 Parmenion P. Tsitsopoulos
Parmenion P. Tsitsopoulos Sami Abu Hamdeh
Sami Abu Hamdeh Niklas Marklund
Niklas Marklund- 1Section of Neurosurgery, Department of Neuroscience, Uppsala University, Uppsala, Sweden
- 2Hippokratio General Hospital, Aristotle University, Thessaloniki, Greece
- 3Department of Clinical Sciences Lund, Neurosurgery, Skåne University Hospital, Lund University, Lund, Sweden
Traumatic brain injury (TBI) is a multidimensional and highly complex disease commonly resulting in widespread injury to axons, due to rapid inertial acceleration/deceleration forces transmitted to the brain during impact. Axonal injury leads to brain network dysfunction, significantly contributing to cognitive and functional impairments frequently observed in TBI survivors. Diffuse axonal injury (DAI) is a clinical entity suggested by impaired level of consciousness and coma on clinical examination and characterized by widespread injury to the hemispheric white matter tracts, the corpus callosum and the brain stem. The clinical course of DAI is commonly unpredictable and it remains a challenging entity with limited therapeutic options, to date. Although axonal integrity may be disrupted at impact, the majority of axonal pathology evolves over time, resulting from delayed activation of complex intracellular biochemical cascades. Activation of these secondary biochemical pathways may lead to axonal transection, named secondary axotomy, and be responsible for the clinical decline of DAI patients. Advances in the neurocritical care of TBI patients have been achieved by refinements in multimodality monitoring for prevention and early detection of secondary injury factors, which can be applied also to DAI. There is an emerging role for biomarkers in blood, cerebrospinal fluid, and interstitial fluid using microdialysis in the evaluation of axonal injury in TBI. These biomarker studies have assessed various axonal and neuroglial markers as well as inflammatory mediators, such as cytokines and chemokines. Moreover, modern neuroimaging can detect subtle or overt DAI/white matter changes in diffuse TBI patients across all injury severities using magnetic resonance spectroscopy, diffusion tensor imaging, and positron emission tomography. Importantly, serial neuroimaging studies provide evidence for evolving axonal injury. Since axonal injury may be a key risk factor for neurodegeneration and dementias at long-term following TBI, the secondary injury processes may require prolonged monitoring. The aim of the present review is to summarize the clinical short- and long-term monitoring possibilities of axonal injury in TBI. Increased knowledge of the underlying pathophysiology achieved by advanced clinical monitoring raises hope for the development of novel treatment strategies for axonal injury in TBI.
Introduction
Traumatic brain injury (TBI) is a significant cause of morbidity and mortality worldwide (1–5). Mortality due to severe TBI can reach 40% with high rates of disability among the survivors (6–8). Cognitive, behavioral, and emotional impairments are common and particularly disabling post-TBI and can persist into the chronic stage (9–12). Widespread injury to the white matter tracts, a key feature of TBI, disrupts neuronal networks and impairs information processing which contributes to the cognitive impairments observed post-TBI (9–11).
Axonal injury was initially described by Strich in 1956, who observed diffuse axonal degeneration at autopsy of severe TBI patients (13). Axonal pathology was later established as a separate TBI entity by Adams and colleagues in 1982 (14). When axonal damage occurs in multiple brain locations in clinical TBI, it is named diffuse axonal injury (DAI) (15–21).
In the preclinical setting, the term traumatic axonal injury has been applied to describe axonal damage, with the term DAI used to express its clinical counterpart (5, 19). Thus, DAI is a clinical entity characterized by radiological and/or histological findings suggestive of axonal pathology at certain predilection sites, particularly the gray/white matter interface, the corpus callosum, and the brain stem (14, 15). DAI remains a challenging clinical entity with a frequently unpredictable course and outcome. To date, there are limited therapeutic options reflecting the incomplete knowledge of the underlying pathophysiology of DAI. However, it has been established that axonal injury results from a highly dynamic process involving a cascade of events that may evolve over time leading to progressive white matter atrophy with variable clinical impact (19, 22–24). Therefore, detection and monitoring of DAI is relevant from the acute to chronic phase, in order to evaluate its severity, seek treatment options and better predict clinical outcome.
In this review, an overview of the current clinical possibilities for monitoring of DAI is provided. A literature search was performed in PubMed, Scopus and ISI Web of Knowledge. Experimental/preclinical studies on axonal injury in TBI were excluded from the overview with the exception of those describing key pathophysiological mechanisms. Articles where the injury type was not mentioned, was unclear or encompassing only focal TBI were also excluded from the analysis. Articles on mild, moderate, and/or severe TBI, DAI, and traumatic axonal injury were extracted, further screened and were included if the investigated mechanisms involved aspects of axonal/white matter injury. The literature on mild TBI, considered a diffuse TBI subtype with features of axonal and white matter pathology, was thus also included in our search.
Biomechanics and Structural Changes in DAI
The predominant mechanism in the development of axonal injury in TBI is mechanical shearing and stretch forces produced by inertial acceleration/deceleration stresses to the head (25–28) (Figure 1). This inertial loading triggers dynamic shear, tensile, and compressive strains within the brain. Consequently, certain parts of the brain move at a slower pace relative to others, leading to deformation of the brain tissue (17, 29). In the preclinical TBI setting, unmyelinated axons sustained more injury compared to myelinated ones, suggesting that axons are unequally vulnerable (30).
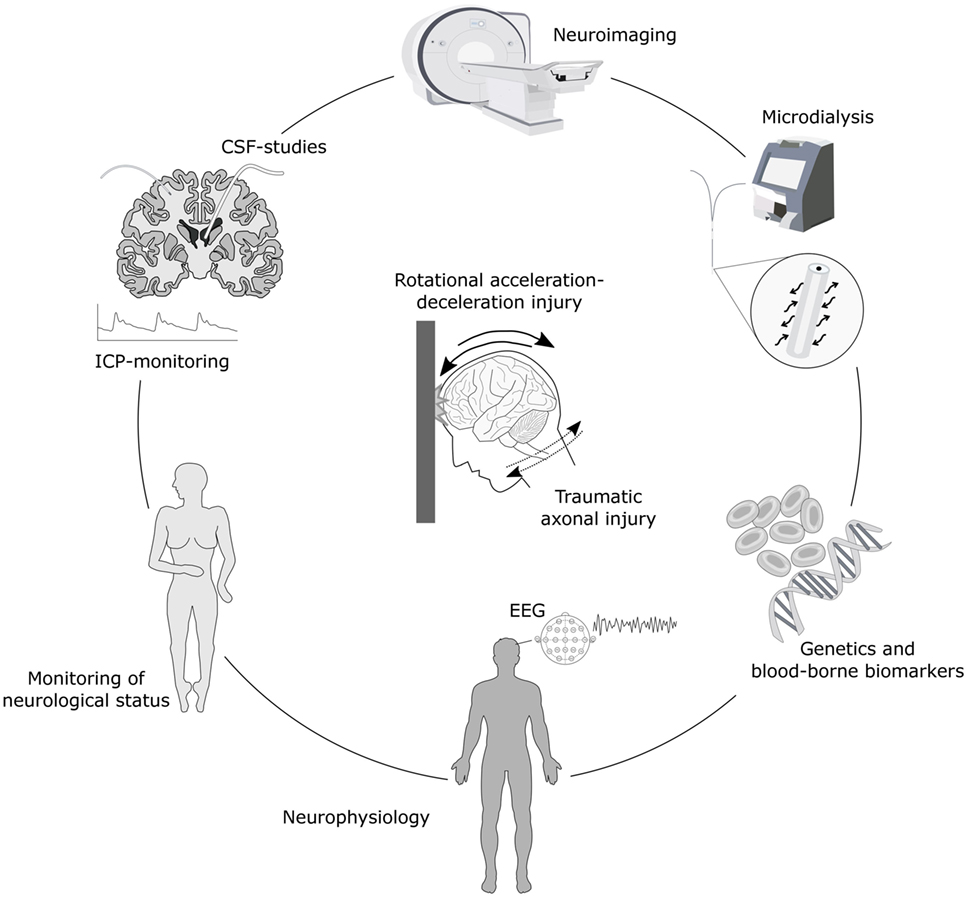
Figure 1. Schematic illustration of monitoring options for axonal injury. Biomechanically, traumatic axonal injury results from head impact with rotational acceleration-deceleration forces. Detection and monitoring of axonal injury is possible with numerous advanced neuroimaging techniques such as magnetic resonance imaging (MRI), including diffusion tensor imaging (DTI) and magnetic resonance spectrometry (MRS), as well as neuromolecular imaging by single-photon emission computed tomography (SPECT) and/or positron emission tomography (PET). Axonal injury also results in the secretion of various biomarkers into the interstitial fluid (ISF), cerebrospinal fluid (CSF) and the bloodstream which can be detected in ISF using microdialysis, in CSF by sampling through an external ventricular drainage or through lumbar puncture, and in serum by blood sampling. These biomarkers provide clues of temporal patterns of axonal injury and ongoing secondary injury processes and may be associated with outcome. Monitoring of axonal injury progression may also be achieved by placement of an intracranial pressure (ICP) monitoring device for continuous surveillance of ICP, neurophysiological methods such as electroencephalography (EEG) and periodic assessments of neurological status including level of consciousness. Furthermore, the genetic profile may add additional information of risk for secondary injury cascades and neurodegenerative development.
Under normal conditions, brain tissue can withstand stretches and easily return to its original geometry without any resulting injury. In contrast, when the strain is rapidly applied, the brain tissue loses its plasticity and acts stiffer, becoming more vulnerable and brittle (31). In particular, axons in the white matter appear poorly prepared to withstand injury from rapid mechanical brain deformation at time of TBI (17), resulting in injury to the axonal cytoskeleton (17, 32, 33). Nevertheless, the development and severity of axonal injury is dependent on both the magnitude and rate of strain during impact (17).
Primary axotomy in humans is rare with secondary (delayed) axotomy being the most likely mechanism leading to axonal disconnection (5, 19, 34, 35). Experimental evidence indicates that mitochondrial dysfunction (5, 36), as well as TBI-induced inflammatory responses, contribute to the secondary axonal injury (5, 37, 38). In addition, impaired axonal transport causes axonal swelling over time post-injury, leading to the accumulation of numerous potential biomarkers which may then be released into the surrounding tissue (see later section of this review).
Clinical Features and Neurocritical Care Monitoring of Axonal Injury
Clinical Characteristics
From a clinical point of view, initial loss of consciousness and coma as well as later features such as prolonged vegetative state or cognitive impairment can be characteristics of both focal TBI and DAI, although may be more frequently observed in the latter (29, 39). The presence of a decreased level of consciousness and coma is commonly a result of axonal injury in the diencephalon and/or the brain stem (29, 40, 41). In DAI survivors, cognitive dysfunction, mood disorders, and behavioral problems are frequent and result in a decreased quality of life (17, 29, 42). In particular, memory impairment and problems in executive functioning are frequent in DAI (43, 44), and so is impaired information–processing speed (45). In addition, motor weakness may be caused by injury to the pyramidal tract (46), which is more frequently encountered in DAI compared to focal TBI (29).
By definition, axonal injury is difficult to diagnose using only clinical signs and symptoms. Some clinical features of axonal injury are presumably to a large extent related to the anatomic distribution of DAI. Based on the limited ability of computed tomography (CT) and standard T1- and T2-weighted magnetic resonance imaging (MRI) sequences to precisely detect the underlying axonal injury in TBI, it has been difficult to confirm and diagnose DAI with certainty (17). However, neuropsychology testing after release from hospital can detect cognitive and memory deficits as well as slowed mental processing, characteristics which, when combined with knowledge of the underlying injury mechanisms (Figure 1) and neuroimaging findings, may be highly suggestive of DAI. These features can help confirm the diagnosis of moderate or severe DAI with a relatively high degree of certainty in TBI patients (29, 47, 48).
In addition, advanced neuroimaging has recently enabled improved visualization of surrogate markers for the histopathological features of DAI, and for subsequent surveillance of secondary injury processes. Injury to white matter tracts interconnecting cortical regions, disrupting large scale brain networks of particular importance for complex cognitive functions (49, 50), are now possible to estimate using modalities like diffusion tensor imaging (DTI) and be correlated with cognitive and behavioral deficits observed using neuropsychology testing (10, 50, 51).
Intracranial Pressure (ICP) Monitoring of DAI
Intracranial pressure monitoring remains a cornerstone in the management of severe TBI patients, although the incidence of raised ICP in DAI is not well established. Maximum ICP has been correlated to the number of identifiable white matter lesions on MRI (52), and a relationship with the Marshall CT classification score and ICP levels was suggested (53, 54). In some studies of severe DAI patients, ICP was not elevated (55), whereas others found increased ICP in most TBI patients (56–58). In an early study of ICP monitoring in TBI patients, of whom 61 had a normal CT scan, no ICP elevations were observed unless the patient was aged >40 years, had unilateral or bilateral motor posturing or episodes of systolic blood pressure <90 mmHg (59). In contrast, another study found elevated ICP despite the absence of mass lesions, midline shift, or compressed basal cisterns on the initial CT scan (60). Later, less ICP elevations were observed in DAI compared to other TBI subtypes and it was suggested that ICP monitoring could be omitted in DAI (55). However, in that particular study, 10% of patients had ICP >20 mmHg, and two patients required treatment for elevated ICP. Similar patterns of transient ICP elevations triggered by neurocritical care events in DAI patients were also observed (61). Recently, ICP was analyzed in MRI-verified DAI patients, and although persistently raised ICP during the first 96 hours of monitoring was not seen, 20% of patients required treatment for transient ICP elevations (62).
Thus, the use of ICP monitoring in DAI is controversial (63). Although it was suggested that individuals with DAI documented by neuroimaging may not require treatment for elevated ICP, high ICP values were still frequently encountered in such patients. The use of ICP monitoring in comatose patients with initial normal CT scan or CT scan with minimal findings has also been questioned and recommended only in the presence of radiological worsening (64). On the other hand, in comatose patients with diffuse TBI with evidence of brain swelling on CT scan, ICP monitoring is indicated in the early post-injury period (64). It should be also noted that DAI patients with effaced basal cisterns on CT scan carry a high risk of increased ICP (58, 65).
In summary, studies evaluating the incidence of elevated ICP in DAI patients are scarce and provide contradictory results. Repeated clinical examinations and neuroimaging may be possible alternatives for monitoring of DAI patients when the initial CT scan is free from or shows only minimal abnormalities, since these patients may have a low risk of intracranial hypertension (63). Nevertheless, although it has not been firmly shown that outcome is improved, ICP monitoring in DAI patients with reduced level of consciousness and pathological findings on CT scan is recommended in the initial post-injury period (64, 66, 67).
Monitoring of Cerebral Blood Flow (CBF) and Brain Oxygenation
Perfusion CT or xenon-enhanced CT (Xe-CT) are both rapid and widely available techniques for the evaluation of CBF. For Xe-CT, a mobile CT scanner enabling bedside measurement of CBF is used (68). Although clinical experience in DAI is still limited, significant CBF alterations seem less frequent than in focal TBI (69–71). These imaging techniques, however, allow only intermittent CBF measurements and transient CBF impairment in the intervals between examinations cannot be established. Continuous monitoring of CBF is possible using thermal diffusion or laser Doppler methods, both requiring insertion of an intraparenchymal probe to assess focal CBF in a small brain volume (72, 73). Clinical experience with these techniques is still limited, and to date, there are no studies specifically evaluating local CBF measurements in DAI.
Cerebral blood flow may also be indirectly estimated using jugular venous oxygen saturation (Sjvo2) and brain tissue oxygenation (PBto2). Sjvo2 can be measured using a fiberoptic probe placed in the jugular bulb and ranges between 55–75% under normal conditions. Low Sjvo2 values may suggest hypoperfusion and ischemia and episodes of desaturation correlate with poor outcome (74). On other hand, high values >75% may represent hyperemia and also correlate with brain infarctions, since oxygen is not extracted from irreversibly injured brain tissue.
PBto2 measurements require a sensor to be inserted in deep white matter, and allow regional measurements of cerebral oxygenation. In the uninjured brain, PBto2 values are >20 mmHg while critical hypoxia may develop with values <10 mmHg. Although reductions of PBto2 have been associated with poor outcome in TBI (75), and current treatment recommendations suggest interventions when PBto2 falls below 15 mmHg (76), no studies have to our knowledge focused on the clinical impact of PBto2 in DAI patients.
Available methods for CBF measurements as well as brain oxygenation cannot be firmly recommended in DAI in view of the limited clinical experience with these methods. Nonetheless, they are expected to play a greater role in the future especially in multifocal/mixed cases with elevated ICP and impaired CPP as a complement to ICP-CPP guided treatment protocols.
Electroencephalography (EEG)
In TBI patients, continuous EEG (cEEG) has been proven useful for the monitoring of seizure activity and the depth of sedation especially in those on barbiturate coma (77, 78). The use of cEEG in TBI is also indicated for the detection and treatment of non-convulsive seizures (NCS), a common risk in severe TBI patients (79, 80). Although only low quality evidence exists, cEEG monitoring may be recommended in TBI patients with unexplained behavioral alterations or sudden changes in mental state and/or altered consciousness, and to rule out NCS especially in penetrating injuries, large intracranial lesions, and depressed skull fractures (79).
There is limited data on the use of EEG/cEEG in the monitoring of DAI. In a study of 90 patients after diffuse TBI, where EEG recording was applied in the early post-injury phase, the EEG patterns correlated with prognosis (81). Specifically, most DAI patients with “benign” EEG patterns (stage 1; normal records with preserved activity, stage 2; reactive with rhythmic theta activity dominant, stage 3; usually reactive spindle coma where sleep patterns of stage 2 demonstrated rhythmic spindles) survived while most patients with “malignant” EEG findings (low amplitude delta activity, burst suppression pattern, alpha pattern coma) died (81). Following blast TBI, typically resulting in a degree of white matter/axonal injury, reduced EEG phase synchrony in the frontal area was associated with axonal injury on DTI (82, 83).
To date, the role of EEG in the monitoring of DAI has not been established. Although there is evidence to support that cEEG monitoring may be useful for the diagnosis of NCS in severe TBI, there is insufficient data in DAI at present. This monitoring modality has also not been shown to improve outcome and/or alter treatment in DAI patients. To date, it should primarily be regarded as a scientific tool awaiting additional studies evaluating its clinical role in the multimodality monitoring of DAI.
Neuroimaging
In recent years, advances in neuroimaging have facilitated DAI diagnostics as well as allowed for more accurate prognosis and monitoring of ongoing, secondary axonal injury. While image acquisition speed, accessibility and accuracy in detecting traumatic intracranial hemorrhages make CT the leading neuroimaging modality in the acute evaluation of TBI patients, its utility in DAI is limited. Although traumatic edema of the brain and petechial hemorrhages in the white matter indicate DAI, CT is generally insensitive for subtle axonal lesions. Hemorrhagic lesions in deep-seated predilection sites for DAI such as the corpus callosum and the rostral brain stem are rarely seen on CT. In addition, non-hemorrhagic lesions, which have been linked to poor outcome (84, 85), are not detectable (86). As discussed previously, there is a degree of axonal injury caused by the initial impact although most axonal pathology in DAI is a delayed secondary event evolving over days to weeks resulting in clinical deterioration of the patient. Thus, the admission CT in combination with the clinical picture following TBI may indicate DAI, but for the confirmation of diagnosis, monitoring of the progression of axonal injury and adequate prognosis, more advanced imaging modalities are needed.
Magnetic Resonance Imaging
Magnetic resonance imaging is a more sensitive modality for visualization of DAI-associated lesions and can detect microscopic amounts of blood, as well as non-hemorrhagic lesions secondary to axonal strain (Figure 2). In addition, MRI provides means to assess and visually reconstruct white matter tracts following DAI, with high sensitivity for lesion detection using DTI. Furthermore, neurochemical alterations following axonal injury can be detected with magnetic resonance spectrometry (MRS).
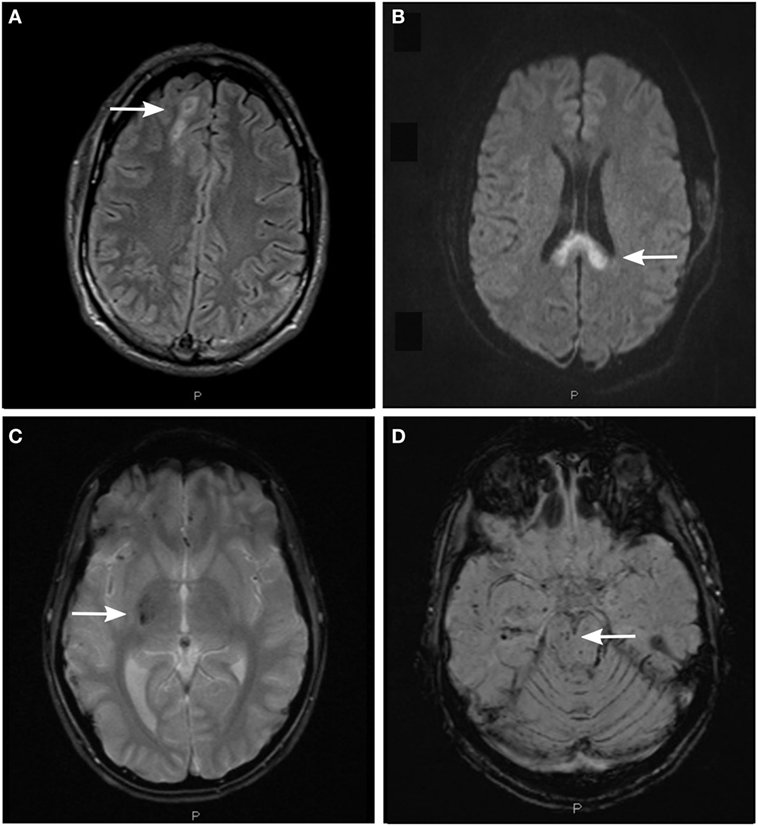
Figure 2. Detection of axonal injury with conventional magnetic resonance imaging (MRI) using different MRI sequences. (A) Fluid-attenuated inversion recovery (FLAIR) image depicting non-hemorrhagic diffuse axonal injury (DAI)-associated lesions in the subcortical white matter of the right cerebral hemisphere (arrow). (B) Diffusion-weighted image (DWI) depicting non-hemorrhagic DAI-associated lesions in the body and splenium of the corpus callosum. (C) T2*-weighted gradient echo (T2*GRE) image depicting hemorrhagic DAI-associated lesions in the right thalamus and putamen (arrow). (D) Susceptibility-weighted image (SWI) depicting hemorrhagic DAI-associated lesions in the right mesencephalon (arrow) and in the white matter of right temporal lobe.
Conventional MRI Sequences for DAI Monitoring
Magnetic resonance imaging sequences sensitive to hemorrhagic lesions include T2*-weighted gradient echo (T2*GRE) and susceptibility-weighted imaging (SWI). Both sequences can detect microhemorrhages, taking advantage of the paramagnetic properties of hemoglobin degradation products. The lesions are typically seen as small hypointense foci in DAI predilection sites and appear larger than their true size due to the magnetic field distortion. Hemorrhagic lesions seem to be fairly stable over time although some reduction of lesion numbers may be seen in the chronic phase following DAI (84, 87). Adding sensitivity of microhemorrhage detection in deep-seated brain regions, SWI has emerged as a preferred MRI sequence (88, 89) particularly in brain regions such as central brain stem previously difficult to assess (62). However, this sequence may be more complicated to interpret, since deoxygenated blood in veins can mimic hemorrhagic lesions. Nonetheless, lesions seen on SWI sequence correlate strongly to outcome (62, 90), in contrast to T2*GRE (62, 87, 91).
The Fluid-Attenuated Inversion Recovery (FLAIR) sequence facilitates detection of non-hemorrhagic lesions adjacent to cerebrospinal fluid (CSF) spaces. This sequence is useful for visualizing axonal injuries in periventricular white matter, the corpus callosum and the brain stem (84, 85). However, its capacity to visualize axonal injury is highly dependent on the timing. Lesions demonstrated in the acute phase represent tissue edema, but some seem to disappear already by 3 months post-injury (84). On the other hand, FLAIR lesions represent encephalomalacia (softening or loss of brain tissue) or tissue gliosis at the long-term, chronic phase in DAI (92).
The diffusion-weighted imaging (DWI) sequence is sensitive to the microscopic motion of water molecules (93), allowing for excellent detection of non-hemorrhagic lesions following axonal shearing. Lesions on DWI seem to correlate with initial glasgow coma scale (GCS) score and coma duration in DAI (91), and are associated with poor outcome in pediatric TBI (94). Specifically, the DWI lesion load in the corpus callosum may be of particular importance (85). However, similar to the FLAIR sequence, timing of the MRI scan is imperative when assessing DWI images (95) with the number of lesions being significantly reduced at 3 months post-TBI (84).
MRI can be difficult to obtain, particularly in the critically ill, since patients’ transfer to the MRI facility may be prevented by clinical instability from intracranial and/or systemic causes. In situations where the time for MRI must for clinical reasons be extended beyond the acute phase, hemorrhagic lesions depicted in particular on SWI sequence seem to have higher prognostic value than other MR sequences. It is notable that conventional MRI sequences may still be insensitive to microstructural damage to axons and injury to white matter tracts of clinical significance can still be missed with this imaging modality (96).
Diffusion Tensor Imaging
By using image acquisition in multiple directions, the anisotropic diffusion of water molecules can be used to create DTI, providing anatomical reconstruction images of white matter tracts and quantitative measurements of axonal injury (97). DTI is more sensitive for DAI than conventional MRI, and can be used to visualize ultrastructural changes. By adding post-processing techniques to the DTI data, diffusion tensor tractography can visualize the three-dimensional anatomy of white matter tracts (98, 99). Reduction of fractional anisotropy (FA) and increased diffusivity are observed following DAI in numerous studies (100–102), they correlate to TBI severity (103, 104) and are strongly associated to cognitive and behavioral deficits in both adult and pediatric patients (10, 105–110). In addition, DTI detection of axonal injury has been cross-validated using microdialysis (MD), where FA reduction correlated to interstitial fluid (ISF) tau levels (111). As for monitoring of axonal integrity, DTI parameters have shown signs of ongoing microstructural changes long after the acute phase (105, 107, 112–114). Longitudinal studies suggest continuous changes of DTI parameters, where FA decreases over time while diffusivity increases following DAI (102, 112–114). Measurable deterioration of white matter integrity continues beyond 24 months post-injury (105, 112–115), but may stabilize thereafter (105, 115).
In summary, DTI is a robust tool to visualize posttraumatic white matter abnormalities. However, variations in data acquisition, analysis techniques, spatial location of investigated structures, lack of correlation with clinical findings, and costs (116) still impede generalized conclusions of its applied utility in DAI. Moreover, DAI lesions seen on DTI are currently used predominantly for diagnostic purposes since patient management remains predominantly symptomatic awaiting the implementation of novel pharmacological treatments.
Magnetic Resonance Spectroscopy
Magnetic resonance spectroscopy takes advantage of the chemical shift, a phenomenon caused by variations of proton resonance due to the local chemical environment. Although MRS has a low spatial resolution in comparison to MRI, it provides a mean of detecting and quantifying neurochemical alterations (117). N-acetyl aspartate (NAA), a marker for neuronal and axonal integrity found in high concentrations in neurons (118), and choline (Cho), which is increased after damage to cell membranes (119), are well-studied metabolites in relation to TBI. In most studies, decreased NAA and increased Cho is observed and in mild TBI, the NAA and Cho levels can appreciate axonal injury undetected by conventional MRI (120–124). These MRS findings are associated with neurocognitive deficits (125–127) and global outcomes (120, 121, 128). Longitudinal studies suggest recovery of decreased NAA levels in patients with mild DAI and/or better outcomes, implying marginal dysfunction of neurons and restoration of function over time (120, 122). However, complete recovery of NAA levels may also be possible following severe DAI (129).
Thus, MRS provides a mean for detecting and monitoring alterations in brain chemistry following DAI, although its clinical utility in DAI patients has still not been well defined. This is likely due to the lack of standardized protocols for measurements and interpretation of metabolite concentrations, a shortcoming that needs to be addressed in future studies.
Neuromolecular Imaging
Single-Photon Emission Computed Tomography
Single-photon emission computed tomography (SPECT) uses radiopharmaceutical agents to produce images of physiologic or pathological processes. For TBI, [99mTc] Hexamethylpropylenamine oxime (HMPAO) and [99mTc] Ethylcisteinate dimer are the most widely used agents for evaluating regional cerebral blood flow (rCBF) and indirectly, regional cerebral metabolism (130). Following DAI, decreased rCBF commonly involving the cingulate gyrus revealed signs of frontal lobe dysfunction, despite the absence of distinct anatomical abnormalities (131, 132). One plausible explanation to these alterations is deafferentation of interconnecting white matter due to widespread axonal damage, causing reduction of metabolic activity and eventually neurocognitive deficits (133, 134).
Although SPECT cannot be independently used in the evaluation of DAI due to limited image resolution and sensitivity, it provides an available and affordable adjunct measure to other anatomical imaging modalities of white matter injury.
Positron Emission Tomography (PET)
Imaging of physiologic and biochemical processes following DAI is also possible by PET, using radiopharmaceutical agents labeled with positron-emitting radioisotopes such as fluorine-18 [18F], carbon-11 [11C], and oxygen-15 [15O]. The [18F]-labeled fluorodeoxyglucose (FDG) PET is widely used in brain imaging to measure local glucose metabolism, and thus regional neuronal activity. Similarly, FDG PET studies of DAI patients have revealed regional hypometabolism in medial frontal lobe structures including the cingulate gyrus (134), findings associated with neuropsychological and cognitive symptoms (134, 135). Additionally, neuroinflammatory alterations can be studied with [11C] PK11195, reflecting microglial activation (136). Using this tracer, widespread neuroinflammation post-TBI was observed in subcortical structures of DAI patients (136, 137). Furthermore, amyloid binding tracers commonly used in Alzheimer’s disease have recently established PET as a method for imaging of amyloid-β (Aβ) also in TBI (138, 139). Aβ retention signals were assessed in nine TBI patients, of which four had DAI, and appeared to peak within the first week post-injury (138) and correlate with white matter damage. However, in the chronic stage, Aβ retention signals were also increased with longer time from the initial injury (139).
Future studies will provide knowledge on the topographical distribution and temporal patterns of Aβ deposition following axonal injury, and their relation to the development of neurodegenerative diseases. Although PET scan is a powerful tool which offers superior image resolution, sensitivity, and quantification of regional radioactivity concentrations compared to SPECT (140), its main disadvantage remains the requirement of an on-site cyclotron which considerably increases cost and limits availability.
Fluid and Structural Biomarkers
Per definition, biomarkers are molecules measurable in biological fluids and structures given that the measurable value is related to a biological or pathological process in the body (141, 142) (Table 1).
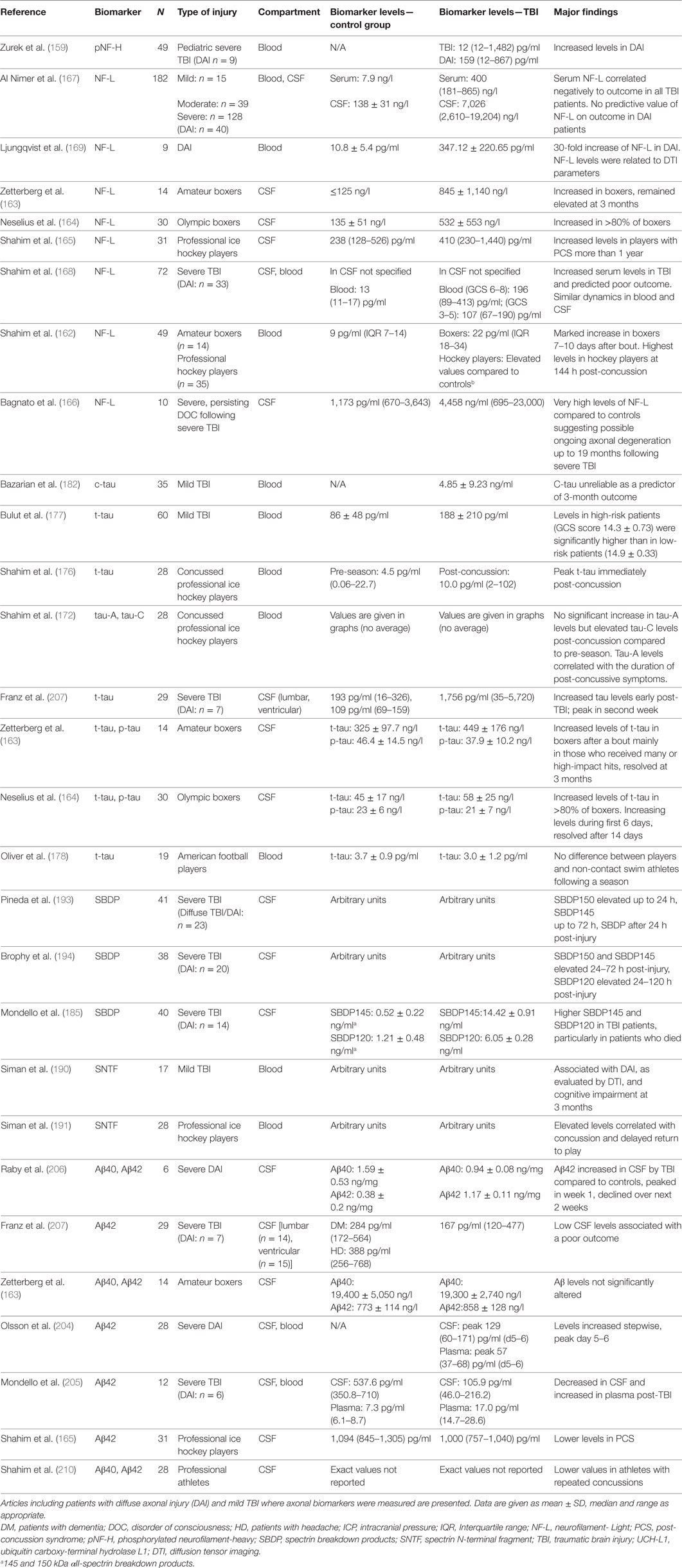
Table 1. Blood and cerebrospinal fluid (CSF) levels of common axonal injury biomarkers (neurofilament, tau, SBDP and amyloid-β) in clinical TBI.
Biomarkers may be subdivided into four categories: diagnostic, prognostic, predictive, and pharmacodynamic and can potentially be used to examine injury severity, monitor pathophysiology of injury, explain adaptive and recovery processes, guide management, predict response to treatment and estimate prognosis following DAI/TBI (141–143). Biomarkers can thus be considered a reflection of the mechanisms resulting in axonal injury, where the underlying structural changes are to a large extent related to the activation of the calpain and caspase enzymes. These enzymes belong to the cysteine protease family and play an important role in cell necrosis and apoptosis (144–150). They are activated by calcium influx leading to cytoskeletal disruption including an impaired axoplasmic transport, axonal swelling and eventually axonal transection/lysis (35, 151–154).
Thus, following TBI and in particular following axonal injury, a delayed axonal transection may occur resulting in the release and accumulation of various biomarkers which can be detected in plasma, CSF, and ISF using cerebral microdialysis (MD) (141, 155) and can, therefore, be used for monitoring. Biomarkers reviewed more extensively below are neurofilaments, tau, Spectrin breakdown products (SBDP) and Aβ and are summarized in Tables 1 and 2.
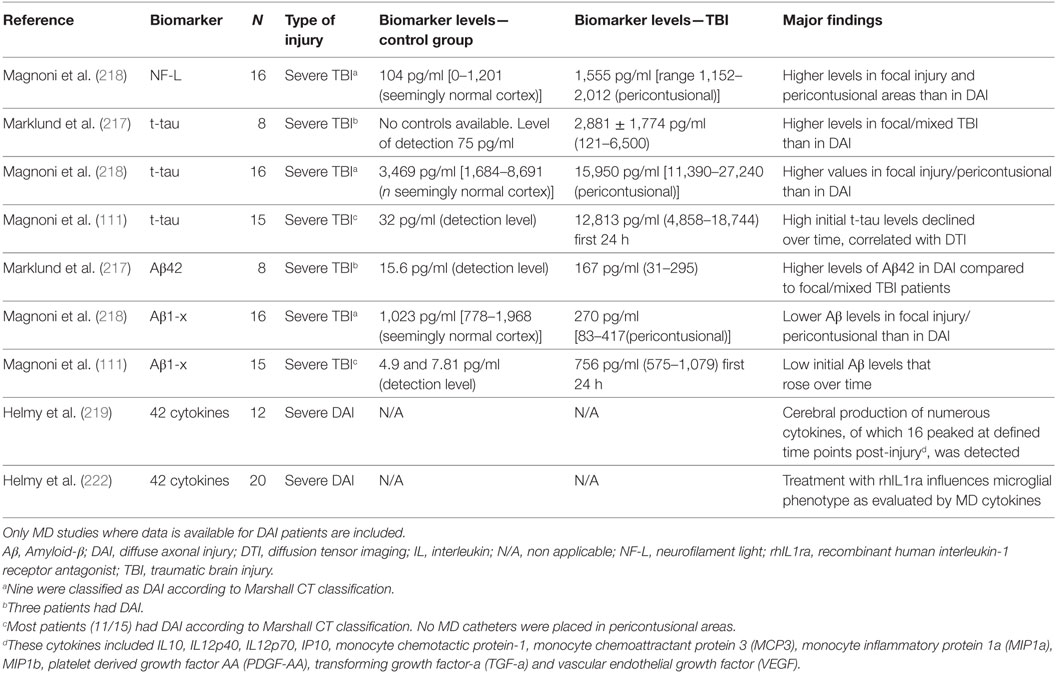
Table 2. Results from cerebral microdialysis (MD) studies of commonly used biomarkers for monitoring axonal injury in clinical DAI.
NFL
Neurofilaments (NF) are important components of the axonal cytoskeleton, mainly involved in synapses and neurotransmission (156). They represent intermediate neuronal filaments and include three major subunits: neurofilament light (NF-L), neurofilament medium and neurofilament heavy chain (NF-H) (156). The latter becomes phosphorylated (pNF-H), likely by TBI-induced calcium influx, which can alter axonal integrity (156). Of the three subunits, NF-L is rapidly degraded following axonal injury (157) making it a rather sensitive and specific biomarker for the detection of injured axons (5, 141, 158). Following axotomy, phosphorylated neurofilaments (pNF-H) are released in CSF and blood, correlating with injury severity and outcome both in the pediatric and adult population (159, 160).
Neurofilament light fragments can also be identified in both blood and CSF in TBI (143, 161, 162). Following mild and repetitive impacts to the head like those occurring in contact sports such as boxing, American football and ice hockey, increased levels of NF-L may be associated primarily with injury to long, myelinated axons (8, 162–165). Recently, very high levels of NF-L compared to controls were found in 10 patients with impaired level of consciousness following TBI, with the samples taken at least 10 months following injury, results suggesting ongoing axonal degeneration (166).
Increased serum and CSF NF-L levels in TBI patients did also correlate with clinical outcome although without any predictive value for DAI (167). In another study that included 72 patients with severe TBI (of which 33 had DAI), initial NF-L levels independently predicted clinical outcome (168). Additionally, in patients with severe DAI, a 30-fold increase in serum NF-L was recently found (169).
Repeated biomarker sampling during the course of the disease as well as the correlation with advanced neuroimaging is expected to better discern the role of neurofilaments in DAI, their contribution in the pathophysiology of DAI and their prognostic value on outcome.
Tau
Tau is a structural protein with six isoforms in humans and is a normal constituent of axons. Four distinct isoforms of tau are usually applied in biomarker studies; total-tau (t-tau), cleaved microtubule-associated tau (c-tau), phosphorylated tau (p-tau), and the recently discovered tau-A (155, 170–172). Tau has been linked to axonal damage following TBI (141, 173). Specifically, the presence of c-tau in CSF is a highly sensitive indicator of axonal injury (35).
In patients with DAI, t-tau and p-tau levels also increase rapidly within hours after injury, especially in CSF (35, 170, 174). Increased CSF levels of t-tau were found in boxers after repetitive head injury, although this increase was modest compared to that of NF-L (163, 164).
The Simoa platform has shown excellent analytical sensitivity for tau in serum (8, 175). Serum c-tau levels are increased but at much lower levels than in CSF and have been used as an indicator of blood–brain barrier damage (35). Compared to off-season levels, serum t-tau levels were elevated in ice hockey players sustaining a concussion, with the highest levels detected immediately after injury (141, 176). In mild TBI, t-tau levels were found to be higher in high-risk patients with greater likelihood for TBI-related complications than in low-risk individuals (177). However, no difference in serum tau levels was recently noted in American football athletes (178).
In DAI, CSF c-tau correlated negatively with the degree of clinical improvement (170, 179, 180). Furthermore, increased serum c-tau levels were associated with poor outcome in patients with mild TBI (181). In contrast, another study found that c-tau is not a reliable predictor for 3-month outcome following mild TBI (182). In concussed professional ice hockey players, the levels of the newly discovered biomarker tau-A correlated with the duration of symptoms post-injury, and may possibly predict return to play (172).
The association of elevated tau levels with axonal damage is well established. Especially in severe DAI, high tau levels are associated with worse outcome. Ongoing and future research efforts need to focus more on its possible correlation with the extent of injury, interaction with other blood and CSF biomarkers, long-term sequelae, and clinical outcome.
Spectrin Breakdown Products
Spectrin is a cytoskeletal protein playing an important role in the cytoskeletal structure and maintenance of plasma membrane (183). In DAI, spectrin is proteolytically cleaved by calpain, resulting in cytoskeletal destruction (184). SBDP are increased in human CSF and blood following severe TBI and may predict injury severity and outcome (5, 185).
In rodents, SBDP are detected within minutes after DAI (186–188). In human TBI, αII-spectrin N-terminal fragment (SNTF) accumulates in injured axons, rises in serum as early as 1 h after mild TBI and correlates with cognitive impairment (141, 189–191). Importantly, SNTF immunoreactive axons have been also identified both in mild and severe TBI (192). Serum SNFT levels were also increased early after concussion in ice hockey players, particularly in more severe injuries (141, 191).
In severe TBI patients, elevated levels of calpain-mediated 150- and 145-kDa SBDP in CSF were found 24–72 h post-injury (193, 194) which were associated with the initial injury severity and 6-month outcome (193). In addition, CSF calpain-mediated SBDP levels correlated positively with the severity of injury, lesion size, and behavioral deficits in severe TBI, suggesting that CSF SBDPs could be used to evaluate the magnitude of axonal injury and predict functional deficits (193).
Spectrin breakdown products are relatively new biomarkers detected in serum and CSF. Therefore, ample evidence on their significance in the clinical setting is lacking. However, available data suggests that they represent promising molecules in determining the extent of axonal injury and its association to outcome.
Amyloid-β Peptides
Axonal injury in TBI has been characterized by amyloid precursor protein (APP) immunohistochemistry, accumulating at sites of axonal transport failure (8, 195, 196). The presence of APP-positive axonal bulbs and grossly swollen axons are main findings in DAI (195), observed within hours in severe TBI patients (197). However, APP is not a specific diagnostic marker of DAI, since it may also be detected in non-traumatic, ischemic, axonal injury and in multiple sclerosis plaques (35, 198–200). APP co-accumulates with the enzymes necessary for its cleavage to Aβ peptides, such as presenelin-1 and beta-site APP-cleaving enzyme (19, 201, 202). Conversely, notable amount of Aβ has been repeatedly found in axonal bulbs (17, 32, 201–203).
By cleaving APP, the Aβ peptides Aβ40 and Aβ42, the substrates for Aβ aggregates/plaques also observed in Alzheimer’s disease, are produced (19). APP and Aβ species are rapidly detectable following TBI in plasma (204, 205), CSF (205–207) and ISF (208, 209). In severe TBI, monomeric Aβ levels in ventricular CSF were increased stepwise until 5–6 days after injury, although not in plasma (204). Conversely, a more recent study using an ultrasensitive digital immunoassay evaluating 12 severe TBI patients of which 6 had DAI, reduced CSF levels of Aβ42 direct after injury with lower levels in patients who died 6 months post-injury were observed. In the same study, plasma levels were increased with lower levels detected in surviving patients (205). The differences in analytical methods may partly explain the discrepancy in the results between these studies. Additionally, the latter study also included patients with focal TBI, although no difference in Aβ levels was observed between TBI subtypes (205). Similarly, lower CSF levels of Aβ40 and Aβ42 were recently detected in professional athletes following concussions (165, 210).
An increased interest in soluble intermediary Aβ oligomers/protofibrils as the pathogenic form of Aβ has emerged since they are likely to contribute to the development of Alzheimer’s disease (211, 212). Aβ oligomers have been detected in lumbar CSF from severe TBI patients, were elevated in patients with poor neurological outcomes and were negatively correlated to CSF Aβ42 (213). Therefore, it is plausible that aggregation of Aβ into oligomers may explain the reduced levels of CSF Aβ seen in TBI. However, the corresponding brain tissue levels of soluble intermediary Aβ species and their role in human DAI remains to be established. In addition, these potentially neurotoxic species could represent a pathophysiologic link between DAI and Alzheimer disease-like dementia.
The association between TBI and the development of neurodegenerative diseases, in particular Alzheimer’s disease, has been repeatedly demonstrated (24, 214). Longitudinal monitoring of Aβ dynamics may provide further knowledge of neurodegenerative processes following DAI. Aβ42 levels, which can be monitored in both CSF and blood are likely the most promising biomarker from the amyloid family for detecting the extent and severity of DAI. In addition, specific monitoring of potentially neurotoxic oligomeric and protofibrillar Aβ species will become possible using newly developed antibody-based PET imaging (215). This will further increase the understanding of the potential link between DAI and neurodegeneration.
Biomarkers and Cerebral Microdialysis
Cerebral MD is a neurocritical care monitoring technique predominantly used in patients with severe TBI and subarachnoid hemorrhage (Table 2). Its main advantage is that it allows continuous neurochemical monitoring of factors located in the extracellular, interstitial fluid (216).
Using MD on 8 severe TBI patients, particularly high ISF Aβ42 values were found in the three DAI patients (217). In another MD study, in nine DAI patients the initial Aβ levels inversely correlated with tau levels in ISF (218), suggesting that low Aβ levels in regions with elevated tau may be due to reduced synaptic activity after axonal injury (218).
Tau was also evaluated by MD in eight severe TBI patients (217). Although mean t-tau levels were clearly above the detection limit in the first days after injury, patients with focal/mixed injury (n = 5) had lower levels compared to those with DAI (n = 3). Conversely, in a previous MD study, higher tau values were observed pericontusionally in focal TBI patients when compared to tau levels obtained from DAI patients with the MD catheter placed in structurally normal frontal cortex. Early tau levels were inversely correlated with the initial Aβ levels (155, 218). In this study, NF-L levels were also higher in pericontusional tissue (218). Further, in a study of 15 patients with severe TBI (11/15 had DAI), initially high t-tau levels in ISF declined over time and a correlation with DTI and reduced brain white matter integrity in the region of MD sampling was observed, suggesting that increased tau levels reflected axonal injury (111).
The cytokine response was evaluated by MD in patients with severe DAI suggesting that cytokine production is highly compartmentalized with significant differences between brain parenchymal and systemic concentrations (219–222). Several cytokines are produced in different phases of the inflammatory response (220). It has been also shown that in DAI patients, treatment with an interleukin receptor-1 antagonist increased microglial activation, altering the cytokine profile to one consistent with an M1 microglial phenotype, providing proof of concept that an anti-inflammatory treatment administered systemically can alter cerebral cytokine productions in human TBI (222). These data suggested that the patterns of cytokine release in ISF are promising targets for biomarker research in DAI.
Following DAI, markers analyzed in MD samples indicating acute and chronic neuroinflammation may potentially be used to guide treatment, as measures for pharmacological response, and/or for tissue outcome. Specifically, MD may aid in detecting factors related to the progression of the disease and in the understanding of the pathophysiology of axonal injury. Therefore, it is likely that data from MD, in combination with widely used measures such as ICP-CPP guided monitoring and protocols, will contribute to the understanding of the pathophysiology of DAI and potentially aid in evaluating novel pharmacological treatments.
However, MD is time consuming, usually used for low-molecular weight molecules and frequently characterized by high variability and lack of standardization. In addition, MD remains a predominantly focal measurement technique. To date, there is insufficient data arguing for MD to be used as a clinical decision tool for DAI patients and should rather be considered an integral part of the multimodality monitoring during neurocritical care as well as a research tool.
Other Biomarkers
There are numerous additional biomarkers associated to CNS injury that can potentially be related to axonal injury. Examples of such biomarkers are Glial Fibrillary Acidic Protein, ubiquitin carboxy-terminal hydrolase L1 myelin basic protein, microtubule-associated protein 2, protein S-100B and neuron-specific enolase, among others (5, 35, 141–143, 223–228). Especially the astrocytic protein S-100B is a promising biomarker across all injury severities, with higher S-100B levels observed in focal compared to diffuse TBI (229, 230). Moreover, it has been shown that S-100B correlates with Marshall CT classification scores (231). Furthermore, following TBI, neuroinflammation may as previously noted play an important role as a key secondary injury factor (141, 232). Specifically, it has been repeatedly shown both in the experimental and clinical setting that cytokines such as Tumor Necrosis Factor-α and Interleukins (ILs) 1β, 6, 8, and 10 are increased following TBI both in blood and CSF (233).
Since the above mentioned biomarkers are not specific for axonal injury, at present, their elevations in CSF or serum should be interpreted with caution from both the diagnostic and predictive perspective with regards to DAI.
Limitations of Biomarkers
Although considerable progress has been made in the recent years in research, the quest for TBI-specific biomarkers continues. The currently used biomarkers commonly have different specificity- and/or sensitivity, limited availability for bedside analysis and use in daily practice as well as variable half-lives. Moreover, some can also be released from other organ systems during different disease or injury processes.
Biomarkers obtained from CSF and ISF are theoretically considered better and more reliable sources compared to blood biomarkers. Therefore, it is preferable to obtain samples from these compartments whenever possible. However, in particular in mild and moderate TBI, there is generally no clinical indication for CSF and, for obvious reasons, ISF sampling by invasive means. On the other hand, blood samples are easily accessible in almost every TBI patient.
Nevertheless, important issues relevant to TBI-related blood biomarkers include their relatively low concentrations, proteolytic degradation, the requirement of carrier proteins and the different permeability across the blood–brain barrier for certain biomarkers (141).
Standardization and validation of biomarker levels are other important issues since different methods of analysis, preparation and sample quality can provide different results among laboratories and centers which can cause problems during the interpretation and comparison of results (234). As the field of biomarker research is expanding, CNS sensitivity and/or specificity increases their importance. Analysis of many currently used biomarkers requires specialized research laboratories which are not available on a daily basis. Additionally, difficulties may be encountered when attempting to assess biomarker half-time, especially in those that are continuously released from the brain following injury and in those with complex elimination or degradation mechanisms (230).
Recommendations for Monitoring Axonal Injury
1. ICP and CPP monitoring as well as ICP-CPP guided therapy are advised in all severe TBI patients with suspected axonal injury and decreased level of consciousness especially in the initial post-injury phase.
2. Conventional MRI scan sequences such as FLAIR, DWI, and SWI should be considered in the first post-injury period following TBI to detect and confirm the presence of DAI.
3. Advanced MRI techniques such as DTI and MRS are useful modalities for further delineation of axonal damage in TBI, particularly in the subacute and chronic phase.
4. Due to high cost and limited availability, PET scanning is recommended solely as a valuable research tool although not to date in clinical management.
5. Biomarkers specific for axonal injury can be analyzed in blood and CSF from the acute to chronic post-injury period in TBI, aiming to aid in the understanding of the axonal injury process, follow the course of the disease, monitor for possible deterioration, estimate the extent of axonal injury and aid in prognostication.
6. Repeated clinical examinations and neuropsychological tests can provide invaluable information on the extent of injury, prognosis and for monitoring possible recovery or exacerbation of cognitive functions and mental status.
Conclusion
Although axonal injury has traditionally been associated with an impaired level of consciousness and poor prognosis, patients with confirmed axonal damage can achieve a good clinical outcome. Using advanced neuroimaging, axonal injury is increasingly recognized also in mild TBI or sports-related concussions. Many tenets of the pathophysiology of axonal injury are being elucidated through major efforts in basic science and medical research. Nonetheless, it remains an exceedingly complex subtype of TBI with many unknown secondary pathological processes. Since the secondary injury cascades are continuing for a considerable time post-injury, monitoring is critically important for clinical as well as research purposes. Advanced imaging techniques such as MRS, DTI and PET show promise in better identifying and quantifying axonal injury and its importance for patient outcome. In addition, both invasive and non-invasive neurocritical care techniques are becoming increasingly important in monitoring axonal injury. Numerous biomarkers with, plausibly, high specificity for axonal damage have been and are being developed. This evolving field of TBI research is promising for the development of bedside, rapid analysis kits for small-volume body fluids. When these novel biomarkers are available for routine use as monitoring tools for axonal injury, they may in the future aid in the detection and prevention of secondary axotomy and atrophy of white matter tracts. They may also be used as secondary outcome measures in DAI, assist in the development of novel therapies, guide treatment, and monitor treatment response. Finally, the short-and long-term monitoring options for axonal pathology and its progression described in this review may become crucial for the prevention of neurodegeneration at the chronic stage in DAI.
Author Contributions
Study concept and design, drafting of the manuscript, analysis and interpretation of data, and critical revision of the manuscript: PT, SAH, and NM. Acquisition of data: PT and SAH. Study supervision: NM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ghajar J. Traumatic brain injury. Lancet (2000) 356(9233):923–9. doi:10.1016/S0140-6736(00)02689-1
2. Park E, Bell JD, Baker AJ. Traumatic brain injury: can the consequences be stopped? CMAJ (2008) 178(9):1163–70. doi:10.1503/cmaj.080282
3. Corrigan JD, Selassie AW, Orman JA. The epidemiology of traumatic brain injury. J Head Trauma Rehabil (2010) 25(2):72–80. doi:10.1097/HTR.0b013e3181ccc8b4
4. Rubiano AM, Carney N, Chesnut R, Puyana JC. Global neurotrauma research challenges and opportunities. Nature (2015) 527(7578):S193–7. doi:10.1038/nature16035
5. Hill CS, Coleman MP, Menon DK. Traumatic axonal injury: mechanisms and translational opportunities. Trends Neurosci (2016) 39(5):311–24. doi:10.1016/j.tins.2016.03.002
6. Murray GD, Teasdale GM, Braakman R, Cohadon F, Dearden M, Iannotti F, et al. The European brain injury consortium survey of head injuries. Acta Neurochir (Wien) (1999) 141(3):223–36. doi:10.1007/s007010050292
7. Cremer OL, Moons KG, van Dijk GW, van Balen P, Kalkman CJ. Prognosis following severe head injury: development and validation of a model for prediction of death, disability, and functional recovery. J Trauma (2006) 61(6):1484–91. doi:10.1097/01.ta.0000195981.63776.ba
8. Blennow K, Brody DL, Kochanek PM, Levin H, McKee A, Ribbers GM, et al. Traumatic brain injuries. Nat Rev Dis Primers (2016) 2:16084. doi:10.1038/nrdp.2016.84
9. Mesulam MM. From sensation to cognition. Brain (1998) 121(Pt 6):1013–52. doi:10.1093/brain/121.6.1013
10. Kinnunen KM, Greenwood R, Powell JH, Leech R, Hawkins PC, Bonnelle V, et al. White matter damage and cognitive impairment after traumatic brain injury. Brain (2011) 134(Pt 2):449–63. doi:10.1093/brain/awq347
11. Sharp DJ, Scott G, Leech R. Network dysfunction after traumatic brain injury. Nat Rev Neurol (2014) 10(3):156–66. doi:10.1038/nrneurol.2014.15
12. Filley CM, Fields RD. White matter and cognition: making the connection. J Neurophysiol (2016) 116(5):2093–104. doi:10.1152/jn.00221.2016
13. Strich SJ. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry (1956) 19(3):163–85. doi:10.1136/jnnp.19.3.163
14. Adams JH. Diffuse axonal injury in non-missile head injury. Injury (1982) 13(5):444–5. doi:10.1016/0020-1383(82)90105-X
15. Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology (1989) 15(1):49–59. doi:10.1111/j.1365-2559.1989.tb03040.x
16. Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma (1994) 11(2):173–86. doi:10.1089/neu.1994.11.173
17. Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. J Head Trauma Rehabil (2003) 18(4):307–16. doi:10.1097/00001199-200307000-00003
18. Li XY, Feng DF. Diffuse axonal injury: novel insights into detection and treatment. J Clin Neurosci (2009) 16(5):614–9. doi:10.1016/j.jocn.2008.08.005
19. Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol (2013) 246:35–43. doi:10.1016/j.expneurol.2012.01.013
20. Armstrong RC, Mierzwa AJ, Marion CM, Sullivan GM. White matter involvement after TBI: clues to axon and myelin repair capacity. Exp Neurol (2016) 275(Pt 3):328–33. doi:10.1016/j.expneurol.2015.02.011
21. Ma J, Zhang K, Wang Z, Chen G. Progress of research on diffuse axonal injury after traumatic brain injury. Neural Plast (2016) 2016:9746313. doi:10.1155/2016/9746313
22. Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol (2002) 103(6):607–14. doi:10.1007/s00401-001-0510-8
23. Ding K, Marquez de la Plata C, Wang JY, Mumphrey M, Moore C, Harper C, et al. Cerebral atrophy after traumatic white matter injury: correlation with acute neuroimaging and outcome. J Neurotrauma (2008) 25(12):1433–40. doi:10.1089/neu.2008.0683
24. Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol (2013) 9(4):211–21. doi:10.1038/nrneurol.2013.29
25. Gennarelli TA, Thibault LE, Adams JH, Graham DI, Thompson CJ, Marcincin RP. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol (1982) 12(6):564–74. doi:10.1002/ana.410120611
27. Reilly PL. Brain injury: the pathophysiology of the first hours. ‘Talk and die revisited’. J Clin Neurosci (2001) 8(5):398–403. doi:10.1054/jocn.2001.0916
28. Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain (2003) 126(Pt 3):515–30. doi:10.1093/brain/awg061
29. Andriessen TM, Jacobs B, Vos PE. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J Cell Mol Med (2010) 14(10):2381–92. doi:10.1111/j.1582-4934.2010.01164.x
30. Reeves TM, Phillips LL, Povlishock JT. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol (2005) 196(1):126–37. doi:10.1016/j.expneurol.2005.07.014
31. Meythaler JM, Peduzzi JD, Eleftheriou E, Novack TA. Current concepts: diffuse axonal injury-associated traumatic brain injury. Arch Phys Med Rehabil (2001) 82(10):1461–71. doi:10.1053/apmr.2001.25137
32. Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci (1999) 19(11):4263–9.
33. Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci (2001) 21(6):1923–30.
34. Shaw G, Yang C, Ellis R, Anderson K, Parker Mickle J, Scheff S, et al. Hyperphosphorylated neurofilament NF-H is a serum biomarker of axonal injury. Biochem Biophys Res Commun (2005) 336(4):1268–77. doi:10.1016/j.bbrc.2005.08.252
35. Li J, Li XY, Feng DF, Pan DC. Biomarkers associated with diffuse traumatic axonal injury: exploring pathogenesis, early diagnosis, and prognosis. J Trauma (2010) 69(6):1610–8. doi:10.1097/TA.0b013e3181f5a9ed
36. Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol (2009) 46(6):821–31. doi:10.1016/j.yjmcc.2009.02.021
37. Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol (2007) 66(11):989–1001. doi:10.1097/NEN.0b013e3181588245
38. Helmy A, De Simoni MG, Guilfoyle MR, Carpenter KL, Hutchinson PJ. Cytokines and innate inflammation in the pathogenesis of human traumatic brain injury. Prog Neurobiol (2011) 95(3):352–72. doi:10.1016/j.pneurobio.2011.09.003
39. Vieira RC, Paiva WS, de Oliveira DV, Teixeira MJ, de Andrade AF, de Sousa RM. Diffuse axonal injury: epidemiology, outcome and associated risk factors. Front Neurol (2016) 7:178. doi:10.3389/fneur.2016.00178
40. Adams JH, Graham DI, Jennett B. The neuropathology of the vegetative state after an acute brain insult. Brain (2000) 123(Pt 7):1327–38. doi:10.1093/brain/123.7.1327
41. Iverson GL. Outcome from mild traumatic brain injury. Curr Opin Psychiatry (2005) 18(3):301–17. doi:10.1097/01.yco.0000165601.29047.ae
42. Bigler ED. Anterior and middle cranial fossa in traumatic brain injury: relevant neuroanatomy and neuropathology in the study of neuropsychological outcome. Neuropsychology (2007) 21(5):515–31. doi:10.1037/0894-4105.21.5.515
43. Wallesch CW, Curio N, Galazky I, Jost S, Synowitz H. The neuropsychology of blunt head injury in the early postacute stage: effects of focal lesions and diffuse axonal injury. J Neurotrauma (2001) 18(1):11–20. doi:10.1089/089771501750055730
44. Scheid R, Walther K, Guthke T, Preul C, von Cramon DY. Cognitive sequelae of diffuse axonal injury. Arch Neurol (2006) 63(3):418–24. doi:10.1001/archneur.63.3.418
45. Hellyer PJ, Leech R, Ham TE, Bonnelle V, Sharp DJ. Individual prediction of white matter injury following traumatic brain injury. Ann Neurol (2013) 73(4):489–99. doi:10.1002/ana.23824
46. Jang SH. Review of motor recovery in patients with traumatic brain injury. NeuroRehabilitation (2009) 24(4):349–53. doi:10.3233/NRE-2009-0489
47. Esbjornsson E, Skoglund T, Sunnerhagen KS. Fatigue, psychosocial adaptation and quality of life one year after traumatic brain injury and suspected traumatic axonal injury; evaluations of patients and relatives: a pilot study. J Rehabil Med (2013) 45(8):771–7. doi:10.2340/16501977-1170
48. Scholten AC, Haagsma JA, Andriessen TM, Vos PE, Steyerberg EW, van Beeck EF, et al. Health-related quality of life after mild, moderate and severe traumatic brain injury: patterns and predictors of suboptimal functioning during the first year after injury. Injury (2015) 46(4):616–24. doi:10.1016/j.injury.2014.10.064
49. Hagmann P, Cammoun L, Gigandet X, Meuli R, Honey CJ, Wedeen VJ, et al. Mapping the structural core of human cerebral cortex. PLoS Biol (2008) 6(7):e159. doi:10.1371/journal.pbio.0060159
50. Hayes JP, Bigler ED, Verfaellie M. Traumatic brain injury as a disorder of brain connectivity. J Int Neuropsychol Soc (2016) 22(2):120–37. doi:10.1017/S1355617715000740
51. Fagerholm ED, Hellyer PJ, Scott G, Leech R, Sharp DJ. Disconnection of network hubs and cognitive impairment after traumatic brain injury. Brain (2015) 138(Pt 6):1696–709. doi:10.1093/brain/awv075
52. Yanagawa Y, Sakamoto T, Takasu A, Okada Y. Relationship between maximum intracranial pressure and traumatic lesions detected by T2*-weighted imaging in diffuse axonal injury. J Trauma (2009) 66(1):162–5. doi:10.1097/TA.0b013e3181469857
53. Miller MT, Pasquale M, Kurek S, White J, Martin P, Bannon K, et al. Initial head computed tomographic scan characteristics have a linear relationship with initial intracranial pressure after trauma. J Trauma (2004) 56(5):967–72; discussion 972–3. doi:10.1097/01.TA.0000123699.16465.8B
54. Hiler M, Czosnyka M, Hutchinson P, Balestreri M, Smielewski P, Matta B, et al. Predictive value of initial computerized tomography scan, intracranial pressure, and state of autoregulation in patients with traumatic brain injury. J Neurosurg (2006) 104(5):731–7. doi:10.3171/jns.2006.104.5.731
55. Lee TT, Galarza M, Villanueva PA. Diffuse axonal injury (DAI) is not associated with elevated intracranial pressure (ICP). Acta Neurochir (Wien) (1998) 140(1):41–6. doi:10.1007/s007010050055
56. Toutant SM, Klauber MR, Marshall LF, Toole BM, Bowers SA, Seelig JM, et al. Absent or compressed basal cisterns on first CT scan: ominous predictors of outcome in severe head injury. J Neurosurg (1984) 61(4):691–4. doi:10.3171/jns.1984.61.4.0691
57. Poca MA, Sahuquillo J, Baguena M, Pedraza S, Gracia RM, Rubio E. Incidence of intracranial hypertension after severe head injury: a prospective study using the traumatic coma data bank classification. Acta Neurochir Suppl (1998) 71:27–30.
58. Yuan Q, Wu X, Cheng H, Yang C, Wang Y, Wang E, et al. Is intracranial pressure monitoring of patients with diffuse traumatic brain injury valuable? An observational multicenter study. Neurosurgery (2016) 78(3):361–8; discussion 368–9. doi:10.1227/NEU.0000000000001050
59. Narayan RK, Kishore PR, Becker DP, Ward JD, Enas GG, Greenberg RP, et al. Intracranial pressure: to monitor or not to monitor? A review of our experience with severe head injury. J Neurosurg (1982) 56(5):650–9. doi:10.3171/jns.1982.56.5.0650
60. O’Sullivan MG, Statham PF, Jones PA, Miller JD, Dearden NM, Piper IR, et al. Role of intracranial pressure monitoring in severely head-injured patients without signs of intracranial hypertension on initial computerized tomography. J Neurosurg (1994) 80(1):46–50. doi:10.3171/jns.1994.80.1.0046
61. Calvi MR, Beretta L, Dell’Acqua A, Anzalone N, Licini G, Gemma M. Early prognosis after severe traumatic brain injury with minor or absent computed tomography scan lesions. J Trauma (2011) 70(2):447–51. doi:10.1097/TA.0b013e3182095e14
62. Abu Hamdeh S, Marklund N, Lannsjo M, Howells T, Raininko R, Wikström J, et al. Extended anatomical grading in diffuse axonal injury using MRI: hemorrhagic lesions in the substantia nigra and mesencephalic tegmentum indicate poor long-term outcome. J Neurotrauma (2017) 34(2):341–52. doi:10.1089/neu.2016.4426
63. Chesnut R, Videtta W, Vespa P, Le Roux P; Participants in the International Multidisciplinary Consensus Conference on Multimodality Monitoring. Intracranial pressure monitoring: fundamental considerations and rationale for monitoring. Neurocrit Care (2014) 21(Suppl 2):S64–84. doi:10.1007/s12028-014-0048-y
64. Stocchetti N, Picetti E, Berardino M, Buki A, Chesnut RM, Fountas KN, et al. Clinical applications of intracranial pressure monitoring in traumatic brain injury: report of the Milan consensus conference. Acta Neurochir (Wien) (2014) 156(8):1615–22. doi:10.1007/s00701-014-2127-4
65. Gennarelli TA, Spielman GM, Langfitt TW, Gildenberg PL, Harrington T, Jane JA, et al. Influence of the type of intracranial lesion on outcome from severe head injury. J Neurosurg (1982) 56(1):26–32. doi:10.3171/jns.1982.56.1.0026
66. Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care, AANS/CNS, Bratton SL, et al. Guidelines for the management of severe traumatic brain injury. VI. Indications for intracranial pressure monitoring. J Neurotrauma (2007) 24(Suppl 1):S37–44. doi:10.1089/neu.2007.9990
67. Roh D, Park S. Brain multimodality monitoring: updated perspectives. Curr Neurol Neurosci Rep (2016) 16(6):56. doi:10.1007/s11910-016-0659-0
68. Rostami E, Engquist H, Enblad P. Imaging of cerebral blood flow in patients with severe traumatic brain injury in the neurointensive care. Front Neurol (2014) 5:114. doi:10.3389/fneur.2014.00114
69. Aygok GA, Marmarou A, Fatouros P, Kettenmann B, Bullock RM. Assessment of mitochondrial impairment and cerebral blood flow in severe brain injured patients. Acta Neurochir Suppl (2008) 102:57–61. doi:10.1007/978-3-211-85578-2_12
70. Bendinelli C, Bivard A, Nebauer S, Parsons MW, Balogh ZJ. Brain CT perfusion provides additional useful information in severe traumatic brain injury. Injury (2013) 44(9):1208–12. doi:10.1016/j.injury.2013.03.039
71. Honda M, Sase S, Yokota K, Ichibayashi R, Yoshihara K, Masuda H, et al. Early cerebral circulation disturbance in patients suffering from different types of severe traumatic brain injury: a xenon CT and perfusion CT study. Acta Neurochir Suppl (2013) 118:259–63. doi:10.1007/978-3-7091-1434-6_49
72. Lam JM, Hsiang JN, Poon WS. Monitoring of autoregulation using laser Doppler flowmetry in patients with head injury. J Neurosurg (1997) 86(3):438–45. doi:10.3171/jns.1997.86.3.0438
73. Soukup J, Bramsiepe I, Brucke M, Sanchin L, Menzel M. Evaluation of a bedside monitor of regional CBF as a measure of CO2 reactivity in neurosurgical intensive care patients. J Neurosurg Anesthesiol (2008) 20(4):249–55. doi:10.1097/ANA.0b013e31817ef487
74. Robertson CS, Gopinath SP, Goodman JC, Contant CF, Valadka AB, Narayan RK. SjvO2 monitoring in head-injured patients. J Neurotrauma (1995) 12(5):891–6. doi:10.1089/neu.1995.12.891
75. Beynon C, Kiening KL, Orakcioglu B, Unterberg AW, Sakowitz OW. Brain tissue oxygen monitoring and hyperoxic treatment in patients with traumatic brain injury. J Neurotrauma (2012) 29(12):2109–23. doi:10.1089/neu.2012.2365
76. Carney N, Totten AM, O’Reilly C, Ullman JS, Hawryluk GW, Bell MJ, et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery (2017) 80(1):6–15. doi:10.1227/neu.0000000000001432
77. Gutling E, Gonser A, Imhof HG, Landis T. EEG reactivity in the prognosis of severe head injury. Neurology (1995) 45(5):915–8. doi:10.1212/WNL.45.5.915
78. Haddad SH, Arabi YM. Critical care management of severe traumatic brain injury in adults. Scand J Trauma Resusc Emerg Med (2012) 20:12. doi:10.1186/1757-7241-20-12
79. Claassen J, Taccone FS, Horn P, Holtkamp M, Stocchetti N, Oddo M, et al. Recommendations on the use of EEG monitoring in critically ill patients: consensus statement from the neurointensive care section of the ESICM. Intensive Care Med (2013) 39(8):1337–51. doi:10.1007/s00134-013-2938-4
80. Gavvala J, Abend N, LaRoche S, Hahn C, Herman ST, Claassen J, et al. Continuous EEG monitoring: a survey of neurophysiologists and neurointensivists. Epilepsia (2014) 55(11):1864–71. doi:10.1111/epi.12809
81. Synek VM. Revised EEG coma scale in diffuse acute head injuries in adults. Clin Exp Neurol (1990) 27:99–111.
82. Sponheim SR, McGuire KA, Kang SS, Davenport ND, Aviyente S, Bernat EM, et al. Evidence of disrupted functional connectivity in the brain after combat-related blast injury. Neuroimage (2011) 54(Suppl 1):S21–9. doi:10.1016/j.neuroimage.2010.09.007
83. Hannawi Y, Stevens RD. Mapping the connectome following traumatic brain injury. Curr Neurol Neurosci Rep (2016) 16(5):44. doi:10.1007/s11910-016-0642-9
84. Moen KG, Skandsen T, Folvik M, Brezova V, Kvistad KA, Rydland J, et al. A longitudinal MRI study of traumatic axonal injury in patients with moderate and severe traumatic brain injury. J Neurol Neurosurg Psychiatry (2012) 83(12):1193–200. doi:10.1136/jnnp-2012-302644
85. Moen KG, Brezova V, Skandsen T, Haberg AK, Folvik M, Vik A. Traumatic axonal injury: the prognostic value of lesion load in corpus callosum, brain stem, and thalamus in different magnetic resonance imaging sequences. J Neurotrauma (2014) 31(17):1486–96. doi:10.1089/neu.2013.3258
86. Lee H, Wintermark M, Gean AD, Ghajar J, Manley GT, Mukherjee P. Focal lesions in acute mild traumatic brain injury and neurocognitive outcome: CT versus 3T MRI. J Neurotrauma (2008) 25(9):1049–56. doi:10.1089/neu.2008.0566
87. Scheid R, Preul C, Gruber O, Wiggins C, von Cramon DY. Diffuse axonal injury associated with chronic traumatic brain injury: evidence from T2*-weighted gradient-echo imaging at 3 T. AJNR Am J Neuroradiol (2003) 24(6):1049–56.
88. Haacke EM, Mittal S, Wu Z, Neelavalli J, Cheng YC. Susceptibility-weighted imaging: technical aspects and clinical applications, part 1. AJNR Am J Neuroradiol (2009) 30(1):19–30. doi:10.3174/ajnr.A1400
89. Mittal S, Wu Z, Neelavalli J, Haacke EM. Susceptibility-weighted imaging: technical aspects and clinical applications, part 2. AJNR Am J Neuroradiol (2009) 30(2):232–52. doi:10.3174/ajnr.A1461
90. Tong KA, Ashwal S, Holshouser BA, Nickerson JP, Wall CJ, Shutter LA, et al. Diffuse axonal injury in children: clinical correlation with hemorrhagic lesions. Ann Neurol (2004) 56(1):36–50. doi:10.1002/ana.20123
91. Schaefer PW, Huisman TA, Sorensen AG, Gonzalez RG, Schwamm LH. Diffusion-weighted MR imaging in closed head injury: high correlation with initial Glasgow coma scale score and score on modified Rankin scale at discharge. Radiology (2004) 233(1):58–66. doi:10.1148/radiol.2323031173
92. Parizel PM, Van Goethem JW, Ozsarlak O, Maes M, Phillips CD. New developments in the neuroradiological diagnosis of craniocerebral trauma. Eur Radiol (2005) 15(3):569–81. doi:10.1007/s00330-004-2558-z
93. Dmytriw AA, Sawlani V, Shankar J. Diffusion-weighted imaging of the brain: beyond stroke. Can Assoc Radiol J (2017) 68(2):131–46. doi:10.1016/j.carj.2016.10.001
94. Galloway NR, Tong KA, Ashwal S, Oyoyo U, Obenaus A. Diffusion-weighted imaging improves outcome prediction in pediatric traumatic brain injury. J Neurotrauma (2008) 25(10):1153–62. doi:10.1089/neu.2007.0494
95. Lansberg MG, Thijs VN, O’Brien MW, Ali JO, de Crespigny AJ, Tong DC, et al. Evolution of apparent diffusion coefficient, diffusion-weighted, and T2-weighted signal intensity of acute stroke. AJNR Am J Neuroradiol (2001) 22(4):637–44.
96. Toth A. Frontiers in neuroengineering magnetic resonance imaging application in the area of mild and acute traumatic brain injury: implications for diagnostic markers? In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton, FL: CRC Press/Taylor & Francis (2015).
97. Mukherjee P, Berman JI, Chung SW, Hess CP, Henry RG. Diffusion tensor MR imaging and fiber tractography: theoretic underpinnings. AJNR Am J Neuroradiol (2008) 29(4):632–41. doi:10.3174/ajnr.A1051
98. Treble A, Hasan KM, Iftikhar A, Stuebing KK, Kramer LA, Cox CS Jr, et al. Working memory and corpus callosum microstructural integrity after pediatric traumatic brain injury: a diffusion tensor tractography study. J Neurotrauma (2013) 30(19):1609–19. doi:10.1089/neu.2013.2934
99. Dennis EL, Jin Y, Villalon-Reina JE, Zhan L, Kernan CL, Babikian T, et al. White matter disruption in moderate/severe pediatric traumatic brain injury: advanced tract-based analyses. Neuroimage Clin (2015) 7:493–505. doi:10.1016/j.nicl.2015.02.002
100. Arfanakis K, Haughton VM, Carew JD, Rogers BP, Dempsey RJ, Meyerand ME. Diffusion tensor MR imaging in diffuse axonal injury. AJNR Am J Neuroradiol (2002) 23(5):794–802.
101. Wilde EA, Chu Z, Bigler ED, Hunter JV, Fearing MA, Hanten G, et al. Diffusion tensor imaging in the corpus callosum in children after moderate to severe traumatic brain injury. J Neurotrauma (2006) 23(10):1412–26. doi:10.1089/neu.2006.23.1412
102. Sidaros A, Engberg AW, Sidaros K, Liptrot MG, Herning M, Petersen P, et al. Diffusion tensor imaging during recovery from severe traumatic brain injury and relation to clinical outcome: a longitudinal study. Brain (2008) 131(Pt 2):559–72. doi:10.1093/brain/awm294
103. Benson RR, Meda SA, Vasudevan S, Kou Z, Govindarajan KA, Hanks RA, et al. Global white matter analysis of diffusion tensor images is predictive of injury severity in traumatic brain injury. J Neurotrauma (2007) 24(3):446–59. doi:10.1089/neu.2006.0153
104. Rutgers DR, Fillard P, Paradot G, Tadie M, Lasjaunias P, Ducreux D. Diffusion tensor imaging characteristics of the corpus callosum in mild, moderate, and severe traumatic brain injury. AJNR Am J Neuroradiol (2008) 29(9):1730–5. doi:10.3174/ajnr.A1213
105. Dinkel J, Drier A, Khalilzadeh O, Perlbarg V, Czernecki V, Gupta R, et al. Long-term white matter changes after severe traumatic brain injury: a 5-year prospective cohort. AJNR Am J Neuroradiol (2014) 35(1):23–9. doi:10.3174/ajnr.A3616
106. Veeramuthu V, Narayanan V, Kuo TL, Delano-Wood L, Chinna K, Bondi MW, et al. Diffusion tensor imaging parameters in mild traumatic brain injury and its correlation with early neuropsychological impairment: a longitudinal study. J Neurotrauma (2015) 32(19):1497–509. doi:10.1089/neu.2014.3750
107. Edlow BL, Copen WA, Izzy S, Bakhadirov K, van der Kouwe A, Glenn MB, et al. Diffusion tensor imaging in acute-to-subacute traumatic brain injury: a longitudinal analysis. BMC Neurol (2016) 16:2. doi:10.1186/s12883-015-0525-8
108. Faber J, Wilde EA, Hanten G, Ewing-Cobbs L, Aitken ME, Yallampalli R, et al. Ten-year outcome of early childhood traumatic brain injury: diffusion tensor imaging of the ventral striatum in relation to executive functioning. Brain Inj (2016) 30(13–14):1635–41. doi:10.1080/02699052.2016.1199910
109. Ressel V, O’Gorman Tuura R, Scheer I, van Hedel HJ. Diffusion tensor imaging predicts motor outcome in children with acquired brain injury. Brain Imaging Behav (2017) 11(5):1373–84. doi:10.1007/s11682-016-9637-z
110. Mohammadian M, Roine T, Hirvonen J, Kurki T, Ala-Seppala H, Frantzen J, et al. High angular resolution diffusion-weighted imaging in mild traumatic brain injury. Neuroimage Clin (2017) 13:174–80. doi:10.1016/j.nicl.2016.11.016
111. Magnoni S, Mac Donald CL, Esparza TJ, Conte V, Sorrell J, Macri M, et al. Quantitative assessments of traumatic axonal injury in human brain: concordance of microdialysis and advanced MRI. Brain (2015) 138(Pt 8):2263–77. doi:10.1093/brain/awv152
112. Moen KG, Haberg AK, Skandsen T, Finnanger TG, Vik A. A longitudinal magnetic resonance imaging study of the apparent diffusion coefficient values in corpus callosum during the first year after traumatic brain injury. J Neurotrauma (2014) 31(1):56–63. doi:10.1089/neu.2013.3000
113. Moen KG, Vik A, Olsen A, Skandsen T, Haberg AK, Evensen KA, et al. Traumatic axonal injury: relationships between lesions in the early phase and diffusion tensor imaging parameters in the chronic phase of traumatic brain injury. J Neurosci Res (2016) 94(7):623–35. doi:10.1002/jnr.23728
114. Ljungqvist J, Nilsson D, Ljungberg M, Esbjörnsson E, Eriksson-Ritzen C, Skoglund T. Longitudinal changes in diffusion tensor imaging parameters of the corpus callosum between 6 and 12 months after diffuse axonal injury. Brain Inj (2017) 31(3):344–50. doi:10.1080/02699052.2016.1256500
115. Farbota KD, Bendlin BB, Alexander AL, Rowley HA, Dempsey RJ, Johnson SC. Longitudinal diffusion tensor imaging and neuropsychological correlates in traumatic brain injury patients. Front Hum Neurosci (2012) 6:160. doi:10.3389/fnhum.2012.00160
116. Hulkower MB, Poliak DB, Rosenbaum SB, Zimmerman ME, Lipton ML. A decade of DTI in traumatic brain injury: 10 years and 100 articles later. AJNR Am J Neuroradiol (2013) 34(11):2064–74. doi:10.3174/ajnr.A3395
117. Croall I, Smith FE, Blamire AM. Magnetic resonance spectroscopy for traumatic brain injury. Top Magn Reson Imaging (2015) 24(5):267–74. doi:10.1097/rmr.0000000000000063
118. Moffett JR, Ross B, Arun P, Madhavarao CN, Namboodiri AM. N-acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol (2007) 81(2):89–131. doi:10.1016/j.pneurobio.2006.12.003
119. Miller BL, Chang L, Booth R, Ernst T, Cornford M, Nikas D, et al. In vivo 1H MRS choline: correlation with in vitro chemistry/histology. Life Sci (1996) 58(22):1929–35. doi:10.1016/0024-3205(96)00182-8
120. Signoretti S, Marmarou A, Aygok GA, Fatouros PP, Portella G, Bullock RM. Assessment of mitochondrial impairment in traumatic brain injury using high-resolution proton magnetic resonance spectroscopy. J Neurosurg (2008) 108(1):42–52. doi:10.3171/jns/2008/108/01/0042
121. Tollard E, Galanaud D, Perlbarg V, Sanchez-Pena P, Le Fur Y, Abdennour L, et al. Experience of diffusion tensor imaging and 1H spectroscopy for outcome prediction in severe traumatic brain injury: preliminary results. Crit Care Med (2009) 37(4):1448–55. doi:10.1097/CCM.0b013e31819cf050
122. Vagnozzi R, Signoretti S, Cristofori L, Alessandrini F, Floris R, Isgro E, et al. Assessment of metabolic brain damage and recovery following mild traumatic brain injury: a multicentre, proton magnetic resonance spectroscopic study in concussed patients. Brain (2010) 133(11):3232–42. doi:10.1093/brain/awq200
123. Kirov II, Tal A, Babb JS, Lui YW, Grossman RI, Gonen O. Diffuse axonal injury in mild traumatic brain injury: a 3D multivoxel proton MR spectroscopy study. J Neurol (2013) 260(1):242–52. doi:10.1007/s00415-012-6626-z
124. Dhandapani S, Sharma A, Sharma K, Das L. Comparative evaluation of MRS and SPECT in prognostication of patients with mild to moderate head injury. J Clin Neurosci (2014) 21(5):745–50. doi:10.1016/j.jocn.2013.07.038
125. Govind V, Gold S, Kaliannan K, Saigal G, Falcone S, Arheart KL, et al. Whole-brain proton MR spectroscopic imaging of mild-to-moderate traumatic brain injury and correlation with neuropsychological deficits. J Neurotrauma (2010) 27(3):483–96. doi:10.1089/neu.2009.1159
126. George EO, Roys S, Sours C, Rosenberg J, Zhuo J, Shanmuganathan K, et al. Longitudinal and prognostic evaluation of mild traumatic brain injury: a 1H-magnetic resonance spectroscopy study. J Neurotrauma (2014) 31(11):1018–28. doi:10.1089/neu.2013.3224
127. Sivak S, Bittsansky M, Grossmann J, Nosal V, Kantorova E, Sivakova J, et al. Clinical correlations of proton magnetic resonance spectroscopy findings in acute phase after mild traumatic brain injury. Brain Inj (2014) 28(3):341–6. doi:10.3109/02699052.2013.865270
128. Shutter L, Tong KA, Holshouser BA. Proton MRS in acute traumatic brain injury: role for glutamate/glutamine and choline for outcome prediction. J Neurotrauma (2004) 21(12):1693–705. doi:10.1089/neu.2004.21.1693
129. Danielsen ER, Christensen PB, Arlien-Soborg P, Thomsen C. Axonal recovery after severe traumatic brain injury demonstrated in vivo by 1H MR spectroscopy. Neuroradiology (2003) 45(10):722–4. doi:10.1007/s00234-003-1068-z
130. Amyot F, Arciniegas DB, Brazaitis MP, Curley KC, Diaz-Arrastia R, Gandjbakhche A, et al. A review of the effectiveness of neuroimaging modalities for the detection of traumatic brain injury. J Neurotrauma (2015) 32(22):1693–721. doi:10.1089/neu.2013.3306
131. Okamoto T, Hashimoto K, Aoki S, Ohashi M. Cerebral blood flow in patients with diffuse axonal injury – examination of the easy Z-score imaging system utility. Eur J Neurol (2007) 14(5):540–7. doi:10.1111/j.1468-1331.2007.01742.x
132. Uruma G, Hashimoto K, Abo M. A new method for evaluation of mild traumatic brain injury with neuropsychological impairment using statistical imaging analysis for Tc-ECD SPECT. Ann Nucl Med (2013) 27(3):187–202. doi:10.1007/s12149-012-0674-4
133. Devinsky O, Morrell MJ, Vogt BA. Contributions of anterior cingulate cortex to behaviour. Brain (1995) 118(Pt 1):279–306. doi:10.1093/brain/118.1.279
134. Nakashima T, Nakayama N, Miwa K, Okumura A, Soeda A, Iwama T. Focal brain glucose hypometabolism in patients with neuropsychologic deficits after diffuse axonal injury. AJNR Am J Neuroradiol (2007) 28(2):236–42.
135. Kawai N, Maeda Y, Kudomi N, Yamamoto Y, Nishiyama Y, Tamiya T. Focal neuronal damage in patients with neuropsychological impairment after diffuse traumatic brain injury: evaluation using (1)(1)C-flumazenil positron emission tomography with statistical image analysis. J Neurotrauma (2010) 27(12):2131–8. doi:10.1089/neu.2010.1464
136. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol (2011) 70(3):374–83. doi:10.1002/ana.22455
137. Folkersma H, Boellaard R, Yaqub M, Kloet RW, Windhorst AD, Lammertsma AA, et al. Widespread and prolonged increase in (R)-(11)C-PK11195 binding after traumatic brain injury. J Nucl Med (2011) 52(8):1235–9. doi:10.2967/jnumed.110.084061
138. Hong YT, Veenith T, Dewar D, Outtrim JG, Mani V, Williams C, et al. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol (2014) 71(1):23–31. doi:10.1001/jamaneurol.2013.4847
139. Scott G, Ramlackhansingh AF, Edison P, Hellyer P, Cole J, Veronese M, et al. Amyloid pathology and axonal injury after brain trauma. Neurology (2016) 86(9):821–8. doi:10.1212/wnl.0000000000002413
140. Rahmim A, Zaidi H. PET versus SPECT: strengths, limitations and challenges. Nucl Med Commun (2008) 29(3):193–207. doi:10.1097/MNM.0b013e3282f3a515
141. Bogoslovsky T, Gill J, Jeromin A, Davis C, Diaz-Arrastia R. Fluid biomarkers of traumatic brain injury and intended context of use. Diagnostics (Basel) (2016) 6(4):E37. doi:10.3390/diagnostics6040037
142. Zetterberg H, Blennow K. Fluid biomarkers for mild traumatic brain injury and related conditions. Nat Rev Neurol (2016) 12(10):563–74. doi:10.1038/nrneurol.2016.127
143. Zetterberg H, Smith DH, Blennow K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat Rev Neurol (2013) 9(4):201–10. doi:10.1038/nrneurol.2013.9
144. Clark RS, Kochanek PM, Chen M, Watkins SC, Marion DW, Chen J, et al. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB J (1999) 13(8):813–21.
145. Wang KK. Calpain and caspase: can you tell the difference? Trends Neurosci (2000) 23(1):20–6. doi:10.1016/S0166-2236(99)01536-2
146. Huang Y, Wang KK. The calpain family and human disease. Trends Mol Med (2001) 7(8):355–62. doi:10.1016/S1471-4914(01)02049-4
147. Saatman KE, Abai B, Grosvenor A, Vorwerk CK, Smith DH, Meaney DF. Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J Cereb Blood Flow Metab (2003) 23(1):34–42. doi:10.1097/01.WCB.0000035040.10031.B0
148. Agoston DV, Gyorgy A, Eidelman O, Pollard HB. Proteomic biomarkers for blast neurotrauma: targeting cerebral edema, inflammation, and neuronal death cascades. J Neurotrauma (2009) 26(6):901–11. doi:10.1089/neu.2008.0724
149. Portelius E, Bogdanovic N, Gustavsson MK, Volkmann I, Brinkmalm G, Zetterberg H, et al. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol (2010) 120(2):185–93. doi:10.1007/s00401-010-0690-1
150. Saatman KE, Creed J, Raghupathi R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics (2010) 7(1):31–42. doi:10.1016/j.nurt.2009.11.002
151. Buki A, Koizumi H, Povlishock JT. Moderate posttraumatic hypothermia decreases early calpain-mediated proteolysis and concomitant cytoskeletal compromise in traumatic axonal injury. Exp Neurol (1999) 159(1):319–28. doi:10.1006/exnr.1999.7139
152. Kaur B, Rutty GN, Timperley WR. The possible role of hypoxia in the formation of axonal bulbs. J Clin Pathol (1999) 52(3):203–9. doi:10.1136/jcp.52.3.203
153. Buki A, Povlishock JT. All roads lead to disconnection? – Traumatic axonal injury revisited. Acta Neurochir (Wien) (2006) 148(2):181–93; discussion 193–4. doi:10.1007/s00701-005-0674-4
154. DiLeonardi AM, Huh JW, Raghupathi R. Impaired axonal transport and neurofilament compaction occur in separate populations of injured axons following diffuse brain injury in the immature rat. Brain Res (2009) 1263:174–82. doi:10.1016/j.brainres.2009.01.021
155. Tsitsopoulos PP, Marklund N. Amyloid-beta peptides and tau protein as biomarkers in cerebrospinal and interstitial fluid following traumatic brain injury: a review of experimental and clinical studies. Front Neurol (2013) 4:79. doi:10.3389/fneur.2013.00079
156. Perrot R, Berges R, Bocquet A, Eyer J. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol Neurobiol (2008) 38(1):27–65. doi:10.1007/s12035-008-8033-0
157. Chen XH, Meaney DF, Xu BN, Nonaka M, McIntosh TK, Wolf JA, et al. Evolution of neurofilament subtype accumulation in axons following diffuse brain injury in the pig. J Neuropathol Exp Neurol (1999) 58(6):588–96. doi:10.1097/00005072-199906000-00003
158. Siedler DG, Chuah MI, Kirkcaldie MT, Vickers JC, King AE. Diffuse axonal injury in brain trauma: insights from alterations in neurofilaments. Front Cell Neurosci (2014) 8:429. doi:10.3389/fncel.2014.00429
159. Zurek J, Bartlova L, Fedora M. Hyperphosphorylated neurofilament NF-H as a predictor of mortality after brain injury in children. Brain Inj (2011) 25(2):221–6. doi:10.3109/02699052.2010.541895
160. Gatson JW, Barillas J, Hynan LS, Diaz-Arrastia R, Wolf SE, Minei JP. Detection of neurofilament-H in serum as a diagnostic tool to predict injury severity in patients who have suffered mild traumatic brain injury. J Neurosurg (2014) 121(5):1232–8. doi:10.3171/2014.7.JNS132474
161. Van Geel WJ, Rosengren LE, Verbeek MM. An enzyme immunoassay to quantify neurofilament light chain in cerebrospinal fluid. J Immunol Methods (2005) 296(1–2):179–85. doi:10.1016/j.jim.2004.11.015
162. Shahim P, Zetterberg H, Tegner Y, Blennow K. Serum neurofilament light as a biomarker for mild traumatic brain injury in contact sports. Neurology (2017) 88(19):1788–94. doi:10.1212/WNL.0000000000003912
163. Zetterberg H, Hietala MA, Jonsson M, Andreasen N, Styrud E, Karlsson I, et al. Neurochemical aftermath of amateur boxing. Arch Neurol (2006) 63(9):1277–80. doi:10.1001/archneur.63.9.1277
164. Neselius S, Brisby H, Theodorsson A, Blennow K, Zetterberg H, Marcusson J. CSF-biomarkers in olympic boxing: diagnosis and effects of repetitive head trauma. PLoS One (2012) 7(4):e33606. doi:10.1371/journal.pone.0033606
165. Shahim P, Tegner Y, Gustafsson B, Gren M, Arlig J, Olsson M, et al. Neurochemical aftermath of repetitive mild traumatic brain injury. JAMA Neurol (2016) 73(11):1308–15. doi:10.1001/jamaneurol.2016.2038
166. Bagnato S, Grimaldi LME, Di Raimondo G, Sant’Angelo A, Boccagni C, Virgilio V, et al. Prolonged cerebrospinal fluid neurofilament light chain increase in patients with post-traumatic disorders of consciousness. J Neurotrauma (2017) 34(16):2475–9. doi:10.1089/neu.2016.4837
167. Al Nimer F, Thelin E, Nyström H, Dring AM, Svenningsson A, Piehl F, et al. Comparative assessment of the prognostic value of biomarkers in traumatic brain injury reveals an independent role for serum levels of neurofilament light. PLoS One (2015) 10(7):e0132177. doi:10.1371/journal.pone.0132177
168. Shahim P, Gren M, Liman V, Andreasson U, Norgren N, Tegner Y, et al. Serum neurofilament light protein predicts clinical outcome in traumatic brain injury. Sci Rep (2016) 6:36791. doi:10.1038/srep36791
169. Ljungqvist J, Zetterberg H, Mitsis M, Blennow K, Skoglund T. Serum neurofilament light protein as a marker for diffuse axonal injury: results from a case series study. J Neurotrauma (2017) 34(5):1124–7. doi:10.1089/neu.2016.4496
170. Zemlan FP, Jauch EC, Mulchahey JJ, Gabbita SP, Rosenberg WS, Speciale SG, et al. C-tau biomarker of neuronal damage in severe brain injured patients: association with elevated intracranial pressure and clinical outcome. Brain Res (2002) 947(1):131–9. doi:10.1016/S0006-8993(02)02920-7
171. Schraen-Maschke S, Sergeant N, Dhaenens CM, Bombois S, Deramecourt V, Caillet-Boudin ML, et al. Tau as a biomarker of neurodegenerative diseases. Biomark Med (2008) 2(4):363–84. doi:10.2217/17520363.2.4.363
172. Shahim P, Linemann T, Inekci D, Karsdal MA, Blennow K, Tegner Y, et al. Serum Tau fragments predict return to play in concussed professional ice hockey players. J Neurotrauma (2016) 33(22):1995–9. doi:10.1089/neu.2014.3741
173. Anderson JM, Hampton DW, Patani R, Pryce G, Crowther RA, Reynolds R, et al. Abnormally phosphorylated tau is associated with neuronal and axonal loss in experimental autoimmune encephalomyelitis and multiple sclerosis. Brain (2008) 131(Pt 7):1736–48. doi:10.1093/brain/awn119
174. Rubenstein R, Chang B, Davies P, Wagner AK, Robertson CS, Wang KK. A novel, ultrasensitive assay for tau: potential for assessing traumatic brain injury in tissues and biofluids. J Neurotrauma (2015) 32(5):342–52. doi:10.1089/neu.2014.3548
175. Randall J, Mortberg E, Provuncher GK, Fournier DR, Duffy DC, Rubertsson S, et al. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation (2013) 84(3):351–6. doi:10.1016/j.resuscitation.2012.07.027
176. Shahim P, Tegner Y, Wilson DH, Randall J, Skillback T, Pazooki D, et al. Blood biomarkers for brain injury in concussed professional ice hockey players. JAMA Neurol (2014) 71(6):684–92. doi:10.1001/jamaneurol.2014.367
177. Bulut M, Koksal O, Dogan S, Bolca N, Ozguc H, Korfali E, et al. Tau protein as a serum marker of brain damage in mild traumatic brain injury: preliminary results. Adv Ther (2006) 23(1):12–22. doi:10.1007/BF02850342
178. Oliver J, Jones M, Anzalone A, Kirk M, Gable D, Repshas J, et al. A season of American football is not associated with changes in plasma tau. J Neurotrauma (2017). doi:10.1089/neu.2017.5064
179. Zemlan FP, Rosenberg WS, Luebbe PA, Campbell TA, Dean GE, Weiner NE, et al. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem (1999) 72(2):741–50. doi:10.1046/j.1471-4159.1999.0720741.x
180. Ost M, Nylen K, Csajbok L, Ohrfelt AO, Tullberg M, Wikkelso C, et al. Initial CSF total tau correlates with 1-year outcome in patients with traumatic brain injury. Neurology (2006) 67(9):1600–4. doi:10.1212/01.wnl.0000242732.06714.0f
181. Ma M, Lindsell CJ, Rosenberry CM, Shaw GJ, Zemlan FP. Serum cleaved tau does not predict postconcussion syndrome after mild traumatic brain injury. Am J Emerg Med (2008) 26(7):763–8. doi:10.1016/j.ajem.2007.10.029
182. Bazarian JJ, Zemlan FP, Mookerjee S, Stigbrand T. Serum S-100B and cleaved-tau are poor predictors of long-term outcome after mild traumatic brain injury. Brain Inj (2006) 20(7):759–65. doi:10.1080/02699050500488207
183. Huh GY, Glantz SB, Je S, Morrow JS, Kim JH. Calpain proteolysis of alpha II-spectrin in the normal adult human brain. Neurosci Lett (2001) 316(1):41–4. doi:10.1016/S0304-3940(01)02371-0
184. Buki A, Okonkwo DO, Wang KK, Povlishock JT. Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci (2000) 20(8):2825–34.
185. Mondello S, Robicsek SA, Gabrielli A, Brophy GM, Papa L, Tepas J, et al. AlphaII-spectrin breakdown products (SBDPs): diagnosis and outcome in severe traumatic brain injury patients. J Neurotrauma (2010) 27(7):1203–13. doi:10.1089/neu.2010.1278
186. Huh JW, Franklin MA, Widing AG, Raghupathi R. Regionally distinct patterns of calpain activation and traumatic axonal injury following contusive brain injury in immature rats. Dev Neurosci (2006) 28(4–5):466–76. doi:10.1159/000094172
187. Thompson SN, Gibson TR, Thompson BM, Deng Y, Hall ED. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol (2006) 201(1):253–65. doi:10.1016/j.expneurol.2006.04.013
188. Serbest G, Burkhardt MF, Siman R, Raghupathi R, Saatman KE. Temporal profiles of cytoskeletal protein loss following traumatic axonal injury in mice. Neurochem Res (2007) 32(12):2006–14. doi:10.1007/s11064-007-9318-9
189. Siman R, Toraskar N, Dang A, McNeil E, McGarvey M, Plaum J, et al. A panel of neuron-enriched proteins as markers for traumatic brain injury in humans. J Neurotrauma (2009) 26(11):1867–77. doi:10.1089/neu.2009.0882
190. Siman R, Giovannone N, Hanten G, Wilde EA, McCauley SR, Hunter JV, et al. Evidence that the blood biomarker SNTF predicts brain imaging changes and persistent cognitive dysfunction in mild TBI patients. Front Neurol (2013) 4:190. doi:10.3389/fneur.2013.00190
191. Siman R, Shahim P, Tegner Y, Blennow K, Zetterberg H, Smith DH. Serum SNTF increases in concussed professional ice hockey players and relates to the severity of postconcussion symptoms. J Neurotrauma (2015) 32(17):1294–300. doi:10.1089/neu.2014.3698
192. Johnson VE, Stewart W, Weber MT, Cullen DK, Siman R, Smith DH. SNTF immunostaining reveals previously undetected axonal pathology in traumatic brain injury. Acta Neuropathol (2016) 131(1):115–35. doi:10.1007/s00401-015-1506-0
193. Pineda JA, Lewis SB, Valadka AB, Papa L, Hannay HJ, Heaton SC, et al. Clinical significance of alphaII-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury. J Neurotrauma (2007) 24(2):354–66. doi:10.1089/neu.2006.003789
194. Brophy GM, Pineda JA, Papa L, Lewis SB, Valadka AB, Hannay HJ, et al. AlphaII-spectrin breakdown product cerebrospinal fluid exposure metrics suggest differences in cellular injury mechanisms after severe traumatic brain injury. J Neurotrauma (2009) 26(4):471–9. doi:10.1089/neu.2008.0657
195. Oppenheimer DR. Microscopic lesions in the brain following head injury. J Neurol Neurosurg Psychiatry (1968) 31(4):299–306. doi:10.1136/jnnp.31.4.299
196. Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ. Staining of amyloid precursor protein to study axonal damage in mild head injury. Lancet (1994) 344(8929):1055–6. doi:10.1016/S0140-6736(94)91712-4
197. Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, et al. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol (2004) 190(1):192–203. doi:10.1016/j.expneurol.2004.06.011
198. Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol (1994) 87(1):55–62. doi:10.1007/BF00386254
199. Gehrmann J, Banati RB, Cuzner ML, Kreutzberg GW, Newcombe J. Amyloid precursor protein (APP) expression in multiple sclerosis lesions. Glia (1995) 15(2):141–51. doi:10.1002/glia.440150206
200. Hayashi T, Ago K, Nakamae T, Higo E, Ogata M. Two different immunostaining patterns of beta-amyloid precursor protein (APP) may distinguish traumatic from nontraumatic axonal injury. Int J Legal Med (2015) 129(5):1085–90. doi:10.1007/s00414-015-1245-8
201. Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol (2007) 208(2):185–92. doi:10.1016/j.expneurol.2007.06.018
202. Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol (2009) 19(2):214–23. doi:10.1111/j.1750-3639.2008.00176.x
203. Smith DH, Chen XH, Iwata A, Graham DI. Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg (2003) 98(5):1072–7. doi:10.3171/jns.2003.98.5.1072
204. Olsson A, Csajbok L, Ost M, Höglund K, Nylén K, Rosengren L, et al. Marked increase of beta-amyloid(1-42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J Neurol (2004) 251(7):870–6. doi:10.1007/s00415-004-0451-y
205. Mondello S, Buki A, Barzo P, Randall J, Provuncher G, Hanlon D, et al. CSF and plasma amyloid-beta temporal profiles and relationships with neurological status and mortality after severe traumatic brain injury. Sci Rep (2014) 4:6446. doi:10.1038/srep06446
206. Raby CA, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, et al. Traumatic brain injury increases beta-amyloid peptide 1-42 in cerebrospinal fluid. J Neurochem (1998) 71(6):2505–9. doi:10.1046/j.1471-4159.1998.71062505.x
207. Franz G, Beer R, Kampfl A, Engelhardt K, Schmutzhard E, Ulmer H, et al. Amyloid beta 1-42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology (2003) 60(9):1457–61. doi:10.1212/01.WNL.0000063313.57292.00
208. Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, et al. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science (2008) 321(5893):1221–4. doi:10.1126/science.1161591
209. Marklund N, Farrokhnia N, Hanell A, Vanmechelen E, Enblad P, Zetterberg H, et al. Monitoring of beta-amyloid dynamics after human traumatic brain injury. J Neurotrauma (2014) 31(1):42–55. doi:10.1089/neu.2013.2964
210. Shahim P, Tegner Y, Marklund N, Hoglund K, Portelius E, Brody DL, et al. Astroglial activation and altered amyloid metabolism in human repetitive concussion. Neurology (2017) 88(15):1400–7. doi:10.1212/WNL.0000000000003816
211. Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci (2012) 15(3):349–57. doi:10.1038/nn.3028
212. Esparza TJ, Wildburger NC, Jiang H, Gangolli M, Cairns NJ, Bateman RJ, et al. Soluble amyloid-beta aggregates from human Alzheimer’s disease brains. Sci Rep (2016) 6:38187. doi:10.1038/srep38187
213. Gatson JW, Warren V, Abdelfattah K, Wolf S, Hynan LS, Moore C, et al. Detection of beta-amyloid oligomers as a predictor of neurological outcome after brain injury. J Neurosurg (2013) 118(6):1336–42. doi:10.3171/2013.2.jns121771
214. Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology (2000) 55(8):1158–66. doi:10.1212/WNL.55.8.1158
215. Sehlin D, Fang XT, Cato L, Antoni G, Lannfelt L, Syvanen S. Antibody-based PET imaging of amyloid beta in mouse models of Alzheimer’s disease. Nat Commun (2016) 7:10759. doi:10.1038/ncomms10759
216. Hutchinson PJ, Jalloh I, Helmy A, Carpenter KL, Rostami E, Bellander BM, et al. Consensus statement from the 2014 International Microdialysis Forum. Intensive Care Med (2015) 41(9):1517–28. doi:10.1007/s00134-015-3930-y
217. Marklund N, Blennow K, Zetterberg H, Ronne-Engstrom E, Enblad P, Hillered L. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg (2009) 110(6):1227–37. doi:10.3171/2008.9.JNS08584
218. Magnoni S, Esparza TJ, Conte V, Carbonara M, Carrabba G, Holtzman DM, et al. Tau elevations in the brain extracellular space correlate with reduced amyloid-beta levels and predict adverse clinical outcomes after severe traumatic brain injury. Brain (2012) 135(Pt 4):1268–80. doi:10.1093/brain/awr286
219. Helmy A, Carpenter KL, Menon DK, Pickard JD, Hutchinson PJ. The cytokine response to human traumatic brain injury: temporal profiles and evidence for cerebral parenchymal production. J Cereb Blood Flow Metab (2011) 31(2):658–70. doi:10.1038/jcbfm.2010.142
220. Helmy A, Antoniades CA, Guilfoyle MR, Carpenter KL, Hutchinson PJ. Principal component analysis of the cytokine and chemokine response to human traumatic brain injury. PLoS One (2012) 7(6):e39677. doi:10.1371/journal.pone.0039677
221. Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J Cereb Blood Flow Metab (2014) 34(5):845–51. doi:10.1038/jcbfm.2014.23
222. Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ. Recombinant human interleukin-1 receptor antagonist promotes M1 microglia biased cytokines and chemokines following human traumatic brain injury. J Cereb Blood Flow Metab (2016) 36(8):1434–48. doi:10.1177/0271678X15620204
223. Barbarese E, Barry C, Chou CH, Goldstein DJ, Nakos GA, Hyde-DeRuyscher R, et al. Expression and localization of myelin basic protein in oligodendrocytes and transfected fibroblasts. J Neurochem (1988) 51(6):1737–45. doi:10.1111/j.1471-4159.1988.tb01153.x
224. Metting Z, Wilczak N, Rodiger LA, Schaaf JM, van der Naalt J. GFAP and S100B in the acute phase of mild traumatic brain injury. Neurology (2012) 78(18):1428–33. doi:10.1212/WNL.0b013e318253d5c7
225. Papa L, Lewis LM, Falk JL, Zhang Z, Silvestri S, Giordano P, et al. Elevated levels of serum glial fibrillary acidic protein breakdown products in mild and moderate traumatic brain injury are associated with intracranial lesions and neurosurgical intervention. Ann Emerg Med (2012) 59(6):471–83. doi:10.1016/j.annemergmed.2011.08.021
226. Okonkwo DO, Yue JK, Puccio AM, Panczykowski DM, Inoue T, McMahon PJ, et al. GFAP-BDP as an acute diagnostic marker in traumatic brain injury: results from the prospective transforming research and clinical knowledge in traumatic brain injury study. J Neurotrauma (2013) 30(17):1490–7. doi:10.1089/neu.2013.2883
227. Diaz-Arrastia R, Wang KK, Papa L, Sorani MD, Yue JK, Puccio AM, et al. Acute biomarkers of traumatic brain injury: relationship between plasma levels of ubiquitin C-terminal hydrolase-L1 and glial fibrillary acidic protein. J Neurotrauma (2014) 31(1):19–25. doi:10.1089/neu.2013.3040
228. Papa L, Brophy GM, Welch RD, Lewis LM, Braga CF, Tan CN, et al. Time course and diagnostic accuracy of glial and neuronal blood biomarkers GFAP and UCH-L1 in a large cohort of trauma patients with and without mild traumatic brain injury. JAMA Neurol (2016) 73(5):551–60. doi:10.1001/jamaneurol.2016.0039
229. Herrmann M, Jost S, Kutz S, Ebert AD, Kratz T, Wunderlich MT, et al. Temporal profile of release of neurobiochemical markers of brain damage after traumatic brain injury is associated with intracranial pathology as demonstrated in cranial computerized tomography. J Neurotrauma (2000) 17(2):113–22. doi:10.1089/neu.2000.17.113
230. Thelin EP, Nelson DW, Bellander BM. A review of the clinical utility of serum S100B protein levels in the assessment of traumatic brain injury. Acta Neurochir (Wien) (2017) 159(2):209–25. doi:10.1007/s00701-016-3046-3
231. Pleines UE, Morganti-Kossmann MC, Rancan M, Joller H, Trentz O, Kossmann T. S-100 beta reflects the extent of injury and outcome, whereas neuronal specific enolase is a better indicator of neuroinflammation in patients with severe traumatic brain injury. J Neurotrauma (2001) 18(5):491–8. doi:10.1089/089771501300227297
232. Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury (2007) 38(12):1392–400. doi:10.1016/j.injury.2007.10.005
233. Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol (2013) 4:18. doi:10.3389/fneur.2013.00018
Keywords: traumatic brain injury, diffuse axonal injury, monitoring, neurocritical care, neuroimaging, biomarkers, microdialysis
Citation: Tsitsopoulos PP, Abu Hamdeh S and Marklund N (2017) Current Opportunities for Clinical Monitoring of Axonal Pathology in Traumatic Brain Injury. Front. Neurol. 8:599. doi: 10.3389/fneur.2017.00599
Received: 05 May 2017; Accepted: 25 October 2017;
Published: 20 November 2017
Edited by:
Denes V. Agoston, Karolinska Institutet, SwedenReviewed by:
William Doster Watson, University of Kentucky, United StatesNamas Chandra, New Jersey Institute of Technology, United States
Copyright: © 2017 Tsitsopoulos, Abu Hamdeh and Marklund. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Niklas Marklund, bmlrbGFzLm1hcmtsdW5kQG5ldXJvLnV1LnNl