 Juanjuan Zhao1
Juanjuan Zhao1 Dongxu Yue1Ya Zhou2Li Jia1Hairong Wang1Mengmeng Guo1Hualin Xu1Chao Chen1Jidong Zhang1
Dongxu Yue1Ya Zhou2Li Jia1Hairong Wang1Mengmeng Guo1Hualin Xu1Chao Chen1Jidong Zhang1 Lin Xu1*
Lin Xu1*
- 1Department of Immunology, Zunyi Medical College, Guizhou, China
- 2Department of Medical Physics, Zunyi Medical College, Guizhou, China
Alzheimer’s disease (AD), with main clinical features of progressive impairment in cognitive and behavioral functions, is the most common degenerative disease of the central nervous system. Recent evidence showed that microRNAs (miRNAs) played important roles in the pathological progression of AD. In this article, we reviewed the promising role of miRNAs in both Aβ deposition and Tau phosphorylation, two key pathological characters in the pathological progression of AD, which might be helpful for the understanding of pathogenesis and the development of new strategies of clinical diagnosis and treatment of AD.
Introduction
It is estimated worldwide that as many as 46.8 million people suffer from Alzheimer’s disease (AD) in 2015. By the year 2050, this number would further exceed 1.315 billion (1). AD is a kind degenerative disease of central nervous system, which is characterized by clinical features such as early stage memory loss, cognitive impairment, and personality change in the early and old age. The main pathological hallmark in brain of AD patients was β-amyloid (amyloid-β, Aβ) deposition-formed extracellular amyloid plaques (senile plaques) and intracellular neurofibrillary tangles, which were mainly composed by the excessive phosphorylation of Tau protein and loss of neurons. Despite the discovery of numerous molecular changes in AD, the pathogenesis of AD is still not entirely clear.
MicroRNAs (miRNAs) are a novel class of short (18–25 nucleotides) and single-stranded non-coding RNAs involved in the posttranscriptional regulation of gene expression. Their mechanism of action is mediated by complement binding to the 3’untranslated region (3’UTR) of mRNA, leading to degradation or translation repression of the target mRNA (2). In recent years, miRNAs have attracted increased research interest and a substantial amount of work has been reported that miRNAs could be used as new targets for diagnosis and treatment of a variety of neurodegenerative diseases (3, 4). Although researches that were focused on the role of miRNAs in AD have only just began (5), there have been already some evidence showing that aberrantly expressed miRNAs such as miR-29b (6), miR-399 (7), miR-106b (8), miR-34a (9), let-7 (10), miR-155 (11), and miR-7 (12) were present in different cell types at different stages of AD. These literatures indicated that multiple miRNAs might be contributed to the development of AD. Especially, functional studies have further shown that these miRNAs were closely related to the changes of both Aβ formation and Tau phosphorylation, which were vital in the pathogenesis of AD (13, 14). Therefore, these observations provided novel and important evidences for successive study on the investigation of underlying mechanism of AD and the development of new targets for clinical therapy.
miRNAs and Aβ
miRNAs and Amyloid Precursor Protein (APP)
Amyloid precursor protein, which is a source of Aβ, is widely present in all histiocytes and especially abundant in neurons and astrocytes. Posttranscriptional alternative splicing resulted in six APP isoforms. Among these isoforms, APP695 is the most dominant type in normal brain, whereas APP751 is found at a higher level in brain of AD patients. Recent studies have shown that some miRNA molecules were involved in the regulation of alternative splicing of APP. Donev et al. reported that neuronal cells transfected with APP695, which was the main expressed subtype of APP mRNA lacking exons 7 and 8, produced significantly less Aβ, than the sequence of full-length APP-transfected neuronal cells did (15). Studies by Smith et al. further showed that APP695 expression was noticeably downregulated and Aβ production significantly increased in neuronal cells from mice with knocked out of Dicer gene, which was deficient for a broad range of miRNAs (16). Successive work reported that the significantly decreased level of miR-124, a representative member of miRNA family, in the brain of AD patients, greatly improved the expression of its corresponding target molecule polypyrimidine tract-binding protein1 (PTBP1), which was critical splicing factor to interrupt the splicing process of APP exons 7 and 8 and subsequently accelerate Aβ generation and AD progression (17). Combining these data demonstrated that specific miRNAs may influence the formation of Aβ through regulating APP splicing.
Prior researches on the regulation of APP expression by miRNAs were mainly focused on direct targeted regulation. For instance, Niwa et al. found that, in C. elegans, the expression of APP homolog—apl-1, an important gene for developmental regulation, was regulated by miRNA family member let-7 (17). In subsequent studies, it was found that APP was a direct target of some members of the miRNA family, such as miR-20a, miR-155, miR-17 and miR-106b (18, 19), miR-135 and miR-200b (20), miR-101 (21), miR-16 (22) and miR-147, miR-153, miR-323-3p, miR-644, and miR-655 (23, 24). Recent studies further revealed that miRNAs could affect the expression of APP by binding to APP 3’UTR cis-regulatory elements. For example, Rck/p54 was a component of a protein complex near the APP stop codon, and its main role was to enhance APP mRNA and protein expression level in neuronal cells through interacting with APP mRNA. Several new studies reveal that miRNAs may be involved in transcription and translation of APP by interacting with Rck/p54 (25, 26). Besides, other research work showed that 81-nucleotide cis-regulatory sequence on APP mRNA was not only the binding target for miR-106b/miR-520c but also consisted of a predicted-binding site for miR-20 family members. However, whether these elements were working in cooperative or competitive manners in the pathogenesis of AD remains to be investigated.
Interestingly, other studies further showed that a single nucleotide polymorphism (SNP) at the 3’UTR of APP mRNA could interfere with the binding of miRNAs to APP mRNA (24), indicating the complexity of regulation of miRNAs on APP expression. Among these SNPs, SNP T171C was found to significantly inhibit the binding of miR-147, which resulted in an increased expression of APP and subsequent generation of Aβ, whereas SNP A454G increased the binding of miR-20a to APP mRNA, thereby reducing APP expression. These data showed that SNPs were involved in the regulation of APP expression by miRNA and may, therefore, be potential risk factors for AD (27). In short, miRNAs could affect APP expression in numerous ways, such as by direct binding to APP 3’UTR, or by interacting with APP mRNA regulatory elements and AD-associated SNPs. However, the exact underlying mechanism remains to be fully elucidated.
miRNAs and BACE1
As the rate-limiting enzyme for Aβ production, β-site APP cleaving enzyme (BACE1) has attracted increased interest in pathogenesis of AD. Numerous studies have found that BACE1 mRNA level were regulated by various miRNA molecules. For example, a recent study has shown that miR-485-5p could inhibit BACE1 translation through binding to BACE1 exon 6 and overexpression of miR-485-5p reduced BACE1 protein level by 30% (28). Kim et al. showed that miR-186 could inhibit BACE1 expression by direct binding its mRNA 3’UTR in neuronal cells, which suggest that miR-186 downregulated might be a risk factor for the development of AD (29). Meanwhile, Hebert et al. have also shown that upregulation of miR-29a, -29b-1, and -29b-9 levels could directly inhibit BACE1 expression in neuronal cells (30). Consistent with these findings, reduced level of miR-29a/b-1 in cultured neuronal cells in vitro significantly increased the expression of Aβ and vice versa. These research data suggested that the miR-29a/b family may potentially inhibit the expression of BACE1 protein level and, therefore, be related to the pathogenesis of AD. In addition, Lei et al. also showed that increased miR-29c expression in vitro directly reduced BACE1 protein level by binding to the 3’UTR of its mRNA (31). These results were consistent with the finding that the amount of brain miR-29c was doubled in BACE1-knockdown mice compared with that in wild-type mice. Moreover, Boissonneault et al. also confirmed that miR-298 and miR-328 regulated BACE1 protein expression in neuronal cells by targeting specific binding sites of the BACE1 3’UTR (32). Finally, during the pathogenesis of AD, BACE1 mRNA levels of neuronal cells negatively correlated with miR-107 level (33). Conversely, increased BACE1 level was accompanied with a decreased level of miR-107 (33). Bioinformatics and luciferase gene reporter assays further confirmed that miR-107 could effectively bind to the 3’UTR sequence of BACE1. In summary, the above data demonstrated that a number of miRNAs could control BACE1 translation by binding to the 3’UTR of BACE1 mRNA, which affected the modification and metabolic processes of the Aβ protein, reflecting the complexity of regulatory mechanisms involved in pathogenesis of AD.
Additional Factors Affecting Aβ Production
It has been demonstrated that Aβ formation could be regulated by membrane ceramide, an important component of lipid rafts and was found at high level in sporadic AD. Moreover, ceramide could promote lipid raft formation via erroneous localization of BACE1 and γ-secretase, thereby increasing Aβ production (34). During that process, the first rate-limiting enzyme of ceramide synthesis was serine palmitoyltransferase (SPT). In a recent study by Geekiyanage and Chan, it was shown that the SPT protein was regulated by miR-181c, miR-137, miR-29a/b-1, and miR-9 (35). These miRNA molecules, which were abnormally downregulated in the AD frontal cortex, may modify the lipid composition and affect the level of Aβ formation through increasing SPF level. The expression of miR-181c, miR-137, and miR-29a/b-1, which were related to growth and development, reached peak levels in adult mice. Conversely, SPT expression negatively correlated with growth, i.e., the SPT level steadily decreased with age. The incidence of AD was higher in women than that in men (36), which also might be related to higher SPT protein level and lower miR-181c, -137, -29a/b-1 levels in the female population (35). Interestingly, it has been reported that a high-fat diet could increase plasma level of ceramides, thereby exacerbating the burden of Aβ deposition in animal models. In this experimental group, the expression levels of miR-181c and miR-137 were abnormally lower. These studies indicated that regulation of SPT by miRNAs provided a novel mechanism of pathogenesis of AD, which was also related to some AD-related risk factors such as age, gender, and intake of high-saturated fat.
Cholesterol homeostasis also has significant impact on Aβ metabolism and deposition. APP, BACE1, and PSEN1 are all membrane-bound proteins, whose hydrolysis and transportation are dependent on membrane fluidity, which is governed by cholesterol. Meanwhile, cholesterol can be transported out of cells by the ATP-binding cassette transporter A1 (ABCA1) to form high-density lipoprotein particles (37), which reduce the risk of AD (38). Recent research work further showed that ABCA1 deficiency in neurons could decrease the cellular cholesterol efflux and subsequently enhance Aβ deposition. Moreover, overexpression of ABCA1 significantly decreased Aβ accumulation (33). Similarly, ABCA1 expression level was found increased in the hippocampus of AD patients and was positively correlated to the severity of cognitive impairment (39). Interestingly, some new studies have demonstrated that, in neurons, miR-33 could directly inhibit the expression of ABCA1 by binding to the 3’UTR of ABCA1 mRNA, which reduced the efflux of cellular cholesterol and increased the level of Aβ, thereby increasing the risk of AD (33).
In summary, miRNAs could affect Aβ metabolism in many ways including through their direct action on 3’UTR of APP, BACE1, ABCA1, and other related genes, as well as indirect regulation through other factors, and played important roles in the pathogenesis of AD.
miRNAs and Tau Protein
Microtubule-associated protein Tau (MAPT) is another major player in the pathogenesis of AD. The disruption of any of the steps in the Tau protein metabolic pathway may exacerbate the progression of AD. Recent studies have shown that conditional knockout of the Dicer gene in the mouse brain created an extensive miRNA deficiency, which resulted in abnormal Tau protein metabolism in mice with AD-like Tau hyperphosphorylation and aberrant splicing of MAPT (40, 41). Similar studies by Bilen et al. showed that when Dicer1 was knocked out, there were increased level of Tau protein and neurodegeneration in the Drosophila brain (42). These results indicated that miRNAs played key roles in the regulation of Tau protein metabolism and subsequently were involved in the pathogenesis of AD.
miRNAs and Tau Protein Editing
Alternative splicing of exons 2, 3, and 10 has resulted in 6 Tau isoforms in the human brain (43). Exon 10 encodes the microtubule-binding repeat regions and is responsible for generating 3R or 4R isoforms. Under normal conditions, the ratio between 4R/3R isoforms is approximately 1:1 and an imbalance of this ratio could cause neurodegenerative diseases, such as dementia (44, 45). An important finding was reported by Smith et al. that many miRNAs, including miR-124, miR-9, miR-132, and miR-137, could affect the 4R/3R ratio in neuronal cells by regulating MAPT splicing (41). Further analysis of miRNA expression profiling of patients with progressive supranuclear palsy, a neurodegenerative disease caused by Tau 4R isoform overexpression, showed that miR-132 level were significantly downregulated in a patients’ brain, whereas the level of polypyrimidine tract-binding protein 2 (PTBP2), a target of miR-132, increased obviously. Importantly, overexpressing miR-132 or suppressing PTBP2 expression could reverse the ratio of 4R/3R isoforms (41). Similarly, in vitro experimental setting, PTBP1 was found closely related to MAPT exon 10 splicing (46). Moreover, its level was upregulated in conditional Dicer-knockout mouse brains (16). In addition, PTBP1/2 level were also significantly altered during the development of diseases associated with alternative splicing (47, 48). In summary, these data showed that specific miRNAs were involved in the regulation of MAPT exon 10 splicing and subsequently contributed to the pathogenesis of AD.
miRNAs and Tau Protein-Associated Kinases
Dysregulation of the balance of protein kinases and phosphatases is the direct cause of Tau protein hyperphosphorylation. It is now known that Tau protein phosphorylation can be mediated by a variety of protein kinases, such as extracellular regulated protein kinases (ERKs) and glycogen synthase kinase-3 (GSK-3). In studies described earlier in this review, Hebert et al. conditionally knocked out Dicer enzyme gene in the mouse brain and found that there were hyperphosphorylation of endogenous Tau protein in pathological regions and increased expression of mitogen-activated protein kinase 3 (MAPK3/ERK1) and GSK-3β. Bioinformation analysis further showed that the 3’UTR of mRNA of these protein kinase molecules could be directly bound by some miRNA mature sequences (40). In vitro studies from this research group also showed that in murine neuronal cells, some members of the miR-15 family, including miR-15a, miR-16, miR-195, and miR-497, could directly interact with the 3’-UTR of ERK1 mRNA (40). Meanwhile, to another protein kinase GSK-3β, which could hyperphosphorylate the PHF1 site of the Tau protein and was associated with both Aβ generation and NFT formation in AD brain (49), Mohamed et al. recently reported that miR-26a could directly regulate GSK-3β gene expression (50). Finally, it would be noticed that other research work further suggested that miR-26a was also involved in the pathogenesis of the AD (19), which was related to its interaction with brain-derived neurotrophic factor, an important neurotrophin for neuronal development and plasticity (51), indicating the complexity of target molecules of distinct miRNA molecules in the development of AD. Collectedly, these literatures suggested that various miRNAs could blind directly to phosphorylated Tau-associated protein kinase molecules or indirectly regulate the level of associated protein kinase molecule, so as to form a complex biological regulatory network of miRNAs regulated phosphorylated Tau-related protein kinase. However, the exact role of different miRNAs in the pathogenesis of AD through the regulation of protein kinases remains to be illuminated.
miRNAs and Tau Protein Clearance
Acetylation at specific sites of the Tau protein can promote its autophosphorylation, exacerbating abnormal Tau protein aggregation and facilitating AD progression. The Tau protein acetylation state is mainly dependent on acetyltransferase p300 (an acetylase) and sirtuin 1 (SIRT1, a deacetylase). With the reduced level of SIRT1 in the AD brain, the acetylation level of Tau protein aggravated correspondingly (52, 53), followed by significant accumulation of phosphorylated Tau protein (54). Recent studies have demonstrated that the SIRT1 gene could be directly inhibited by miR-9, miR-212, and miR-181c, respectively (55, 56). In addition, co-chaperone BCL2-associated athanogene 2 (BAG2) could preferentially and efficiently degrade insoluble and phosphorylated Tau, primarily through forming a complex with Hsp70 on microtubules by capturing and delivering Tau protein to the non-ubiquitin-independent proteasomal pathway for degradation. During this process, miR-128a, as an intrinsic regulator, could fine-tune tau protein level in neuronal cells by acting directly on 3’-UTR of BAG2 mRNA (57). Combing these literatures suggested that miRNAs may regulate the edition and clearance of Tau protein, as well as accumulation of phosphorylated Tau protein. However, whether Tau protein itself could induce the degradation of distinct miRNA molecules and subsequently affect the pathogenesis of AD remain to be further investigated.
Summary
To date, some active advances have been made on the potential value of miRNAs expression on early detection of AD. For example, some recent studies showed that the levels of miR-126b and miR-27a were significantly reduced, whereas levels of miR-9, miR-125b, miR-15, and miR-138 were significantly increased in the cerebrospinal fluid of clinical AD patients (58, 59). Similarly, the serum levels of various other miRNAs, such as miR-15a, let-7d, let-7g, miR-142, miR-191, miR-301a, miR-545, and miR-342-3p in clinical AD patients were significantly different from those in normal population (54, 55). Moreover, other studies further showed that the expression of miR-34a and miR-181b were also significantly increased in peripheral mononuclear blood cells of AD patients (52). Importantly, it has been found that the altered expression of distinct miRNAs in AD patients appeared earlier than Aβ deposition and Tau phosphorylation, which were as biomarkers for early diagnosis of AD currently (16). These studies suggested that the altered expression of specific miRNAs might be used as important promising biomarkers for AD prediction and/or diagnosis.
In summary, recent researches have proven that miRNAs could be participated in the pathogenesis of AD by regulating multiple targets (see Table 1; Figure 1). However, there are still a large number of scientific questions worthy to further exploration. For example, although some studies have shown that adding exogenous Aβ42 peptides to rat hippocampal neurons could lead to rapid downregulation of multi-specific miRNAs (60), it was still a question whether or not abnormal expression of miRNAs was an accompanied phenomenon or/and induction factor for the pathogenesis of AD. Another question was how can a concerted subnetwork of potential target genes of miRNAs be set up, in which miRNAs specifically aimed to its target genes in AD. Brain degeneration in AD patients is a heterogeneous process, therefore, the pathological changed in some brain regions was earlier and more severe than that in other brain regions, making it more challenging for researchers to decipher the exact roles of miRNAs in the complex process. Together, further in-depth miRNA research and discovery will provide novel promising ideas and strategies for elucidating the pathological mechanisms of AD and for developing effective methods for early diagnosis and treatment of AD.
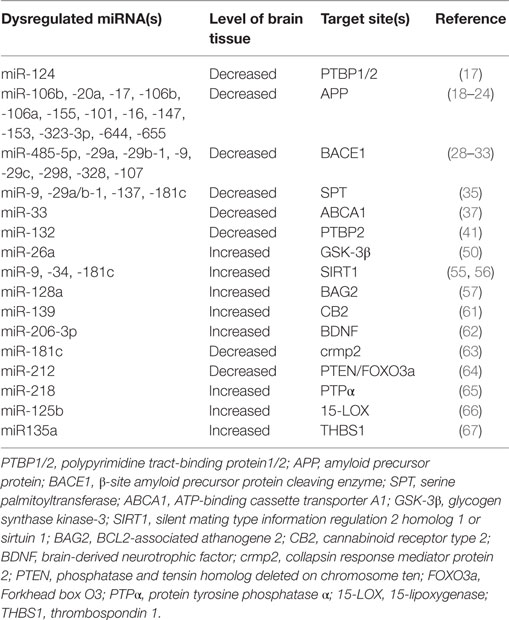
Table 1. microRNAs (miRNAs) in Alzheimer’s disease.
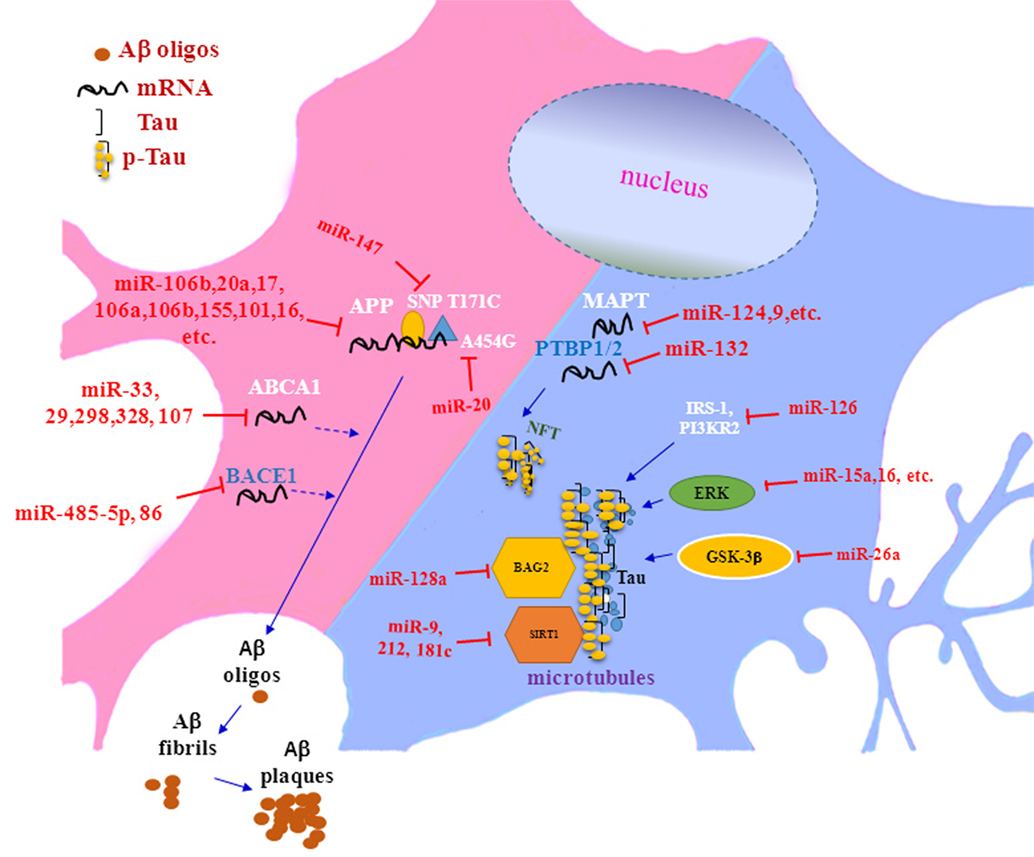
Figure 1. The role of microRNAs in Aβ deposition and tau phosphorylation in pathogenesis of Alzheimer’s disease.
Author Contributions
JZ, DY, YZ, LJ, HW, MG, HX, CC, and JZ carried out reading the related literatures and drafting the manuscript. LX and JZ designed the outline of manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
All authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Meng Zhang of Xinxiang Medical University for the help of revising the manuscript.
Funding
This manuscript was supported by Program for New Century Excellent Talents in University, Ministry of Education of China (NCET-12-0661), National Natural Science foundation of China (31370918), Program for High level innovative talents in Guizhou Province (QKH-RC-2016-4031), Program for Excellent Young Talents of Zunyi Medical University (15ZY-001), and Program for Science and Technology Joint Fund Project in Zunyi Science and Technology Bureau and Zunyi Medical University (ZSKH-SZ-2016-38).
References
1. Shah H, Albanese E, Duggan C, Rudan I, Langa KM, Carrillo MC, et al. Research priorities to reduce the global burden of dementia by 2025. Lancet Neurol (2016) 15:1285–94. doi:10.1016/S1474-4422(16)30235-6
2. Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol (2016) 16:279–94. doi:10.1038/nri.2016.40
3. Pignataro G. Editorial: mechanisms of innate neuroprotection. Front Neurol (2016) 7:80. doi:10.3389/fneur.2016.00080
4. Hill JM, Pogue AI, Lukiw WJ. Pathogenic microRNAs common to brain and retinal degeneration; recent observations in Alzheimer’s disease and age-related macular degeneration. Front Neurol (2015) 6:232. doi:10.3389/fneur.2015.00232
5. Lukiw WJ. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport (2007) 18:297–300. doi:10.1097/WNR.0b013e3280148e8b
6. Pereira PA, Tomás JF, Queiroz JA, Figueiras AR, Sousa F. Recombinant pre-miR-29b for Alzheimer’s disease therapeutics. Sci Rep (2016) 6:19946. doi:10.1038/srep19946
7. Long JM, Ray B, Lahiri DK. MicroRNA-339-5p down-regulates protein expression of β-site amyloid precursor protein-cleaving enzyme 1 (BACE1) in human primary brain cultures and is reduced in brain tissue specimens of Alzheimer disease subjects. J Biol Chem (2014) 2898:5184–8. doi:10.1074/jbc.M113.518241
8. Wang H, Liu J, Zong Y, Xu Y, Deng W, Zhu H, et al. miR-106b aberrantly expressed in a double transgenic mouse model for Alzheimer’s disease targets TGF-β type II receptor 8. Brain Res (2010) 1357:166–74. doi:10.1016/j.brainres.2010.08.023
9. Dickson JR, Kruse C, Montagna DR, Finsen B, Wolfe MS. Alternative polyadenylation and miR-34 family members regulate Tau expression. J Neurochem (2013) 127:739–49. doi:10.1111/jnc.12437
10. Kong Y, Wu J, Yuan L. MicroRNA expression analysis of adult-onset Drosophila Alzheimer’s disease model. Curr Alzheimer Res (2014) 111:882–91. doi:10.2174/1567205011666141001121416
11. Guedes JR, Santana I, Cunha C, Duro D, Almeida MR, Cardoso AM, et al. MicroRNA deregulation and chemotaxis and phagocytosis impairment in Alzheimer’s disease. Alzheimers Dement (Amst) (2015) 3:7–17. doi:10.1016/j.dadm.2015.11.004
12. Zhao J, Chen C, Guo M, Tao Y, Cui P, Zhou Y, et al. MicroRNA-7 deficiency ameliorates the pathologies of acute lung injury through elevating KLF4. Front Immunol (2016) 7:389. doi:10.3389/fimmu.2016.00389
13. Bekris LM, Leverenz JB. The biomarker and therapeutic potential of miRNA in Alzheimer’s disease. Neurodegener Dis Manag (2015) 5:61–74. doi:10.2217/nmt.14.52
14. Wang X, Liu P, Zhu H, Xu Y, Ma C, Dai X, et al. miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits bcl2 translation. Brain Res Bull (2009) 80:268–73. doi:10.1016/j.brainresbull.2009.08.006
15. Donev R, Newall A, Thome J, Sheer D. A role for SC35 and hnRNPA1 in the determination of amyloid precursor protein isoforms. Mol Psychiatry (2007) 12(7):681–90. doi:10.1038/sj.mp.4001971
16. Smith P, Al Hashimi A, Girard J, Delay C, Hébert SS. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J Neurochem (2011) 116:240–7. doi:10.1111/j.1471-4159.2010.07097.x
17. Niwa R, Zhou F, Li C, Slack FJ. The expression of the Alzheimer’s amyloid precursor protein-like gene is regulated by developmental timing microRNAs and their targets in Caenorhabditis elegans. Dev Biol (2008) 315:418–25. doi:10.1016/j.ydbio.2007.12.044
18. Hébert SS, Horré K, Nicolaï L, Bergmans B, Papadopoulou AS, Delacourtem A, et al. MicroRNA regulation of Alzheimer’s amyloid precursor protein expression. Neurobiol Dis (2009) 33:422–8. doi:10.1016/j.nbd.2008.11.009
19. Cheng XR, Cui XL, Zheng Y, Zhang GR, Li P, Huang H, et al. Nodes and biological processes identified on the basis of network analysis in the brain of the senescence accelerated mice as an Alzheimer’s disease animal model. Front Aging Neurosci (2013) 5:65. doi:10.3389/fnagi.2013.00065
20. Liu CG, Wang JL, Li L, Xue LX, Zhang YQ, Wang PC. MicroRNA-135a and -200b, potential biomarkers for Alzheimer’s disease, regulate β secretase and amyloid precursor protein. Brain Res (2014) 1583:55–64. doi:10.1016/j.brainres.2014.04.026
21. Barbato C, Pezzola S, Caggiano C, Antonelli M, Frisone P, Ciotti MT, et al. A lentiviral sponge for miR-101 regulates RanBP9 expression and amyloid precursor protein metabolism in hippocampal neurons. Front Cell Neurosci (2014) 8:37. doi:10.3389/fncel.2014.00037
22. Parsi S, Smith PY, Goupil C, Dorval V, Hébert SS. Preclinical evaluation of miR-15/107 family members as multifactorial drug targets for alzheimer’s disease. Mol Ther Nucleic Acids (2015) 4:e256. doi:10.1038/mtna.2015.33
23. Delay C, Calon F, Mathews P, Hébert SS. Alzheimer-specific variants in the 3’UTR of Amyloid precursor protein affect microRNA function. Mol Neurodegener (2011) 6:70. doi:10.1186/1750-1326-6-70
24. Liang C, Zhu H, Xu Y, Huang L, Ma C, Deng W, et al. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res (2012) 1455:103–13. doi:10.1016/j.brainres.2011.10.051
25. Broytman O, Westmark PR, Gurel Z, Malter JS. Rck/p54 interacts with APP mRNA as part of a multi-protein complex and enhances APP mRNA and protein expression in neuronal cell lines. Neurobiol Aging (2009) 30:1962–74. doi:10.1016/j.neurobiolaging.2008.02.011
26. Chu CY, Rana TM. Translation repression in human cells by microRNA-induced gene silencing requires RCK/p54. PLoS Biol (2006) 4:e210. doi:10.1371/journal.pbio.0040210
27. Glinsky GV. An SNP-guided microRNA map of fifteen common human disorders identifies a consensus disease phenocode aiming at principal components of the nuclear import pathway. Cell Cycle (2008) 7:2570–83. doi:10.4161/cc.7.16.6524
28. Faghihi MA, Zhang M, Huang J, Modarresi F, Van der Brug MP, Nalls MA, et al. Evidence for natural antisense transcript-mediated inhibition of microRNA function. Genome Biol (2010) 11:R56. doi:10.1186/gb-2010-11-5-r56
29. Kim J, Yoon H, Chung DE, Brown JL, Belmonte KC, Kim J. miR-186 is decreased in aged brain and suppresses BACE1 expression. J Neurochem (2016) 137:436–45. doi:10.1111/jnc.13507
30. Roshan R, Ghosh T, Gadgil M, Pillai B. Regulation of BACE1 by miR-29a/b in a cellular model of spinocerebellar ataxia 17. RNA Biol (2012) 9(6):891–9. doi:10.4161/rna.19876
31. Yang G, Song Y, Zhou X, Deng Y, Liu T, Weng G, et al. MicroRNA-29c targets β-site amyloid precursor protein-cleaving enzyme 1 and has a neuroprotective role in vitro and in vivo. Mol Med Rep (2015) 12(2):3081–8. doi:10.3892/mmr.2015.3728
32. Boissonneault V, Plante I, Rivest S, Provost P. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem (2009) 284(4):1971–81. doi:10.1074/jbc.M807530200
33. Jiao Y, Kong L, Yao Y, Li S, Tao Z, Yan Y, et al. Osthole decreases beta amyloid levels through up-regulation of miR-107 in Alzheimer’s disease. Neuropharmacology (2016) 108:332–44. doi:10.1016/j.neuropharm.2016.04.046
34. Puglielli L, Ellis BC, Saunders AJ, Kovacs DM. Ceramide stabilizes β-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid β-peptide biogenesis. J Biol Chem (2003) 27:19777–83. doi:10.1074/jbc.M300466200
35. Geekiyanage H, Chan C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid β, novel targets in sporadic Alzheimer’s disease. J Neurosci (2011) 31:14820–30. doi:10.1523/JNEUROSCI.3883-11.2011
36. Liu KP, Kuo MC, Tang KC, Chau AW, Ho IH, Kwok MP, et al. Effects of age, education and gender in the consortium to establish a registry for the Alzheimer’s disease (CERAD)-neuropsychological assessment battery for cantonese-speaking Chinese elders. Int Psychogeriatr (2011) 23:1575–81. doi:10.1017/S1041610211001153
37. Nordestgaard LT, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement (2015) 11:1430–8. doi:10.1016/j.jalz.2015.04.006
38. Stukas S, Robert J, Wellington CL. High-density lipoproteins and cerebrovascular integrity in Alzheimer’s disease. Cell Metab (2014) 19:574–91. doi:10.1016/j.cmet.2014.01.003
39. Akram A, Schmeidler J, Katsel P, Hof PR, Haroutunian V. Increased expression of cholesterol transporter ABCA1 is highly correlated with severity of dementia in AD hippocampus. Brain Res (2010) 1318:167–77. doi:10.1016/j.brainres.2010.01.006
40. Hébert SS, Papadopoulou AS, Smith P, Galas MC, Planel E, Silahtaroglu AN, et al. Genetic ablation of dicer in adult forebrain neurons results in abnormal Tau hyperphosphorylation and neurodegeneration. Hum Mol Genet (2010) 19(20):3959–69. doi:10.1093/hmg/ddq311
41. Smith PY, Delay C, Girard J, Papon MA, Planel E, Sergeant N, et al. MicroRNA-132 loss is associated with Tau exon 10 inclusion in progressive supranuclear palsy. Hum Mol Genet (2011) 20(20):4016–24. doi:10.1093/hmg/ddr330
42. Bilen J, Liu N, Burnett BG, Pittman RN, Bonini NM. MicroRNA pathways modulate polyglutamine-induced neurodegeneration. Mol Cell (2006) 24:157–63. doi:10.1016/j.molcel.2006.07.030
43. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein Tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron (1989) 3(4):519–26. doi:10.1016/0896-6273(89)90210-9
44. D’Souza I, Schellenberg GD. Regulation of Tau isoform expression and dementia. Biochim Biophys Acta (2005) 1739:104–15. doi:10.1016/j.bbadis.2004.08.009
45. Caffrey TM, Joachim C, Paracchini S, Esiri MM, Wade-Martins R. Haplotype-specific expression of exon 10 at the human MAPT locus. Hum Mol Genet (2006) 15(24):3529–37. doi:10.1093/hmg/ddl429
46. Wang J, Gao QS, Wang Y, Lafyatis R, Stamm S, Andreadis A. Tau exon 10, whose missplicing causes frontotemporal dementia, is regulated by an intricate interplay of cis elements and trans factors. J Neurochem (2004) 88(5):1078–90. doi:10.1046/j.1471-4159.2003.02232.x
47. Tollervey JR, Wang Z, Hortobágyi T, Witten JT, Zarnack K, Kayikci M, et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res (2011) 21(10):1572–82. doi:10.1101/gr.122226.111
48. Sterne-Weiler T, Howard J, Mort M, Cooper DN, Sanford JR. Loss of exon identity is a common mechanism of human inherited disease. Genome Res (2011) 21:1563–71. doi:10.1101/gr.118638.110
49. Maqbool M, Mobashir M, Hoda N. Pivotal role of glycogen synthase kinase-3: a therapeutic target for Alzheimer’s disease. Eur J Med Chem (2016) 107:63–81. doi:10.1016/j.ejmech.2015.10.018
50. Mohamed JS, Lopez MA, Boriek AM. Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3β. J Biol Chem (2010) 285:29336–47. doi:10.1074/jbc.M110.101147
51. Caputo V, Sinibaldi L, Fiorentino A, Parisi C, Catalanotto C, Pasini A, et al. Brain derived neurotrophic factor (BDNF) expression is regulated by microRNAs miR-26a and miR-26b allele-specific binding. PLoS One (2011) 6:e28656. doi:10.1371/journal.pone.0028656
52. Gan L, Mucke L. Paths of convergence: sirtuins in aging and neurodegeneration. Neuron (2008) 58:10–4. doi:10.1016/j.neuron.2008.03.015
53. Rodriguez-Ortiz CJ, Baglietto-Vargas D, Martinez-Coria H, LaFerla FM, Kitazawa M. Upregulation of miR-181 decreases c-Fos and SIRT-1 in the hippocampus of 3xTg-AD mice. J Alzheimers Dis (2014) 42:1229–38. doi:10.3233/JAD-140204
54. Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, et al. Acetylation of Tau inhibits its degradation and contributes to tauopathy. Neuron (2010) 67:953–66. doi:10.1016/j.neuron.2010.08.044
55. Schonrock N, Humphreys DT, Preiss T, Götz J. Target gene repression mediated by miRNAs miR-181c and miR-9 both of which are down-regulated by amyloid-β. J Mol Neurosci (2012) 46:324–35. doi:10.1007/s12031-011-9587-2
56. Weinberg RB, Mufson EJ, Counts SE. Evidence for a neuroprotective microRNA pathway in amnestic mild cognitive impairment. Front Neurosci (2015) 9:430. doi:10.3389/fnins.2015.00430
57. Carrettiero DC, Hernandez I, Neveu P, Papagiannakopoulos T, Kosik KS. The cochaperone BAG2 sweeps paired helical filament-insoluble Tau from the microtubule. J Neurosci (2009) 29:2151–61. doi:10.1523/JNEUROSCI.4660-08.2009
58. Kumar S, Reddy PH. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim Biophys Acta (2016) 1862:1617–27. doi:10.1016/j.bbadis.2016.06.001
59. Yılmaz ŞG, Erdal ME, Özge AA, Sungur MA. Can peripheral MicroRNA expression data serve as epigenomic (upstream) biomarkers of Alzheimer’s disease? OMICS (2016) 20:456–61. doi:10.1089/omi.2016.0099
60. Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis (2008) 14:27–41. doi:10.3233/JAD-2008-14103
61. Tang Y, Bao JS, Su JH, Huang W. MicroRNA-139 modulates Alzheimer’s-associated pathogenesis in SAMP8 mice by targeting cannabinoid receptor type 2. Genet Mol Res (2017) 16(1). doi:10.4238/gmr16019166
62. Wang CN, Wang YJ, Wang H, Song L, Chen Y, Wang JL, et al. The anti-dementia effects of Donepezil involve miR-206-3p in the hippocampus and cortex. Biol Pharm Bull (2017) 40:465–72. doi:10.1248/bpb.b16-00898
63. Zhou H, Zhang R, Lu K, Yu W, Xie B, Cui D, et al. Deregulation of miRNA-181c potentially contributes to the pathogenesis of AD by targeting collapsin response mediator protein 2 in mice. J Neurol Sci (2016) 367:3–10. doi:10.1016/j.jns.2016.05.038
64. Wong HK, Veremeyko T, Patel N, Lemere CA, Walsh DM, Esau C, et al. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer’s disease. Hum Mol Genet (2013) 22:3077–92. doi:10.1093/hmg/ddt164
65. Xiong YS, Liu FF, Liu D, Huang HZ, Wei N, Tan L, et al. Opposite effects of two estrogen receptors on Tau phosphorylation through disparate effects on the miR-218/PTPA pathway. Aging Cell (2015) 14:867–77. doi:10.1111/acel.12366
66. Zhao Y, Bhattacharjee S, Jones BM, Hill J, Dua P, Lukiw WJ. Regulation of neurotropic signaling by the inducible, NF-kB-sensitive miRNA-125b in Alzheimer’s disease (AD) and in primary human neuronal-glial (HNG) cells. Mol Neurobiol (2014) 50:97–106. doi:10.1007/s12035-013-8595-3
Keywords: Alzheimer’s disease, microRNAs, amyloid-beta, amyloid precursor protein, Tau protein
Citation: Zhao J, Yue D, Zhou Y, Jia L, Wang H, Guo M, Xu H, Chen C, Zhang J and Xu L (2017) The Role of MicroRNAs in Aβ Deposition and Tau Phosphorylation in Alzheimer’s Disease. Front. Neurol. 8:342. doi: 10.3389/fneur.2017.00342
Received: 20 April 2017; Accepted: 30 June 2017;
Published: 18 July 2017
Edited by:
Massimiliano Caiazzo, Utrecht University, NetherlandsReviewed by:
Scott Edward Counts, Michigan State University, United StatesJames David Doecke, Commonwealth Scientific and Industrial Research Organisation (CSIRO), Australia
Copyright: © 2017 Zhao, Yue, Zhou, Jia, Wang, Guo, Xu, Chen, Zhang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Xu, eHVsaW56aG91eWFAMTYzLmNvbQ==