 Eliana Baez1
Eliana Baez1 Valentina Echeverria2
Valentina Echeverria2 Ricardo Cabezas1
Ricardo Cabezas1 Marco Ávila-Rodriguez1
Marco Ávila-Rodriguez1 Luis Miguel Garcia-Segura3*
Luis Miguel Garcia-Segura3* George E. Barreto1,4*
George E. Barreto1,4*
- 1Departamento de Nutrición y Bioquimica, Facultad de Ciencias, Pontificia Universidad Javeriana, Bogotá D.C., Colombia
- 2Facultad de Ciencias de la Salud, Universidad San Sebastián, Concepción, Chile
- 3Instituto Cajal, CSIC, Madrid, Spain
- 4Instituto de Ciencias Biomédicas, Universidad Autónoma de Chile, Santiago, Chile
Astrocytes play an important role in physiological, metabolic, and structural functions, and when impaired, they can be involved in various pathologies including Alzheimer, focal ischemic stroke, and traumatic brain injury. These disorders involve an imbalance in the blood flow and nutrients such as glucose and lactate, leading to biochemical and molecular changes that cause neuronal damage, which is followed by loss of cognitive and motor functions. Previous studies have shown that astrocytes are more resilient than neurons during brain insults as a consequence of their more effective antioxidant systems, transporters, and enzymes, which made them less susceptible to excitotoxicity. In addition, astrocytes synthesize and release different protective molecules for neurons, including neuroglobin, a member of the globin family of proteins. After brain injury, neuroglobin expression is induced in astrocytes. Since neuroglobin promotes neuronal survival, its increased expression in astrocytes after brain injury may represent an endogenous neuroprotective mechanism. Here, we review the role of neuroglobin in the central nervous system, its relationship with different pathologies, and the role of different factors that regulate its expression in astrocytes.
Introduction
The brain has about 170 billion cells (1), which consume an average of 516 kcal of energy per day, representing 22% of total energy demand of an organism (2). This energy demand is required to carry out essential functions such as synaptic transmission, uptake and metabolism of neurotransmitters, and maintenance of ion gradients (3). For this reason, it is of pivotal importance to maintain optimal conditions of the intra- and extracellular environment targeting nerve cells needs. However, in diseases such as ischemic and traumatic brain injuries, an energy imbalance induced by the interruption of blood flow leads to metabolic stress, ionic disturbance, and activation of a complex cascade of biochemical and molecular events that can cause neuronal death (4). Moreover, there are other diseases such as hypoglycemia and diabetes, in which a misbalance in glucose levels can trigger brain damage (5, 6). In this context, traumatic brain injury has become a global public health problem, and it is the leading cause of death in individuals under 45 years of age and recurrent in young people, adolescents, and elders (7). Brain trauma induces cognitive and motor dysfunction (8). In a study reported by Quijano et al., cognitive abilities were assessed in subjects who suffered moderate head trauma and a control non-injured group. The results revealed significant differences in orientation, attention, memory, language, reading, and writing abilities (9). Despite the enormous efforts and progress in research, treatment strategies for traumatic brain injury are still limited, and currently, there are no effective treatments against their consequences. It has been described that after injury, the first phase involves focal hematoma and diffuse edema that results in white matter damage. The second phase involves additional pathological cellular and molecular events such as the abnormal release of neurotransmitters, the generation of free radicals, Ca2+-signaling abnormalities, apoptotic factors activation, and mitochondrial dysfunction leading to neuronal damage, neuroinflammation, and brain dysfunction (10). Moreover, changes that occur in this second phase trigger the death of neurons and astrocytic reactivity. Therefore, it is necessary to search for therapeutic alternatives to prevent further neurological damage and restore CNS homeostasis upon injury.
Neurons are usually affected to a greater extent during injuries, since they have less antioxidant mechanisms than astrocytes being more affected by increased excitotoxicity than glial cells (11). Astrocytes have an active and critical role in the nervous system under normal and pathological conditions. During brain injury and neurodegenerative conditions, astrocytes participate in the removal of toxic molecules and promote neuronal survival throughout the release of trophic factors and antioxidant molecules (12). For example, astrocytes produce various antioxidant molecules, such as glutathione transferase (GSH), superoxide synthase (SOD), and catalase, providing further antioxidant protection to neurons (13). Also, astrocytes integrate the blood–brain barrier (BBB) (13, 14), thus providing active support in the formation of neural connections and brain activity (15). These cells are key regulators of neuronal energy by providing them with lactate (16–18). For all these reasons, astrocytes are vital to restore brain function after injury. In different neurodegenerative conditions such as ischemia–reperfusion injury, a key role is played by mitochondria in the generation of reactive oxygen species (ROS), dysfunctional energy metabolism, and mitochondria-induced apoptosis (19). Likewise, the disruption of synaptic regulation by astroglia seems to play an important role in neurodegeneration and brain damage (20).
It is considered that a transient or permanent impairment of astrocytic functions may negatively impact neurons during pathological conditions. For this reason, it is important to expand the knowledge about the neuroprotective mechanisms mediated by astrocytes during brain injury to find alternatives to prevent altered responses affecting neuroprotection and recovery (21). In this context, neuroglobin (Ngb), a protein expressed astrocytes and neurons, of the central nervous system (CNS) and the peripheral nervous system (PNS) (22, 23) has often been linked to neuroprotection in different neuropathological conditions (24, 25) through antioxidant and antiapoptotic mechanisms (26, 27). The expression of Ngb is induced in human astrocytes during brain injury, possibly as a neuroprotective mechanism (28). Interestingly, Ngb is expressed in astrocytes and neurons of whales and seals as a mechanism to withstand long periods of hypoxia (29). However, more research is needed to completely address the importance of Ngb protective mechanisms and its relationship with astrocyte functions. In the present review article, we explore the role of Ngb in the CNS, focusing on astrocytes and its relationship with different pathologies.
Astrocytes and Brain Pathologies
Astrocytes are responsible for glucose uptake and release of lactate to neurons (16, 17, 30), which are involved in memory and cognition, glutamate recycling, and synthesis of antioxidant glutathione (31). Furthermore, astrocytes have a unique cellular structure that allows them to detect any change in the environment and dynamically respond to extracellular changes or metabolic requirements, providing sources of energy from the glucose taken from blood flow (32) or from energy reserves such as glycogen (33). In addition to glutathione, astrocytes have a special antioxidant system that includes glutathione peroxidase, heme oxygenase I, and catalase, which are able to detoxify ROS in the brain (32, 34). Astrocytes are also considered polyfunctional cells because they also contribute to the elimination of glutamate (Glu), the major excitatory neurotransmitter in the CNS (35). Moreover, Bergmann glia from the cerebellum express the EAAT1 and EAAT2 transporters (36). In this respect, the EAAT2 (GLT1) is responsible for 90% of glutamate uptake through the astrocyte endfeet that make direct contact with the synapses (37). However, this mechanism of Glu uptake and transport becomes affected during brain pathologies, and the increased levels of Glu in the extracellular space might induce excitotoxicity and the severity of brain injury (38, 39). Other astrocytic functions include the remodeling of the blood brain barrier (14) and production of growth factors (18, 40, 41), which in turn promote cell repair during episodes of injury. Faced with an insult or injury, astrocytes adopt a reactive metabolic phenotype (16, 17, 42–44). This phenotype has a beneficial effect on the preservation of neural tissue and in the restriction of moderate focal inflammation (17, 45, 46). However, when this response is maintained and generalized, it can become counterproductive because astrocytic efforts are redirected toward defensive and repair tasks at the expense of providing adequate metabolic support to neurons and also by blocking axonal regeneration (47). Despite this controversial harmful role of reactive astrocytes, a recent study indicated that astrocyte scar formation might help axon regeneration by augmenting multiple axon-growth-supporting molecules (48), demonstrating that inhibiting glial scar might reduce axon regrowth and worsen CNS damage.
According to Sofroniew (49–51), reactive astrogliosis covers some key characteristics: (1) molecular, cellular, and functional changes in astrocytes related to the severity of injury in the CNS; (2) changes are regulated by specific context of molecules via inter- and intracellular signaling; and (3) astrogliosis can exert an alteration in normal astrocytic activities, which in turn can lead to positive or harmful effects in surrounding cells. Additionally, astrocyte gap junctions can remain open after brain injury (52), allowing the entrance of pro-apoptotic factors and immune cells that exacerbate cellular injury (53).
Astrocytes, as other CNS cells, are affected by decreased levels of ATP. This decrease in ATP levels is associated with two fundamental aspects: (i) decreased cerebral blood flow to the range of 100 g−1 (5–8.5 ml min−1), which leads to irreversible tissue damage by the small amount of glucose and oxygen available (8), and (ii) increased intracellular calcium that leads to damaging calcium levels in mitochondria (54). These facts suggest the importance of astrocytic protection as a potential therapeutic target for neuroprotection and preservation of CNS functions following injury.
Mitochondrial Function and Dysfunction in Astrocytes
Mitochondria are essential organelles to sustain life and the physiological function of cells under normal conditions by maintaining energy balance through substrate oxidation, modulation of calcium levels, and redox balance (13, 30, 55). However, these organelles are also the main target of oxidative stress (30) by an imbalance between the production of oxidative molecules, such as hydrogen peroxide (H2O2), superoxide radical (), and the hydroxyl radical (OH), and the ability of the cell to defend against these radicals (56). Because there is a close relationship between mitochondrial dysfunction and brain injury, mitochondrial protection has become a main therapeutic strategy for treating multiple neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis, among others (13, 30, 40, 57–59).
As reviewed by Kubik and Philbert (60), from a total of 12,614 mitochondrial investigations in cells of the nervous system, only 1,214 were directed to astrocytic mitochondria. This fact overshadows that mitochondria in astrocytes provide the metabolic substrates necessary for neural function and are essential to maintain the energetic balance of the brain and the production of antioxidants (61, 62). For example, according to Voloboueva et al. (63), the inhibition of mitochondria during glucose deprivation conditions induces functional changes in astrocytes related to a decrease in ATP levels, depolarization of the plasma membrane, and reduced glutamate uptake, without a significant loss of their viability. Therefore, this evidence strongly suggests that the damage to the astrocytic mitochondrial function may be the start of brain lesions and neuronal death (61).
Studies in animal models of PD showed that the administration of 1 and 10 μg of either vascular endothelial growth factor (VEGF) or glial-derived neurotrophic factor (GDNF) increased the expression of mitochondrial genes (64), suggesting that these growth factors may have a role in mitochondrial protection. Similarly, in vitro administration of platelet-derived growth factor BB (PDGF-BB) preserved mitochondrial function in astrocytes treated with rotenone (40). Furthermore, another study reported that the transmembrane protein TrkB (a receptor for BDNF) was co-localized with mitochondria in astrocytes (63), suggesting that astrocytes’ mitochondria have the potential to directly interact with neurotrophic factors and other protective proteins such as Ngb (65, 66). Finally, other substances studied in mitochondrial protection are CoQ10 (Coenzyme Q10) and conditioned medium from mesenchymal stem cells (67–69). CoQ10 is a ubiquinone with multiple functions such as decreasing the production of ROS, stabilizing mitochondrial membrane potential, improving mitochondrial respiration, inhibiting mitochondria-mediated pathway of cell death, and activating the mitochondrial biogenesis (69). On the other hand, conditioned medium from mesenchymal stem cells has been shown as a protective substance, which contains proangiogenic and antiapoptotic factors, immunomodulators, antioxidants, and neuronal differentiation factors among others, that improve the mitochondrial protection during injuries (68). However, further studies are necessary in order to find new methodologies for the protection of astrocytic mitochondria.
Neuroglobin in Brain Pathologies
Oxygen depletion is one of the more detrimental conditions for the CNS, inducing irreversible damage and as a result loss of cognitive functions. Oxygen depletion is underlying several CNS diseases such as ischemia or TBI.
As stated above, aquatic mammals such as whales and seals (29) withstand conditions of severe hypoxia without damage; they are unique models to investigate neuroprotective mechanisms. The comparison of Ngb protein sequence between terrestrial and aquatic mammals revealed minor differences in its sequence of only two or three amino acids, which did not give rise to confer functional differences between both groups. However, Ngb mRNA expression levels were 4–15 times higher in the brain of seals and whales than in those from terrestrial mammals, suggesting that higher Ngb levels in aquatic mammals can be a neuroprotective mechanism against brain hypoxia and ROS production (70). Similarly, in a behavioral study in transgenic mice overexpressing Ngb under normoxia and hypoxia, it was shown that Ngb promotes survival in vivo and may play an important role in countering the adverse effects of a hypoxic ischemic stroke. No significant behavioral differences were detected between control and Ngb overexpressing mice at 3 months of age, but transgenic mice showed a superior behavioral performance than control mice at 1 year of age (71).
Traumatic brain injury is another major pathology of the brain, which affects world population. Basic features include bleeding, cell death, increased production of β-amyloid, basic fibroblast growth factor (FGF-2), and increased expression of Ngb that remains upregulated until the sixth day post-injury (72). In this respect, other studies have demonstrated an increased expression of Ngb with stroke, hypoxia, and ischemia (73, 74). In an experimental model of TBI, overexpression of Ngb correlated with a significant reduction in sensorimotor deficits compared with a control group that did not overexpressed Ngb. The immunohistochemical analysis of injured cortex and hippocampus revealed that Ngb is mainly expressed in neurons and glial cells (75).
Human studies have correlated the genetic polymorphisms of the Ngb with susceptibility to neurodegeneration. One of these studies showed that decreased expression of Ngb in the elderly is associated with an increased risk of AD (76). In a preclinical study using transgenic AD mice, it was found that intracerebroventricular injection of Ngb decreased the formation of Aβ peptides, and the mitochondrial dysfunction, apoptosis, and neuronal death in the AD brains. In addition, other studies suggested that the neuroprotective effects of Ngb involved the inhibition of caspase-3 and 9, the activation of the PI3K/Akt pathway (77), and the removal of proteins aggregates (78). Finally, other study has demonstrated that Ngb is related to the neurotoxic effect produced by CNS 1-bromopropane (1-BP), a volatile organic compound implicated in damage to both the ozone layer of the atmosphere and the CNS. This compound is used as cleaning agent for metal, electronics, and optical instruments, as well as a substrate for the synthesis of pharmaceuticals and insecticides (79). The frequent use of this compound may cause health problems in mammals. For example, a previous study showed that exposure to 1-BP resulted in cognitive deficits and increased levels of 4-HNE and MDA modified proteins in rats (80). Also, rats exposed to 1-BP showed elongated astrocytic processes, a decreased number of oligodendrocytes, suggesting possible negative effects on myelination and degeneration of granular and Purkinje cells in the cerebellum (81). Similarly, a previous study showed that occupational exposure to 1-BP resulted in CNS adverse effects and peripheral neuropathy (82). Some evidence suggests that Ngb dysfunction may be involved with the toxic effects of 1-BP on CNS. For example, 1-BP correlated in a dose-dependent manner with a decrease in Ngb, cognitive dysfunction in rats, and a significant loss of neurons in layer V of the prelimbic cortex. These results suggest that the decreased expression of Ngb probably plays an important role in CNS neurotoxicity induced by 1-BP (83).
It is important to highlight that the presence of Ngb in vivo or in vitro depends on specific astrocytes (84) and the brain region affected. For example, in one study, Ngb-positive astrocytes were mostly found in the rhinencephalon region severely damaged in terms of hemorrhage (85, 86). However, Ngb was not detected in astrocytes from healthy mouse brains, suggesting that Ngb may have cytoprotective properties with the potential to be a therapeutic agent for intervention. However, the potential neuroprotective effect of Ngb in astrocytes (28) during ischemia (84) has neither been characterized nor the role of Ngb in neurogenesis and glial scar (28). In addition, whether Ngb is secreted by astrocytes as a neuroprotective agent has not been explored, and it requires further investigations.
Neuroprotective Potential of Neuroglobin
Until recently, hemoglobin (Hb) was the most studied member of the globin family of proteins because of its oxygen-binding affinity in blood. Nevertheless, recent studies have revealed expression of other globin proteins in erythrocytes of vertebrates, including myoglobin (Mb), cytoglobin (CYGB), globin E (GbE), globin Y (GBY), and Ngb (87). Ngb is a 17-kDa monomeric protein, which shows a classic folded structure adapted to hold the heme-hexa-Fe-type HisF8 HisE7 in both the ferric and ferrous forms (26). According to homology studies, it was found that Ngb sequence is highly conserved between species, accounting for almost 76% of sequence conservation between humans and amphibians (88). Ngb is not only expressed in the nervous system (22) but also in the eyes, intestine, and ovary; however, no expression has been detected in kidney liver, heart, and skeletal muscle (89). In the brain, Ngb has been found in different regions including the cortex, thalamus, cerebellum, hippocampus, and hypothalamus (90). These areas are important in the processing of sensations, memory, and learning, and are often affected in hypoxic and ischemic insult or traumatic injuries.
Ngb, with its molecular properties, has been characterized as a protein responsible for O2 transport and scavenging of ROS and as O2-sensor and oxygen transporter (91). These functions suggest that the presence of Ngb is a key factor to brain homeostasis. At present, there are several studies addressing the role of Ngb in different pathologies such as focal ischemia and hypoxic–ischemic injuries (92–95). These studies suggest that Ngb serves as a sensor to hypoxic stress and has a protective effect. For example, Tiso et al. (96) reported that a nitrite reductase activity of Ngb inhibited mitochondrial respiration in presence of nitrite in vitro and suppressed oxygen consumption and ROS production. Current studies have shown a direct functional relationship between mitochondrial integrity and Ngb in vivo (74). For example, Ngb is associated with reduced oxidative damage induced by either reactive nitrogen species (RNS) or ROS (97). Moreover, Ngb structure was found to be extremely stable, in which its holoprotein was able to support temperatures exceeding 100°C and low pH values of up to 2.0 before denaturation (98). Finally, this protein may be involved in enhancing G-protein signal transduction by inhibiting the dissociation of guanosine diphosphate from the G-α subunit (73, 99, 100) and therefore involved in cell signaling processes (66).
Initially, Ngb was considered to be exclusively a cytoplasmic protein (101), but recent confocal microscopy studies have shown that it is also located in mitochondria and nucleus (102, 103). In this respect, it was shown that Ngb is associated with microdomains of lipid rafts and α-subunits of heterotrimeric protein G and becomes activated during oxidative stress, undergoing structural changes that lead to neuroprotection (103). On the other hand, the mitochondrial expression of Ngb becomes increased after oxygen-glucose deprivation (OGD) in primary cultures of mouse cortical neurons (104, 105). Furthermore, mitochondrial Ngb has interactions with cytochrome c and the voltage-dependent anion channel (VDAC), suggesting the importance of Ngb in mitochondrial function and neuroprotection (73, 106, 107). Additionally, it has been shown the influence of thyroid hormones on Ngb expression (97, 108). In one of these studies, the authors evaluated Ngb expression in different areas of the rat brain after T3 (100 L/100 g) administration. The authors found that T3 increased Ngb expression in the hippocampus and cerebellum; however, in cerebellum, Ngb expression was only detected at 120 min, 6 h, 12 h, and 24 h after T3 injection (97).
Other studies have shown that Ngb promoter is regulated by the transcription factors NFκB, SP1, and CREB (109), the hypoxia-inducible factor 1-α (HIF-1α), erythropoietin (EPO), and the VEGF (84, 110). In this respect, it was observed that VEGF2 increased the expression of Ngb by stimulating the Flk1 receptor, which in turn induced the expression of HIF-α (111).
The mechanisms of neuroprotection by Ngb have not been completely elucidated. Various stimuli can affect the expression of Ngb in different tissues, including the CNS. For example, in the cardiac tissue, which is also affected by ischemia, oxidative stress, and reperfusion, Ngb has been involved in the protection against cardiac hypertrophy induced by oxidation in cardiomyocytes, preventing them from cell death by ROS and therefore can be a clinical candidate for the treatment of heart diseases (112). On the other hand, it has been shown that cochlear oxidative stress is the main cause of sensorineural hearing loss and for which so far there is no treatment. In this aspect, Ngb is highly expressed in the cochlear nuclei and the superior olivary complex (SOC). Moreover, it was reported that Ngb is colocalized with the antioxidant neuronal protein nitric oxide synthase (NOS) in the SOC, suggesting the importance of Ngb in oxygen homeostasis and energetic metabolism in the auditory nervous system (113) (see Table 1). Ngb has been also involved in calcium homeostasis (27), ATP storage, inhibition of actin assembly, and response against increased hydrogen peroxide ion levels. This evidence suggests that Ngb is also involved in the maintenance of the integrity of the cytoskeleton, cell viability, neuroprotection, and glutamate removal (114).
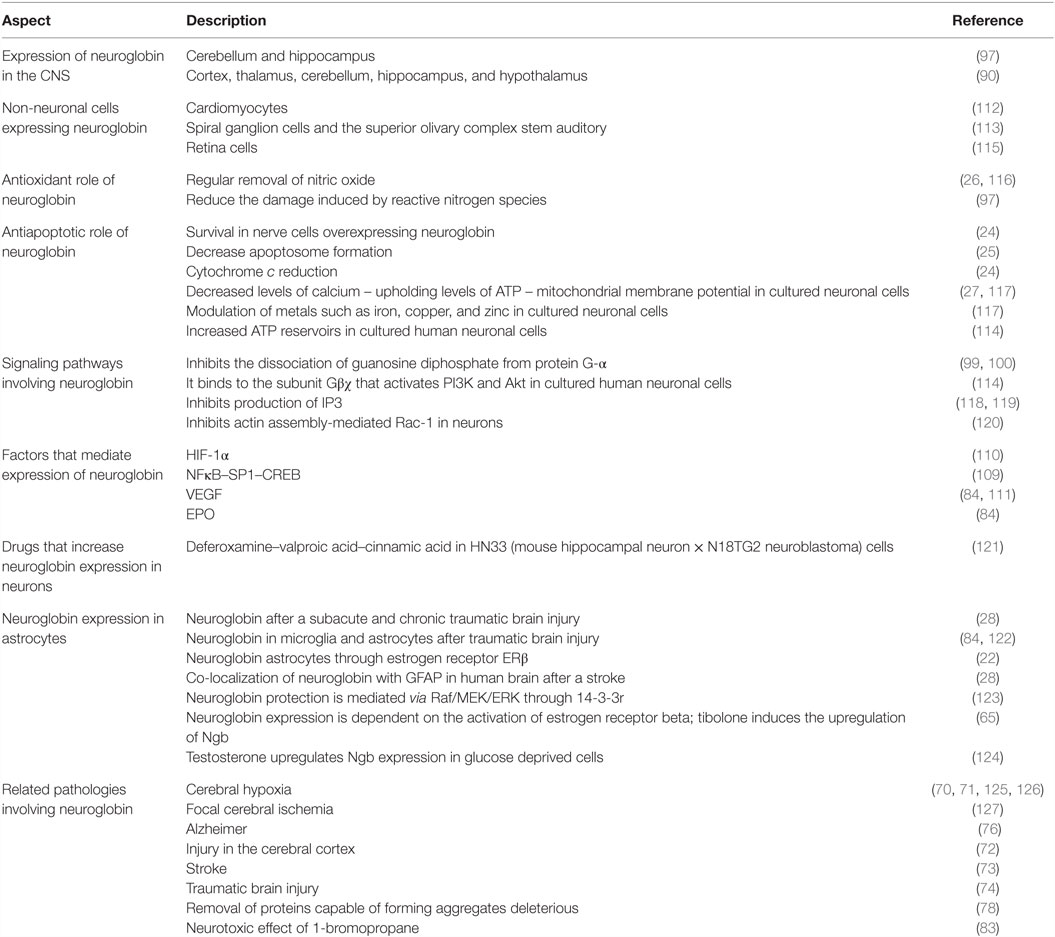
Table 1. Description of the fundamental aspects and biological effects of neuroglobin.
Recent studies have used different drugs to increase Ngb expression in neurons as a therapeutic neuroprotective agent in traumatic brain injury. Among the drugs studied are deferoxamine, an iron chelator, valproic acid, and cinnamic acid (121). Further in vivo studies are needed in order to determine both the induction levels of Ngb by these drugs and also if they have adverse effects in the sensorimotor or cognitive recovery after traumatic brain injury or brain ischemic injury.
Antioxidant Effect
Ngb has very little affinity for oxygen, and the oxygenated species formed with Ngb are unstable; thus, it does not provide a stable source of oxygen, due to the low concentration of Ngb in neurons (128). In fact, it is recognized that one of the roles of Ngb is basically its affinity for NO (26, 129), and this action can be related to the clearance of this gaseous ligand. Moreover, Ngb has been shown to act as a ROS and RNS scavenger in different models (84, 112, 122), although its antioxidant activity is lower than that of N-acetyl cysteine, glutathione, and vitamin C (115). Finally, Ngb has interactions with many antioxidant-related proteins such as Cyt c, Thio, AIF, Prdx3, 4, and 6, Thop1, and Dj1, among others. However, further research is needed in order to address the relevance of the antioxidant effects of Ngb in CNS diseases.
Antiapoptotic Effect of Ngb
The process of apoptosis is complex, and few have investigated the action of Ngb. However, some studies used computational modeling to determine the mechanism of Ngb in cell death. For example, data obtained using computational modeling suggest that Ngb reduces the formation of the apoptosome by a redox reaction with cytochrome c (24), causing the blocking of initiator pro-caspase 9 activation and thus significantly blocking the triggering of apoptosis. In the same study, the authors simulated the interaction between Ngb and cytochrome c and validated their results using an in vitro approach. The authors found a very rapid reaction between reduced (ferrous) neuroglobin and oxidized (ferric) cytochrome c (24), suggesting that Ngb might affect the initiation of apoptosis by interacting with cytochrome c. Interestingly, under normal conditions, these molecules do not interact with each other, but under stress, Ngb prevents cytochrome c release from mitochondria, thus protecting the cell from apoptosis (115). In this respect, it was reported that after reducing cytochrome c, Ngb, in the ferric form, binds to two receptors coupled to G protein subunits (GPCR). This fact is interesting, as the G-α subunit can cause an inhibition in the production of IP3 (inosine triphosphate) and reduce cytosolic calcium release (118, 119). On the other hand, Ngb binds to the Gβχ subunit, activating PI3K and Akt and thus promoting cell viability (114). Also, as mentioned before, Ngb inhibits Rac-1, Pak1 kinase, and actin assembly, preventing cytoskeletal rearrangement and avoiding the initiation of death signaling (120). Moreover, Ngb expression has been found to be higher in metabolically active cells, such as neurons and retinal cells, which have some features in common such as high cytosolic calcium levels, which can trigger apoptosis (115). This aspect has already been validated experimentally, showing that increased Ngb expression is directly linked to calcium homeostasis and maintenance of both mitochondrial membrane potential and ATP levels (27). Ngb may be also important in the modulation of metallic ions such as Fe, Cu, and Zn, which are increased in neurons under hypoxic conditions. These ions can induce inflammation, mitochondrial damage, ROS production, and the release of neurotransmitters, leading to neuronal death excitotoxicity (117). All these findings support the role of Ngb in apoptotic regulation, a subject that merits further research.
Neuroglobin Expression in Astrocytes
The expression of Ngb is evident in neurons and the protective role of Ngb in neuronal cells has been well documented; however, the function of Ngb in astrocytes is less well studied (84). In 2000, a study reinforced the expression of Ngb mRNA in spinal cord funicles, hippocampal alveus, and cerebellar medulla of rodents (101). Until recently, only few studies have reported the expression and function of Ngb in astrocytes (28, 84, 122, 130). Previously, in 2005, a study reported that Ngb mRNA was detected in primary cultures of cortical astrocytes and transfection of these astrocytes with anti-sense for Ngb led to a 2.5-fold increase in apoptotic cells when compared to controls, suggesting a possible protective role of Ngb expression in astrocytes against insult (131).
Consistently, Avivi et al. reported that the subterranean mole rat (Spalax) expresses Ngb in neurons and astrocytes isolated from the corpus callosum (125). More recently, Lechauve et al. reported that Ngb was detected in astrocytes processes optic nerve under physiological conditions in vivo (132). Indeed, it is important to mention that Ngb expression has been also observed in astrocytes under pathological conditions (e.g., reactive astrocytes). For example, the expression of Ngb was found upregulated in reactive astrocytes located in the proximity of a penetrating cortical injury in vivo (22, 133), in Müller cells during reactive gliosis (132) or located in regions associated with the most severe pathology and the astroglial scar in murine models (134). Moreover, Ngb was also detected in astrocytic tumors such as rat astrocytoma cells (C6) and human astrocytoma cells (U251) (135, 136), thus confirming the existence of Ngb in tumoral cell lines.
A previous study evaluated Ngb expression in astrocytes after brain trauma and reported that Ngb expression is present in subacute and chronic injuries but not acute trauma (28). Moreover, in other studies, it was found that Ngb is expressed in microglia and astrocytes specifically during conditions such as traumatic brain injury (84, 122) and that estradiol regulates the expression of Ngb in astrocytes (66) through the estrogen receptor β (ERβ) (22), while ERα is involved in the regulation of Ngb in neurons (137). Interestingly, tibolone, a synthetic hormone with estrogenic, progestogenic, and androgenic actions, has also been reported to induce Ngb expression in astrocytic-like cells in vitro under glucose deprivation (65) (Figure 1). This expression was dependent on the activation of ERβ. Moreover, inhibition of Ngb by siRNA significantly affected the protective effects of tibolone in glucose-deprived cells, suggesting that its actions might be mediated by ERβ and Ngb upregulation (65). Similarly, testosterone also induced the expression of Ngb in astrocytes subjected to glucose deprivation, indicating that estrogenic and androgenic compounds might play a protective role via induction of Ngb (124). Furthermore, a previous report (130) showed co-localization of Ngb and GFAP in glia from human brains after stroke (28). It has been postulated that neuroprotection by Ngb in astrocytes is mediated by Raf/MEK/ERK pathway through 14-3-3r, which has the ability to bind to multiple signaling proteins as kinases, phosphatases, or transmembrane receptors (123). Controversially, Ngb has not been detected in astrocytes by conventional immunohistochemistry or fluorescent immunostaining in normal mice brains (134). Indeed, a strong correlation between the cellular expression of Ngb and the neuronal marker NeuN, but not the astroglial marker GFAP, has been found (130). These results indicate that is debatable whether Ngb is expressed in astrocytes and others glial cells under non-pathological conditions (84). Nevertheless, Ngb is detected in hypoxic mice brains (138), brain tissues in stroke patients, and specialized glial cells such as pituicytes (101). Therefore, it is clear that further studies are necessary in order to determine the role of Ngb in astrocyte-mediated neuroprotection.
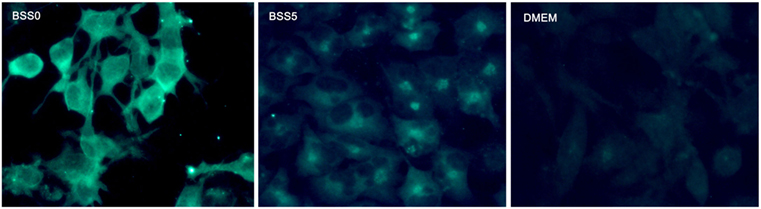
Figure 1. Representative microphotographs of astrocyte-like cells (T98G cell line) expressing neuroglobin. Data generated in our group showed that in cells subjected to metabolic insult by adding a balanced salt solution devoid of glucose (BSS0), neuroglobin expression is enhanced and homogeneously distributed in the cytoplasm (left). The control condition (BSS5) was the same as BSS0, but adding 5-mM glucose; in this case, neuroglobin expression was decreased in comparison with BSS0 and located in proximity of the cell nucleus (center). Under basal culture conditions with DMEM medium, Ngb expression was similar to that of BSS5.
Conclusion and Perspectives
The studies reviewed here show that Ngb is a molecule with antioxidant and antiapoptotic properties acting on mitochondrial and cytosol mechanism of pathology. Therefore, Ngb can be considered as a potential target to decrease neural damage, and its enhanced expression after brain injury probably reflects endogenous mechanisms of neuroprotection. Ngb is upregulated by pharmacological compounds, such as estrogenic molecules. Overexpressing Ngb by gene therapy or its pharmacological induction may represent a potential therapeutic approach for the treatment of traumatic brain injury.
Author Contributions
EB, GEB, VE, and LG-S wrote the manuscript; RC revised the manuscript; and MA-R provided the figure.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
GB’s work is supported by Pontificia Universidad Javeriana.
References
1. Azevedo FAC, Carvalho LRB, Grinberg LT, Farfel JM, Ferretti REL, Leite REP, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol (2009) 513:532–41. doi:10.1002/cne.21974
2. Carmody RN, Wrangham RW. The energetic significance of cooking. J Hum Evol (2009) 57:379–91. doi:10.1016/j.jhevol.2009.02.011
3. Magistretti P, Ransom B. Astrocytes. In: Davis KL, Charney D, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: The Fifth Generation of Progress. American College of Neuropsychopharmacology (2002).
4. Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab (2004) 24:133–50. doi:10.1097/01.WCB.0000111614.19196.04
5. Bree AJ, Puente EC, Daphna-Iken D, Fisher SJ. Diabetes increases brain damage caused by severe hypoglycemia. Am J Physiol Endocrinol Metab (2009) 297:E194–201. doi:10.1152/ajpendo.91041.2008
6. Zhou D, Qian J, Liu CX, Chang H, Sun RP. Repetitive and profound insulin-induced hypoglycemia results in brain damage in newborn rats: an approach to establish an animal model of brain injury induced by neonatal hypoglycemia. Eur J Pediatr (2008) 167:1169–74. doi:10.1007/s00431-007-0653-2
7. Bruns J, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia (2003) 44:2–10. doi:10.1046/j.1528-1157.44.s10.3.x
8. Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth (2007) 99:4–9. doi:10.1093/bja/aem131
9. Quijano MC, Arango JC, Cuervo MT, Aponte M. Neuropsicología del trauma craneoencefálico en Cali, Colombia. Rev Cienc Salud (2012) 10:21–31.
10. Maas AIR, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol (2008) 7:728–41. doi:10.1016/S1474-4422(08)70164-9
11. Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med (2004) 4:193–205. doi:10.2174/1566524043479185
12. Rappold PM, Tieu K. Astrocytes and therapeutics for Parkinson’s disease. Neurotherapeutics (2010) 7:413–23. doi:10.1016/j.nurt.2010.07.001
13. Cabezas R, Avila M, Gonzalez J, El-Bacha RS, Baez E, Garcia-Segura LM, et al. Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci (2014) 8:211. doi:10.3389/fncel.2014.00211
14. Posada-Duque RA, Barreto GE, Cardona-Gomez GP. Protection after stroke: cellular effectors of neurovascular unit integrity. Front Cell Neurosci (2014) 8:231. doi:10.3389/fncel.2014.00231
15. Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci (2003) 26:523–30. doi:10.1016/j.tins.2003.08.008
16. Barreto G, Gonzalez J, Torres Y, Morales L. Astrocytic-neuronal crosstalk: implications for neuroprotection from brain injury. Neurosci Res (2011) 71:107–13. doi:10.1016/j.neures.2011.06.004
17. Barreto G, White RE, Ouyang Y, Xu L, Giffard RG. Astrocytes: targets for neuroprotection in stroke. Cent Nerv Syst Agents Med Chem (2011) 11:164–73. doi:10.2174/187152411796011303
18. Cabezas R, Avila-Rodriguez M, Vega-Vela NE, Echeverria V, Gonzalez J, Hidalgo OA, et al. Growth factors and astrocytes metabolism: possible roles for platelet derived growth factor. Med Chem (2016) 12:204–10. doi:10.2174/1573406411666151019120444
19. Hu Y, Deng H, Xu S, Zhang J. MicroRNAs regulate mitochondrial function in cerebral ischemia-reperfusion injury. Int J Mol Sci (2015) 16:24895–917. doi:10.3390/ijms161024895
20. Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol (2006) 2:679–89. doi:10.1038/ncpneuro0355
21. Gouix E, Buisson A, Nieoullon A, Kerkerian-Le Goff L, Tauskela JS, Blondeau N, et al. Oxygen glucose deprivation-induced astrocyte dysfunction provokes neuronal death through oxidative stress. Pharmacol Res (2014) 87:8–17. doi:10.1016/j.phrs.2014.06.002
22. De Marinis E, Fiocchetti M, Acconcia F, Ascenzi P, Marino M. Neuroglobin upregulation induced by 17β-estradiol sequesters cytocrome c in the mitochondria preventing H2O2-induced apoptosis of neuroblastoma cells. Cell Death Dis (2013) 4:e508–508. doi:10.1038/cddis.2013.30
23. De Marinis E, Ascenzi P, Pellegrini M, Galluzzo P, Bulzomi P, Arevalo MA, et al. 17beta-estradiol – a new modulator of neuroglobin levels in neurons: role in neuroprotection against H(2)O(2)-induced toxicity. Neurosignals (2010) 18:223–35. doi:10.1159/000323906
24. Raychaudhuri S, Skommer J, Henty K, Birch N, Brittain T. Neuroglobin protects nerve cells from apoptosis by inhibiting the intrinsic pathway of cell death. Apoptosis (2010) 15:401–11. doi:10.1007/s10495-009-0436-5
25. Raychaudhuri S, Willgohs E, Nguyen T-N, Khan EM, Goldkorn T. Monte Carlo simulation of cell death signaling predicts large cell-to-cell stochastic fluctuations through the type 2 pathway of apoptosis. Biophys J (2008) 95:3559–62. doi:10.1529/biophysj.108.135483
26. Lee VY, McClintock DS, Santore MT, Budinger GR, Chandel NS. Hypoxia sensitizes cells to nitric oxide-induced apoptosis. J Biol Chem (2002) 277:16067–74. doi:10.1074/jbc.M111177200
27. Liu J, Yu Z, Guo S, Lee S-R, Xing C, Zhang C, et al. Effects of neuroglobin overexpression on mitochondrial function and oxidative stress following hypoxia/reoxygenation in cultured neurons. J Neurosci Res (2009) 87:164–70. doi:10.1002/jnr.21826
28. Chen X, Liu Y, Zhang L, Zhu P, Zhu H, Yang Y, et al. Long-term neuroglobin expression of human astrocytes following brain trauma. Neurosci Lett (2015) 606:194–9. doi:10.1016/j.neulet.2015.09.002
29. Meir JU, Ponganis PJ. High-affinity hemoglobin and blood oxygen saturation in diving emperor penguins. J Exp Biol (2009) 212:3330–8. doi:10.1242/jeb.033761
30. Cabezas R, El-Bachá R, González J, Barreto G. Mitochondrial functions in astrocytes: neuroprotective implications from oxidative damage by rotenone. Neurosci Res (2012) 74:80–90. doi:10.1016/j.neures.2012.07.008
31. Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem (2003) 384:505–16. doi:10.1515/BC.2003.059
32. Bélanger M, Magistretti PJ. The role of astroglia in neuroprotection. Dialogues Clin Neurosci (2009) 11:281–95.
33. Pellerin L, Magistretti PJ. Sweet sixteen for ANLS. J Cereb Blood Flow Metab (2012) 32:1152–66. doi:10.1038/jcbfm.2011.149
34. Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci (2003) 23:3394–406.
35. Lee M-C, Yasuda R, Ehlers MD. Metaplasticity at single glutamatergic synapses. Neuron (2010) 66:859–70. doi:10.1016/j.neuron.2010.05.015
36. Medina-Ceja L, Guerrero-cazares H, Canales-aguirre A, Morales-Villagrán A, Feria-Velasco A. Características estructurales y funcionales de los transportadores de glutamato: su relación con la epilepsia y el estrés oxidativo. Rev Neurol (2007) 45:341–52.
37. Haugeto O, Ullensvang K, Levy LM, Chaudhry FA, Honore T, Nielsen M, et al. Brain glutamate transporter proteins form homomultimers. J Biol Chem (1996) 271:27715–22. doi:10.1074/jbc.271.44.27715
38. Yi J-H, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int (2006) 48:394–403. doi:10.1016/j.neuint.2005.12.001
39. Zou J, Wang Y-X, Dou F-F, Lü H-Z, Ma Z-W, Lu P-H, et al. Glutamine synthetase down-regulation reduces astrocyte protection against glutamate excitotoxicity to neurons. Neurochem Int (2010) 56:577–84. doi:10.1016/j.neuint.2009.12.021
40. Cabezas R, Avila MF, Gonzalez J, El-Bacha RS, Barreto GE. PDGF-BB protects mitochondria from rotenone in T98G cells. Neurotox Res (2015) 27:355–67. doi:10.1007/s12640-014-9509-5
41. Chen Y, Swanson RA. Astrocytes and brain injury. J Cereb Blood Flow Metab (2003) 23:137–49. doi:10.1097/01.WCB.0000044631.80210.3C
42. Barreto GE, Santos-Galindo M, Garcia-Segura LM. Selective estrogen receptor modulators regulate reactive microglia after penetrating brain injury. Front Aging Neurosci (2014) 6:132. doi:10.3389/fnagi.2014.00132
43. Barreto GE, Gonzalez J, Capani F, Morales L. Neuroprotective agents in brain injury: a partial failure? Int J Neurosci (2012) 122:223–6. doi:10.3109/00207454.2011.648292
44. Iglesias J, Morales L, Barreto GE. Metabolic and inflammatory adaptation of reactive astrocytes: role of PPARs. Mol Neurobiol (2016). doi:10.1007/s12035-016-9833-2
45. Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain (2006) 129:2761–72. doi:10.1093/brain/awl165
46. Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci (2015) 16:249–63. doi:10.1038/nrn3898
47. Steele ML, Robinson SR. Reactive astrocytes give neurons less support: implications for Alzheimer’s disease. Neurobiol Aging (2012) 33:423.e1–e13. doi:10.1016/j.neurobiolaging.2010.09.018
48. Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, et al. Astrocyte scar formation aids central nervous system axon regeneration. Nature (2016) 532:195–200. doi:10.1038/nature17623
49. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol (2010) 119:7–35. doi:10.1007/s00401-009-0619-8
50. Pekny M, Pekna M, Messing A, Steinhauser C, Lee JM, Parpura V, et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol (2016) 131:323–45. doi:10.1007/s00401-015-1513-1
51. Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol (2015) 7:a020420. doi:10.1101/cshperspect.a020420
52. Ohsumi A, Nawashiro H, Otani N, Ooigawa H, Toyooka T, Shima K. Temporal and spatial profile of phosphorylated Connexin43 after traumatic brain injury in rats. J Neurotrauma (2010) 27:1255–63. doi:10.1089/neu.2009.1234
53. Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, et al. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci (1998) 1:494–500. doi:10.1038/2210
54. Prins M, Greco T, Alexander D, Giza CC. The pathophysiology of traumatic brain injury at a glance. Dis Model Mech (2013) 6:1307–15. doi:10.1242/dmm.011585
55. Duchen MR, Szabadkai G. Roles of mitochondria in human disease: Figure 1. Essays Biochem (2010) 47:115–37. doi:10.1042/bse0470115
56. LeDoux SP, Druzhyna NM, Hollensworth SB, Harrison JF, Wilson GL. Mitochondrial DNA repair: a critical player in the response of cells of the CNS to genotoxic insults. Neuroscience (2007) 145:1249–59. doi:10.1016/j.neuroscience.2006.10.002
57. Avila Rodriguez M, Garcia-Segura LM, Cabezas R, Torrente D, Capani F, Gonzalez J, et al. Tibolone protects T98G cells from glucose deprivation. J Steroid Biochem Mol Biol (2014) 144(Pt B):294–303. doi:10.1016/j.jsbmb.2014.07.009
58. Jurado-Coronel JC, Avila-Rodriguez M, Capani F, Gonzalez J, Moran VE, Barreto GE. Targeting the nicotinic acetylcholine receptors (nAChRs) in astrocytes as a potential therapeutic target in Parkinson’s disease. Curr Pharm Des (2016) 22:1305–11. doi:10.2174/138161282210160304112133
59. Patel VP, Chu CT. Nuclear transport, oxidative stress, and neurodegeneration. Int J Clin Exp Pathol (2011) 4:215–29.
60. Kubik LL, Philbert MA. The role of astrocyte mitochondria in differential regional susceptibility to environmental neurotoxicants: tools for understanding neurodegeneration. Toxicol Sci (2015) 144:7–16. doi:10.1093/toxsci/kfu254
61. Dugan LL, Kim-Han J-S. Astrocyte mitochondria in in vitro models of ischemia. J Bioenerg Biomembr (2004) 36:317–21. doi:10.1023/B:JOBB.0000041761.61554.44
62. Greenamyre JT, Betarbet R, Sherer TB. The rotenone model of Parkinson’s disease: genes, environment and mitochondria. Parkinsonism Relat Disord (2003) 9(Suppl 2):S59–64. doi:10.1016/S1353-8020(03)00023-3
63. Voloboueva LA, Suh SW, Swanson RA, Giffard RG. Inhibition of mitochondrial function in astrocytes: implications for neuroprotection. J Neurochem (2007) 102:1383–94. doi:10.1111/j.1471-4159.2007.04634.x
64. Yue X, Hariri DJ, Caballero B, Zhang S, Bartlett MJ, Kaut O, et al. Comparative study of the neurotrophic effects elicited by VEGF-B and GDNF in preclinical in vivo models of Parkinson’s disease. Neuroscience (2014) 258:385–400. doi:10.1016/j.neuroscience.2013.11.038
65. Avila-Rodriguez M, Garcia-Segura LM, Hidalgo-Lanussa O, Baez E, Gonzalez J, Barreto GE. Tibolone protects astrocytic cells from glucose deprivation through a mechanism involving estrogen receptor beta and the upregulation of neuroglobin expression. Mol Cell Endocrinol (2016). doi:10.1016/j.mce.2016.05.024
66. Acaz-Fonseca E, Avila Rodriguez M, Garcia-Segura LM, Barreto GE. Regulation of astroglia by gonadal steroid hormones under physiological and pathological conditions. Prog Neurobiol (2016). doi:10.1016/j.pneurobio.2016.06.002
67. Torrente D, Avila MF, Cabezas R, Morales L, Gonzalez J, Samudio I, et al. Paracrine factors of human mesenchymal stem cells increase wound closure and reduce reactive oxygen species production in a traumatic brain injury in vitro model. Hum Exp Toxicol (2014) 33(7):673–84. doi:10.1177/0960327113509659
68. Cantinieaux D, Quertainmont R, Blacher S, Rossi L, Wanet T, Noël A, et al. Conditioned medium from bone marrow-derived mesenchymal stem cells improves recovery after spinal cord injury in rats: an original strategy to avoid cell transplantation. PLoS One (2013) 8:e69515. doi:10.1371/journal.pone.0069515
69. Jing L, He MT, Chang Y, Mehta SL, He QP, Zhang JZ, et al. Coenzyme Q10 protects astrocytes from ROS-induced damage through inhibition of mitochondria-mediated cell death pathway. Int J Biol Sci (2015) 11:59–66. doi:10.7150/ijbs.10174
70. Schneuer M, Flachsbarth S, Czech-Damal NU, Folkow LP, Siebert U, Burmester T. Neuroglobin of seals and whales: evidence for a divergent role in the diving brain. Neuroscience (2012) 223:35–44. doi:10.1016/j.neuroscience.2012.07.052
71. Van Leuven W, Van Dam D, Moens L, De Deyn PP, Dewilde S. A behavioural study of neuroglobin-overexpressing mice under normoxic and hypoxic conditions. Biochim Biophys Acta (2013) 1834:1764–71. doi:10.1016/j.bbapap.2013.04.015
72. Purushothuman S, Stone J. The reaction of cerebral cortex to a nearby lesion: damage, survival, self-protection. Brain Res (2015) 1601:52–63. doi:10.1016/j.brainres.2015.01.003
73. Yu Z, Liu N, Liu J, Yang K, Wang X. Neuroglobin, a novel target for endogenous neuroprotection against stroke and neurodegenerative disorders. Int J Mol Sci (2012) 13:6995–7014. doi:10.3390/ijms13066995
74. Zhao S, Yu Z, Zhao G, Xing C, Hayakawa K, Whalen MJ, et al. Neuroglobin-overexpression reduces traumatic brain lesion size in mice. BMC Neurosci (2012) 13:67. doi:10.1186/1471-2202-13-67
75. Taylor JM, Kelley B, Gregory EJ, Berman NEJ. Neuroglobin overexpression improves sensorimotor outcomes in a mouse model of traumatic brain injury. Neurosci Lett (2014) 577:125–9. doi:10.1016/j.neulet.2014.03.012
76. Lin Y, Fang L, Xue X-H, Murong S-X, Wang N, Wu Z-Y. Association between Ngb polymorphisms and ischemic stroke in the Southern Chinese Han population. BMC Med Genet (2008) 9:110. doi:10.1186/1471-2350-9-110
77. Li Y, Dai Y-B, Sun J-Y, Xiang Y, Yang J, Dai S-Y, et al. Neuroglobin attenuates beta amyloid-induced apoptosis through inhibiting caspases activity by activating PI3K/Akt signaling pathway. J Mol Neurosci (2016) 58(1):28–38. doi:10.1007/s12031-015-0645-z
78. Lechauve C, Rezaei H, Celier C, Kiger L, Corral-Debrinski M, Noinville S, et al. Neuroglobin and prion cellular localization: investigation of a potential interaction. J Mol Biol (2009) 388:968–77. doi:10.1016/j.jmb.2009.03.047
79. Shelby M, Portier C, Goldman L, Moore J, Iannucci A, Jahnke G, et al. NTP-CERHR Expert Panel report on the reproductive and developmental toxicity of 1-bromopropane. Reprod Toxicol (2004) 18:157–87. doi:10.1016/j.reprotox.2004.01.001
80. Zhong Z, Zeng T, Xie K, Zhang C, Chen J, Bi Y, et al. Elevation of 4-hydroxynonenal and malondialdehyde modified protein levels in cerebral cortex with cognitive dysfunction in rats exposed to 1-bromopropane. Toxicology (2013) 306:16–23. doi:10.1016/j.tox.2013.01.022
81. Mohideen SS, Ichihara S, Subramanian K, Huang Z, Naito H, Kitoh J, et al. Effects of exposure to 1-bromopropane on astrocytes and oligodendrocytes in rat brain. J Occup Health (2013) 55:29–38. doi:10.1539/joh.12-0118-OA
82. Wang TH, Wu ML, Wu YH, Tsai WJ, Lin KP, Wang CL, et al. Neurotoxicity associated with exposure to 1-bromopropane in golf-club cleansing workers. Clin Toxicol (Phila) (2015) 53:823–6. doi:10.3109/15563650.2015.1064939
83. Guo Y, Yuan H, Jiang L, Yang J, Zeng T, Xie K, et al. Involvement of decreased neuroglobin protein level in cognitive dysfunction induced by 1-bromopropane in rats. Brain Res (2015) 1600:1–16. doi:10.1016/j.brainres.2014.12.046
84. Qiu XY, Chen XQ. Neuroglobin – recent developments. Biomol Concepts (2014) 5:195–208. doi:10.1515/bmc-2014-0011
85. Lackner P, Beer R, Heussler V, Goebel G, Rudzki D, Helbok R, et al. Behavioural and histopathological alterations in mice with cerebral malaria. Neuropathol Appl Neurobiol (2006) 32:177–88. doi:10.1111/j.1365-2990.2006.00706.x
86. Wiese L, Kurtzhals JA, Penkowa M. Neuronal apoptosis, metallothionein expression and proinflammatory responses during cerebral malaria in mice. Exp Neurol (2006) 200:216–26. doi:10.1016/j.expneurol.2006.02.011
87. Gotting M, Nikinmaa M. More than hemoglobin – the unexpected diversity of globins in vertebrate red blood cells. Physiol Rep (2015) 3:e12284–12284. doi:10.14814/phy2.12284
88. Hankeln T, Ebner B, Fuchs C, Gerlach F, Haberkamp M, Laufs TL, et al. Neuroglobin and cytoglobin in search of their role in the vertebrate globin family. J Inorg Biochem (2005) 99:110–9. doi:10.1016/j.jinorgbio.2004.11.009
89. Fuchs C, Burmester T, Hankeln T. The amphibian globin gene repertoire as revealed by the Xenopus genome. Cytogenet Genome Res (2006) 112:296–306. doi:10.1159/000089884
90. Wystub S, Laufs T, Schmidt M, Burmester T, Maas U, Saaler-Reinhardt S, et al. Localization of neuroglobin protein in the mouse brain. Neurosci Lett (2003) 346:114–6. doi:10.1016/S0304-3940(03)00563-9
91. Simon R. Neuroglobin: neuroprotection and neurogenesis. Neurosci Lett (2013) 549:1–2. doi:10.1016/j.neulet.2013.05.016
92. Shang A, Yang Y, Wang H, Wang J, Hang X, Wang Z, et al. Upregulation of neuroglobin expression and changes in serum redox indices in a rat model of middle cerebral artery occlusion. Mol Med Rep (2015) 12:1693–8. doi:10.3892/mmr.2015.3593
93. Song X, Xu R, Xie F, Zhu H, Zhu J, Wang X. Hemin offers neuroprotection through inducing exogenous neuroglobin in focal cerebral hypoxic-ischemia in rats. Int J Clin Exp Pathol (2014) 7:2163–71.
94. Raida Z, Hundahl CA, Nyengaard JR, Hay-Schmidt A. Neuroglobin over expressing mice: expression pattern and effect on brain ischemic infarct size. PLoS One (2013) 8:e76565. doi:10.1371/journal.pone.0076565
95. Raida Z, Hundahl CA, Kelsen J, Nyengaard JR, Hay-Schmidt A. Reduced infarct size in neuroglobin-null mice after experimental stroke in vivo. Exp Transl Stroke Med (2012) 4:15. doi:10.1186/2040-7378-4-15
96. Tiso M, Tejero J, Basu S, Azarov I, Wang X, Simplaceanu V, et al. Human neuroglobin functions as a redox-regulated nitrite reductase. J Biol Chem (2011) 286:18277–89. doi:10.1074/jbc.M110.159541
97. Oliveira KC, da Conceição RR, Piedade GC, de Souza JS, Sato MA, de Barros Maciel RM, et al. Thyroid hormone modulates neuroglobin and cytoglobin in rat brain. Metab Brain Dis (2015) 30(6):1401–8. doi:10.1007/s11011-015-9718-5
98. Picotti P, Dewilde S, Fago A, Hundahl C, De Filippis V, Moens L, et al. Unusual stability of human neuroglobin at low pH – molecular mechanisms and biological significance. FEBS J (2009) 276:7027–39. doi:10.1111/j.1742-4658.2009.07416.x
99. Burmester T, Hankeln T. What is the function of neuroglobin? J Exp Biol (2009) 212:1423–8. doi:10.1242/jeb.000729
100. Yu Z, Fan X, Lo EH, Wang X. Neuroprotective roles and mechanisms of neuroglobin. Neurol Res (2009) 31:122–7. doi:10.1179/174313209X389866
101. Reuss S, Saaler-Reinhardt S, Weich B, Wystub S, Reuss MH, Burmester T, et al. Expression analysis of neuroglobin mRNA in rodent tissues. Neuroscience (2002) 115:645–56. doi:10.1016/S0306-4522(02)00536-5
102. Hundahl CA, Allen GC, Hannibal J, Kjaer K, Rehfeld JF, Dewilde S, et al. Anatomical characterization of cytoglobin and neuroglobin mRNA and protein expression in the mouse brain. Brain Res (2010) 1331:58–73. doi:10.1016/j.brainres.2010.03.056
103. Watanabe S, Takahashi N, Uchida H, Wakasugi K. Human neuroglobin functions as an oxidative stress-responsive sensor for neuroprotection. J Biol Chem (2012) 287:30128–38. doi:10.1074/jbc.M112.373381
104. Yu Z, Liu J, Guo S, Xing C, Fan X, Ning M, et al. Neuroglobin-overexpression alters hypoxic response gene expression in primary neuron culture following oxygen glucose deprivation. Neuroscience (2009) 162:396–403. doi:10.1016/j.neuroscience.2009.04.055
105. Yu Z, Liu N, Li Y, Xu J, Wang X. Neuroglobin overexpression inhibits oxygen-glucose deprivation-induced mitochondrial permeability transition pore opening in primary cultured mouse cortical neurons. Neurobiol Dis (2013) 56:95–103. doi:10.1016/j.nbd.2013.04.015
106. Yu Z, Poppe JL, Wang X. Mitochondrial mechanisms of neuroglobin’s neuroprotection. Oxid Med Cell Longev (2013) 2013:756989–756989. doi:10.1155/2013/756989
107. Yu Z, Xu J, Liu N, Wang Y, Li X, Pallast S, et al. Mitochondrial distribution of neuroglobin and its response to oxygen–glucose deprivation in primary-cultured mouse cortical neurons. Neuroscience (2012) 218:235–42. doi:10.1016/j.neuroscience.2012.05.054
108. Sharlin DS, Gilbert ME, Taylor MA, Ferguson DC, Zoeller RT. The nature of the compensatory response to low thyroid hormone in the developing brain. J Neuroendocrinol (2010) 22:153–65. doi:10.1111/j.1365-2826.2009.01947.x
109. Liu N, Yu Z, Li Y, Yuan J, Zhang J, Xiang S, et al. Transcriptional regulation of mouse neuroglobin gene by cyclic AMP responsive element binding protein (CREB) in N2a cells. Neurosci Lett (2013) 534:333–7. doi:10.1016/j.neulet.2012.11.025
110. Woo SK, Kwon MS, Geng Z, Chen Z, Ivanov A, Bhatta S, et al. Sequential activation of hypoxia-inducible factor 1 and specificity protein 1 is required for hypoxia-induced transcriptional stimulation of Abcc8. J Cereb Blood Flow Metab (2012) 32:525–36. doi:10.1038/jcbfm.2011.159
111. Jin K, Mao X, Xie L, Greenberg DA. Interactions between vascular endothelial growth factor and neuroglobin. Neurosci Lett (2012) 519:47–50. doi:10.1016/j.neulet.2012.05.018
112. Liu Z-F, Zhang X, Qiao Y-X, Xu W-Q, Ma C-T, Gu H-L, et al. Neuroglobin protects cardiomyocytes against apoptosis and cardiac hypertrophy induced by isoproterenol in rats. Int J Clin Exp Med (2015) 8:5351–60.
113. Reuss S, Banica O, Elgurt M, Mitz S, Disque-Kaiser U, Riemann R, et al. Neuroglobin expression in the mammalian auditory system. Mol Neurobiol (2015) 53(3):1461–77. doi:10.1007/s12035-014-9082-1
114. Antao ST, Duong TTH, Aran R, Witting PK. Neuroglobin overexpression in cultured human neuronal cells protects against hydrogen peroxide insult via activating phosphoinositide-3 kinase and opening the mitochondrial K ATP channel. Antioxid Redox Signal (2010) 13:769–81. doi:10.1089/ars.2009.2977
115. Brittain T, Skommer J, Henty K, Birch N, Raychaudhuri S. A role for human neuroglobin in apoptosis. IUBMB Life (2010) 62:878–85. doi:10.1002/iub.405
116. Jayaraman T, Tejero J, Chen BB, Blood AB, Frizzell S, Shapiro C, et al. 14-3-3 binding and phosphorylation of neuroglobin during hypoxia modulate six-to-five heme pocket coordination and rate of nitrite reduction to nitric oxide. J Biol Chem (2011) 286:42679–89. doi:10.1074/jbc.M111.271973
117. Duong TTH, Witting PK, Antao ST, Parry SN, Kennerson M, Lai B, et al. Multiple protective activities of neuroglobin in cultured neuronal cells exposed to hypoxia re-oxygenation injury. J Neurochem (2009) 108:1143–54. doi:10.1111/j.1471-4159.2008.05846.x
118. Dewilde S, Kiger L, Burmester T, Hankeln T, Baudin-Creuza V, Aerts T, et al. Biochemical characterization and ligand binding properties of neuroglobin, a novel member of the globin family. J Biol Chem (2001) 276:38949–55. doi:10.1074/jbc.M106438200
119. Fago A, Mathews AJ, Dewilde S, Moens L, Brittain T. The reactions of neuroglobin with CO: evidence for two forms of the ferrous protein. J Inorg Biochem (2006) 100:1339–43. doi:10.1016/j.jinorgbio.2006.03.009
120. Khan AA, Mao XO, Banwait S, DerMardirossian CM, Bokoch GM, Jin K, et al. Regulation of hypoxic neuronal death signaling by neuroglobin. FASEB J (2008) 22:1737–47. doi:10.1096/fj.07-100784
121. Jin K, Mao XO, Xie L, John V, Greenberg DA. Pharmacological induction of neuroglobin expression. Pharmacology (2011) 87:81–4. doi:10.1159/000322998
122. Yu ZL, Qiu S, Chen XC, Dai ZH, Huang YC, Li YN, et al. Neuroglobin – a potential biological marker of retinal damage induced by LED light. Neuroscience (2014) 270:158–67. doi:10.1016/j.neuroscience.2014.04.013
123. Zhao J, Meyerkord CL, Du Y, Khuri FR, Fu H. 14-3-3 proteins as potential therapeutic targets. Semin Cell Dev Biol (2011) 22:705–12. doi:10.1016/j.semcdb.2011.09.012
124. Toro N, Garcia-Segura LM, Echeverria V, Barreto GE. Testosterone protects mitochondrial function and regulates neuroglobin expression in astrocytic cells exposed to glucose deprivation. Front Aging Neurosci (2016) 8:152. doi:10.3389/fnagi.2016.00152
125. Avivi A, Gerlach F, Joel A, Reuss S, Burmester T, Nevo E, et al. Neuroglobin, cytoglobin, and myoglobin contribute to hypoxia adaptation of the subterranean mole rat Spalax. Proc Natl Acad Sci U S A (2010) 107:21570–5. doi:10.1073/pnas.1015379107
126. Greenberg DA, Jin K, Khan AA. Neuroglobin: an endogenous neuroprotectant. Curr Opin Pharmacol (2008) 8:20–4. doi:10.1016/j.coph.2007.09.003
127. Sun Y, Jin K, Peel A, Mao XO, Xie L, Greenberg DA. Neuroglobin protects the brain from experimental stroke in vivo. Proc Natl Acad Sci U S A (2003) 100:3497–500. doi:10.1073/pnas.0637726100
128. Fago A, Hundahl C, Dewilde S, Gilany K, Moens L, Weber RE. Allosteric regulation and temperature dependence of oxygen binding in human neuroglobin and cytoglobin. Molecular mechanisms and physiological significance. J Biol Chem (2004) 279:44417–26. doi:10.1074/jbc.M407126200
129. Brunori M, Giuffrè A, Nienhaus K, Nienhaus GU, Scandurra FM, Vallone B. Neuroglobin, nitric oxide, and oxygen: functional pathways and conformational changes. Proc Natl Acad Sci U S A (2005) 102:8483–8. doi:10.1073/pnas.0408766102
130. Jin K, Mao Y, Mao X, Xie L, Greenberg DA. Neuroglobin expression in ischemic stroke. Stroke (2010) 41:557–9. doi:10.1161/STROKEAHA.109.567149
131. Chen XQ, Qin LY, Zhang CG, Yang LT, Gao Z, Liu S, et al. Presence of neuroglobin in cultured astrocytes. Glia (2005) 50:182–6. doi:10.1002/glia.20147
132. Lechauve C, Augustin S, Roussel D, Sahel JA, Corral-Debrinski M. Neuroglobin involvement in visual pathways through the optic nerve. Biochim Biophys Acta (2013) 1834:1772–8. doi:10.1016/j.bbapap.2013.04.014
133. De Marinis E, Acaz-Fonseca E, Arevalo MA, Ascenzi P, Fiocchetti M, Marino M, et al. 17beta-oestradiol anti-inflammatory effects in primary astrocytes require oestrogen receptor beta-mediated neuroglobin up-regulation. J Neuroendocrinol (2013) 25:260–70. doi:10.1111/jne.12007
134. DellaValle B, Hempel C, Kurtzhals JA, Penkowa M. In vivo expression of neuroglobin in reactive astrocytes during neuropathology in murine models of traumatic brain injury, cerebral malaria, and autoimmune encephalitis. Glia (2010) 58:1220–7. doi:10.1002/glia.21002
135. Gorr TA, Wichmann D, Pilarsky C, Theurillat JP, Fabrizius A, Laufs T, et al. Old proteins – new locations: myoglobin, haemoglobin, neuroglobin and cytoglobin in solid tumours and cancer cells. Acta Physiol (Oxf) (2011) 202:563–81. doi:10.1111/j.1748-1716.2010.02205.x
136. Qin H, Guo Y, Zhang C, Zhang L, Li M, Guan P. The expression of neuroglobin in astrocytoma. Brain Tumor Pathol (2012) 29:10–6. doi:10.1007/s10014-011-0066-9
137. Guglielmotto M, Reineri S, Iannello A, Ferrero G, Vanzan L, Miano V, et al. E2 regulates epigenetic signature on neuroglobin enhancer-promoter in neuronal cells. Front Cell Neurosci (2016) 10:147. doi:10.3389/fncel.2016.00147
Keywords: astrocytes, neuroglobin, mitochondria, neuroprotection, brain injury
Citation: Baez E, Echeverria V, Cabezas R, Ávila-Rodriguez M, Garcia-Segura LM and Barreto GE (2016) Protection by Neuroglobin Expression in Brain Pathologies. Front. Neurol. 7:146. doi: 10.3389/fneur.2016.00146
Received: 09 June 2016; Accepted: 29 August 2016;
Published: 12 September 2016
Edited by:
Firas H. Kobeissy, University of Florida, USAReviewed by:
Bridgette D. Semple, University of Melbourne, AustraliaHala Darwish, American University of Beirut, Lebanon
Copyright: © 2016 Baez, Echeverria, Cabezas, Ávila-Rodriguez, Garcia-Segura and Barreto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis Miguel Garcia-Segura, bG1nc0BjYWphbC5jc2ljLmVz;
George E. Barreto, Z3NhbXBhaW9AamF2ZXJpYW5hLmVkdS5jbw==