005) 18:421–31. doi:10.1016/j.nbd.2004.09.022
61. Donovan V, Bianchi A, Hartman R, Bhanu B, Carson MJ, Obenaus A. Computational analysis reveals increased blood deposition following repeated mild traumatic brain injury. Neuroimage Clin (2012) 1:18–28. doi:10.1016/j.nicl.2012.08.001
62. Donovan V, Kim C, Anugerah AK, Coats JS, Oyoyo U, Pardo AC, et al. Repeated mild traumatic brain injury results in long-term white-matter disruption. J Cereb Blood Flow Metab (2014) 34:715–23. doi:10.1038/jcbfm.2014.6
63. Slemmer JE, Weber JT, De Zeeuw CI. Cell death, glial protein alterations and elevated S-100 beta release in cerebellar cell cultures following mechanically induced trauma. Neurobiol Dis (2004) 15:563–72. doi:10.1016/j.nbd.2003.11.027
64. Laurer HL, Bareyre FM, Lee VM, Trojanowski JQ, Longhi L, Hoover R, et al. Mild head injury increasing the brain’s vulnerability to a second concussive impact. J Neurosurg (2001) 95:859–70. doi:10.3171/jns.2001.95.5.0859
65. Mouzon B, Chaytow H, Crynen G, Bachmeier C, Stewart J, Mullan M, et al. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotrauma (2012) 29:2761–73. doi:10.1089/neu.2012.2498
66. Bailes JE, Petraglia AL, Omalu BI, Nauman E, Talavage T. Role of subconcussion in repetitive mild traumatic brain injury. J Neurosurg (2013) 119:1235–45. doi:10.3171/2013.7.JNS121822
67. Aungst SL, Kabadi SV, Thompson SM, Stoica BA, Faden AI. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab (2014) 34:1223–32. doi:10.1038/jcbfm.2014.75
68. Olsson Y, Rinder L, Lindgren S, Stalhammar D. Studies on vascular permeability changes in experimental brain concussion. 3. A comparison between the effects of single and repeated sudden mechanical loading of the brain. Acta Neuropathol (1971) 19:225–33. doi:10.1007/BF00684599
69. Raghupathi R, Mehr MF, Helfaer MA, Margulies SS. Traumatic axonal injury is exacerbated following repetitive closed head injury in the neonatal pig. J Neurotrauma (2004) 21:307–16. doi:10.1089/089771504322972095
70. DeRoss AL, Adams JE, Vane DW, Russell SJ, Terella AM, Wald SL. Multiple head injuries in rats: effects on behavior. J Trauma (2002) 52:708–14. doi:10.1097/00005373-200204000-00017
71. Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology (2000) 55:1158–66. doi:10.1212/WNL.55.8.1158
72. DeFord SM, Wilson MS, Rice AC, Clausen T, Rice LK, Barabnova A, et al. Repeated mild brain injuries result in cognitive impairment in B6C3F1 mice. J Neurotrauma (2002) 19:427–38. doi:10.1089/08977150252932389
73. Manley GT, Rosenthal G, Lam M, Morabito D, Yan D, Derugin N, et al. Controlled cortical impact in swine: pathophysiology and biomechanics. J Neurotrauma (2006) 23:128–39. doi:10.1089/neu.2006.23.128
74. Long JB, Bentley TL, Wessner KA, Cerone C, Sweeney S, Bauman RA. Blast overpressure in rats: recreating a battlefield injury in the laboratory. J Neurotrauma (2009) 26:827–40. doi:10.1089/neu.2008.0748
75. Budde MD, Shah A, McCrea M, Cullinan WE, Pintar FA, Stemper BD. Primary blast traumatic brain injury in the rat: relating diffusion tensor imaging and behavior. Front Neurol (2013) 4:154. doi:10.3389/fneur.2013.00154
76. Turner RC, Naser ZJ, Logsdon AF, Dipasquale KH, Jackson GJ, Robson MJ, et al. Modeling clinically relevant blast parameters based on scaling principles produces functional & histological deficits in rats. Exp Neurol (2013) 248:520–9. doi:10.1016/j.expneurol.2013.07.008
77. Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature (2014) 505:223–8. doi:10.1038/nature12808
78. LaPlaca MC, Thibault LE. Dynamic mechanical deformation of neurons triggers an acute calcium response and cell injury involving the N-methyl-D-aspartate glutamate receptor. J Neurosci Res (1998) 52:220–9. doi:10.1002/(SICI)1097-4547(19980415)52:2<220::AID-JNR10>3.3.CO;2-V
79. Mukhin AG, Ivanova SA, Allen JW, Faden AI. Mechanical injury to neuronal/glial cultures in microplates: role of NMDA receptors and pH in secondary neuronal cell death. J Neurosci Res (1998) 51:748–58. doi:10.1002/(SICI)1097-4547(19980315)51:6<748::AID-JNR8>3.0.CO;2-B
80. Sieg F, Wahle P, Pape HC. Cellular reactivity to mechanical axonal injury in an organotypic in vitro model of neurotrauma. J Neurotrauma (1999) 16:1197–213. doi:10.1089/neu.1999.16.1197
81. Morrison B III, Cater HL, Benham CD, Sundstrom LE. An in vitro model of traumatic brain injury utilising two-dimensional stretch of organotypic hippocampal slice cultures. J Neurosci Methods (2006) 150:192–201. doi:10.1016/j.jneumeth.2005.06.014
82. LaPlaca MC, Cullen DK, McLoughlin JJ, Cargill RS II. High rate shear strain of three-dimensional neural cell cultures: a new in vitro traumatic brain injury model. J Biomech (2005) 38:1093–105. doi:10.1016/j.jbiomech.2004.05.032
83. Hamm RJ, Jenkins LW, Lyeth BG, White-Gbadebo DM, Hayes RL. The effect of age on outcome following traumatic brain injury in rats. J Neurosurg (1991) 75:916–21. doi:10.3171/jns.1991.75.6.0916
84. Maughan PH, Scholten KJ, Schmidt RH. Recovery of water maze performance in aged versus young rats after brain injury with the impact acceleration model. J Neurotrauma (2000) 17:1141–53. doi:10.1089/neu.2000.17.1141
85. Hoane MR, Lasley LA, Akstulewicz SL. Middle age increases tissue vulnerability and impairs sensorimotor and cognitive recovery following traumatic brain injury in the rat. Behav Brain Res (2004) 153:189–97. doi:10.1016/j.bbr.2003.11.012
86. Anderson J, Sandhir R, Hamilton ES, Berman NE. Impaired expression of neuroprotective molecules in the HIF-1alpha pathway following traumatic brain injury in aged mice. J Neurotrauma (2009) 26:1557–66. doi:10.1089/neu.2008-0765
87. Wali B, Sayeed I, Stein DG. Improved behavioral outcomes after progesterone administration in aged male rats with traumatic brain injury. Restor Neurol Neurosci (2011) 29:61–71. doi:10.3233/RNN-2011-0579
88. Mehan ND, Strauss KI. Combined age- and trauma-related proteomic changes in rat neocortex: a basis for brain vulnerability. Neurobiol Aging (2012) 33:1857–73. doi:10.1016/j.neurobiolaging.2011.09.029
89. Timaru-Kast R, Luh C, Gotthardt P, Huang C, Schafer MK, Engelhard K, et al. Influence of age on brain edema formation, secondary brain damage and inflammatory response after brain trauma in mice. PLoS One (2012) 7:e43829. doi:10.1371/journal.pone.0043829
90. Turner RC, Seminerio MJ, Naser ZJ, Ford JN, Martin SJ, Matsumoto RR, et al. Effects of aging on behavioral assessment performance: implications for clinically relevant models of neurological disease. J Neurosurg (2012) 117:629–37. doi:10.3171/2012.5.JNS112224
91. Turner RC, Vangilder RL, Naser ZJ, Lucke-Wold BP, Bailes JE, Matsumoto RR, et al. Elucidating the severity of preclinical traumatic brain injury models: a role for functional assessment? Neurosurgery (2014) 74:382–94. doi:10.1227/NEU.0000000000000292
92. Van Den Heuvel C, Thornton E, Vink R. Traumatic brain injury and Alzheimer’s disease: a review. Prog Brain Res (2007) 161:303–16. doi:10.1016/S0079-6123(06)61021-2
93. Magnoni S, Brody DL. New perspectives on amyloid-beta dynamics after acute brain injury: moving between experimental approaches and studies in the human brain. Arch Neurol (2010) 67:1068–73. doi:10.1001/archneurol.2010.214
94. Sivanandam TM, Thakur MK. Traumatic brain injury: a risk factor for Alzheimer’s disease. Neurosci Biobehav Rev (2012) 36:1376–81. doi:10.1016/j.neubiorev.2012.02.013
95. Bower JH, Maraganore DM, Peterson BJ, McDonnell SK, Ahlskog JE, Rocca WA. Head trauma preceding PD: a case-control study. Neurology (2003) 60:1610–5. doi:10.1212/01.WNL.0000068008.78394.2C
96. Omalu BI, Dekosky ST, Hamilton RL, Minster RL, Kamboh MI, Shakir AM, et al. Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery (2006) 59:1086–92.
97. Omalu BI, Bailes J, Hammers JL, Fitzsimmons RP. Chronic traumatic encephalopathy, suicides and parasuicides in professional American athletes: the role of the forensic pathologist. Am J Forensic Med Pathol (2010) 31:130–2. doi:10.1097/PAF.0b013e3181ca7f35
98. Omalu BI, Fitzsimmons RP, Hammers J, Bailes J. Chronic traumatic encephalopathy in a professional American wrestler. J Forensic Nurs (2010) 6:130–6. doi:10.1111/j.1939-3938.2010.01078.x
99. Omalu BI, Hamilton RL, Kamboh MI, Dekosky ST, Bailes J. Chronic traumatic encephalopathy (CTE) in a National Football League Player: case report and emerging medicolegal practice questions. J Forensic Nurs (2010) 6:40–6. doi:10.1111/j.1939-3938.2009.01064.x
100. Gavett BE, Cantu RC, Shenton M, Lin AP, Nowinski CJ, McKee AC, et al. Clinical appraisal of chronic traumatic encephalopathy: current perspectives and future directions. Curr Opin Neurol (2011) 24:525–31. doi:10.1097/WCO.0b013e32834cd477
101. Omalu B, Hammers JL, Bailes J, Hamilton RL, Kamboh MI, Webster G, et al. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus (2011) 31:E3. doi:10.3171/2011.9.FOCUS11178
102. Montenigro PH, Baugh CM, Daneshvar DH, Mez J, Budson AE, Au R, et al. Clinical subtypes of chronic traumatic encephalopathy: literature review and proposed research diagnostic criteria for traumatic encephalopathy syndrome. Alzheimers Res Ther (2014) 6:68. doi:10.1186/s13195-014-0068-z
103. Omalu B. Chronic traumatic encephalopathy. Prog Neurol Surg (2014) 28:38–49. doi:10.1159/000358761
104. Kasturi BS, Stein DG. Progesterone decreases cortical and sub-cortical edema in young and aged ovariectomized rats with brain injury. Restor Neurol Neurosci (2009) 27:265–75. doi:10.3233/RNN-2009-0475
105. Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging (2013) 34:1397–411. doi:10.1016/j.neurobiolaging.2012.11.013
106. Theadom A, Parmar P, Jones K, Barker-Collo S, Starkey N, McPherson K, et al. Frequency and impact of recurrent traumatic brain injury in a population-based sample. J Neurotrauma (2015) 32:674–81. doi:10.1089/neu.2014.3579
107. Kobeissy F, Mondello S, Tumer N, Toklu HZ, Whidden MA, Kirichenko N, et al. Assessing neuro-systemic & behavioral components in the pathophysiology of blast-related brain injury. Front Neurol (2013) 4:186. doi:10.3389/fneur.2013.00186
108. Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci (2014) 34:16180–93. doi:10.1523/JNEUROSCI.3020-14.2014
109. Cernak I. Blast injuries and blast-induced neurotrauma: overview of pathophysiology and experimental knowledge models and findings. In: Kobeissy FHP, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton, FL: CRC Press (2015).
110. Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res (2011) 2:492–516. doi:10.1007/s12975-011-0125-x
111. Kovesdi E, Kamnaksh A, Wingo D, Ahmed F, Grunberg NE, Long JB, et al. Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front Neurol (2012) 3:111. doi:10.3389/fneur.2012.00111
112. Salazar AM, Grafman J. Post-traumatic epilepsy: clinical clues to pathogenesis and paths to prevention. Handb Clin Neurol (2015) 128:525–38. doi:10.1016/B978-0-444-63521-1.00033-9
113. Koliatsos VE, Xu L. The problem of neurodegeneration in cumulative sports concussions: emphasis on neurofibrillary tangle formation. In: Kobeissy FHP, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton, FL: CRC Press (2015).
114. Ponsford J, McLaren A, Schonberger M, Burke R, Rudzki D, Olver J, et al. The association between apolipoprotein E and traumatic brain injury severity and functional outcome in a rehabilitation sample. J Neurotrauma (2011) 28:1683–92. doi:10.1089/neu.2010.1623
115. Saulle M, Greenwald BD. Chronic traumatic encephalopathy: a review. Rehabil Res Pract (2012) 2012:816069. doi:10.1155/2012/816069
116. Laskowitz DT, Dawson HN. Genetic influences and neuropathological sequelae of repetitive brain injury. Ann Neurol (2014) 75:617–8. doi:10.1002/ana.24057
117. Maroon JC, Winkelman R, Bost J, Amos A, Mathyssek C, Miele V. Chronic traumatic encephalopathy in contact sports: a systematic review of all reported pathological cases. PLoS One (2015) 10:e0117338. doi:10.1371/journal.pone.0117338
118. Kors EE, Terwindt GM, Vermeulen FL, Fitzsimons RB, Jardine PE, Heywood P, et al. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol (2001) 49:753–60. doi:10.1002/ana.1031
119. Sabirzhanov B, Zhao Z, Stoica BA, Loane DJ, Wu J, Borroto C, et al. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neurosci (2014) 34:10055–71. doi:10.1523/JNEUROSCI.1260-14.2014
120. Niesman IR, Schilling JM, Shapiro LA, Kellerhals SE, Bonds JA, Kleschevnikov AM, et al. Traumatic brain injury enhances neuroinflammation and lesion volume in caveolin deficient mice. J Neuroinflammation (2014) 11:39. doi:10.1186/1742-2094-11-39
121. Sharma P, Su YA, Barry ES, Grunberg NE, Lei Z. Mitochondrial targeted neuron focused genes in hippocampus of rats with traumatic brain injury. Int J Crit Illn Inj Sci (2012) 2:172–9. doi:10.4103/2229-5151.100931
122. Lu J, Moochhala S, Kaur C, Ling E. Changes in apoptosis-related protein (p53, Bax, Bcl-2 and Fos) expression with DNA fragmentation in the central nervous system in rats after closed head injury. Neurosci Lett (2000) 290:89–92. doi:10.1016/S0304-3940(00)01307-0
123. Carlson SW, Madathil SK, Sama DM, Gao X, Chen J, Saatman KE. Conditional overexpression of insulin-like growth factor-1 enhances hippocampal neurogenesis and restores immature neuron dendritic processes after traumatic brain injury. J Neuropathol Exp Neurol (2014) 73:734–46. doi:10.1097/NEN.0000000000000092
124. Ke C, Poon WS, Ng HK, Pang JC, Chan Y. Heterogeneous responses of aquaporin-4 in oedema formation in a replicated severe traumatic brain injury model in rats. Neurosci Lett (2001) 301:21–4. doi:10.1016/S0304-3940(01)01589-0
125. Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M, et al. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem (2004) 90:1068–75. doi:10.1111/j.1471-4159.2004.02547.x
126. Lipsky RH, Lin M. Genetic predictors of outcome following traumatic brain injury. Handb Clin Neurol (2015) 127:23–41. doi:10.1016/B978-0-444-52892-6.00003-9
127. Bailey ZS, Grinter MB, De La Torre Campos D, Vandevord PJ. Blast induced neurotrauma causes overpressure dependent changes to the DNA methylation equilibrium. Neurosci Lett (2015) 604:119–23. doi:10.1016/j.neulet.2015.07.035
128. Bailey ZS, Sujith Sajja VS, Hubbard WB, Vandevord PJ. Blast induced neurotrauma leads to changes in the epigenome. Biomed Sci Instrum (2015) 51:423–30.
129. Haghighi F, Ge Y, Chen S, Xin Y, Umali MU, De Gasperi R, et al. Neuronal DNA methylation profiling of blast-related traumatic brain injury (TBI). J Neurotrauma (2015) 32(16):1200–9. doi:10.1089/neu.2014.3640
130. Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3beta/PTEN/Akt axis. Proc Natl Acad Sci U S A (2015) 112:2853–8. doi:10.1073/pnas.1501441112
131. Almli LM, Stevens JS, Smith AK, Kilaru V, Meng Q, Flory J, et al. A genome-wide identified risk variant for PTSD is a methylation quantitative trait locus and confers decreased cortical activation to fearful faces. Am J Med Genet B Neuropsychiatr Genet (2015) 168B:327–36. doi:10.1002/ajmg.b.32315
132. Bramlett HM, Dietrich WD. Neuropathological protection after traumatic brain injury in intact female rats versus males or ovariectomized females. J Neurotrauma (2001) 18:891–900. doi:10.1089/089771501750451811
133. Kadyan V, Mysiw WJ, Bogner JA, Corrigan JD, Fugate LP, Clinchot DM. Gender differences in agitation after traumatic brain injury. Am J Phys Med Rehabil (2004) 83:747–52. doi:10.1097/01.PHM.0000140790.30468.F4
134. Bruce-Keller AJ, Dimayuga FO, Reed JL, Wang C, Angers R, Wilson ME, et al. Gender and estrogen manipulation do not affect traumatic brain injury in mice. J Neurotrauma (2007) 24:203–15. doi:10.1089/neu.2006.0163
135. Farace E, Alves WM. Do women fare worse: a metaanalysis of gender differences in traumatic brain injury outcome. J Neurosurg (2000) 93:539–45. doi:10.3171/jns.2000.93.4.0539
136. Simpkins JW, Green PS, Gridley KE, Singh M, De Fiebre NC, Rajakumar G. Role of estrogen replacement therapy in memory enhancement and the prevention of neuronal loss associated with Alzheimer’s disease. Am J Med (1997) 103:19S–25S. doi:10.1016/S0002-9343(97)00260-X
137. Suzuki S, Gerhold LM, Bottner M, Rau SW, Dela Cruz C, Yang E, et al. Estradiol enhances neurogenesis following ischemic stroke through estrogen receptors alpha and beta. J Comp Neurol (2007) 500:1064–75. doi:10.1002/cne.21240
138. Jia J, Guan D, Zhu W, Alkayed NJ, Wang MM, Hua Z, et al. Estrogen inhibits Fas-mediated apoptosis in experimental stroke. Exp Neurol (2009) 215:48–52. doi:10.1016/j.expneurol.2008.09.015
139. Li J, Siegel M, Yuan M, Zeng Z, Finnucan L, Persky R, et al. Estrogen enhances neurogenesis and behavioral recovery after stroke. J Cereb Blood Flow Metab (2011) 31:413–25. doi:10.1038/jcbfm.2010.181
140. Gatson JW, Liu MM, Abdelfattah K, Wigginton JG, Smith S, Wolf S, et al. Estrone is neuroprotective in rats after traumatic brain injury. J Neurotrauma (2012) 29:2209–19. doi:10.1089/neu.2011.2274
141. Day NL, Floyd CL, D’Alessandro TL, Hubbard WJ, Chaudry IH. 17beta-estradiol confers protection after traumatic brain injury in the rat and involves activation of G protein-coupled estrogen receptor 1. J Neurotrauma (2013) 30:1531–41. doi:10.1089/neu.2013.2854
142. Brown CM, Suzuki S, Jelks KA, Wise PM. Estradiol is a potent protective, restorative, and trophic factor after brain injury. Semin Reprod Med (2009) 27:240–9. doi:10.1055/s-0029-1216277
143. Wagner AK, Willard LA, Kline AE, Wenger MK, Bolinger BD, Ren D, et al. Evaluation of estrous cycle stage and gender on behavioral outcome after experimental traumatic brain injury. Brain Res (2004) 998:113–21. doi:10.1016/j.brainres.2003.11.027
144. Wagner AK, Bayir H, Ren D, Puccio A, Zafonte RD, Kochanek PM. Relationships between cerebrospinal fluid markers of excitotoxicity, ischemia, and oxidative damage after severe TBI: the impact of gender, age, and hypothermia. J Neurotrauma (2004) 21:125–36. doi:10.1089/089771504322778596
145. Garringer JA, Niyonkuru C, McCullough EH, Loucks T, Dixon CE, Conley YP, et al. Impact of aromatase genetic variation on hormone levels and global outcome after severe TBI. J Neurotrauma (2013) 30:1415–25. doi:10.1089/neu.2012.2565
146. Bell KR, Pepping M. Women and traumatic brain injury. Phys Med Rehabil Clin N Am (2001) 12:169–82.
147. Farace E, Alves WM. Do women fare worse? A metaanalysis of gender differences in outcome after traumatic brain injury. Neurosurg Focus (2000) 8:e6. doi:10.3171/foc.2000.8.1.152
148. Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, et al. Traumatic brain injury and time to onset of Alzheimer’s disease: a population-based study. Am J Epidemiol (1999) 149:32–40. doi:10.1093/oxfordjournals.aje.a009724
149. McCauley SR, Boake C, Levin HS, Contant CF, Song JX. Postconcussional disorder following mild to moderate traumatic brain injury: anxiety, depression, and social support as risk factors and comorbidities. J Clin Exp Neuropsychol (2001) 23:792–808. doi:10.1076/jcen.23.6.792.1016
150. Khan F, Baguley IJ, Cameron ID. 4: Rehabilitation after traumatic brain injury. Med J Aust (2003) 178:290–5.
151. Wu A, Ying Z, Gomez-Pinilla F. Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J Neurotrauma (2004) 21:1457–67. doi:10.1089/neu.2004.21.1457
152. Mills JD, Bailes JE, Sedney CL, Hutchins H, Sears B. Omega-3 fatty acid supplementation and reduction of traumatic axonal injury in a rodent head injury model. J Neurosurg (2011) 114:77–84. doi:10.3171/2010.5.JNS08914
153. Mills JD, Hadley K, Bailes JE. Dietary supplementation with the omega-3 fatty acid docosahexaenoic acid in traumatic brain injury. Neurosurgery (2011) 68:474–81. doi:10.1227/NEU.0b013e3181ff692b
154. Bailes JE, Patel V. The potential for DHA to mitigate mild traumatic brain injury. Mil Med (2014) 179:112–6. doi:10.7205/MILMED-D-14-00139
155. Mills JD, Bailes JE, Turner RC, Dodson SC, Sakai J, Maroon JC. Anabolic steroids and head injury. Neurosurgery (2012) 70:205–9. doi:10.1227/NEU.0b013e3182250918
156. Hu SL, Hu R, Li F, Liu Z, Xia YZ, Cui GY, et al. Hyperbaric oxygen preconditioning protects against traumatic brain injury at high altitude. Acta Neurochir Suppl (2008) 105:191–6. doi:10.1007/978-3-211-09469-3_37
157. Talavage TM, Nauman EA, Breedlove EL, Yoruk U, Dye AE, Morigaki KE, et al. Functionally-detected cognitive impairment in high school football players without clinically-diagnosed concussion. J Neurotrauma (2014) 31:327–38. doi:10.1089/neu.2010.1512
Keywords: chronic traumatic encephalopathy, preclinical models, neurodegeneration, hyperphosphorylated tau, neurotrauma
Citation: Turner RC, Lucke-Wold BP, Logsdon AF, Robson MJ, Lee JM, Bailes JE, Dashnaw ML, Huber JD, Petraglia AL and Rosen CL (2015) Modeling chronic traumatic encephalopathy: the way forward for future discovery. Front. Neurol. 6:223. doi: 10.3389/fneur.2015.00223
Received: 06 July 2015; Accepted: 09 October 2015;
Published: 26 October 2015
Edited by:
Kenneth Curley, US Army Medical Research and Materiel Command, USAReviewed by:
Firas H. Kobeissy, University of Florida, USAKarim A. Sarhane, Johns Hopkins University, USA
Copyright: © 2015 Turner, Lucke-Wold, Logsdon, Robson, Lee, Bailes, Dashnaw, Huber, Petraglia and Rosen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles L. Rosen, Y3Jvc2VuQGhzYy53dnUuZWR1
†Denotes equal authorship.
‡Denotes co-senior authorship.