 Carlos R. Camara-Lemarroy
Carlos R. Camara-Lemarroy Luanne Metz1,2
Luanne Metz1,2 Eric E. Smith
Eric E. Smith V. Wee Yong
V. Wee Yong- 1Department of Clinical Neurosciences, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 2Hotchkiss Brain Institute, Cumming School of Medicine, University of Calgary, Calgary, AB, Canada
- 3UANL School of Medicine and University Hospital, Monterrey, Mexico
Ischemia-Reperfusion and Ischemic Preconditioning
We found the recent review paper by Chen et al. (1) on remote ischemic-preconditioning (ReIP) fascinating. They describe ReIP as an attractive low-cost, low-risk therapy for ischemia. But why stop there?
Ischemia-reperfusion, characterized by cascade of deleterious biochemical processes that increase target organ/systemic injury, is involved in the pathophysiology of stroke, myocardial infarction, and solid organ transplantation. Over 3 decades ago, investigators found that experimentally inducing short periods of cyclical tissue ischemia confer subsequent protection against ischemia-reperfusion-injury. This became known as Ischemic-Preconditioning (2–4). The rationale behind ischemic-preconditioning is that these short periods of ischemia do not cause irreversible injury, but instead induce an endogenous protective environment. Although playing out at different time periods, the concept is reminiscent of acclimatization to high-altitude conditions by alpinists, a process involving short term exposure to gradually increasing altitudes that induce endogenous mechanisms optimizing oxygen metabolism and conditioning for an eventual full-scale ascent (5).
The mechanisms involved in ischemic-preconditioning are complex. Beneficial changes in intracellular, anti-inflammatory, antioxidant pathways, and in gene expression have all been described (1, 3, 6). It has been shown to be protective in almost every solid organ ischemia-reperfusion-injury model. When investigators discovered that ischemic-preconditioning in one organ or limb could confer protection to a remote organ, this started a new era in ischemia-reperfusion research. This phenomenon was called ReIP (1).
The mechanisms involved in ReIP are also quite complex, although many are shared with standard ischemic-preconditioning (1). There is evidence for both humoral and neurogenic mechanisms as being the signal carriers that mediate protection, and the precise molecular pathways have yet to be elucidated (1, 3, 6, 7). Typical ReIP induced protection is known to occur in an early stage (2–3 h) and a late stage lasting up to 72 h (1, 3, 6). Long-term repeated ReIP has also been studied and shown that it can play its protective roles consistently; if applied once-daily for an extended period, the different phases of protection can be at play simultaneously or successively (8, 9). ReIP is less invasive (no need to access the target organs vasculature) and could theoretically protect any organ in the body. In the brain, ReIP before a local ischemic event is able to modulate cytokine production, maintain blood-brain barrier integrity, and promote the expression of protective intracellular molecules (1, 3, 6). In humans, ReIP of a limb (usually rendered transiently ischemic by a blood-pressure cuff) is a safe and tolerable procedure, and large trials evaluating its efficacy in acute coronary syndromes, heart surgery, organ transplantation, and stroke have been published (1, 4, 6). In cardiology, results have been mixed, with some evidence for organ-protection but inconsistent effects over clinically relevant outcomes (10–12). In neurology, there has been great interest in harnessing ReIP's protective properties in stroke research and in the elusive promise of clinically relevant neuroprotection (1). We will introduce ReIP as preconditioning to improve stroke outcome and introduce the opinion that the time is right to expand ReIP trials to other neurological disorders including MS treatment.
ReIP in Neurology: Encouraging Results
A promising clinical candidate for using ReIP is acute ischemic stroke. Conceivably, one would perform ReIP early after stroke onset in an attempt to protect from further ischemic injury. In an early study, investigators randomized 443 patients with acute ischemic stroke to receive pre-hospital ReIP or sham procedure before thrombolysis, and results showed a trend toward less tissue infarction although overall clinical and imaging endpoints were negative (13). In a recent, but smaller trial, involving 26 patients with acute ischemic stroke, ReIP showed a favorable trend toward improving clinical outcomes at 3 months as well as an increase in the circulating levels of a putative biomarker, heat shock protein-27 (14). Unfortunately, definitive results on hard clinical endpoints are lacking.
Investigators aiming to treat chronic conditions have evaluated the clinical feasibility of long term, repeated ReIP. In one such trial involving 68 patients, 300 days of consecutive, bilateral arm ReIP, led to reduced stroke recurrence in patients with intracranial artery stenosis (15). In another study, 17 patients with small-vessel ischemic disease underwent ReIP of their upper limbs 2 times per day for 1 full year, and investigators found improved middle cerebral artery hemodynamics and reduced white-matter lesion volumes (16). In a similar study, 30 patients with small-vessel disease and cognitive impairment underwent ReIP daily for a year, resulting in reduced white-matter lesion volume and improvements in cognitive testing (17).
In all trials, ReIP was found to be safe and tolerable, and long-term data suggests it can be used on a daily basis for years, and potentially in chronic, degenerative conditions. Furthermore, since many of the proposed endogenous protective mechanisms involved in ReIP are non-specific (antioxidant, anti-inflammatory, immune modulating) to ischemia-reperfusion-injury (1, 3, 6), it might prove efficacious in protecting against non-ischemic injury as well. In support, the wide ranging neuroprotective effects of ReIP have been demonstrated in various rodent models of non-ischemic injury, such as optic nerve transection (18), traumatic brain injury (19), and ketamine-induced neuronal apoptosis (20). Could there be a role for ReIP in the treatment of Multiple Sclerosis (MS)?
ReIP in MS: Some Pathophysiological Links
There are two main ways ReIP could be relevant in MS. On the one hand, MS pathophysiology might involve ischemic/hypoxic mechanisms, and on the other, some of the pathways beneficially modulated by ReIP could be protective against inflammatory demyelination/neurodegeneration. A recent review summarizes multiple lines of evidence suggesting that there is generalized cerebral hypo-perfusion and chronic hypoxia in patients with MS, and that this could contribute to neurodegeneration (21). Furthermore, the white-matter regions more commonly affected in demyelinating disease are similar to those affected by small-vessel disease. Oligodendrocytes are very susceptible to hypoxia and in animal models of demyelination, demyelination is prevented if adequate oxygenation is maintained (22). A recent study was able to show in vivo that there is reduced cortical microvascular oxygenation in patients with MS (23). Nevertheless, the precise role of hypoxia/ischemia in MS remains unknown.
There is also evidence suggesting that ReIP can modulate some of the mechanisms involved in the pathogenesis of Experimental Autoimmune Encephalomyelitis (EAE), the prototypical animal model of MS. In mice studies, hind-limb ReIP increased serum and mRNA expression of erythropoietin (EPO) and hypoxia-inducible factor-1alpha (HIF1-alpha) in the brain (24, 25). EPO is known to be neuroprotective in the mouse EAE model (26), and HIF1-alpha is increased in EAE, acting as a transcription factor for IL-17-triggered cytokine production (27). Heat shock protein (Hsp)70 is also one of the principal mediators of ReIP-induced neuroprotection, by mediating chaperone-cytoprotective effects, blocking multiple steps in the apoptosis pathway and immune-modulation (25, 28). In EAE, Hsp70 is upregulated, and Hsp70 knockdown alters the immune response associated with demyelination (29, 30). Other shared mechanisms in ReIP-induced protection and in EAE pathophysiology are the mTOR kinase (involved in regulation of immune cell function) and nitric oxide signaling (31–33).
Studies also point toward hypoxia being associated with EAE pathophysiology. Investigators found direct and indirect evidence of tissue hypoxia in the spinal cord of a rat model (34), and the cortex and cerebellum of a mouse model (35), as well as a protective effect of oxygen therapy (34). Mice kept in hypoxic conditions (hypoxic preconditioning) show decreased numbers of (CD)4+ T cells and a delayed Th17-specific cytokine response in the spinal cord after EAE induction, as well as increased numbers of Treg cells and Interleukin (IL)-10 (36). Another study found that hypoxic preconditioning delayed the onset of EAE, and when hypoxic preconditioning was established after the onset of clinical symptoms, spinal cord pathology, and inflammation decreased (37).
ReIP can also alter cellular and humoral immune responses classically thought to be key regulators of MS pathogenesis. Ischemic-preconditioning can induce an increase in CD4(+)CD25(+)FoxP3(+) and CD4(+)CD25(+)IL-10(+) Tregs in peripheral organs (38, 39). Tregs are essential to resolve immune responses in EAE (40). In mouse models of stroke, ReIP was able to modulate the populations of peripheral T and B cells (41) and inhibit the galectin-9/Tim-3 inflammatory cell signaling pathway, which induces cell death in lymphocytes (42). Resident CNS cells such as microglia and astrocytes also respond to ischemic-preconditioning by a shift in function toward provision of trophic support and neuroprotection (31). In fact, activation of inflammatory pathways may be necessary for the induction of ischemic tolerance by ReIP, eventually leading to subdued immune activation (43).
We have summarized molecular/inflammatory pathways involved in MS pathophysiology that appear to be modulated by ReIP. However, other pathways involved in ReIP-induced protection, such as autophagy, potassium channels, adenosine, and neurogenic signals are neuroprotective (1, 3, 6), and might also be of benefit in the context of inflammatory demyelination/neurodegeneration.
ReIP might then stimulate an endogenously protective milieu in the brain that could theoretically reduce inflammatory demyelination/neurodegeneration through its non-specificity (Figure 1A). Interestingly, there is evidence suggesting that the brain is already attempting to use this strategy in MS. In a pathology study on normally appearing white-matter from patients with MS, investigators found an upregulation of genes associated to ischemic-preconditioning mechanisms such as HIF-1alpha, PI3K/Akt signaling among others (44). The authors of this study suggested that these molecular changes might reflect an adaptation of cells to the chronic progressive pathophysiology of MS. In a model of cultured glia treated with Th1 and Th2 cytokines, investigators found that many of the changes in gene expression were similar to those seen in ischemic-preconditioning and EAE (genes related to mitochondrial function, neurotransmission, vitamin D metabolism, and a variety of transcription factors) (45).
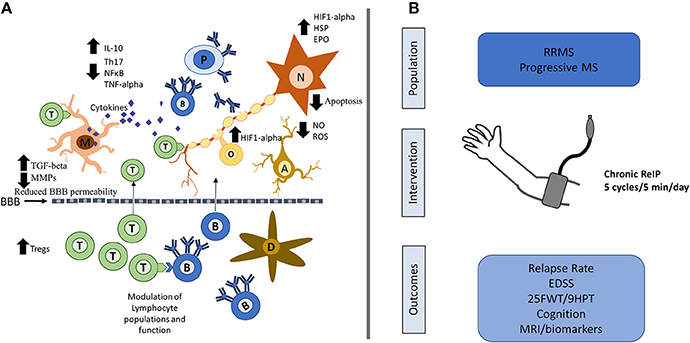
Figure 1. Possible neuroprotective mechanisms of ReIP in MS and proposed intervention. (A) ReIP can modulate several of the typical pathogenic mechanisms involved in MS. In the periphery, ReIP can alter lymphocyte populations and may also maintain blood brain barrier (BBB) permeability through reduction in matrix metalloproteinases (MMPs) and increases in transforming growth factor beta (TGF-beta). In the central nervous system, ReIP increases the expression of hypoxia inducible factor-1 alpha (HIF1-alpha), heat shock proteins (HSP) and erythropoietin (EPO), conferring neurons and glia protection against inflammatory insults and apoptosis. ReIP also reduces the production of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-alpha) and increases levels of anti-inflammatory cytokines such as interleukin (IL)-10. ReIP ameliorates oxidative stress by reducing the production of reactive oxygen species (ROS) and nitric oxide (NO). (B) Patients with relapsing remitting MS (RRMS) or progressive forms of MS could be randomized to receive daily, chronic arm ReIP for 1–2 years or a sham procedure. Relevant outcomes would include relapse rate for RRMS, and for all patients, measures of disability such as the expanded disability status score (EDSS), 25 foot-walking test (25FWT), the 9-hole peg test (9HPT) and measures of cognition. MRI biomarkers looking at lesion load and atrophy as well as plausible biomarkers could be of use as well. T, T-lymphocytes; B, B-lymphocytes; D, dendritic cell; M, microglia; N, neuron; A, astrocyte; O, oligodendrocyte; P, plasma cell; NF-κB, nuclear factor kappa-B.
Although the mechanisms involved might differ from ReIP, an exciting area of research from where further encouragement might be derived is the neuroprotective potential of hypoxic preconditioning (46, 47). In a study involving patients with chronic incomplete spinal cord injuries, a regimen of intermittent hypoxia (short term inhalation of a low-oxygen mixture through a breathing mask) was shown to increase volitional strength in specific muscle groups, albeit transiently and immediately after hypoxia (48). In a randomized, double-blind, placebo-controlled trial, 15, 90-s exposures to hypoxia for 5 days led to increased walking speed distances in patients with incomplete spinal cord injury, with a sustained effect over 2 weeks (49). A recent trial using a similar design confirmed the benefits of repetitive intermittent hypoxia in patients with incomplete spinal cord injury, and showed a sustained effect over 5 weeks in walking speed and endurance (50).
ReIP: a Proposal in MS
ReIP induces protective changes in the central nervous system; it is safe, tolerable, and can be induced for virtually indefinite periods of time; it may also ameliorate both acute inflammatory insults and chronic neurodegenerative processes. We suggest that ReIP could potentially be of therapeutic value in MS (Figure 1B). However, several issues would have to be addressed before a trial is designed. MS is a heterogeneous disease, and it is unclear what sub-population would be ideally suited for a clinical trial. Long-term functional endpoints, and cognitive measures in particular, may be realistic goals. Chronic ReIP induced in the upper extremities, such as that described in trials of cerebral small vessel disease for progressive-forms of MS may be favored. ReIP could also be conceived as an add-on therapy, with the benefit that it has no known or theorized pharmacological interactions. Imaging biomarkers, including white-matter lesion and brain atrophy measures would be ideal secondary endpoints, and putative mechanistic biomarkers such as HIF1-alpha and HSPs could be readily analyzed.
Conclusion
Recent research on the underlying mechanisms of white-matter pathology have found curious similarities between ischemic and non-ischemic disorders (51). Furthermore, the CNS appears to use redundant, endogenous protective mechanisms against different types of insults. Even if the precise mechanism involved in ReIP-induced neuroprotection remain elusive, we are optimistic in the prospects of expanding its indications in neurology beyond ischemic conditions. There is currently one trial aiming to evaluate the safety and tolerability of ReIP in patients with MS, with the aim of showing short-term benefits on exercise tolerance (52). While we await the results of this trial with great interest, we believe that ReIP might be a way to harness these mechanisms to protect against inflammatory demyelination and associated neurodegeneration. A clinical trial will be the only way to evaluate the merits of this proposal.
Author Contribution
CC-L: conceptualization and writing of original draft; LM, JD, and ES: writing, review, and editing; VY: supervision, writing, review, and editing.
Funding
CC-L holds the Lejoie-Lake Fellowship awarded by the Hotchkiss Brain Institute.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors' research has been funded by Alberta Innovates—Health Solutions CRIO Team program.
References
1. Chen G, Thakkar M, Robinson C, Doré S. Limb remote ischemic conditioning: mechanisms, anesthetics, and the potential for expanding therapeutic options. Front Neurol. (2018) 9:40. doi: 10.3389/fneur.2018.00040
2. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation (1986) 74:1124–36. doi: 10.1161/01.CIR.74.5.1124
3. Meller R, Simon RP. A critical review of mechanisms regulating remote preconditioning-induced brain protection. J Appl Physiol. (2015) 119:1135–42. doi: 10.1152/japplphysiol.00169.2015
4. Le Page S, Prunier F. Remote ischemic conditioning: current clinical perspectives. J Cardiol. (2015) 66:91–6. doi: 10.1016/j.jjcc.2015.01.009
5. Luo Y, Wang Y, Lu H, Gao Y. 'Ome' on the range: update on high-altitude acclimatization/adaptation and disease. Mol Biosyst. (2014) 10:2748–55. doi: 10.1039/c4mb00119b
6. Stokfisz K, Ledakowicz-Polak A, Zagorski M, Zielinska M. Ischaemic preconditioning-Current knowledge and potential future applications after 30 years of experience. Adv Med Sci. (2017) 62:307–16. doi: 10.1016/j.advms.2016.11.006
7. Aulakh AS, Randhawa PK, Singh N, Jaggi AS. Neurogenic pathways in remote ischemic preconditioning induced cardioprotection: evidences and possible mechanisms. Korean J Physiol Pharmacol. (2017) 21:145–52. doi: 10.4196/kjpp.2017.21.2.145
8. Thijssen DH, Maxwell J, Green DJ, Cable NT, Jones H. Repeated ischaemic preconditioning: a novel therapeutic intervention and potential underlying mechanisms. Exp Physiol. (2016) 101:677–92. doi: 10.1113/EP085566
9. Zhao W, Li S, Ren C, Meng R, Ji X. Chronic remote ischemic conditioning may mimic regular exercise: perspective from clinical studies. Aging Dis. (2018) 9:165–171. doi: 10.14336/AD.2017.1015
10. Davies WR, Brown AJ, Watson W, McCormick LM, West NE, Dutka DP, et al. Remote ischemic preconditioning improves outcome at 6 years after elective percutaneous coronary intervention: the CRISP stent trial long-term follow-up. Circ Cardiovasc Interv. (2013) 6:246–51. doi: 10.1161/CIRCINTERVENTIONS.112.000184
11. Cho YJ, Lee EH, Lee K, Kim TK, Hong DM, Chin JH, et al. Long-term clinical outcomes of Remote Ischemic Preconditioning and Postconditioning Outcome (RISPO) trial in patients undergoing cardiac surgery. Int J Cardiol. (2017) 231:84–89. doi: 10.1016/j.ijcard.2016.12.146
12. Zarbock A, Kellum JA, Van Aken H, Schmidt C, Küllmar M, Rosenberger P, et al. Long-term effects of remote ischemic preconditioning on kidney function in high-risk cardiac surgery patients: follow-up results from the RenalRIP Trial. Anesthesiology (2017) 126:787–98. doi: 10.1097/ALN.0000000000001598
13. Hougaard KD, Hjort N, Zeidler D, Sørensen L, Nørgaard A, Hansen TM, et al. Remote ischemic perconditioning as an adjunct therapy to thrombolysis in patients with acute ischemic stroke: a randomized trial. Stroke (2014) 45:159–67. doi: 10.1161/STROKEAHA.113.001346
14. England TJ, Hedstrom A, O'Sullivan S, Donnelly R, Barrett DA, Sarmad S, et al. RECAST (Remote Ischemic Conditioning After Stroke Trial): a Pilot Randomized Placebo Controlled Phase II Trial in Acute Ischemic Stroke. Stroke (2017) 48:1412–5. doi: 10.1161/STROKEAHA.116.016429
15. Meng R, Asmaro K, Meng L, Liu Y, Ma C, Xi C, et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology (2012) 79:1853–61. doi: 10.1212/WNL.0b013e318271f76a
16. Mi T, Yu F, Ji X, Sun Y, Qu D. The interventional effect of remote ischemic preconditioning on cerebral small vessel disease: a pilot randomized clinical trial. Eur Neurol. (2016) 76:28–34. doi: 10.1159/000447536
17. Wang Y, Meng R, Song H, Liu G, Hua Y, Cui D, et al. Remote ischemic conditioning may improve outcomes of patients with cerebral small-vessel disease. Stroke (2017) 48:3064–72. doi: 10.1161/STROKEAHA.117.017691
18. Liu X, Sha O, Cho EY. Remote ischemic postconditioning promotes the survival of retinal ganglion cells after optic nerve injury. J Mol Neurosci. (2013) 51:639–46. doi: 10.1007/s12031-013-0036-2
19. Pérez-Pinzón MA, Alonso O, Kraydieh S, Dietrich WD. Induction of tolerance against traumatic brain injury by ischemic preconditioning. Neuroreport (1999) 10:2951–4.
20. Ma W, Cao YY, Qu S, Ma SS, Wang JZ, Deng LQ, et al. Remote ischemic preconditioning provides neuroprotection: impact on ketamine-induced neuroapoptosis in the developing rat brain. Eur Rev Med Pharmacol Sci. (2016) 20:4972–9. Available online at: http://www.europeanreview.org/article/11864
21. D'haeseleer M, Hostenbach S, Peeters I, Sankari SE, Nagels G, De Keyser J, et al. Cerebral hypoperfusion: a new pathophysiologic concept in multiple sclerosis? J Cereb Blood Flow Metab. (2015) 35:1406–10. doi: 10.1038/jcbfm.2015.131
22. Martinez Sosa S, Smith KJ. Understanding a role for hypoxia in lesion formation and location in the deep and periventricular white matter in small vessel disease and multiple sclerosis. Clin Sci. (2017) 131:2503–24. doi: 10.1042/CS20170981
23. Yang R, Dunn JF. Reduced cortical microvascular oxygenation in multiple sclerosis: a blinded, case-controlled study using a novel quantitative near-infrared spectroscopy method. Sci Rep. (2015) 5:16477. doi: 10.1038/srep16477
24. Oba T, Yasukawa H, Nagata T, Kyogoku S, Minami T, Nishihara M, et al. Renal nerve-mediated erythropoietin release confers cardioprotection during remote ischemic preconditioning. Circ J. (2015) 79:1557–67. doi: 10.1253/circj.CJ-14-1171
25. Xia M, Ding Q, Zhang Z, Feng Q. Remote limb ischemic preconditioning protects rats against cerebral ischemia via HIF-1α/AMPK/HSP70 Pathway. Cell Mol Neurobiol. (2017) 37:1105–14. doi: 10.1007/s10571-016-0444-2
26. Shin T, Ahn M, Moon C, Kim S. Erythropoietin and autoimmune neuroinflammation: lessons from experimental autoimmune encephalomyelitis and experimental autoimmune neuritis. Anat Cell Biol. (2012) 45:215–20. doi: 10.5115/acb.2012.45.4.215
27. Shan K, Pang R, Zhao C, Liu X, Gao W, Zhang J, et al. IL-17-triggered downregulation of miR-497 results in high HIF-1α expression and consequent IL-1β and IL-6 production by astrocytes in EAE mice. Cell Mol Immunol. (2017) 14:909–23. doi: 10.1038/cmi.2017.12
28. Selimoglu O, Ugurlucan M, Basaran M, Gungor F, Banach M, Cucu O, et al. Efficacy of remote ischaemic preconditioning for spinal cord protection against ischaemic injury: association with heat shock protein expression. Folia Neuropathol. (2008) 46:204–12. Available online at: https://www.termedia.pl/Journal/-20/Artykul-11045
29. Mansilla MJ, Montalban X, Espejo C. Heat shock protein 70: roles in multiple sclerosis. Mol Med. (2012) 18:1018–28. doi: 10.2119/molmed.2012.00119
30. Mansilla MJ, Costa C, Eixarch H, Tepavcevic V, Castillo M, Martin R, et al. Hsp70 regulates immune response in experimental autoimmune encephalomyelitis. PLoS ONE (2014) 9:e105737. doi: 10.1371/journal.pone.0105737
31. McDonough A, Weinstein JR. Neuroimmune response in ischemic preconditioning. Neurotherapeutics (2016) 13:748–61. doi: 10.1007/s13311-016-0465-z
32. Dello Russo C, Lisi L, Feinstein DL, Navarra P. mTOR kinase, a key player in the regulation of glial functions: relevance for the therapy of multiple sclerosis. Glia (2013) 61:301–11. doi: 10.1002/glia.22433
33. Willenborg DO, Staykova M, Fordham S, O'Brien N, Linares D. The contribution of nitric oxide and interferon gamma to the regulation of the neuro-inflammation in experimental autoimmune encephalomyelitis. J Neuroimmunol. (2007) 191:16–25. doi: 10.1016/j.jneuroim.2007.09.007
34. Davies AL, Desai RA, Bloomfield PS, McIntosh PR, Chapple KJ, Linington C, et al. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann Neurol. (2013) 74:815–25. doi: 10.1002/ana.24006
35. Johnson TW, Wu Y, Nathoo N, Rogers JA, Wee Yong V, Dunn JF. Gray matter hypoxia in the brain of the experimental autoimmune encephalomyelitis model of multiple sclerosis. PLoS ONE (2016) 11:e0167196. doi: 10.1371/journal.pone.0167196
36. Esen N, Katyshev V, Serkin Z, Katysheva S, Dore-Duffy P. Endogenous adaptation to low oxygen modulates T-cell regulatory pathways in EAE. J Neuroinflamm. (2016) 13:13. doi: 10.1186/s12974-015-0407-4
37. Dore-Duffy P, Wencel M, Katyshev V, Cleary K. Chronic mild hypoxia ameliorates chronic inflammatory activity in myelin oligodendrocyte glycoprotein (MOG) peptide induced experimental autoimmune encephalomyelitis (EAE). Adv Exp Med Biol. (2011) 701:165–73. doi: 10.1007/978-1-4419-7756-4_23
38. Kinsey GR, Huang L, Vergis AL, Li L, Okusa MD. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int. (2010) 77:771–80. doi: 10.1038/ki.2010.12
39. Cho WY, Choi HM, Lee SY, Kim MG, Kim HK, Jo SK. The role of Tregs and CD11c(+) macrophages/dendritic cells in ischemic preconditioning of the kidney. Kidney Int. (2010) 78:981–92. doi: 10.1038/ki.2010.266
40. Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflamm. (2017) 14:117. doi: 10.1186/s12974-017-0892-8
41. Liu ZJ, Chen C, Li XR, Ran YY, Xu T, Zhang Y, et al. Remote ischemic preconditioning-mediated neuroprotection against stroke is associated with significant alterations in peripheral immune responses. CNS Neurosci Ther. (2016) 22:43–52. doi: 10.1111/cns.12448
42. Wei D, Ren C, Chen X, Zhao H. The chronic protective effects of limb remote preconditioning and the underlying mechanisms involved in inflammatory factors in rat stroke. PLoS ONE (2012) 7:e30892. doi: 10.1371/journal.pone.0030892
43. Garcia-Bonilla L, Benakis C, Moore J, Iadecola C, Anrather J. Immune mechanisms in cerebral ischemic tolerance. Front Neurosci. (2014) 8:44. doi: 10.3389/fnins.2014.00044
44. Graumann U, Reynolds R, Steck AJ, Schaeren-Wiemers N. Molecular changes in normal appearing white matter in multiple sclerosis are characteristic of neuroprotective mechanisms against hypoxic insult. Brain Pathol. (2003) 13:554–73. doi: 10.1111/j.1750-3639.2003.tb00485.x
45. Lisak RP, Benjamins JA, Bealmear B, Nedelkoska L, Studzinski D, Retland E, et al. Differential effects of Th1, monocyte/macrophage and Th2 cytokine mixtures on early gene expression for molecules associated with metabolism, signaling and regulation in central nervous system mixed glial cell cultures. J Neuroinflamm. (2009) 6:4. doi: 10.1186/1742-2094-6-4
46. Li S, Hafeez A, Noorulla F, Geng X, Shao G, Ren C, et al. Preconditioning in neuroprotection: from hypoxia to ischemia. Prog Neurobiol. (2017) 157:79–91. doi: 10.1016/j.pneurobio.2017.01.001
47. Speer R, Ratan RR. Hypoxic adaptation in the nervous system: promise for novel therapeutics for acute and chronic neurodegeneration. Adv Exp Med Biol. (2016) 903:221–43. doi: 10.1007/978-1-4899-7678-9_16
48. Trumbower RD, Jayaraman A, Mitchell GS, Rymer WZ. Exposure to acute intermittent hypoxia augments somatic motor function in humans with incomplete spinal cord injury. Neurorehabil Neural Repair (2012) 26:163–72. doi: 10.1177/1545968311412055
49. Hayes HB, Jayaraman A, Herrmann M, Mitchell GS, Rymer WZ, Trumbower RD. Daily intermittent hypoxia enhances walking after chronic spinal cord injury: a randomized trial. Neurology (2014) 82:104–13. doi: 10.1212/01.WNL.0000437416.34298.43
50. Navarrete-Opazo A, Alcayaga J, Sepúlveda O, Rojas E, Astudillo C. Repetitive intermittent hypoxia and locomotor training enhances walking function in incomplete spinal cord injury subjects: a randomized, triple-blind, placebo-controlled clinical trial. J Neurotrauma (2017) 34:1803–12. doi: 10.1089/neu.2016.4478
51. Fern RF, Matute C, Stys PK. White matter injury: ischemic and nonischemic. Glia (2014) 62:1780–9. doi: 10.1002/glia.22722
52. ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Identifier NCT03153553, Ischemic Preconditioning, Exercise Tolerance and Multiple Sclerosis. Available online at: https://clinicaltrials.gov/ct2/show/NCT03153553 (Accessed March 5, 2018).
Keywords: multiple sclerosis, ischemic preconditioning, remote ischemic preconditioning, neuroprotection, neuroimmunology
Citation: Camara-Lemarroy CR, Metz L, Smith EE, Dunn JF and Yong VW (2018) Expanding the Potential Therapeutic Options for Remote Ischemic Preconditioning: Use in Multiple Sclerosis. Front. Neurol. 9:475. doi: 10.3389/fneur.2018.00475
Received: 11 April 2018; Accepted: 01 June 2018;
Published: 19 June 2018.
Edited by:
Fabienne Brilot, University of Sydney, AustraliaReviewed by:
Yoshihiko Kakinuma, Nippon Medical School, JapanCopyright © 2018 Camara-Lemarroy, Metz, Smith, Dunn and Yong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos R. Camara-Lemarroy, YXJsb3MuY2FtYXJhLWxlbWFycm95QGFsYmVydGFoZWFsdGhzZXJ2aWNlcy5jYQ==