 Nir Giladi1,2*
Nir Giladi1,2* Anat Mirelman
Anat Mirelman Avner Thaler
Avner Thaler- 1Laboratory for Early Markers of Neurodegeneration, Department of Neurology, Tel-Aviv Sourasky Medical Center, Tel Aviv University, Tel Aviv, Israel
- 2Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
- 3Genetic Institute, Tel Aviv Sourasky Medical Center, Tel Aviv, Israel
While the phenotype of Parkinson disease (PD) is heterogeneous, treatment approaches are mostly uniform. Personalized medicine aims to treat diseases with targeted therapies based on cumulative variables, including genotype. We believe that sufficient evidence has accumulated to warrant the initiation of personalized medicine in PD based on subjects genotype and provide examples for our reasoning from observations of GBA and LRRK2 mutations carriers. While PD patients who carry the G2019S mutation in the LRRK2 gene seem to develop relatively mild disease with more frequent postural instability gait disturbance phenotype, carriers of mutations in the GBA gene tend to have an early onset, rapidly deteriorating disease, with more pronounced cognitive and autonomic impairments. These characteristics have significant implications for treatment and outcome and should be addressed from an early stage in the attempt to improve the patient’s quality of life.
Introduction
Parkinson disease (PD) is a complex neurodegenerative disorder affecting older adults. The etiology of PD is heterogeneous, genetic, and multi-factorial, resulting in a highly variable clinical course, spanning from a slowly progressive, benign course to a rapidly progressive, disabling disease (1, 2).
Parkinson disease is a highly variable disease; it can start early in the 3rd or 4th decade of life, or as late as the 10th decade, the predominant symptom can be tremor, or the disease phenotype might be non-tremulous with gait, postural impairments, or rigidity as the main features. PD can involve late or early cognitive and behavioral changes, early or late autonomic disturbances, and various manifestation of pain or sleep disturbances (3–5). However, in spite of this large diversity in clinical features, the therapeutic approach, as reflected in therapeutic guidelines, is uniform and symptom oriented, with the exception of age and cognitive state as factors influencing clinical decisions (6, 7). Recent understanding of the significant contribution of multiple gene loci to the risk of developing PD, as well as to the disease phenotypic clinical course, raise the possibility of tailoring an individualized therapeutic strategy based on the patients’ genetic and personal background (8).
Ashkenazi Jews (AJ) constitute a unique population to start implementing such genetic-based personalized approach since more than one-third of AJ-PD patients have a known mutation in either the GBA (eight mutations) or the LRRK2 (G2019S) genes (9, 10). Herein, we will highlight some clinical features of GBA or LRRK2-associated PD among the AJ population and propose a new vision for personalized medicine based on genetic background. The immediate consequence of such genetic-based personalized approach is the need to genetically test for GBA and LRRK2 mutations all recently diagnosed AJ-PD patients, patients entering clinical trials and those referred to deep brain stimulation (DBS). It is important to stress that the clinical genetic correlation we report here is based mostly on retrospective data and the proposed clinical approach has never been examined in prospective long-term care. We hope this paper will lead a scientific discussion and encourage prospective studies and will be the first step in the development of a personalized approach in the development of therapeutic strategies for patients with PD.
LRRK2-G2019S PD
LRRK2-G2019S PD has been reported to manifest with a mild phenotype with less cognitive impairments (11, 12), higher frequencies of sleep onset insomnia, and equal frequencies of daytime sleepiness but less REM sleep behavior disorders (RBD) compared to idiopathic PD (iPD) (13, 14). LRRK2-G2019S PD patients seem to express better olfactory function compared to iPD patients (15) and appear to present more frequently with the postural instability gait difficulty (PIGD) sub-type (16), although in another publication tremor was identified as a distinguished feature of LRRK2-PD (17). Some discrepancies appear in the literature relating to the presence of higher prevalence of psychiatric symptoms in LRRK2-G2019S PD patients (18, 19) while other studies could not detect such a trend (11). Both higher and lower rates of depression in LRRK2-G2019S PD patients have been reported compared to iPD (17, 20).
GBA-PD
Studies reported that heterozygous, homozygous, and compound-heterozygote GBA-PD patients present with more severe cognitive decline compared to iPD (21–23). The association between mutations in the GBA gene and early cognitive decline has recently received much support by observations that GBA carriers frequently develop Lewy Body Dementia (LBD), a syndrome that is tightly associated with PD and is one of the classical synucleopathies, defined by the development of clinically significant cognitive decline prior to, or at about the same time as, the appearance of parkinsonism (24). In addition, higher incidence of RBD, a known marker for cognitive decline in PD, was reported in GBA-PD patients (25) as well as more frequent psychiatric symptoms than in iPD (26). Carriers of severe GBA mutations were found to have an earlier onset PD symptoms (27) and more autonomic disturbances at PD diagnosis and as the disease progressed, reflecting earlier and more sever autonomic degeneration (27) (Table 1).
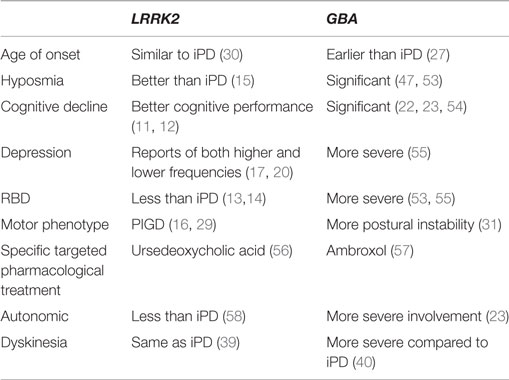
Table 1. Differences in symptomatology based on genetic status as compared to iPD.
How PD Differs in Patients with LRRK2 or GBA Mutations?
Early in the Clinical Course
Gait
Early postural instability and gait disturbances in PD is known to be associated with earlier falls, more freezing of gait (FOG) and more frequent cognitive decline (28). A large proportion of LRRK2-G2019S-PD patients were found to present with PIGD phenotype that is associated with higher risk of falls (29, 30). Similarly, GBA-PD mutation carriers demonstrated increased gait impairments and FOG, which has been associated with disturbed executive functions (31). Such decline in functional performance is an indirect marker of decreased motor-cognitive reserve, which should be taken into account while developing treatment strategies.
Parkinson disease patients, with either LRRK2-G2019S mutation or any mutation in the GBA gene, should be informed about their higher risk for earlier falls or FOG, and treatment should be offered to try and delay or prevent these serious motor complications. Such recommendations will include using extra caution when offering drugs that increase fall risk, such as anti-depressive drugs, benzodiazepines, hypnotics, anti-cholinergics and possibly dopamine agonists (DA) because of their effect on alertness (32, 33). In conjunction, physical or cognitive therapy should be recommended and encouraged as early as possible and reinforced in every visit. Prevention of falls and FOGs should be a priority in order to avoid the need for treatment of their destructive consequences.
Early Cognition–Mood Changes
The tendency for earlier, faster, and more clinically meaningful cognitive decline in GBA-PD should be a major consideration in treatment strategy. In view of the negative impact of all anti-parkinsonian drugs on cognition, alertness, sleep, and the risk of psychosis (34), drug choices or drug combinations should be prescribed more cautiously than when treating iPD patients. In addition, cognitive state, sleep quality, mood (depression and anxiety), hallucinations, or delusions should be assessed in every visit of a GBA-PD patient, in an effort to establish early detection and to prevent sever psychosis, a leading cause for hospitalization and institutionalization (35). Intensive exercise programs and multi-domain cognitive enhancing interventions should be recommended early to enhance capacity and reserve, and the above knowledge should be used to increase adherence to both short-term and long-term behavioral modifications.
Such variability in disease course and in the risk of developing significant complications, as well as drug-induced side effects in GBA-PD, must be taken into consideration when including patients into any clinical trial, but especially in studies testing disease modifying drugs.
Increased Risk of Experiencing Autonomic Disturbances
The higher rates of autonomic symptoms presented by GBA-PD patients (23) has major importance in treatment strategies since all anti-parkinsonian drugs can worsen or cause orthostatic hypotension while some can trigger urinary retention, delayed gastric emptying time, constipation, erectile dysfunction, delayed ejaculation, and lymphatic edema (36). The increased risk of early occurrence of clinically significant autonomic disturbances or treatment-induced autonomic complications should influence the therapeutic approach as well. Careful monitoring for early signs of autonomic dysfunctions in GBA-PD patients should be a recommended practice. Furthermore, the increased vulnerability of the autonomic nervous system should be taken into account when such patients are considered for participating in any drug or interventional clinical trials.
Later Disease Stage Features
Dyskinesia
One of the most significant motor complication of dopaminergic treatment and especially levodopa is dyskinesia (37). Involuntary movements are frequently the biggest hurdle for effective dopaminergic treatment and the strategy to delay levodopa and use less effective substitutes, such as DA, was initiated specifically to delay these complications (38). LRRK2-PD patients were found to have equal frequencies of levodopa-induced dyskinesia compared to iPD (39). By contrast, it has been reported that GBA-PD patients are more sensitive to levodopa, developing dyskinesia earlier in the course of the disease (40). Combined with the earlier age of disease onset in GBA-PD (27), which is known to be associated with earlier and more aggressive Dopa-induced dyskinesia (41). GBA-PD patients should be considered from disease diagnosis to have increased risk for early disabling dyskinesia, lower dosages of levodopa and possibly, delaying levodopa should be common practice, even more than in the general PD population, in this group of GBA-PD patients.
Should Mutation Status Influence Referrals to Deep Brain Stimulation?
The role of genotype in the outcome of DBS has not been well studied but it is possible that the risk to develop cognitive decline or gait disturbances, the two most common serious and long-term complications of DBS (42, 43) could be influenced by PD-associated mutations. Angeli et al. identified earlier cognitive decline in GBA-PD patients compared with controls (44), AJ LRRK2-PD patients had a similar outcome as iPD patients after subthalamic nucleus (STN) DBS (45), while a study in North African Arabs identified better outcome for LRRK2-PD patients after STN-DBS compared to iPD patients (46). Based on current knowledge, it could be speculated that more frequent behavioral and cognitive changes as well as earlier gait disturbances could be observed in PD patients, carriers of mutation in either the GBA or LRRK2 genes after DBS.
To the best of our knowledge, the effect of DBS on gait was not explored in the context of patients’ genetic status. Because of its cardinal clinical consequences, it is important to explore retrospective cohorts and retrieve information about the short- and long-term clinical outcomes of DBS to the globus pallidus interna and STN in patients with iPD as well as those with known mutations. Until such data are available, referring PD patients with mutations in the GBA or the LRRK2 genes to DBS should be done after special consideration and after sharing the potential risks with the patients and their families.
Parkinsonism as the Manifesting Symptom of GD
Recent investigation into the association of Gaucher disease (GD) and PD has indicated that parkinsonism could be the first neurological symptom of GD (47). In addition, our group has recently assessed 1100 AJ-PD patients and found 12 subjects who were either homozygous or compound-heterozygous for mutations in the GBA gene, half of which were not aware of any GD symptoms (ahead of print). This has important implications regarding the potential for reduced bone density and increased fractures following falls, which is a further reason for genetic screening of the AJ population.
Therapeutic Approach based on Genetic Testing
Throughout the course of PD, many pharmacological options as well as non-pharmacological interventions are available for the treatment of disease symptoms. Decisions should be made regarding the first anti-parkinsonian drug to prescribe, whether to add DA or levodopa or rather start treatment with levodopa right away, how to treat depression, anxiety, and sleep disturbances and how to monitor autonomic dysfunctions. Later on, questions regarding the combination of drugs, the use of anti-cholinergic drugs, or cholinesterase inhibitors for treating cognitive decline or sleep impairments and RBD, and the use of atypical dopamine-blocking agents, such as quetiapine or clozapine, should be addressed. In addition, the intensity and quality of physiotherapy, cognitive enhancement therapies, and falls prevention strategies are all available but, currently, not uniformly recommended and implemented. Lastly, a recent epidemiological study exploring environmental and behavioral habits, such as caffeine consumption, found a possible risk-reducing interaction between caffeine consumption and LRRK2-PD (48). Yet, patients’ engagement in life-style modification strategies is highly variable, highly associated with the level of knowledge and commitment of patients and care-givers to the recommended non-pharmacological interventions.
We believe that the present knowledge about the different course of PD among AJ and non-Jewish patients based on GBA and LRRK2 mutations’ status and the extremely high frequency of mutation carriers in the AJ-PD patients (35%) justifies early genetic screening and tailoring treatment approach based on patients’ genotypes. However, the reduced penetrance rates for these mutations (49, 50) suggest that other modifier genes may be responsible for both increased and reduced risk of developing PD in this unique population (51, 52) as well as other aspects of the disease and demand further elucidation. We further recommend that the genotype needs to be considered when recruiting patients into clinical trials especially when examining the efficacy of novel drugs. Such approach could foster the recruitment of a smaller sample size in fewer centers. Moreover, exploring efficacy of a drug with specific biological target, such as on lysosomal activity should likely be first tested in patients with mutations in the GBA gene.
In the present paper, we lumped together our discussion on GBA mutation carriers although we believe that specific mutations may influence disease course and also the effect of medications. Currently, there is only limited information to support splitting the recommendations according to individual mutations, or as a group of “mild” or “severe” mutations. However, future studies will likely lead to refinement of this approach also within GBA-PD.
In conclusion, we believe that the available evidence in regard to the specific course of GBA or LRRK2 mutations’ associated PD is sufficient to recommend genotyping new PD patient (especially those with AJ background). Such genotyping should influence the therapeutic approach throughout the course of the disease and should also be considered when recruiting patients into clinical trials.
Author Contributions
NG – conceptualization and writing, AM – writing, AT – writing, AO-U – writing. All authors agree to be accountable for the content of this work. All co-authors have read and approved the content of this manuscript. This manuscript is not under review of any other journal.
Conflict of Interest Statement
NG serves as a member of the Editorial Board for the Journal of Parkinson’s disease. He serves as consultant to Teva-Lundbeck, IntecPharma, NeuroDerm, Armon Neuromedical Ltd\Dexel, Monfort and Lysosomal Therapeutic Inc. He received payment for lectures at Teva-Lundbeck, Novartis, UCB, Abviee, Shaier, and Genzyme. Prof. NG received research support from the Michael J Fox Foundation, the National Parkinson Foundation, and the Israel Science Foundation. AM – nothing to disclose. AT – nothing to disclose. AO-U – nothing to disclose.
References
1. Lawton M, Baig F, Rolinski M, Ruffman C, Nithi K, May MT, et al. Parkinson’s disease subtypes in the Oxford Parkinson Disease Centre (OPDC) discovery cohort. J Parkinsons Dis (2015) 5:269–79. doi: 10.3233/JPD-140523
2. Selikhova M, Williams DR, Kempster PA, Holton JL, Revesz T, Lees AJ. A clinico-pathological study of subtypes in Parkinson’s disease. Brain (2009) 132:2947–57. doi:10.1093/brain/awp234
3. Jankovic J, McDermott M, Carter J, Gauthier S, Goetz C, Golbe L, et al. Variable expression of Parkinson’s disease: a base-line analysis of the DATATOP cohort. The Parkinson Study Group. Neurology (1990) 40:1529–34.
4. Wickremaratchi MM, Knipe MD, Sastry BS, Morgan E, Jones A, Salmon R, et al. The motor phenotype of Parkinson’s disease in relation to age at onset. Mov Disord (2011) 26:457–63. doi:10.1002/mds.23469
5. van Rooden SM, Colas F, Martinez-Martin P, Visser M, Verbaan D, Marinus J, et al. Clinical subtypes of Parkinson’s disease. Mov Disord (2011) 26:51–8. doi:10.1002/mds.23346
6. Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord (2015) 30:1591–601. doi:10.1002/mds.26424
7. Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA (2014) 311:1670–83. doi:10.1001/jama.2014.3654
8. Korczyn AD, Hassin-Baer S. Can the disease course in Parkinson’s disease be slowed? BMC Med (2015) 13:295. doi:10.1186/s12916-015-0534-x
9. Thaler A, Ash E, Gan-Or Z, Orr-Urtreger A, Giladi N. The LRRK2 G2019S mutation as the cause of Parkinson’s disease in Ashkenazi Jews. J Neural Transm (2009) 116:1473–82. doi:10.1007/s00702-009-0303-0
10. Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology (2008) 70:2277–83. doi:10.1212/01.wnl.0000304039.11891.29
11. Somme JH, Molano Salazar A, Gonzalez A, Tijero B, Berganzo K, Lezcano E, et al. Cognitive and behavioral symptoms in Parkinson’s disease patients with the G2019S and R1441G mutations of the LRRK2 gene. Parkinsonism Relat Disord (2015) 21:494–9. doi:10.1016/j.parkreldis.2015.02.019
12. Srivatsal S, Cholerton B, Leverenz JB, Wszolek ZK, Uitti RJ, Dickson DW, et al. Cognitive profile of LRRK2-related Parkinson’s disease. Mov Disord (2015) 30:728–33. doi:10.1002/mds.26161
13. Saunders-Pullman R, Alcalay RN, Mirelman A, Wang C, Luciano MS, Ortega RA, et al. REM sleep behavior disorder, as assessed by questionnaire, in G2019S LRRK2 mutation PD and carriers. Mov Disord (2015) 30(13):1834–9. doi:10.1002/mds.26413
14. Pont-Sunyer C, Iranzo A, Gaig C, Fernández-Arcos A, Vilas D, Valldeoriola F, et al. Sleep disorders in parkinsonian and nonparkinsonian LRRK2 mutation carriers. PLoS One (2015) 10:e0132368. doi:10.1371/journal.pone.0132368
15. Saunders-Pullman R, Mirelman A, Wang C, Alcalay RN, San Luciano M, Ortega R, et al. Olfactory identification in LRRK2 G2019S mutation carriers: a relevant marker? Ann Clin Transl Neurol (2014) 1:670–8. doi:10.1002/acn3.95
16. Alcalay RN, Mejia-Santana H, Mirelman A, Saunders-Pullman R, Raymond D, Palmese C, et al. Neuropsychological performance in LRRK2 G2019S carriers with Parkinson’s disease. Parkinsonism Relat Disord (2015) 21:106–10. doi:10.1016/j.parkreldis.2014.09.033
17. Marras C, Schüle B, Munhoz RP, Rogaeva E, Langston JW, Kasten M, et al. Phenotype in parkinsonian and nonparkinsonian LRRK2 G2019S mutation carriers. Neurology (2011) 77:325–33. doi:10.1212/WNL.0b013e318227042d
18. Goldwurm S, Zini M, Di Fonzo A, De Gaspari D, Siri C, Simons EJ, et al. LRRK2 G2019S mutation and Parkinson’s disease: a clinical, neuropsychological and neuropsychiatric study in a large Italian sample. Parkinsonism Relat Disord (2006) 12:410–9. doi:10.1016/j.parkreldis.2006.04.001
19. Belarbi S, Hecham N, Lesage S, Kediha MI, Smail N, Benhassine T, et al. LRRK2 G2019S mutation in Parkinson’s disease: a neuropsychological and neuropsychiatric study in a large Algerian cohort. Parkinsonism Relat Disord (2010) 16:676–9. doi:10.1016/j.parkreldis.2010.09.003
20. Ben Sassi S, Nabli F, Hentati E, Nahdi H, Trabelsi M, Ben Ayed H, et al. Cognitive dysfunction in Tunisian LRRK2 associated Parkinson’s disease. Parkinsonism Relat Disord (2012) 18:243–6. doi:10.1016/j.parkreldis.2011.10.009
21. Alcalay RN, Mejia-Santana H, Tang MX, Rakitin B, Rosado L, Ross B, et al. Self-report of cognitive impairment and mini-mental state examination performance in PRKN, LRRK2, and GBA carriers with early onset Parkinson’s disease. J Clin Exp Neuropsychol (2010) 32:775–9. doi:10.1080/13803390903521018
22. Alcalay RN, Caccappolo E, Mejia-Santana H, Tang M-, Rosado L, Orbe Reilly M, et al. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology (2012) 78:1434–40. doi:10.1212/WNL.0b013e318253d54b
23. Brockmann K, Srulijes K, Hauser AK, Schulte C, Csoti I, Gasser T, et al. GBA-associated PD presents with nonmotor characteristics. Neurology (2011) 77:276–80. doi:10.1212/WNL.0b013e318225ab77
24. Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O, Escott-Price V, et al. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet (2014) 23:6139–46. doi:10.1093/hmg/ddu334
25. Gan-Or Z, Mirelman A, Postuma RB, Arnulf I, Bar-Shira A, Dauvilliers Y, et al. GBA mutations are associated with rapid eye movement sleep behavior disorder. Ann Clin Transl Neurol (2015) 2:941–5. doi:10.1002/acn3.228
26. Oeda T, Umemura A, Mori Y, Tomita S, Kohsaka M, Park K, et al. Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol Aging (2015) 36:3306–13. doi:10.1016/j.neurobiolaging.2015.08.027
27. Gan-Or Z, Amshalom I, Kilarski LL, Bar-Shira A, Gana-Weisz M, Mirelman A, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology (2015) 84:880–7. doi:10.1212/WNL.0000000000001315
28. Jankovic J, Kapadia AS. Functional decline in Parkinson disease. Arch Neurol (2001) 58:1611–5. doi:10.1001/archneur.58.10.1611
29. Mirelman A, Heman T, Yasinovsky K, Thaler A, Gurevich T, Marder K, et al. Fall risk and gait in Parkinson’s disease: the role of the LRRK2 G2019S mutation. Mov Disord (2013) 28:1683–90. doi:10.1002/mds.25587
30. Alcalay RN, Mirelman A, Saunders-Pullman R, Tang MX, Mejia Santana H, Raymond D, et al. Parkinson disease phenotype in Ashkenazi jews with and without LRRK2 G2019S mutations. Mov Disord (2013) 28:1966–71. doi:10.1002/mds.25647
31. Wang C, Cai Y, Gu Z, Ma J, Zheng Z, Tang BS, et al. Clinical profiles of Parkinson’s disease associated with common leucine-rich repeat kinase 2 and glucocerebrosidase genetic variants in Chinese individuals. Neurobiol Aging (2014) 35(725):e721–6. doi:10.1016/j.neurobiolaging.2013.08.012
32. Nonnekes J, Snijders AH, Nutt JG, Deuschl G, Giladi N, Bloem BR. Freezing of gait: a practical approach to management. Lancet Neurol (2015) 14:768–78. doi:10.1016/S1474-4422(15)00041-1
33. Bloem BR, Hausdorff JM, Visser JE, Giladi N. Falls and freezing of gait in Parkinson’s disease: a review of two interconnected, episodic phenomena. Mov Disord (2004) 19:871–84. doi:10.1002/mds.20115
34. Emre M, Ford PJ, Bilgic B, Uc EY. Cognitive impairment and dementia in Parkinson’s disease: practical issues and management. Mov Disord (2014) 29:663–72. doi:10.1002/mds.25870
35. Low V, Ben-Shlomo Y, Coward E, Fletcher S, Walker R, Clarke CE. Measuring the burden and mortality of hospitalisation in Parkinson’s disease: a cross-sectional analysis of the English Hospital Episodes Statistics database 2009-2013. Parkinsonism Relat Disord (2015) 21:449–54. doi:10.1016/j.parkreldis.2015.01.017
36. Park A, Stacy M. Dopamine-induced nonmotor symptoms of Parkinson’s disease. Parkinsons Dis (2011) 2011:485063. doi:10.4061/2011/485063
37. Rascol O, Perez-Lloret S, Ferreira JJ. New treatments for levodopa-induced motor complications. Mov Disord (2015) 30:1451–60. doi:10.1002/mds.26362
38. Poewe W, Mahlknecht P, Jankovic J. Emerging therapies for Parkinson’s disease. Curr Opin Neurol (2012) 25:448–59. doi:10.1097/WCO.0b013e3283542fde
39. Yahalom G, Kaplan N, Vituri A, Cohen OS, Inzelberg R, Kozlova E, et al. Dyskinesias in patients with Parkinson’s disease: effect of the leucine-rich repeat kinase 2 (LRRK2) G2019S mutation. Parkinsonism Relat Disord (2012) 18:1039–41. doi:10.1016/j.parkreldis.2012.05.014
40. Lesage S, Anheim M, Condroyer C, Pollak P, Durif F, Dupuits C, et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum Mol Genet (2011) 20:202–10. doi:10.1093/hmg/ddq454
41. Bjornestad A, Forsaa EB, Pedersen KF, Tysnes OB, Larsen JP, Alves G. Risk and course of motor complications in a population-based incident Parkinson’s disease cohort. Parkinsonism Relat Disord (2016) 22:48–53. doi:10.1016/j.parkreldis.2015.11.007
42. Benabid AL, Chabardes S, Torres N, Piallat B, Krack P, Fraix V, et al. Functional neurosurgery for movement disorders: a historical perspective. Prog Brain Res (2009) 175:379–91. doi:10.1016/S0079-6123(09)17525-8
43. St George RJ, Nutt JG, Burchiel KJ, Horak FB. A meta-regression of the long-term effects of deep brain stimulation on balance and gait in PD. Neurology (2010) 75:1292–9. doi:10.1212/WNL.0b013e3181f61329
44. Angeli A, Mencacci NE, Duran R, Aviles-Olmos I, Kefalopoulou Z, Candelario J, et al. Genotype and phenotype in Parkinson’s disease: lessons in heterogeneity from deep brain stimulation. Mov Disord (2013) 28:1370–5. doi:10.1002/mds.25535
45. Greenbaum L, Israeli-Korn SD, Cohen OS, Elincx-Benizri S, Yahalom G, Kozlova E, et al. The LRRK2 G2019S mutation status does not affect the outcome of subthalamic stimulation in patients with Parkinson’s disease. Parkinsonism Relat Disord (2013) 19:1053–6. doi:10.1016/j.parkreldis.2013.07.005
46. Sayad M, Zouambia M, Chaouch M, Ferrat F, Nebbal M, Bendini M, et al. Greater improvement in LRRK2 G2019S patients undergoing subthalamic nucleus deep brain stimulation compared to non-mutation carriers. BMC Neurosci (2016) 17:6. doi:10.1186/s12868-016-0240-4
47. Saunders-Pullman R, Hagenah J, Dhawan V, Stanley K, Pastores G, Sathe S, et al. Gaucher disease ascertained through a Parkinson’s center: imaging and clinical characterization. Mov Disord (2010) 25:1364–72. doi:10.1002/mds.23046
48. Kumar PM, Paing SS, Li H, Pavanni R, Yuen Y, Zhao Y, et al. Differential effect of caffeine intake in subjects with genetic susceptibility to Parkinson’s Disease. Sci Rep (2015) 5:15492. doi:10.1038/srep15492
49. Marder K, Wang Y, Alcalay RN, Mejia-Santana H, Tang MX, Lee A, et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology (2015) 85:89–95. doi:10.1212/WNL.0000000000001708
50. Trinh J, Guella I, Farrer MJ. Disease penetrance of late-onset parkinsonism: a meta-analysis. JAMA Neurol (2014) 71:1535–9. doi:10.1001/jamaneurol.2014.1909
51. Gan-Or Z, Bar-Shira A, Mirelman A, Gurevich T, Giladi N, Orr-Urtreger A. The age at motor symptoms onset in LRRK2-associated Parkinson’s disease is affected by a variation in the MAPT locus: a possible interaction. J Mol Neurosci (2012) 46:541–4. doi:10.1007/s12031-011-9641-0
52. Gan-Or Z, Bar-Shira A, Dahary D, Mirelman A, Kedmi M, Gurevich T, et al. Association of sequence alterations in the putative promoter of RAB7L1 with a reduced parkinson disease risk. Arch Neurol (2012) 69:105–10. doi:10.1001/archneurol.2011.924
53. Beavan M, McNeill A, Proukakis C, Hughes DA, Mehta A, Schapira AH. et al. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol (2015) 72:201–8. doi:10.1001/jamaneurol.2014.2950
54. Zokaei N, McNeill A, Proukakis C, Beavan M, Jarman P, Korlipara P, et al. Visual short-term memory deficits associated with GBA mutation and Parkinson’s disease. Brain (2014) 137:2303–11. doi:10.1093/brain/awu143
55. McNeill A, Duran R, Hughes DA, Mehta A, Schapira AH. A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J Neurol Neurosurg Psychiatry (2012) 83:853–4. doi:10.1136/jnnp-2012-302402
56. Mortiboys H, Johansen KK, Aasly JO, Bandmann O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology (2010) 75:2017–20. doi:10.1212/WNL.0b013e3181ff9685
57. McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain (2014) 137:1481–95. doi:10.1093/brain/awu020
Keywords: Parkinson’s disease, GBA, LRRK2, phenotype, personalized medicine
Citation: Giladi N, Mirelman A, Thaler A and Orr-Urtreger A (2016) A Personalized Approach to Parkinson’s Disease Patients Based on Founder Mutation Analysis. Front. Neurol. 7:71. doi: 10.3389/fneur.2016.00071
Received: 13 March 2016; Accepted: 25 April 2016;
Published: 10 May 2016
Edited by:
Antonio Pisani, Università di Roma Tor Vergata, ItalyReviewed by:
Graziella Madeo, University of Rome Tor Vergata, ItalyFrancesca Morgante, University of Messina, Italy
Kathrin Grundmann, University Hospital of Tuebingen, Germany
Copyright: © 2016 Giladi, Mirelman, Thaler and Orr-Urtreger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nir Giladi, bmlyZ0B0bHZtYy5nb3YuaWw=