 Qingxian Wen1
Qingxian Wen1 Cun Yang
Cun Yang- 1Department of Neurology, Jining No. 1 People's Hospital, Jining, China
- 2Department of Reproductive Medicine, Jining No. 1 People's Hospital, Jining, China
- 3Department of Pediatrics, Jinning No. 1 People's Hospital, Jining, China
- 4Department of Neurology, Washington Institute of Clinical Research, Vienna, VA, United States
Background: Electrophysiological examination plays an important role in the diagnosis of X-linked Charcot-Marie-Tooth disease (CMTX1) with transient central nervous system deficits. However, the electrophysiological features are seldom reported.
Methods: We reviewed and analyzed published reports to determine the electrophysiological features of CMTX1 patients with transient central nervous system deficits.
Results: A total of 21 CMTX1 patients with transient central nervous system deficits were found in 17 published case reports/series. The age of onset ranged from 0.5 to 18 years (mean 12.02 ± 0.78 years). All patients were male. Recurrent episodes of central nervous system deficits were reported in all 21 cases and resolved in periods ranging from several minutes to 3 days. All 20 patients who had MRIs at presentation had bilaterally symmetrical abnormal T2/Diffusion signals in the white matter without enhancement of gadolinium. All subsequent MRIs showed improvement or were within normal limits. The median motor nerve conduction velocity (MNCV), motor latencies, and compound muscle action potential (CMAP) amplitude were the most commonly measurable electrophysiological parameters (85.7%). All cases that had MNCV at presentation had slower and significantly decreased MNCV compared with the normal value (34.1 ± 1.10 m/s vs. 46.8±2.05 m/s, P < 0.0001; 95% CI, −17.4 to −7.92). The average variations of MNCV in median nerve, ulnar nerve, peroneal nerve, and tibial nerve were 22.0 ± 5.96%, 27.6 ± 11.9%, 25.9 ± 4.36%, and 27.3 ± 4.30%, respectively. All cases with measured sensory nerve conduction velocity (SNCV) at presentation had slower and significantly decreased SNCV compared with the normal value (35.3 ± 1.33 m/s vs. 47.7 ± 2.40 m/s, P < 0.001; 95% CI −18.2 to −6.46). The average variations of SNCV in median nerve, ulnar nerve, and sural nerve were 19.9 ± 8.24%, 25.2 ± 7.75%, and 23.2 ± 3.95%, respectively.
Conclusion: This case series serves as a reminder that electrophysiological examination should be included in the diagnosis of recurrent and episodic neurological deficit with white matter lesions. Median MNCV is a sensitive and valuable parameter to support the diagnosis of CMTX1 with transient central nervous system deficits.
Introduction
Charcot-Marie-Tooth disease (CMT), also known as hereditary motor and sensory neuropathy or peroneal muscular atrophy, is a genetically and clinically heterogeneous group of inherited neuropathies (1). Clinically, it is characterized by progressive muscle weakness and atrophy, and distal sensory loss (2). X-linked CMT (CMTX1) is the second most common form of CMT, and it is caused by gap junction protein beta-1 (GJB1, also known as Cx32) gene mutations. The phenotype of CMTX1 is typically characterized by a mixture of demyelinating and axonal features (3).
Transient central nervous system deficits associated with white matter abnormalities on magnetic resonance imaging (MRI) have been reported in patients with CMTX1 (4, 5). Electrophysiological examination plays an important role in diagnosis of CMTX1, particularly when there is a need to differentiate it from the diagnosis of stroke-like acute onset neurological deficits with white matter lesions (6) or episodic acute demyelinating encephalomyelitis (ADEM)-like illness (7). However, electrophysiological assessment in CMTX1 with transient central nervous system deficits is rare, and thus the electophysiological features are seldom reported.
Here we report a large series of CMTX1 patients with transient central nervous system deficits who had electrophysiological examination. We reviewed and analyzed the published reports with a primary goal of determining the electrophysiological features of CMTX1 patients with transient central nervous system deficits and discussing aids in diagnosis.
Methods
Search Strategy and Study Screening
A systematic literature search was conducted on March 21, 2018 on PubMed (from January 1966) and Embase databases (from 1947). Key search terms used were: Charcot-Marie-Tooth, X-linked, gap junction or GJB1 or connexin, white matter or encephalomyel* or central nervous or leukoencephal*, electrophysiol* OR electromyo* OR EMG OR motor nerve conduction velocity (MNCV) OR distal motor latencies OR compound muscle action potential (CMAP). A manual search of relevant review papers was also performed.
The current investigation was performed and reported according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (8). All citations and selected articles were read in full and rated on quality by two independent reviewers (Wen and Yang).
Inclusion Criteria, and Exclusion Criteria
The inclusion criteria were as follows: (1) X-linked Charcot-Marie-Tooth with transient central nervous system deficits; (2) electrophysiological examination was performed; (3) case report or case series. The exclusion criteria were as follows: (1) no adequate data reported or obtained by contacting the authors; (2) duplicate publications.
Data Extraction
Search strategy and study selection are presented in Figure 1. A data extraction sheet was used to record study and patient demographic variables; family history; genetic mutation; and clinical, MRI and electrophysiological findings. Data were extracted and verified by two investigators (Wen and Cao).
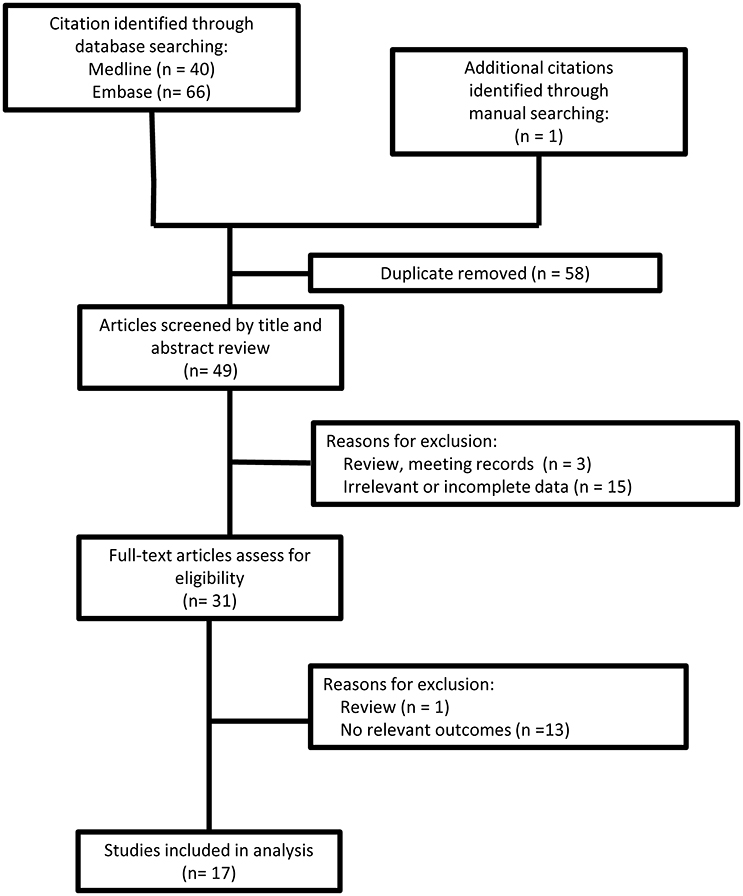
Figure 1. Flowchart of search strategy and study selection.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA) and SPSS Statistics 13 (SPSS Corporation, Chicago, IL). The continuous variables are presented as means with 95% confidence intervals (CI). A two-tailed comparison with P < 0.05 was considered statistically significant. Differences between groups were analyzed by the independent sample t-test or the Mann-Whitney U-test (for nonparametric comparisons), for the continuous variables.
Results
Search Results and Demographic Characteristics
A total of 21 CMTX1 patients with transient central nervous system deficits were found in 17 case reports/series published between 2001 and 2016 (4–7, 9–21). Demographic, clinical, MRI, genetic, and electrophysiological data of 21 cases from the literature were reviewed (see Tables 1–3, Supplement Table 1).
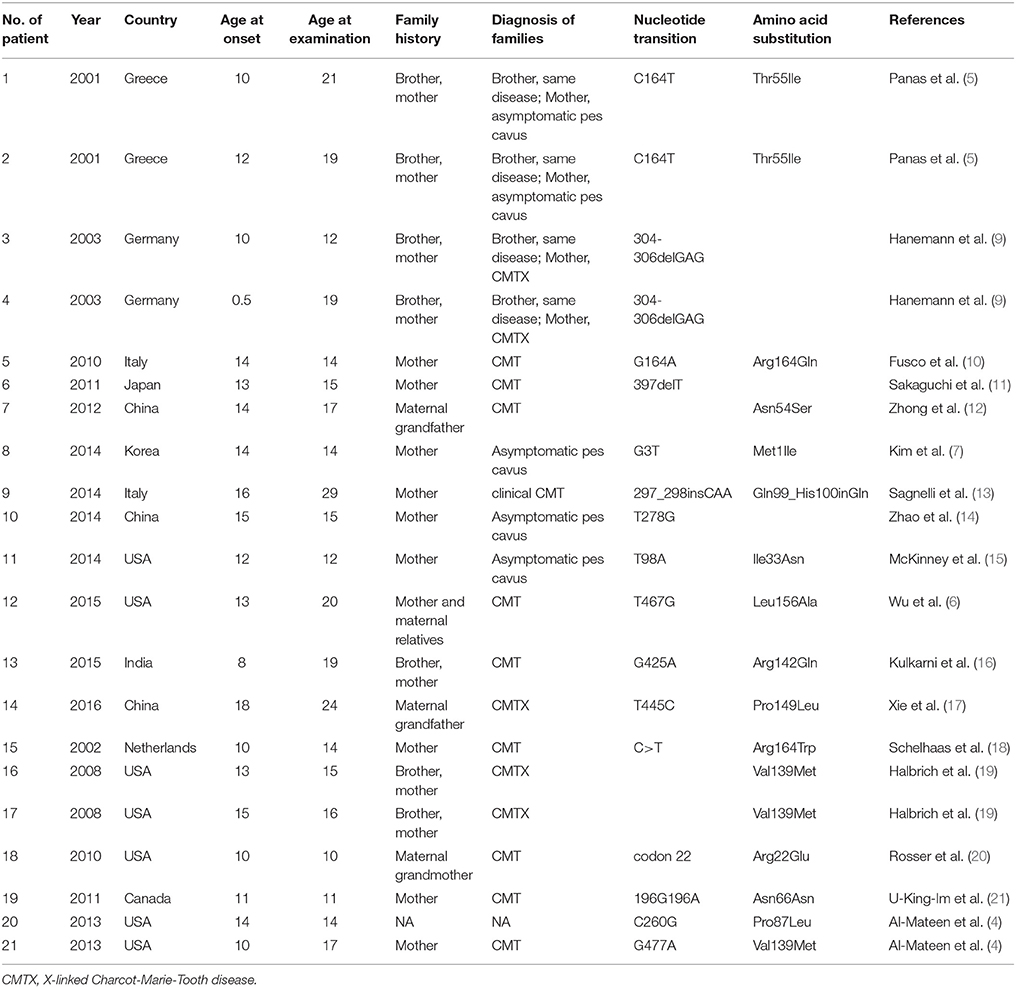
Table 1. Demographic characteristics, family history, and mutation in CMTX1 with transient central nervous system deficits.
Demographic Characteristics, Family History, and Mutation
Demographic characteristics, family history, and mutation in CMTX1 with transient central nervous system deficits are shown in Table 1. The age of CNS disturbance onset ranged from 0.5 to 18 years old with a mean of 12.02 ± 0.78 years. The age range of examination was 10–29 years old, with a mean ± standard deviation (SD) of 16.52 ± 0.99 years. All patients were male. Seven patients were from Europe, six from Asia, and eight from North America. Family histories were found in all cases. Two brothers were reported as CMTX1 with transient central nervous system deficits with the same mutation in GJB1 gene in 3 families. All the other families had proband's mother or maternal relatives with CMT or asymptomatic pes cavus.
Clinical and MRI Features
Clinical and MRI features in CMTX1 with transient central nervous system deficits are shown in Table 2. Recurrent episodes of the central nervous system deficits were reported in all 21 cases and resolved in periods ranging from several minutes to 3 days. All 21 patients (100%) had transient weakness of limbs, 17 patients (80.95%) had transient dysarthria (10/21), dysphagia (3/21), or both (4/21); 6 patients (28.57%) had numbness, and 2 patients (9.52%) had ataxia. The transient central nervous system deficits presented after a fever in 9 cases (42.86%), exercise in 4 patients (19.05%), travel to high altitude in 2 patients (9.52%), and vomiting and diarrhea in 1 patient (4.76%). All 20 patients who had MRI at presentation had bilateral abnormal T2/Diffusion signal in the white matter. The other patients without MRI presented with recurrent episodes of transient neurological dysfunction on two consecutive days. Investigations included normal results for brain computed tomography (CT) scan, CT angiogram, lumber puncture (n = 19, Tables 1, 2).
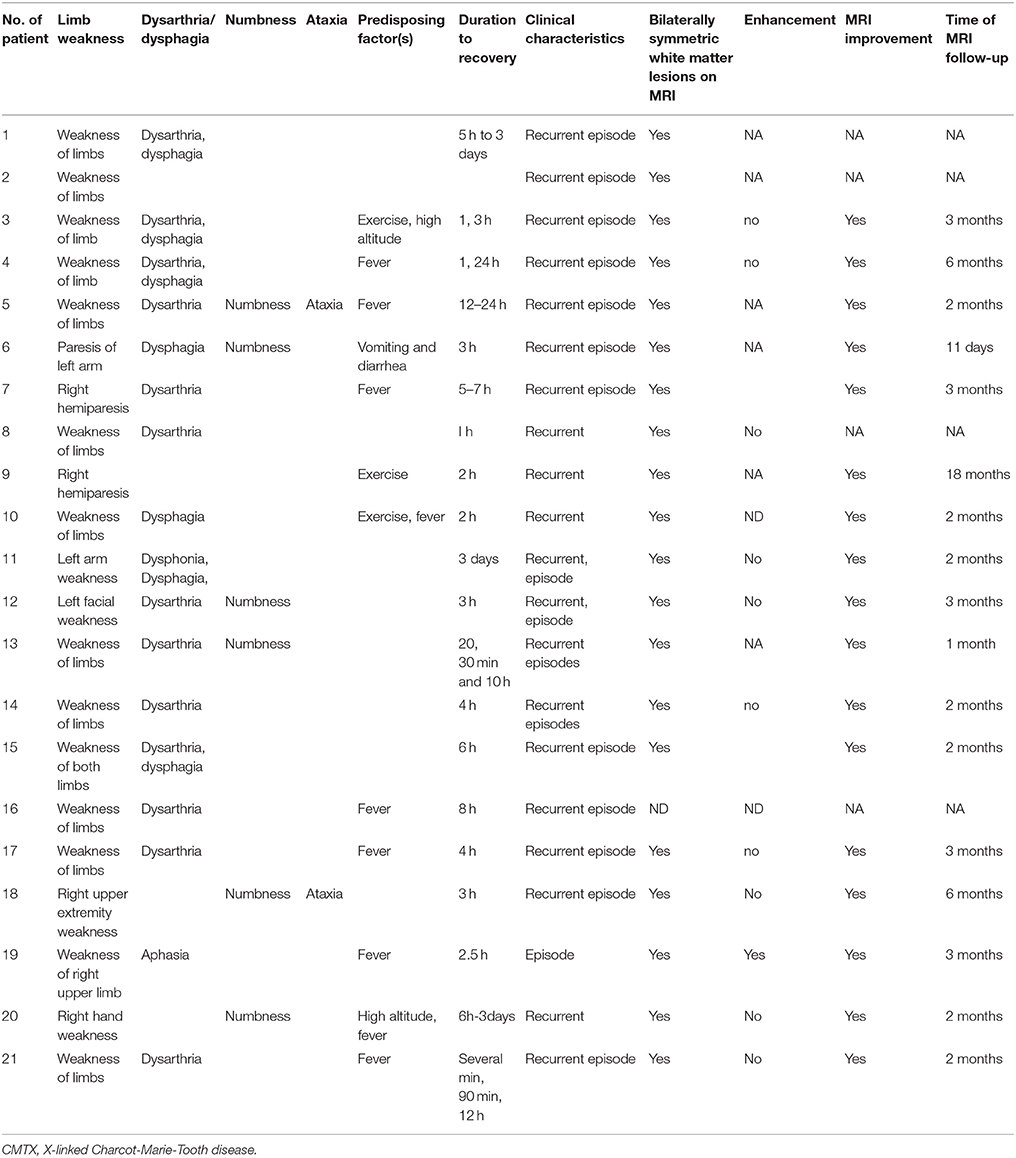
Table 2. clinical and MRI features in CMTX1 with transient central nervous system deficits.
All 11 cases with administration of gadolinium during the brain MRI reported no enhancement. Subsequent MRI results in 17 cases were within normal limits or showed improvement. The follow-up MRIs were performed from 11 days to 18 months after the episode of the disease, median 2 months.
Electrophysiological Features
Of the 21 electrophysiological examinations, 14 cases had detectable electrophysiological parameters, and 7 cases had normal laboratory values (9–12, 14, 15). Supplementary Table 1 summarizes 21 electrophysiological examinations in CMTX1 patients with transient central nervous system deficits. The changes in electrophysiological parameters were compared with the normal values, and the variation with respect to normal values is shown in Table 3.
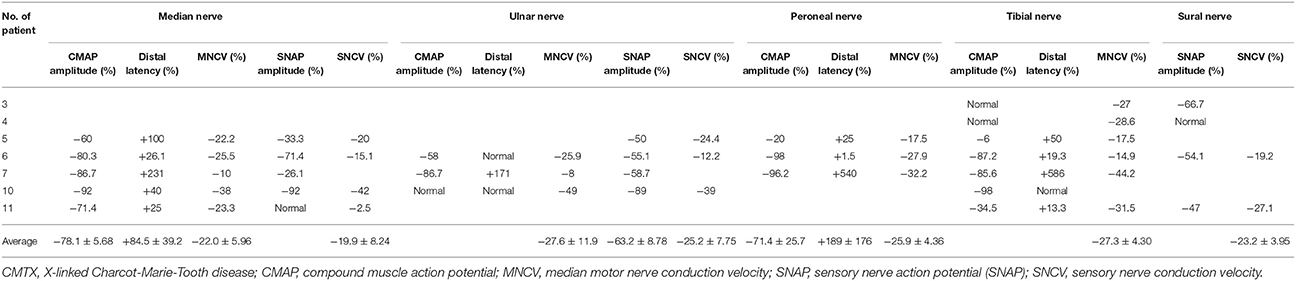
Table 3. Variations of electrophysiological examination in CMTX1 with transient central nervous system deficits.
All nerve conduction examinations performed at presentation showed marked slowing of conduction velocity. The median MNCV, CMAP amplitude, and motor latencies were the most commonly measurable electrophysiological parameters (85.7%). Median MNCVs were all below the normal value, and mean ± SD MNCVs of the 12 cases were 37.7 ± 1.22 m/s (range 32–48.5 m/s).
All cases that had MNCV at presentation had slower and significantly decreased MNCV compared with the normal value (34.1 ± 1.10 m/s vs. 46.8 ± 2.05 m/s, P < 0.0001; 95% CI, −17.4 to −7.92). The average variations of MNCV in median nerve, ulnar nerve, peroneal nerve and tibial nerve were 22.0 ± 5.96%, 27.6 ± 11.9%, 25.9 ± 4.36%, 27.3 ± 4.30%, respectively (Table 3). All cases that had SNCV at presentation had slower and significantly decreased SNCV compared with the normal value (35.3 ± 1.33 m/s vs. 47.7 ± 2.40 m/s, P < 0.001; 95% CI −18.2 to −6.46). The average variations of SNCV in median nerve, ulnar nerve and sural nerve were 19.9 ± 8.24%, 25.2 ± 7.75%, and 23.2 ± 3.95%, respectively.
Distal latency was observed. All cases that had distal latency for median nerve and peroneal nerve had prolonged latency compared with the normal value, and mean changes were 84.5 ± 39.2% and 189 ± 176%, respectively. There was normal distal latency for the ulnar nerve in 2 cases (11, 14) and for tibial nerve in 1 case (14).
For the CMAP amplitude, all cases that had median nerve and peroneal nerve examination had reduced amplitude compared with normal values. Mean changes were 78.1 ± 5.68% and 71.4 ± 25.7%, respectively. However, the tibial nerve in 2 cases (9) and the ulnar nerve in 1 case had normal CMAP amplitude (14).
Most cases had reduced sensory nerve action potential (SNAP) amplitude, except 1 case in the median nerve (15) and 1 case in the sural nerve (9).
Brainstem Auditory Evoked Potentials, Visual Evoked Potentials, and Somatosensory Evoked Potentials
Eight cases had detectable Brainstem auditory evoked potentials, with 6 had prolonged or loss of later waves (5, 9, 10, 14, 21) and 2 had the normal response (13, 16). Visual evoked potentials (VEP) were performed in 3 cases and all reported prolonged VEP (5, 16). Somatosensory evoked potentials (SEP) was performed in 2 cases and all reported prolonged SEP (13, 14).
Discussion
To the best of our knowledge, this is the first systematic investigation of electrophysiological features in CMTX1 with transient central nervous deficits. In the present study, we found that all cases had significant but variable degrees of slowed nerve conduction. The median MNCV was the most frequently detected and valuable parameter.
Onset of CMTX1 with transient central nervous system deficits occurs most often in early childhood (4). Typical features of CMTX1 disease may not be recognized until after presentation with central nervous system manifestations, such as recurrent and transient weakness, dysarthria, and dysphagia (4). CMTX1 is usually caused by mutation in the GJB1 gene encoding the gap junction beta 1 protein connexin 32 (Cx 32) (3). Proposed mechanisms include loss of Cx32 function affecting the gap junctions in the myelin sheath and causing CMTX1 peripheral manifestations or gain of function affecting the central nervous system (2). The pathological mechanism by which GJB1 mutations cause the CNS deficit in patients with CMTX1 is not well understood, but may involve a decrease in the number of functioning gap junctions between oligodendrocytes and astrocytes, disrupting gap junction communication and leading to abnormalities in the ability of these cells to regulate intercellular exchange of ions and small molecules (2). It is important to consider CMTX1 in previously healthy children who present with symptoms suggestive of recurrent or transient central nervous system deficits and to conduct thorough examination for features of CMT disease and electrophysiological testing (4).
Median MNCV can be a sensitive parameter to support the diagnosis of CMTX1 with transient central nervous system deficits. In the current study, NCVs at presentation were slower and characterized as intermediate (30–40 m/s) or mild (>40 m/s) slowing in CMTX1 patients with transient central nervous system deficits. Transient paralysis in children may also represent a feature of mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS), ADEM, adrenoleukodystrophy, moya moya disease, and younger transient ischemic attack (TIA). Electrodiagnostic irregularities noted at presentation with central nervous system deficits are expected in a chronic hereditary motor and sensory neuropathy, as documented in the present analysis (4).
Uniform slowing of NCVs is suggestive of hereditary demyelinating neuropathy. So NCVs are valuable to separate demyelinating and axonal damage in CMTX1. Theoretically, CMTX1 is characterized by sensory-motor neuropathy with mixed axonal and demyelinating pathology. In the present study, electrophysiological findings during the episode of acute nervous system deficits showed a diffuse and symmetrical slowing of MNCV and SNCV consistent with demyelinating-type neuropathy (6). Reduced CMAP and SNAP amplitudes and prolonged distal latencies were also present. One month following the episode, the MNCV, SNCV, and distal latencies were unchanged, showing evidence of peripheral involvement during the chronic disease (6).
Prolonged brainstem auditory evoked potentials, VEP and SEP were found in most CMTX1 patients with transient central nervous deficits. It would be interesting to know the presence of subclinical alterations of the central white matter before the acute event.
Conclusions
This case series serves as a reminder that the differential diagnosis of acute lesions of cerebral white matter should include the possibility of CMTX disease. Electrophysiological examination might be helpful in the diagnosis of recurring and intermittent neurological deficits with white matter lesion and median MCV is a sensitive and valuable parameter.
Author Contributions
QW design, acquisition of data, analysis and interpretation of data, drafting and revision of article, final approval. LC acquisition of data, analysis and interpretation of data, final approval. YX design, drafting and revision of article, final approval. CY design, acquisition of data, analysis and interpretation of data, drafting and revision of article, final approval.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2018.00461/full#supplementary-material
References
1. Berciano J, García A, Gallardo E, Peeters K, Pelayo-Negro AL, Álvarez-Paradelo S, et al. Intermediate charcot-marie-tooth disease: an electrophysiological reappraisal and systematic review. J Neurol. (2017) 264:1655–77. doi: 10.1007/s00415-017-8474-3
2. Wang Y, and Yin F A review of X-linked charcot-marie-tooth disease. J Child Neurol. (2016) 31:761–72. doi: 10.1177/0883073815604227
3. Liu L, Li XB, Hu ZHM, Zi XH, Zhao X, Xie YZ, et al. Phenotypes and cellular effects of GJB1 mutations causing CMT1X in a cohort of 226 Chinese CMT families. Clin Genet. (2017) 91:881–91. doi: 10.1111/cge.12913
4. Al-Mateen M, Craig AK, and Chance PF The central nervous system phenotype of X-linked charcot-marie-tooth disease: a transient disorder of children and young adults. J Child Neurol. (2014) 29:342–8. doi: 10.1177/0883073812474343
5. Panas M, Kalfakis N, Karadimas C, and Vassilopoulos D Episodes of generalized weakness in two sibs with the C164T mutation of the connexin 32 gene. Neurology (2001) 57:1906–8. doi: 10.1212/WNL.57.10.1906
6. Wu N, Said S, Sabat S, Wicklund M, and Stahl MC Recurrent episodes of stroke-like symptoms in a patient with charcot-marie-tooth neuropathy X type 1. Case Rep Neurol. (2015) 7:247–52. doi: 10.1159/000442410
7. Kim GH, Kim KM, Suh SI, Ki CS, and Eun BL Charcot-marie-tooth disease masquerading as acutedemyelinating encephalomyelitis-like illness. Pediatrics (2014) 134:e270-3. doi: 10.1542/peds.2012-3243
8. Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JP, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. J Clin Epidemiol. (2009) 62:e1–34. doi: 10.1136/bmj.b2700
9. Hanemann CO, Bergmann C, Senderek J, Zerres K, and Sperfeld AD Transient, recurrent, white matter lesions in X-linked Charcot-Marie-Tooth disease with novel connexin 32 mutation. Arch Neurol. (2003) 60:605–9. doi: 10.1001/archneur.60.4.605
10. Fusco C, Frattini D, Pisani F, Spaggiari F, Ferlini A, and Della Giustina E Coexistent central and peripheral nervous system involvement in a Charcot-Marie-Tooth syndrome X-linked patient. J Child Neurol. (2010) 25:759–63. doi: 10.1177/0883073809344119
11. Sakaguchi H, Yamashita S, Miura A, Hirahara T, Kimura E, Maeda Y, et al. A novel GJB1 frameshift mutation produces a transient CNSsymptom of X-linked Charcot-Marie-Tooth disease. J Neurol. (2011) 258:284–90. doi: 10.1007/s00415-010-5752-8
12. Zhong L, Yan K, Liu C, Xue J, Wu L, and Yin F Clinical reasoning: a young man with reversible paralysis, cerebral white matter lesions, and peripheral neuropathy. Neurology (2012) 79:e70-2. doi: 10.1212/WNL.0b013e3182661eca
13. Sagnelli A, Piscosquito G, Chiapparini L, Ciano C, Salsano E, Saveri P, et al. X-linked Charcot-Marie-Tooth type 1: stroke-like presentation of a novel GJB1 mutation. J Peripher Nerv Syst. (2014) 19:183–6. doi: 10.1111/jns5.12070
14. Zhao Y, Xie Y, Zhu X, Wang H, Li Y, and Li J Transient, recurrent, white matter lesions in x-linked Charcot-Marie-tooth disease with novel mutation of gap junction proteinbeta 1 gene in China: a case report. BMC Neurol. (2014) 14:156. doi: 10.1186/s12883-014-0156-5
15. McKinney JL, De Los Reyes EC, Lo WD, and Flanigan KM Recurrent central nervous system white matter changes in charcot-Marie-tooth type X disease. Muscle Nerve (2014) 49:451–4. doi: 10.1002/mus.24108
16. Kulkarni GB, Mailankody P, Isnwara PP, Prasad C, and Mustare V Episodic neurological dysfunction in hereditary peripheral neuropathy. Ann Indian Acad Neurol. (2015) 18:111–4. doi: 10.4103/0972-2327.144314
17. Xie C, Zhou X, Zhu D, Liu W, Wang X, Yang H, et al. CNS involvement in CMTX1 caused by a novel connexin 32mutation: a 6-year follow-up in neuroimaging and nerveconduction. Neurol Sci. (2016) 37:1063–70. doi: 10.1007/s10072-016-2537-6
18. Schelhaas HJ, Van Engelen BG, Gabreëls-Festen AA, Hageman G, Vliegen JHR, van der Knaap MS, et al. Transient cerebral white matter lesions in a patient with connexin32 missense mutation. Neurology (2002) 59:2007–8. doi: 10.1212/01.WNL.0000038390.29853.46
19. Halbrich M, Barnes J, Bunge M, and Joshi C A V139M mutation also causes the reversible CNS phenotype in CMTX. Can J Neurol Sci. (2008) 35:372–4. doi: 10.1017/S0317167100008994
20. Rosser T, Muir J, Panigrahy A, Baldwin EE, and Boles RG Transient leukoencephalopathy associated with X-linkedCharcot-Marie-Tooth disease. J Child Neurol. (2010) 25:1013–6. doi: 10.1177/0883073809352378
Keywords: X-linked Charcot-Marie-Tooth disease, electrophysiological, white matter, transient, GJB1
Citation: Wen Q, Cao L, Yang C and Xie Y (2018) The Electrophysiological Features in X-Linked Charcot-Marie-Tooth Disease With Transient Central Nervous System Deficits. Front. Neurol. 9:461. doi: 10.3389/fneur.2018.00461
Received: 03 April 2018; Accepted: 30 May 2018;
Published: 27 June 2018.
Edited by:
Paola Sandroni, Mayo Clinic, United StatesReviewed by:
Giuseppe Piscosquito, Fondazione Salvatore Maugeri, Telese (IRCCS), ItalyFiore Manganelli, Università degli Studi di Napoli Federico II, Italy
Copyright © 2018 Wen, Cao, Yang and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cun Yang, eWFuZ2N1bmpuQDE2My5jb20=
Yanchen Xie, eWFuY2hlbnhpZUB3aWNsaW5pY2FscmVzZWFyY2gub3Jn