 Jordi A. Matías-Guiu1*
Jordi A. Matías-Guiu1* Celia Oreja-Guevara1
Celia Oreja-Guevara1 María Nieves Cabrera-Martín2Teresa Moreno-Ramos1José Luis Carreras2
María Nieves Cabrera-Martín2Teresa Moreno-Ramos1José Luis Carreras2 Jorge Matías-Guiu1
Jorge Matías-Guiu1
- 1Department of Neurology, Hospital Clínico San Carlos, San Carlos Institute for Health Research (IdISSC), Complutense University of Madrid, Madrid, Spain
- 2Department of Nuclear Medicine, Hospital Clínico San Carlos, San Carlos Institute for Health Research (IdISSC), Complutense University of Madrid, Madrid, Spain
Thioflavin T derivatives are used in positron-emission tomography (PET) studies to detect amyloid protein deposits in patients with Alzheimer disease. These tracers bind extensively to white matter, which suggests that they may be useful in studies of multiple sclerosis (MS), and that proteins resulting from proteolytic processing of the amyloid precursor protein (APP) may contribute to MS. This article reviews data from both clinical and preclinical studies addressing the role of these proteins, whether they are detected in CSF studies or using PET imaging. APP is widely expressed in demyelinated axons and may have a protective effect in MS and in experimental allergic encephalomyelitis in animals. Several mechanisms associated with this increased expression may affect the degree of remyelination in MS. Amyloid-PET imaging may help determine the degree of demyelination and provide information on the molecular changes linked to APP proteolytic processing experienced by patients with MS.
Background
Multiple sclerosis (MS) is an autoimmune inflammatory disease of the central nervous system (CNS) that causes inflammatory lesions in the brain and spinal cord and ruptures the blood–brain barrier, leading to demyelination and axonal damage. In normal practice, MS is diagnosed based on clinical symptoms, exclusion of other causes, and findings in cerebrospinal fluid (CSF) and magnetic resonance imaging studies. From a pathogenic point of view, MS is characterized by demyelination, which is attributed to inflammatory mechanisms and followed by neurodegeneration. In most cases, the disease initially presents a relapsing-remitting pattern (RRMS). Patients with this type of MS experience relapses followed by periods of partial or total recovery associated with incomplete remyelination. Remyelinating capacity decreases with time, especially in the secondary progressive form of the disease (1, 2).
Although β-amyloid protein (Aβ) is mainly linked to Alzheimer disease (AD), recent review articles suggest a connection between Aβ and MS (3, 4). One reason that led researchers to associate Aβ with MS was that white matter exhibits significant uptake of the PET tracers binding to this protein (5, 6), whereas white matter lesions associated with AD display lower uptake (7, 8). Myelin loss and breakdown of myelin basic protein (MBP) in AD patients and animal models of AD are associated with aging, the ApoE4 allele, or head injury, all of which are risk factors for AD, as well as with increases in Aβ peptides (9). Several pathology studies of AD have found decreased expression of MBP in the areas presenting Aβ deposition, and decreased Aβ deposition in white matter areas exhibiting greater expression of MBP. MBP has not been detected in amyloid plaques in AD patients (10, 11).
Amyloid-PET in MS
Positron-emission tomography using different amyloid tracers [Pittsburgh Compound-B (PiB), florbetapir, florbetaben, flutemetamol, and others under study] can detect fibrillar Aβ deposits with high sensitivity and specificity; fibrillar Aβ is therefore considered a biomarker for AD along with levels of Aβ in CSF. This technique enables an in vivo pathological and molecular diagnosis, and it is currently included in clinical trial protocols for early detection of AD. Amyloid-PET findings have been proven to correlate well with fibrillar Aβ in neuropathology studies (12). Assessing amyloid tracer uptake in gray matter is a technique for diagnosing AD and for differential diagnosis of neurodegenerative cognitive disorders. Most studies using amyloid-PET aim to assess this imaging technique’s utility for confirming AD diagnosis and predicting progression of mild cognitive impairment to dementia (13, 14). It is also used to diagnose other pathologies presenting with cognitive impairment and which are not linked to Aβ exclusively (15–17). However, changes in amyloid-PET images may also be indicative of other neurological diseases (18). These tracers are thioflavin T derivatives and have been proven more specific than previous compounds based on Congo red and whose chemical basis was the styrylbenzene molecule or Chrysamine G, a derivative of Congo red (19). Thioflavin T analogs bind to amyloid fibrils, unlike Congo red derivatives, which also bind to tau fibrils. Several molecules have been developed by modifying the original structure, giving rise to other tracers that may have different affinities for certain tissues (20–23). Other molecules now being developed may have an even greater affinity for yelin (24).
Molecules currently in use derive from Pittsburgh Compound-A (25), an alternative name for BTA-1 (26), which resulted in PiB. This compound was used to develop three different radioligands: (1) SB1, which gave rise to 18F-florbetaben (AV1) and subsequently 18F-florbetapir (AV45); (2) 18F-flutemetamol; and (3) AZD2184, and subsequently AZD4694 (renamed NAV4694). At present, PiB, florbetaben, florbetapir, and flutemetamol have been tested in clinical trials, and the last three tracers are approved and available for clinical use.
Amyloid tracers detect decreased activity in black hole areas in T1-weighted MR images (27) and in white matter lesions in T2-weighted MR images (28, 29), in both the relapsing-remitting and the progressive forms of MS (Tables 1 and 2; Figure 1). These results showed that amyloid tracers bind extensively to white matter and that uptake decreases with demyelination. This inevitably leads us to question whether the usefulness of amyloid tracers in MS is due to their non-specific binding to white matter, or whether there may be a connection between Aβ and myelination.

Table 1. Studies of amyloid-related measurements in MS.

Table 2. MRI correlations with measurements related to the amyloid cascade in MS.
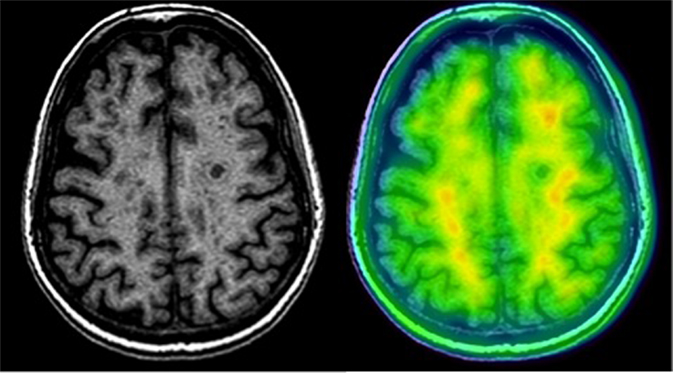
Figure 1. Amyloid-PET and MRI image of a patient with RRMS using 18F-florbetaben. Note the decreased uptake of the tracer in white matter lesions.
Biomarkers of APP Proteolytic Processing in CSF in Patients with MS
Different studies evaluating Aβ levels in CSF in patients with clinically isolated syndrome (CIS) or MS have yielded divergent results (30, 31, 36, 37). However, it seems that levels of intermediate products of proteolysis of the amyloid precursor protein (APP), such as soluble α-APP and β-APP, and one of the final products, Aβ1–42, are reduced in patients with both the RR and the primary progressive forms of MS (34, 35, 38, 39). Likewise, there is an inverse correlation between Aβ levels and presence of gadolinium-enhancing lesions. Low activity of β-site APP-cleaving enzyme 1 (BACE1), the enzyme participating in amyloidogenic APP proteolysis, has also been demonstrated in CSF in patients with MS (32). However, these data are challenging to interpret, since CSF Aβ levels fluctuate throughout the day. This biomarker is therefore difficult to assess and extrapolating changes observed in CSF to demyelinating plaques is not always possible (Table 1). Altered Aβ CSF levels seem to be linked to situations of lower activity as shown by gadolinium uptake in MR images. These findings are not correlated with a greater degree of atrophy (Table 2).
Effects of APP Proteolytic Processing in MS
In patients with MS, β-APP accumulates in damaged axons (40). Experimental allergic encephalomyelitis (EAE), an experimental model for MS, is more severe in association with a genetic deletion of APP. Pathology studies have found increased Aβ expression in demyelinating plaques (41–43), which may even provide protection from damage (44); in fact, treatment with either Aβ42 or Aβ40 reduces motor paralysis and brain inflammation and suppresses lymphocyte activation in animals with EAE. Similarly, decreased levels of pro-inflammatory cytokines and chemokines have been found in mice with EAE receiving Aβ peptides. Although these findings suggest that Aβ peptides are beneficial, we should not forget that they are neurotoxic and neuroinflammatory, and that APP proteolytic processing may provoke the opposite effect in demyelinated axons (45). This idea is consistent with studies describing increased Aβ42 levels in lesions and damaged axons. Several experimental studies report similar results: mice immunized with Aβ1–42 peptide experience symptoms whose presentation and pathological basis resemble those associated with EAE (46); Aβ injection in mice may damage the white matter (47) and induce oligodendrocyte death (48); and Aβ decreases the number of neurons in the subventricular zone and hippocampus and inhibits neurogenesis in the dentate gyrus of hippocampus, but not in the subventricular zone (49).
Amyloid precursor protein is extensively expressed in humans. Functions attributed to APP include neurite outgrowth and synaptogenesis, protein trafficking along axons, cell adhesion, calcium metabolism, and signal transduction (50). Due to the activity of several successive proteolytic processes involving α- and β-secretases (depending on whether the process is amyloidogenic), and subsequently γ-secretase, APP gives rise to soluble extracellular domains (sAPPα or sAPPβ) and the APP intracellular domain (AICD). Aβ is a protein with a great capacity to generate fibrils: it initially forms soluble monomers, and then oligomers, which remain soluble, until it ends up forming insoluble fibrils. Intracellular cascade of soluble peptides (β peptides, especially Aβ40 and Aβ42), which derive from APP proteolysis, may form oligomers and insoluble fibrillar deposits that become amyloid plaques (51). Another important fact is that APP is not an isolated protein, but rather one with two homologs: amyloid-like proteins 1 and 2, or APLP1 and APLP2 (52). Although genetic deletion of APP in mice provokes minor impairment (53), triple-knockout mice show such problems as perinatal death, cranial abnormalities, and cortical dysplasia (54, 55). The above suggests that APP family proteins fulfill essential yet partially redundant functions that can compensate for each other when several family members are present.
Although information on APP proteolytic processing in MS is scarce, we currently know that it is upregulated in damaged axons, which suggests that it may constitute a reliable marker of axon demyelination (56). Increased APP expression has been observed following compression injury in spinal cord white matter in rats (57). In APP knockout mice, nodal length is greater, and sodium channels are clustered. Spinal cord myelin sheaths are thinner in both APP knockout and APP-overexpressing transgenic mice (58). The potential impact of APP on MS may be related to coexpressing proteins. In fact, APP aggregates have been found in nodes of Ranvier, where APP expression colocalizes with tenascin-R, near the juxtaparanodal potassium channels. Tenascin-R is an extracellular matrix glycoprotein of the tenascin family that is exclusive to the CNS. It acts on cell differentiation, migration, and adhesion. Tenascin-R expression increases following microglial activation (59). It is upregulated by platelet-derived growth factor (PDGF) and participates in oligodendrocyte differentiation and consequently in remyelination (60). Tenascin-R has been studied in connection with MS due to its role in myelination (61), and expression has been shown to be reduced in chronic demyelinating plaques and present in acute and subacute plaques. Some studies therefore suggest that Tenascin-R inhibits remyelination (62) and prevents repair (63). APP has also been associated with Tau and αB-crystallin proteins in MS lesions, and αB-crystallin (HspB5) and Aβ peptides appear to be beneficial in EAE (64). A small heat-shock protein, αB-crystallin is highly immunogenic and associated with MS (65). It forms part of amyloid fibrils and improves EAE symptoms when administered systemically (66, 67). Other proteins that form part of amyloid fibrils are also beneficial, including Aβ A4, tau, amylin, and serum amyloid P (SAP). APP, αB-crystallin, and tau have been found in amyloid deposits in MS and they have demonstrated anti-inflammatory properties in MS animal models. The benefits of αB-crystallin are believed to be due to this protein’s ability to bind to pro-inflammatory proteins, and this ability increases in inflammatory processes. This activity takes place in a region of the molecule corresponding to the peptide that includes residues 73–92: in fact, this region alone is involved in EAE, and its activity is similar to that of the whole protein, which does not occur with other regions of the protein (68). This peptide can also form part of amyloid fibrils (69). At the same time, APP, αB-crystallin, SAP, and tau deficiencies in mice exacerbate EAE (70, 71). Furthermore, administration of the hexapeptide complex comprising the proteins included in amyloid fibrils rapidly decreases plasma levels of such pro-inflammatory cytokines as IL-6 and IL-2 (72).
Another relevant enzyme is BACE1, a membrane-bound aspartyl protease (73). It is the only enzyme that directly breaks down APP to generate Aβ (74), and it accumulates in AD brains (75–79). BACE1-knockout mice also lack Aβ (80–82). Genetic deletion of BACE1 during development leads to hypomyelination in the central and peripheral nervous systems (83, 84), and the enzyme is necessary for sciatic nerve remyelination after an injury (85). The role of BACE1 in myelination may be explained by the fact that it processes neuregulin-1 and -3 (NRG1, NRG3) (86). Members of the NRG family of proteins are neurotrophic factors that act on ErbB receptors and trigger a biochemical cascade regulating several functions, including myelination. Decreased activity in this signaling pathway reduces myelin sheath thickness (87–90). This suggests that β secretase may play a crucial role in remyelination in MS.
On the other hand, Aβ peptides can trigger microglial activation (91–93). Microglial activation induced by Aβ in vivo is accompanied by decreased CD200 neuronal expression. The CD200 protein controls microglia and assists in inflammatory processes (94, 95).
Proteins Involved in APP Proteolytic Processing in Demyelination
The role of APP and its homologs in demyelination may be due to APP proteolytic processing via substrates and enzymes. Both β- and γ-secretase are located in the lipid raft of the cell membrane, which contains sphingolipids and cholesterol (96). This lipid composition of the membrane influences β- and γ-secretase activity (97–99). The potential role of lipid components in APP proteolytic processing has been extensively reviewed (100); Aβ production is modulated by sphingolipids. Demyelination leads to a release of myelin proteins (101): Nogo, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein inhibit neuronal regeneration via Nogo and PirB receptors (102, 103), and MBP causes damage since it acts directly on the neuronal membrane (104). This protein, which has been regarded as one of the antigens for MS, performs many functions: it is involved in Aβ aggregation and inhibits Aβ fibril assembly (105), which affects Aβ levels. In experimental models, brain tissue inflammation followed by ischemia produces axonal and myelin damage with myelin aggregates that colocalize with APP and Aβ. In the 5XFAD mouse model, Aβ plaques were observed to colocalize with myelin aggregates (106). As shown by in vitro studies, MBP inhibits Aβ fibril assembly via residues 1–64 (107), a fragment known as MBP1 (108). MBP1 has been proven to reduce pathological Aβ accumulation and clinical alterations in the 5XFAD mouse (109). This occurs in control animal models and has also been observed in models presenting mutant forms of Aβ (Dutch- and Iowa-type Aβ) that are responsible for cerebral amyloid angiopathy, in which MBP inhibits fibril formation (105). Although MBP1 may have a protective role in AD, it may be harmful in MS since it reduces amyloid fibril production, which favors the detrimental effect of Aβ peptides.
Conclusion
Tracer uptake in white matter in amyloid PET imaging studies has raised questions about its utility as a biomarker of demyelination, specifically in white matter diseases such as MS. Several studies have aimed to determine how remyelination and MS are affected by APP and the proteins expressed via APP proteolytic processing, and whether amyloid-PET can provide an in vivo molecular diagnosis of this process. Although further research on APP in MS is necessary, recent studies have demonstrated that (1) APP does play a role in MS; (2) APP proteolytic processing occurs as a result of demyelination, due to the action of myelin protein or lipid detritus; and (3) APP is involved in remyelination to a greater or lesser extent. In conclusion, amyloid-PET may serve as a tool for determining the degree of demyelination and remyelination as well as a means of studying molecular changes linked to remyelination in MS in vivo.
Author Contributions
All authors listed, have made substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewers, CE and RD, and the handling Editor, declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
References
1. Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Brück W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain (2008) 131:1749–58. doi: 10.1093/brain/awn096
2. Lassmann H, Brück W, Lucchinetti C, Rodriguez M. Remyelination in multiple sclerosis. Mult Scler (1997) 3:133–6. doi:10.1177/135245859700300213
3. Chandra A. Role of amyloid from a multiple sclerosis perspective: a literature review. Neuroimmunomodulation (2015) 22(6):343–6. doi:10.1159/j.nrl.2012.03.015
4. Gentile A, Mori F, Bernardini S, Centonze D. Role of amyloid-β CSF levels in cognitive deficit in MS. Clin Chim Acta (2015) 449:23–30. doi:10.1016/j.cca.2015.01.035
5. Niccolini F, Su P, Politis M. PET in multiple sclerosis. Clin Nucl Med (2015) 40:e46–52. doi:10.1097/RLU.0000000000000359
6. Fodero-Tavoletti MT, Rowe CC, McLean CA, Leone L, Li QX, Masters CL, et al. Characterization of PiB binding to white matter in Alzheimer disease and other dementias. J Nucl Med (2009) 50:198–204. doi:10.2967/jnumed.108.057984
7. Glodzik L, Kuceyeski A, Rusinek H, Tsui W, Mosconi L, Li Y, et al. Reduced glucose uptake and Aβ in brain regions with hyperintensities in connected white matter. Neuroimage (2014) 100:684–91. doi:10.1016/j.neuroimage.2014.06.060
8. Brun A, Englund E. A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol (1986) 19:253–62. doi:10.1002/ana.410190306
9. Roher AE, Weiss N, Kokjohn TA, Kuo YM, Kalback W, Anthony J, et al. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry (2002) 41:11080–90. doi:10.1021/bi026173d
10. Mitew S, Kirkcaldie MT, Halliday GM, Shepherd CE, Vickers JC, Dickson TC. Focal demyelination in Alzheimer’s disease and transgenic mouse models. Acta Neuropathol (2010) 119:567–77. doi:10.1007/s00401-010-0657-2
11. Ou-Yang MH, Van Nostrand WE. The absence of myelin basic protein promotes neuroinflammation and reduces amyloid β-protein accumulation in Tg-5xFAD mice. J Neuroinflammation (2013) 10:134. doi:10.1186/1742-2094-10-134
12. Driscoll I, Troncoso JC, Rudow G, Sojkova J, Pletnikova O, Zhou Y, et al. Correspondence between in vivo (11)C-PiB-PET amyloid imaging and postmortem, region-matched assessment of plaques. Acta Neuropathol (2012) 124:823–31. doi:10.1007/s00401-012-1025-1
13. Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain (2007) 130:2837–44. doi:10.1093/brain/awm238
14. Resnick SM, Sojkova J, Zhou Y, An Y, Ye W, Holt DP, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology (2010) 74:807–15. doi:10.1212/WNL.0b013e3181d3e3e9
15. Marchant NL, Reed BR, Sanossian N, Madison CM, Kriger S, Dhada R, et al. The aging brain and cognition: contribution of vascular injury and aβ to mild cognitive dysfunction. JAMA Neurol (2013) 70:488–95. doi:10.1001/2013.jamaneurol.405
16. Jack CR Jr, Wiste HJ, Vemuri P, Weigand SD, Senjem ML, Zeng G, et al. Brain beta-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer’s disease. Brain (2010) 133:3336–48. doi:10.1093/brain/awq277
17. Matías-Guiu JA, Cabrera-Martín MN, Moreno-Ramos T, Valles-Salgado M, Fernández-Matarrubia M, Carreras JL, et al. Amyloid and FDG-PET study of logopenic primary progressive aphasia: evidence for the existence of two subtypes. J Neurol (2015) 262:1463–72. doi:10.1007/s00415-015-7738-z
18. Catafau AM, Bullich S. Amyloid PET imaging: applications beyond Alzheimer’s disease. Clin Transl Imaging (2015) 3:39–55. doi:10.1007/s40336-014-0098-3
19. Mathis CA, Mason NS, Lopresti BJ, Klunk WE. Development of positron emission tomography β-amyloid plaque imaging agents. Semin Nucl Med (2012) 42:423–32. doi:10.1053/j.semnuclmed.2012.07.001
20. Flaherty DP, Kiyota T, Dong Y, Ikezu T, Vennerstrom JL. Phenolic bis-styrylbenzenes as β-amyloid binding ligands and free radical scavengers. J Med Chem (2010) 53:7992–9. doi:10.1021/jm1006929
21. Nakazono M, Obayashi K, Sasamoto K, Tomiyoshi K, Suenaga G, Ando Y. Novel styrylbenzene derivatives for detecting amyloid deposits. Clin Chim Acta (2014) 436:27–34. doi:10.1016/j.cca.2014.04.028
22. Furukawa K, Ikeda S, Okamura N, Tashiro M, Tomita N, Furumoto S, et al. Cardiac positron-emission tomography images with an amyloid-specific tracer in familial transthyretin-related systemic amyloidosis. Circulation (2012) 125:556–7. doi:10.1161/CIRCULATIONAHA.111.045237
23. Chen W, Dilsizian V. Molecular imaging of amyloidosis: will the heart be the next target after the brain? Curr Cardiol Rep (2012) 14:226–33. doi:10.1007/s11886-011-0239-5
24. Stankoff B, Wang Y, Bottlaender M, Aigrot MS, Dolle F, Wu C, et al. Imaging of CNS myelin by positron-emission tomography. Proc Natl Acad Sci U S A (2006) 103:9304–9. doi:10.1073/pnas.0600769103
25. Klunk WE, Mathis CA. Whatever happened to Pittsburgh compound-A? Alzheimer Dis Assoc Disord (2008) 22:198–203. doi:10.1097/WAD.0b013e318188c0c8
26. Mathis CA, Bacskai BJ, Kajdasz ST, McLellan ME, Frosch MP, Hyman BT, et al. A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorg Med Chem Lett (2002) 12:295–8. doi:10.1016/S0960-894X(01)00734-X
27. Stankoff B, Freeman L, Aigrot MS, Chardain A, Dolle F, Williams A, et al. Imaging central nervous system myelin by positron emission tomography in multiple sclerosis using [methyl-(1)(1)C]-2-(4′-methylaminophenyl)-6-hydroxybenzothiazole. Ann Neurol (2011) 69(4):673–80. doi:10.1002/ana.22320
28. Bodini B, Veronese M, Garcia-Lorenzo D, Freeman L, Papeix C, Zalc B, et al. Positron emission tomography with [11C]-PIB: a clinically relevant tool for voxel-wise myelin quantification in multiple sclerosis. Mult Scler (2013) 19(Suppl):174–5.
29. Matias-Guiu JA, Cabrera-Martín MN, Matías-Guiu J, Oreja-Guevara C, Riola-Parada C, Moreno-Ramos T, et al. Amyloid PET imaging in multiple sclerosis: an (18)F-florbetaben study. BMC Neurol (2015) 15:243. doi:10.1186/s12883-015-0502-2
30. Valis M, Talab R, Stourac P, Andrys C, Masopust J. Tau protein, phosphorylated tau protein and beta-amyloid42 in the cerebrospinal fluid of multiple sclerosis patients. Neuro Endocrinol Lett (2008) 29:971–6.
31. Hein Née Maier K, Köhler A, Diem R, Sättler MB, Demmer I, Lange P, et al. Biological markers for axonal degeneration in CSF and blood of patients with the first event indicative for multiple sclerosis. Neurosci Lett (2008) 436:72–6. doi:10.1016/j.neulet.2008.02.064
32. Mattsson N, Axelsson M, Haghighi S, Malmeström C, Wu G, Anckarsäter R, et al. Reduced cerebrospinal fluid BACE1 activity in multiple sclerosis. Mult Scler (2009) 15:448–54. doi:10.1177/1352458508100031
33. Mitosek-Szewczyk K, Gordon-Krajcer W, Flis D, Stelmasiak Z. Some markers of neuronal damage in cerebrospinal fluid of multiple sclerosis in relapse. Folia Neuropathol (2011) 49:191–6.
34. Mori F, Rossi S, Sancesario G, Codecà C, Mataluni G, Monteleone F, et al. Cognitive and cortical plasticity deficits correlate with altered amyloid-β CSF levels in multiple sclerosis. Neuropsychopharmacology (2011) 36:559–68. doi:10.1038/npp.2010.187
35. Mai W, Hu X, Lu Z, Peng F, Wang Y. Cerebrospinal fluid levels of soluble amyloid precursor protein and β-amyloid 42 in patients with multiple sclerosis, neuromyelitis optica and clinically isolated syndrome. J Int Med Res (2011) 39:2402–13. doi:10.1177/147323001103900641
36. Sladkova V, Mareš J, Lubenova B, Zapletalova J, Stejskal D, Hlustik P, et al. Degenerative and inflammatory markers in the cerebrospinal fluid of multiple sclerosis patients with relapsing–remitting course of disease and after clinical isolated syndrome. Neurol Res (2011) 33:415–20. doi:10.1179/016164110X12816242542535
37. Szalardy L, Zadori D, Simu M, Bencsik K, Vecsei L, Klivenyi P. Evaluating biomarkers of neuronal degeneration and neuroinflammation in CSF of patients with multiple sclerosis-osteopontin as a potential marker of clinical severity. J Neurol Sci (2013) 331:38–42. doi:10.1016/j.jns.2013.04.024
38. Augutis K, Axelsson M, Portelius E, Brinkmalm G, Andreasson U, Gustavsson MK, et al. Cerebrospinal fluid biomarkers of β-amyloid metabolism in multiple sclerosis. Mult Scler (2013) 19:543–52. doi:10.1177/1352458512460603
39. David MA, Tayebi M. Detection of protein aggregates in brain and cerebrospinal fluid derived from multiple sclerosis patients. Front Neurol (2014) 5:251. doi:10.3389/fneur.2014.00251
40. Mangiardi M, Crawford DK, Xia X, Du S, Simon-Freeman R, Voskuhl RR, et al. An animal model of cortical and callosal pathology in multiple sclerosis. Brain Pathol (2011) 21:263–78. doi:10.1111/j.1750-3639.2010.00444.x
41. Lassmann H. Mechanisms of neurodegeneration shared between multiple sclerosis and Alzheimer’s disease. J Neural Transm (2011) 118:747–52. doi:10.1007/s00702-011-0607-8
42. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med (1998) 338:278–85. doi:10.1056/NEJM199801293380502
43. Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain (1997) 120:393–9. doi:10.1093/brain/120.3.393
44. Sun SW, Nishioka C, Labib W, Liang HF. Axonal terminals exposed to amyloid-β may not lead to pre-synaptic axonal damage. J Alzheimers Dis (2015) 45:1139–48. doi:10.3233/JAD-142154
45. Hohlfeld R, Wekerle H. β-Amyloid: enemy or remedy? Sci Transl Med (2012) 4:145fs24. doi:10.1126/scitranslmed.3004586
46. Furlan R, Brambilla E, Sanvito F, Roccatagliata L, Olivieri S, Bergami A, et al. Vaccination with amyloid-β peptide induces autoimmune encephalomyelitis in C57/BL6 mice. Brain (2003) 126:285–91. doi:10.1093/brain/awg031
47. Sun SW, Liang HF, Mei J, Xu D, Shi WX. In vivo diffusion tensor imaging of amyloid-β-induced white matter damage in mice. J Alzheimers Dis (2014) 38:93–101. doi:10.3233/JAD-130236
48. Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, et al. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol (2004) 164:123–31. doi:10.1083/jcb.200307017
49. Li X, Zuo P. Effects of Abeta25-35 on neurogenesis in the adult mouse subventricular zone and dentate gyrus. Neurol Res (2005) 27:218–22. doi:10.1179/016164105X35585
50. Small DH, Nurcombe V, Moir R, Michaelson S, Monard D, Beyreuther K, et al. Association and release of the amyloid protein precursor of Alzheimer’s disease from chick brain extracellular matrix. J Neurosci (1992) 12:4143–50.
51. Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci (2009) 12:1567–76. doi:10.1038/nn.2433
52. Jacobsen KT, Iverfeldt K. Amyloid precursor protein and its homologues: a family of proteolysis-dependent receptors. Cell Mol Life Sci (2009) 66:2299–318. doi:10.1007/s00018-009-0020-8
53. Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rülicke T, et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci (2000) 20:7951–63.
54. Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, et al. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J (2004) 23:4106–15. doi:10.1038/sj.emboj.7600390
55. Von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, et al. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging (1997) 18:661–9. doi:10.1016/S0197-4580(97)00151-6
56. Moore S, Khalaj AJ, Patel R, Yoon J, Ichwan D, Hayardeny L, et al. Restoration of axon conduction and motor deficits by therapeutic treatment with glatiramer acetate. J Neurosci Res (2014) 92:1621–36. doi:10.1002/jnr.23440
57. Ward RE, Huang W, Kostusiak M, Pallier PN, Michael-Titus AT, Priestley JV. A characterization of white matter pathology following spinal cord compression injury in the rat. Neuroscience (2014) 260:227–39. doi:10.1016/j.neuroscience.2013.12.024
58. Xu DE, Zhang WM, Yang ZZ, Zhu HM, Yan K, Li S, et al. Amyloid precursor protein at node of Ranvier modulates nodal formation. Cell Adh Migr (2014) 8:396–403. doi:10.4161/cam.28802
59. Angelov DN, Walther M, Streppel M, Guntinas-Lichius O, Neiss WF, Probstmeier R, et al. Tenascin-R is antiadhesive for activated microglia that induce downregulation of the protein after peripheral nerve injury: a new role in neuronal protection. J Neurosci (1988) 18:6218–29.
60. Pesheva P, Gloor S, Schachner M, Probstmeier R. Tenascin-R is an intrinsic autocrine factor for oligodendrocyte differentiation and promotes cell adhesion by a sulfatide-mediated mechanism. J Neurosci (1997) 17:4642–51.
61. Gutowski NJ, Newcombe J, Cuzner ML. Tenascin-R and C in multiple sclerosis lesions: relevance to extracellular matrix remodelling. Neuropathol Appl Neurobiol (1999) 25:207–14. doi:10.1046/j.1365-2990.1999.00176.x
62. Czopka T, Von Holst A, Schmidt G, Ffrench-Constant C, Faissner A. Tenascin C and tenascin R similarly prevent the formation of myelin membranes in a RhoA-dependent manner, but antagonistically regulate the expression of myelin basic protein via a separate pathway. Glia (2009) 7:1790–801. doi:10.1002/glia.20891
63. Pesheva P, Probstmeier R. Association of tenascin-R with murine brain myelin membranes: involvement of divalent cations. Neurosci Lett (2000) 283:165–8. doi:10.1016/S0304-3940(00)00900-9
64. Kurnellas M, Adams CM, Sobel RA, Steinman L, Rothbard JB. Amyloid fibrils composed of hexameric peptides attenuate neuroinflammation. Sci Transl Med (2013) 5:179ra42. doi:10.1126/scitranslmed.3005681
65. Van Noort JM, van Sechel AC, Bajramovic JJ, el Ouagmiri M, Polman CH, Lassmann H, et al. The small heat-shock protein alpha B-crystallin as candidate autoantigen in multiple sclerosis. Nature (1995) 375:798–801. doi:10.1038/375798a0
66. Ousman SS, Tomooka BH, Van Noort JM, Wawrousek EF, O’Connor KC, Hafler DA, et al. Protective and therapeutic role for αB-crystallin in autoimmune demyelination. Nature (2007) 448:474–9. doi:10.1038/nature05935
67. Han MH, Hwang S, Roy DB, Lundgren DH, Price JV, Ousman S, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature (2008) 451:1076–81. doi:10.1038/nature06559
68. Kurnellas MP, Brownell SE, Su L, Malkovskiy AV, Rajadas J, Dolganov G, et al. Chaperone activity of small heat shock proteins underlies therapeutic efficacy in experimental autoimmune encephalomyelitis. J Biol Chem (2012) 287:36423–34. doi:10.1074/jbc.M112.371229
69. Tanaka N, Tanaka R, Tokuhara M, Kunugi S, Lee YF, Hamada D. Amyloid fibril formation and chaperone-like activity of peptides from alphaA-crystallin. Biochemistry (2008) 47:2961–7. doi:10.1021/bi701823g
70. Ji Z, Ke ZJ, Geng JG. SAP suppresses the development of experimental autoimmune encephalomyelitis in C57BL/6 mice. Immunol Cell Biol (2012) 90:388–95. doi:10.1038/icb.2011.51
71. Weinger JG, Davies P, Acker CM, Brosnan CF, Tsiperson V, Bayewitz A, et al. Mice devoid of tau have increased susceptibility to neuronal damage in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J Neuropathol Exp Neurol (2012) 71:422–33. doi:10.1097/NEN.0b013e3182540d2e
72. Steinman L, Rothbard JB, Kurnellas MP. Janus faces of amyloid proteins in neuroinflammation. J Clin Immunol (2014) 34:S61–3. doi:10.1007/s10875-014-0034-3
73. Yan R, Han P, Miao H, Greengard P, Xu H. The transmembrane domain of the Alzheimer’s β-secretase (BACE1) determines its late Golgi localization and access to β-amyloid precursor protein (APP) substrate. J Biol Chem (2001) 276:36788–96. doi:10.1074/jbc.M104350200
74. Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science (1999) 286:735–41. doi:10.1126/science.286.5440.735
75. Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, et al. Membrane-anchored aspartyl protease with Alzheimer’s disease β-secretase activity. Nature (1999) 402:533–7. doi:10.1038/990107
76. Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, et al. Identification of a novel aspartic protease (Asp 2) as β-secretase. Mol Cell Neurosci (1999) 14:419–27. doi:10.1006/mcne.1999.0811
77. Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature (1999) 402:537–40. doi:10.1038/990114
78. Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the β-secretase site of β-amyloid precursor protein. Proc Natl Acad Sci U S A (2000) 97:1456–60. doi:10.1073/pnas.97.4.1456
79. Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med (2003) 9:3–4. doi:10.1038/nm0103-3
80. Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, et al. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci (2001) 4:233–4. doi:10.1038/85064
81. Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, et al. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci (2001) 4:231–2. doi:10.1038/nn1101-1158
82. Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum Mol Genet (2001) 10:1317–24. doi:10.1093/hmg/10.12.1317
83. Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, et al. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci (2006) 9:1520–5. doi:10.1038/nn1797
84. Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, et al. Control of peripheral nerve myelination by the beta-secretase BACE1. Science (2006) 314:664–6. doi:10.1126/science.1132341
85. Hu X, He W, Diaconu C, Tang X, Kidd GJ, Macklin WB, et al. Genetic deletion of BACE1 in mice affects remyelination of sciatic nerves. FASEB J (2008) 22:2970–80. doi:10.1096/fj.08-106666
86. Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR, et al. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci (2005) 25:2386–95. doi:10.1523/JNEUROSCI.3089-04.2005
87. Nave KA, Salzer JL. Axonal regulation of myelination by neuregulin 1. Curr Opin Neurobiol (2006) 16:492–500. doi:10.1016/j.conb.2006.08.008
88. Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, et al. Axonal neuregulin-1 regulates myelin sheath thickness. Science (2004) 304:700–3. doi:10.1126/science.1095862
89. Taveggia C, Thaker P, Petrylak A, Caporaso GL, Toews A, Falls DL, et al. Type III neuregulin-1 promotes oligodendrocyte myelination. Glia (2008) 56:284–93. doi:10.1002/glia.20612
90. Luo X, Prior M, He W, Hu X, Tang X, Sheng W, et al. Cleavage of neuregulin-1 by BACE1 or ADAM10 produces differential effects on myelination. J Biol Chem (2011) 286:23967–74. doi:10.1074/jbc.M111.251538
91. Lyons A, Griffin RJ, Costelloe CE, Clarke RM, Lynch MA. IL-4 attenuates the neuroinflammation induced by amyloid-beta in vivo and in vitro. J Neurochem (2007) 101:771–81. doi:10.1111/j.1471-4159.2006.04370.x
92. Hensley K. Neuroinflammation in Alzheimer’s disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis (2010) 21:1–14. doi:10.3233/JAD-2010-1414
93. Combs CK, Karlo JC, Kao SC, Landreth GE. β-amyloid stimulation of microglia and monocytes results in TNF α-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci (2001) 21:1179–88.
94. Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, Lynch MA. CD200 ligand-receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci (2007) 27:8309–13. doi:10.1523/JNEUROSCI.1781-07.2007
95. Lyons A, Downer EJ, Costello DA, Murphy N, Lynch MA. Dok2 mediates the CD200Fc attenuation of Aβ-induced changes in glia. J Neuroinflammation (2012) 9:107. doi:10.1186/1742-2094-9-107
96. Vetrivel KS, Thinakaran G. Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim Biophys Acta (2010) 1801:860–7. doi:10.1016/j.bbalip.2010.03.007
97. Kalvodova L, Kahya N, Schwille P, Ehehalt R, Verkade P, Drechsel D, et al. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem (2005) 280:36815–23. doi:10.1074/jbc.M504484200
98. Osenkowski P, Ye W, Wang R, Wolfe MS, Selkoe DJ. Direct and potent regulation of γ-secretase by its lipid microenvironment. J Biol Chem (2008) 283:22529–40. doi:10.1074/jbc.M801925200
99. Holmes O, Paturi S, Ye W, Wolfe MS, Selkoe DJ. The effects of membrane lipids on the activity and processivity of purified γ-secretase. Biochemistry (2012) 51:3565–75. doi:10.1021/bi300303g
100. Grimm MO, Rothhaar TL, Hartmann T. The role of APP proteolytic processing in lipid metabolism. Exp Brain Res (2012) 217:365–75. doi:10.1007/s00221-011-2975-6
101. Gendelman HE, Pezeshkpour GH, Pressman NJ, Wolinsky JS, Quarles RH, Dobersen MJ, et al. A quantitation of myelin-associated glycoprotein and myelin basic protein loss in different demyelinating diseases. Ann Neurol (1985) 18:324–8. doi:10.1002/ana.410180309
102. Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci (2006) 7:617–27. doi:10.1038/nrn1956
103. Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, et al. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science (2008) 322:967–70. doi:10.1126/science.1161151
104. Zhang J, Sun X, Zheng S, Liu X, Jin J, Ren Y, et al. Myelin basic protein induces neuron-specific toxicity by directly damaging the neuronal plasma membrane. PLoS One (2014) 9:e108646. doi:10.1371/journal.pone.0108646
105. Hoos MD, Ahmed M, Smith SO, Van Nostrand WE. Inhibition of familial cerebral amyloid angiopathy mutant amyloid β-protein fibril assembly by myelin basic protein. J Biol Chem (2007) 282:9952–61. doi:10.1074/jbc.M603494200
106. Zhan X, Cox C, Ander BP, Liu D, Stamova B, Jin LW, et al. Inflammation combined with ischemia produces myelin injury and plaque-like aggregates of myelin, amyloid-β and AβPP in adult rat brain. J Alzheimers Dis (2015) 46:507–23. doi:10.3233/JAD-143072
107. Kotarba AME, Aucoin D, Hoos MD, Smith SO, Van Nostrand WE. Fine mapping of the amyloid β-protein binding site on myelin basic protein. Biochemistry (2013) 52:2565–73. doi:10.1021/bi4001936
108. Liao MC, Hoos MD, Aucoin D, Ahmed M, Davis J, Smith SO, et al. Amino terminal domain of myelin basic protein inhibits amyloid β-protein fibril assembly. J Biol Chem (2010) 285:35590–8. doi:10.1074/jbc.M110.169599
Keywords: multiple sclerosis, amyloid PET, biomarkers, white matter, amyloid precursor protein, amyloid, myelin basic protein, positron emission tomography
Citation: Matías-Guiu JA, Oreja-Guevara C, Cabrera-Martín MN, Moreno-Ramos T, Carreras JL and Matías-Guiu J (2016) Amyloid Proteins and Their Role in Multiple Sclerosis. Considerations in the Use of Amyloid-PET Imaging. Front. Neurol. 7:53. doi: 10.3389/fneur.2016.00053
Received: 01 July 2015; Accepted: 22 March 2016;
Published: 31 March 2016
Edited by:
Björn Tackenberg, Philipps University of Marburg, GermanyReviewed by:
Richard Dodel, Phillipps University of Marburg, GermanyChristian Eienbröker, Philipps University of Marburg, Germany
Copyright: © 2016 Matías-Guiu, Oreja-Guevara, Cabrera-Martín, Moreno-Ramos, Carreras and Matías-Guiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jordi A. Matías-Guiu, am9yZGltYXRpYXNndWl1QGhvdG1haWwuY29t, aW5jLmhjc2NAc2FsdWQubWFkcmlkLm9yZw==